 Ikenna Onyido
Ikenna Onyido Onyeka F. Obumselu
Onyeka F. Obumselu Chinyelu I. Egwuatu
Chinyelu I. Egwuatu- Department of Pure and Industrial Chemistry, Nnamdi Azikiwe University, Awka, Nigeria
Organophosphorus esters fulfil many industrial, agricultural, and household roles. Nature has deployed phosphates and their related anhydrides as energy carriers and reservoirs, as constituents of genetic materials in the form of DNA and RNA, and as intermediates in key biochemical conversions. The transfer of the phosphoryl (PO3) group is thus a ubiquitous biological process that is involved in a variety of transformations at the cellular level such as bioenergy and signals transductions. Significant attention has been paid in the last seven decades to understanding the mechanisms of uncatalyzed (solution) chemistry of the phospho group transfer because of the notion that enzymes convert the dissociative transition state structures in the uncatalyzed reactions into associative ones in the biological processes. In this regard, it has also been proposed that the rate enhancements enacted by enzymes result from the desolvation of the ground state in the hydrophobic active site environments, although theoretical calculations seem to disagree with this position. As a result, some attention has been paid to the study of the effects of solvent change, from water to less polar solvents, in uncatalyzed phospho transfer reactions. Such changes have consequences on the stabilities of the ground and the transition states of reactions which affect reactivities and, sometimes, the mechanisms of reactions. This review seeks to collate and evaluate what is known about solvent effects in this domain, especially their effects on rates of reactions of different classes of organophosphorus esters. The outcome of this exercise shows that a systematized study of solvent effects needs to be undertaken to fully understand the physical organic chemistry of the transfer of phosphates and related molecules from aqueous to substantially hydrophobic environments, since significant knowledge gaps exist.
1 Introduction
1.1 Importance and classes of organophosphorus esters
Organophosphorus esters are widely used as pesticides, flame retardants and plasticizers in a number of household and industrial products, and as additives for lubricants and hydraulic fluids (Johnson and Henschler, 1975; Marklund et al., 2005; Wan et al., 2017). The transfer of the phosphoryl group (PO3) from phosphate esters to nucleophilic entities and fragments at the cellular level is a fundamental process in biology, which plays indispensable roles in the biochemistry of life, such as bioenergy transductions, signal transductions, and in genome processing (Knowles, 1980; Westheimer, 1987; Cleland and Hengge, 2006; Lassila, et al., 2011). It is the mechanistic aspects of the solution chemistry of these compounds, which are uncatalyzed reactions, that attention has been focussed on in our laboratories (Omakor, et al., 2001; Buncel et al., 2004; Onyido, et al., 2005a; Onyido, et al., 2005b; Kalu, et al., 2021; Kalu, et al., 2022), which forms the base for this review.
In this review, we are concerned with how solvents have influenced reactivities and mechanistic outcomes and their nuances in the reactions of organophosphorus esters with oxygen and nitrogen nucleophiles. The structures of the three classes of organophosphorus esters–phosphates and phosphorothioates (1–3), phosphonates and phosphonothioates (4), and phosphinates and phosphinothioates (5) are shown below. For phosphate esters, the alkylation state of the substrate determines whether it is a mono-, di-, or triester. Notice that, structurally, the compounds that end with -ate are P=O bonded (X = O), while those with -thioate ending are P=S bonded, i.e., one of the non-bridging oxygen atoms of the phosphoryl group has been replaced by sulphur (X = S).
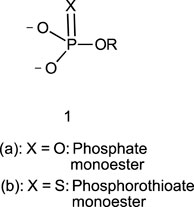

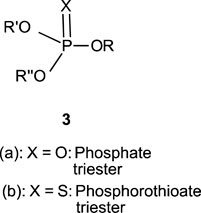

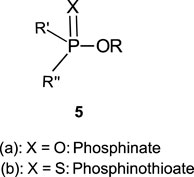
1.2 Phospho group transfer
Although the term “phosphoryl group transfer” refers specifically to the transfer of the phosphoryl (PO3) group to a nucleophilic entity in the reaction of organophosphates (1a-3a, X = O) with nucleophiles, it is loosely applied in the literature also to the transfer to nucleophiles of the other groups, PO2R’ (phosphonoyl group) and POR’R’’ (phosphinoyl group) from organophosphonates (4a, X = O) and organophosphinates (5a, X = O), respectively, to nucleophiles. In the same vein, the term “thiophosphoryl group transfer” for the transfer of the thiophosphoryl (PSO2) group from organophosphorothioates (1b–3b, X = S) to nucleophiles has also been loosely attached to the transfer of the other groups, PSOR’ and PSR’R″, from organophosphonates (4b, X = S) and organophosphinates (5b, X = S), respectively. The transferable groups from the organophosphorus esters listed as 1–5 above are shown in Table 1. It is clear that the distribution of charges in the transferable groups is different in all the esters shown as 1–5. For this reason, we prefer to use the all-embracing term “phospho” group transfer to refer to the transfer of any of the transferable groups to nucleophiles in the span of compounds represented as 1–5 and reserve the term “phosphoryl” for transfers from any of the phosphates represented as 1–3.
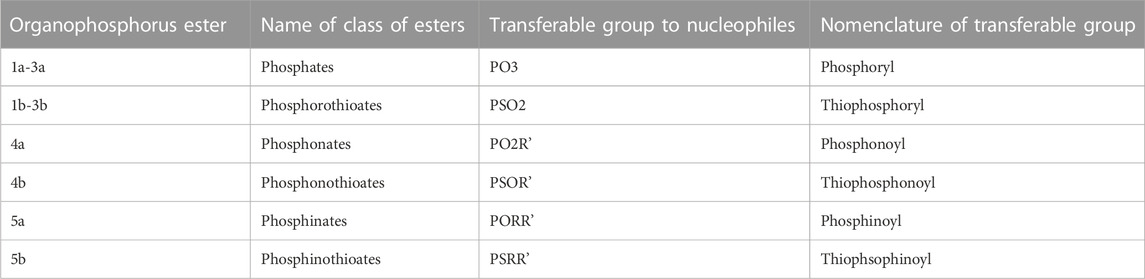
TABLE 1. Nomenclature of the transferable groups in organophosphorus esters 1–5
1.3 General and specific solvent effects
It has been long recognized that solvents can affect the course of chemical reactions conducted in them in fundamental ways (Reichardt, 1982; Buncel, et al., 2003; Reichardt and Welton, 2011). They may alter the kinetics and thermodynamics of reactions or alter product selectivities significantly by exerting substantial influences on the characteristics of the dissolved solutes (Reichardt, 2007). The origins of such interventions, and the computational tools to unravel them, have been discussed in some detail recently (Varghese and Mushrif, 2019).
As far back as 1935, Hughes and Ingold put forward their theory of solvent action (Hughes and Ingold, 1935; Ingold, 1969) as a qualitative predictive tool, based on solvent type and reactants’ charge-type, for discussing the effect of solvent polarity on the rates of bimolecular nucleophilic substitution (SN2) reactions. Bickelhaupt’s group (Hamlin et al., 2018) has shown that the contours of the potential energy surface (PES) of SN2-type processes are altered by solvation in ways that depend on the polarity of the solvent and the patterns of charge dispersal over the interacting entities. Such solvents effects, which depend on the interactions of the solutes with their environment, the solvent, and in which the solvent acts as a dielectric continuum to alter the free energies of activation (
There exists in the literature a number of parameters by which rates of chemical reactions are correlated with various solvent properties using linear free energy relationships (Williams, 2003; Anslyn and Dougherty, 2006; Onyido, 2018). General solvent effects have been empirically parameterized (Catalán, 2009) by Kosower’s Z scale (Kosower, 1958a; 1958b; 1958c), Kamlet, Abboud and Taft’s dipolarity and polarizability
Significant information regarding solution reactivities has become available from studies of solvent effects on diverse chemical reactions. A solvent could profoundly influence the
1.4 Mechanisms of phospho transfer
The range of mechanisms (Pathways A-C) by which organophosphorus esters of the types shown in 1-5 react with nucleophiles is shown in Figure 1. The substrate in this scheme is specifically a phosphate/phosphorothioate triester (3a/3b). Pathway A is the stepwise associative [AN + DN] mechanism which involves a pentacoordinate intermediate whose formation or decomposition may be rate-limiting. Pathway B is a one-step, concerted [ANDN] SN2-like mechanism involving concerted bond-making and bond-rupture. Pathway C is a stepwise, dissociative [DN + AN] mechanism in which the substrate undergoes rate-limiting ionization to yield a metaphosphate-type intermediate which reacts with the nucleophile in a fast step to yield the products.
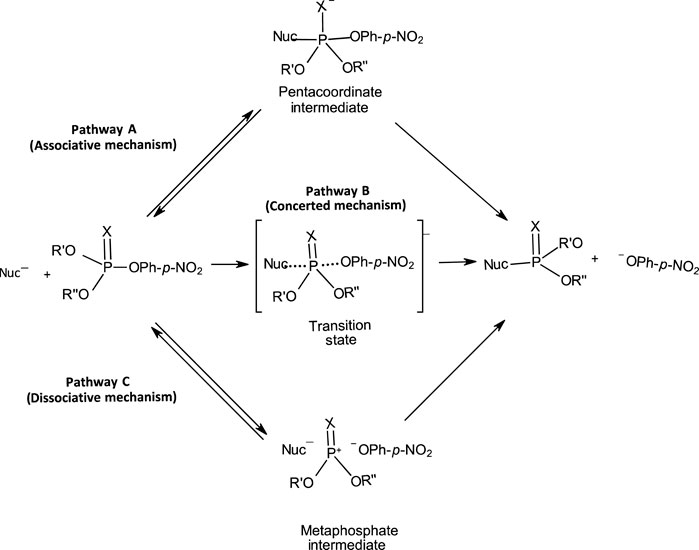
FIGURE 1. The three possible mechanisms for the reactions of nucleophiles with organophosphorus esters.
These three mechanisms are linked graphically in Figure 2 in a reaction map, otherwise known as the More O’Ferrall-Jencks diagram (More O’Ferrall, 1970; Jencks, 1972). In this diagram, bond fission to the leaving group is shown along the horizontal axis, while bond formation between the nucleophile and the substrate is displayed along the vertical axis. Accordingly, the stepwise mechanisms of Pathways A and C progress through the upper left and lower right corners of the diagram, respectively, while the concerted pathway (Pathway B) is defined by the diagonal linking the reactant (lower left corner) and product (upper right corner) ends of the diagram. The transition states of concerted mechanisms are located in the interior of the diagram, depending on the relative synchronicity of leaving group departure and nucleophile attack. The symmetrical concerted TS with 50% bond-making and 50% bond-breaking is located at the intersection of the synchronous route and the tightness diagonal in a wholly synchronous mechanism. The TS for the rate-limiting formation of the pentacoordinate intermediate would lie on the vertical arm of (A) while the TS for its rate-limiting decomposition would lie on the horizontal arm of (A). Route (C) is the pathway for the formation of the metaphosphate-like intermediate; its TS can only lie on the horizontal arm of (C).
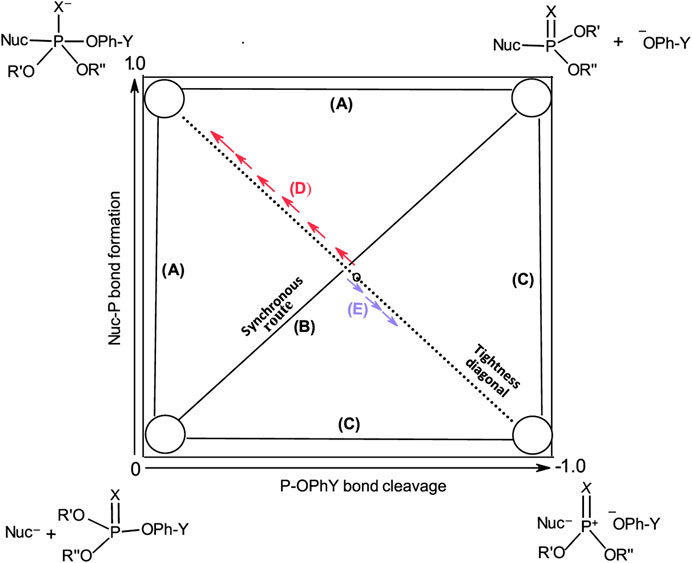
FIGURE 2. A More O’Ferrall-Jencks diagram for the reaction of a nucleophile with a hypothetical phosphate/phosphorothioate triester (3a/3b) showing the three mechanistic pathways in Figure 1 as (A–C).
1.5 Scope of this review
Some recent work, especially from our laboratories (Buncel et al., 2004; Onyido, et al., 2005a; Onyido, et al., 2005b; Kalu, et al., 2021; Kalu, et al., 2022), discussed in greater detail in Section 3 below, complementing earlier results in the literature (Haake et al., 1968; Douglas and Williams, 1976; Williams and Naylor, 1976; Eliseeva, et al., 1978; Istomin et al., 1982; Cook and Rahhal-Arabi, 1985; Bourne, et al., 1988; Dunn and Buncel, 1989; Dunn et al., 1990; Pregel, et al., 1995; etc.), demonstrates a change in the mechanism of phospho transfer from aryl phosphinates and phosphinothioates of the types shown as 6 to oxyanionic nucleophiles (HO− and PhO−), from a concerted mechanism to an associative one, when solvent is changed from water to the less polar ethanol. Lowering the polarity of the medium by the incremental addition of ethanol to the aqueous solution (Kalu et al., 2021; Kalu et al., 2022) enabled the tracking of the loosening of the concerted TS to looser TS structures, until a solvent-independent concerted TS was attained in ethanol-rich aqueous ethanol solvents. The fact that the reactions proceed via two different mechanisms in water or aqueous ethanol solvents on the one hand and in pure ethanol solvent on the other hand, shows that the potential energy surfaces are different in the two solvent systems. In addition, the reactions became slower by factors of 102–103 in aqueous ethanol and pure ethanol solvents.
Parenthetically, it has been advocated (Herschlag and Jencks, 1987; Nikolic-Hughes et al., 2004; Leiros et al., 2004; Lassila et al., 2011) that enzymes catalyse phosphoryl group transfers from organophosphorus ester substrates by altering the dissociative structures in chemical systems to associative ones in biological processes which take advantage of induced intra-molecularity for enzymatic catalysis. This proposal is equivalent to the alteration of the PES of the reaction by the enzyme. In surveying solvent effects on mechanisms of solution phospho transfer processes as undertaken in this review, an underlying motivation was to probe the extent solvent changes influence PES changes in the reactions of organophosphorus esters with nucleophiles. It was thought that such streamlining of information could be a useful contribution to present and future discussions of the mechanisms of biological phosphoryl transfers and the related debate about the strategy through which enzymes achieve the high rate enhancements associated with biological transformations.
2 Solvent effects on nucleophilic reactions of phosphates and phosphinates
2.1 Phosphates and phosphorothioates
2.1.1 Phosphate monoesters
Phosphate monoesters occur, and have recognized functions, in genetic constituents, in coenzymes, and as energy pools; they are involved in energy production and signal transduction and occur as intermediates in biochemical conversions. As products of protein phosphorylation, they play key functions in the regulation of a variety of cellular processes (Cleland and Hengge, 2006; Kamerlin et al., 2008a).
2.1.1.1 Reactions with oxygen nucleophiles
Considerable attention has been attached to the study of the hydrolysis reactions of phosphate monoesters, across the decades (Kirby and Jencks, 1965b; Westheimer, 1987; Friedman et al., 1988; Lassila et al., 2011). Studies have shown that phosphate monoesters, reacting either as monoanions or dianions, hydrolyse via dissociative mechanisms (Benkovic and Schray, 1978; Schwartz and Murray, 2011). The phosphate monoester dianion reaction occurs via a concerted mechanism with a highly dissociative TS (see Figure 3). This TS is a loose one, in which there is extensive bond fission to the leaving group and a small amount of bond formation to the nucleophile. The characterization of the mechanism of the aqueous hydrolysis of phosphate monoester dianions as a concerted one with a highly dissociative TS is supported by small dependencies of rate on nucleophile basicity with
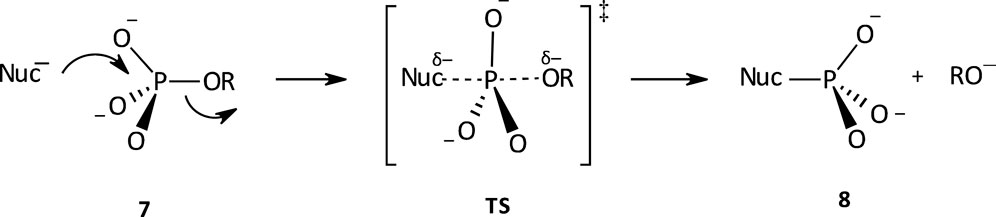
FIGURE 3. A concerted mechanism with a dissociative TS for the aqueous hydrolysis of phosphate monoester dianions.
A facilitating role has been ascribed (Hoff and Hengge, 1998; Medeiros et al., 2013) to the presence of the three non-bridging O atoms of the PO32- group in the monoester dianion; the internal electron donation interactions available in the dianion, involving the three O atoms as shown in Figure 4, provide a nucleophilic “push” which results in the elongation and consequent weakening of the P-OR bond. These interactions are thought to be responsible for the high sensitivity of the hydrolysis rates of monoester dianions to leaving group p
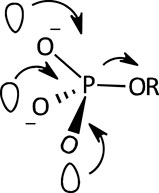
FIGURE 4. An illustration of the multiple n-σ*P-O interactions which assist the rupture of the P-OR bond in phosphate monoester dianions (Medeiros et al., 2013).
The hydrolysis of the phosphate monoester monoanion 9 proceeds by a dissociative mechanism (see Figure 5) with a negative value of
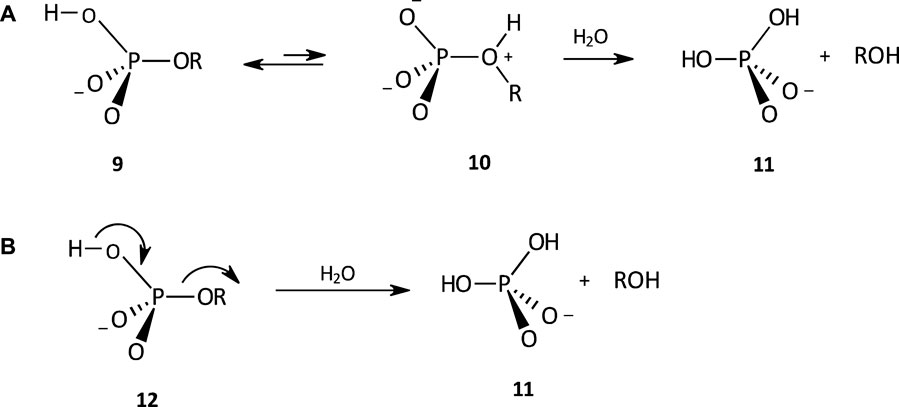
FIGURE 5. The hydrolysis of a phosphate monoester monoanion involving (A) an internal proton transfer pre-equilibrium to generate the dipolar species 10, from which ROH, a powerful leaving group, is expelled, and (B) simultaneous proton transfer and leaving group departure, to yield 11.
Theoretical calculations by Warshel’s group reproduce linear free energy relationships (LFER) results for the solution hydrolysis of phosphate monoester dianions leaving group (Klӓhn et al., 2006). The results demonstrate that decreasing the leaving group
In terms of relative reactivities in the hydrolysis of aryl phosphate monoester dianions and monoanions, dianions with very good leaving groups react faster than their corresponding monoanions. However, as the leaving group pKa increases, i.e., the leaving group becomes poorer, the hydrolysis of the monoanions becomes faster due to the protonation of the leaving group by the proton from the phosphoryl group. Thus, there is an intersection of the reactivity of the dianion and monoanion at pKa = 5.5 at 39°C (Di Sabato and Jencks, 1961; Kirby and Varvoglis, 1967; Forconi, 2015). By contrast, the monoanions of alkyl phosphates react faster in water than their dianion counterparts by a factor of
Solvent effects on the rates of the solvolysis of phosphate monoester monoanion and dianion have been reported. Solvolysis of the monoanion 9 is 14 and 16 times faster in water (Kirby and Varvoglis, 1967) than in the less polar tert-butyl alcohol and tert-amyl alcohol (Hoff and Hengge, 1998). The dielectric constants,
On the other hand, solvolysis of the dianion 7 is 7.5 × 103 and 8.8 × 103 times faster in tert-butyl alcohol and tert-amyl alcohol (Hoff and Hengge, 1998), respectively, than in water (Kirby and Varvoglis, 1967). Such rate enhancements may be ascribed to the reduced capacity for hydrogen bonding by the alcohol solvents when compared with water. The huge magnitudes of the solvent effects suggest that the dianion reacts by a different mechanism in the significantly less polar alcohol solvents. The activation entropy for the solvolysis of the dianion in water is 3.5 e.u. The higher values of activation entropy of 24.5 and 23.0 e.u. in tert-butyl alcohol and tert-amyl alcohol, respectively, are consistent with a change in mechanism, from ANDN (bimolecular, SN2(P)-type) to DN + AN [a two-step, unimolecular, SN1(P)-type] mechanism in which there is no nucleophilic participation in the rate-determining step, with change in solvent. The change in mechanism is consistent with an altered PES, caused by a change in solvent polarity.
The case for a symmetrically solvated, diffusible monomeric metaphosphate intermediate in the reaction of the dianion of phosphate monoester was made by the study of Knowles and co-workers (Friedman et al., 1988). This was shown in the solvolysis of the dianion of chiral p-nitrophenyl (R)-[16O, 17O, 18O]phosphate in tert-butyl alcohol, which gave completely racemic tert-butyl [16O, 17O, 18O]phosphate as the product. Heavy atom kinetic isotope effect measurements 15k, 18kbridge, and 18knon-bridge indicate similar late TS structures with little or no change in bond order between P and the non-bridge O atoms for the reaction of the dianion in water and in tert-butyl alcohol (Hengge et al., 1994). This may seem confusing at first when it is realized that reactions in the two solvents occur by different mechanisms, ANDN in water and DN + AN in tert-butyl alcohol. Such misconception is easily reconciled by the fact that the ANDN mechanism in water has a highly dissociative TS with small P-nucleophile bond formation,
There is information on the effect of dipolar aprotic solvents as co-solvents on the rates of phosphoryl transfer from the monoanion and dianion of phosphate monoesters. For example, use of >95% aqueous dimethyl sulphoxide (DMSO) instead of water as solvent causes about 106–107-fold increase in the hydrolysis rate of the dianion of p-nitrophenyl phosphate monoester (Abell and Kirby, 1986). The dipolar aprotic solvent hexamethylphosphoramide (HMPA) produces similar accelerations. Such dramatic accelerations have been ascribed to significant desolvation of the phosphoryl group in the ground state and TS of the reaction due to the reduction in hydrogen bonding when this group binds. These GS and TS effects combine to reduce the activation free energy of the reaction significantly. Reduced solvation of the substrate is also thought to lead to a weakening of the P-O bond. The combined effect of desolvation of the ground state, stabilization of the TS, and the weakening of the P-O bond is responsible for the dramatic rate accelerations reported. In a related study, Beltran et al. (1985) showed that the hydrolysis of p-nitrophenyl phosphate dianion in acetonitrile (MeCN) containing 0.02 M water occurs >106 times faster than the same reaction in pure water. This large rate enhancement is entropic in origin. There are other examples in which the hydrolysis of aryl phosphate dianions in water is significantly accelerated by the addition of a number of organic cosolvents which includes alcohols, acetone, dioxane, and polar aprotic solvents such as MeCN, DMSO, formamide and dimethylformamide (Kirby and Varvoglis, 1967). Although no logical relationship was found between solvent polarity parameters such as dielectric constant or the Grunwald-Winstein Y value and the magnitudes of the rate enhancement, the effects due to dipolar aprotic solvents were more dramatic.
The effect of added DMSO on the hydrolysis of phosphate monoesters was investigated in greater detail by Hengge and co-workers (Grzyska et al., 2002). Activation parameter and heavy atom kinetic isotope effect measurements and theoretical calculations were undertaken for the hydrolysis of a variety of phosphate monoesters. Accelerations by DMSO were restricted to phosphate monoesters dianions with pKas < pKa of phenol. Loss of solvation by the anionic phosphoryl group led to the weakening P-O bond; the latter was corroborated by the lower activation enthalpies and the heavy atom kinetic isotope effects (18kbridge and 15k) measured in the study. These kinetic isotope effects show that the TS is loose in both solvent systems, supporting the fact that the observed rate accelerations in DMSO have no mechanistic change component and implications.
The response of the hydrolysis of the monoanions of phosphate monoesters to the presence of dipolar aprotic cosolvents is different from that of the dianions, as shown in the rate of the hydrolysis of the monoanion of p-nitrophenyl phosphate in water which becomes slower in 80% DMSO-20% aqueous formate buffer, pH = 3.2 (Abell and Kirby, 1986). In this instance, acceleration of the breakdown of the dipolar intermediate 10 by dipolar aprotic solvents is outweighed by the unfavourable proton transfer preequilibrium which is disfavoured in such solvents. It should be pointed out, however, that only the rates were affected in the examples cited above; the mechanisms of hydrolysis remained the same in pure water and in the aqueous dipolar aprotic solvents.
The hydrolysis of neopentyl phosphate monoester, an unactivated alkyl substrate when compared with the aryl phosphate monoesters discussed above, was investigated by Wolfenden in the non-polar aprotic solvent cyclohexane (Stockbridge and Wolfenden, 2009). Hydrolysis of the dianion of neopentyl phosphate monoester, present in wet cyclohexane as its tetrabutylammonium salt, proceeds with the second-order rate constant of 8.8 × 10−10 M−1s−1. Comparison with the rate constant for its hydrolysis in water (3.6 × 10−22 M−1s−1) shows that neopentyl phosphate monoester dianion reacts faster in cyclohexane than in water by a factor of 2.5 × 1012. The origin of this remarkable rate acceleration is entirely entropic in nature, as seen by a comparison of the activation parameters in water (
Forconi (2015) has correlated rate accelerations of the hydrolysis of alkyl and aryl phosphate monoester dianions in protic and aprotic solvents by plotting the log of rate acceleration versus solvent dielectric constant as shown in Figure 6. The plot is interestingly linear, despite the structural differences in the substrate-types. The point for tert-butyl alcohol shows significant deviation from the straight line, and this is thought to arise from the fact that this solvent, though non-polar, is able to form hydrogen bonds with the reactive species, which is not possible with the other solvents apart from water. The correlation seems to provide evidence for catalysis by the more hydrophobic solvents due to the differences between the electrostatic properties of the ground and transition states of the reactions. A comment is made below (see Section 2.2.1) on the similar plot (Figure 2B) for phosphate diesters.
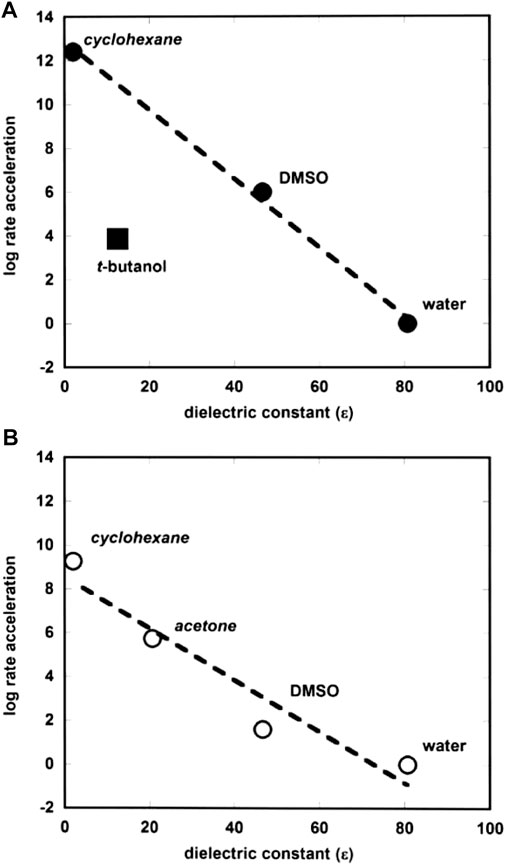
FIGURE 6. Plots of log rate acceleration versus dielectric constant (
2.1.1.2 Reactions with nitrogen nucleophiles
Reactions of phosphate monoesters with amine nucleophiles have been reported. Kirby and Jencks (1965a) studied the reactions of a variety amine nucleophiles with p-nitrophenyl phosphate dianion in aqueous solutions. A wide range of reactivity behaviour toward this substrate was found in the amines investigated, ranging from very high reactivity, including the manifestation of an α-effect, through moderate reactivity, to no reactivity at all. An important mechanistic information obtained from this study is the very small dependence of the rate on the basicity of the attacking amine,
The situation is different with phosphate monoester monoanions. Phosphorylation of amine nucleophiles, such as pyridine, by simple phosphate monoester monoanions does not occur, whereas fluoride ion reacts with the monoanion of acetyl phosphate to give fluorophosphate; the cleavage of the P-O bond in the latter compound is catalysed by tertiary amines (Di Sabato, and Jencks, 1961). The reactions of nicotinamide with a series of monoester monoanions give a Bronsted-type plot with
On the other hand, Ramirez and Marecek (1979); Ramirez and Marecek (1980) have shown that the reactions of 2,4-ditrophenyl phosphate monoanion with water and alcohols in MeCN solutions undergo phosphoryl transfer by the addition-elimination, associative [AN + DN] mechanism (Pathway A in Figure 1) involving a pentacoordinate intermediate and may be uncatalyzed or nucleophilic amine-catalysed. The reactions are slow. Steric hindrance in ROH decreases the rate of formation of alkyl phosphates significantly. There is no formation of tert-butyl phosphate when the reaction is performed in tert-butyl alcohol. Reactions of the dianion, on the other hand, occur by the elimination-addition, dissociative [ANDN] mechanism (Pathway C in Figure 1) involving a metaphosphate intermediate and are not subject to nucleophilic catalysis by added amine. These reactions are relatively fast and the rates of formation of alkyl phosphates are insensitive to structural variations in ROH. Reaction in the presence of tert-butyl alcohol produces tert-butyl phosphate. Thus, changing the solvent from water to MeCN changes the mechanistic profiles of the reactions of the monoanion and dianion of 2,4-dinitrophenyl phosphate.
2.1.2 Phosphorothioate monoesters
The replacement of the non-bridging oxygen in phosphate esters with sulphur to obtain phosphorothioate esters changes rates of reactions. This change in rate following the change of the non-bridging O to S is termed the thio effect, expressed as the kinetic ratio ko/ks, where ko and ks are the rate constants for the reactions of the oxygen and thio substrates, respectively. Several explanations have been advanced for the difference in the reactivity of phosphorothioate and phosphate esters. These include the different electron-releasing abilities of O and S (Chlebowski and Coleman, 1974; Zhang et al., 1999), changes in polarization of P=O and P=S bonds (Ketelaar et al., 1952), the relative electrophilicity of the P atom in phosphoryl versus thiophosphoryl groups (Teichmann and Hilgetag, 1967; Neimysheva, et al., 1968), and structural differences between the two compounds (Cheng et al., 2002). Carvalho et al. (2015) have emphasized the interplay between nucleophile charge and TS solvation in defining reactivities, including thio effects, in the SN2(P) reactions of simple phosphoryl transfer systems.
The solvolyses of chiral O-aryl phosphorothioate monoester dianions in water and ethanol proceed by the SN1-type (DN + AN) mechanism involving rate-limiting formation of free thiometaphosphate as an intermediate (Cullis and Iagrossi, 1986; Domanico et al., 1986; Cullis et al., 1987; Burgess et al., 1988). This conclusion was supported by the large degree of racemization observed in the reactions. The value of
Activation parameters for the hydrolysis of p-nitrophenyl phosphorothioate dianion were measured as ∆H‡ = 37.0 kcal mol−1, ∆S‡ = +29 e.u., which amount to ∆G‡ = 27.9 kcal mol−1 at 39°C (Catrina and Hengge, 1999); these may be compared with the values of
p-Nitrophenyl phosphorothioate monoester monoanion hydrolyses by the same mechanism as the phosphorothioate monoester dianion, involving the formation of a free thiometaphosphate intermediate. Activation parameters are
2,4-Dinitrophenyl phosphorothioate dianion undergoes hydrolysis in aqueous solution with an activation volume of 11 cm3 mol−1 (Burgess et al., 1988), as against the value of −4.8 cm3 mol−1 measured for the hydrolysis of 2,4-dinitrophenyl phosphate (Ramirez et al., 1986). These results are consistent with an SN1 mechanism with the formation of a free thiometaphosphate anion intermediate for the phosphorothioate substrate and a concerted mechanism for its oxygen analogue.
Solvent effects on the hydrolysis of the monoanion and dianion of p-nitrophenyl phosphorothioate were elucidated in aqueous DMSO solutions (Catrina and Hengge, 1999). These workers found that the log of the rate constant (s-1) for the hydrolysis of p-nitrophenyl phosphorothioate dianion was curvilinearly dependent on % DMSO in a concave upward fashion. The reaction was accelerated by more than 6 orders of magnitude in 95% DMSO compared with the reaction in water. Measured activation parameters in this solvent system of
Sorensen-Stowell and Hengge (2006) investigated the thermodynamic origin for the accelerated hydrolysis rate of the dianions of phosphate and phosphorothioate esters in aqueous DMSO solvent. In 0.6 (mol) aqueous DMSO (60 mol% water in DMSO),
2.1.3 Phosphate diesters
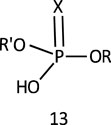
Phosphate diesters occur as nature’s genetic material in the form of DNA and RNA (Westheimer, 1987) and in phosphatidyl choline, one of the major constituents of the cell membrane (Sung, 1994). The resistance of the phosphodiester bond in DNA to hydrolysis in the absence of enzymes is well recognized. The neutral forms of this ester, 13, whose pKas are typically in the range of 1-2, exist as the monoanion 2 in the entire pH range. The negative charge of the monoanion is shared between two equivalent oxygens, thus making the P centre in 2 less electrophilic than in phosphate triesters. The absence of a proton, as found in the monoanion of phosphate monoesters, ensures that there are no other ionic forms of 2 around neutral pH (Kirby et al., 2015). The presence of enzymes, structural endowments in uncatalyzed reactions such as coordination of the phosphate diester substrate to metal ion complexes (Humphry et al., 2004; Berreau, 2006; Tirel et al., 2014), linkage of the two RO groups via 5-membered rings which induces the stereoelectronic effect (Taira et al., 1984) or ring strain (Westheimer, 1968; Kluger and Taylor, 1990), advantageously located nucleophiles (Livieri et al., 2007; Mancin and Tecilla, 2007), and inherently very good leaving groups (Kirby and Younas, 1970a) all enhance the reactivity of phosphate diester monoanions. Generally speaking, in the absence of intervening structural factors, the reactions of phosphate diester substrates occur mostly by the concerted SN2(P)-type mechanism (Hengge and Onyido, 2005).
Zalatan and Herschlag (2006) have shown that both the solution (non-enzymatic) and the AP-catalyzed (enzymatic) hydrolysis of a series of methyl phenyl phosphate diesters proceed via identical concerted transition states, i.e., the TS for the enzyme-catalyzed hydrolysis is indistinguishable from that of its solution counterpart. Rather than alter the TS of the solution reaction, as advocated to explain the high rate enhancements delivered by enzymes (see Section 1.3 above), the enzyme enacts catalysis by stabilizing the solution TS.
2.1.3.1 Reactions with oxygen nucleophiles
The hydrolysis of bis(p-nitrophenyl) phosphate, a substrate with a weakly basic leaving group, in neutral water at 50°C is too slow for direct measurement. However, measuring the rates at 80°C, 90°C and 100°C, gave the first-order rate constants as 1.0 × 10−8 s−1, 2.5 × 10−8 s−1, and 6.5 × 10−8 s−1, respectively (Chin et al., 1989). From these rate constants, the activation parameters for the reaction were obtained as
The alkaline hydrolysis of bis(p-nitrophenyl) phosphate was studied in aqueous DMSO, dioxane- and MeCN-water mixtures (Gomez-Tagle et al., 2006). In all the solvent mixtures investigated, the reaction rate showed a steady decline in the 0–70 vol% region of organic cosolvent, to reach about 50% of its value in water; thereafter the reaction rate increased sharply as the organic solvent content increased. The response of the rate constant to increasing per cent of the organic cosolvent was found to be qualitatively similar for the three organic solvents. The solvent effect in this system was satisfactorily correlated using a simplified stepwise solvent-exchange model, which focuses on the equilibrium replacement of solvent molecules in a solute’s coordination sphere in the treatment of the free energies of transfer of solutes to a mixed solvent (Waghorne, 1993; Gomez-Tagle et al., 2006). In 94% DMSO the rate constant for the hydrolysis of bis(p-nitrophenyl) phosphate was accelerated
Solvent effects on the hydrolysis of the unactivated phosphate diester dineopentyl phosphate in water were studied (Stockbridge and Wolfenden, 2010) by determining the rates of hydrolysis in the non-polar aprotic solvents acetone and cyclohexane and comparing them with the rate in water (Schroeder et al., 2006). The reactions in water and these solvents proceed wholly via P-O bond cleavage. The reactions proceed much more rapidly in cyclohexane and acetone than in water: at 25°C, the second-order rate constant for the hydrolysis of the substrate is increased by factors of 2 × 109 and 5 × 105 in cyclohexane and acetone, respectively. These results clearly show that the rate acceleration is dependent on solvent polarity, since the dielectric constants,
2.1.4 Phosphate triesters
Although phosphate esters are not naturally occurring, their worldwide deployment as pesticides (Wan et al., 2017) and their existence as stockpiles of nerve agents (Kim et al., 2011) make the study of the strategies and mechanisms for their detoxification and degradation an important chemical enterprise. For the reason that phosphate triesters are constitutionally neutral and do not repel nucleophiles as their monoester and diester counterparts do, these substrates are the most reactive among the three classes of phosphate esters (Hengge and Onyido, 2005). Although LFER studies show that the reactivity of phosphate triesters depends on the nature of the nucleophile and the leaving group, the nature of the two non-leaving groups, called spectator groups, can also be an important factor (Kirby et al., 2011; Mora et al., 2012; Kirby et al., 2013). There is evidence that triesters react by mainly associative mechanisms involving the phosphorane intermediate (Cleland and Hengge, 1995; Kirby and Nome, 2015), although concerted mechanisms have been reported with less nucleophilic bases such as phenoxides, secondary alicyclic amines and pyridines (Ba-Saif et al., 1990; Omakor et al., 2001; Castro et al., 2011). A theoretical study of the alkaline hydrolysis of dimethyl phosphate triesters showed that substrates with poor leaving groups (
2.1.4.1 Reactions with oxygen and nitrogen nucleophiles
Under nucleophilic catalysis by imidazole, diethyl 2,4-dinitrophenyl phosphate undergoes hydrolysis in water, first to give the phosphorylimidazole intermediate 14 in a concerted reaction, which then decomposes to products (Orth et al., 2011). The corresponding intermediate with 1-methylimidazole was unstable but its presence was inferred by NMR and mass spectral analysis (Campos et al., 2015). For the reactions of both imidazoles in DMSO-water mixtures, four distinct regions are discernible in the log rate versus mol% DMSO plot. The rate first decreased in the 0–10 mol% DMSO range, remained constant in the 10–20 mol% DMSO range, increased rapidly in the 20–40 mol% DMSO range and then less rapidly in the 40–100 mol% DMSO range. The rates were 103-fold and 2 × 102-fold higher in 100% DMSO for imidazole and 1-methylimidazole, respectively, relative to rate in pure water. The solvent effect recorded in this study (Orth et al., 2011) was satisfactorily correlated with Catalan’s SA, SB and SPP solvent parameters (Catalán et al., 2001; Gomez-Tagle et al., 2006; Catalán, 2009) which showed that decreased solute-solvent interactions in the wholly organic solvent or predominantly organic solvent in the aqueous organic solvent, caused by decreased hydrogen bonding, led to higher rates in DMSO-rich and pure DMSO solvents.
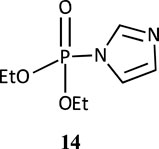
The second-order rate constant for the alkaline hydrolysis of p-nitrophenyl diphenyl phosphate in 95% water-5% dioxane (v/v) at 25°C is 3.30 × 10−1 M−1s−1 (Bunton et al., 1968). In 60% water-40% dioxane, under the same conditions, this rate constant was measured as 2.15 × 10−1 M−1s−1, showing decreased rate in the presence of dioxane. This suggests that the TS for the reaction is strongly hydrated, consistent with the low
Conversely, de Souza and Ionescu (1999) reported the values of the second-order rate constant as 9.6 × 10−1 M−1s−1 and 24.3 M−1 s−1 in 50% water-50% DMSO and 10% water-90% DMSO (v/v), respectively, clearly showing the accelerating influence of added DMSO. The rate-solvent mixture profile showed very little effect of added DMSO between the 0%–50% DMSO range; there was a steady increase in rate from 50% DMSO to reach 24.3 M−1 s−1 in 90% DMSO. None of these authors supplied the value of the second-order rate constant in 100% water. From the data of Bunton et al. (1968), a rough extrapolation could be made to give the rate constant in pure water as 2.9 × 10−1 M−1s−1; this value agrees with the value of 2.6 × 10−1 M−1s−1 measured in the laboratories of Williams (Ba-Saif et al., 1990) for reaction in pure water. This implies that the rate of reaction undergoes ca. 4-fold and 100-fold acceleration in 50% water-50% DMSO and 10% water-90% DMSO, respectively; the rate increase in 90% DMSO is therefore modest, when compared to the effects of this solvent in the hydrolysis of phosphate monoester dianions considered earlier. The activation parameters (
On the other hand, incremental addition of quantities of MeCN or tert-butyl alcohol to water retards the rate of the alkaline hydrolysis of p-nitrophenyl diphenyl phosphate across the entire range of organic solvent-water mixtures studied to give rate constants that are lower in these water-organic solvent mixtures than in pure water (Bunton et al., 1996), a clearly different scenario from the behaviour in DMSO (de Souza and Ionescu, 1999). Data in water-MeCN and water-tert-butyl alcohol mixtures obtained in the study by Bunton et al. (1996), plotted in Figure 7, show that the second-order rate constant, k2, in both solvent mixtures go through a minimum to attain values in ca. 84% organic solvent which are 44% (water-MeCN) and 56% (water-tert-butyl alcohol) of the value of k2 in pure water. Bunton et al. have explained that the rate decrease is caused by the stabilization of the phosphate ester substrate through the decrease of its activity coefficient on the addition of the organic solvent to water. This GS stabilization is partially offset by the stabilization of the anionic TS by the organic component of the solvent mixture. The increase in rate in the less aqueous solvent mixtures heralds the destabilization of the GS in this region due to the partial desolvation of the anionic nucleophile. This concept is considered further below (see Section 2.2.4) where solvent effects on the reactivities of phosphate and phosphinothioate esters are discussed.
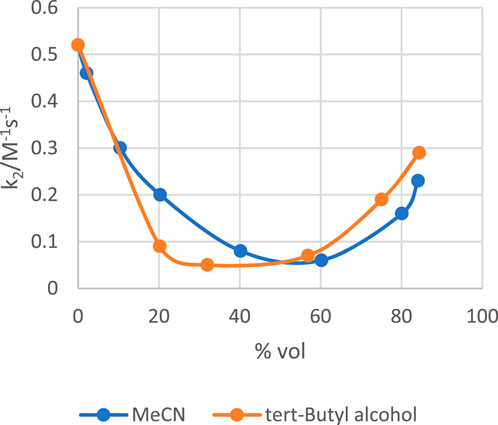
FIGURE 7. Plots of the second-order rate constant (k2/M-1s-1) versus % volume for the alkaline hydrolysis of p-nitrophenyl diphenyl phosphate in water-MeCN and water-tert-butyl alcohol mixtures. Data taken from the work of Bunton et al. (1996).
Although not definitively proven, the accepted mechanism for the hydrolysis of p-nitrophenyl diphenyl phosphate in water and predominantly aqueous water-organic solvent mixtures involves the attack of the nucleophile on the substrate to form a pentacoordinate intermediate from which p-nitrophenoxide, the leaving group, is expelled (Bunton et al., 1968; Mackay et al., 1987). The reaction of this substrate with the less basic nucleophile, phenoxide, has been shown to be concerted (Ba-Saif et al., 1990). The activation parameters of
2.2 Phosphinates and phosphinothioates
Although there are no known naturally occurring phosphinate and phosphinothioate esters, these synthetic compounds are structurally related to phosphate and phosphonate esters that have found use as pesticides, neurotoxins, and other biologically active and important substances (Toy and Walsh, 1987). A number of suitably substituted phosphinic acids function as metalloprotease inhibitors (Abdou, 2020). Understanding the mechanisms of phospho group transfer reactions involving phosphinates and their thio analogues has often helped to elucidate the constitutional and environmental factors at play in the reactions of phosphonates and phosphates and, by extension, in biological phosphoryl transfer reactions (Hengge and Onyido, 2005).
2.2.1 Reactions with oxygen nucleophiles
Phosphinates (5a) and phosphinothioates (5b) as esters of phosphinic acid (15) represent the simplest structural framework among organophosphorus esters. The proton in the OH group in 15 is replaced by an alkyl group or fragment or an aryl group to form the ester 5.
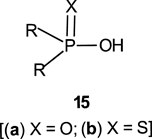
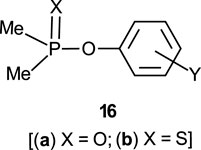
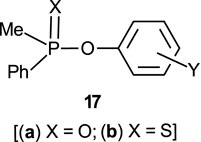
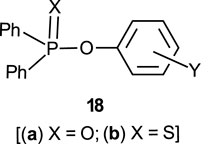
The reactions of phosphinates and phosphinothioates (16–18) with nucleophiles in a variety of solvents has been the subject of detailed mechanistic studies. Table 1, adapted from our earlier review article (Hengge and Onyido, 2005), summarizes the Brönsted and/or Hammett selectivity parameters reported in the literature for the reactions of oxygen nucleophiles with different phosphinate and phosphinothioate ester substrates with oxygen nucleophiles in different solvents. These selectivity parameters form the basis for the mechanisms assigned to these reactions. Broad mechanistic behaviours are discernible from the body of data summarized in Table 1, depending on the basicity of the attacking nucleophiles and the polarity of the reaction medium.
2.2.2 Resolving mechanistic ambiguities
Two mechanistic ambiguities in the literature which have now been satisfactorily resolved should be pointed out, to aid a discussion of the mechanistic categorizations mentioned above. The first ambiguity concerns entry 13a in Table 1 for the hydrolysis of the aryl dimethylphosphinothioates 16b in water, first studied by Istomin et al. (Eliseeva et al., 1979) who concluded that the mechanism of the reaction was associative on the basis of a positive value of Hammett
The second ambiguity which surrounds entry 15a in Table 1, the hydrolysis of the same substrate 16b in 50% water-50% ethanol, arose exactly the same way as outlined above for the reaction in water. The reaction was first investigated by the same group, Istomin and Eliseeva (1981) to obtain a Hammett
2.2.3 Reactivity trends
There are three trends that are embedded in or discernible from the data collated in Table 2, which are of mechanistic significance; these are now discussed.
TABLE 2. Hammett and/or Brönsted selectivity parameters for the reactions of phosphinate and phosphinothioate esters with oxyanionic nucleophiles in different solventsa.
First, reactions of the same phosphinate ester substrate with the same nucleophile are slower in ethanol and aqueous ethanol solvents than in pure water, contrary to the prediction of the Hughes-Ingold theory of solvent effects for charge-type 1 SN2-type reactions of neutral substrates and negatively charged nucleophiles. For such reactions, destabilization of the nucleophile by desolvation by the less polar organic solvent makes it more reactive, thereby decreasing the reaction activation energy, which results in significant rate increases (Hughes and Ingold, 1935; Ingold, 1969; Parker, 1969; Parker, et al., 1978). Desolvation involves the partial or complete removal, from the nucleophilic site, of solvent molecules associated with the nucleophile in the GS, as illustrated in the equilibria shown in Figure 8. This GS destabilization makes the nucleophile more reactive. The trend for the nucleophilic displacement reactions on phosphorus esters is different because non-polar solvents stabilize the neutral substrates to greater extents than they destabilize the negatively charged nucleophiles (Bunton et al., 1996), leading to rate reductions in such solvents. The means by which less polar solvents stabilize neutral phosphorus esters is discussed further below.
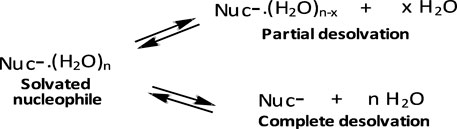
FIGURE 8. Desolvation of a solvated nucleophile molecule in the GS, giving rise to a more reactive nucleophile; the desolvation may be partial or complete.
Second, all reactions in water involving phenoxide ion and its weakly basic analogues, all of which are weaker nucleophiles when compared with hydroxide ion, react with the same substrates by a concerted mechanism. The corresponding reactions with the much more basic hydroxide ion as nucleophile occur via the associative mechanism. Table 2 lists the pKa values showing the relative basicities in water, ethanol and some water-ethanol solvent mixtures of the anionic oxygen nucleophiles employed in the reactions. The same point was made earlier in a study of the reaction of fenitrothion ([O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothioate], 19) with a variety of oxyanionic nucleophiles–hydroxide, alkoxide, and phenoxides–in water (Omakor et al., 2001) and appears to be general for the reactions of phenoxides and other weakly basic oxyanions with a diversity of (thio)phosphates and (thio)phosphinates for which kinetic data are available. Thus, we have a mechanistic dichotomy in water in which the strongly basic nucleophile hydroxide ion reacts by an associative mechanism, while the weaker nucleophiles react by a concerted mechanism. The question that arises then is, what is responsible for this difference in behaviour of the two oxyanionic nucleophile types which is denominated by nucleophilic strength?
The third trend in Table 2 is that all reactions which proceed by the concerted mechanism in water proceed by the associative mechanism when the solvent is changed to ethanol. This change of mechanism has been explained (Buncel et al., 2004) on the basis of the large changes in the basicity of the nucleophiles, as measured by their
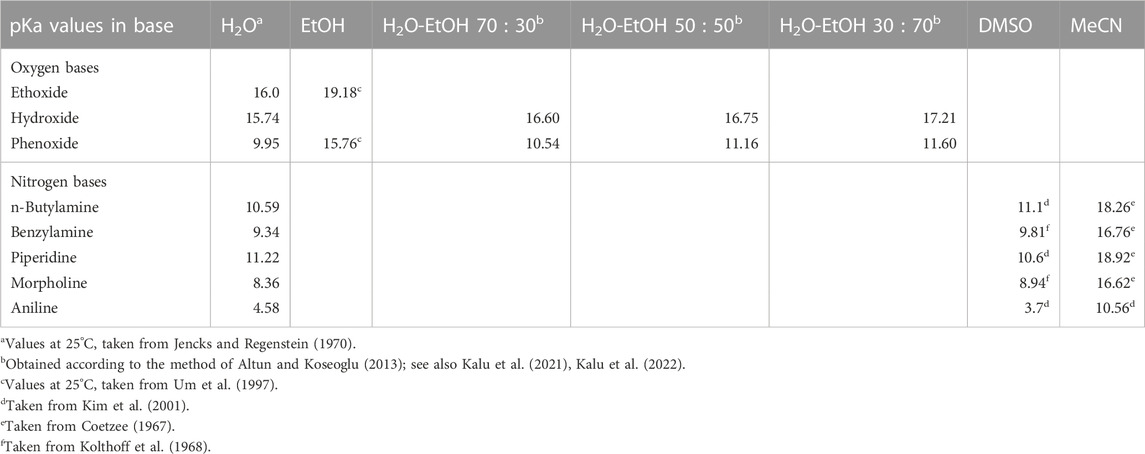
TABLE 3. The pKa values of selected oxygen and nitrogen bases in water, ethanol, various H2O-EtOH solvent mixtures, dimethyl sulphoxide, and acetonitrile.
The dielectric constant,
The above reasoning to explain the change in the mechanistic character observed in the reactions collected in Table 2 with solvent change from water to ethanol is tantamount to the assumption that, for any particular concerted reaction, e.g., the hydrolysis of the phosphinothioate esters Me2P (=S)-OPhX (entry 13b) for which
This path to associative structures is quantified by a vector r, which is the resultant of the TS movement due to changes in nucleophile basicity (quantified by vector p) and that of the TS movement due to changes in leaving group basicity (quantified by vector q). Vector p is the resultant vector of the Hammond and anti-Hammond movements of the TS resulting from changes in nucleophile basicity (vectors k and l, respectively, Figure 9), while vector q is the resultant vector of the Hammond and anti-Hammond movements of the TS resulting from changes in leaving group basicity (vectors m and n, respectively, Figure 9). Figure 9 gives a diagrammatic representation of how these vectors interact to effect TS movements as a consequence of nucleophile and leaving group basicity changes, in tandem with solvent polarity changes (see below).
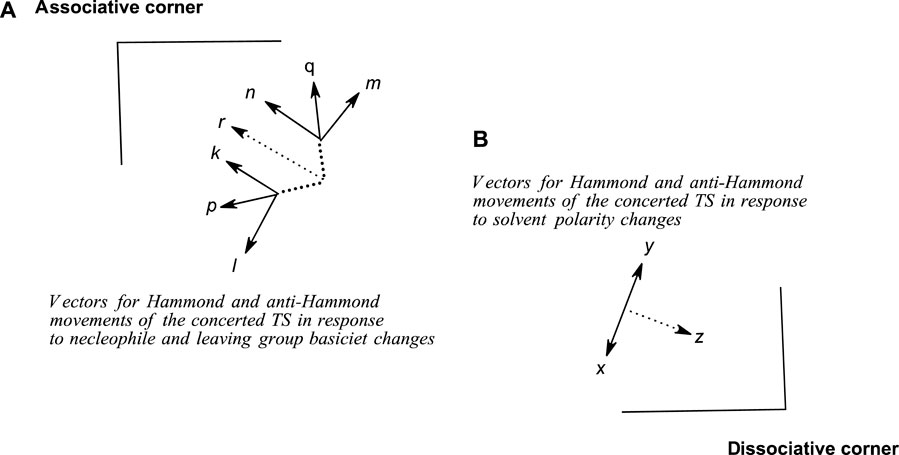
FIGURE 9. A schematic diagram showing Hammond and anti-Hammond vectorial movements of the concerted TS in response to changes in (A) nucleophile and leaving group base strengths and (B) solvent polarity.
An examination of the combined influence of the change in the base strengths of the nucleophile and leaving group and change of solvent polarity as the solvent is changed from water to ethanol on the structure of the concerted TS was undertaken by studying the same reaction in a series of water-ethanol mixtures in which the solvent polarity was decreased by sequentially increasing the proportion of the ethanol component (Onyido, et al., 2005a; Kalu et al., 2021; Kalu et al., 2022). The outcome of this study was counterintuitive: the TS, rather than move towards the associative end of Figure 2, moved in the opposite direction, towards the dissociative end, up to a point Y where further decreases of solvent polarity had no effect on TS structure. This movement towards the dissociative end of Figure 2 is shown by the trajectory (E) defined by the arrows in blue and quantified by a vector z, which is the anti-Hammond response to the movement of the concerted TS along the reaction coordinate according to the arrow x
As can be seen from the above analysis, increasing the base strength of the nucleophile and leaving group moves the concerted TS, along vector r, towards associative TS structures, while a decrease in solvent polarity effectuates TS movement along vector z, towards the dissociative end of the diagram. For the situation in which both nucleophile and leaving group basicity is increased and solvent polarity is decreased at the same time by the incremental enhancement of the proportion of ethanol in the aqueous ethanol solvent, the observed movement of the TS toward more dissociative structures is the balance between two effects. The various scenarios inherent in the relative magnitudes of the two opposing vectors r and z are summarized in Table 4.
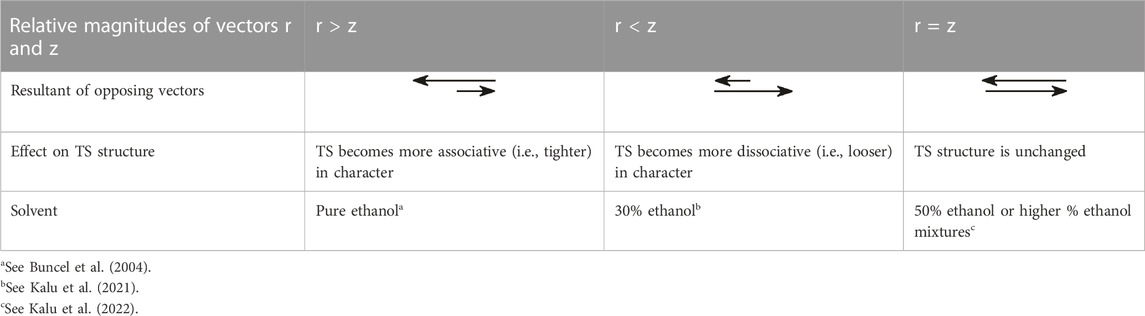
TABLE 4. The relative magnitudes of the vectors r and z in Figure 2 and their effects on the structure of the concerted TS for the reaction of 8b with HO- and PhO- nucleophiles in aqueous ethanol solvents (entries 12b, 13, 14b, and 15 in Table 1). From Kalu et al. (2022), reproduced by permission of the Royal Society of Chemistry.
2.2.4 The origin of observed solvent polarity effects on reaction rates
As pointed out above, SN2-like reactions of phosphinates (Cook et al., 1986b; Blásko et al., 1991; Bunton et al., 1996) and some other organophosphorus esters (Blásko et al., 1991) with negatively charged nucleophiles in water become slower when conducted in less polar solvents. This is contrary to the general solvent effects encountered in the reactions of carbon-based SN2 reactions which proceed faster in less polar solvents than in water, in accord with the Hughes-Ingold theory of solvent action. This behaviour, which was investigated in some depth (Blásko et al., 1991; Bunton et al., 1996), has been attributed to the stabilization of the neutral ester substrate by the less polar solvent, an inhibitory effect that more than compensates the rate-increasing destabilization of anionic nucleophiles due to desolvation by the organic solvent. Figure 10 shows the reaction coordinate diagrams for (A) a hypothetical SN2 reaction which obeys the Hughes-Ingold theory of solvent action, in which the organic solvent destabilizes the initial state by desolvating the nucleophile, and for (B) a hypothetical SN2(P) reaction in which the organic solvent stabilizes the initial state by decreasing the activity coefficient of the neutral organic substrate (see below).
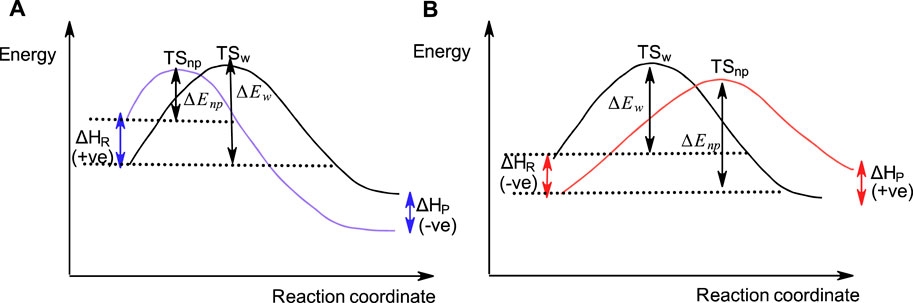
FIGURE 10. A hypothetical reaction coordinate diagram showing qualitatively the SN2-type reactions in water and a less polar organic solvent involving an anionic nucleophile with (A) a carbon-based substrate, and (B) a phosphinate ester substrate. In (A), the initial state is destabilized in the less polar solvent (
Bunton et al. (1996), in their study of the rates of hydrolysis of diverse organophosphorus ester substrates in two binary aqueous organic solvents (water-MeCN and water-tert-butyl alcohol), demonstrated linearity with a negative slope in plots of substrate activity coefficient,
This qualitative treatment, which can be extended to other binary aqueous organic solvents in which the organic solvent is less polar than, and miscible, with water, is sufficient to rationalize why organophosphorus esters behave differently from alkyl substrates in their SN2 reactions with anionic nucleophiles.
2.2.5 Reactions with nitrogen nucleophiles
In their study of the reactions of a number of aryl phosphinates and aryl phosphinothioates with an array of primary, secondary and diamine nucleophiles in acetonitrile, Cook et al. (1986a) demonstrated the nucleophilic reactivity order of diamine > primary amine > secondary amine. While the diamines and secondary amines obeyed a rate law which is first-order in amine, the kinetics of the only primary amine studied, n-butylamine, obeyed a two-term rate law according to Eq. 3, the first and second terms being first-order and second-order, respectively, in amine. This kinetic behaviour is consistent with the mechanism shown in Figure 11, a conclusion that is reinforced by the observed leaving group effects, solvent effects and activation parameters. In this mechanism, Pathway A is followed by the reactions involving the diamines and secondary amines in which the zwitterionic pentacoordinate intermediate Z decomposes unimolecularly to give the reaction products. Although both amine types obey the same rate law, the intermediates arising from them differ in their decomposition modes to obtain the products. For the secondary amines, rate-limiting dissociation of Z followed by proton loss, or rate-limiting intramolecular proton transfer to X− which is in concert with leaving group expulsion, are relatable to the measured activation entropy in the region of −59 e.u. (for piperidine, as an example) and a good correlation of
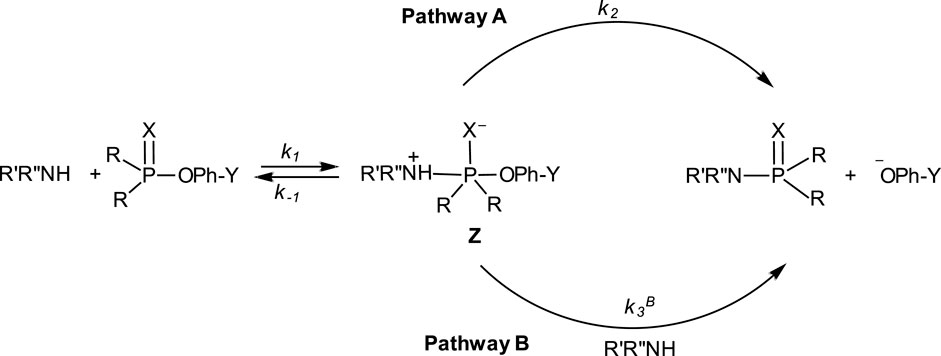
FIGURE 11. The mechanisms for the aminolysis of aryl phosphinates and aryl phosphinothioates by a diversity of amine types–primary, secondary, and di-amines–in acetonitrile. See Cook et al. (1986a).
The reactions of anilines with aryl dimethyl, methyl phenyl, and diphenyl phosphinates (21a-c) in DMSO occur with normal primary deuterium kinetic isotope effects (
Contrary to the above results in acetonitrile and DMSO, Um et al. (2006); Um et al. (2007); Um et al. (2009) have reported that the reactions of 2,4-dinitrophenyl diphenylphosphinate and some other aryl diphenylphosphinate substrates and their thio analogues with secondary amines in the predominantly aqueous binary solvent 80 mol% water-20 mol% DMSO proceed through a concerted mechanism, on the basis of the Brönsted and Yukawa-Tsuno parameters,
The dielectric constants,
This phenomenon, in which the mechanism of the reactions of nitrogen nucleophiles with phosphinates and phosphinothioates changes from concerted in water and predominantly aqueous DMSO solvent to associative in the less polar solvents MeCN and DMSO, was also noted and extensively discussed for the reactions of oxygen nucleophiles above, where the change of solvent is from water to the less polar ethanol. Overall, the potential energy surface (PES) involving concerted TS structures should be markedly different from the PES involving associative TS structures. Noting that even though ethanol, MeCN and DMSO all favour the associative mechanism for the reactions of these substrates with oxygen and nitrogen nucleophiles, MeCN and DMSO are dipolar aprotic solvents while ethanol is a hydroxylic one, it appears that the choice of the PES in these reactions is more solvent polarity-driven and is independent of solvent type.
3 Discussion
In order to assess the status of information on solvent and solvation effects on reactivities and mechanisms in the phospho group transfers considered in this paper, with the objective of determining future directions for research in this domain, we have summarized available information and data discussed in the preceding sections in Table 5.
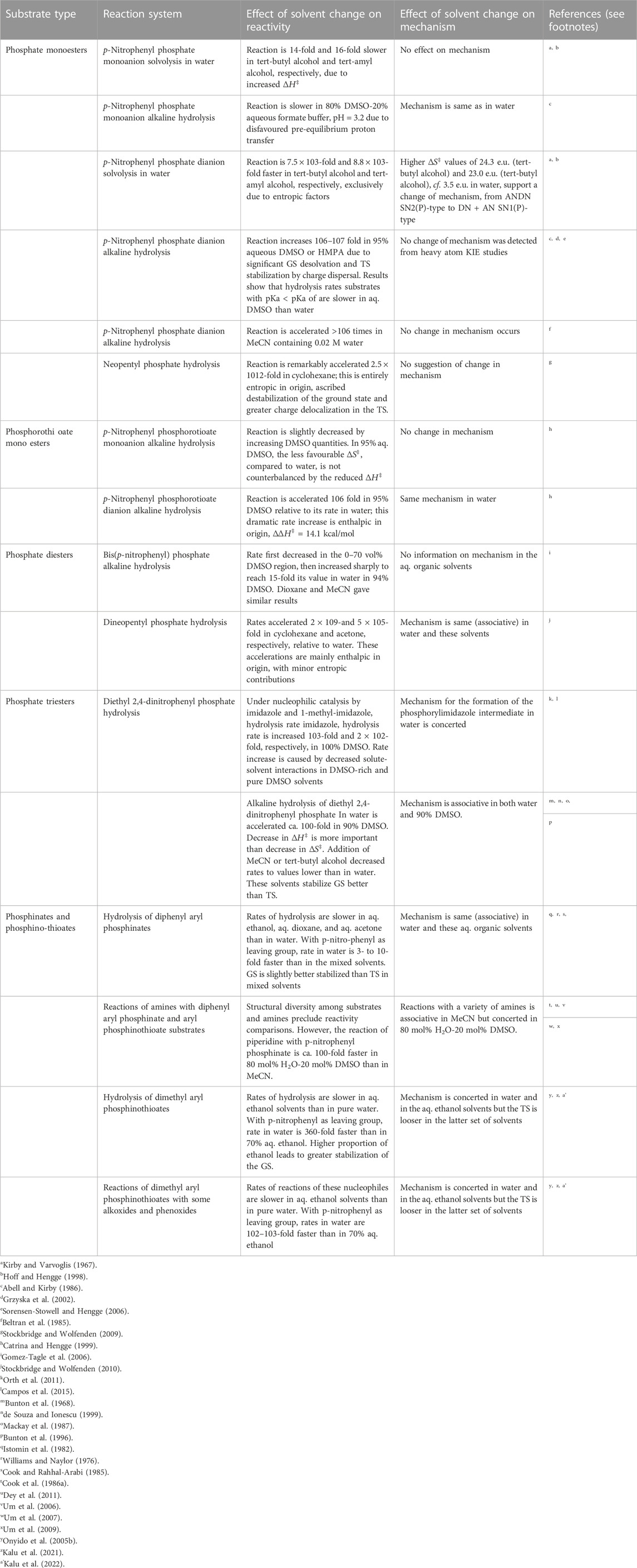
TABLE 5. A summary of the effects of solvent change on reactivities and mechanism in some of the different reaction systems encountered in phospho group transfers.
A number of general points emerge from a careful scrutiny of Table 5.
1. For the same nucleophile, the effect on reactivity of solvent change from water to dipolar aprotic solvent (DMSO or HMPA or MeCN or acetone)-water mixtures or to neat dipolar aprotic solvents follows the following order of substrates: phosphate monoester dianions >> phosphate monoester monoanion
2. For the same substrate, the effect of a change from a polar solvent to less polar one is by far greater for charged nucleophiles such as HO− (e.g., the 106–107-fold increase in the rate of the alkaline hydrolysis of phosphate monoester dianions in aq. DMSO or HMPA) than for uncharged nucleophiles such as water (e.g., the
3. The greatest effects are observed when the solvent is changed from water to the much less polar cyclohexane, as found in the hydrolysis of neopentyl phosphate dianion (1012-fold rate increase) and dineopentyl phosphate monoanion (109-fold rate increase) in cyclohexane saturated with water. An estimation of the magnitude of the rate enhancement experienced by phospho transfer reactions involving specific substrate types (phosphate monoanion or dianion) as functions of solvent polarity as measured by, e.g., solvent dielectric constant, will therefore be an important contribution to the understanding of solvent effects in these processes.
4. With phosphate or phosphorothioate monoester monoanions as substrates for the alkaline hydrolysis reactions, the effects of solvent change, from water to dipolar aprotic solvents or their aqueous mixtures, on reactivity are quite modest, usually in the range of 10- to 20-fold increase in rates.
5. Rate increases which are also modest, in the range of 102-103-fold, are induced by solvent change, from water to less polar solvents, when the substrates are neutral esters, as encountered in the hydrolysis of phosphate triesters and in the reactions of phosphinate/phosphinothioate esters with amines.
6. All reactions of the nucleophiles hydroxide, alkoxides and phenoxides with phosphinate and phosphinothioate esters are slower in water-ethanol mixtures than in water.
7. It is not in all cases that the activation parameters for the reactions in the solvents of interest were determined in the studies summarized in Table 5. The determination of activation parameters and the thermodynamic parameters for the transfer of solutes from water to the less polar solvents are minimum but vital pieces of information which solvent effect studies should seek to convey.
8. It is instructive to note that for systems for which definitive information about their mechanisms of reaction is available, only in a few cases have the observed solvent effects on reactivities delivered changes in mechanism as well.
9. This review has revealed that scant attention has been paid to solvent effects on catalyzed reactions. It will be an important endeavour to investigate the relationship between the effect of catalysts and solvents on reactivity and mechanisms when compared with uncatalyzed systems.
From the foregoing, it is clear that solvent effects, which are deserving of attention because the rate enhancements they deliver approach or surpass those found in enzymatic reactions, have been encountered only in a few of the phospho group transfers considered in this review. Such interesting systems involve a few phosphate monoester dianions and phosphate diester monoanions as substrates while the solvents utilized are aprotic solvents such as cyclohexane, DMSO and a few other dipolar aprotic solvents which are, to different extents, less polar than water. The extension of such studies to more phosphate ester substrates of different activation statuses is critical to characterising a good range of responses to solvent changes, which is a prerequisite to the proper understanding of the causative factors that deliver large rate enhancements. The combination of negative charges in the GS with the hydrophobic environment which solvents with reduced dielectric constants relative to water provide, in order to obtain significant rate enhancements, suggests the importance of GS desolvation and the stabilization of the more charge delocalized TS in the enactment of “enzyme”-type rates in such systems. Such charge redistribution, which is intensely promoted in the non-polar active sites of enzymes through preferential charge destabilization in the GS and stabilization of the redistributed charges in the TS, is behind the proposal that desolvation contributes to rate enhancements by enzymes.
4 Concluding remarks
The authoritative computational work by Warshel’s group (Warshel et al., 2006) does not support desolvation as a mechanism by which enzymes enact tremendous rate enhancements. Their work demonstrated that enzyme active sites supply preorganized environments with electrostatic endowments for TS stabilization that far outweigh the TS stabilization which obtains in water. Polar interactions are promoted in the hydrophobic environment of the enzyme, compared to water, by the enhanced strength of these interactions which is known to increase with the reciprocal of the effective dielectric constant of the active site environment (Richard et al., 2014). Nevertheless, it is important for solution experimentalists to undertake detailed evaluations of solvent effects in the reactions of activated and less activated substrates so as to systematize and quantify GS desolvation and TS stabilization effects by solvents in order to fully understand what happens when the reactants and transition states are transferred from water to less polar environments. Such studies will be important contributions to the physical organic chemistry of phospho group transfer. Such studies will also seek to provide answers to the important question posed by Stockbridge and Wolfenden (2009) about what changes occur in the TS of a phospho transfer reaction and the degree to which water molecules continue to be associated with the phosphate ester substrate when it is transferred to more hydrophobic environments.
Author contributions
IO is the senior author who conceptualized the Project and contributed 40% of the work. OO, CE, and NO contributed equally to the remaining 60% of the work. All authors read the draft manuscripts and approved the final version produced by the senior author.
Acknowledgments
The authors are indebted to Emmanuel C. Ohaekenyem for assistance with some of the graphics.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdou, M. M. (2020). Synopsis of recent synthetic methods and biological applications of phosphinic acid derivatives. Tetrahedron 76, 131251. doi:10.1016/j.tet.2020.131251
Abell, K. W. Y., and Kirby, A. J. (1986). Acceleration of P_O cleavage reactions of phosphate monoester dianions in dipolar aprotic solvents. Tetrahedron Lett. 27, 1085–1088. doi:10.1016/s0040-4039(00)84184-2
Abraham, M. H. (1986). Solvent effects on reaction rates. Pure Appl. Chem. 57, 1055–1064. doi:10.1351/pac198557081055
Admiraal, S. J., and Herschlag, D. (2000). The substrate-assisted general base catalysis model for phosphate monoester hydrolysis. Evaluation using reactivity comparisons. J. Am. Chem. Soc. 122, 2145–2148. doi:10.1021/ja993942g
Altun, Y., and Koseoglu, F. (2013). Influence of solute-solvent interaction on acid strength of some substituted phenols in ethanol-water media. J. Solut. Chem. 42, 1691–1705. doi:10.1007/s10953-013-0056-0
Anslyn, E. V., and Dougherty, D. A. (2006). Modern physical organic chemistry. Melville, N.Y.: University Science Books.
Arnett, E. M., Joris, L., Mitchell, E., Murty, T. S. S. R., Gorrie, T. M., and Schleyer, P. V. R. (1976). Hydrogen-bonded complex formation. III. Thermodynamics of complexing by infrared spectroscopy and calorimetry. J. Am. Chem. Soc. 92, 2365–2377. doi:10.1021/ja00711a029
Arnett, E. M., Mitchell, E. J., and Murty, T. S. S. R. (1974). Basicity. Comparison of hydrogen bonding and proton transfer to some Lewis bases. J. Am. Chem. Soc. 96, 3875–3891. doi:10.1021/ja00819a028
Ba-Saif, S. A., Waring, M. A., and Williams, A. (1990). Single transition state in the transfer of a neutral phosphoryl group between phenoxide ion nucleophiles in aqueous solution. J. Am. Chem. Soc. 112, 8115–8120. doi:10.1021/ja00178a040
Beltran, A. M., Klaebe, A., and Perie, J. J. (1985). Etude cinetique de l’hydrolyse du paranitrophenylphosphate dans l’acetonitrile faiblement aqueux. Process. Dissociatif. Tetrahedron Lett. 26, 1711–1712. doi:10.1016/s0040-4039(00)98318-7
Benkovic, S. J., and Schray, K. J. (1978). “The mechanism of phosphoryl transfer,” in Transition states of biochemical processes. Editors R. D. Gandor, and R. L. Schowen (New York: Plenum Press), 493–521.
Berreau, L. (2006). Kinetic and mechanistic studies of the reactivity of Zn-OHn (n = 1 or 2) species in small molecule analogues of zinc-containing metalloenzymes. Adv. Phys. Org. Chem. 41, 79–181.
Blásko, A., Bunton, C. A., Hong, Y. S., Mhala, M. M., Moffat, J. R., and Wright, S. (1991). Micellar rate effects on reactions of hydroxide ion with phosphinate and thiophosphinate esters: Micellar rate effects on reactions OF OH−WITH phosphinate. J. Phys. Org. Chem. 4, 618–628. doi:10.1002/poc.610041006
Bourne, N., Chrystiuk, E., Davis, A. M., and Williams, A. (1988). A single transition state in the reaction of aryl diphenylphosphinate esters with phenolate ions in aqueous solution. J. Am. Chem. Soc. 110, 1890–1895. doi:10.1021/ja00214a037
Brooker, L. G. S., Craig, A. C., Heseltine, D. W., Jenkins, P. W., and Lincoln, L. L. (1965). Color and constitution. XIII.1 merocyanines as solvent property Indicators2. J. Am. Chem. Soc. 87, 2443–2450. doi:10.1021/ja01089a025
Buchwald, S. L., Friedman, J. M., and Knowles, J. R. (1984). Stereochemistry of nucleophilic displacement on two phosphoric monoesters and a phosphoguanidine: The role of metaphosphate. J. Am. Chem. Soc. 106, 4911–4916. doi:10.1021/ja00329a047
Buncel, E., Albright, K., and Onyido, I. (2004). Nucleophilic displacement on 4-nitrophenyl dimethyl phosphinate by ethoxide ion: Alkali metal ion catalysis and mechanism. Org. Biomol. Chem. 2, 601–610. doi:10.1039/b314886f
Buncel, E., and Rajagopal, S. (1989). Solvatochromic studies of novel azo merocyanine dyes. The.pi.*azo scale of solvent polarity. J. Org. Chem. 54, 798–809. doi:10.1021/jo00265a017
Buncel, E., and Rajagopal, S. (1990). Solvatochromism and solvent polarity scales. Acc. Chem. Res. 23, 226–231. doi:10.1021/ar00175a004
Buncel, E., Stairs, R., and Wilson, H. (2003). The role of solvents in chemical reactions. Oxford: Oxford University Press. Oxford Chemistry Masters Series.
Bunton, C. A., Farber, S. J., and Fendler, E. J. (1968). Hydrolysis of p-nitrophenyl diphenyl phosphate. J. Org. Chem. 33, 29–33. doi:10.1021/jo01265a006
Bunton, C. A., Gillit, N. D., and Kumar, A. (1996). Solvent effects on the reactions of hydroxide ion with phosphorus (V) esters: A quantitative treatment. J. Phys. Org. Chem. 9, 145–151. doi:10.1002/(sici)1099-1395(199603)9:3<145::aid-poc763>3.0.co;2-e
Burgess, J., Blundell, N., Cullis, P. M., Hubbard, C. D., and Misra, R. (1988). Evidence for free monomeric thiometaphosphate anion in aqueous solution. J. Am. Chem. Soc. 110, 7900–7901. doi:10.1021/ja00231a064
Butcher, W. W., and Westheimer, F. H. (1955). The lanthanum hydroxide gel promoted hydrolysis of phosphate esters. J. Am. Chem. Soc. 77, 2420–2424. doi:10.1021/ja01614a018
Cainelli, G., Galletti, P., and Giacomini, D. (2009). Solvent effects on stereoselectivity: More than just an environment. Chem. Soc. Rev. 38, 990–1001. doi:10.1039/b802815j
Campos, R. B., Santos, E. H., Oliveira, A. R. M., Marins Ocampos, F. M., Souza, B. S., Barison, A., et al. (2015). Reactivity of imidazole derivatives toward phosphate triester in DMSO/water mixtures: A comprehensive study on the solvent effect. J. Org. Chem. 80, 7572–7580. doi:10.1021/acs.joc.5b01152
Carvalho, A. T. P., O’Donoghue, A. C., Hodgson, D. R. V., and Kamerlin, S. C. L. (2015). Understanding thio-effects in simple phosphoryl systems: Role of solvent effects and nucleophile charge. Org. Biomol. Chem. 13, 5391–5398. doi:10.1039/c5ob00309a
Castro, E. A., Ugarte, D., Rojas, F. M., Pavez, P., and Santos, J. G. (2011). Nucleophilic substitution reactions of diethyl 4-nitrophenyl phosphate triester: Kinetics and mechanism. Int. J. Chem. Kinet. 43, 708–714. doi:10.1002/kin.20605
Catalán, J., Diaz, C., and Garcia-Blanco, F. (2001). Characterization of binary solvent mixtures of DMSO with water and other cosolvents. J. Org. Chem. 66, 5846–5852. doi:10.1021/jo010415i
Catalán, J. (2009). Toward a generalized treatment of the solvent effect based on four empirical scales: Dipolarity (SdP, a new scale), polarizability (SP), acidity (SA), and basicity (SB) of the medium. J. Phys. Chem. B 119, 5951–5960. doi:10.1021/jp8095727
Catrina, I. E., and Hengge, A. C. (1999). Comparisons of phosphorothioate and phosphate monoester transfer reactions: Activation parameters, solvent effects and the effect of metal ions. J. Am. Chem. Soc. 121, 2156–2163. doi:10.1021/ja983862x
Cevasco, G., and Thea, S. (1993). The quest for carbanion-promoted dissociative pathways in the hydrolysis of aryl phosphinates. J. Chem. Soc., Perkin Trans. 2, 1103–1106.
Cheng, H., Nikolic-Hughes, I., Wang, J. H., Deng, H., O’Brien, P. J., Wu, L., et al. (2002). Environmental effects on phosphoryl group bonding probed by vibrational spectroscopy: Implications for understanding phosphoryl transfer and enzymatic catalysis. J. Am. Chem. Soc. 124, 11295–11306.
Chin, J., Banaszczyk, M., Jubian, V., and Zou, X. (1989). Cobalt(III) complex-promoted hydrolysis of phosphate diesters: Comparison in reactivity of rigid cis-diaquo(tetraaza)cobalt(III) complexes. J. Am. Chem. Soc. 111, 186–190. doi:10.1021/ja00183a029
Chlebowski, J. F., and Coleman, J. E. (1974). Mechanisms of hydrolysis of O-phosphorothioates and inorganic thiophosphate by Escherichia coli alkaline phosphatase. J. Biol. Chem. 249, 7192–7202. doi:10.1016/s0021-9258(19)42092-9
Cleland, W. W., and Hengge, A. C. (2006). Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 106, 3252–3278. doi:10.1021/cr050287o
Cleland, W. W., and Hengge, A. C. (1995). Mechanisms of phosphoryl and acyl transfer. FASEB J. 9, 1585–1594. doi:10.1096/fasebj.9.15.8529838
Cook, R. D., Daouk, W. A., Haji, A. N., Kabbani, A., Kurku, A., Samaha, M., et al. (1986a). The aminolysis of phosphinates: The kinetics and mechanism of the aminolysis of phosphinate esters in acetonitrile. Can. J. Chem. 64, 213–219. doi:10.1139/v86-037
Cook, R. D., Farah, S., Gwahi, L., Itani, A., and Rahil, J. (1986b). The influence of the changing of P=O to P=S and P-O-R to P-S-R on the reactivity of phosphinate esters under alkaline hydrolysis conditions. Can. J. Chem. 64, 1630–1637. doi:10.1139/v86-269
Cook, R. D., and Rahhal-Arabi, L. (1985). The kinetics of the alkaline hydrolysis of aryl diphenyl phosphinothioates; the significance for the mechanism of displacement at phosphorus. Tetrahedron Lett. 26, 3147–3150. doi:10.1016/s0040-4039(00)98642-8
Cullis, P. M., and Iagrossi, A. (1986). Thiophosphoryl transfer reactions: Stereochemical course of solvolysis of p-nitrophenyl thiophosphate in protic solvent and the possible role of thiometaphosphate. J. Am. Chem. Soc. 108, 7870–7871. doi:10.1021/ja00284a083
Cullis, P. M., Misra, R., and Wilkins, D. J. (1987). Free monomeric thiometaphosphate in protic solvents: Complete racemisation at phosphorus in the ethanolysis of 4-nitrophenyl thiophosphate. J. Chem. Soc. Chem. Commun., 1594–1596. doi:10.1039/c39870001594
Das, A. K., and Tembe, B. L. (1999). Molecular dynamics simulations of sodium chloride solutions in water-dimethyl sulphoxide mixtures: Potentials of mean force and solvation structures. J. Chem. Phys. 111, 7526–7536. doi:10.1063/1.480079
de Souza, E. F., and Ionescu, L. G. (1999). Micellar-catalyzed reactions of a phosphate ester: Effect of the dielectric constant of the medium. Colloids Surf. A 149, 609–615. doi:10.1016/s0927-7757(98)00670-0
Dey, N. K., Kim, C. K., and Lee, H. W. (2011). Kinetics and mechanism of the anilinolyses of aryl dimethyl, methyl phenyl and diphenyl phosphinates. Org. Biomol. Chem. 9, 717–724. doi:10.1039/c0ob00517g
Di Sabato, G., and Jencks, W. P. (1961). Mechanism and catalysis of reactions of acyl phosphates. II. Hydrolysis. J. Am. Chem. Soc. 83, 4400–4405. doi:10.1021/ja01482a025
Dimroth, K., Reichardt, C., Siepmann, T., and Bolhmann, F. (1963). Über Pyridinium-N-phenol-betaine und ihre verwendung zur charakterisierung der polarität von lösungsmitteln. Justus Liebigs Ann. Chem. 661, 1–37. doi:10.1002/jlac.19636610102
Domanico, P., Mizrahi, V., and Benkovic, S. J. (1986). “Observations on the chemistry of phosphorothioate transfer,” in Mechanisms of enzymatic reactions: Stereochemistry. Editor P. A. Frey (New York: Elsevier), 127–137.
Douglas, K. T., and Williams, A. (1976). Nucleophilic versus general base catalysis in phosphyl (PV) group transfer: Application to α-chymotrypsin action. J. Chem. Soc. Perkin Trans. 2, 515–521. doi:10.1039/p29760000515
Dunn, E. J., and Buncel, E. (1989). Metal ion catalysis in nucleophilic displacement reactions at carbon, phosphorus and sulphur centres. I. Catalysis by metal ions in the reaction of p-nitrophenyl diphenylphosphinate with ethoxide. Can. J. Chem. 67, 1440–1448. doi:10.1139/v89-220
Dunn, E. J., Moir, R. Y., Buncel, E., Purdon, J. G., and Bannard, R. A. B. (1990). Metal ion catalysis in nucleophilic displacement reactions at carbon, phosphorus, and sulfur centers. II. Metal ion catalysis in the reaction of p-nitrophenyl diphenylphosphinate with alkali metal phenoxides in ethanol. Can. J. Chem. 68, 1837–1845. doi:10.1139/v90-286
Eliseeva, G. D., Istomin, B. I., and Kalabina, A. V. (1978). Kinetics of the alkaline hydrolysis of O-(m- and p-substituted phenyl) diphenylphosphinothioates. J. Gen. Chem. U.S.S.R. 48, 1735.
Eliseeva, G. D., Istomin, B. I., and Kalabina, A. V. (1979). Kinetics of the alkaline hydrolysis of p-substituted O-phenyl dimethylphosphinothioates. J. Gen. Chem. U.S.S.R. 49, 1684–1685.
Epley, T. D., and Drago, R. S. (1967). Calorimetric studies on some hydrogen-bonded adducts. J. Am. Chem. Soc. 89, 5770–5773. doi:10.1021/ja00999a006
Forconi, M. (2015). Medium effects in biologically relevant catalysis. Adv. Phys. Org. Chem. 49, 57–101.
Friedman, J. M., Freeman, S., and Knowles, J. R. (1988). The quest for free metaphoshate in solution: Racemization at phosphorus in the transfer of the phospho group from aryl phosphate monoesters to tert-butyl alcohol in acetonitrile or in tert-butyl alcohol. J. Am. Chem. Soc. 110, 1268–1275. doi:10.1021/ja00212a040
Gomez-Tagle, P., Vargas-Zúñiga, I., Taran, O., and Yatsimirski, A. K. (2006). Solvent effects and alkali metal ion catalysis in phosphodiester hydrolysis. J. Org. Chem. 71, 9713–9722. doi:10.1021/jo061780i
Gorenstein, D. G., Lee, Y.-G., and Kar, D. (1977). Kinetic isotope effects in the reactions of aryl-18O-2,4-dinitrophenyl dibenzyl phosphate and aryl-18O-2,4-dinitrophenyl phosphate. Evidence for monomeric metaphosphate. J. Am. Chem. Soc. 99, 2264–2267. doi:10.1021/ja00449a039
Gregory, A. P., and Clarke, R. N. (2005). Traceable measurements of the static permittivity of dielectric reference liquids over the temperature range of 5-50oC. Meas. Sci. Technol. 16, 1506–1516. doi:10.1088/0957-0233/16/7/013
Grzyska, P. K., Czyryca, P. G., Golightly, J., Small, K., Larsen, P., Hoff, R. H., et al. (2002). Generality of solvation effects on the hydrolysis rates of phosphate monoesters and their possible relevance to enzymatic catalysis. J. Org. Chem. 67, 1214–1220. doi:10.1021/jo016104p
Haake, P., Diebert, C. E., and Marmor, R. S. (1968). Phosphinic acids and derivatives. 5. Displacement at phosphorus studied through alkaline hydrolysis of alkyl diphenylphosphinates. Effect of orientation of the phenyl rings on rates of displacement. Tetrahedron Lett. 9, 5247–5250. doi:10.1016/s0040-4039(00)89833-0
Hamlin, T. A., van Beek, B., Wolters, L. P., and Bickelhaupt, F. M. (2018). Nucleophilic substitution in solution: Activation strain analysis of weak and strong solvent effects. Chem. Eur. J. 24, 5927–5938. doi:10.1002/chem.201706075
Harnett, S. P., and Lowe, G. (1987) Stereochemical evidence for monomeric thiometaphosphate as an intermediate in the hydrolysis of (Rp) and (Sp)-deoxyadenosine 5’-[β-17O]-β-thiodiphosphate. J. Chem. Soc. Chem. Commun. 1416–1418. doi:10.1039/c39870001416
Harris, J. M., Shafer, S. G., Moffat, J. R., and Becker, A. R. (1979). Prediction of SN2 transition state variation by the use of More O’Ferrall plots. J. Am. Chem. Soc. 101, 3295–3300. doi:10.1021/ja00506a026
Henchman, M., Viggiano, A. A., Paulson, J. F., Freedman, A., and Wormhoudt, J. (1985) Thermodynamic and kinetic properties of the metaphosphate anion, PO3-, in the gas phase. J. Am. Chem. Soc. 107, 1453–1455. doi:10.1021/ja00291a081
Hengge, A. C., and Cleland, W. W. (1991). Phosphoryl transfer reactions of phosphodiester: characterization of transition states by heavy-atom isotope effects. J. Am. Chem. Soc. 113, 5835–5841.
Hengge, A. C., Edens, W. A., and Elsing, (1994). H. Transition state structures for phosphoryl-transfer reactions of p-nitrophenyl phosphate. J. Am. Chem. Soc. 116, 5045–5049. doi:10.1021/ja00091a003
Hengge, A. C., and Onyido, I. (2005). Physical organic perspectives on phospho group transfer from phosphates and phosphinates. Curr. Org. Chem. 9, 61–74. doi:10.2174/1385272053369349
Herschlag, D., and Jencks, W. P. (1989a). Evidence that metaphosphate monoanion is not an intermediate in solvolysis reactions in aqueous solution. J. Am. Chem. Soc. 111, 7579–7586. doi:10.1021/ja00201a047
Herschlag, D., and Jencks, W. P. (1989b). Phosphoryl transfer to anionic oxygen nucleophiles. Nature of the transition state and electrostatic repulsion. J. Am. Chem. Soc. 111 (111), 7587–7596. doi:10.1021/ja00201a048
Herschlag, D., and Jencks, W. P. (1987). The effect of divalent metal ions on the rate and transition-state structure of phosphoryl-transfer reactions. J. Am. Chem. Soc. 109, 4665–4674. doi:10.1021/ja00249a033
Hoff, R. H., and Hengge, A. C. (1998). Entropy and enthalpy contributions to solvent effects on phosphate monoester solvolysis. The importance of entropy effects in the dissociative transition state. J. Org. Chem. 63, 6680–6688. doi:10.1021/jo981160k
Hollfelder, F., and Herschlag, D. (1995). The nature of the transition state for enzyme-catalyzed phosphoryl transfer. Hydrolysis of O-aryl phosphorothioates by alkaline phosphatase. Biochemistry 34, 12255–12264. doi:10.1021/bi00038a021
Hughes, E. D., and Ingold, C. K. (1935). 55. Mechanism of substitution at a saturated carbon atom. Part IV. A discussion of constitutional and solvent effects on the mechanism, kinetics, velocity, and orientation of substitution. J. Chem. Soc., 244–255. doi:10.1039/jr9350000244
Humphry, T., Forconi, M., Williams, N. H., and Hengge, A. C. (2004). Altered mechanisms of reactions of phosphate esters bridging a dinuclear metal center. J. Am. Chem. Soc. 126, 11864–11869. doi:10.1021/ja047110g
Ingold, C. K. (1969). Structure and mechanism in organic chemistry. Ithaca, N.Y.: Cornell University Press.
Istomin, B. I., and Eliseeva, G. D. (1981). Effect of structural factors, temperature, and medium on the kinetics of the alkaline hydrolysis of phenyl esters of thionephosphinic acids in aqueous ethanol. J. Gen. Chem. U.S.S.R. 51, 2063–2075.
Istomin, B. I., Sukhorukova, N. A., Kalabina, A. V., and Sukhorkov, I. (1982). Kinetics of the bimolecular alkaline cleavage of m- and p-substituted phenyl diphenylphosphinates in water and water-ethanol mixtures. J. Gen. Chem. U.S.S.R. 52, 1787–1795.
Jencks, D. A., and Jencks, W. P. (1977). The characterization of transition states by structure-reactivity coefficients. J. Am. Chem. Soc. 99, 7948–7960. doi:10.1021/ja00466a030
Jencks, W. P. (1985). A primer for the Bema Hapothle. An empirical approach to the characterization of changing transition state structures. Chem. Rev. 85, 511–527. doi:10.1021/cr00070a001
Jencks, W. P. (1972). General acid-base catalysis of complex reactions in water. Chem. Rev. 72, 705–718. doi:10.1021/cr60280a004
Jencks, W. P. (1981). How does a reaction choose its mechanism? Chem. Soc. Rev. 10, 345–375. doi:10.1039/cs9811000345
Jencks, W. P., and Regenstein, J. (1970). “Ionization constants of acids and bases,” in Handbook of biochemistry. Editor H. A. Sober 2nd Edn. (Cleveland, OH: The Chemical Rubber Co.). Section J-187.
Jencks, W. P. (1980). When is an intermediate not an intermediate? Enforced mechanisms of general acid-base, catalysed, carbocation, carbanion, and ligand exchange reaction. Acc. Chem. Res. 13, 161–169. doi:10.1021/ar50150a001
Johnson, M. K., and Henschler, D. (1975). The delayed neuropathy caused by some organophosphorus esters: Mechanism and challenge. CRC Crit. Rev. Toxicol. 3, 289–316. doi:10.3109/10408447509079861
Kalu, G. I., Ubochi, C. I., and Onyido, I. (2022). Mapping transition state structures for thiophosphinoyl group transfer between oxyanionic nucleophiles in water and aqueous ethanol solvents. New. J. Chem. 46, 12981–12993. doi:10.1039/d2nj02008d
Kalu, G. I., Ubochi, C. I., and Onyido, I. (2021). Reactions of aryl dimethylphosphinothioate esters with anionic oxygen nucleophiles: Transition state structure in 70% water-30% ethanol. RSC Adv. 11, 8833–8845. doi:10.1039/d0ra10759j
Kamerlin, S. C., Florián, J., and Warshel, A. (2008a). Associative versus dissociative mechanisms of phosphate monoester hydrolysis: On the interpretation of activation entropies. ChemPhysChem 9, 1767–1773. doi:10.1002/cphc.200800356
Kamerlin, S. C., Williams, N. H., and Warshel, A. (2008b). Dineopentyl phosphate hydrolysis: Evidence for stepwise water attack. J. Org. Chem. 73, 6960–6969. doi:10.1021/jo801207q
Kamlet, M. J., and Taft, R. A. (1976b). The solvatochromic comparison method. 2. The.alpha.-scale of solvent hydrogen-bond donor (HBD) acidities. J. Am. Chem. Soc. 98, 2886–2894. doi:10.1021/ja00426a036
Kamlet, M. J., and Taft, R. A. (1976a). The solvatochromic comparison method. I. The.beta.-scale of solvent hydrogen-bond acceptor (HBA) basicities. J. Am. Chem. Soc. 98, 377–383. doi:10.1021/ja00418a009
Kelly, C. P., Cramer, C. J., and Truhlar, D. G. (2007). Single-ion solvation free energies and the normal hydrogen electrode potential in methanol, acetonitrile, and dimethyl sulfoxide. J. Phys. Chem. B 111, 408–422. doi:10.1021/jp065403l
Ketelaar, J. A., Gresmann, H. R., and Koopmans, K. (1952). The rate of hydrolysis of some p-nitrophenol esters of ortho-phosphoric and thio-phosphoric acid. Rec. Trav. Chim. Pays-Bas. 71, 1253–1258. doi:10.1002/recl.19520711214
Kim, H. S., Chung, T. D., and Kim, H. (2001). Voltametric determination of the pKa of various acids in polar aprotic solvents using 1,4-benzoquinone. J. Electroanal. Chem. 498, 209–215. doi:10.1016/s0022-0728(00)00413-7
Kim, K., Tsay, O. G., Atwood, D. A., and Churchill, D. G. (2011). Destruction and detection of chemical warfare agents. Chem. Rev. 111, 5345–5403. doi:10.1021/cr100193y
Kirby, A. J., and Jencks, W. P. (1965b). Base catalysis of the reaction of secondary amines with p-nitrophenyl phosphate: Kinetic evidence for an addition intermediate in nucleophilic aromatic substitution. J. Am. Chem. Soc. 87, 3217–3224. doi:10.1021/ja01092a037
Kirby, A. J., and Jencks, W. P. (1965a). The reactivity of nucleophilic reagents toward the p-nitrophenyl phosphate dianion. J. Am. Chem. Soc. 87, 3209–3216. doi:10.1021/ja01092a036
Kirby, A. J., Medeiros, M., Oliveira, P. S. M., Orth, E. S., Brandão, T. A. S., Wanderlind, E. H., et al. (2011). Activating water: Important effects of non-leaving groups on the hydrolysis of phosphate triesters. Chem. Eur. J. 17, 14996–15004. doi:10.1002/chem.201101926
Kirby, A. J., Mora, J. R., and Nome, F. (2013). New light on phosphate transfer from triesters. Biochim. Biophys. Acta 1834, 454–463. doi:10.1016/j.bbapap.2012.04.010
Kirby, A. J., and Nome, F. (2015). Fundamentals of phosphate transfer. Acc. Chem. Res. 48, 1806–1814. doi:10.1021/acs.accounts.5b00072
Kirby, A. J., Souza, B. S., and Nome, F. (2015). Structure and reactivity of phosphate diesters. Dependence on the non-leaving group. Can. J. Chem. 93, 422–427. doi:10.1139/cjc-2014-0358
Kirby, A. J., and Varvoglis, A. G. (1968). The reactivity of phosphate esters: Reactions of monoesters with nucleophiles. Nucleophilicity independent of basicity in a bimolecular substitution reaction. J. Chem. Soc. B 135-141, 135. doi:10.1039/j29680000135
Kirby, A. J., and Varvoglis, A. G. (1967). The reactivity of phosphate esters. Monoester hydrolysis. J. Am. Chem. Soc. 89, 415–423. doi:10.1021/ja00978a044
Kirby, A. J., and Younas, M. (1970a). The reactivity of phosphate esters. Diester hydrolysis. J. Chem. Soc. B, 510–513. doi:10.1039/j29700000510
Kirby, A. J., and Younas, M. (1970b). The reactivity of phosphate esters. Reactions of diesters with nucleophiles. J. Chem. Soc. B, 1165–1172. doi:10.1039/j29700001165
Klӓhn, M., Rosta, E., and Warshel, A. (2006). On the mechanism of hydrolysis of phosphate monoesters dianions in solutions and proteins. J. Am. Chem. Soc. 128, 15310–15323. doi:10.1021/ja065470t
Kluger, R., and Taylor, S. D. (1990). On the origins of enhanced reactivity of five-membered cyclic phosphate esters. The relative contributions of enthalpic and entropic factors. J. Am. Chem. Soc. 112, 6669–6671. doi:10.1021/ja00174a033
Knowles, J. R. (1980). Enzyme-catalyzed phosphoryl transfer reactions. Annu. Rev. Biochem. 49, 877–919. doi:10.1146/annurev.bi.49.070180.004305
Kolthoff, I. M., Chantooni, M. K., and Bhowmik, S. (1968). Dissociation constants of uncharged and monovalent cation acids in dimethyl sulfoxide. J. Am. Chem. Soc. 90, 23–28. doi:10.1021/ja01003a005
Kosower, E. M. (1958a) Effect of solvent on spectra. I. A new empirical measure of solvent polarity: Z values. J. Am. Chem. Soc. 80, 3253–3260. doi:10.1021/ja01546a020
Kosower, E. M. (1958b). Effect of solvent on spectra. II. Correlation of spectral absorption data with Z values. J. Am. Chem. Soc. 80, 3261–3267. doi:10.1021/ja01546a021
Kosower, E. M. (1958c). Effect of solvent on spectra. III. The use of Z values in connection with kinetic data. J. Am. Chem. Soc. 80, 3267–3270. doi:10.1021/ja01546a022
Lad, C., Williams, N. H., and Wolfenden, R. (2003). The rate of hydrolysis of phosphomonoester dianions and the exceptional catalytic proficiencies of protein and inositol phosphatases. Proc. Natl. Acad. Sci. U. S. A. 100, 5607–5610. doi:10.1073/pnas.0631607100
Laidler, K. J., and Eyring, H. (1939). The effect of solvents on reaction rates. Ann. N.Y. Acad. Sci. 39, 303–340. doi:10.1111/j.1749-6632.1939.tb55380.x
Laidler, K. J., and Landskroener, P. A. (1956). The influence of the solvent on reaction rates. Trans. Faraday Soc. 52, 200–210. doi:10.1039/tf9565200200
Lassila, J. K., Zalatan, J. G., and Herschlag, D. (2011). Biological phosphoryl transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem. 80, 669–702. doi:10.1146/annurev-biochem-060409-092741
Leiros, I., McSweeney, S., and Hough, E. (2004). The reaction mechanism of phospholipase D from Streptomyces sp. strain PMF. Snapshots along the reaction pathway reveal a pentacoordinate reaction intermediate and an unexpected final product. J. Mol. Biol. 339, 805–820.
Livieri, M., Mancin, F., Saielli, G., Chin, J., and Tonellato, U. (2007). Mimicking enzymes: Cooperation between organic functional groups and metal ions in the cleavage of phosphate diesters. Chem. Eur. J. 13, 2246–2256. doi:10.1002/chem.200600672
Luzar, A., and Stefan, J. (1990). Dielectric behaviour of DMSO-water mixtures. A hydrogen-bonding model. J. Mol. Liq. 46, 221–238. doi:10.1016/0167-7322(90)80056-p
Mackay, R. A., Longo, F. R., Knier, B. L., and Durst, H. D. (1987). Iodosobenzoate catalyzed hydrolysis of 4-nitrophenyl diphenyl phosphate in a CTAB microemulsion. J. Phys. Chem. 91, 861–864. doi:10.1021/j100288a020
Mancin, F., and Tecilla, P. (2007). Zinc(ii) complexes as hydrolytic catalysts of phosphate diester cleavage: From model substrates to nucleic acids. New J. Chem. 31, 800–817. doi:10.1039/b703556j
Marklund, A., Andersson, B., and Haglund, P. (2005). Organophosphorus flame retardants and plasticizers in Swedish sewage treatment plants. Environ. Sci. Technol. 39, 7423–7429. doi:10.1021/es051013l
Maryott, A. A., and Smith, E. R. (1951). Table of dielectric constants of pure liquids, 514. U.S. Department of Commerce. National Bureau of Standards Circular.
Medeiros, M., Manfredi, A. M., Kirby, A. J., and Nome, F. (2013). The spontaneous hydrolysis of 2-pyridyl phosphate is a good model for the special mechanism for the hydrolysis of phosphate monoester monoanions. J. Phys. Org. Chem. 26, 1044–1047. doi:10.1002/poc.3165
Meyerson, S., Harvan, D. J., Hass, J. R., Ramirez, F., and Maracek, J. (1984). The monomeric metaphosphate anion in negative-ion chemical-ionization mass spectra of phosphotriesters. J. Am. Chem. Soc. 106, 6877–6883. doi:10.1021/ja00335a001
Moelwyn-Hughes, E. A. (1936a). A theory of electro-kinetic effects in solution: Reactions between ions. Proc. R. Soc. A 155, 308–315.
Moelwyn-Hughes, E. A. (1936b). A theory of electro-kinetic effects in solution: Reactions between ions and polar molecules. Proc. R. Soc. A 157, 667–679.
Mora, J. R., Kirby, A. J., and Nome, F. (2012). Theoretical study of the importance of the spectator groups on the hydrolysis of phosphate triesters. J. Org. Chem. 77, 7061–7070. doi:10.1021/jo301380v
More O’Ferrall, R. A. (1970). Relationships between E2 and E1cB mechanisms of β-elimination. J. Chem. Soc. B 0, 274–277. doi:10.1039/j29700000274
Neimysheva, A. A., Savchik, V. I., Ermolaeva, M. V., and Knunyants, I. L. (1968). Change in the reactivity and spectral characteristics from phosphoryl to thionephosphoryl compounds. Bull. Acad. Sci. USSR. Div. Chem. Sci. Engl. Transl. 17, 2104–2108. doi:10.1007/bf00904023
Nikolic-Hughes, I., Rees, D. C., and Herschlag, D. (2004). Do electrostatic interactions with positively charged active site groups tighten the transition state for enzymatic phosphoryl transfer? J. Am. Chem. Soc. 126, 11814–11819. doi:10.1021/ja0480421
O’Brien, P. J., and Herschlag, D. (2002). Alkaline phosphatase revisited: Hydrolysis of alkyl phosphates. Biochemistry 41, 3207–3225. doi:10.1021/bi012166y
Omakor, J. E., Onyido, I., vanLoon, G. W., and Buncel, E. (2001). Mechanisms of abiotic degradation and soil-water interactions of pesticides and other hydrophobic organic compounds. Part 3. Nucleophilic displacement at the phosphorus centre of the pesticide fenitrothion [O,O-dimethyl O-(3-methyl-4-nitrophenyl) phosphorothioate] by oxygen nucleophiles in aqueous solution: α-Effect and mechanisms. J. Chem. Soc. Perkin Trans. 2, 324–330. doi:10.1039/b008615k
Onyido, I., Albright, K., and Buncel, E. (2005a). Catalysis of the ethanolysis of aryl methyl phenyl phosphinate esters by alkali metal ions: Transition state structures for uncatalyzed and metal ion-catalyzed reactions. Org. Biomol. Chem. 3, 1468–1475. doi:10.1039/b501537e
Onyido, I. (2018). Catalysis, structure and reactivity in chemistry and the molecular life Sciences. Ibadan, Nigeria: University Press Plc.
Onyido, I., Swierczek, K., Purcell, J., and Hengge, A. C. (2005b). A concerted mechanism for the transfer of the thiophosphinoyl group from aryl dimethylphosphinothioate esters to oxyanionic nucleophiles in aqueous solution. J. Am. Chem. Soc. 127, 7703–7711. doi:10.1021/ja0501565
Orth, E. S., Wanderlind, E. H., Medeiros, M., Oliveira, P. S. M., Vaz, B. G., Eberlin, M. N., et al. (2011). Phosphorylimidazole derivatives: Potentially biosignaling molecules. J. Org. Chem. 76, 8003–8008. doi:10.1021/jo2017394
Parker, A. J., Mayer, U., Schmid, R., and Gutman, V. (1978). Correlation of solvent effects on rates of solvolysis and SN2 reactions. J. Org. Chem. 43, 1843–1854. doi:10.1021/jo00404a001
Parker, A. J. (1969). Protic-dipolar aprotic solvent effects on rates of bimolecular reactions. Chem. Rev. 69, 1–32. doi:10.1021/cr60257a001
Parker, A. J. (1962). The effects of solvation on the properties of anions in dipolar aprotic solvents. Quart. Rev. 16, 163–187. doi:10.1039/qr9621600163
Pregel, M. J., Dunn, E. J., Nagelkerke, R., Thatcher, G. R. J., and Buncel, B. (1995). Alkali–metal lon catalysis and inhibition in nucleophilic displacement reaction of phosphorus–sulfur–and carbon–based esters. Chem. Soc. Rev. 24, 449–455. doi:10.1039/cs9952400449
Ramirez, F., and Marecek, J. F. (1979). Amine catalysis in phosphoryl transfer from 2,4-dinitrophenyl phosphate in aprotic and protic solvents. Tetrahedron 35, 1581–1589. doi:10.1016/0040-4020(79)80019-8
Ramirez, F., and Marecek, J. F. (1980). Phosphoryl transfer from phosphomonoesters in aprotic and protic solvents. Pure Appl. Chem. 52, 1021–1045. doi:10.1351/pac198052041021
Ramirez, F., Marecek, J., Minore, J., Srivastava, S., and le Noble, W. J. (1986). On the freeness of the metaphosphate anion in aqueous solution. J. Am. Chem. Soc. 108, 348–349. doi:10.1021/ja00262a056
Reichardt, C. (1994). Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 94, 2319–2358. doi:10.1021/cr00032a005
Reichardt, C. (1982). Solvent effects on chemical reactivity. Pure Appl. Chem. 54, 1867–1884. doi:10.1351/pac198254101867
Reichardt, C. (2007). Solvents and solvent effects: An introduction. Org. Process Res. Dev. 11, 105–113. doi:10.1021/op0680082
Reichardt, C., and Welton, T. (2011). Solvents and solvent effects in organic chemistry. Weinheim: Wiley-VCH Verlag GmbH & Co.
Richard, J. P., Amyes, T. L., Goryanova, B., and Zhai, X. (2014). Enzyme architecture: On the importance of being in a protein cage. Curr. Opin. Chem. Biol. 21, 1–10. doi:10.1016/j.cbpa.2014.03.001
Ritchie, C. D., Skinner, G. A., and Badding, V. G. (1967). Solvent effects on the reactions of stabilized carbonium ions with nucleophiles. J. Am. Chem. Soc. 89, 2063–2071. doi:10.1021/ja00985a017
Roesky, H. W., Ahlrichs, R., and Brode, S. (1986). Trithiometaphosphate PS3- – An anion with phosphorus of coordination number 3. Angew. Chem. Int. Ed. Engl. 25, 82–83. doi:10.1002/anie.198600821
Schroeder, G. K., Lad, C., Wyman, P., Williams, N. H., and Wolfenden, R. (2006). The time required for water attack at the phosphorus atom of simple phosphodiesters and of DNA. Proc. Natl. Acad. Sci. U. S. A. 103, 4052–4055. doi:10.1073/pnas.0510879103
Schwartz, P. A., and Murray, B. W. (2011). Protein kinase biochemistry and drug discovery. Bioorg. Chem. 39, 192–210. doi:10.1016/j.bioorg.2011.07.004
Simonsen, D. R., and Washburn, E. R. (1946). The ternary system: t-Butyl alcohol, benzene and water at 25o. J. Am. Chem. Soc. 68, 235–237. doi:10.1021/ja01206a025
Sorensen-Stowell, K., and Hengge, A. C. (2006). Thermodynamic origin of the increased rate of hydrolysis of phosphate and phosphorothioate esters in DMSO/water mixtures. J. Org. Chem. 71, 7180–7184. doi:10.1021/jo060896b
Stockbridge, R. B., and Wolfenden, R. (2009). Phosphate monoester hydrolysis in cyclohexane. J. Am. Chem. Soc. 131, 18248–18249. doi:10.1021/ja907967y
Stockbridge, R. B., and Wolfenden, R. (2010). The hydrolysis of phosphate diesters in cyclohexane and acetone. Chem. Commun. 46, 4306–4308. doi:10.1039/c0cc00229a
Sung, N.-D. (1994). Hydrolysis of phosphate diesters as nucleic acid model. Agric. Chem. Biotechnol. 37, 447–450.
Taft, R. W., Abboud, J. L. M., Kamlet, M. J., and Abraham, M. H. (1985). Linear solvation energy relations. J. Sol. Chem. 14, 153–186. doi:10.1007/bf00647061
Taira, K., Fanni, T., and Gorenstein, D. G. (1984). Stereoelectronic effects in the hydrolysis of ethyl and methyl ethylene phosphates. J. Org. Chem. 49, 4531–4536. doi:10.1021/jo00197a041
Tarrat, N. (2010). Alkaline hydrolysis of phosphate triesters in solution: Stepwise or concerted? A theoretical study. J. Mol. Struct. Theochem. 941, 56–60. doi:10.1016/j.theochem.2009.11.001
Teichmann, H., and Hilgetag, G. (1967). Nucleophilic reactivity of the thiophosphoryl group. Angew. Chem. Int. Ed. Engl. 6, 1013–1023. doi:10.1002/anie.196710131
Tirel, E. Y., Bellamy, Z., Adams, H., Lebrun, V., Duarte, F., and Williams, N. H. (2014). Catalytic zinc complexes for phosphate diester hydrolysis. Angew. Chem. Int. Ed. 53, 8246–8250. doi:10.1002/anie.201400335
Toy, A. D. F., and Walsh, E. N. (1987). Phosphorus in Everyday living. Washington, DC: American Chemical Society.
Turan, H. T., Brickel, S., and Meuwly, S. (2022). Solvent effects on the Menshutkin reaction. J. Phys. Chem. B 126, 1951–1961. doi:10.1021/acs.jpcb.1c09710
Um, I.-H., Hong, Y.-J., and Kwon, D.-S. (1997). Acid dissociation constants of phenols and reaction mechanism for the reactions of substituted phenyl benzoates with phenoxide anions in absolute ethanol. Tetrahedron 53, 5073–5082. doi:10.1016/s0040-4020(97)00227-5
Um, I. H., Akhtar, K., Shin, Y. H., and Han, J. Y. (2007). Aminolyses of aryl diphenylphosphinates and diphenylphosphinoates: Effect of modification of electrophilic centre from P=O to P=S. J. Org. Chem. 72, 3823–3829. doi:10.1021/jo070171n
Um, I. H., Han, J. Y., and Shin, Y. H. (2009). Aminolysis of X-substituted phenyl diphenylphosphinates: Effect of amine nature on reactivity and transition-state structure. J. Org. Chem. 74, 3073–3078. doi:10.1021/jo900219t
Um, I. H., Shin, Y. H., Han, J. Y., and Mishima, M. (2006). Aminolysis of Y-substituted phenyl diphenylphosphinates and benzoates: Effect of modification of the electrophilic centre from C=O to P=O. J. Org. Chem. 71, 7715–7720. doi:10.1021/jo061308x
Varghese, J. J., and Mushrif, S. H. (2019). Origins of complex solvent effects on chemical reactivity and computational tools to investigate them: A review. React. Chem. Eng. 4, 165–206. doi:10.1039/c8re00226f
Waghorne, W. E. (1993). Thermodynamics of solvation in mixed solvents. Chem. Soc. Rev. 22, 285–292. doi:10.1039/cs9932200285
Wan, W., Huang, H., Lv, J., Han, R., and Zhang, S. (2017). Uptake, translocation, and biotransformation of organophosphorus esters in wheat (Triticum aestivum L.). Environ. Sci. Technol. 51, 13649–13658. doi:10.1021/acs.est.7b01758
Warshel, A., Sharma, P. K., Kato, M., Xiang, Y., Liu, H., and Olsson, M. H. M. (2006). Electrostatic basis for enzyme catalysis. Chem. Rev. 106, 3210–3235. doi:10.1021/cr0503106
Westheimer, F. H. (1968). Pseudo-rotation in the hydrolysis of phosphate esters. Acc. Chem. Res. 1, 70–78. doi:10.1021/ar50003a002
Westheimer, F. H. (1987). Why nature chose phosphates. Science 235, 1173–1178. doi:10.1126/science.2434996
Williams, A. (2003). Free energy relationships in organic and biorganic chemistry. Cambridge, England: Royal Society of Chemistry.
Williams, A., and Naylor, R. A. (1976). Hydrolysis of phosphinic esters: General base catalysis by imidazole. J. Chem. Soc. B, 1967–1972. doi:10.1039/j29710001967
Zalatan, J. G., and Herschlag, D. (2006). Alkaline phosphatase mono- and diesterase reactions: Comparative transition state analysis. J. Am. Chem. Soc. 128, 1293–1303. doi:10.1021/ja056528r
Zhang, Y. L., Hollfelder, F., Gordon, S. J., Chen, L., Keng, Y. F., Wu, L., et al. (1999). Impaired transition state complementarity in the hydrolysis of O-arylphosphorothioates by protein-tyrosine phosphatases. Biochemistry 38, 12111–12123. doi:10.1021/bi990836i
Keywords: phospho group transfer, mechanism, solvent effects, desolvation, transition state stabilization, rate enhancements, solvent polarity
Citation: Onyido I, Obumselu OF, Egwuatu CI and Okoye NH (2023) Solvent and solvation effects on reactivities and mechanisms of phospho group transfers from phosphate and phosphinate esters to nucleophiles. Front. Chem. 11:1176746. doi: 10.3389/fchem.2023.1176746
Received: 28 February 2023; Accepted: 03 April 2023;
Published: 27 April 2023.
Edited by:
Tieqiao Chen, Hainan University, ChinaReviewed by:
Akiya Ogawa, Osaka Prefecture University, JapanKatie R. Mitchell-Koch, Wichita State University, United States
Copyright © 2023 Onyido, Obumselu, Egwuatu and Okoye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ikenna Onyido, aWtlbm5hb255aWRvQHlhaG9vLmNvbQ==, aS5vbnlpZG9AdW5pemlrLmVkdS5uZw==