 Yuxiao Meng1,2
Yuxiao Meng1,2 Yongyong Cao
Yongyong Cao- 1State Key Laboratory Breeding Base of Green−Chemical Synthesis Technology, College of Chemical Engineering, Institute of Industrial Catalysis, Zhejiang University of Technology, Hangzhou, China
- 2College of Biological Chemical Science and Engineering, Jiaxing University, Jiaxing, Zhejiang, China
Excess of carbon dioxide (CO2) in the atmosphere poses a significant threat to the global climate. Therefore, the electrocatalytic carbon dioxide reduction reaction (CO2RR) is important to reduce the burden on the environment and provide possibilities for developing new energy sources. However, highly active and selective catalysts are needed to effectively catalyze product synthesis with high adhesion value. Single-atom catalysts (SACs) and double-atom catalysts (DACs) have attracted much attention in the field of electrocatalysis due to their high activity, strong selectivity, and high atomic utilization. This review summarized the research progress of electrocatalytic CO2RR related to different types of SACs and DACs. The emphasis was laid on the catalytic reaction mechanism of SACs and DACs using the theoretical calculation method. Furthermore, the influences of solvation and electrode potential were studied to simulate the real electrochemical environment to bridge the gap between experiments and computations. Finally, the current challenges and future development prospects were summarized and prospected for CO2RR to lay the foundation for the theoretical research of SACs and DACs in other aspects.
1 Introduction
Electrochemical technology shows significant promise for energy storage and addressing environmental issues. The massive consumption of fossil fuels has enormously increased carbon dioxide (CO2) in the atmosphere, accelerating the deterioration of the environment (Gao et al., 2016a; Cao et al., 2020a; Bai et al., 2022). Electrocatalytic carbon dioxide reduction reaction (CO2RR) directly converts CO2 into high-value chemicals, providing a favorable way to solve energy and environmental issues. However, the efficient activation of CO2 is a critical issue depending on the interaction between the catalyst surface and the CO2 molecules (Gong et al., 2019; Han et al., 2020a; He et al., 2020a). Therefore, various metal-based catalysts have been developed to accelerate the electrocatalytic CO2RR and achieve efficient CO2 conversion. Although bulk noble metal-based catalysts exhibit high catalytic activity, their large-scale production and practical applications are limited due to high cost and poor stability (Wu et al., 2016; Wang et al., 2019; Li et al., 2021a; Yang et al., 2022a). When the metal nanoparticles are reduced to nano clusters or even single atoms, the supported metal catalyst reaches an idealized state, which can greatly reduce the production cost of the catalyst and maximize the utilization rate of the precious metal (Ling et al., 2017; Lininger et al., 2021). Therefore, single-atom catalysts (SACs) have been used in a variety of reactions to promote the synthesis of high-value compounds and improve significant catalytic performance due to their unique electronic structure, high selectivity, homogeneous active sites and near 100% atomic utilization (Wang et al., 2020; Wan et al., 2022; Xu et al., 2022). However, the surface free energy of the SACs increases sharply with increased specific surface area, leading to easy agglomeration coupling and the formation of large clusters during their preparation and reaction. Therefore, double-atom catalysts (DACs) were proposed to make up for the deficiency of SACs. The increase of atomic load in the DACs results in multiple active central sites, and the synergistic effect, spacing enhancement effect and electron effect between diatomic sites help to regulate the electron distribution of the active sites, and effectively improve the catalytic performance (Song et al., 2020; Zhang et al., 2020; Hu et al., 2022). Therefore, SACs and DACs are increasingly popular in oxygen reduction reaction (ORR) (Cao et al., 2020b; Talib et al., 2021; Zhao and Liu, 2021; Chen et al., 2022), oxygen evolution reaction (OER) (Zhou et al., 2020a; Zeng et al., 2020; Xue et al., 2021), nitrogen reduction reaction (NRR) (Lv et al., 2021; Xu et al., 2021; Niu et al., 2022), hydrogen evolution reaction (HER) (Gao et al., 2016b; Zhang et al., 2017), and CO2RR due to their unique properties (Cao et al., 2022a; Chou et al., 2022; Sun et al., 2023).
Density functional theory (DFT) is a powerful tool to understand materials and catalytic reaction elementary steps and mechanisms at the atomic scale. Additionally, DFT can explore the electronic structure of the catalyst and directly identify the active sites compared with the experimental method (Li et al., 2020a; Lininger et al., 2021; Yang et al., 2022b). Furthermore, it reveals the intrinsic nature of the activated CO2 molecule to understand the CO2RR mechanism. The influence of solvation and ionic effects on CO2RR at the solid-liquid interface is relatively complex and relies on a first-principles approach to provide theoretical insights into its mechanism. Therefore, the theoretical computational methods provide fundamental guidance and predictions to rationalize catalyst models.
Various studies and reviews on the synthesis, characterization, and electrocatalytic applications of SACs and DACs have been published. This review discussed the unique internal performance of SACs and DACs and reviewed their catalytic mechanism on CO2RR from the perspective of theoretical calculations. This review explored the influence of solvent and surface charge on electrocatalytic CO2RR. Finally, the challenges and perspectives for future studies on CO2RR were also provided.
2 SACs
In 2011, Zhang et al. synthesized the Pt1/FeOx catalyst and first proposed the concept of SAC (Qiao et al., 2011). In the case of the supported catalysts, the catalytic activity of metal catalysts is closely related to their particle size. The specific surface area increases dramatically when the dispersion of nanoparticles reaches the single atom size, maximizing the utilization of metals and significantly reducing the cost of precious metal catalysts (Hossain et al., 2020; Pu et al., 2020). Multiple active sites are exposed on the catalyst surface with uniform contact between the active site and the support, significantly improving the activity and selectivity of the catalytic reaction (Lu et al., 2019). SACs often exhibit different activity and selectivity compared to conventional nano-catalysts due to the coordination number of central atoms and the electronegativity of neighboring atoms (Cepitis et al., 2021; Huang et al., 2023; Li et al., 2023). The strong interaction between the isolated atoms and the carrier can induce charge redistribution on the carrier surface, improving the reactional intrinsic activity and stability of the catalyst. SACs can modify the adsorption and desorption selectivity of the active components of the catalyst in different molecules, affecting the reaction kinetics.
2.1 Strong covalent metal–support interaction
Numerous efforts have been made to increase the surface area by reducing the size of metallic nanomaterials to atomic levels, adequately exposing many active sites, and improving the electrocatalytic performance (Konsolakis and Ioakeimidis, 2014; He et al., 2023). However, due to the extremely high surface energy of single metallic atoms, they are highly susceptible to migration and aggregation under synthetic and catalytic conditions. Therefore, the migration of single atoms is often confined using the strong interactions between the isolated metal atom and the support. Different properties of the support materials affect the coordination number, steric environment, and chemical bonding, resulting in SACs with different electronic and morphological structures (Yang et al., 2020). Most single metal atoms can be immobilized on the carbon material by coordination with nitrogen atoms (M-Nx), which maximizes the atomic utilization of the metal atoms (Figure 1). Moreover, the low coordination environment allows firm anchorage of metal atoms on the carrier with high stability and electrocatalytic activity (Tang et al., 2022). Cao et al. designed a series of M-N-C SACs with macrocycle cucurbit (He et al., 2020a) uril (CB (He et al., 2020a)) self-assembly as carbon precursors. The Fe-loaded N-doped holey carbon single-atom electrocatalyst (Fe-NHC) exhibited higher stability compared to SACs of cobalt (Co) or nickel (Ni) and showed higher activity for ORRs under alkaline conditions (Figure 1A) (Zhang et al., 2021a). Feng et al. synthesized carbon nanosheets embedded with isolated copper atoms coordinated with N(Cu-N-C-800). The catalysts exhibited excellent stability after 20 consecutive cycles and facilitated the conversion of nitrate to ammonia (NH3) and nitrogen (Zhu et al., 2020). MXene materials (two-dimensional) with a graphene-like structure can control their structure and properties by changing the ratio of M and X elements, regulating their electrical conductivity and carrier mobility, and the MXene materials have high mechanical stability. They improve the activity of SACs as catalyst carriers and exhibit strong catalytic reduction abilities (Zhang et al., 2021b). Wang et al. showed that single platinum (Pt) atoms occupied the Mo vacancies in MXene, and it were connected with the carbon (C) atoms to form three Pt-C bonds (Zhang et al., 2018). The strong covalent interactions between the positively charged Pt atoms and MXene improved the stability of the catalyst and the catalytic ability for HER with low overpotentials of 30 and 77 mV to achieve 10 and 100 mA cm−2 (Figure 1B). SACs based on oxide supports are also used in various reactions (Zhou et al., 2020c; Han et al., 2021). The defect sites and -OH groups on the surface of oxides provide the possibility of anchoring single atoms. The electron transfer makes the strong interaction between oxides and single atoms, which improves the mechanical and thermal stability of SACs. It was conducive to the application in electrocatalytic reactions (Han et al., 2020b; Lang et al., 2020) In Y2-xCoxRu2O7−δ, the introduction of Co atoms leads to a redistribution of charge between Ru and Co atoms, resulting in ultra-high OER activity. In addition, Co substitution also creates oxygen vacancy, which leads to the rapid transfer of OER charge. The strengthening of the bond hybridization between the d orbitals of Y and Ru and the 2p orbitals of O further enhances the chemical stability of the catalyst (Qin and Su, 2021). Murayama et al. reported a nickel oxide-supported single gold atom catalyst (Au1/NiO) for carbon monoxide (CO) oxidation reaction (Mochizuki et al., 2022). The adsorption energies of gold (Au) atoms dispersed on nickel oxide (NiO) surfaces and Au atoms dispersed on NiO surfaces with Ni vacancies were calculated using DFT. Subsequently, it was observed that the formation of single Au atoms was more favorable on the NiO surfaces with Ni vacancies than on clean NiO surfaces, single Au atoms were cationic on the NiO surfaces. The results were consistent with the experimental observations. Besides, Au1/NiO showed high catalytic stability to the CO oxidation reaction (Figure 1C). Zhang et al. used Mo atoms to regulate the oxidation state of transition metals in perovskite oxides by substituting Ni sites as highly selective and active catalysts for efficient 2e− ORR production of H2O2 (Han et al., 2023). Cerium oxide (CeO2) can stabilize single metal atoms due to its high vacancy density. Su et al. used DFT to calculate all the configurations of the single-atom Pt/CeO2 (110) catalyst (Qin and Su, 2021). They found high stability for structural models where cerium cations were doped with one or two Pt atoms, which was ascribed to the formation of square-planar oxygen coordination (Figure 1D). Therefore, rationally selecting the support and designing catalysts with stably anchored isolated atoms are crucial for electrocatalytic reactions in the future.
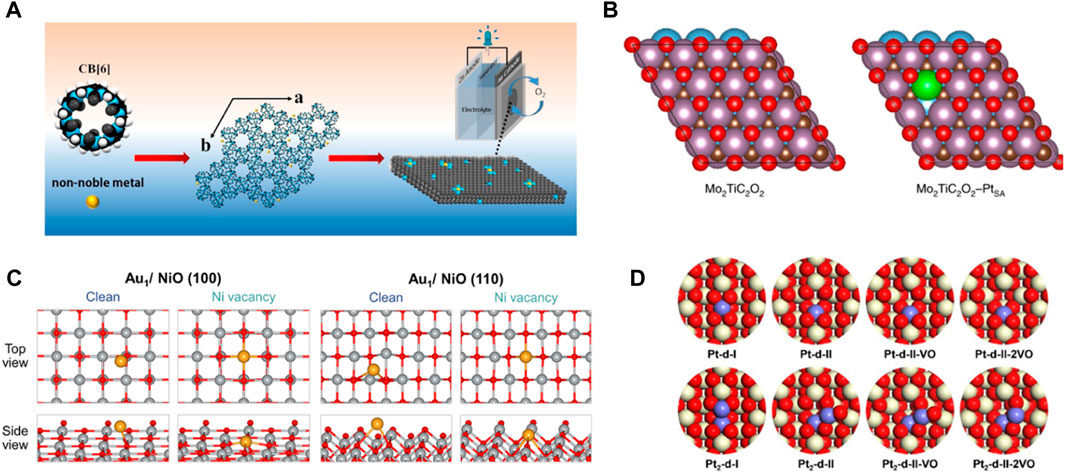
FIGURE 1. | The interaction of metals with different supported materials. (A) Schematic diagram of non-noble metal atom mixed with CB (He et al., 2020a) for ORR electrocatalysis. (Figure used with permission from Cao et al., 2021) (B) Top view of Mo2TiC2O2 and Mo2TiC2O2-PtSA catalyst configurations. Color codes: Ti, blue; Mo, purple; Pt, green; C, brown; O, red. (Figure used with permission from Wang et al., 2018) (C) Geometry of Au single atoms deposited on clean and defective NiO(100) or NiO(110) planes. Gray, Red and Yellow represent Ni, O, Au atoms, respectively. (Figure used with permission from Murayama et al., 2022) (D) Local structure of Pt atoms doped into CeO2 (110). White, blue, Red and lattice represent Ce, O, Pt atoms, respectively. (Figure used with permission from Su et al., 2021).
2.2 Activity of SACs
Improving catalytic activity has been a key problem that has been studied by many researchers. Although the intrinsic activity of the catalyst can be affected by the interaction between the carrier and single atoms, the abundance of active sites on the catalyst surface is an important factor for effectively activating the reactants (Hou et al., 2023; Liu et al., 2023). Isolated metal atoms in SACs are often considered the main active sites (Figure 2). However, increasing the density of metal atoms and the number of active sites to achieve the target chemisorption of the reactants is an important indicator for improving the catalytic reaction activity (Zhang et al., 2021c). Theoretical studies have indicated that the activity of Ni−N−C for CO2RR changes as the coordination elements (N, C) vary, such as Ni−N4, Ni−N3C1, Ni−N2C2, Ni−N1C3, and Ni−C4 (Hossain et al., 2020). The CO stretch for CO bound to Ni was 1985 cm−1 at −1.0 V on the Ni–N2C2 site, which agrees best with the experimental results. It showed the best catalytic activity and selectivity. Furthermore, single metal atoms embedded into the carrier affect the surrounding carrier atomic sites and can act as active sites to play a synergistic catalytic role and improve the catalytic activity (Figure 2A). Zeng et al. found that isolated rhodium (Rh) atoms aggregated clusters of different atomicity by increasing Rh mass loading. Rh/CoO promoted the adsorption and activation of propylene in the hydroformylation of propylene (Wang et al., 2016). Due to the structural reconstruction of Rh single atoms in Rh/CoO, propylene and CO were trapped by Rh1, and the neighboring oxygen atoms of CoO provided adsorption sites for hydrogen (H) atoms, synergistically catalyzing the promotion of the hydroformylation of propylene (Figure 2B). Theoretical computations revealed that an increased content of single Pt atoms loaded on molybdenum disulfide (MoS2) affects the activity of the adjacent surface S atoms, increasing the activity of the catalytic reaction (Figure 2C) (Deng et al., 2015). In single-atom alloy (SAA) catalysts, composed of bimetallic and polymetallic complexes, one type of metal atom is atomically dispersed on the other metal material (Zhang et al., 2021d). The active sites are generated by the strong metal interaction (alloying bonding) between the isolated-single atom and the metal carrier material. Luo et al. found that ensembles composed of the Cu (111) surface and the palladium (Pd) atoms subsurface were considered active sites to effectively reduce the energy barrier of H2 dissociation through theoretical calculations (Fu and Luo, 2013). Subsequently, this can be employed to adjust the catalytic activity of SACs by doping other atoms in the subsurface layer (Figure 2D). In a newly reported study, it was suggested that the SAA catalyst with Rh atoms dispersed in Cu had a very low kinetic barrier in the process of dehydrogenation and formation of propylene (Hannagan et al., 2021). Additionally, it showed strong activation ability for the C-H bond. Therefore, the activity of SACs directly affects the reaction route and different activation mechanisms (Figure 2E). It guides the further designing of SACs by increasing the density of single-atom metals and the number of active sites.
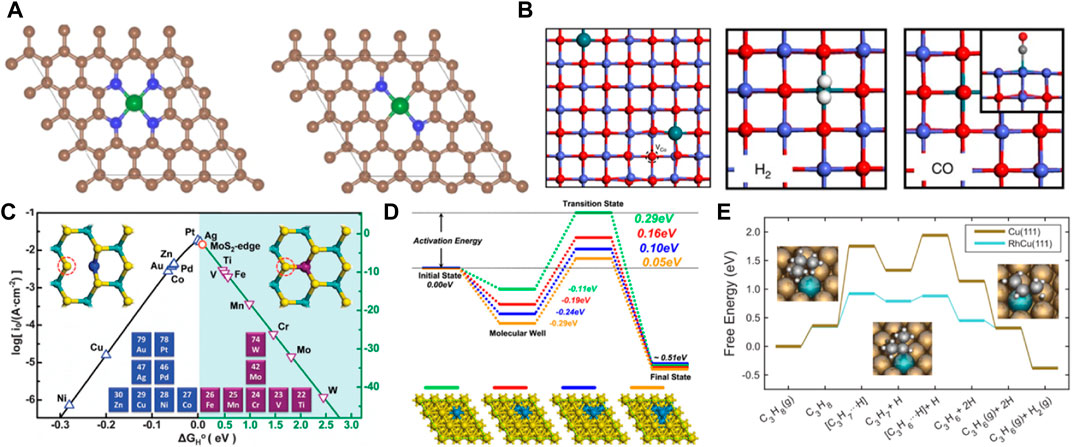
FIGURE 2. (A) Ni–SAC model diagram containing Ni–N4 and Ni–N2C2 moieties as active sites for CO2RR (figure used with permission from Luo et al., 2020). (B) The model of Rh1/CoO and the adsorption configurations of H2 and CO on Rh1/CoO (figure used with permission from Zeng et al., 2016) (C) The volcano curve relation between currents [log(i0)] and △GH。. The inserted diagram points to different doped MoS2 configurations (figure used with permission from Luo et al., 2013). (D) Energy profiles of H2 dissociation and structure diagram of Pd-doped Cu(111) surfaces (figure used with permission from Luo et al., 2013). (E) Free-energy diagram of RhCu (111) SAA and Cu(111) for the formation of propylene. (Figure used with permission from Sykes et al., 2021).
2.3 Selectivity of SACs
The reaction process is often accompanied by the occurrence of side reactions. Therefore, product selectivity is an important parameter in evaluating catalyst performance. Product selectivity is affected by the characteristics and conditions of the reactions. In addition, it is related to the catalyst (Li et al., 2017a; Ying et al., 2021; Zhang et al., 2022a). SACs exhibit high catalyst selectivity due to the unsaturated coordination of the active centers. The theoretical calculations showed that the charges were redistributed by replacing a B site in the B12N12 nanocage with a Pd atom (Gong and Kang, 2018). Moreover, the electrons were found to accumulate around the Pd atom in B11N12Pd. H2 was adsorbed and dissociated on single Pd atoms to form the B11N12Pd(2H) dihydride complex and then hydrogenated with C2H2 on the B11N12Pd SACs. The energy barrier was as low as 26.55 kcal mol−1, and it had higher selectivity than many bimetallic alloy monatomic catalysts (Figures 3A, B). Zhao et al. constructed 11 kinds of SACs by modulating the coordination environment of Mn atom and graphene substrate for NRR (Gao et al., 2020). MnSA@Vs-N1 promoted the conversion of N2 to NH3 at 0.77 eV by distal mechanism, inhibiting the occurrence of HER (Figures 3C, D). The lower reaction free energy in the HCOO* formation (ΔGHCOO*) made it the main product on the atomically dispersed Inδ+−N4 (Shang et al., 2020) and Sb-N4 SACs (Jiang et al., 2020). However, the HER pathway had much higher free energy than HCOO* formation, exhibiting excellent selectivity. The catalyst selectivity is evaluated with appropriate descriptors. Considering that H2 evolution was the most competitive in CO2RR, the difference between thermodynamic limiting potentials for CO2RR and H2 evolution was a proposed descriptor to evaluate the high selectivity of CO2RR to CO on Ni-N4 catalyst (Figures 3E, F) (Li et al., 2017b). Therefore, constructing correlations between selectivity and electronegativity, overpotential, and charge number are very useful for evaluating catalytic performance. Additionally, the theoretical designs provide a great basis for developing the experiments.
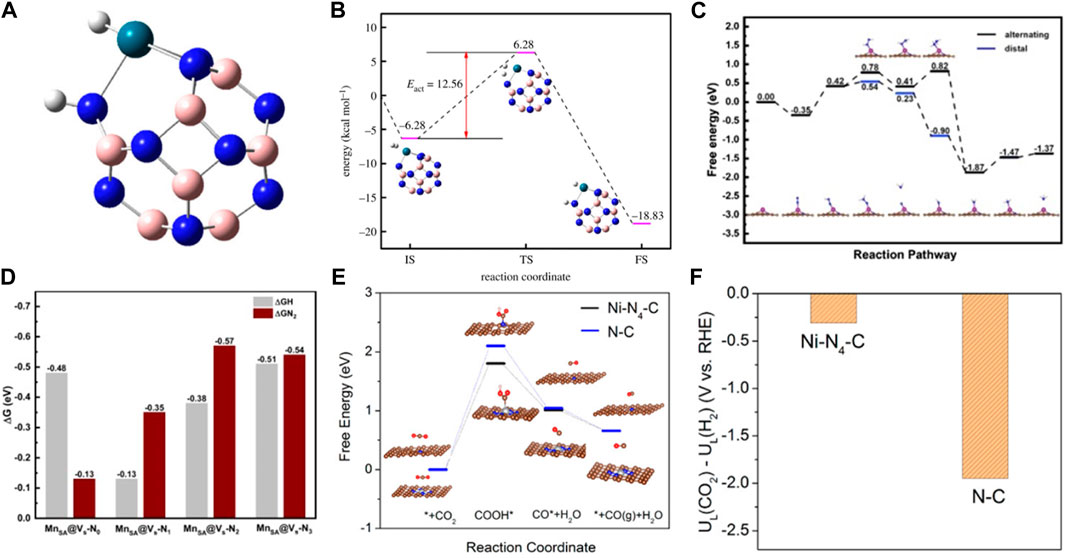
FIGURE 3. (A) The stable structures of H2 absorbed on B11N12Pd SAC. (B) Energy barrier diagram of the B11N12Pd SAC for hydrogenation reaction and the schematic diagrams of initial (IS), transition (TS) and final (FS) states. White spheres: H atoms, pink spheres: B atoms, blue spheres: N atoms, blue-green spheres: Pd. (Figure used with permission from Kang et al., 2017) (C) Free-energy diagrams for the NRR on Fe-N/C. (D) The difference between the free energy of H and N2 atom (figure used with permission from Zhao et al., 2020) (E) (a) The free energy diagram for CO2RR. (F) Limiting potential difference for CO2RR and HER on Ni-N4 catalyst (figure used with permission from Xie et al., 2017).
3 Applications of SACs for CO2RR
3.1 Noble metal SACs
Noble metal SACs offer the benefits of high conductivity, a large active surface area, vacancies in the d orbitals, and the ability to adsorb/stabilize reactants and intermediates. Therefore, noble metal SACs are extensively utilized in electrocatalytic CO2RR. Chen et al. incorporated atomic Pd into N-doped carbon support to improve the CO2RR activity (He et al., 2020b). The binding energy of the intermediates, *H and *HOCO, on Pt-N4 were weaker than on Pd/PdH. However, the formation of *HOCO was more favorable than *H through the difference computed in their binding energies. Furthermore, in a realistic electrocatalytic environment, the intermediate *HOCO can enhance its stability on Pd-N4 by forming H bonds with water molecules. Therefore, Pt-N4 was the most likely to obtain CO without the formation of palladium hydride (PdH). The atomically dispersed Pd facilitated the desorption of weakly bound *CO and enhanced electrocatalytic CO2 reduction capability (Figures 4A, B). Since a single Pt atom can remarkably improve the CO2RR activity of graphene, it was anchored on defective graphene with double vacancies to form an atomically coordinated Pt@dv-Gr catalyst, achieving high selectivity and activity of CO2RR to form CH3OH (Back et al., 2017). Moreover, the weak binding energy of *CO on Pt@dv-Gr promotes protonation to form *CHO species. Pt fills the bonding orbital via orbital mixing due to single occupancy in the dxz orbital that can interact with the C(pz) orbital of *CHO during the *CHO binding, leading to CO2 reduction to produce CH3OH at the low limit potential (UL) of −0.27 eV on Pt@dv-Gr (Figure 4C). Zhao et al. reported single Pt atoms supported on MoS2 nanolayers as CO2RR catalyst by DFT calculations (Ren et al., 2022). Pt@MoS2 were selective catalysts for CO2RR to produce CH4 with a UL of −0.50 eV (Figures 4D, E). Poater et al. reported that single atom Ag was anchored on g-C3N4 support (Ag1@ g-C3N4) as a SACs, showing different activity for Cu1@ g-C3N4 (Posada-Pérez et al., 2023). The CO formation process requires to overcome the thermodynamic barrier with a limiting potential of −0.55 V. The desorption energy of CO was 0.56 eV, lower than that of Cu1@g-C3N4 catalyst. The most favored reaction pathway was the formation of HCOOH product with a low potential of −0.37 V. In addition, Hads on the catalyst Ag1@ g-C3N4 are very unstable compared to CO2 adsorption. Therefore, Ag1@ g-C3N4 catalyst promotes the generation of HCOOH. Liu et al. study the structure of Ru/In2O3 catalyst and the intermediates and transition states of CO2 hydrogenation to methanol by DFT. The results not only confirm that the strong metal-support interaction improves the catalyst stability, but also show obvious charge transfer and rearrangement between Ru and In2O3 (Wu et al., 2021). The CO2 molecule was first activated at the oxygen site of the metal support interface, and then directly dissociated into CO*, the CO2 molecule was gradually hydrogenated until methanol was formed, and finally desorbed from the catalyst surface. The Ru/In2O3 catalyst showed high catalytic activity and stability. Lu et al. found that electron promoter Na+ enhances the electron interaction between the neighboring Rh1 and ZrO2 carriers and promotes electron transfer from Rh to ZrO2 carriers (Li et al., 2022a). It not only weakens the adsorption of CO on Rh1/ZrO2, but also reduces the activation ability of H2. This can promote the desorption of the reaction product CO, avoid the deep hydrogenation of CO products to CH4, and achieve high CO selectivity (Figures 4F, G).
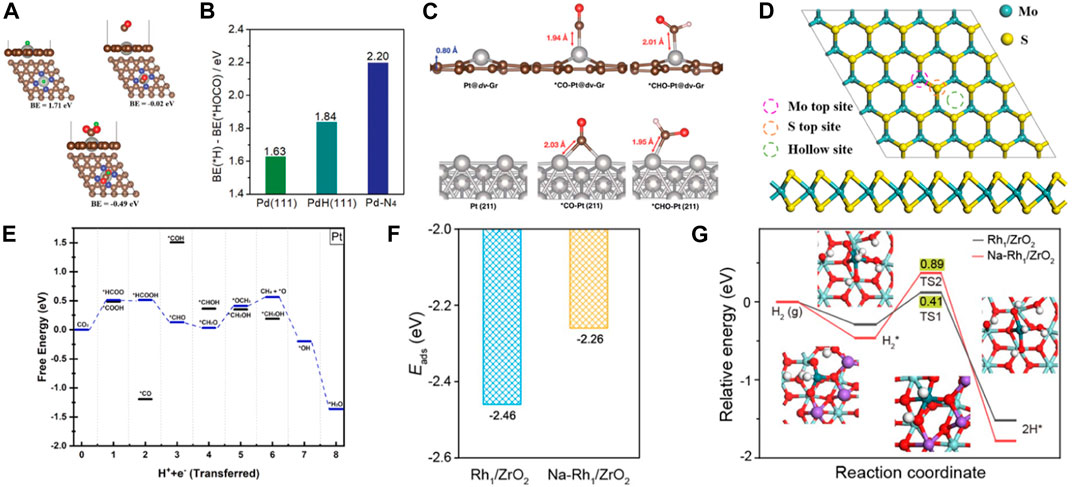
FIGURE 4. (A) The optimized structures and calculated binding energies of *H, *CO, and *HOCO adsorbed on a Pd-N4 catalytic by DFT calculations. Gray, Pd; brown, C; blue, N; red, O; green, H. (B) The binding energies difference of *H and *HOCO on Pd(111), PdH(111), and Pd-N4 (figure used with permission from Chen et al., 2020a). (C) The optimized configurations of *CO and *CHO adsorbed on Pt@dv-Gr and Pt(211) (figure used with permission from Jung et al., 2017). (D) The top views and side views of the MoS2 nanolayers. (E) Free energy diagrams for the CO2RR on Pt@MoS2. (Figure used with permission from Zhao et al., 2022a). (F) The adsorption energies of CO on Rh1/ZrO2 and Na-Rh1/ZrO2. (Figure used with permission from Lu et al., 2022). (G) The energy profiles of H2 reaction and the structures of the optimized intermediates on Rh1/ZrO2 and Na-Rh1/ZrO2. (Figure used with permission from Lu et al., 2022).
3.2 Non-noble metal SACs
Non-noble metal catalysts with the transition metal coordination on carrier via defect engineering have also been reported for CO2RR with excellent catalytic performance. In a previously reported study, seven metals were atomically dispersed in a tungsten ditelluride monolayer (M@WTe2) for CO2RR using DFT calculations (Zhang et al., 2022b). Subsequently, Ni@WTe2 had the highest adsorption capacity for *CO and *HCOOH near the Fermi level and a suppressed competing reaction which led to higher activity and selectivity. The Gibbs free energy of CO2RR also demonstrated that the thermodynamic energy barrier of Ni@WTe2 was only 0.11 eV. Therefore, Ni@WTe2 was a promising electrocatalytic material for CO2RR. Sun et al. constructed the Ni–N3 (pyridinic) and Ni–N3 (pyrrolic) structures incorporated in different N-doped carbon supports to achieve high selectivity and activity of CO2RR to produce CO (Fan et al., 2019). DFT calculations showed that Ni–N3 (pyridinic) had low free energy for *COOH and *CO formation. The active site of Ni@N3 (pyridinic) was easily poisoned by *CO due to strong *CO adsorption. On the contrary, Ni-N3 (pyrrolic) was more likely to obtain CO. The free energy value for *COOH formation on Ni@N3 (pyrrolic) was lower than that reported for Ni-N4. Therefore, the catalytic activity for the Ni@N3 (pyrrolic) site may be better than the Ni@N4 site (Figures 5A, B). Li et al. designed atomically dispersed Co-N5 sites anchored on polymer-derived hollow N-doped porous carbon spheres (HNPCSs) for efficient CO2 reduction to produce CO (Pan et al., 2018). The Co-N5 site was the active center for CO2 reduction to produce CO. The thermodynamic barrier for the key COOH* formation was −0.28 eV on Co-N5/HNPCSs, and CO desorption ability was increased than that of the CoPc catalyst, exhibiting a higher CO2RR activity. Moreover, single Fe atoms can also significantly increase the CO2RR activity (Figure 5C). Tang et al. reported an atomically dispersed Fe-coordinated N-doped carbon catalyst that enhanced CO2 reduction to produce CO with high activity (Takele Menisa et al., 2022). The neighboring graphitic N transferred more electrons to the intermediate COOH*, increasing COOH* adsorption strength. However, the neighboring graphitic N transferred a reduced number of electrons between CO and the catalyst, promoting CO desorption. Therefore, the origin of the high activity was attributed to the synergistic effects between FeN4 and the neighboring graphitic N. The Fe-N5 sites were more conducive to producing CO than FeN4 (Figure 5D). Wu et al. reported the incorporation of Fe–N5 sites in the defect-rich porous carbon nanofiber (Fe-N5/DPCF) catalyst for CO2RR (Li et al., 2022b). The coupling of Fe-N5 with defect carbon (DC) substrate provided active sites for CO2 adsorption and activation. DC, Fe-N5/C, Fe-N4/C, and Fe-N5/DC were also studied to explore the activity and selectivity of CO2RR. The energy barrier of CO desorption on Fe-N5/DC was significantly lower than that of Fe-N4/C, suggesting high CO2RR activity. Moreover, the positive limiting potential differences between CO2RR and HER indicated the highest selectivity of Fe-N5/DPCF. Furthermore, Cu-based catalysts are unique catalysts capable of reducing CO2 to a range of hydrocarbon compounds with excellent selectivity. Zhu et al. performed DFT calculations and found that the N and O-coordinated single Cu atom catalyst (CuN2O2) had high selectivity and activity for CO2 reduction to produce CH4 (Cai et al., 2021). The positive charge of Cu in CuN2O2 was lower than in CuN4, resulting in a low-valence oxidation state of Cu. The energy barriers of intermediates *COOH and *COH in CuN2O2 were reduced, and HER was well-inhibited (Figures 5E, F). Zheng et al. demonstrated that the binding energies of CO in the two adjacent Cu–N2 sites were stronger than at the Cu-N4 sites, which was conducive to the formation of C4H4 through C-C coupling (Guan et al., 2020a). However, the isolated single-atomic Cu sites also had high activity and selectivity for CH4. It was found that the single tungsten (W) atom could improve the CO2RR activity of In2O3 (Zou et al., 2022). The single W atom was embedded on the In2O3 (111) substrate containing oxygen vacancies to form an atomically coordinated W-In2O3_D model. The oxygen vacancy could provide more electrons for activating CO2. Additionally, W induced a higher degree of electron distribution in oxygen vacancies, resulting in the easier formation of the HCOO* intermediate and breaking of the C-O bond in the H2COOH* intermediate. Therefore, the generation of methanol products was significantly easier on the W-In2O3_D catalyst. Xu et al. introduced single vanadium (V) atom into bismuth oxide (Bi2O3) to enhance CO2 conversion to formic acid (Zhang et al., 2022c). The redistribution of electrons on the Bi2O3 surface promoted CO2 adsorption and activity due to the introduction of the V atom, improving the selectivity of formic acid.
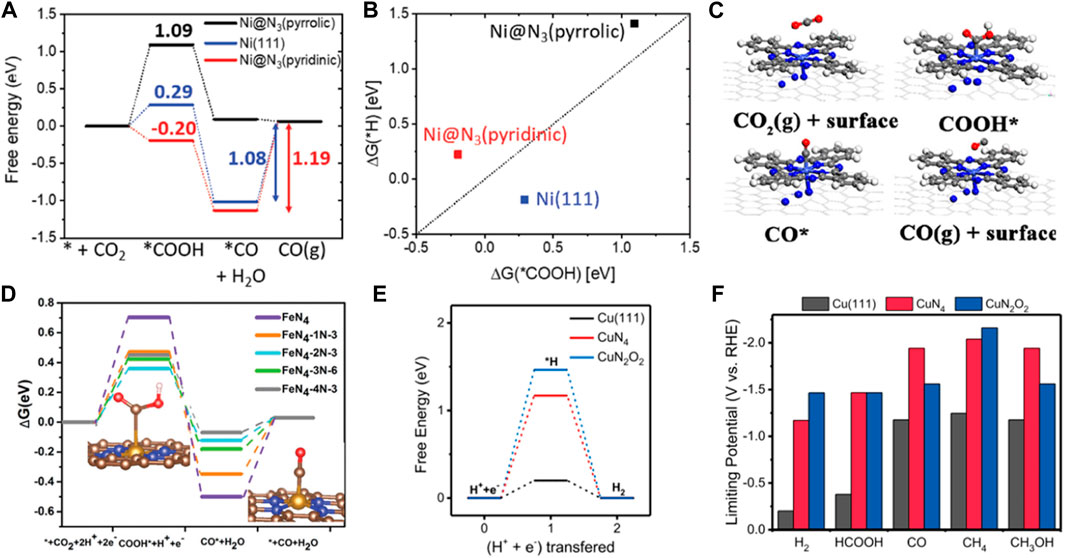
FIGURE 5. (A) Free energy diagram of electrochemical CO2 reduction to CO on Ni@N3 (pyridinic), Ni(111) and Ni@N3(pyrrolic). (B) The linear relationship between ΔG(*H) and ΔG(*COOH) (figure used with permission from Sun et al., 2019) (C) Optimized structures of intermediates for CO2RR (figure used with permission from Li et al., 2018) (D) Free energy diagram and intermediates geometries for electrochemical CO2 reduction to CO on Fe–N4 moieties embedded on carbon sheets. Gray, C; Orange, Fe; Blue, N; Red, O; light white, H. (Figure used with permission from Tang et al., 2022) (E) Free energy diagram of hydrogen evolution on the CuN2O2(Cu-CDs), CuN4, and Cu(111). (F) The limiting potentials of the all products on the CuN2O2, CuN4, and Cu(111) (figure used with permission from Zhu et al., 2021).
3.3 Metal-free SACs
Metal catalysts have developed rapidly in electrocatalysis since Hori et al. used metals for electrocatalytic reactions. However, there are some limitations, such as poor tolerance of metal catalysts in acidic and alkaline environments. In recent years, metal-free materials have emerged as a new class of electrocatalytic materials. Gu et al. demonstrated the potential of producing CO using an N-rich (11.0 wt%) graphene-like carbon electrocatalyst (NG-1000). The C atom next to the graphite-N atom in NG-1000 was the main active species of CO2RR. The absorbed COOH* species was considered the potential-limiting step with an overpotential of 0.82 eV (Figure 6A) (Li et al., 2021b). Phani et al. found that the adsorption energy for CO2 molecules was higher on boron (B)-doped graphene. Additionally, it was observed that the catalyst promoted the reduction of CO2 to formic acid. DFT theoretical calculation showed that B doping led to an asymmetric spin density and activated inert carbon atoms, resulting in higher electrocatalytic activity for the catalyst than graphene (Figure 6B) (Sreekanth et al., 2015). Einaga et al. reported that B-doped diamond (BDD) could catalyze CO2 reduction to HCOOH and HCHO. BDD had high stability, and its activity originated from the low binding energy of the *COOH intermediate (Natsui et al., 2018). Scheier et al. studied charge-dependent CO2 adsorption in C60 and found that C60+ achieved the highest CO2 adsorption capacity by exposing C60 and CO2 to the ionizing free electrons by molecular dynamics simulations (Ralser et al., 2016). Cai et al. systematically studied the effect of pyridine N-doped graphene on the electrocatalytic CO2RR performance (Liu et al., 2016). The activity of CO2 improved as N-doping changed the electronic properties of graphene. The adsorption energies of the COOH species on N-doped graphene were higher than on pristine graphene. However, the weak adsorption energy of CO or HCOOH made desorption easy to produce CO and HCOOH. PyrroN3 possesses the best catalytic performance for CO2RR to produce HCOOH due to its lower overpotential of 0.24 V (Figures 6C, D). Borophene is used for electrocatalytic CO2RR to produce CH4 with a limiting potential of −0.27 V (Qin et al., 2020). The electron-deficient borophene can transfer electrons to CO2 and activate it by breaking the π bond. Borophene showed strong activity for CO2 with an adsorption energy of −1.02 eV (Figure 6E). Smith et al. calculated the adsorption energy of CO2 on borophene nanosheets by adjusting charge density (Tan et al., 2017). The results indicated that the CO2 molecule was chemisorbed on the B3 site of the neutral and 3.5 e–negatively charged borophene. CO2 molecules had weak physical adsorption on neutral borophene with adsorption energy in the range of −0.15 eV to −0.19 eV. However, the adsorption energy of CO2 molecules on 3.5e–negatively charged borophene was in the range of −0.4 eV to −0.8 eV. It was shown that negatively charged borophene could promote CO2 molecule capture (Figure 6F). Sun et al. adopted the metal-free B2S catalyst to convert CO2 into CH3OH by DFT calculations (Tang et al., 2020). The thermodynamic and kinetic energy barriers were −0.12 eV and 0.43 eV, respectively. The free energy barrier of the catalyst was about 1/3 of Cu in the metal Cu (211) catalyst, demonstrating high catalytic activity (Figures 6G, H).
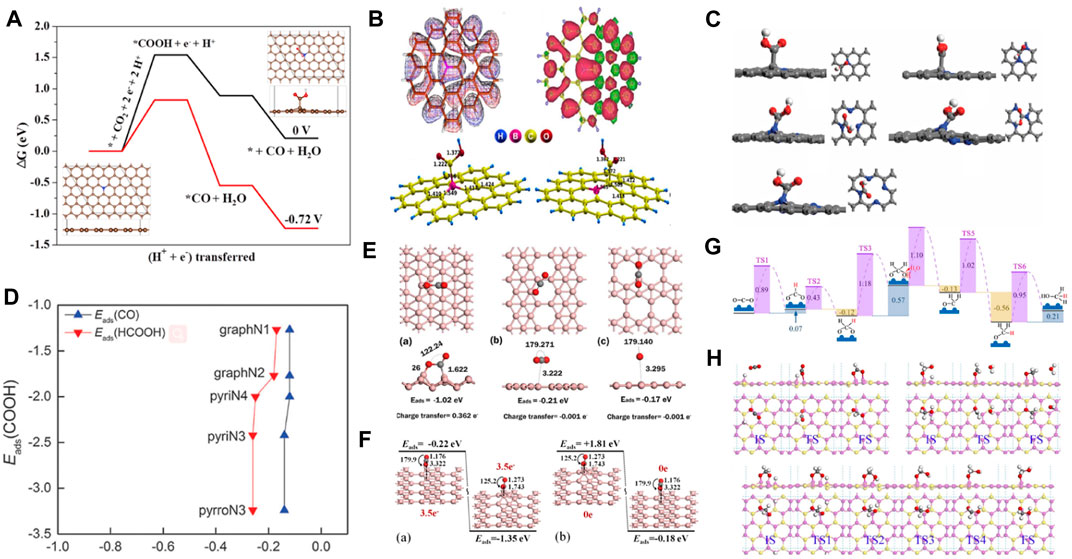
FIGURE 6. (A) The calculated free energy diagram for CO2RR to CO on NG-1000 catalyst at 0 V and −0.72 V vs. RHE. (Figure used with permission from Gu et al., 2021). (B) Electron configuration of C41BH16 and adsorption configuration of *COOH on different sites. (Figure used with permission from Phani et al., 2013). (C) Structure of COOH species on N-doped graphene. (D) The correlation of the adsorption energies of the intermediate species on various N-doped graphenes. (Figure used with permission from Cai et al., 2016). (E) The adsorption energy and charge transfer (e−) in the most stable configuration of CO2 adsorption on borophene, β12 and χ3 boron sheets (figure used with permission from Smith et al., 2019). (F) Energy change of CO2 on borophene by injection and removal of 3.5 extra electrons (figure used with permission from Smith et al., 2017). (G) Free energy diagram for “Formate” path and the models of intermediates and the transition states. (H) The initial, transition, and final states of the OCHO*, OCH2* and OCH2OH* on B2S (figure used with permission from Sun et al., 2020).
In summary, metal SACs and metal-free SACs exhibit high activity, stability, and selectivity in electrocatalytic CO2RR compared to typical metal catalysts, significantly reducing the catalytic cost. However, SACs frequently aggregate and form huge clusters due to the high surface free energy. Therefore, SACs have high requirements for their loaded carriers. Therefore, DACs have been introduced in recent years. DACs maintain high atomic utilization and stability (Guan et al., 2020b; Li and Tang, 2021). Moreover, the synergy between two metal atoms significantly improves the catalytic activity. DACs have significant development prospects in electrocatalytic reduction.
4 DACs
DACs are two metal atoms (homonuclear or heteronuclear) supported on a carrier to create dimers that catalyze the CO2RR and form hydrocarbons or oxygen-containing compounds through interatomic synergy (Das and Arunan, 2019). They increase the active sites and enhance the catalytic activity and selectivity of the process, significantly reducing the theoretical limits of SACs (Zhou et al., 2020b; Liu et al., 2022). The synergistic DACs require selective activation of the reactants on the carrier, the highly active substances degrade or produce by-products if the activation rate is not constant, reducing the efficiency of CO2RR (Ying et al., 2021; Ling et al., 2022). DACs are primarily classified as homonuclear DACs or heteronuclear DACs. Due to the compatibility of redox reactions between two metals and changes in the kinetics of the catalytic cycle, heteronuclear DACs exhibit superior catalytic performance than homonuclear DACs in electrocatalysis, resulting in the emerging research on heteronuclear double atoms (Zheng et al., 2020). The structures of DACs are difficult to be accurately regulated in experiments. Therefore, DFT is often used to simulate the active sites of atoms, deduce the route of the catalytic reaction, and calculate the free energy barrier of the reaction. The catalytic performance of CO2RR was analyzed to judge the catalyst quality. Therefore, DFT plays a pivotal role in double-atom electrocatalysis.
4.1 Homonuclear DACs
Homonuclear metal atoms have similar energy and the same atomic orbitals. In DACs, two identical metal atoms are bonded by a chemical bond, solving the agglomeration phenomenon of SACs due to large surface free energy. DACs increase the content of metal atoms and improve the stability of the catalyst. Zhou et al. (2020c) successfully prepared a double-atom Ag2/graphene catalyst (Ag2-G) for CO2RR (Li et al., 2020b). The two AgN3-AgN3 sites were firmly anchored to the graphene matrix by the Ag-C bond. The CO2 was adsorbed on the two active sites of Ag2-G. The formation barrier of *COOH was also reduced, promoting the conversion of CO2 into CO with a limit potential of 0.7 eV (Figure 7A). Compared with the Ag1-G monoatomic catalyst, DAC showed higher selectivity and excellent catalytic performance. Moreover, the competitive HER was almost completely suppressed. Cao et al. achieved controlled growth of double Ni metal by the metal-N combination strategy in the pyrolysis process (Sun et al., 2020). The DACs reduced the possibility of metal cluster polymerization and showed good activity for electrocatalytic CO2 reduction at low Ni content. The electrocatalytic reduction of CO2 to CO was effectively achieved by the catalyst (Figure 7B). Zeng et al. systematically studied the synergistic effect of adjacent Pt metal atoms on the catalytic performance of CO2 hydrogenation (Li et al., 2018). They prepared Pt/MoS2 by replacing Mo of MoS2 nanocrystals with Pt atoms. The oxidation rate of Pt gradually decreased with the increased amount of Pt, enhancing the activity of the Pt/MoS2 catalyst. DFT calculations indicated that COOH* was the main intermediate obtained. (Figure 7C). Therefore, the Pt2/MoS2 catalyst can promote CO2 conversion to formic acid and methanol. Zhou et al. systematically investigated a series of DACs with various double transition metal atoms supported on defective BN monolayers (TM2@BN) (Huang et al., 2020). The onset potentials for CO2RR to CH4 on various TM2@BN nanosheets were built. It was found that Fe2@BN and Ni2@BN possessed lesser negative onset potentials, and their catalytic performance was better or similar to the Cu (100) and Cu (211) catalysts. In addition, the Fe2@BN and Ni2@BN exhibited high CO2RR activity to produce CH4 with limiting potentials of −0.47 and −0.39 V, respectively (Figure 7D). The Ni2-N4-C2 catalyst was synthesized for electrocatalytic CO2 reduction to CO with superior activity due to the unique metal-metal bridging structure and proper N coordination number (Cao et al., 2022b). The DFT calculations verified that the d-band center of the catalyst was close to the Fermi level, and more electrons were transferred from the catalyst to the CO2 molecules, which was more conducive to CO2 adsorption and desorption of *CO intermediates (Figures 7E, F). Double-atoms were anchored on graphitic carbon nitride (g-CN) to investigate their structure stability, CO2RR mechanism, and performance (Bui et al., 2021). It was found that Fe2@g-CN was a superior electrocatalyst for CO2RR with UL of 0.58 and 0.54 V for C1 and C2 products, respectively. The introduction of Fe13 clusters into Fe2@g-CN caused charge redistribution, which enhanced CO2 adsorption and reduced the limiting potentials of CH4 formation. Consequently, Fe2@g-CN showed higher CO2RR activity and selectivity. Lu et al. reported uniformly anchored Ni2 dual-atoms on N-doped carbon nanotubes (Ni2-NCNT) by modulating the ligands of the precursors for CO2RR (Liang et al., 2023). The Ni2-N4 and Ni2-N3 DACs models were developed to reveal the activities of the catalysts. A bridged *COOH intermediate at the Ni2 catalytic site was formed on the Ni2–N3 structure, the synergistic effect between two Ni atoms could stabilize the *COOH intermediate, leading to a lower reaction free energy, and Ni2–N3 exhibited superior CO2 reduction activity (Figure 7G). A nitrogen-doped porous carbon anchored homonuclear Fe2N6 diatomic electrocatalyst has been reported to effectively reduce CO2 to CO (Zhao et al., 2022a). The activation of CO2 was promoted by the Fe-Fe dual sites. The synergistic effect of two Fe centers further reduces the energy barrier of CO desorption reaction, and the largest energy barrier of CO production was 0.90 eV, which was significantly lower than that at FeN4 site (1.03 eV). Therefore, Fe2N6 was more conducive to the formation of CO products.
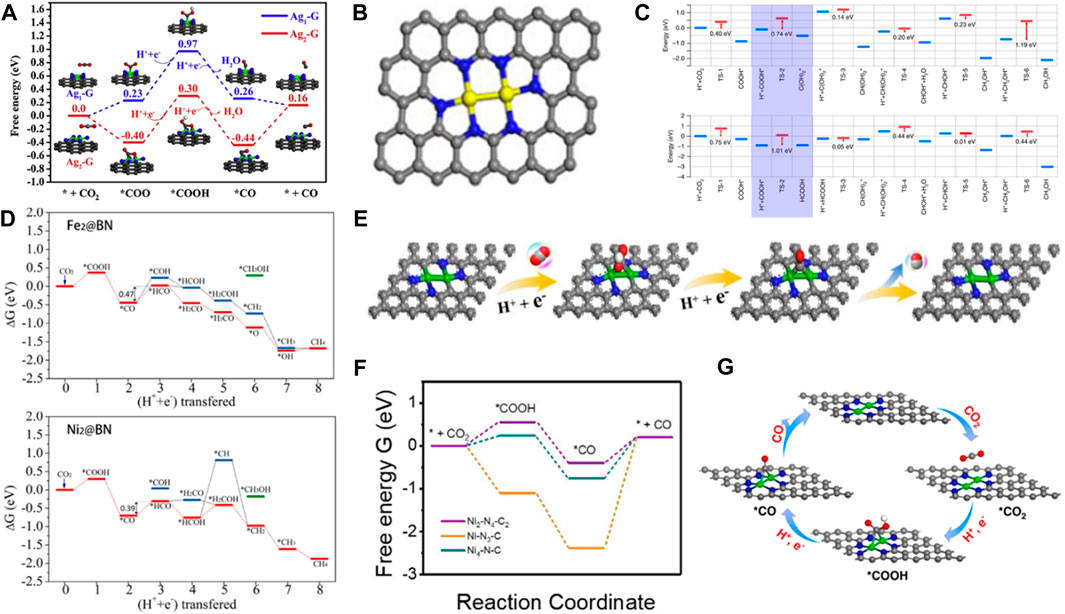
FIGURE 7. (A) Free energy diagrams of CO2 reduction on Ag1-G and Ag2-G catalyst (figure used with permission from Li et al., 2020a). (B)The di-atomic models used in EXAFS spectrum fitting (figure used with permission from Cao et al., 2020). (C) The CO2 reaction paths for Pt1/MoS2 and Pt2iii/MoS2 (figure used with permission from Li et al., 2018) (D) Free energy profiles of CO2 hydrogenation on Fe2@BN and Ni2@BN nanosheet (figure used with permission from Zhou et al., 2020a) (E) Reaction paths for CO2 reaction to CO on Ni2-N4-C2 catalyst. Gray, C; red, O; blue, N; while, H; green, Ni. (F) Free-energy diagram of CO2RR to CO (figure used with permission from Zhang et al., 2022c). (G) Scheme diagram of CO2 electroreduction to CO on Ni2–N3 (Ni2-NCNT) (figure used with permission from Lu et al., 2023).
4.2 Heteronuclear DACs
Heteronuclear DACs have unique structures and electronic properties due to the asymmetric active sites. The symmetry-breaking active centers can cause charge redistribution and more effectively overlap atomic orbitals when activating small fractions. It can strengthen the binding of the reactant molecules at the active sites. Therefore, the catalysts are more conducive to CO2 adsorption and activation (He et al., 2022). Luo et al. systematically studied 15 kinds of DACs (ten heteronuclear DACs and five homonuclear DACs) supported on graphene using DFT and dynamic simulation (Luo et al., 2020). Additionally, they screened out three heteronuclear catalysts with high CO2RR activity compared to the Au (211), Ag (211), and Cu (211) catalysts. The free energy calculations of the *OH species showed that the active sites of 9 kinds of catalysts were not occupied by the *OH species when the free energy was lower than −0.8 eV (Figure 8A). The adsorption of *COOH and *CO showed that the free energy values of the *COOH intermediate on Cu/Mn, Ni/Mn, and Ni/Fe catalysts were lower than 0.7 eV, exhibiting strong *COOH adsorption. Further hydrogenation and desorption to obtain CO on the catalyst surface were easy. Wang et al. designed 21 kinds of heteronuclear double atoms supported on monolayer C2N as effective DACs for CO2RR (Ouyang et al., 2020). The analysis of the electronic structures and adsorption configurations of CuMn/C2N, CuCr/C2N, FeCr/C2N, and MnCr/C2N showed that they were the most probable catalysts to produce CH4 with low overpotential and high selectivity. The C-affinity and O-affinity active sites were formed on the heteronuclear DACs, breaking the transition-metal scaling relations (Figures 8B, C). CuCr/C2N and CuMn/C2N exhibited the best performance for CH4 formation with UL of −0.37 V and −0.32 V, respectively. Furthermore, CO2 reduction to CO was proposed on the Ni-Cu dual active sites supported on MOF-templated porous carbon (Cheng et al., 2021). The DFT calculations showed that the electronic redistribution and band gap narrowing caused by coordinating Cu with the Ni site enhanced the electron conductivity and decreased the energy barrier for CO2 activation. The *COOH formation was the rate-limiting step due to the strengthened interactions between *COOH intermediates and the Ni centers (Figures 8D, E). DACs have been reported in the literature for synthesizing C2 products via C-C coupling. Fe-Co@C2N was predicted as a promising DAC for producing C2H4 (Liu et al., 2021). The synergistic effect of dual-metal-atom active sites and C2N substrate can promote CO2 activation (Figure 8F). The study on the CO2RR performance of seven DACs found that the FeCo@C2N exhibited the lowest limiting potential of −0.63 V for CO2RR to produce C2H4 by *CO+*CO co-binding (Figure 8G). It was revealed that C-affinity was beneficial to the coupling of the C–C bond and C2H4 formation. Chen et al. reported Cu-Fe atomic pairs anchored on nitrogenized carbon monolayers for electrocatalysis of CO2 reduction to C2 products with high activity and selectivity (Xie et al., 2021). The coordinated CuFe dimer on the nitrogenized graphene substance prevented metal agglomeration (Figure 8H). It showed strong stability and significantly improved activity for CO2 reduction to C2 products. Especially, the synergistic effects of metals on CuFe@C2N could significantly increase the adsorption ability of the ∗CO species intermediates, exhibiting high activity and selectivity for the CO2 reduction to produce C2H5OH (Figure 8I). Goddard et al. reported a similar study where it was found that the heteronuclear FeCu−grafiN6 promoted efficient electron transport and generated C2 products at a low limiting potential of −0.68 V (Chen et al., 2020a). Chen et al. conducted a systematic computational screening of double transition metal embedded N-doped graphene for electrocatalysis of CO2 reduction to C2H4 and C2H5OH (Figure 8J) (Chen et al., 2020b). It was demonstrated that 6 DACs had the stability and electrocatalytic activity for CO2RR to produce C2 by screening the reaction paths and the limiting potentials for 21 DACs (Figure 8K). Moreover, it was verified that the energy barrier of C-C coupling of the CuCr DAC was much lower than the threshold value (+0.75 eV), exhibiting the lowest limiting potential of −0.58 V for CO2RR to obtain C2 products. Chen et al. used the heteronuclear diatomic Mo-Se catalyst to explore the reaction of CO2 conversion to CO. The formation of COOH* was thermodynamic favorable on MoSA. However, the formation of CO* has a high free energy barrier, and the strongly adsorbed CO* may poisoning the catalyst. After the introduction of SeSA, the free energy of CO* decreased from 1.28 (MoSA) to 1.15 eV (MoSA-SeSA), indicating that the synergistic effect of MoSA-SeSA can significantly promote the generation of CO (Sun et al., 2022). The potential of B, P doped C2N monolayer as metal-free catalyst for reducing CO to C2H4 was explored (Wang et al., 2022). It was shown that B&P/C2N catalysts contribute to C-C coupling and prevent catalyst poisoning due to suitable adsorption energy for CO and lower ethylene desorption energy. In addition, B&P/C2N catalysts showed high stability. *COCO intermediates were captured by B-N center and transferred to B-P center for C-C coupling and hydrogenation to produce C2H4. Hydrogen evolution reaction further demonstrated the high selectivity of catalyst for C2H4 synthesis.
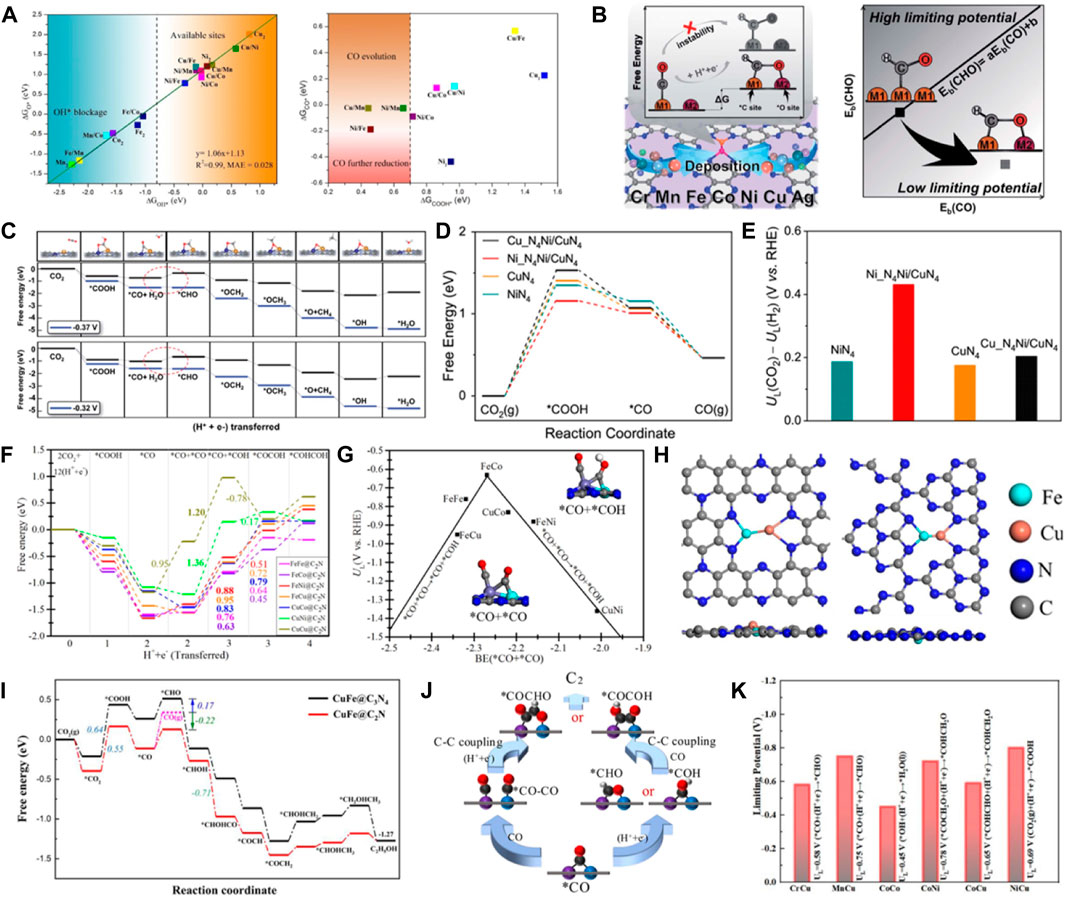
FIGURE 8. (A) Linear diagram of adsorption energy between ΔGOH* and ΔGO* on various DMSCs. COOH* (ΔGCOOH*) and CO* (ΔGCO*) on 9 DMSCs. (Figure used with permission from Li et al., 2020). (B) Design of DACs to reduce free energy variation by stabilizing *CHO. (C) Simplified schematic of decoupling the scaling relations between binding energies of *CHO and *CO. (figure used with permission from Wang et al., 2020). (D) Free energy profiles of the CO2RR for four catalysts. (E) Difference in limiting potentials for the CO2RR and HER (figure used with permission from Lu et al., 2023). (F) Gibbs free energy profiles for CO2RR toward C2 product. (G) Volcano curve between limiting potential (UL) and binding energy (BE) of co-adsorbed *CO+*CO (figure used with permission from Liu et al., 2021). (H) Optimized structures of CuFe@C2N and CuFe@C3N4. (I) Free energy profile of CO2 reduction to various C2 products. (Figure used with permission from Chen et al., 2021). (J) The general two pathway from *CO to C2. (K) Limiting potential for the 6 DACs (figure used with permission from Singh et al., 2020).
Although DACs have been reported in electrocatalysis in an infinite number of ways, there are significant difficulties in experimental research, the studies on the catalytic mechanism are more one-sided, and the role of the synergy between double atoms at the atomic scale is unknown. Therefore, future studies must focus on efficiently handling experimental obstacles and employing theoretical calculations to investigate synergy mechanisms.
5 Electrode potential and solvent effect on electrocatalytic CO2RR
The traditional computational hydrogen electrode (CHE) model assumes that the catalyst is charge-neutral for calculating the energy barrier of a reaction (Andrew et al., 2010). However, the electrochemical reaction usually occurs at the gas-liquid-solid phase interface with a constant electrode potential. The surface of the catalyst is charged, and the electron exchange between the catalyst and the electrode reaches an equilibrium state (Heenen et al., 2020; Zhao and Liu, 2020; Zhao et al., 2022b). The surface charge can significantly change the Fermi level and the electronic structure of the catalyst. Additionally, more electrons accumulate due to the increased charge capacity, significantly favoring the electrochemical reaction (Cao et al., 2023). Therefore, the influence of surface charge and constant potential on catalytic reactions cannot be ignored in heterogeneous electrochemistry. Nørskov et al. determined the constant potential reaction energetics by calculating a single potential barrier in an electrochemical environment and corresponding surface charges in the initial, transition, and final states (Figure 9A) (Chan and Nørskov, 2015). It was also demonstrated that the potential dependence of the activation energy was related to the partial charge transferred in the transition state and predicted by calculating a single constant charge (Chan and Norskov, 2016). Furthermore, the electrode potential significantly affects the elementary steps of proton-electron transfer for electrochemically reducing CO2 to CO on the Cu (100) catalyst (Sheng and Sun, 2017). The potential barrier of the electrochemical step decreased with an increase in the electrode potential (Figures 9B, C). Wang et al. probed the CO2 proton-electron transfer processes at different potentials using constrained MD sampling and thermodynamic integration (Cao et al., 2022c). And the electrode potential of the system was adjusted by introducing anions and cations (Na+, Cl−). Subsequently, it revealed the ET-PT decoupling and H bond-assisted CO2 activation mechanism. The adsorption of CO2 was significantly coupled with the electron transfer from the substrate (Figure 9D). And the electrode potential was essential to achieve efficient activation of CO2 adsorption. In addition to the electrode potential, the solvent effect is shown to be another important factor influences the reaction. The H bonding network between the water solvent molecules interacts strongly with the key species adsorbed on the catalyst surface affecting the intermediate stability. The water molecules promote the transfer of H protons and accelerate the reaction. Zhou et al. (2020b) studied the facilitative effect of axial O on CO2 activation on NiN4 SACs using constrained ab initio molecular dynamics (AIMD) with the explicit solvent model. 36 H2O molecules and an additional hydrogen atom were introduced into the system to construct the explicit solvent model (Figure 9E) (Hu et al., 2021). Additionally, the last 1,000 samples from the 3–6 ps AIMD simulations were collected to get the potential of mean force to obtain the free energy profile of CO2RR and the structures of explicitly solvated intermediates. The constant potential correction method proposed by Chan and Nørskov was used to determine the constant potential of −0.90 V vs. SHE. It was found that the H atoms in the solvent environment bonded with the O atoms in the CO2 molecule to form *COOH intermediates. Furthermore, the solvent formed H bonds with the adsorbates, the C-O bond was completely broken, and the dissociated OH group was protonated by water molecules, promoting the protonation of CO2 to form CO (Figure 9F). Liu et al. investigated six possible active sites on Ni-NxCy catalysts for reducing CO2 to CO, including Ni embedded in graphene-single vacancies (SV), and x N atoms coordinated with the Ni atom embedded in a divacancy (x = 0, 1, 2, 3, and 4) (Zhao and Liu, 2020). The solvent effect and the applied electrode potential were simulated using six layers of water molecules and net charge, respectively, resolving the catalytic origin due to both factors. The kinetic barriers were evaluated using AIMD and a “slow-growth” sampling approach. The calculations revealed that the multiple H bonds between the solvent water molecules and the polar intermediates improved the intermediate stability (Figure 9G). Moreover, the water molecules facilitated the transfer of protons from water to the intermediates, improving the selectivity of the reaction. Different net charges were introduced at −0.65 V vs. RHE. The 0 N, 1 N, and SV sites had nearly two negative charges (2e−). However, the 4 N site had nearly one negative charge (1e–) for the adsorption of CO2. Moreover, the formation barrier for *COOH was lower on 0 N, 1 N, and SV sites. After *COOH formation, 0 N and 1 N sites still had nearly two negative charges (2e−) (Figures 9H, I). However, the SV site had only ∼1.2e–, resulting in a large formation barrier for the *CO species on the SV site. Therefore, the barriers of electrochemical reactions can be significantly lowered by excessive charge.
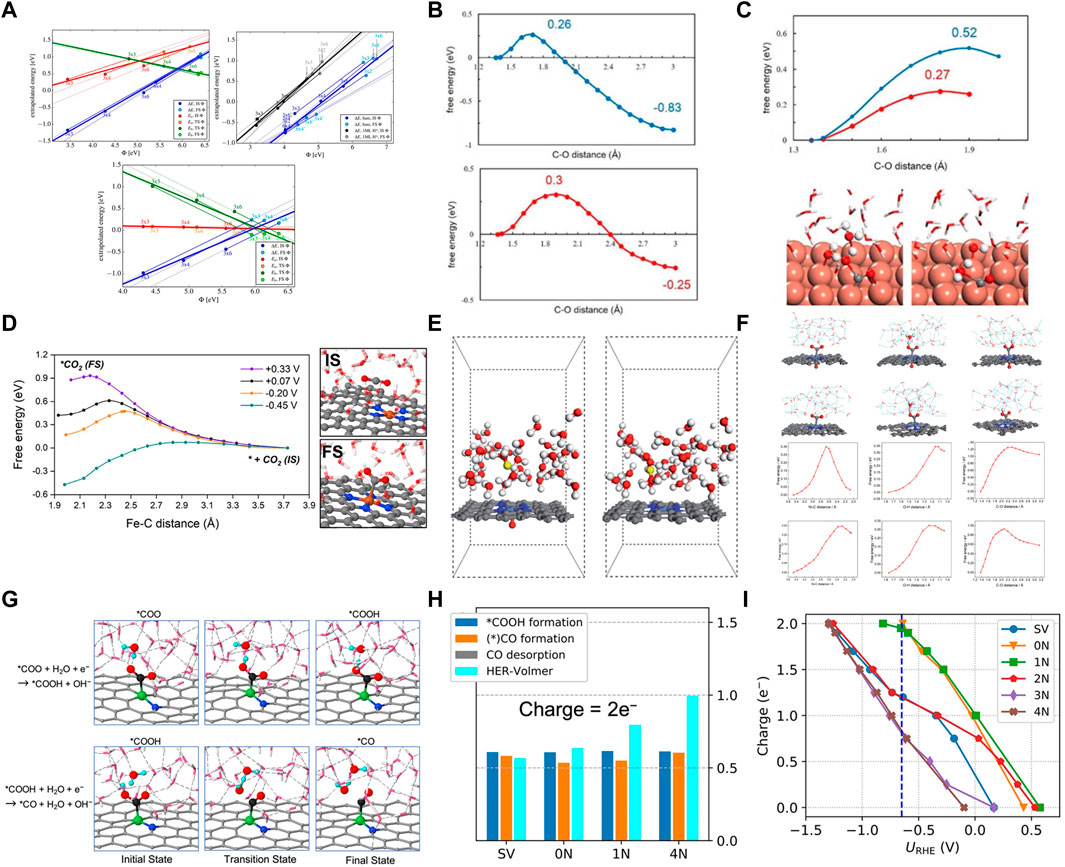
FIGURE 9. (A) Each constant charge barrier/reaction energy is represented by thin solid lines; thin dotted lines stand for beyond these energies. (Figure used with permission from Nørskov et al., 2016) (B) Free energy profiles for the C-O1 (in blue) and C-O2 bond breaking (in red). (C) Free energy profiles for the C-O1 and C-O2 bond breaking and key structures in the C-O1 bond breaking. (Figure used with permission from Sun et al., 2012) (D) Free energy profiles of CO2 adsorption under different potential energy potentials at Fe-N4-C/water interface. Reproduced with permission. (figure used with permission from Wang et al., 2022) (E) Geometric configuration of NiN4–O/C and NiN4/C in an aqueous solution. Grey, C; blue, N; light blue, Ni; red, O; white, H; yellow, hydronium. (F) Snapshots and reaction energies of initial, transition and final states for the *COOH formation on NiN4–O/C and NiN4/C by AIMD simulations. (Figure used with permission from Zhou et al., 2021) (G) Reaction evolution snapshots of *COOH and *CO formation on 1 N site. Color codes: Ni, Green; C, black; O, red; H, cyan spheres. Dashed lines represent hydrogen bonds. (H) Charge = 2e− and (I) Charge capacity for *COOH at different sites under the potentials. (Figure used with permission from Liu et al., 2020).
6 Summary and outlook
SACs have attracted wide interest and have undergone continuous development recently in electrochemical catalysis due to maximum atom utilization and homogeneous active sites. SACs can convert CO2 into more valuable products with high electrocatalytic activity and selectivity. Furthermore, developing non-precious metal SACs is highly attractive in reducing reliance on precious metal catalysts. It not only reduces electrochemical costs, but also changes the monolithic nature of precious metal CO2RR products, which have been intensively explored by researchers. Recently, metal-free catalysts have been applied to CO2RR, exhibiting high activity, stability, and selectivity. However, due to the high free energy of SACs, they can form agglomerates. Therefore, the synergetic effect of DAC circumvents the conventional linear scaling relationship to break through the limitations of SACs. Additionally, DACs show a very high potential to replace SACs in electrocatalysis. Although computational breakthroughs for both SACs and DACs have been made in recent years, there are still many bottlenecks to overcome despite the computational breakthroughs made for SACs and DACs in recent years.
Under practical catalytic conditions, explicit solvation effects and electrode potentials non-negligible factors in catalytic reactions. Therefore, it is necessary to construct an explicit aqueous solvent model and introduce the electrode potential. The kinetic barriers of the reaction were fitted using AIMD and “slow-growth” approaches. They revealed the interaction between the H bond networks and the reactive species and the structure-dependent charge states in the presence of electrode potential, bridging the gap between experiments and computations. In addition, theoretical calculations can be utilized to examine catalytic mechanisms and determine the reaction routes or pathways when experimental methods are insufficient to address related issues. DFT describes the electronic structure and free energy of adsorption for the catalyst, which can be used to determine catalytic performance and reaction potential. DFT has strong directional relevance for mechanism research and performance.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work is financially supported by the National Natural Science Foundation of China (grant no. 22108093), and the Innovation Jiaxing Elite Leadership Plan and Technology Development Project of Jiaxing University (70518040).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andrew, F. A. P., Peterson, A., Studt, F., Rossmeisl, J., and Nørskov, J. K. (2010). How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315. doi:10.1039/C0EE00071J
Back, S., Lim, J., Kim, N. Y., Kim, Y. H., and Juang, Y. (2017). Single-atom catalysts for CO2 electroreduction with significant activity and selectivity improvements. Chem. Sci. 8, 1090–1096. doi:10.1039/C6SC03911A
Bai, X., Zhao, X., Zhang, Y., Ling, C., Zhou, Y., Wang, J., et al. (2022). Dynamic stability of copper single-atom catalysts under working conditions. J. Am. Chem. Soc. 144, 17140–17148. doi:10.1021/jacs.2c07178
Bui, H. T. D., Bui, V. Q., Kim, S. G., Kawazoe, Y., and Lee, H. (2021). Revealing well-defined cluster-supported bi-atom catalysts for enhanced CO2 electroreduction: A theoretical investigation. Phys. Chem. Chem. Phys. 23, 25143–25151. doi:10.1039/D1CP03854K
Cai, Y., Fu, J., Zhou, Y., Chang, Y. C., Min, Q., Zhu, J. J., et al. (2021). Insights on forming N,O-coordinated Cu single-atom catalysts for electrochemical reduction CO2 to methane. Nat. Commun. 12, 586. doi:10.1038/s41467-020-20769-x
Cao, C., Ma, D. D., Gu, J. F., Xie, X., Zeng, G., Li, X., et al. (2020a). Metal-organic layers leading to atomically thin bismuthene for efficient carbon dioxide electroreduction to liquid fuel. Angew. Chem. Int. Ed. Engl. 59, 15014–15020. doi:10.1002/anie.202005577
Cao, Y., Zhang, Z., Chen, J. W., and Wang, Y. G. (2022c). Potential-dependent free energy relationship in interpreting the electrochemical performance of CO2 reduction on single atom catalysts. ACS Catal. 12, 6606–6617. doi:10.1021/acscatal.2c01470
Cao, W., Xia, G. J., Yao, Z., Zeng, K. H., Qiao, Y., and Wang, Y. G. (2023). Aldehyde hydrogenation by Pt/TiO2 catalyst in aqueous phase: Synergistic effect of oxygen vacancy and solvent water. JACS Au 3, 143–153. doi:10.1021/jacsau.2c00560
Cao, Y., Zhao, L., Wulan, B., Tan, D., Chen, Q., Ma, J., et al. (2022b). Atomic bridging structure of nickel-nitrogen-carbon for highly efficient electrocatalytic reduction of CO2. Angew. Chem. Int. Ed. Engl. 61, e202113918. doi:10.1002/anie.202113918
Cao, Y., Meng, Y., Wu, Y., Huang, H., Zhong, W., Shen, Z., et al. (2022a). Metal-free boron nanosheet as “buffer electron pool” for urea and ethanol synthesis via C–N and C–C coupling. J. Mat. Chem. A 10, 23843–23853. doi:10.1039/D2TA06739K
Cao, C., Zhao, C., Fang, Q., Zhong, X., Zhuang, G., Deng, S., et al. (2020b). Hydrogen peroxide electrochemical synthesis on hybrid double-atoms (Pd-Cu) doped N vacancy g-C3N4: A novel design strategy for electrocatalysts screening. J. Mat. Chem. A 8, 2672–2683. doi:10.1039/C9TA12468C
Cepitis, R., Kongi, N., Grozovski, V., Ivaništšev, V., and Lust, E. (2021). Multifunctional electrocatalysis on single-site metal catalysts: A computational perspective. Catalysts 11, 1165. doi:10.3390/catal11101165
Chan, K., and Nørskov, J. K. (2015). Electrochemical barriers made simple. J. Phy. Chem. Lett. 6, 2663–2668. doi:10.1021/acs.jpclett.5b01043
Chan, K., and Norskov, J. K. (2016). Potential dependence of electrochemical barriers from ab initio calculations. J. Phys. Chem. Lett. 7, 1686–1690. doi:10.1021/acs.jpclett.6b00382
Chen, D., Chen, Z., Lu, Z., Tang, J., Zhang, X., and Singh, C. V. (2020a). Computational screening of homo and hetero transition metal dimer catalysts for reduction of CO2 to C2 products with high activity and low limiting potential. J. Mat. Chem. A 8, 21241–21254. doi:10.1039/D0TA05212D
Chen, J. W., Zhang, Z., Yan, H. M., Xia, G. J., Cao, H., and Wang, Y. G. (2022). Pseudo-adsorption and long-range redox coupling during oxygen reduction reaction on single atom electrocatalyst. Nat. Commun. 13, 1734. doi:10.1038/s41467-022-29357-7
Chen, D., Yuan, H., Morozov, S. I., Ge, L., Li, L., Xu, L., et al. (2020b). Design of a graphene nitrene two-dimensional catalyst heterostructure providing a well-defined site accommodating one to three metals, with application to CO2 reduction electrocatalysis for the two-metal case. J. Phys. Chem. Lett. 11, 2541–2549. doi:10.1021/acs.jpclett.0c00642
Cheng, H., Wu, X., Feng, M., Li, X., Lei, G., Fan, Z., et al. (2021). Atomically dispersed Ni/Cu dual sites for boosting the CO2 reduction reaction. ACS Catal. 11, 12673–12681. doi:10.1021/acscatal.1c02319
Chou, L. C., Mohamed, M. G., Kuo, S. W., Nakamura, Y., and Huang, C. F. (2022). Synthesis of multifunctional poly(carbamoyl ester)s containing dual-cleavable linkages and an AIE luminogen via Passerini-type multicomponent polymerization. Chem. Commun. 58, 12317–12320. doi:10.1039/D2CC03829C
Das, A., and Arunan, E. (2019). Chemical bonding in period 2 homonuclear diatomic molecules: A comprehensive relook. J. Chem. Sci. 131, 120. doi:10.1007/s12039-019-1707-5
Deng, J., Li, H., Xiao, J., Tu, Y., Deng, D., Yang, H., et al. (2015). Triggering the electrocatalytic hydrogen evolution activity of the inert two-dimensional MoS2 surface via single-atom metal doping. Energy Environ. Sci. 8, 1594–1601. doi:10.1039/C5EE00751H
Fan, Q., Hou, P., Choi, C., Wu, T. S., Hong, S., Li, F., et al. (2019). Activation of Ni particles into single Ni–N atoms for efficient electrochemical reduction of CO2. Adv. Energy Mat. 10, 1903068. doi:10.1002/aenm.201903068
Fu, Q., and Luo, Y. (2013). Active sites of Pd-doped flat and stepped Cu(111) surfaces for H2 dissociation in heterogeneous catalytic hydrogenation. ACS Catal. 3, 1245–1252. doi:10.1021/cs400267x
Gao, G., Jiao, Y., Waclawik, E. R., and Du, A. (2016a). Single atom (Pd/Pt) supported on graphitic carbon nitride as an efficient photocatalyst for visible-light reduction of carbon dioxide. J. Am. Chem. Soc. 138, 6292–6297. doi:10.1021/jacs.6b02692
Gao, G., O’Mullane, A. P., and Du, A. (2016b). 2D MXenes: A new family of promising catalysts for the hydrogen evolution reaction. ACS Catal. 7, 494–500. doi:10.1021/acscatal.6b02754
Gao, Z., Huang, H., Xu, S., Li, L., Yan, G., Zhao, M., et al. (2020). Regulating the coordination environment through doping N atoms for single-atom Mn electrocatalyst of N2 reduction with high catalytic activity and selectivity: A theoretical study. Mol. Catal. 493, 111091. doi:10.1016/j.mcat.2020.111091
Gong, Q., Ding, P., Xu, M., Zhu, X., Wang, M., Deng, J., et al. (2019). Structural defects on converted bismuth oxide nanotubes enable highly active electrocatalysis of carbon dioxide reduction. Nat. Commun. 10, 2807. doi:10.1038/s41467-019-10819-4
Gong, W., and Kang, L. (2018). BNPd single-atom catalysts for selective hydrogenation of acetylene to ethylene: A density functional theory study. R. Soc. Open Sci. 5, 171598. doi:10.1098/rsos.171598
Guan, A., Chen, Z., Quan, Y., Peng, C., Wang, Z., Sham, T. K., et al. (2020a). Boosting CO2 electroreduction to CH4 via tuning neighboring single-copper sites. ACS Energy Lett. 5, 1044–1053. doi:10.1021/acsenergylett.0c00018
Guan, A., Ciston, J., Bare, S. R., Runnebaum, R. C., Katz, A., Kulkarni, A., et al. (2020b). Supported metal pair-site catalysts. ACS Catal. 10, 9065–9085. doi:10.1021/acscatal.0c02000
Han, L., Song, S., Liu, M., Yao, S., Liang, Z., Cheng, H., et al. (2020a). Stable and efficient single-atom Zn catalyst for CO2 reduction to CH4. J. Am. Chem. Soc. 142, 12563–12567. doi:10.1021/jacs.9b12111
Han, N., Feng, S., Liang, Y., Wang, J., Zhang, C., Fransaer, J., et al. (2023). Achieving efficient electrocatalytic oxygen evolution in acidic media on yttrium ruthenate pyrochlore through cobalt incorporation. Adv. Funct. Mat. 2023, 2208399. doi:10.1002/adfm.202208399
Han, N., Guo, X., Cheng, J., Liu, P., Zhang, S., Huang, S., et al. (2021). Inhibiting in situ phase transition in Ruddlesden-Popper perovskite via tailoring bond hybridization and its application in oxygen permeation. Matter 4, 1720–1734. doi:10.1016/j.matt.2021.02.019
Han, L., Wang, S., Yao, Z., Zhang, W., Zhang, X., Zeng, L., et al. (2020b). Superior three-dimensional perovskite catalyst for catalytic oxidation. EcoMat 2, e12044. doi:10.1002/eom2.12044
Hannagan, R. T., Giannakakis, G., Réocreux, R., Schumann, J. L., Finzel, J., Wang, Y. C., et al. (2021). First-principles design of a single-atom–alloy propane dehydrogenation catalyst. Science 372, 1444–1447. doi:10.1126/science.abg8389
He, Q., Lee, J. H., Liu, D., Liu, Y., Lin, Z., Xie, Z., et al. (2020b). Accelerating CO2 electroreduction to CO over Pd single-atom catalyst. Adv. Funct. Mat. 30, 2000407. doi:10.1002/adfm.202000407
He, T., Santiago, A. R. P., Kong, Y., Ahsan, M. A., Luque, R., Du, A., et al. (2022). Atomically dispersed heteronuclear dual-atom catalysts: A new rising star in atomic catalysis. Small 18, e2106091. doi:10.1002/smll.202106091
He, Q., Zhang, L., Kour, G., and Du, A. (2020a). Electrochemical reduction of carbon dioxide on precise number of Fe atoms anchored graphdiyne. J. CO2 Util. 37, 272–277. doi:10.1016/j.jcou.2019.12.025
He, Y., Zhang, J., Polo-Garzon, F., and Wu, Z. (2023). Adsorbate-induced strong metal-support interactions: Implications for catalyst design. J. Phys. Chem. Lett. 14, 524–534. doi:10.1021/acs.jpclett.2c03391
Heenen, H. H., Gauthier, J. A., Kristoffersen, H. H., Ludwig, T., and Chan, K. (2020). Solvation at metal/water interfaces: An ab initio molecular dynamics benchmark of common computational approaches. J. Chem. Phys. 152, 144703. doi:10.1063/1.5144912
Hossain, M. D., Huang, Y., Yu, T. H., Goddard, W. A., and Luo, Z. (2020). Reaction mechanism and kinetics for CO2 reduction on nickel single atom catalysts from quantum mechanics. Nat. Commun. 11, 2256. doi:10.1038/s41467-020-16119-6
Hou, P., Huang, Y., Ma, F., Wei, X., Du, R., Zhu, G., et al. (2023). S and N coordinated single-atom catalysts for electrochemical CO2 reduction with superior activity and selectivity. Appl. Surf. Sci. 619, 156747. doi:10.1016/j.apsusc.2023.156747
Hu, X., Yao, S., Chen, L. T., Zhang, X., Jiao, M., Lu, Z. Y., et al. (2021). Understanding the role of axial O in CO2 electroreduction on NiN4 single-atom catalysts via simulations in realistic electrochemical environment. J. Mat. Chem. A 9, 23515–23521. doi:10.1039/D1TA07791K
Hu, Y., Li, Z., Li, B., and Yu, C. (2022). Recent progress of diatomic catalysts: General design fundamentals and diversified catalytic applications. Small 18, 2203589. doi:10.1002/smll.202203589
Huang, B., Wu, Y., Luo, Y., and Zhou, N. (2020). Double atom-anchored defective boron nitride catalyst for efficient electroreduction of CO2 to CH4: A first principles study. Chem. Phy. Lett. 756, 137852. doi:10.1016/j.cplett.2020.137852
Huang, Y., Zhu, C., Liao, J., Gu, X. K., and Li, W. X. (2023). First-principles study of the effect of the local coordination environment on the electrochemical activity of Pd1-CxNy single atom catalysts. Chem. Eng. Sci. 270, 118551. doi:10.1016/j.ces.2023.118551
Jiang, Z., Wang, T., Pei, J., Shang, H., Zhou, D., Li, H., et al. (2020). Discovery of main group single Sb–N4 active sites for CO2 electroreduction to formate with high efficiency. Energy Environ. Sci. 13, 2856–2863. doi:10.1039/D0EE01486A
Konsolakis, M., and Ioakeimidis, Z. (2014). Surface/structure functionalization of copper-based catalysts by metal-support and/or metal–metal interactions. Appl. Surf. Sci. 320, 244–255. doi:10.1016/j.apsusc.2014.08.114
Lang, R., Du, X., Huang, Y., Jiang, X., Zhang, Q., Guo, Y., et al. (2020). Single-atom catalysts based on the metal-oxide interaction. Chem. Rev. 120, 11986–12043. doi:10.1021/acs.chemrev.0c00797
Li, F., and Tang, Q. (2021). Understanding trends in the activity and selectivity of bi-atom catalysts for the electrochemical reduction of carbon dioxide. J. Mat. Chem. A 9, 8761–8771. doi:10.1039/D1TA01120K
Li, H., Wang, L., Dai, Y., Pu, Z., Lao, Z., Chen, Y., et al. (2018). Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation. Nat. Nanotechnol. 13, 411–417. doi:10.1038/s41565-018-0089-z
Li, J., Zan, W. Y., Kang, H., Dong, Z., Zhang, X., Lin, Y., et al. (2021b). Graphitic-N highly doped graphene-like carbon: A superior metal-free catalyst for efficient reduction of CO2. Appl. Catal. B Environ. 298, 120510. doi:10.1016/j.apcatb.2021.120510
Li, L., Chang, X., Lin, X., Zhao, Z. J., and Gong, J. (2020a). Theoretical insights into single-atom catalysts. Chem. Soc. Rev. 49, 8156–8178. doi:10.1039/D0CS00795A
Li, M., Wu, S., Yang, X., Hu, J., Peng, L., Bai, L., et al. (2017a). Highly efficient single atom cobalt catalyst for selective oxidation of alcohols. Appl. Catal. A Gen. 543, 61–66. doi:10.1016/j.apcata.2017.06.018
Li, J., Nagarajan, A. V., Alfonso, D. R., Sun, M., Kauffman, D. R., Mpourmpakis, G., et al. (2021a). Boosting CO2 electrochemical reduction with atomically precise surface modification on gold nanoclusters. Angew. Chem. Int. Ed. Engl. 60, 6351–6356. doi:10.1002/anie.202016129
Li, S., Xu, Y. X., Wang, H. W., Teng, B. T., Liu, Q., Li, Q. H., et al. (2022a). Tuning the CO2 hydrogenation selectivity of rhodium single-atom catalysts on zirconium dioxide with alkali ions. Angew. Chem. Int. Ed. 62, e202218167. doi:10.1002/anie.202218167
Li, T., Ren, S., Zhang, C., Qiao, L., Wu, J., He, P., et al. (2023). Cobalt single atom anchored on N-doped carbon nanoboxes as typical single-atom catalysts (SACs) for boosting the overall water splitting. Chem. Eng. J. 458, 141435. doi:10.1016/j.cej.2023.141435
Li, M., Bi, W., Chen, M., Sun, Y., Ju, H., Yan, W., et al. (2017b). Exclusive Ni-N4 sites realize near-unity CO selectivity for electrochemical CO2 reduction. J. Am. Chem. Soc. 139, 14889–14892. doi:10.1021/jacs.7b09074
Li, L., Chen, C., Cao, R., Pan, Z., He, H., and Zhou, K. (2020b). Dual-atom Ag2/graphene catalyst for efficient electroreduction of CO2 to CO. Appl. Catal. B Environ. 268, 118747. doi:10.1016/j.apcatb.2020.118747
Li, S., Jiang, J., Liu, X., Zhu, Z., Wang, J., He, Q., et al. (2022b). Coupling atomically dispersed Fe-N(5) sites with defective N-doped carbon boosts CO2 electroreduction. Small 18, e2203495. doi:10.1002/smll.202203495
Liang, X. M., Wang, H. J., Zhang, C., Zhong, D. C., and Lu, T. B. (2023). Controlled synthesis of a Ni2 dual-atom catalyst for synergistic CO2 electroreduction. Appl. Catal. B Environ. 322, 122073. doi:10.1016/j.apcatb.2022.122073
Ling, C., Shi, L., Ouyang, Y., Zeng, X. C., and Wang, J. (2017). Nanosheet supported single-metal atom bifunctional catalyst for overall water splitting. Nano. Lett. 17, 5133–5139. doi:10.1021/acs.nanolett.7b02518
Ling, Y., Ge, H., Chen, J., Zhang, Y., Duan, Y., Liang, M., et al. (2022). General strategy toward hydrophilic single atom catalysts for efficient selective hydrogenation. Adv. Sci. 9, e2202144. doi:10.1002/advs.202202144
Lininger, C. N., Gauthier, J. A., Li, W. L., Rossomme, E., Welborn, V. V., Lin, Z., et al. (2021). Challenges for density functional theory: Calculation of CO adsorption on electrocatalytically relevant metals. Phys. Chem. Chem. Phys. 23, 9394–9406. doi:10.1039/D0CP03821K
Liu, H., Huang, Q., An, W., Wang, Y., Men, Y., and Liu, S. (2021). Dual-atom active sites embedded in two-dimensional C2N for efficient CO2 electroreduction: A computational study. J. Energy Chem. 61, 507–516. doi:10.1016/j.jechem.2021.02.007
Liu, H., Rong, H., and Zhang, J. (2022). Synergetic dual-atom catalysts: The next boom of atomic catalysts. Chem. Sus Chem. 15, e202200498. doi:10.1002/cssc.202200498
Liu, T., Zhao, X., Liu, X., Xiao, W., Luo, Z., Wang, W., et al. (2023). Understanding the hydrogen evolution reaction activity of doped single-atom catalysts on two-dimensional GaPS4 by DFT and machine learning. J. Energy Chem. 81, 93–100. doi:10.1016/j.jechem.2023.02.018
Liu, Y., Zhao, J., and Cai, Q. (2016). Pyrrolic-nitrogen doped graphene: A metal-free electrocatalyst with high efficiency and selectivity for the reduction of carbon dioxide to formic acid: A computational study. Phys. Chem. Chem. Phys. 18, 5491–5498. doi:10.1039/C5CP07458D
Lu, M., Wang, C., Ding, Y., Peng, M., Zhang, W., Li, K., et al. (2019). Fe-N/C single-atom catalysts exhibiting multienzyme activity and ROS scavenging ability in cells. Chem. Commun. 55, 14534–14537. doi:10.1039/c9cc07408b
Luo, G., Yu, J., and Li, Y. (2020). Rational design of dual-metal-site catalysts for electroreduction of carbon dioxide. J. Mat. Chem. A 8, 15809–15815. doi:10.1039/D0TA00033G
Lv, L., Shen, Y., Liu, J., Meng, X., Gao, X., Zhou, M., et al. (2021). Computational screening of high activity and selectivity TM/g-C3N4 single-atom catalysts for electrocatalytic reduction of nitrates to ammonia. J. Phys. Chem. Lett. 12, 11143–11150. doi:10.1021/acs.jpclett.1c03005
Mochizuki, C., Inomata, Y., Yasumura, S., Lin, M., Taketoshi, A., Honma, T., et al. (2022). Defective NiO as a stabilizer for Au single-atom catalysts. ACS Catal. 12, 6149–6158. doi:10.1021/acscatal.2c00108
Natsui, K., Iwakawa, H., Ikemiya, N., Nakata, K., and Einaga, Y. (2018). Stable and highly efficient electrochemical production of formic acid from carbon dioxide using diamond electrodes. Angew. Chem. Int. Ed. Engl. 57, 2639–2643. doi:10.1002/anie.201712271
Niu, K., Chi, L., Rosen, J., and Bjork, J. (2022). Termination-accelerated electrochemical nitrogen fixation on single-atom catalysts supported by MXenes. J. Phys. Chem. Lett. 13, 2800–2807. doi:10.1021/acs.jpclett.2c00195
Ouyang, Y., Shi, L., Bai, X., Li, Q., and Wang, J. (2020). Breaking scaling relations for efficient CO2 electrochemical reduction through dual-atom catalysts. Chem. Sci. 11, 1807–1813. doi:10.1039/C9SC05236D
Pan, Y., Lin, R., Chen, Y., Liu, S., Zhu, W., Cao, X., et al. (2018). Design of single-atom Co-N(5) catalytic site: A robust electrocatalyst for CO2 reduction with nearly 100% CO selectivity and remarkable stability. J. Am. Chem. Soc. 140, 4218–4221. doi:10.1021/jacs.8b00814
Posada-Pérez, S., Vidal-Lopez, A., Solà, M., and Poater, A. (2023). 2D carbon nitride as a support with single Cu, Ag, and Au atoms for carbon dioxide reduction reaction. Phys. Chem. Chem. Phys. 2023, 392. doi:10.1039/D3CP00392B
Pu, Z., Amiinu, I. S., Cheng, R., Wang, P., Zhang, C., Mu, S., et al. (2020). Single-atom catalysts for electrochemical hydrogen evolution reaction: Recent advances and future perspectives. Nanomicro. Lett. 12, 21. doi:10.1007/s40820-019-0349-y
Qiao, B., Wang, A., Yang, X., Allard, L. F., Jiang, Z., Cui, Y., et al. (2011). Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641. doi:10.1038/nchem.1095
Qin, G., Cui, Q., Du, A., and Sun, Q. (2020). Borophene: A metal-free and metallic electrocatalyst for efficient converting CO2 into CH4. Chem. Cat. Chem. 12, 1483–1490. doi:10.1002/cctc.201902094
Qin, Y. Y., and Su, Y. Q. (2021). A DFT study on heterogeneous Pt/CeO2 (110) single atom catalysts for CO oxidation. Chem. Cat. Chem. 13, 3857–3863. doi:10.1002/cctc.202100643
Ralser, S., Kaiser, A., Probst, M., Postler, J., Renzler, M., Bohme, D. K., et al. (2016). Experimental evidence for the influence of charge on the adsorption capacity of carbon dioxide on charged fullerenes. Phys. Chem. Chem. Phys. 18, 3048–3055. doi:10.1039/C5CP06587A
Ren, Y., Sun, X., Qi, K., and Zhao, Z. (2022). Single atom supported on MoS2 as efficient electrocatalysts for the CO2 reduction reaction: A DFT study. Appl. Surf. Sci. 602, 154211. doi:10.1016/j.apsusc.2022.154211
Shang, H., Wang, T., Pei, J., Jiang, Z., Zhou, D., Wang, Y., et al. (2020). Design of a single-atom indium(delta+) -N4 interface for efficient electroreduction of CO2 to formate. Angew. Chem. Int. Ed. Engl. 59, 22465–22469. doi:10.1002/anie.202010903
Sheng, T., and Sun, S. G. (2017). Electrochemical reduction of CO2 into CO on Cu(100): A new insight into the C-O bond breaking mechanism. Chem. Commun. 53, 2594–2597. doi:10.1039/C6CC08583K
Song, X., Li, N., Zhang, H., Wang, L., Yan, Y., Wang, H., et al. (2020). Graphene-supported single nickel atom catalyst for highly selective and efficient hydrogen peroxide production. ACS Appl. Mat. Interfaces 12, 17519–17527. doi:10.1021/acsami.0c01278
Sreekanth, N., Nazrulla, M. A., Vineesh, T. V., Sailaja, K., and Phani, K. L. (2015). Metal-free boron-doped graphene for selective electroreduction of carbon dioxide to formic acid/formate. Chem. Commun. 51, 16061–16064. doi:10.1039/C5CC06051F
Sun, G. D., Cao, Y. Y., Li, D. Q., Hu, M. Z., Liang, X. H., Wang, Z., et al. (2023). Dual-atom Cu2/N-doped carbon catalyst for electroreduction of CO2 to C2H4. Appl. Catal. A Gen. 651, 119025. doi:10.1016/j.apcata.2023.119025
Sun, K., Yu, K., Fang, J., Zhuang, Z., Tan, X., Wu, Y., et al. (2022). Nature-inspired design of molybdenum–selenium dual-single-atom electrocatalysts for CO2 reduction. Adv. Mat. 34, 2206478. doi:10.1002/adma.202206478
Sun, M. J., Gong, Z. W., Yi, J. D., Zhang, T., Chen, X., and Cao, R. (2020). A highly efficient diatomic nickel electrocatalyst for CO2 reduction. Chem. Commun. 56, 8798–8801. doi:10.1039/D0CC03410J
Takele Menisa, L., Cheng, P., Qiu, X., Zheng, Y., Huang, X., Gao, Y., et al. (2022). Single atomic Fe-N(4) active sites and neighboring graphitic nitrogen for efficient and stable electrochemical CO2 reduction. Nanoscale Horiz. 7, 916–923. doi:10.1039/D2NH00143H
Talib, S. H., Lu, Z., Yu, X., Ahmad, K., Bashir, B., Yang, Z., et al. (2021). Theoretical inspection of M1/PMA single-atom electrocatalyst: Ultra-high performance for water splitting (HER/OER) and oxygen reduction reactions (OER). ACS Catal. 11, 8929–8941. doi:10.1021/acscatal.1c01294
Tan, X., Tahini, H. A., and Smith, S. C. (2017). Borophene as a promising material for charge-modulated switchable CO2 capture. ACS Appl. Mater Interfaces 9, 19825–19830. doi:10.1021/acsami.7b03676
Tang, M., Shen, H. M., Xie, H. H., and Sun, Q. (2020). Metal-free catalyst B2S sheet for effective CO2 electrochemical. Chem. Cat. Chem. 21, 779–784. doi:10.1002/cphc.202000006
Tang, T., Wang, Z., and Guan, J. (2022). Optimizing the electrocatalytic selectivity of carbon dioxide reduction reaction by regulating the electronic structure of single-atom M-N-C materials. Adv. Funct. 32, 2111504. doi:10.1002/adfm.202111504
Wan, J., Zheng, J., Zhang, H., Wu, A., and Li, X. (2022). Single atom catalysis for electrocatalytic ammonia synthesis. Catal. Sci. Technol. 12, 38–56. doi:10.1039/D1CY01442K
Wang, H., Ren, G., Zhao, Y., Sun, L., Sun, J., Yang, L., et al. (2022). In silico design of dual-doped nitrogenated graphene (C2N) employed in electrocatalytic reduction of carbon monoxide to ethylene. J. Mat. Chem. A 10, 4703–4710. doi:10.1039/d1ta09847k
Wang, L., Zhang, W., Wang, S., Gao, Z., Luo, Z., Wang, X., et al. (2016). Atomic-level insights in optimizing reaction paths for hydroformylation reaction over Rh/CoO single-atom catalyst. Nat. Commun. 7, 14036. doi:10.1038/ncomms14036
Wang, Y., Cao, L., Libretto, N. J., Li, X., Li, C., Wan, Y., et al. (2019). Ensemble effect in bimetallic electrocatalysts for CO2 reduction. J. Am. Chem. Soc. 141, 16635–16642. doi:10.1021/jacs.9b05766
Wang, Y., Su, H., He, Y., Li, L., Chen, S., Wu, G., et al. (2020). Advanced electrocatalysts with single-metal-atom active sites. Chem. Rev. 120, 12217–12314. doi:10.1021/acs.chemrev.0c00594
Wu, J., Liu, M., Sharma, P. P., Yadav, R. M., Ma, L., Yang, Y., et al. (2016). Incorporation of nitrogen defects for efficient reduction of CO2 via two-electron pathway on three-dimensional graphene foam. Nano. Lett. 16, 466–470. doi:10.1021/acs.nanolett.5b04123
Wu, Q. L., Shen, C. Y., Rui, N., Sun, K. H., and Liu, C. J. (2021). Experimental and theoretical studies of CO2 hydrogenation to methanol on Ru/In2O3. J. CO2 Util. 53, 101720. doi:10.1016/j.jcou.2021.101720
Xie, H., Wang, F., Liu, T., Wu, Y., Lan, C., Chen, B., et al. (2021). Copper−iron dimer for selective C–C coupling in electrochemical CO2 reduction. Electrochimica Acta 380, 138188. doi:10.1016/j.electacta.2021.138188
Xu, H., Zhao, Y., Wang, Q., He, G., and Chen, H. (2022). Supports promote single-atom catalysts toward advanced electrocatalysis. Coord. Chem. Rev. 451, 214261. doi:10.1016/j.ccr.2021.214261
Xu, L., Yang, L. M., and Ganz, E. (2021). Electrocatalytic reduction of N2 using metal-doped borophene. ACS Appl. Mater Interfaces 13, 14091–14101. doi:10.1021/acsami.0c20553
Xue, Z., Zhang, X., Qin, J., and Liu, R. (2021). TMN4 complex embedded graphene as bifunctional electrocatalysts for high efficiency OER/ORR. J. Energy Chem. 55, 437–443. doi:10.1016/j.jechem.2020.07.018
Yang, T. T., Tian, N., Liu, X. H., and Zhang, X. (2022a). CuNi alloy nanoparticles embedded in N-doped carbon framework for electrocatalytic reduction of CO2 to CO. J. All. Comp. 904, 164042. doi:10.1016/j.jallcom.2022.164042
Yang, W., Zhao, M., Ding, X., Ma, K., Wu, C., Gates, I. D., et al. (2020). The effect of coordination environment on the kinetic and thermodynamic stability of single-atom iron catalysts. Phys. Chem. Chem. Phys. 22, 3983–3989. doi:10.1039/C9CP05349B
Yang, T. T., Wang, J., Shu, Y., Ji, Y., Dong, H., and Li, Y. (2022b). Significance of density functional theory (DFT) calculations for electrocatalysis of N2 and CO2 reduction reactions. Phys. Chem. Chem. Phys. 24, 8591–8603. doi:10.1039/D1CP05442B
Ying, Y., Luo, X., Qiao, J., and Huang, H. (2021). “More is different:” synergistic effect and structural engineering in double-atom catalysts. Adv. Funct. Mat. 31, 2007423. doi:10.1002/adfm.202007423
Zeng, H., Liu, X., Chen, F., Chen, Z., Fan, X., and Lau, W. (2020). Single atoms on a nitrogen-doped boron phosphide monolayer: A new promising bifunctional electrocatalyst for ORR and OER. ACS Appl. Mater Interfaces 12, 52549–52559. doi:10.1021/acsami.0c13597
Zhang, B., Liu, J., Wang, J., Ruan, Y., Ji, X., Xu, K., et al. (2017). Interface engineering: The Ni(OH)2/MoS2 heterostructure for highly efficient alkaline hydrogen evolution. Nano Energy 37, 74–80. doi:10.1016/j.nanoen.2017.05.011
Zhang, Y., Zheng, X., Cui, X., Wang, J., Liu, J., Chen, J., et al. (2022c). Doping of vanadium into bismuth oxide nanoparticles for electrocatalytic CO2 reduction. ACS Appl. Nano Mat. 5, 15465–15472. doi:10.1021/acsanm.2c03503
Zhang, J., Huang, Q., Wang, J., Wang, J., Zhang, J., and Zhao, Y. (2020). Supported dual-atom catalysts: Preparation, characterization, and potential applications. Chin. J. Catal. 41, 783–798. doi:10.1016/S1872-2067(20)63536-7
Zhang, J., Zhao, Y., Guo, X., Chen, C., Dong, C. L., Liu, R. S., et al. (2018). Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction. Nat. Catal. 1, 985–992. doi:10.1038/s41929-018-0195-1
Zhang, M., Lai, C., Li, B., Liu, S., Huang, D., Xu, F., et al. (2021b). MXenes as superexcellent support for confining single atom: Properties, synthesis, and electrocatalytic applications. Small 17, e2007113. doi:10.1002/smll.202007113
Zhang, M., Yang, W., Liang, Y., Yang, X., Cao, M., and Cao, R. (2021a). Template-free synthesis of non-noble metal single-atom electrocatalyst with N-doped holey carbon matrix for highly efficient oxygen reduction reaction in zinc-air batteries. Appl. Catal. B 285, 119780. doi:10.1016/j.apcatb.2020.119780
Zhang, M., Walsh, A. G., Yu, J., and Zhang, P. (2021d). Single-atom alloy catalysts: Structural analysis, electronic properties and catalytic activities. Chem. Soc. Rev. 50, 569–588. doi:10.1039/d0cs00844c
Zhang, M., Fu, Q., Luo, Q., Sheng, L., and Yang, J. (2021c). Understanding single-atom catalysis in view of theory. JACS Au 1, 2130–2145. doi:10.1021/jacsau.1c00384
Zhang, Y., Yang, J., Ge, R., Zhang, J., Cairney, J. M., Li, Y., et al. (2022a). The effect of coordination environment on the activity and selectivity of single-atom catalysts. Coord. Chem. Rev. 461, 214493. doi:10.1016/j.ccr.2022.214493
Zhang, Y., Yang, R., Li, H., and Zeng, Z. (2022b). Boosting electrocatalytic reduction of CO2 to HCOOH on Ni single atom anchored WTe2 monolayer. Small 18, e2203759. doi:10.1002/smll.202203759
Zhao, X., Levell, Z. H., Yu, S., and Liu, Y. (2022b). Atomistic understanding of two-dimensional electrocatalysts from first principles. Chem. Rev. 122, 10675–10709. doi:10.1021/acs.chemrev.1c00981
Zhao, X., and Liu, Y. (2021). Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc. 143, 9423–9428. doi:10.1021/jacs.1c02186
Zhao, X., and Liu, Y. (2020). Unveiling the active structure of single nickel atom catalysis: Critical roles of charge capacity and hydrogen bonding. J. Am. Chem. Soc. 142, 5773–5777. doi:10.1021/jacs.9b13872
Zhao, X., Zhao, K., Liu, Y., Su, Y., Chen, S., Yu, H., et al. (2022a). Highly efficient electrochemical CO2 reduction on a precise homonuclear diatomic Fe−Fe catalyst. ACS Catal. 12, 11412–11420. doi:10.1021/acscatal.2c03149
Zheng, G., Li, L., Hao, S., Zhang, X., Tian, Z., and Chen, L. (2020). Double atom catalysts: Heteronuclear transition metal dimer anchored on nitrogen-doped graphene as superior electrocatalyst for nitrogen reduction reaction. Adv. Theory Simul. 3, 2000190. doi:10.1002/adts.202000190
Zhou, X., Liu, X., Zhang, J., Zhang, C., Yoo, S. J., Kim, J. G., et al. (2020a). Highly-dispersed cobalt clusters decorated onto nitrogen-doped carbon nanotubes as multifunctional electrocatalysts for OER, HER and ORR. Carbon 166, 284–290. doi:10.1016/j.carbon.2020.05.037
Zhou, X., Song, E., Chen, W., Segre, C. U., Zhou, J., Lin, Y. C., et al. (2020b). Dual-metal interbonding as the chemical facilitator for single-atom dispersions. Adv. Mater 32, e2003484. doi:10.1002/adma.202003484
Zhou, X., Xu, Z., Xu, M., Zhou, X., and Wu, K. (2020c). A perspective on oxide-supported single-atom catalysts. Nanoscale Adv. 2, 3624–3631. doi:10.1039/D0NA00393J
Zhu, T., Chen, Q., Liao, P., Duan, W., Liang, S., Yan, Z., et al. (2020). Single-atom Cu catalysts for enhanced electrocatalytic nitrate reduction with significant alleviation of nitrite production. Small 16, e2004526. doi:10.1002/smll.202004526
Keywords: single-atom catalysts, double-atom catalysts, theoretical calculations, CO2RR, electrode potential, solvent effect
Citation: Meng Y, Huang H, Zhang Y, Cao Y, Lu H and Li X (2023) Recent advances in the theoretical studies on the electrocatalytic CO2 reduction based on single and double atoms. Front. Chem. 11:1172146. doi: 10.3389/fchem.2023.1172146
Received: 23 February 2023; Accepted: 13 March 2023;
Published: 28 March 2023.
Edited by:
Huayang Zhang, University of Adelaide, AustraliaReviewed by:
Ning Han, KU Leuven, BelgiumShifei Kang, University of Shanghai for Science and Technology, China
Copyright © 2023 Meng, Huang, Zhang, Cao, Lu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongyong Cao, Y3l5QHpqeHUuZWR1LmNu; Hanfeng Lu, bHVoZkB6anV0LmVkdS5jbg==; Xi Li, eGlsaTIxQG1haWwuemp4dS5lZHUuY24=