 Maciej Spiegel
Maciej Spiegel Giada Ciardullo
Giada Ciardullo Tiziana Marino
Tiziana Marino Nino Russo
Nino Russo- 1Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Rende, Cosenza, Italy
- 2Department of Pharmacognosy and Herbal Medicines, Wroclaw Medical University, Wroclaw, Poland
In the present work, we report a computational study on some important chemical properties of the flavonoid isorhamnetin, used in traditional medicine in many countries. In the course of the study we determined the acid-base equilibria in aqueous solution, the possible reaction pathways with the •OOH radical and the corresponding kinetic constants, the complexing capacity of copper ions, and the reduction of these complexes by reducing agents such as superoxide and ascorbic anion by using density functional level of theory Density Functional Theory. Finally, the non-covalent inhibition ability of the SARS-CoV-2 main protease enzyme by isorhamnetin was examined by molecular dynamics (MD) and docking investigation.
1 Introduction
A commonly accepted definition of oxidative stress is an imbalance between the production of reactive free radicals and the ability of the organism to inactivate thembefore their excessive production becomes harmful (Sies, 1985; Sies et al., 2017). In other words, it is defined when the action of oxidizing agents is not effectively counteracted by molecules present in our body that have antioxidant capacities.
Oxidative stress damages cells and organs, and is a direct or indirect cause of many conditions, ranging from cancer to atherosclerosis, neurodegenerative disorders (e.g., Alzheimer’s and others) and pulmonary diseases (Forman and Zhang, 2021). For these reasons, an enormous amount of scientific work in recent decades has concerned both the specific damage caused by oxidative stress and the chemical mechanisms of action underlying oxidative processes, as well as compounds capable of preventing them.
Considerable attention has been paid to understanding the structures, chemical-physical properties and mechanisms of action of both natural and synthetic antioxidant compounds (Leopoldini et al., 2011; Galano et al., 2016a; Apak et al., 2016). Many antioxidants originate from the plant kingdom (e.g., fruits, cereals, vegetables, and plants) and have been used in traditional medicinal systems of European, African, Asian, and American medicine for many centuries. In particular, flavonoid compounds of natural origin have proven to be powerful antioxidant systems, suitable for combating oxidative stress. Their actions are multidirectional, but the most important can be outlined as:
1) direct scavenging of free radicals, also known as primary antioxidation, which essentially consists of a reaction between a powerful free radical and an antioxidant with the formation of a new less active radical, and reaction can proceed until the formation of a neutral specie (Milenković et al., 2020). This reaction can occur through different mechanisms such as transferring a hydrogen atom or an electron or both from the antioxidant to the free radical (HAT and SET), or sequential proton-loss electron transfer (SPLET), or by bonding the radical to the structure of the antioxidant (RAF) (Galano et al., 2016b; Spiegel, 2022):
2) the capacity of the compound to chelate copper and iron ions responsible for the production of free radicals through the Fenton’s reaction. It is also referred to as a secondary antioxidant effect and is also relevant to the possible treatment of Alzheimer’s disease based on complexation of these metals (Sharma et al., 2013; Spiegel et al., 2022a):
In addition, some antioxidants are effective inhibitor of a number of enzymes implicated in various diseases, including SARS-CoV-2 (Yu et al., 2012; Puttaswamy et al., 2020; Xu et al., 2020).
In this article, we report a theoretical (DFT) and computations (MD and docking) investigation that can contribute to a better understanding of the mechanisms of action of isorhamnetin antioxidant flavonoid (H4Iso), which contains hydroxyl groups in its backbone, which are the main structural elements on which the antioxidant capacity of polyphenols rely. Isorhamnetin is present in the juice of Chinese sea buckthorme (Pengfei et al., 2009), and can also be extracted from the fruits, flowers and leaves of Ginkgo biloba, A. roxburghii, and Hippophae rhmnoides L. (Teng and Luan, 2016; Gong et al., 2020). Furthermore, the great importance of isorhamnetin is due to the fact that it is a metabolite of quercetin, converted by the enzyme catechol-O-metyltransferase. This means that isorhamnetin concentrations in human plasma high, even when only quercetin is taken (Manach et al., 1997).
In order to highlight the performance of theoretical methods in reproducing and predicting antioxidant properties and related mechanisms in environments simulating physiological ones (aqueous and lipid), the paper will examine: 1) the acid-base equilibria in water solvent, 2) the reactions between the •OOH radical and isorhamnetin, 3) the chelating capacity, and 4) the inhibition process of the SARS-CoV-2 main protease enzyme.
2 Methods and computational details
All the computations were performed using Density Functional Theory (DFT) implemented in Gaussian 09 code (Frisch et al., 2014) and following the QM-ORSA (Quantum Mechanics-Based Test for Overall Free Radical Scavenging Activity) computational protocol (Galano and Alvarez-Idaboy, 2013). We used the M06-2X (Zhao et al., 2006) exchange-correlation functional, the 6-311+G (d,p) basis set and the solvation model based on density (SMD) (Marenich et al., 2009), previously tested and applied to a series of molecules with antioxidant properties (Belcastro et al., 2006; Alberto et al., 2013; Ngo et al., 2019; Castaneda-Arriaga et al., 2020; Parise et al., 2021a; Reina et al., 2021; Spiegel et al., 2021; Spiegel, 2022). Pentylethanoate (ε = 4.7) and water (ε = 78.4) was selected to simulate the physiological relevant environments. The geometries of minima (reactants and products) and transition states (TS) located along the considered reaction pathways were optimized and characterized by computing vibrational frequencies and establishing the intrinsic reaction coordinates (IRC).
For the open-shell systems, the unrestricted procedure was used. The pKa values and molar fractions for neutral and charged species were determined according to the methodology proposed and tested earlier (Galano et al., 2016a).
To compute kinetic rate constants conventional transition state theory ((Evans and Polanyi, 1935; Truhlar et al., 1996) was used, and for reactions close to the diffusion limit, the Collins–Kimball theory was applied (Collins and Kimball, 1949). For SET reactions, energy barriers were calculated adopting the Marcus theory (Marcus, 1993). The intrinsic reactivity indices such as bond dissociation energy (BDE), ionization potential (IP), proton affinity (PA) and proton desorption energy (PDE) were estimated under the adiabatic approximation with the following values of solvation enthalpies of H+ (ΔH(H+) = 1,055.7 kJ/mol) and electron (ΔH (e−) = 77.5 kJ/mol. (Markovic et al., 2016). To visualize the structures the MarvinSketch version 21.15.0 software (ChemAxon) was used.
The initial unbound structure of the SARS-CoV-2 main protease (Mpro) has been obtained using the crystal structure of main protease bound to non-covalent inhibitor (PDB code 6W63) (https://www.wwpdb.org/pdb?id=pdb_00006w63, 2020), removing the inhibitor from the crystallographic structure and adding the hydrogen atoms by using H++ (Anandakrishnan et al., 2012) and to calculate the protonation states of titratable residues at pH 7.4. Both the protonation states of the His41 catalytic residue neutral, with the hydrogen on the Nδ and Nε side chain have been considered. 300 ns MDs were performed, by using AMBER16 code (Case et al., 2017) and the FF14SB force field (Maier et al., 2021), for the two protonation states of the unbound Mpro considering a solvated orthorhombic box with a buffer of 10 Å, using TIP3P water model and the following other conditions: integration step of 2 fs coupling SHAKE algorithm; NPT ensemble at 1 bar pressure using the Berendsen barostat (Berendsen et al., 1998) with a time constant τp = 2.0 ps. The Particle mesh Ewald summation method (Darden et al., 1998) has been employed for the electrostatic potential long-range interactions with a 12 Å cutoff distance. In order to select different representative conformations of the system, root-mean square deviation (RMSD) based clustering of the whole trajectory has been performed using the agglomerative bottom-up approach available in Amber16 tools. After removing overall rotations and translations by RMS fitting the Cα atoms’ positions of the trajectory, the average linkage clustering algorithm has been applied, identifying 10 representative conformations of the protein. The complete MD analysis of the unbound Mpro has been reported in our previous study (Parise et al., 2021b).
The representative structure of the unbound Mpro, having the hydrogen on the Nδ of His41, and with the highest percentage of population was considered for the molecular docking approach performed by using AutoDock Vina (version 4.2) (Trott and Olson, 2010). The ligand and the SARS-CoV-2 Mpro models were processed using the AutoDock tools (ADT) to obtain the PDBQT (Protein Data Bank, Partial Charge (Q), and Atom Type (T)) coordinate files containing the information, namely polar hydrogen atoms, partial charges, correct atom types, and information on the articulation of flexible molecules. In particular, Gasteiger-Marsili charges were loaded in ADT. The substrate has been docked into a section of the crystal representing the minimal unit of ligand (37.5 × 15.2 × 37.6 Å) adopted in the simulations. The dimension box of 40 × 40 × 40 Å has been chosen to abundant cover the active site.
Before MD simulations of the Mpro:H4Iso/Mpro:H3Iso− complexes, it was necessary to obtain H4Iso and H3Iso− parameters. At this purpose gas-phase geometry optimization has been carried out using B3LYP/6-31G*. Atomic charges were derived by fitting the electrostatic potential according to the Merz–Singh–Kollman scheme, (Bayly et al., 1993), using the RESP procedure. Antechamber and parmchk modules of Amber16 (Case et al., 2017) have been employed to generate preparatory files to perform molecular mechanics (MM) relaxation of the complexes. 4 Na+ counter ions were added to neutralize the system for Mpro:H4Iso and 5 Na+ for Mpro:H3Iso−. The production step of 100 ns for the both complexes (Mpro:H4Iso and Mpro:H3Iso−) was performed using the same procedure of the apo-form simulation.
RMSD-based clustering of the entire trajectories was performed according to the relaxed complex scheme (RCS) protocol implemented in Amber 16 (Case et al., 2017) to provide a sampled and energetically accessible conformational ensemble. After removing the overall rotations and translations by RMS fitting of the positions of the Cα atoms of the trajectory, the average binding clustering algorithm implemented in cpptraj was applied to identify 10 clusters of representative conformations of the protein, described in Supplementary Table S4.
The resulting MD trajectories were used to assess the magnitude of structural changes in terms of root mean square deviation (RMSD), propensity of a given residue or region to shift, and root mean square fluctuation (RMSF).
The binding free energies between the ligand (H4Iso or H3Iso−) and Mpro were calculated by solving the linearised Poisson-Boltzman equation using the MM-PBSA (Molecular mechanics-Poisson Boltzman surface area) method, as implemented in the Amber code 16 (Case et al., 2017). The igb flag value of five associated with a salt concentration of 0.1 M was used. For the calculations, 100 frames of each MD trajectory over the last 50 ns were analysed.
3 Results and discussion
3.1 Chemical equilibria in water
In understanding the chemical behavior of substances in aqueous solution, it is fundamental to know the relative acid-base equilibria. Sometimes the very low solubility in water of various antioxidants makes experimental measurements difficult. Modern computational chemistry offers a suitable alternative to such situations (Galano et al., 2016b). Figure 1 shows possibly present neutral and charged species of isorhamnetin in water solution, reported along with their pKa values. The distribution diagram of isorhamnetin species as a function of pH is given in the supporting information section (Supplementary Figure S1).
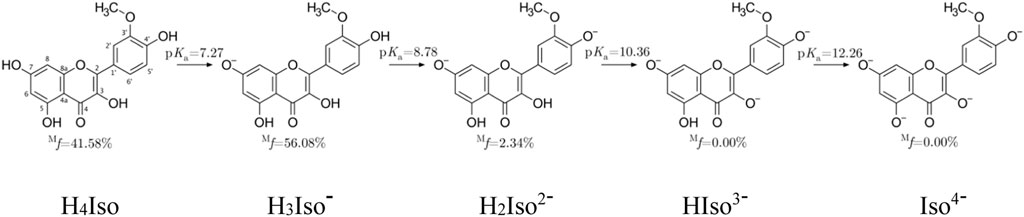
FIGURE 1. Dissociation constants and molar fractions for isorhamnetin species at pH = 7.4.
The lowest pKa value (7.27) was associated with the deprotonation of the–OH group located at the C7 position, followed by C4' (8.78), C3 (10.36), and C5 (12.26). Also, for structurally similar scutellarein, chrysin and quercetin, deprotonation at the C7 position is preferred. Another important data useful for studying antioxidant properties in water is the molar distribution at physiological pH. Figure 1 evidences that the neutral (41.58%) and mono-anion (56.08%) species are found in the highest molar fractions. Contrary to these, the di-anion form is present at much lower, yet still non-negligible amount (2.34%). Therefore, these three species must be considered in the investigation.
3.2 Thermodynamic descriptors
Some indication on the occurrence of possible reaction pathways between radicals and antioxidant compounds can be obtained from adiabatic energy computations of certain molecular indicators, such as:
1) ionization energy (IP):
2) proton affinity (PA):
3) bond dissociation energy (BDE):
4) proton desorption energy (PDE):
The obtained values are collected in Table 1.
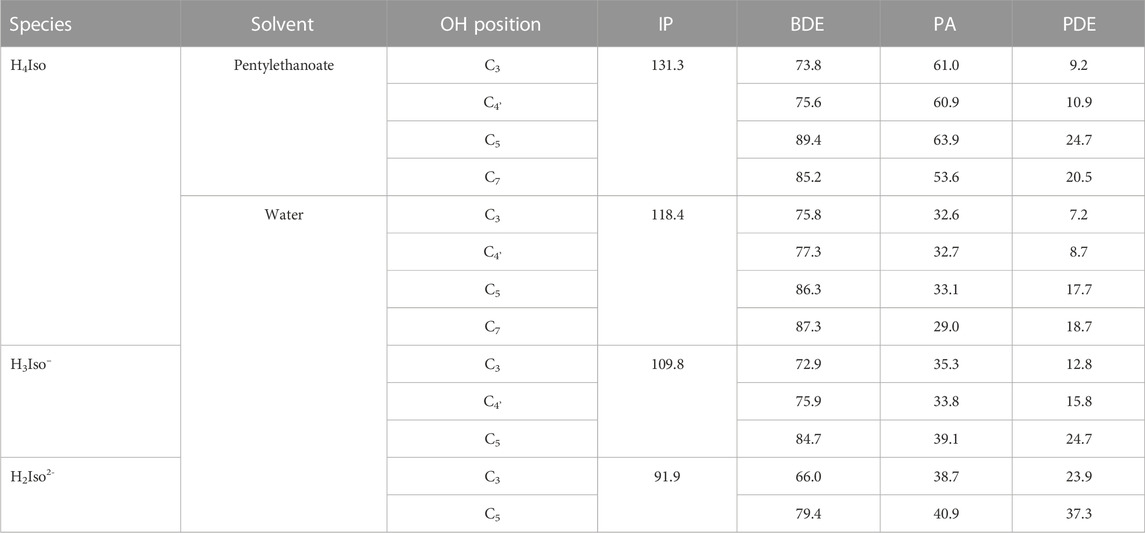
TABLE 1. IP, BDE, PA, and PDE values calculated for neutral and charged isorhamnetin species in water and pentylethanoate solvents. All values are in kcal/mol.
In the pentylethanoate solvent, commonly chosen to mimic a lipid-like environment, the BDE values indicate that the preferred site of dehydrogenation is the one involving the–OH group at the C3 position. The same behavior is observed for all the species present in water. The ionization potential in water decrease when moving from neutral to deprotonated forms, as do PA and PDE. Similar values and trends were also found in previous work done at the DFT level, but using a different exchange-correlation functional (B3LYP) (Thong et al., 2019).
3.3 Reactions in lipid-like and aqueous environments
The computed Gibbs free energies of reaction (ΔG) and activation (ΔG‡) for the considered mechanisms—HAT, SET, and RAF—of the reaction between the ∙OOH radical and isorhamnetin in the two considered environments are shown in Table 2. For RAF processes, we report only those with ΔG values less than 10 kcal/mol, since they are relevant from both thermodynamic and kinetic viewpoints.
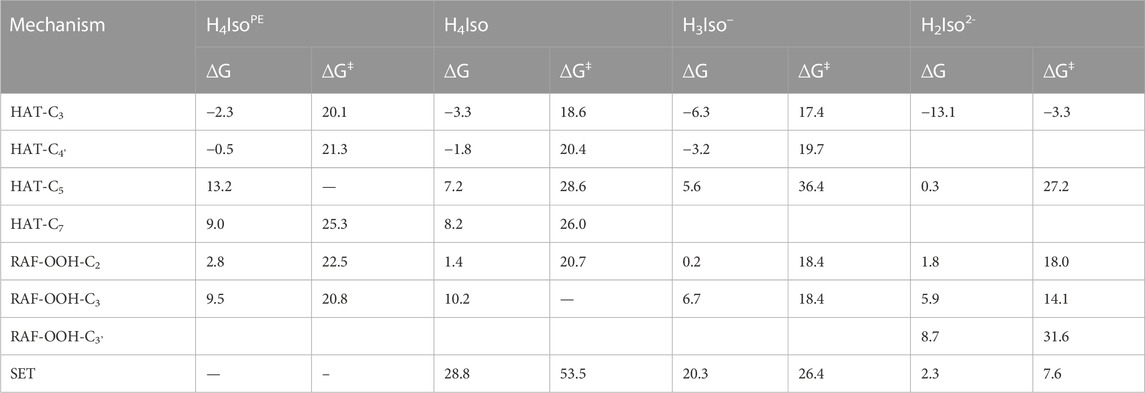
TABLE 2. Gibbs free energies of reaction (ΔG) and activation (∆G‡) at 298.15 K in aqueous solution and pentylethanoate (indicated byPE apex) for neutral and charged isorhamnetin species. All values are in kcal/mol.
From Table 2, we see that the more favored thermodynamic process is the hydrogen atom transfer from the C3 position of neutral and charged isorhamnetin species to the ∙OOH radical in both lipid-like and aqueous environments. In particular, the latter cases provides a noteworthy observation on how the exergonic character of the process increases with the transition from neutral to mono- and di-anion forms. Also, the HAT processes from the C4' position show negative Gibbs free energies, with a trend similar to HAT at the C3 site. The ∆G values for the mechanisms of radical addition and electron transfer reactions allows us to hypothesize that they can occur in both considered environments. Concerning the activation energies of the HAT-C3 and HAT-C4' processes in water solvent, we observe that the corresponding values decrease with the increasing ionic character of the species.
The rate constants and the branching ratios for the considered reaction mechanisms are reported in Table 3, while the transition state structures for HAT and RAF mechanisms are depicted in Figure 2.
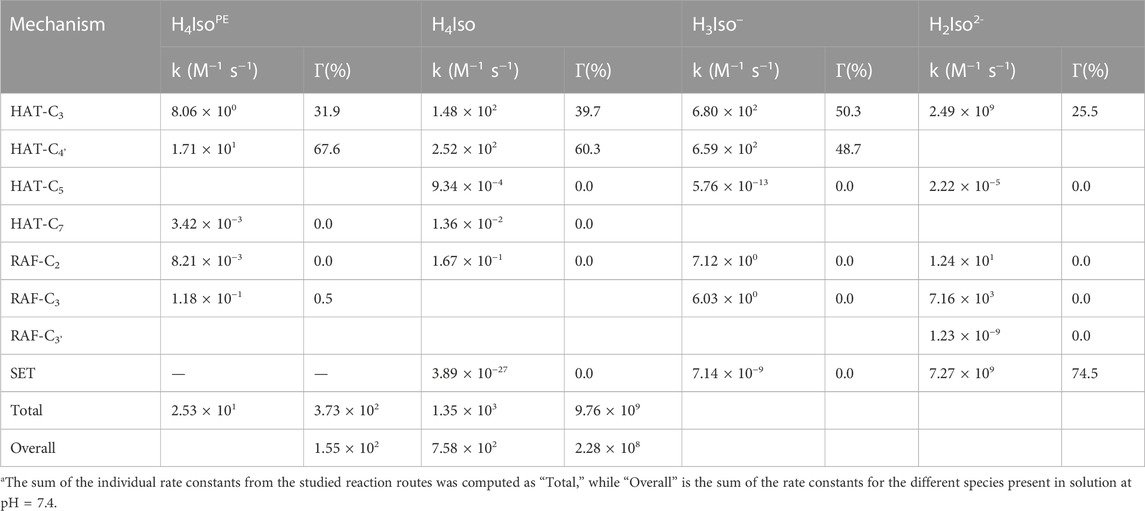
TABLE 3. Rate constants (k)a and branching ratios (Γ) of the reaction between isorhamnetin and *OOH radical in 1:1 ratio, computed at 298.15 K, for the different species present in solution at pH = 7.4
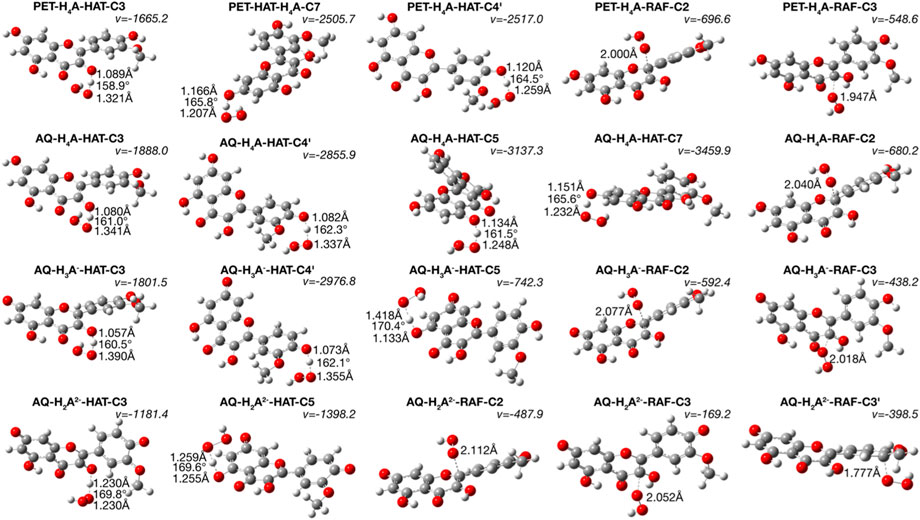
FIGURE 2. TSs structures for HAT and RAF mechanism. Distances are in Å and angles in degrees.
In the lipid-like phase, the total rate constant is 2.53 × 101 and indicates that the process taking place mainly follows the HAT mechanism from the C4'-OH position. Due the presence of different charged and neutral species, on the other hand, the situation in water solution is different. In fact, for the H4Iso form the hydrogen abstraction process at the C4' position is kinetically favored (k = 2.52 × 102), followed by the one at the C3 site (1.48 × 102). The total rate constant take into account the branching ratio that is 31.9% and 67.6% for C3 and C4' site, respectively. In the mono-anion species, the antioxidant properties are essentially due to the SET mechanism as indicated by the k (8.20 × 109) and the molar fraction values (74.5%). The same mechanism is favored by the H3Iso–form, but now, although the SET still gives the highest kinetic constant, the HAT mechanism from the C3 position also contributes considerably (Γ = 25.5%) to the total kinetic constant value. Considering the overall kinetic constant values, Table 3 clearly indicate that the isorhamnetin molecule has a high potential to scavenge ∙OOH radical (k = 4.60 × 109), essentially owing to the mono- and di-anionic forms, underlining the importance of considering all species present at physiological pH.
Inspecting the TSs structures (Figure 2) of the attack of ∙OOH radical on one of the hydroxyl hydrogens provides reliable data on the breakage of the isorhamnetin-OH bonds and the formation of a new one with the radical, observed as the generation of H2O2 molecule. The presence of imaginary frequencies confirm this phenomenon.
A rationalization of the kinetic behaviors can be done also considering the electron spin densities of the radicals obtained after the abstraction of the hydrogens in the different positions. As shown in Supplementary Figure S2, the abstraction of a proton in C3 position induces an electron spin delocalization that involve the entire molecular structure, in both neutral and charged species and in both considered solvents, that stabilize the radicals. On the contrary, the hydrogen loss in position C5 gives a radical in which the electronic spin density is more concentrated in one side of the structure.
A comparison with some other antioxidants of similar structure, studied previously using very similar or identical computational protocols, is possible by examining the data reported in Table 4.
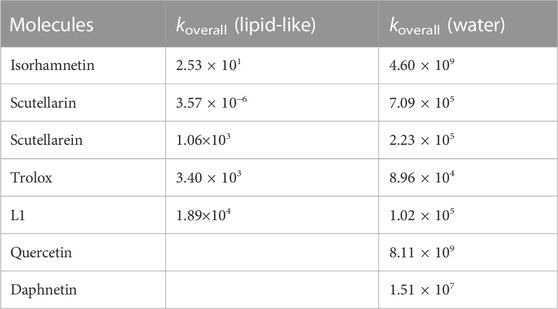
TABLE 4. Koverall values of Isorhamnetin and other structurally similar antioxidant against the OOH radical. Value are in M−1 s−1.
In pentylethanoate, the scavenging activity of isorhamnetin against the ∙OOH radical is relatively low and lower than that of the other compounds tested except for scutellarin (Spiegel et al., 2022b). On the contrary, in aqueous solution, its kinetic constant takes on a very high value (4.60 × 109 M−1 s−1), comparable to that of quercetin (k = 8.11 × 109 M−1 s−1) (Castaneda-Arriaga et al., 2020) and daphnetin (1.51 × 107) (Boulebd and Khodja, 2021) and about five orders of magnitude higher than the corresponding value of Trolox (8.96 × 104 M−1 s−1) (Alberto et al., 2013), which is generally used as a comparison to determine the antioxidant power of a molecule. On the other hand, isorhamnetin is a less efficient scavenger with respect to [4-(benzo[d]thiazol-2-yl)-2-((4,7-dimethyl-1,4,7-triazonan-1-yl)-methyl)-6-methoxyphenol] (L1 in Table 4) in the lipid-like phase, but more efficient in water solution (Spiegel et al., 2022a).
3.4 Copper chelating ability
The computations of the Gibbs energies (ΔGf) for the following reactions:
made it possible to establish the Cu2+chelating power of isorhamnetin. The relative apparent equilibrium constants (Kapp) were calculated using the following expressions (Perez-Gonzalez et al., 2020):
where
We considered three different chelating sites, which include oxygens at the C3'C4', C3C4 and C4C5 positions. The results for a 1:1 M ratio are reported in Table 5, and the related structures are displayed in Figure 3. For neutral and mono-anionic isorhamnetin, the most stable complex is the one with copper being coordinated at the C3-C4 site, while for H2Iso2– the preferred one is C3'C4'. In all cases, the thermodynamic stability of the complexes increases with the ionic character of the ligand.
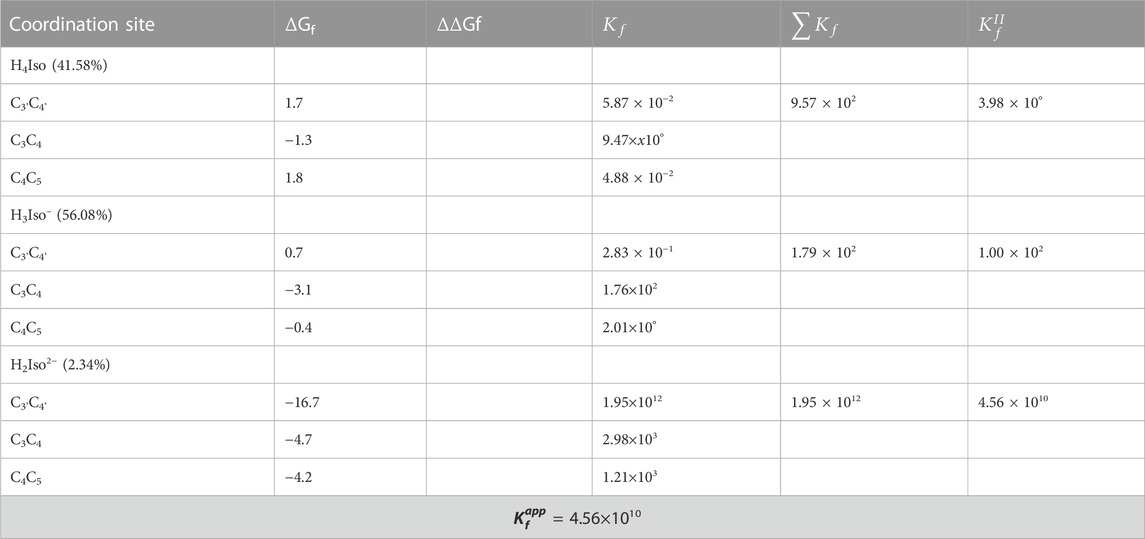
TABLE 5. Gibbs formation energies (ΔGf), their differences (ΔΔGf), both in kcal/mol, and kinetic constants (
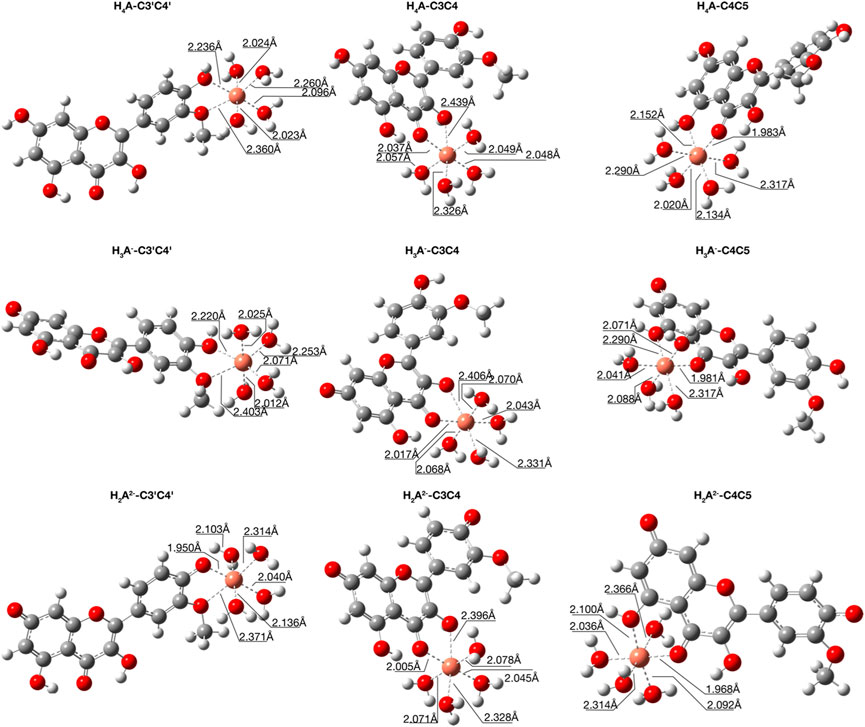
FIGURE 3. Structures for the most stable 1:1 complexes between Cu2+ and the neutral and charged species of isorhamnetin in water solvent.
The computed
From Figure 3, where the optimized structures and main geometrical parameters are reported, we note the distorted tetrahedral topology around the Cu2+ ion, which is coordinated with two ligand oxygens and two water molecules with Cu-O distances that range from about 1.9 Å to 2.1 Å. The other two water molecules act as a micro-solvation sphere.
We also explored what happens when the Cu2+ ion interacts with isorhamnetin at molar ratios of 1: 2. The results are summarized in Table 6. For all considered ligand species, the preferred coordination site is C3C4, and the most stable complex is the one in which the copper cation is coordinated by two H2A2− forms (ΔGf = −14.9 kcal/mol). From Figure 4, it can be proven that the structural topologies are different: the coordination with two charged ligands results in a butterfly-like structure, with the coordinated antioxidant oxygens distances that are different. As in the case of the 1:1 ratio, three coordinate H2O strongly interact with the copper center.
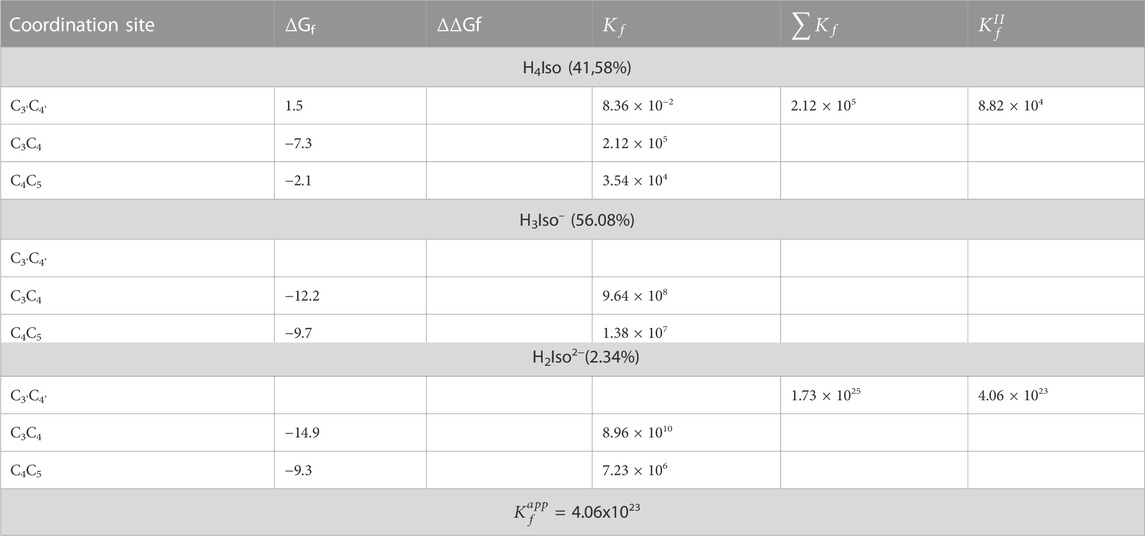
TABLE 6. The Gibbs formation energies (ΔGf), their differences (ΔΔGf), both in kcal/mol, and kinetic constants (
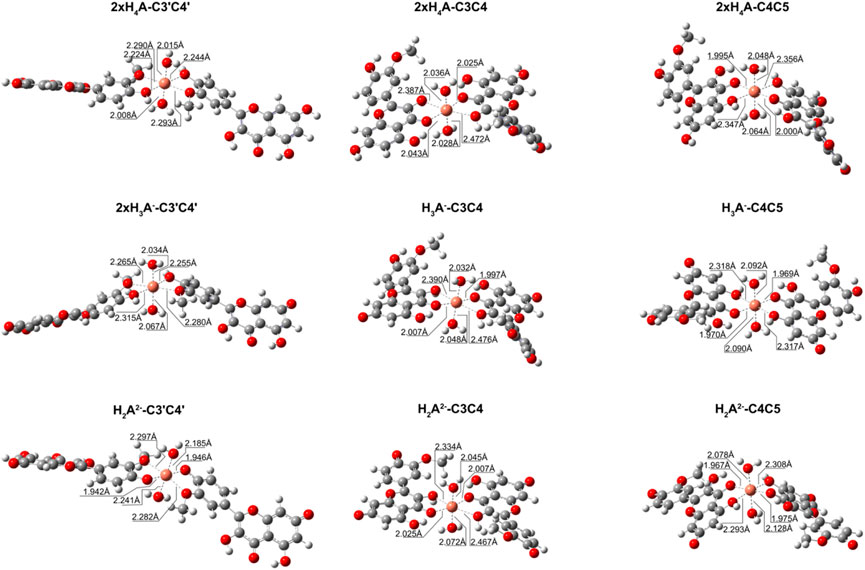
FIGURE 4. Structures for the most stable 1:2 complexes between Cu2+ and the neutral and charged species of isorhamnetin.
Comparing the
In order to explore the spectroscopic changes upon Cu2+ complexation, we calculated the excitation energies for both the isolated isorhamnetin and its complexes with copper (see Supplementary Table S1). The lowest energy transition (S1) for the bare molecule, characterized by high oscillator strength and being of HOMO- > LUMO in nature (more than 90%), undergo a sensible bathochromic shift starting from H4Iso (325.4 nm), to H3Iso− (343.2 nm) and H2Iso2− (373.6 nm). After Cu2+ complexation, the peak that undergoes the major shift at higher wavelengths is related to the C5C6 coordination site in all the species and different molar ratios. In particular, the highest wavelength (410 nm) is observed for the C5C6 coordination site with the isorhamnetin mono-anion in the 1:1 metal-ligand molar ratio. The excited energies in the complexes continue to be characterized by HOMO- > LUMO transitions.
3.5 Reduction mechanism with O2•− and Asc−
The pro-oxidant activity can be theoretically evaluated by studying the Cu(II)- > Cu(I) reduction reaction of the given metal-antioxidant complex (Fenton’s reaction) that leads to the production of *OH radicals. For this purpose, the reduction reaction of the studied complexes (in molar ratios of 1:1 and 1:2) with the reducing agents present in the physiological environment, such as superoxide (O2−) and ascorbic acid (Asc−), were studied. Thermodynamic and kinetic results are reported in Table 7 (1:1 M ratio complexes) and Supplementary Table S2 (1:2 M ratio complexes), along with the results of an analogous reactions but with a solvated copper ion only, which were taken as a reference.
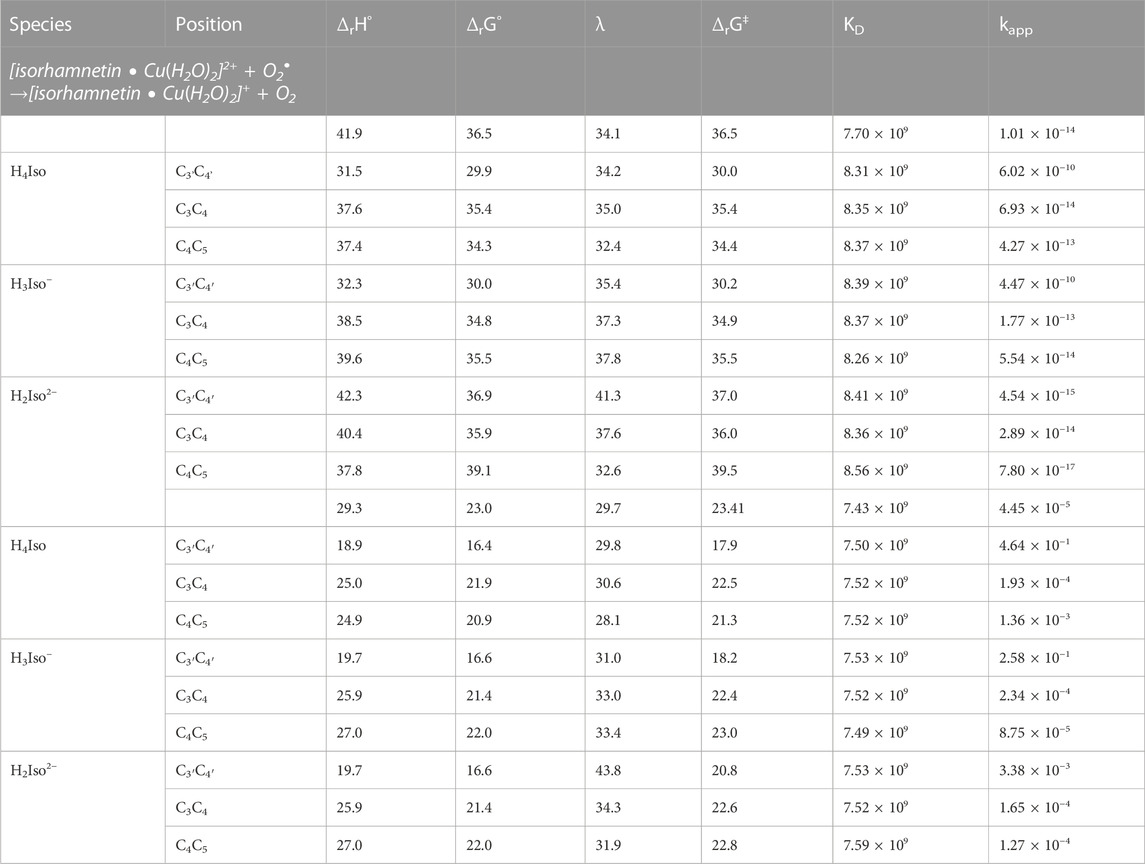
TABLE 7. The standard enthalpy (ΔrH°, kcal/mol), Gibbs free energy of reaction (ΔrG°, kcal/mol), reorganization energy (λ, kcal/mol), Gibbs free energy of activation (ΔrG‡, kcal/mol), diffusion rate constant (kD, M−1 s−1), TST thermal rate constant (kT, M−1 s−1), diffusion-corrected apparent rate constant (kapp, M−1 s−1) calculated at 298.15 K for the redox reaction between the copper complexes and two reducing agents (O2•− and Asc−) in water.
In all the studied systems, the reaction with the superoxide is more exergonic than the corresponding one with the ascorbic acid anion, as also noted previously for the pyridoxal antioxidant (Ngo et al., 2022).
Regarding the reaction with O2−, we found its feasibility to be greatest among neutral and mono-anionic forms of isorhamnetin with the C3’C4', and then the C4C5 coordination sites occupied. The relative kinetic constants indicate that the reduction reaction of these kind of complexes favors the reduction of the copper ion to a lower oxidation state.
Turning to the study of what happens when the Asc anion is used to reduce the copper ion in the Fenton’s reaction, we first note that the Gibbs reaction energies are much less exothermic than for the reactions with the superoxide ion. Indeed, they fall in the range of 16.4–22.0 kcal/mol for the complexes with molar ratio 1:1 and 15.3–29.0 kcal/mol (excluding the value of 53.8 kcal/mol found for the C3'C4' coordination site) for the complexes with a stoichiometry of 1:2.
From the values of the kinetic constants, it can be seen that the formation of complexes promotes the process of copper reduction towards lower oxidation states. In general, we can support the fact that the presence of ascorbic acid causes a greater pro-oxidant hazard than the reaction with the superoxide radical does, as previously noted for other antioxidants (Ngo et al., 2022).
3.6 Inhibitory activity
Flavonoids have been proven to be effective inhibitors of several enzymes involved in several biological and medical processes. In particular, isorhamnetin has been proposed as a promising inhibitor of the cyclooxygenase-2 (Seo et al., 2014), lactate dehydrogenase, adenosine diphosphate and other enzymes. Recently, some flavonoids have been proposed as non-covalent inhibitors of the main protease (Mpro) protein which plays an important role in SARS-CoV-2 main protease enzyme infection (Abian et al., 2020; Puttaswamy et al., 2020; Rizzuti et al., 2021).
Mpro explains its action in the cleavage of polyproteins at many sites generating non-structural proteins, relevant in the replication process of the virus (Anand et al., 2003), such as endo- and exo-ribonuclease as well as RNA polymerase. So, it is an important target for the development of new anticoronavirus therapeutic agents (Yang et al., 2005; Pillaiyar et al., 2016). From a structural point of view, SARS-CoV-2 Mpro is an homodimer and each protomer is characterized by three domains connected by a loop region. The catalytic center contain two crucial residues (His41 and Cys145) and catalyze the cleaving the of the polyprotein, translated from the viral RNA at different positions, generating proteins that contribute in the arresting process of the viral replication cycle (Ullrich and Nitsche, 2020).
Since the global health emergency generated by coronavirus disease 2019 is still in progress, we considered interesting to verify whether the isorhamnetin, coming from plants spread in various continents, can be a substrate capable to inhibit the Mpro.
Our docking analysis shows as isorhamnetin and its mono-anionic form establish many hydrophobic interactions (HI) and hydrogen bonds (H-bond) with different aminoacids in the enzyme catalityc pocket (see Figure 5 and Supplementary Table S3). For H4Iso two HI are established with the Thr24 and Leu27 while for H3Iso− only the interaction with Glu166 is present. Five H-bonds, with Cys44, Thr25, Gly143, and Glu166, are present in the binding of isorhamnetin with Mpro. In the case of H3Iso− they involve the Phe140, Glu166, and Gln189. In both the systems their lenhgts range from 2.91 to 3.99 Å (see Figure 5).
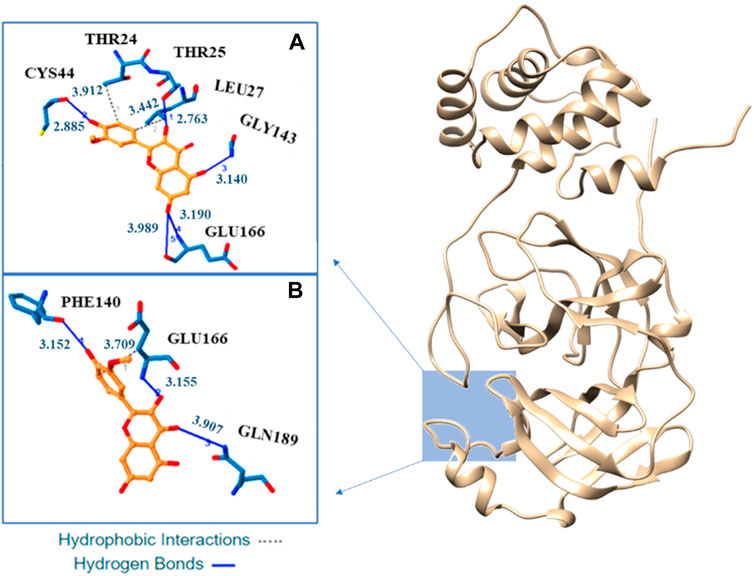
FIGURE 5. Three-dimensional representation of the best docking pose for H4Iso (A) and H3Is− (B) species. The residues that have crucial contacts with the compounds are shown in the square windows (A,B), for H4Iso and H3Is respectively.
Albeit with different amino acid residues, the binding energy values for both the neutral and anionic forms (Supplementary Table S3) result to be very close. The found value for the best pose (−6.1 kcal/mol) well agrees with that proposed for quercetin thoughout isothermal titration calorimetry that result to be −7.6 kcal/mol (Abian et al., 2020) and with the computed binding energy between eugenol and Mpro (Rizzuti et al., 2021). Furthermore, our computed binding energy is sligtly higher than that obtained for Ginkgetin (−9.5 kcal/mol), Delphinidin (−9.4 kcal/mol), Cyanidin 3,5-diglucoside, (−9.4 kcal/mol) and Amentofavone (−9.7 kcal/mol) antioxidants with similar structures (Puttaswamy et al., 2020). Finally, a comparison can be made also with the binding energies of quercetin and its anion with furin protein that result to be −7.8 and −7.7 kcal/mol, respectively (Milanovic et al., 2021).
The molecular docking examination induced us to perform MD silmulations with the aim to observe the dynamic behaviour of the complexes Mpro:H4Iso and Mpro:H3Iso. The analysis of their MD trajectories indicated a different behaviour of the two tested molecules, despite starting from the docked pose where both the ligands were located in proximity of catalytic site. During the simulation time in correspondence of ∼20 ns, the anionic form goes to a distal area from the catalytic pocket generating a different RMSD trend (Supplementary Figure S3) from that obtained in the case of Mpro:H4Iso. In particular, the H3Iso-is placed in a site defined by Ile213, Pro252-Leu253, Gln256, Val296-Val297, Cys300-Gly302 residues. The Supplementary Figure S4 related to RMSF value evidenced a different fluctuation in the related region. This site corresponds to the named site #3, one of the six allosteric sites experimentally proposed (Douangamath et al., 2020; El BabaLutomski et al., 2020; Günther et al., 2021) and then in silico observed (Alzyoud et al., 2022).
In fact, the Mpro enzyme includes several pockets on its surface believed important for its catalytic activity; some of them exist in distal areas from the main catalytic pocket. The site #3 located at the dimer interface showed poor druggability due to its very small and shallow cavity that is significantly less hydrophilic. The protease movement during the MD of Mpro:H3Iso− makes more exposed such distal sites, making them more accessible. This does not take place during the MD of Mpro:H4Iso as it can be evinced from a Figure 6 where the superposition of the most representative structure of the complexes Mpro:H4Iso and Mpro:H3Iso-is reported. Furthermore the ligand affinity to the Mpro, evaluated in terms of ΔG binding, calculated via MMPBSA method proposed also the neutral form, H4Iso, as the most thermodynamically favorable. (see Supplementary Table S5) This outcome better clarifies the different nature of the interactions generated by the two ligands in the two different sites.
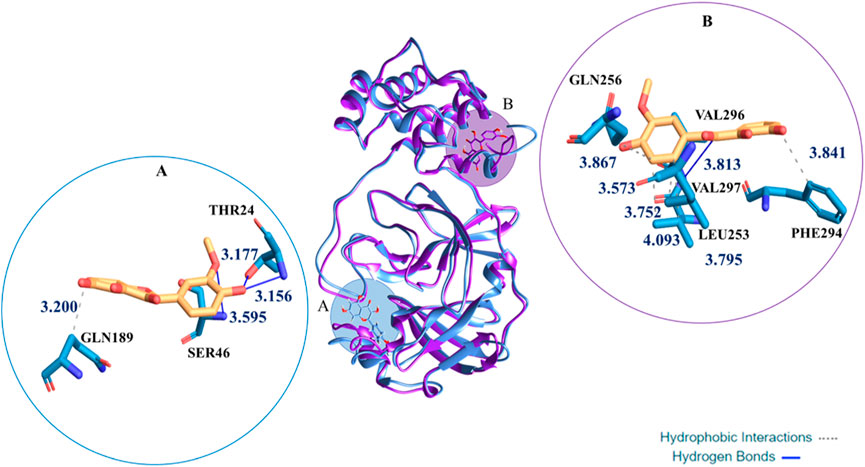
FIGURE 6. Superimposition of the most representative cluster from 100 ns of MD for the Mpro:H4Iso and Mpro:H3Iso−. In round windows complexes three-dimensional representation of the clustered structures with the residues that have crucial contacts with the compounds, in (A) (catalytic site) the H4Iso, in (B) (site #3) the H3Iso− one.
The possible coexistence of the two forms (neutral and anionic), at physiological pH, could be helpful to enhance the antiviral response of the isorhamnetin molecule providing provide a good (natural) starting point for lead optimization chemistry to disactivate the SARS CoV-2 Mpro.
4 Conclusion
In the present work, we have shown, by examining the isorhamnetin molecule, how quantum mechanics methods based on density functional theory can provide useful and reliable information on geometric and electronic structures, chemical equilibria in solution, and reaction mechanisms of antioxidant systems. The results show that.
• isorhamnetin in aqueous solution and at physiological pH (7.4) exists in neutral, mono- and di-anionic forms (molar fractions equal 41.58%, 56.08%, and 2.34%, respectively) rendering them important to consider in calculations aiming to provide reliable kinetics of the reactivity towards *OOH radical;
• it is important to consider different molar ratios (1:1, 1:2) to determine the chelating capacity towards copper ions;
• the reduction process of the obtained complexes with the reducing agents present in the physiological environment (superoxide anion and absorbic acid anion) are essential for predicting their chemical behavior in Fenton’s reactions;
• isorhamnetin has good non-covalent inhibitory potency towards Mpro, which is a pharmacological target for the treatment of SARS-CoV-2 infection.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.rcsb.org/- PDB code 6W63.
Author contributions
NR conceived of the presented idea and supervised the project. MS and GC performed the computations. NR, TM, and MS verified and analysed the results. NR wrote the manuscript. GC, MS, TM, and NR contributed to the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors thank the Dipartimento di Chimica e Tecnologie Chimiche of Università della Calabria for their financial support. The computing resources and the related technical support used for this work have been provided by: the supercomputer Marconi 100 from ISCRA (project code: IsC92) for computer simulations and by resources provided by Poznan Supercomputing and Networking Center (grant number 467).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1122880/full#supplementary-material
References
Abian, O., Ortega-Alarcon, D., Jimenez-Alesanco, A., Ceballos-Laita, L., Vega, S., Reyburn, H. T., et al. (2020). Structural stability of SARS-CoV-2 3CLpro and identification of quercetin as an inhibitor by experimental screening. Int. J. Biol. Macromol. 164, 1693–1703. doi:10.1016/j.ijbiomac.2020.07.235
Alberto, M. E., Grand, A., Russo, N., and Galano, A. (2013). A physicochemical examination of the free radical scavenging activity of Trolox: Mechanism, kinetics and influence of the environment. Phys. Chem. Chem. Phys. 15, 4642. doi:10.1039/c3cp43319f
Alzyoud, L., Ghattas, M. A., and Atatreh, N. (2022). Allosteric binding sites of the SARS-CoV-2 main protease: Potential targets for broad-spectrum anti-coronavirus agents. Drug Des. Dev. Ther. 16, 2463–2478. doi:10.2147/dddt.s370574
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R., and Hilgenfeld, R. (2003). Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 300, 1763–1767. doi:10.1126/science.1085658
Anandakrishnan, R., Aguilar, B., and Onufriev, A. V. (2012). H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, 537–541. doi:10.1093/nar/gks375
Apak, R., Özyürek, M., Kubilay, G., and Çapanoğlu, E. (2016). Antioxidant activity/capacity measurement. 1. Classification, physicochemical principles, mechanisms, and electron transfer (ET)-Based assays. J. Agric. Food Chem. 64, 997–1027. doi:10.1021/acs.jafc.5b04739
Bayly, C. I., Cieplak, P., Cornell, W., and Kollman, P. A. A. (1993). A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 97, 10269–10280. doi:10.1021/j100142a004
Belcastro, M., Marino, T., Russo, N., and Toscano, M. (2006). Structural and electronic characterization of antioxidants from marine organisms. Theor. Chem. Acc. 115, 361–369. doi:10.1007/s00214-006-0077-5
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1998). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. doi:10.1063/1.448118
Boulebd, H., and Khodja, I. A. (2021). A detailed DFT-based study of the free radical scavenging activity and mechanism of daphnetin in physiological environments. Phytochemistry 189, 112831. doi:10.1016/j.phytochem.2021.112831
Case, D. A., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., Cheatham, T. E., Cruzeiro, V. W. D., et al. (2017). AMBER 2017. San Francisco: University of California.
Castaneda-Arriaga, R., Marino, T., Russo, N., Alvarez-Idaboy, J. R., and Galano, A. (2020). Chalcogen effects on the primary antioxidant activity of chrysin and quercetin. New J. Chem. 44, 9073–9082. doi:10.1039/d0nj01795g
Collins, F. C., and Kimball, G. E. (1949). Diffusion-controlled reaction rates. J. Colloid. Sci. 4, 425–437. doi:10.1016/0095-8522(49)90023-9
Darden, T., York, D., and Pedersen, L. (1998). Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J.of Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
Douangamath, A., Fearon, D., Gehrtz, P., Krojer, T., Lukacik, P., Owen, C. D., et al. (2020). Crystallographic and electrophilic fragment screening of the SARSCoV-2 main protease. Nat. Commun. 11 (1), 5047. doi:10.1038/s41467-020-18709-w
El-Baba, T. J. A., Lutomski, C., Kantsadi, A. L., Malla, T. R., John, T., Mikhailov, V., et al. (2020). Allosteric inhibition of the SARS-CoV-2 main protease: Insights from mass spectrometry based assays**. Angew. Chem. Int. Ed. 59, 23544–23548. doi:10.1002/anie.202010316
Evans, M. G., and Polanyi, M. (1935). Some applications of the transition state method to the calculation of reaction velocities, espe-cially in solution. Trans. Faraday Soc. 31, 875–882. doi:10.1039/tf9353100875
Forman, H. J., and Zhang, H. (2021). Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. 20, 689–709. doi:10.1038/s41573-021-00233-1
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2014). Gaussian 09, revision D.01.
Galano, A., Perez-Gonzalez, A., Castaneda-Arriaga, R., Munoz-Rugeles, L., Mendoza-Sarmiento, G., Romero-Silva, A., et al. (2016b). Empirically fitted parameters for calculating pKa values with small deviations from experiments using a simple computational strategy. J. Chem. Inf. Model. 56, 1714–1724. doi:10.1021/acs.jcim.6b00310
Galano, A., and Alvarez-Idaboy, J. R. (2013). A computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. J. Comput. Chem. 34, 2430–2445. doi:10.1002/jcc.23409
Galano, A., Mazzone, G., Alvarez-Diduk, R., Marino, T., Alvarez-Idaboy, J. R., and Russo, N. (2016a). Food antioxidants: Chemical insights at the molecular level. Annu. Rev. Food Sci. Technol. 7, 335–352. doi:10.1146/annurev-food-041715-033206
Gong, G., Guan, Y.-Y., Zhang, Z.-L., Rahman, K., Wang, S.-J., Zhou, S., et al. (2020). Isorhamnetin: A review of pharmacological effects. Biomed. Pharmacother. 128, 110301. doi:10.1016/j.biopha.2020.110301
Günther, S., Reinke, P. Y. A., Fernández-Garciá, Y., Lieske, J., Lane, T. J., Ginn, H. M., et al. (2021). X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 7 (6542), 372642–372646. doi:10.1126/science.abf7945
Leopoldini, M., Russo, N., and Toscano, M. (2011). The molecular basis of working mechanism of natural polyphenolic antioxidants. Food Chem. 125, 288–306. doi:10.1016/j.foodchem.2010.08.012
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2021). ff14SB, Available at: https://pubs.acs.org/doi/full/10.1021/acs.jctc.5b00255, (accessed 13 January 2021).
Manach, C., Morand, C., Demigne, C., Texier, O., Regerat, F., and Remesy, C. (1997). Bioavailability of rutin and quercetin in rats. FEBS Lett. 409, 12–16. doi:10.1016/s0014-5793(97)00467-5
Marcus, R. A. (1993). Electron transfer reactions in chemistry: Theory and experiment (Nobel lecture). Angew. Chem. Int. Ed. Engl. 32, 1111–1121. doi:10.1002/anie.199311113
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396. doi:10.1021/jp810292n
Marković, Z., Tošović, J., Milenković, D., and Marković, S. (2016). Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput. Theor. Chem. 1077, 11–17. doi:10.1016/j.comptc.2015.09.007
Milanovic, Z. B., Antonijevic, M. R., Amic, A. D., Avdovic, E. H., Dimic, D. S., Milenkovic, D. A., et al. (2021). Inhibitory activity of quercetin, its metabolite, and standard antiviral drugs towards enzymes essential for SARS-CoV-2: The role of acid–base equilibria. RSC Adv. 11, 2838–2847. doi:10.1039/d0ra09632f
Milenković, D. A., Dimić, D. S., Avdović, E. H., Amić, A. D., Dimitrić Marković, J. M., and Marković, Z. S. (2020). Advanced oxidation process of coumarins by hydroxyl radical: Towards the new mechanism leading to less toxic products. Chem. Eng. J. 395, 124971. doi:10.1016/j.cej.2020.124971
Ngo, T. C., Nguyen, T. H., and Dao, D. Q. (2019). Radical scavenging activity of natural-based cassaine diterpenoid amides and amines. J. Chem. Inf. Model. 59, 766–776. doi:10.1021/acs.jcim.8b00847
Ngo, T. C., Truong, D. H., Nguyen, T. T. N., Quang, D. T., and Dao, D. Q. (2022). On the free radical scavenging and metallic ion chelating activities of pyridoxal - could the pro-oxidant risk be competitive? Phytochemistry 192, 113176. doi:10.1016/j.phytochem.2022.113176
Parise, A., RomeoRusso, I. N., and Marino, T. (2021b). The Se-S bond formation in the covalent inhibition mechanism of SARS-CoV-2 main protease by ebselen-like inhibitors: A computational study. Int. J. Mol. Sci. 22 (18), 9792. doi:10.3390/ijms22189792
Parise, A., De Simone, B. C., Marino, T., Toscano, M., and Russo, N. (2021a). Quantum mechanical predictions of the antioxidant capability of moracin C isomers. Front. Chem. 9, 666647. doi:10.3389/fchem.2021.666647
Pengfei, L., Tiansheng, D., Xianglin, H., and Jianguo, W. (2009). Antioxidant properties of isolated isorhamnetin from the sea buckthorn marc. Plant Foods Hum. Nutr. 64, 141–145. doi:10.1007/s11130-009-0116-1
Pérez-González, A., Prejanò, M., Russo, N., Marino, T., and Galano, A. (2020). Capsaicin, a powerful •OH-inactivating ligand. Antioxidants 9, 1247. doi:10.3390/antiox9121247
Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y., and Jung, S.-H. (2016). An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 59, 6595–6628. doi:10.1021/acs.jmedchem.5b01461
Puttaswamy, H., Gowtham, H. G., Ojha, M. D., Yadav, A., Choudhir, G., Raguraman, V., et al. (2020). In silico studies evidenced the role of structurally diverse plant secondary metabolites in reducing SARS-CoV-2 pathogenesis. Sci. Rep. 10, 20584. doi:10.1038/s41598-020-77602-0
Reina, M., Guzman-Lopez, E. G., Romeo, I., Marino, T., Russo, N., and Galano, A. (2021). Computationally designed p-coumaric acid analogs: Searching for neuroprotective antioxidants. New J. Chem. 45, 14369–14380. doi:10.1039/d1nj01235e
Rizzuti, B., Ceballos-Laita, L., Ortega-Alarcon, D., Jimenez-Alesanco, A., Vega, S., Grande, F., et al. (2021). Sub-micromolar inhibition of SARS-CoV-2 3CLpro by natural compounds. Pharmaceuticals 14, 892. doi:10.3390/ph14090892
Seo, K., Yang, J. H., Kim, S. C., Ku, S. K., Ki, S. H., and Shin, S. M. (2014). The antioxidant effects of isorhamnetin contribute to inhibit COX-2 expression in response to inflammation: A potential role of HO-1. Inflammation 37, 712–722. doi:10.1007/s10753-013-9789-6
Sharma, A. K., Pavlova, S. T., Kim, J., Kim, J., and Mirica, L. M. (2013). The effect of Cu2+ and Zn2+ on the Aβ42 peptide aggregation and cellular toxicity. Metallomics 5, 1529–1536. doi:10.1039/c3mt00161j
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi:10.1146/annurev-biochem-061516-045037
Spiegel, M. (2022). Current trends in computational quantum chemistry studies on antioxidant radical scavenging activity. J. Chem. Inf. Model. 62 (11), 2639–2658. doi:10.1021/acs.jcim.2c00104
Spiegel, M., Gamian, A., and Sroka, Z. (2021). Antiradical activity of beetroot (Beta vulgaris L.) betalains. Molecules 26, 2439. doi:10.3390/molecules26092439
Spiegel, M., Marino, T., Prejanò, M., and Russo, N. (2022a). Antioxidant and copper-chelating power of new molecules suggested as multiple target agents against Alzheimer's disease. A theoretical comparative study. Phys. Chem. Chem. Phys. 24, 16353–16359. doi:10.1039/d2cp01918c
Spiegel, M., Marino, T., Prejanò, M., and Russo, N. (2022b). Primary and secondary antioxidant properties of scutellarin andscutellarein in water and lipid-like environments: A theoretical investigation. J. Mol. Liq. 366, 120343. doi:10.1016/j.molliq.2022.120343
Teng, D., and Luan, X. (2016). Research progress of isorhamnetin in pharma codynamics. Clin. J. Tradit. Chin. Med. 28, 593–596.
Thong, N. M., Vo, Q. V., Huyen, T. L., Bay, M. V., Tuan, D., and Cam Nam, P. (2019). Theoretical study for exploring the diglycoside substituent effect on the antioxidative capability of isorhamnetin extracted from anoectochilus roxburghii. ACS Omega 4 (12), 14996–15003. doi:10.1021/acsomega.9b01780
Trott, O., and Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. doi:10.1002/jcc.21334
Truhlar, D. G., Garrett, B. C., and Klippenstein, S. J. (1996). Current status of transition-state theory. J. Phys. Chem. 100, 12771–12800. doi:10.1021/jp953748q
Ullrich, S., and Nitsche, C. (2020). The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 30, 127377. doi:10.1016/j.bmcl.2020.127377
Xu, Z., Yang, L., Zhang, X., Zhang, Q., Yang, Z., Liu, Y., et al. (2020). Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Front. Mol. Biosci. 7, 556481. doi:10.3389/fmolb.2020.556481
Yang, H., Xie, W., Xue, X., Yang, K., Ma, J., Liang, W., et al. (2005). Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3, e324. doi:10.1371/journal.pbio.0030324
Yu, M-S., Lee, J., Lee, J. M., Kim, Y., Chin, Y. W., Jee, J.-G., et al. (2012). Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg. Med. Chem. Lett. 22, 4049–4054. doi:10.1016/j.bmcl.2012.04.081
Keywords: antioxidants, chemical equilibria, reaction mechanisms, kinetic constant, copper complexes, Fenton’s reaction, main protease inhibition
Citation: Spiegel M, Ciardullo G, Marino T and Russo N (2023) Computational investigation on the antioxidant activities and on the Mpro SARS-CoV-2 non-covalent inhibition of isorhamnetin. Front. Chem. 11:1122880. doi: 10.3389/fchem.2023.1122880
Received: 13 December 2022; Accepted: 09 January 2023;
Published: 24 January 2023.
Edited by:
Lalith Perera, National Institute of Environmental Health Sciences (NIH), United StatesReviewed by:
Zoran S. Markovic, University of Kragujevac, SerbiaRenjith Thomas, Mahatma Gandhi University, India
Nesimi Uludag, Namik Kemal University, Türkiye
Copyright © 2023 Spiegel, Ciardullo, Marino and Russo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nino Russo, bmluby5ydXNzb0B1bmljYWwuaXQ=