
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 19 June 2023
Sec. Chemical Physics and Physical Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fchem.2023.1028008
 Maria Pintea1*
Maria Pintea1* Nigel Mason1
Nigel Mason1 Anna Peiró-Franch1Ewan Clark1
Anna Peiró-Franch1Ewan Clark1 Kushal Samanta1
Kushal Samanta1 Cristiano Glessi2Inga Lena Schmidtke2Thomas Luxford3
Cristiano Glessi2Inga Lena Schmidtke2Thomas Luxford3With the use of proton-NMR and powder XRD (XRPD) studies, the suitability of specific Au-focused electron beam induced deposition (FEBID) precursors has been investigated with low electron energy, structure, excited states and resonances, structural crystal modifications, flexibility, and vaporization level. 4,5-Dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) is a compound that is a uniquely designed precursor to meet the needs of focused electron beam-induced deposition at the nanostructure level, which proves its capability in creating high purity structures, and its growing importance in other AuImx and AuClnB (where x and n are the number of radicals, B = CH, CH3, or Br) compounds in the radiation cancer therapy increases the efforts to design more suitable bonds in processes of SEM (scanning electron microscopy) deposition and in gas-phase studies. The investigation performed of its powder shape using the XRPD XPERT3 panalytical diffractometer based on CoKα lines shows changes to its structure with change in temperature, level of vacuum, and light; the sensitivity of this compound makes it highly interesting in particular to the radiation research. Used in FEBID, though its smaller number of C, H, and O atoms has lower levels of C contamination in the structures and on the surface, it replaces these bonds with C–Cl and C–N bonds that have lower bond-breaking energy. However, it still needs an extra purification step in the deposition process, either H2O, O2, or H jets.
Increased importance of gold(I) compounds in the pharmaceutical drug development and cancer studies is observed, and thus the gold(I) compounds are developed from the synthesis step for quantum simulations and molecular dynamics analysis, as in our case, or for catalyzed reactions and reductive elimination/migratory insertion reactions (Gil-Rubio and Vicente, 2015). The most worth mentioning application of the gold(I) compounds is the inhibition of bacteria, such as Escherichia coli (Galassi et al., 2015), through bonding of the DHFR (dihydrofolate reductase) to C, F, or P of the gold compound, where a high reduction in the level of DHFR in solution with gold(I) compared to non-gold(I) is observed. A level of reduction of 0.1 from 2.2 to 2.1 of denaturated DHFR and 1.2 to 1 of the native DHFR is observed with inhibitory constants of 2.25 μM, 1.1 μM, and 8.63 μM for 4-benzoic acid-diphenyl-phosphene gold(I) chloride, 2-benzoic acid-diphenyl-phosphane gold(I) chloride, and 4,5-dichloroimidazolato-N-triphenylphosphine–gold(I), respectively, extending the use of the gold(I) compounds to the treatment of inflammatory infections, pneumonia, E. coli, and cancer. In the cancer research, the developments of the gold (III) and gold(I) compounds have opened new ways of treating, inhibiting, and preventing cancer development through the design of new precursors. The clinical trials (Malet-Martino and Martino, 2002) from 1994 of the aurofin compound (2,3,4,6-tetra-O-acetyl-b-1-D-thipyranosato-S-(triphenylphosphine)gold(I)) initially used in the treatment of rheumatoid arthritis (Mirzadeh et al., 2019) revealed new ways of inhibiting cysteine (Cys), seleno-cysteine (Sec), glutathione reductase (GR), mitochondrial thioredoxin reductase (TrxR2), and cytosolic thioredoxin reductase (TrxR1) and opened new paths in the design of new monodentate phosphine precursors. Heterometallic fac-[Re (bipy) (CO3) (L-AuPPh3)]+, where L = imidazole, alkynyl-imidazole, and alkynyl-pyridine in human lung cancer cell treatments (Fernández-Moreira et al., 2014) show values of IC50 of ∼50 times lower than the Re(I) only complexes, obtaining a shift in the localization from the mitochondria to the nucleus with the increase in the concentration of the compound to μM, removing the cytoplasmatic staining with the accumulation in the mitochondria. Other studies involving the usage of coinage metal complexes (Schuh et al., 2012; Tan et al., 2010; Nobili et al., 2010) study the use of Au(I) NHC complexes against ovarian cancer cells (A2780S) and cisplatin resistant cells (A278R) compared to the non-tumoral kidney HEK-293T cells with obtained inhibition of cell growth IC50 values from 2 to 30 μM. Compared to the aurofin precursor, the Au(I) NHC compounds do not lead to the oxidation of the HEK-293T cells targeting only the TrxR1 and TrxR2 enzymes. A high number of Au precursors have been studied as inhibitors in cancer treatment (Mármol et al., 2019; González-Rubio et al., 2022; Zou et al., 2015) over the past few years. Compounds such as Au (xant)PEt3 and Au (dedc)PEt3 (Mármol et al., 2019) present higher inhibition rates compared to the aurofin and AuCl(PR3) exhibits values of IC50 between 4.2 and 5.2 μM in targeting colon carcinoma cells. The bis-chelated gold(I) bisphosphane (2,3-bis(tert-butyl (methyl) phosphino) quinoxaline) (Mármol et al., 2019) has been found to be a promising drug for cancer therapy to inhibit cysteine (Cys), seleno-cysteine (Sec), glutathione reductase (GR), and TrxR. Au compounds containing H2TPP (5,10,15,20-tetraphenyl porphyrin) and dppe (1,2-bis (diphenyl phosphane) ethane), Au (TPP)Cl and Au (dppe)2Cl (Zou et al., 2015) are targeting cancer through reduction of Au(III)—Au(I).
Through the use of the velocity map imaging technique and dissociative electron attachment (DEA) mass spectroscopy studies, employed in multiple analyses involving Au compounds or compounds of gold substrates, we determine the fragmentation pathways with implications to focused electron beam deposition. At 157 nm, the velocity map imaging study of diatomic gold in combination with the density functional theory (DFT) and ab initio calculations brings insight into the dynamics of the Au–Au vibrational and excitational modes, bonding between species with d-electrons valence and the branching ratios for Au 5d96s2 (2D3/2) and Au 5d96s2 (2D5/2) (Gil-Rubio and Vicente, 2015). The optical absorption spectra of Au in vapor form show the allowed transition states between 211 and 229 nm from 1Πu (II) to X1Σg+ and isolates two dissociation processes, first one at a photon energy of 2.301–2.311 eV for Au 5d106s2 (2D5/2) and the second one at 3.437–3.447 eV for Au 5d106s2(2S1/2) + Au 5d96s2 (2D5/2), showing particularity for gold cluster processes and the presence of the 6 s orbitals combined with the relativistic effects of the s electrons.
In nanotechnology applications, the assisted deposition of Au compounds has been performed successfully by Shawrav et al. (2016) with H2O as oxidative enhancer resulting pure Au nanostructures with a resistivity of 8.8 μΩcm with 91% purity of the structure. The Au content of the nanostructures resulting from the focused electron beam-induced deposition of Me2Au (tfac) was improved to reach values of 72% through the refining of the electron beam parameters and further to hit high purity levels of ∼90% through the plasma-assisted structure post-processing (Belić et al., 2017). Chien et al. (2021) report a carbon content of up to 60% in their Au-deposited nanostructures through their newly developed localized surface plasmon resonance measurement (built to enhance structure content reading) and a reduction of 20% of the carbon content through the H2O treatment of the nanostructures. In the normal non-assisted deposition of CF3-Au containing compounds (Carden et al., 2018; Carden et al., 2019), values of the Au content in the deposits of 22% in the case of CF3AuCNMe and 14% for CF3AuCNBu (Carden et al., 2019) were obtained with values of decomposition and sublimation temperatures of the two compounds evaluated at 51°C and 80°C (CF3AuCNMe) and 39°C and 126°C (CF3AuCNBu). CF3–Au containing precursors are known to have very good sublimation and decomposition temperatures becoming highly sought precursors for FEBID deposition (Hagen et al., 2008; Utke et al., 2008; Botman et al., 2009; Thorman et al., 2015), though the lower levels of the Au content and high C contamination (>60 at%) are indications of the need of a post-processing treatment or assisted deposition. Me2Au(Acac) presents comparable results when deposited and annealed at 100°C–300°C, forming structures close to 14 nm in size (Puydinger dos Santos et al., 2018), but at the same time reducing the carbon content at 300°C under H2 jet to almost 0% and removing it out of the lattice through heating. Kuhness et al. (2021) report the growth of AuCx nanopillar results of FEBID of Me2Au (acac) with a height of 2 µm for the development of 3D plasmonic gold nanoantennae, as one of the many applications of induced chemistry at the nanoscale. The focus is indeed on the composition of the nanopillars that are further annealed (300°C) and purified using H2O jets at room temperature. The growth of nanoantennae and nanopillars have based a new lithographic method on the focused electron beam-induced process (FEBIP) by cooling the substrate and thin films to lower than 0°C and further irradiated using e-beams to form structures (Zhao et al., 2019), or more sophisticated methods, such as GIS (gas injection systems) and computer-assisted deposition for the creation of highly complex and accurate 3D nanostructures (Fowlkes et al., 2018). The same methods have been applied to growing carbon nanotubes (Brintlinger et al., 2005), carbene nanostructures (Furst and Cazin, 2010; Johnson, 2016; Glessi et al., 2021), and cold ice organic nanostructures (Zhao et al., 2019). Nanostructures have been printed by Magén et al. (2021) using Au-based compounds in reactive atmospheres (Winkler et al., 2017) with very successful outcomes.
The experimental equipment consists of a high-vacuum chamber with pressures in the range of 10−6mbar helped by an Alcatel vacuum pump backed by a Pfeiffer Duo 6 backing pump. An electron gun is mounted on the top flange of the chamber intersecting at 90° of the molecular beam and in-line with the electron gun, and a three-plate Chevron pattern microchannel plate (MCP) detector. Puller, pusher, and flight tube assemblies (Figure 1) are connected with the detector for guiding the negatively charged ions to the phosphor screen. A charged-coupled device (CCD) industrial camera is used for capturing the ions accelerated at different velocities to the phosphor screen. A pair of Helmholtz coils is placed on the top and bottom of the chamber with the purpose of creating a magnetic field with values up to 80 Gauss that controls the guide path of the particles (ions/molecular fragments and electrons). The simple assembly, electron gun, the detector assembly (MCP), the flight tube, the phosphor screen, and the CCD camera for imaging the negative fragments, is helped by a 200-ns extractor/slicer that would physically select the inner slice of the Newton sphere of ions of a specific time length that are imaged by using the camera and detected by using MCP data acquisition modules for velocity discrimination. Similar set-ups (Prabhudesai et al., 2014; Bull et al., 2014; Gope et al., 2016) have been used at Tata Institute, India, and J. Heyrovský Institute of Physical Chemistry, Czech Republic (Nag et al., 2019), for imaging negative ions.
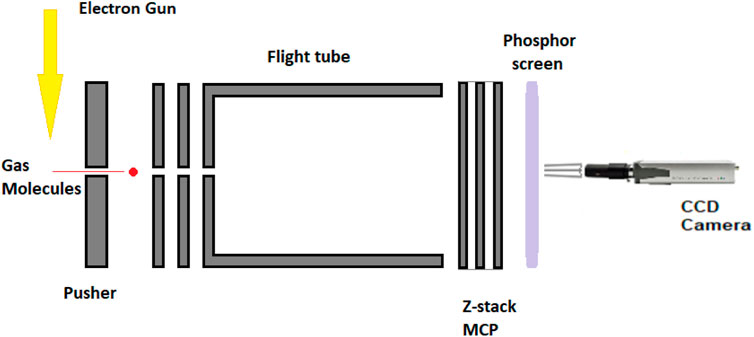
FIGURE 1. Velocity sliced map imaging (VsMI) detector assembly.
The increase in the number of detector’s plates reduces the aberration of the equipment and improves the energy range. The velocity sliced map imaging (VsMI) in Figure 1 assembly uses an energy range of 0–50 eV, specifically for low electron energy applications. The phosphor screen is employing a thin tungsten with a >98% transmission rate foil mounted on a brass ring. The detection of the negative ions is calibrated against a system of two molecules, namely, O2 at 6.5 eV and CO2, while the electron energy scale is zeroed with the signal and current calibration. The kinetic energy spectrum and angular distribution follow the same rules and should be less than 0.1% of the O2 and CO2 spectra. More detailed presentation of equipment and functioning has been given by Nag et al. (2019).
To determine the structural characteristics of the crystalline sample XRD, measurements were taken using a XPERT3 panalytical diffractometer based on CoKα with a time step of 150 s/step and a step size of 0.0167. The angle of diffraction is 5–80 ⁰ at a rate of 40 kV and 40 mA. The measurements were acquired over the duration of 1.5 h. The diffraction-specific wavelength is set to a value of 1.5406 Å.
10 mg of the 4,5-dichloro 1,3-diethyl imidazolylidene trifluoromethyl gold(I) complex (Figure 2) was dissolved in dichloromethane (20 mg/mL), followed by slow addition of pentane/hexane (dichloromethane: pentane/hexane is 5:1 vol/vol) onto the dichloromethane layer. The layered solutions were kept in the dark for crystallization for over 2 weeks at room temperature. Plate-shaped crystals of the trifluoromethyl gold(I) complex were obtained, hand-picked, and subjected to structure determinations by X-ray diffraction analysis.
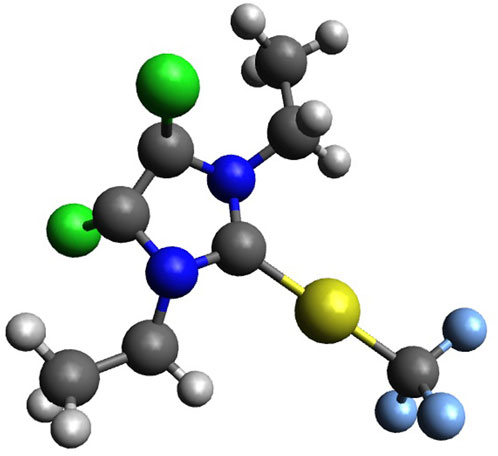
FIGURE 2. 4,5-Dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) (Au, yellow; N, blue; H, white; C, grey; F, light blue; and Cl, green).
The proton NMR (1H NMR) data acquisition was carried out using a JEOL ECS 400 MHz NMR spectrometer at 25°C with a sensitivity of 280 (0.1% ethyl benzene) for 1H and 19F, with an automatic Bruker SampleXpress sample charger run by using a 500 MHz electric DC motor having a 60 sample carousel controlled using ICON-NMR software and equipped with barcode reader registration, with the samples being kept at a temperature between 5 and 30°C and a separate cryo-fit mounting kit for sample cooling. The sample charger and sample unit were both controlled by the Bruker Avance III 400 MHz controller unit.
The sample (∼5 mg) was dissolved in 1-cm3 wet CDCl3 under atmospheric conditions; no special precautions were taken other than that, and the sample was initially transferred into the NMR tube in an Ar glove box.
Gold(I) NHC complexes are a class of compounds that are widely known and studied in chemistry for their versatility, among others in catalysis (Marion and Nolan, 2008), biomedicine (Porchia et al., 2018), and photochemistry (Longevial et al., 2016). The most important characteristic of the NHC ligand is the presence of carbene carbon, which is stabilized by two neighboring nitrogen atoms (Hopkinson et al., 2014). Due to their popularity, several ways for the synthesis of gold(I) NHC complexes have been reported (Wang and Lin, 1998), which makes these systems easily accessible and adaptable to required needs. The gold(I) NHC complex (Glessi et al., 2021) investigated in this work was synthesized following a reported literature procedure, which is illustrated in Figure 3. Starting from 4,5-chloroimidazole, the desired NHC ligand precursor was obtained as a salt in a yield of 94% through two sequential alkylation reactions using ethyl iodide (Solovyev et al., 2010). By reacting the NHC ligand precursor with silver oxide, the respective silver complex was formed in situ, which underwent a trans-metalation reaction upon the addition of one equivalent of the gold precursor Au(SMe2)Cl (Levchenko et al., 2020). The resulting gold NHC chloride complex was isolated in a yield of 92%. In the last step, the title compound was synthesized through another silver-mediated trans-metalation reaction. The active silver species AgCF3 is formed in situ from the reaction of silver fluoride with Me3SiCF3 and exchanges the chloro ligand with a CF3 group when Au(NHC)Cl is added, yielding the desired Au(NHC)CF3 complex as a colorless solid (66%) (Blaya et al., 2014a).
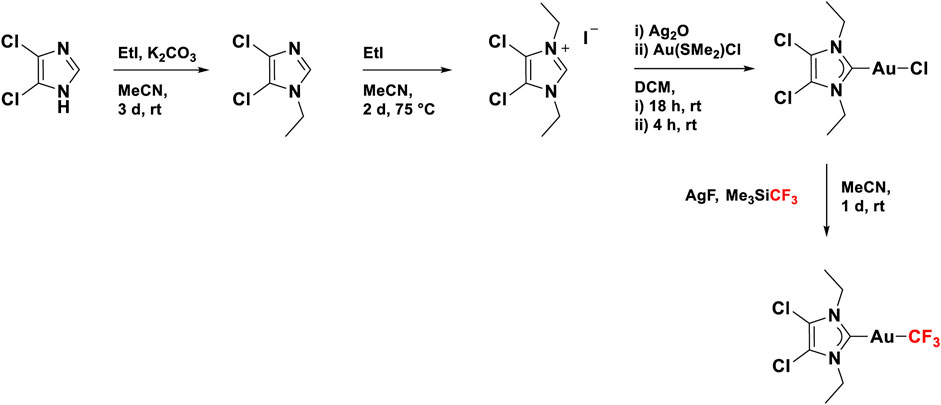
FIGURE 3. Synthesis route for 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I).
The simulations of the structure of C8H10Cl2N2AuCF3 have been run at the DFT/B3LYP level, making use of the full orbital populations and natural bond orbitals using a B3LYP/Def2-TZVPP basis set. The excited state calculations have been run using TDDFT. The crystal structure and slab for XRD simulations were built using Vesta and Avogadro software, and the single crystal structure modeling was carried out using OLEX2 crystallographic software based on the experimental data input.
4,5-Dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) is a gold compound synthesized by the Chemistry Department of the University of Oslo. The empirical formula of the compound is C8H10Cl2N2AuF3, and it has a mass of 459.05. The schematic of the compound is illustrated in Figure 4.
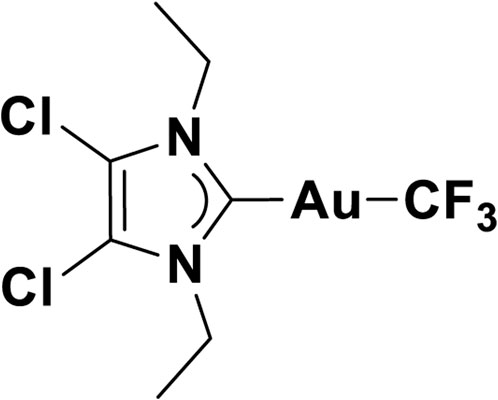
FIGURE 4. Schematics of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I).
A very important characteristic of our molecular dynamic simulations and cross-checking of our experimental results is the bond’s distance to C, Cl, N, and H and bond angles to C, Cl, N, and H. Multiple sources of trifluoromethyl gold(I) (Hasan et al., 1999; Chang et al., 2015; Ye et al., 2018) present the bond distances of the gold(I) compounds as 0.05 Å higher than the gold (II) compounds. In the study by Gil-Rubio and Vicente (2015), the bond distances for the most common gold(I) compounds are experimentally determined with values in the range of ∼2.04 Å as presented in Table 1.
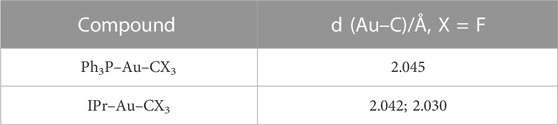
TABLE 1. L–Au–CF3 bond distances (Gil-Rubio and Vicente, 2015).
The characteristics of trifluoromethyl complexes come into a higher bond distance MC–F than C–F, as well as a decrease of F–C–F bond angle and increase in the M–C–F bond angle to the tetrahedral symmetry point group (Wozniak et al., 2019; Péres-Britrián et al., 2017). The bond distance Au–CF3 is shorter than Au–CH3 and Au–C/Au–Cl, where the Au–Cl bond distance is in the range of ∼2.27 Å (Chang et al., 2015) for [AuCl]-. Similar to Au(iii) anion [AuCl4]− in scattering processes, vibrational bands have weak Au 2p3/2 to 5D transitions, the so called white line, for [AuCl2]- ions (Chang et al., 2015), and these weak transitions are the result of a transition from Au 6s/5D hybrid partially occupied to the highest energy level occupied HOMO orbital. For our standard compound, we obtain the HOMO and LUMO orbitals as orbitals 76 and 77, while the total SCF (self-consistent field) density would contain a number of 683 occupied and unoccupied orbitals. Both HOMO and LUMO of 4,5-dichloro–1,3-diethyl–imidazolylidene trifluoromethyl gold(I) are illustrated in Figure 5.

FIGURE 5. HOMO/LUMO orbitals of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) (F, yellow; C, pink; H, blue; N, orange; Cl, green; and Au, magenta). (A) HOMO (orbital energy 6.62 eV); (B) LUMO (orbital energy 1.17 eV).
The bond distances calculated using B3LYP/Def2-TZVPP are longer than the free methyl radical bond distances and more imbalanced ranging from 1.087 Å to 2.076 Å. The 3 C atoms of the methyl radical have the bond lengths of 1.089/1.090 Å, equally spaced in all directions with an angle < HCH of 108.1°. The angle characteristics to the methyl radicals in our compound are 106.02° (<HCH) and 112.71° (<CCH). The bond lengths and angles of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) are presented in Table 2. We report a bond distance for C–Au of 2.065 Å from C1–Au1 and a second bond distance for CF3, Au1–C8 of 2.076 Å, the affinity to CF3 being higher than to CN2 in our case. Lower bond distances of Au–C ligands have been reported by Blaya et al. (2014b) with values declared for the Au–CF3 bond of all gold(I) trifluoromethyl complexes to be between 2.031 Å and 2.046 Å. Both the bond distances to CN2, C2–N1, and C3–N2 have values of 1.384 Å and angles to the axial plane of 125.3°, while the C4–Cl1 and C3–Cl2 bond distances are 1.702Å set at equally spaced angles of 129°. All C–F bond lengths (Figure 7A; Table 2) from CF3 have values of 1.372 Å balanced with an angle of 104.6°, longer than in the free trifluoromethyl radical with distances of the C–F bond of 1.318 Å and an angle of 110.76°, 6.16° lower than in our calculations. The simple ethyl radicals C5–H (from CH2 radical) and C5–H (from CH2 radical) have bond lengths of 1.087 Å and 1.089 Å, respectively.
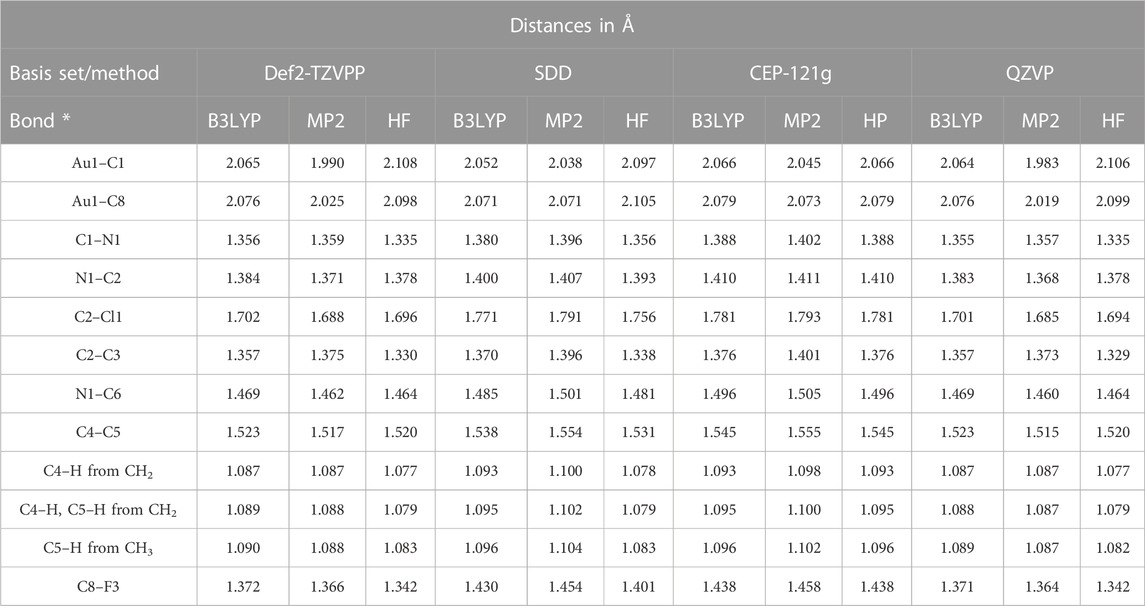
TABLE 2. Bond lengths of ImEtAuCl2CF3 optimized at multiple levels of the theory.
The highest values of the Au1–C1 bonds are for the B3LYP/CEP-121 basis set of 2.066 Å, 0.001 Å higher than the calculations at the B3LYP/Def2-TZVPP level of the theory and 0.002 Å higher than the calculations at B3LYP/QZVP and 0.014 Å; the highest discrepancy is obtained using the SDD basis set with the lowest bond length value of 2.052 Å. The 4,5-dichloro–1,3-diethyl–imidazolylidene trifluoromethyl gold(I) compound has the Au–C and C–Au bond lengths 0.4Å higher than the declared value for the Au–C bond in AuIm2 (Liu et al., 2013; Benitez et al., 2009) of 1.7 Å–2.06 Å by Liu et al. (2013) and Benitez et al. (2009). The accuracy of MP2 (Møller–Plesset 2 perturbation theory) methods compared to the B3LYP and HF (Hartree-Fock method) ones is very low for very complex molecules containing a high number of atoms or organic parts (peptides and alanine) (Kaminskya et al., 2008). Kaminskya et al. (2008) calculated the error of the MP2 methods with the basis set as being 20–30 kJ/mol in the electronic energy calculations; the values we report for the MP2 with QZVP basis set are the shortest distances Au1–C1 to the Cl2-phenyl ring; the calculations and results of Def2-TZVPP have values higher with 0.007 Å of the Au1–C1 distance. All MP2 level calculations for all basis-sets have shorter bond length values and MP2/SDD Au1–C1 has a value of 2.038 Å, while MP2/CEP-121G has a value slightly higher with 2.045 Å, but still lower than B3LYP calculations with the same basis set with values of 2.052 Å and 2.066 Å, respectively.
The chlorine atoms to the phenyl ring bond lengths range from 1.685 Å calculated at MP2/QZVP to 1.793 Å at MP2/CEP-121G, both Cl atoms being placed symmetrically at 129.22° to the C2 and C3 atoms of the phenyl ring. Overall, the structure is balanced at the central symmetry plane formed by two C atoms and Au, C1–Au1–C8, while 110.83⁰ to C (in CH2) and 90⁰ to the symmetry plane is obtained for the two ethyl–methyl groups.
The ImEtAuCl2CF3 crystal simulation was performed in the Vesta software on a pre-optimized molecular structure in Gaussian 16 at the B3LYP/Def2-TZVPP level of the theory using density functional theory calculations with full orbital populations and triple-ζ frequency calculations (Figures 6A–E). A number of bond lengths and the unit cell have been optimized by trial and error to obtain the slab of the crystal with different γ-orientations of the crystal planes.
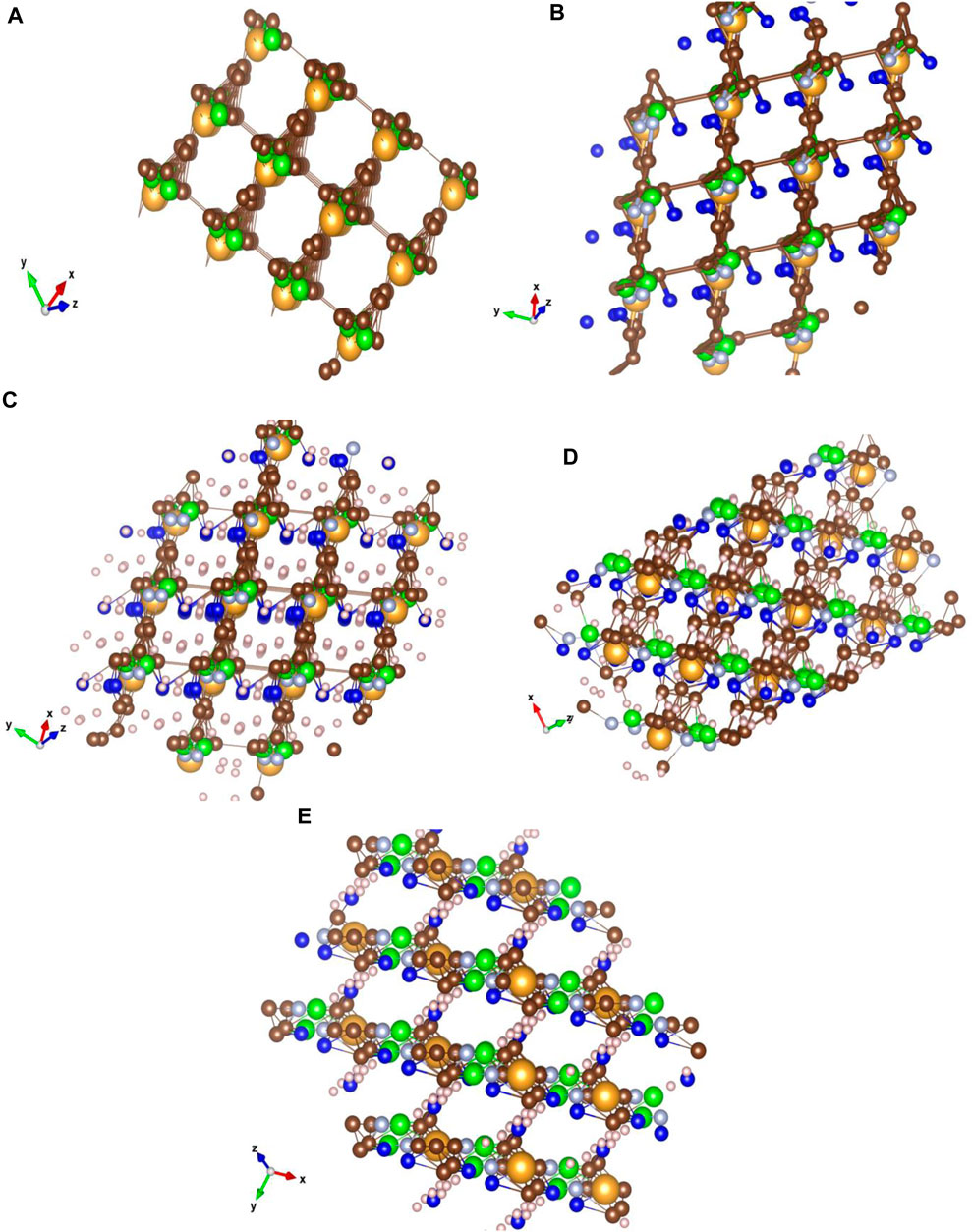
FIGURE 6. Crystal structure simulations of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) for nanomaterial characterization. Characteristic to the ImEtAuCl2CF3 crystal matrix is the bonding at 60° of the carbon basket surrounding the Au atom to the next C-ring structure through a fluorine-hydrogenated C bond, while nitrogen would have a higher noble metal affinity bonding to the Au–C structure forming AuNCl− sites with high electron affinity compared to the C lattice surrounding the Au atoms. The structure was optimized using Gaussian 16 and Avogadro software and analyzed using the VESTA software (Cl, green; F, dark blue; N, light blue; H, white; C, brown; and Au, yellow). (A) Crystal structure of ImEtAuCl2CF3 optimized at the B3LYP/Def2-TZVPP level of the theory with only Au, Cl, and C atoms and characteristic bond lengths of the Au–C bond from 0 to 2.065 Å and C–C between 0 and 1.358 Å, forming a carbonaceous matrix that surrounds the Au and Cl atoms in a square matrix; (B) the same type of matrix with similar bond lengths of Au–C and C–C with the addition of C–N and C–F bonds to the carbonaceous matrix; no bond between F and C is observed. The F atom places itself in the middle of the matrix next to the Au atom; H bond and atoms were removed for clarity; (C) YXZ view of the crystal with focus on the fluorine-hydrogenated bonds between the molecules; (D) XYZ view with focus on the carbon matrix of the crystal; (E) ZXY view of the crystal lattice with focus on the carbon rings and the fluorine-hydrogenated bonds. The Cl− atoms and N atoms gather around the Au atoms creating a basket, while the F− atoms were involved in creating a bond at 90° with the next molecules in a homogenous structure. The crystal lattice was built using 2 × 2 × 2 (a × b × c) Miller indices with a lattice unit cell of 4.275 Å on X and Y and 80 Å on Z at an angle of 60⁰ on z-axis and 90⁰ on x-axis and y-axis. The crystal matrix on all the planes has an inhomogeneous structure, the exceptions being YXZ at 20⁰, XYZ at −10⁰ to −70⁰ and ZXY at −10⁰ planes, where a coherent pattern can be seen between all molecules. Characteristic to the Au-fluorinated carbonaceous matrix is the presence of the Au atom in the center of a carbon ring bonded with a fluorine-hydrogenated carbonaceous bond with other layer molecules. The chlorine and nitrogen atoms follow Au closely through very strong interatomic forces. The lattice allows the presence of four molecules into a unit cell at 60⁰ plane angle.
The crystals belong to the monoclinic space group P21/c (β = 98.188°, V = 1219.69 Å2), and the unit cell is composed of a very weak aurophilic dimer with rather longer Au–Au short contact distance (3.772 Å), compared to the previously reported similar dimeric carbene–Au(I)–CF3 complexes (Blaya et al., 2014b). The coordination geometry of the Au(I) complex is linear (angle C1–Au(I)–CF3 = 176.93°) and slightly distorted with a tad bit shorter Au–CF3 distance (dAu–CF3 = 2.029 Å) compared to previously reported distances found in Au(I) trifluoromethyl complexes containing carbene, isonitrile, or C6F5 ligands (dAu–CF3 = 2.031–2.046 Å) (Blaya et al., 2014b). The molecule has been found to exist in antiparallel conformation with another monomer so as to facilitate closer distance between two Au centers, while minimizing steric repulsion between CF3 and carbene center, as shown in Figure 7B. It is noteworthy to mention that the two methyl groups attached to carbene NCH2 moiety of a molecule exist in the cis configuration to each other, thereby allowing a close approach between two Au centers and widening the distance from the adjacent molecule in the next row. Detailed list of bond angles and bond distances have been summarized in Table 3 taking into account van der Waals forces and short contact angles.
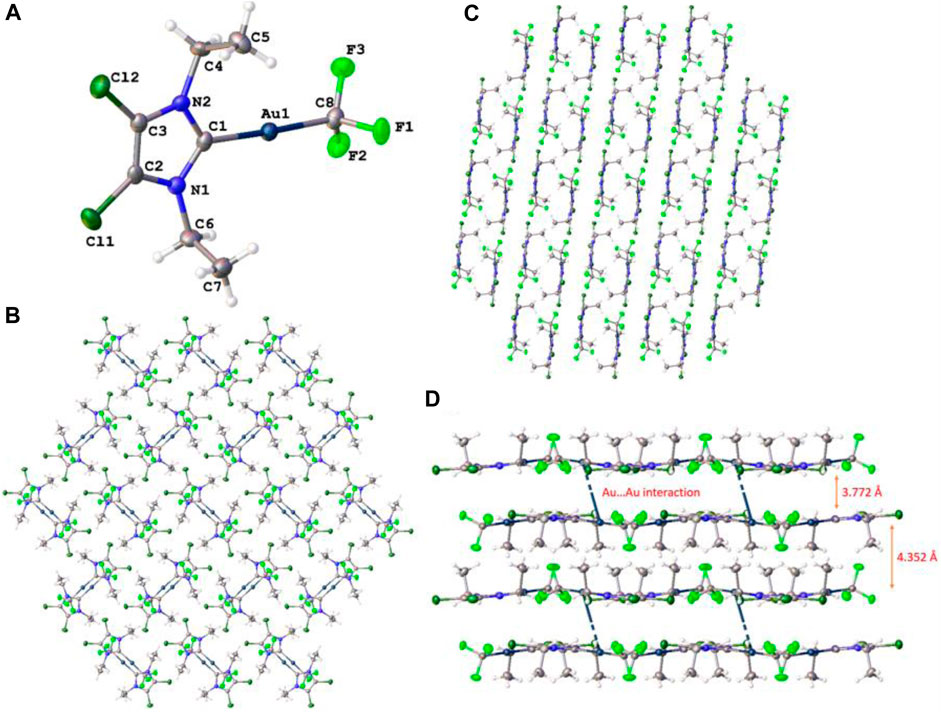
FIGURE 7. (A) Crystal structure of the 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) complex; ellipsoids have been drawn at 50% probability. Packing and interlayer distance (B) viewed along the a-axis, (C) b-axis, and (D) c-axis has been shown.
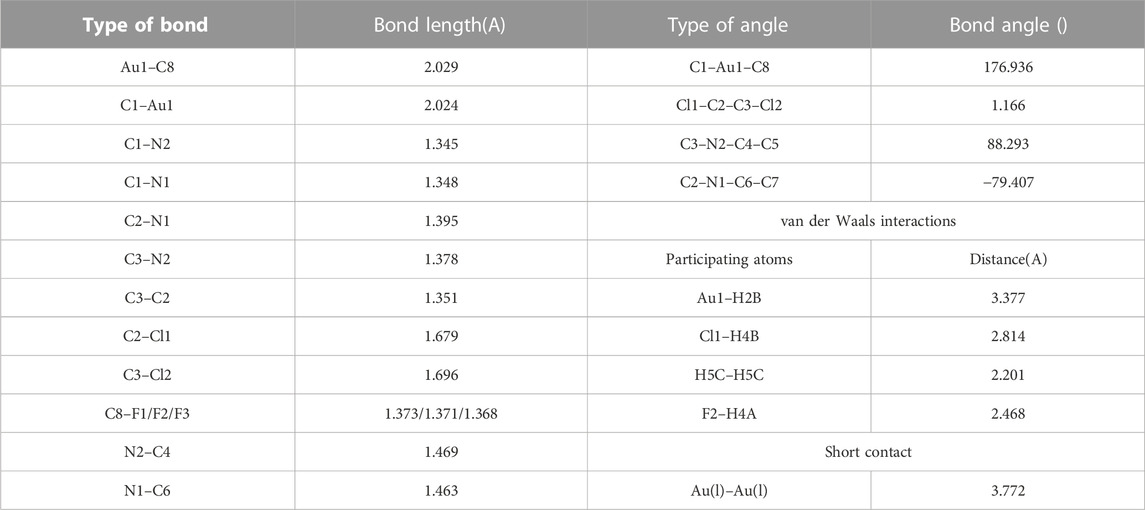
TABLE 3. List of bond lengths, bond angles, torsional angles, van der Waals interactions, and short contact obtained from single crystal XRD structural analysis.
Detailed investigation of the packing of the crystal structure (Figures 7B–D) revealed the presence of various weak non-covalent interactions that exist between adjacent rows of molecules. Apart from aurophilic interactions, each molecule engages with four surrounding molecules via four different van der Waals forces, which are summarized in Table 3. Among these interactions, each molecule engages with two adjacent molecules in the same row via F2···H4A and Cl1···H4B van der Waals forces and two molecules in the next row via Au1···H5B and H5C···H5C interactions. Interestingly, one Au center engages with another nearby Au center via well-known aurophilic interaction (short contact), albeit weak and with a methylene proton of another molecule, positioned in the opposite row to the first one, thereby resulting in different interlayer distances. The interlayer distances have been shown in Figure 7C, where the packing has been viewed along the c-axis.
A set of eight experiments at different temperatures (0–298 K) and pressures have been run using the powder X-ray diffraction method (Doumeng et al., 2021; Khan et al., 2020; Zhou et al., 2018) to determine the crystallinity and structure of the sample. For the characterization of nanomaterials and deposited complexes at the nanoscale, combinations of tools such as TEM (transmission electron microscopy), EXAFS (extended X-ray absorption fine structure), and XRD (Holder and Schaak, 2019; Gao and Lowry, 2018) are run to obtain particle size distributions and interlayer plane distances using TEM for the localized nanostructure size and powder XRD for an average nanostructure size. Further measurements can be carried out for structural characterization of the as-deposited nanomaterials using synchrotron radiation (X-ray absorption near edge structure (XANES) and X-ray small angle scattering (XSAS)) (Gao and Lowry, 2018; Amidani et al., 2021).
Two measurements have been performed to verify the stability and induced chemistry of the ligands of the compound (Figure 8) when exposed to air. Both NMR and XRD equipment work by having the sample handled in air (atmospheric pressure and room temperature). Its stability is an important characteristic of an FEBID precursor. The measurement in vacuum at 10−12mbar compared to the atmospheric pressure and 25 °C presents sharper peaks with reduced noise and a reduced width of the peaks.
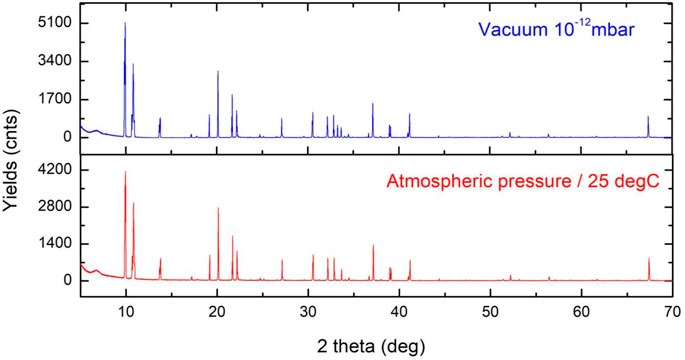
FIGURE 8. XRD of the Au compound under vacuum condition (10−12mbar) and at room temperature and atmospheric pressure. The two graphs show very small differences under two different conditions, and the highest changes are observed between 30 and 35 2 θ(°).
At 2θ (°) 30–35 °, we observe the presence of a third peak corresponding to the Au–CF3 bond increasing the bond distance with the change of pressure. With change in the pressure and temperature, CF3 as the most volatile radical of the compound changes the bond length in the range of 10–1 Å.
The size of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) grains (Figure 9) is calculated using Formula (1), where λ = 1,54060 Å and β is the FWHM width of the peak in radians and θ position of the peaks/2 in radians:
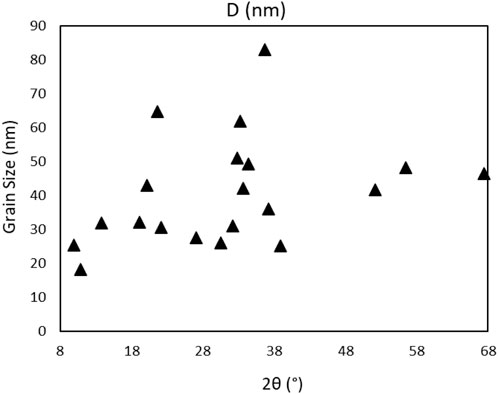
FIGURE 9. Powder grain size distribution of the Au compound.
The highest grain size batch is of 83 nm at 0.64 rad θ, with an average grain size of all peak batch of 41 nm. Further analysis of XRD powder data of the compound gives information on the crystallinity by using the position of the 2θ peaks on the XRD spectrum, the spectrum recorded under the vacuum was used as cleaner and without H2O presence. The crystallinity of the precursor is calculated using Eq. 2 (Zhong et al., 2009) with a value of 54.427%:
A crystallinity of up to 55% is expected, with the grain size limited to an average of 41 nm, and the highest grain of 82.86 nm, rather small compared to a grown crystal structure or multiple grown crystals in the powder structure. The complex is in the amorphous phase mixed with small grains in the form of nanostructures.
An increase in the peak amplitude is observed for the 298 K spectrum compared to the rest, a behavior expected at RT, while a separate increase in the singular peak amplitude is observed for 273K and 278K at 2θ(°) 26 corresponding to the C (002) phase, 298K at 2θ(°) 62 corresponding to Au–chloride (220) body-centered cubic (bcc) plane of the crystalline powder at 293 K at 2θ(°) 66.5 corresponding to the (400) bcc Au–chloride plane and phase (Figure 10). A separate view of the planes and phases of the crystalline powder and temperature is illustrated in Figure 10, where phases of bcc are combined with face-centered cubic (fcc), and an intermediate highly hydrogenated layer creating the interspacing of the atom and network positioned between fcc and bcc is observed, Au surrounding itself with chlorine atoms.
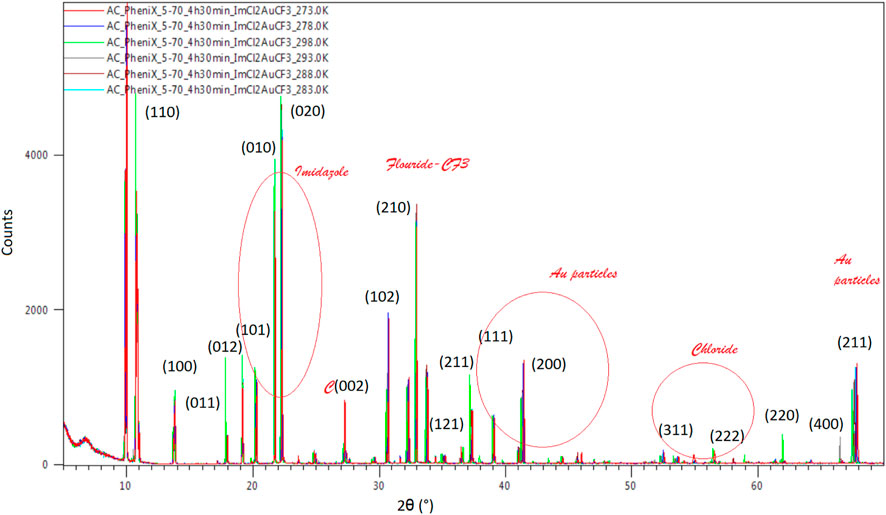
FIGURE 10. Temperature dependence of XRD data of the Au compound to 2θ (°). A normalized behavior with increasing amplitude and constant delay is observed by changing the temperature from 273 to 298K.
A number of six gold(I)-containing ions (Figure 11) are the results of fragmentation of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I), as the only anion that contains a metal atom, found in the dissociative electron attachment (DEA) process (Shuman et al., 2011; Sauer et al., 2002) of the compound. The two higher mass fragments, namely, C5H10N2F2AuCl2− (m/z 389) and C7H10N2FAuCl2− (m/z 334), are rather noisy with low cross sections, both having one resonance peak with the highest value before 1 eV. With higher cross-section values, the C7H10N2AuCl2− anion has an average count value of 150 counts at the maximum of the resonance, falling at 0.86 eV, with an average width of the resonance peak at 2.03 eV. A lower value of the cross-section with maximum counts of two counts characterizes the anion C5H9NFAuCl− (m/z 334), presenting similar shape to the higher mass anion at m/z 389. The electron energy characteristic to the resonance of the anion (m/z 334) is 0.84 eV, with a width of 1.81 eV. In the normal dissociation process of C8H10Cl2N2AuF3, the parent is excited by the collision with an electron and dissociates into an anion and a neutral fragment, but at kinetic energies lower than 0.1 eV, a process of thermal decomposition is the path followed in the dissociation of the precursor, a process observed in the formation of C7H10N2FAuCl2− at kinetic energies as low as 0.03 eV. A transition from the π HOMO state to LUMO in π* takes place at the resonance incident energy for a bond dissociation energy of 0.89 eV with the loss of CF2 in the neutral state.
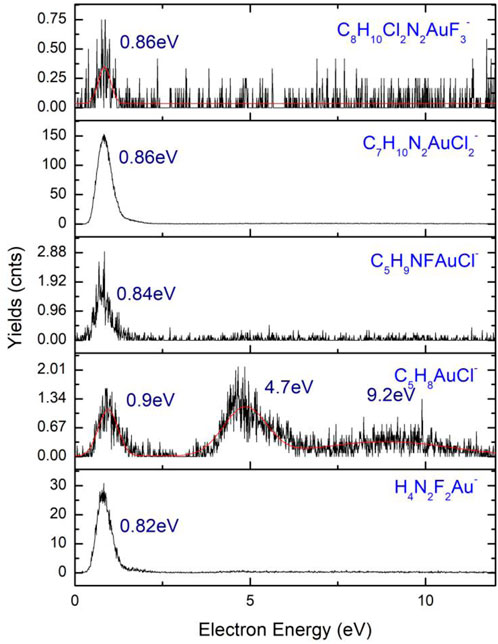
FIGURE 11. Anions of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) m/z 267 (H4N2F2Au−) to m/z 458 (C8H10Cl2N2AuF3−).
At m/z 300, characterized by a very low cross-section value, the C5H8AuCl− anion is found. The three peak resonances of the C5H8AuCl− anion are at electron energies of 0.9 eV, 4.7 eV, and 9.2 eV, with high characteristic widths and a noisier shape. The widths of the three resonances are in the range of ∼2–3 eV, with values of 1.53 eV (0.9 eV), 3.38 eV (4.7 eV), and 3.3 eV (9.2 eV). The smaller mass anion of the six gold(I)-containing ions is H4N2F2Au− characterized by the presence of nitrogen and fluoride atoms in its composition and a high cross-section. The peak of the highest resonance is found at 0.82 eV with a maximum of the peak of 32 counts and a width of 1.67 eV. The smaller amplitude resonance falls at 4.6 eV having a width of 8 eV and a number of counts lower than two counts. Another Au-containing anion is H4N2F2Au− with its resonance peaking at 0.82 eV characterized by a peak width of 0.18 eV. The bond dissociation energy of the ion formation has a value of 0.021 eV with a maximum kinetic energy of 3. 75 eV. A π to σ* transition is characterized by the HOMO to LUMO transition in the dissociation process for a C2v symmetry of the formed H4N2F2Au− anion.
The parent anion C8H10Cl2N2AuF3− is present with a resonance peaking at 0.86 eV, showing a very low cross-section with a reduced number of counts (<0.75 counts). At the excitation of the C8H10Cl2N2AuF3, a temporary negative ion is formed with a maximum kinetic energy of 1.14 eV, where conservation of the C2v symmetry state is observed for the excited parent anion in the transition from π to π*.
An equal number of organic anions (Figure 12) are found as a result of DEA fragmentation of the precursor, the most abundant being Cl− and CH4N2Cl−. These five lower mass fragments are particularly interesting because of the lack of any metal atom in their composition, depositing, and releasing as a result of collision and ionization in the interaction with a secondary electron having only volatile fragments and organic materials, increasing the level of contamination of the FEBID structures. Another very abundant anion is the C2H6NCl− ion having only one resonance peak at 0.84 eV with a width of 1.7 eV and 24 counts and a relatively high cross-section compared to the rest of the ions presenting values under 10 counts.
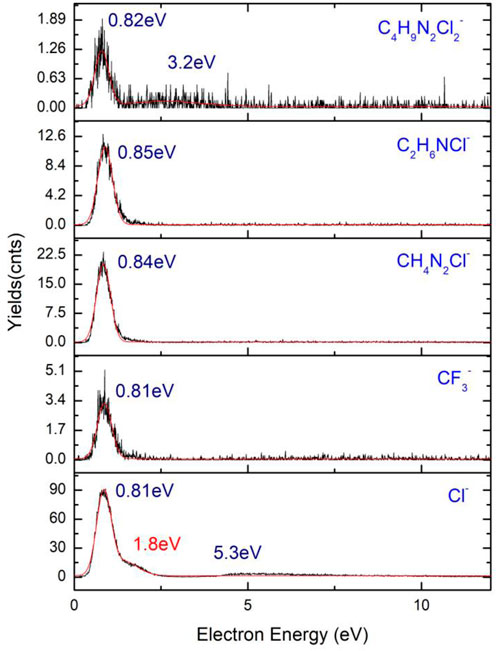
FIGURE 12. Organic parts and volatile fragment anions of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I).
The Cl− ion corresponding to an m/z of 35 presents a resonance peak at 0.85 eV with high cross-section values, being the most abundant anion result of the fragmentation of the precursor. A shoulder corresponding to the same resonance is observed at 1.8 eV, while a wider resonance peak with a width of almost 4 eV is observed at 5.3 eV. The formation of the Cl− ion is a transition from the π to σ* process characterized by a bond dissociation energy (BDE) of 0.59 eV. A maximum of the kinetic energy of the Cl anion is obtained with a value of 0.26 eV. Fragmentation of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) follows the steps in relation (1) to the result of a chloride anion and of a higher mass neutral fragment, C8H10Cl2N2AuF3 + e− → C8H10Cl2N2AuF3− → C8H10ClN2AuF3 + Cl− (1). Xuan et al. (2014) presents the fragmentation of 1,2-dichlorobenzene at low energy DEA studies by ion mass spectroscopy, time-of-flight, and VMI on the fragmentation of the compound, resulting in a Cl− ion with two resonances, 1.2 eV and 6 eV, the latter being a wider resonance of the anion possibly corresponding to the two isotopes of chloride, namely, 35Cl− and 37Cl−.
The chloride anion is not an atypical ion in the fragmentation process of the compounds containing Cl, a majority of them forming the chloride anion as a product of reaction in the induced chemistry during the interaction of the molecule with electrons. In the fragmentation of diatomic molecules, 1,2-dichlorobenzene (1,2 - C6H4Cl2) undergoes a transition from σ to σ* that further initiates the fragmentation of the molecule with the resulting fragments being C6H4–Cl+Cl−, with the chloride anion in the 1∑g* excited state and Oh geometry. A study of four chlorine-containing compounds (CCl4, CH2Cl2, CH3Cl, and CHCl3) (Scheunemann et al., 1980) at DEA fragmentation exhibits the presence of the chloride anion at energies close to 0 eV. The positions of the resonances of the four anions are presented in Table 4. Each of the ion has the highest amplitude peak close to 0 eV at an electron energy of 0.0 ± 0.05 eV, and the second resonance peak between 6 eV and 8 eV. Cl− from CCl4 has a shoulder of the first resonance of chloride falling at 0.75 ± 0.05 eV, with a value of the bond dissociation energy of C–Cl ligand of 3.3 ± 0.3 eV and a characteristic electron affinity of EA (Cl2) = 2.35 ± 0.1 eV. The electron affinity reported by NIST Database is in good agreement with the values of 2.5 ± 0.2 eV reported by Scheunemann et al. (1980).
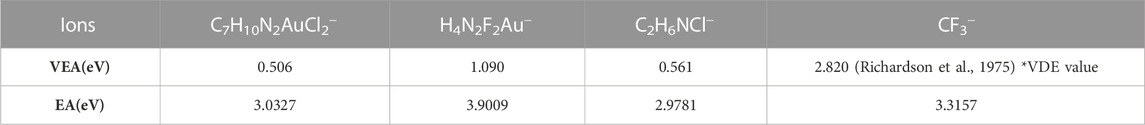
TABLE 4. Electron affinities of ions including C7H10N2AuCl2−(m/z 389), H4N2F2Au−(m/z 267), C2H6NCl− (m/z 79), and CF3− (m/z 69).
The two lower value cross-section fragments, namely, CF3− and C4H9N2Cl2− lacking the presence of any metal atom, have amplitudes in the range of ∼2–5 counts, with the highest amplitude peak for C4H9N2Cl2− falling at 0.82 eV having a width of the resonance of 1.26 eV, while the second peak of the same resonance has its maximum at 3.2 eV with the width of the peak of 2.16 eV. The CF3− anion has its maximum amplitude of the first resonance peak at 0.81 eV with a width of 1.5 eV and the second peak maximum at 7.2 eV characterized by a width of 6.6 eV. BDE of the Cl− formation is 0.59 eV and has a specific kinetic energy of 0.26 eV, where the HOMO to LUMO transition is from π to σ* with the anion having Cs symmetry.
Manaa (2017) defines the value of the calculated electron affinity from Gaussian 4 simulations at the CCSD(T) (coupled cluster single-double and perturbative triple) level of the theory as the sum of the values of energy Ee for the neutral and the anion with added zero-point corrections of the two values. (2) EA = [Ee (optimized neutral) + ZPE (neutral)]–[Ee (anion) + ZPE (anion)]. A similar relation is used for the cation ionization potential at ZPE (zero-point energy); (3) IP = [Ee (cation) + ZPE (cation)]–[Ee (optimized neutral) + ZPE (optimized neutral)]. In the dissociative electron attachment calculations of VEA and EA run at the DFT level, the values of the transitions from HOMO to LUMO orbitals are related to excitation energies with the formation of a temporary negative ion (TNI). The electron affinity and electronegative potential (absolute electronegativity or absolute hardness) of the anion results of the DEA process of 4,5-dichloro–1,3-diethyl-imidazolylidene trifluoromethyl gold(I) follow (2) with resulting values in the range of 2.9–3.9 eV (see Table 4). The electron affinities (EA) of the products have been calculated at room temperature (298.15 K), the zero-point energy (ZPE) corrections being highly sensitive to the input temperature. The highest value of the electron affinity is obtained for mass m/z 267 corresponding to H4N2F2Au− with an EA value of 3.9 eV (∼0.143357 Hartree). Lower electron affinity values are calculated for the Cl− ion and C7H10N2AuCl2− with values of 3.32 eV (∼0.12185 Hartree) and 3.03 eV (∼0.11145 Hartree), respectively. The VEA (vertical electron affinities) values from our Gaussian 16 simulations at the B3LYP/LANL2DZ level of the theory are calculated using the natural bond orbital populations (NBO) and pole p3+ calculations (Table 4). The vertical electron affinities (VEA) (Jiao et al., 2016; Li et al., 2002) are calculated for each ion with high cross-sectional values ranging from 0.51 eV to 1.09 eV. VEA of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) has a value of 0.24 eV, 0.15 eV higher than the excited anion parent with a value of VEA of 0.096 eV, similar to other compounds containing C–H bonds; example of CH3−, SiH3−, and CHCH2− (Amati et al., 2020). The anion parent could not be determined experimentally as it is characterized by a short-lived life and instability in the 2A1’ state.
In the fragmentation of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I), multiple pathways are possible, described by relations (4) and (5), with the formation of the CF3− (m/z 69) anion with C7H10Cl2N2Au as the neutral fragment, while the second fragmentation pathway results in the formation of the C7H10N2Cl2Au− (m/z 389) anion and CF3 as a neutral fragment: C8H10N2Cl2AuF3 + e− → C8H10Cl2N2AuF3− → C7H10N2Cl2Au− + CF3 (4) and C8H10N2Cl2AuF3 + e− → C8H10Cl2N2AuF3− →C7H10N2Cl2Au + CF3− (5). While CF3 in its ground state, 2A1 has a C3v geometry; for the CF3− anion, the symmetry point group conserves (C3v), but the excited state of the anion transitions to 1A1 presenting two peaks of the resonance, namely, at 0.81 eV and 7.2 eV; other higher excited states of the anion correspond to E″1 and E″2. BDE of the CF3− formation is 0.53 eV with a maximum kinetic energy of 0.47 eV. The CF3− anion from 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) is observed to behave similarly to CF3− in 5-trifluoromethanesulfonyl-uracil (Ameixa et al., 2018; Bald et al., 2007), which is used extensively in the research of cancer therapy as a potential radiosensitizer, reducing the amount of radiation needed for the treatment of the cancer cells. Defined by an electron affinity (EA) of 1.69 eV, the fragmentation of 5-trifluoromethanesulfonyl-uracil (OTfU) (Ameixa et al., 2018) in a neutral fragment and CF3− takes place at an electron energy characterized by four resonance peaks, namely, at 0.01 eV, 2.35 eV, 4.75 eV, and 8.45 eV, which is the result of dissociation of the S–CF3 ligand. The CF3− ion in the hexafluoroacetone azine ((CF3)2C = NN = C(CF3)2) (Bald et al., 2007) reaction has its resonances falling at higher energies representing typically a bond cleavage with the fragmentation of a C–CF3−, a C bonded to CF3 having a lower bond dissociation energy than the Au–CF3 bond cleavage for 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I). Values of 1.61 eV are reported for the affinity of CF3− (Bald et al., 2007) with the DEA resonance peaking at two energies, namely, 3.8 eV with the highest amplitude and 7.3 eV resonance with a lower amplitude, with less than <10 counts. Calculations of the vertical electron affinities (VEA) and bond dissociation energies show higher bond strength of the Au–CF3 ligand compared to the organic ligands in ImCl2EtCF3Au.
The proton NMR measurements and water NMR measurements are rather simple measurements used often in the pharmacology industry for stability analysis of complex drugs and compounds and the degradation study of these compounds in specified conditions (pressure, temperature, and luminosity). The applications of the proton NMR studies are not limited to only stability analysis but also have implications in the study of the structure and bonding of complex compounds to certain proteins (RNA signal assignment and validation (Barton et al., 2013), probing metallic-aromaticity (Bass et al., 2020), or structural changes (n-membrane lactones isomerism (Marell et al., 2014). The most common example of the use of this type of measurement is the proton NMR studies to proteins (Taraban et al., 2017) in different storage, transportation, and daily-use conditions. The proton NMR and water NMR offered in these circumstances provide a comprehensive view on the stability of the compound and the time it takes for the chemical complex to degrade or to form new bonds as a result of transition processes to a new form or a chemical reaction induced by temperature or changes in the environmental conditions (pressure or light).
A set of 1H (Figure 13) and 19F (Figure 14) spectra were queued such that each element was monitored over 80 min. 1H NMR spectra are referenced to residual protio-solvent. The 1H spectra show no significant change over 81 min (time in the instrument, ∼5 min between sample creation and injection into the instrument). There was a slight drift in linewidths, and the resonance at δ1.5 ppm caused by water contamination broadens and shifts from 1.565 to 1.569 ppm, i.e., negligibly consistent with changes in H2O and HCl concentration. The image below shows the stacked spectra and a blown-up portion arising from the CH3 groups.

FIGURE 13. Chemical shift of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) in CDCl3 solution.
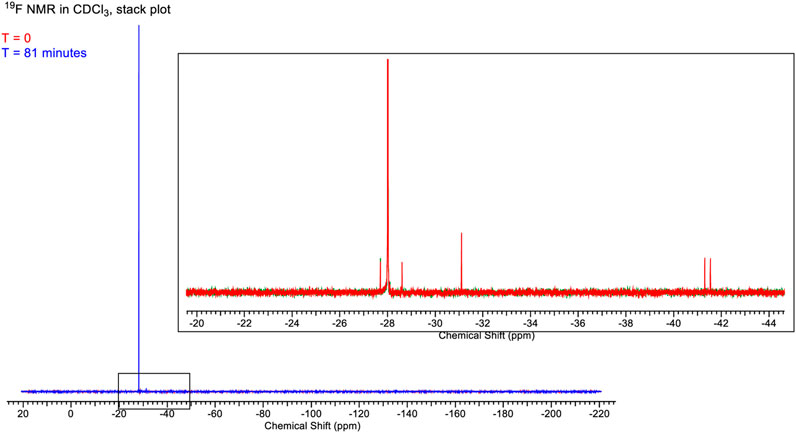
FIGURE 14. Full spectrum of the chemical shift of 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) in CDCl3 solution.
The 19F spectra also show minimal change over the time period. The stackplot shows only the first and last spectra recorded for clarity. The two spectra are essentially identical to save for the disappearance of two minor peaks at δ-41.5 ppm and δ-41.8 ppm. In the initial spectrum, these account for ∼2% of the total integrable intensity.
The Gibbs free energy (Salike and Bhatt, 2020; Lee et al., 2015; Martins and Cabral, 2019) of formation and reaction has been used multiple times to analyze the suitability of different complexes and organic or protein material for drug resistance (Tahir et al., 2020), equilibrium calculations of emulsion systems (Hosseini and Mohammadi, 2020), enthalpy of DNA formation (Lomzov et al., 2015), calculations of different mineral formation (Olivotos and Economou-Eliopoulos, 2016), and nanolithography (the present study). The Gibbs free energy of a compound is explained in terms of enthalpy (H), temperature (T), and entropy (S) by the relation: ΔG = ΔH–TΔS. The values of the enthalpy and entropy are taken from simulation data for each of the products and reactants. For a reaction, the Gibbs free energy is obtained (Ochterski, 2000) from relation (4) ΔG = G (products)–G (reactants), while for molecular complexes, relation (5) ΔG = G (species)–∑G (elements) suffices. The Gaussian software works in calculating the corrections to the enthalpy and entropy of formation or reaction based on total energy and contributions from vibrational, rotation, translational, or electronic motion; according to this, the enthalpy correction is explained by the relation (6): HCorr = Etot + kBT (Ye et al., 2018), where Etot is the total energy. In a similar manner, the entropy is defined by Stot = St + Sr + Sv + Se (7) (Ye et al., 2018), where St, Sr, Sv, and Se are translational, rotational, vibrational, and electronic contributions, respectively.
Materials in powder form are usually analyzed for the moisture content mostly in the food industry (Lüttge, 2006; Cano-Higuita et al., 2015; López-Vidaña et al., 2021) and to obtain the dissolution rates of complexes (Zhang et al., 2019) through the calculation of the Gibbs free energy, but the use of Gibbs free energy still remains the means to determine the stability of a compound using simulation obtained values of entropies and enthalpies. In order to analyze the suitability of the Cl2ImEtCF3Au precursor, the Gibbs free energy from DFT calculations has been used. More industrial oriented applications to pipeline transport industry (water, gas, oil, and steam) (Jäger and Span, 2012) are by the analysis of Gibbs free energy of solid-state CO2 in the transport of CO2 in carbon capture and storage (CCS). The Gibbs free energy is the entity that defines the probability of a reaction to take place, the volatility and stability of the compound. The values of ε0, εZPE, HCorr, and GCorr are calculated from the thermochemistry of Cl2ImEtCF3Au at the DFT level using B3LYP with a Def2-TZVPP basis set, where ε0 is the electronic energy, εZPE is the zero-point energy, HCorr is the enthalpy correction, and GCorr is the Gibbs free energy correction. The sum of the electronic and enthalpy energy, the sum of the electronic and Gibbs free energy, and the sum of the electronic and zero-point energy are used as ε0 + HCorr, ε0 + GCorr, and ε0 + εZPE. The calculated thermochemistry values are presented in Table 5 with the values obtained from the Gaussian calculations. The calculations have been performed at a temperature of 298.15 K and a pressure of 1 × 10-4 Pa. The reaction with the result of an anion and a neutral fragment formation follows the pathway Cl2ImEtCF3Au* → Cl2ImEtAu− (m/z 389) + CF3 (m/z 69) for which the reaction energy and enthalpy are calculated to obtain the bond dissociation energy (BDE) and bond dissociation free energy (BDFE) of the reactants into products of reaction. Calculations of Gibbs free energy at the atomistic level with great results in modeling of crystal defects are reported by Cheng and Ceriotti (2018). Though at defect sites, the model predicts energies 300% higher than evaluated, and at non-defect sites, it predicts energies 10% higher than reported for the evaluated model. The higher defect estimated value of the Gibbs free energy is presented as a result of the anharmonicity at the defect sites with the transition at higher temperatures (>298 K). BDE (Fukaya et al., 2001; Luo, 2007; Woldu and Mai, 2012) and BDFE (Parr and Pearson, 1983; Moroz, 2011) are calculated for the chemical reaction, taking into account the ε0 correction to electronic energy and the enthalpy and Gibbs free energy corrections HCorr and GCorr at 298.15K, obtaining the change in enthalpy (4) and Gibbs free energy (3) with the reaction.
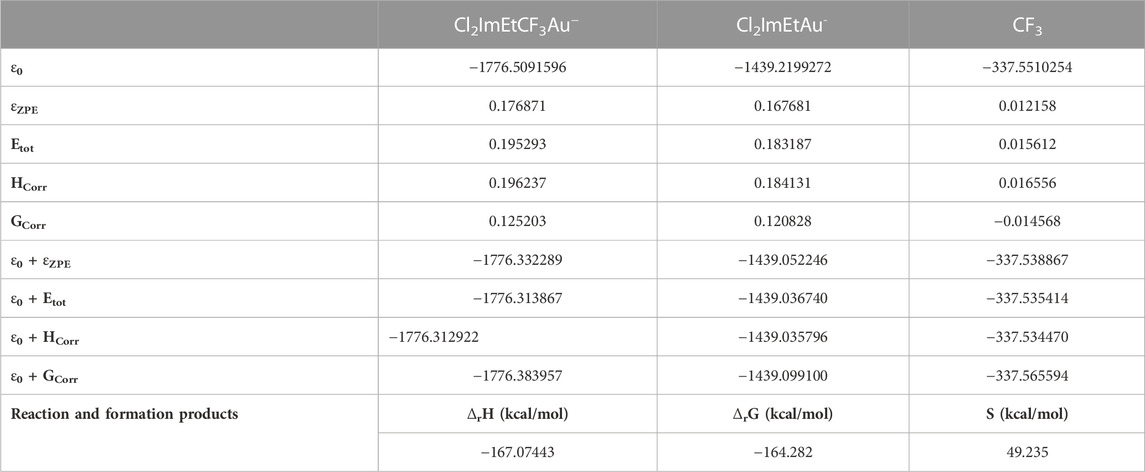
TABLE 5. Free Gibbs energy correction, enthalpy correction, and zero-point corrections of Cl2ImEtCF3Au−, Cl2ImEtAu−, and CF3, respectively, which are products of formation and products of reaction.
BDEs and BDFEs can be obtained for the formation reaction of the products of reaction, the anion and the neutral fragment ΔfH (5) and ΔfG (6):
The results of the calculations are presented in Table 5 with a reaction BDE of −0.2618 (Hartree: 164.282 kcal/mol).
The accuracy of the B3LYP/Def2-TZVPP basis set is related to spin-orbit coupling effects (Armbruster et al., 2006), where large errors are obtained in total atomic energies and atomization energies for heavy atoms (as is the case of the Au). The effective core potential (ECP) addition to the basis sets would reduce the effects induced in the 5p and 6p elements and the relativistic effects. The use of two component approaches is the most common method discussed in detail by Xu and Truhlar (2011), where (2p2s) polarization functions are added to the triple-ζ basis sets to reduce the aforementioned presented errors. A difference of 23.868 kcal/mol is obtained in the BDE values, from the reaction energy and DFT calculation. Large errors of over 10–12 kcal/mol (Armbruster et al., 2006; Xu and Truhlar, 2011) are known to be produced by high basis sets, such as TZVP, LANL2DZ, and Def2-XYVP (where XY = TZ, QZ, and so on).
The metal–ligand bond between Au(I) and CF3 presents higher BDE for Cl2ImEtCF3Au (189.15 kcal/mol) than for Au(I)–CF3 in CF3AuCO of 151.4 kcal/mol, Au(I)–Cl in ClAuPMe3 of 77.9 kcal/mol or Au(I)–Me in MeAuPMe3 of 43.4 kcal/mol reported by Marashdeh et al. (2017). The value reported from our calculations of −164.282 kcal/mol is specific for an exergonic process releasing energy, though it is not characterized by a high cross-section value for the elimination of the Au–CF3 ligand. Lower ΔfG would mean that the molecule is unstable, making it hard to work with and difficult to transfer from the vial through the gas line inside the vacuum chamber. Values as low as +16.5 kcal/mol for ClAuPF3 have been reported by Marashdeh et al. (2017), rendering the ClAuPF3 compound as one of the compounds with low vaporization pressure. Not stable in air and at room temperature, Cl2ImEtCF3Au has similar behavior to AuCF3CO (Marashdeh et al., 2017; Martínez-Salvador et al., 2011) that darkens in the presence of heat and light, a sign of the oxidation process. Cl2ImEtCF3Au is not to be kept at temperatures higher than 5°C as it spontaneously breaks ligands and degrades, while the presence of air would intensify the process of degradation and oxidation.
4,5-Dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I) was analyzed for its suitability as a FEBID precursor. As a newly designed compound specifically for the deposition of nanoscale structure, its vaporization pressure, stability in air, and volatility have been studied using proton NMR and Gibbs free energy of reaction.
A good volatility value was obtained for the compound and a high stability in air with very low modifications of the structure during exposure. Its fragmentation, resonances, and anions at low electron energies and DEA have been obtained using velocity map imaging studies with great success. The structure, packing, orientation of the planes, and grain size have been run making use of powder XRD diffractometer data, and the VESTA simulation software has offered reliable insights into the crystalline vs. amorphous structure of the compound.
The data presented in the study are deposited in https://www.ccdc.cam.ac.uk/structures/. The 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(i) compound was registered in the Cambridge Structural Database with the CCDC number 2223878.
All authors listed have made a substantial, direct, and intellectual contribution to the manuscript and approved it for publication.
We want to thank MP for receiving funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no 722149, and the work of our partner institutions J. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences and University of Oslo. We also thank David S. Wragg for measuring, solving, and refining the single-crystal X-ray structure, and we thank for the use of the Norwegian National Centre for X-ray diffraction and scattering (RECX).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Amati, M., Stoia, S., and Baerends, E. J. (2020). The electron affinity as the highest occupied anion orbital energy with a sufficiently accurate approximation of the exact Kohn–Sham potential. J. Chem. Theory Comput. 16 (1), 443–452. doi:10.1021/acs.jctc.9b00981
Ameixa, J., Arthur-Baidoo, E., Meiβner, R., Makurat, S., Kozak, W., Butowska, K., et al. (2018). Low-energy electron-induced decomposition of 5-trifluoromethanesulfonyl-uracil: A potential radiosensitizer. J. J. Chem. Phys. 149, 164307. doi:10.1063/1.5050594
Amidani, L., Vaughan, G. B. M., Plakhova, T. V., Yu Romanchuk, A., Gerber, E., Svetogorov, R., et al. (2021). The application of HEXS and HERFD XANES for accurate structural characterisation of actinide nanomaterials: The case of ThO 2. Eur. J. 27, 252–263. doi:10.1002/chem.202003360
Armbruster, M. K., Kloppera, W., and Weigend, F. (2006). Basis-set extensions for two-component spin–orbit treatments of heavy elements. Phys. Chem. Chem. Phys. 8, 4862–4865. doi:10.1039/b610211e
Bald, I., Dabkowska, I., Illenberger, E., and Ingólfsson, F. (2007). Energy selective excision of CN− following electron attachment to hexafluoroacetone azine ((CF3)2CN–NC(CF3)2). Phys. Chem. Chem. Phys. 9, 2983–2990. doi:10.1039/b702482g
Barton, S., Heng, X., Johnson, B. A., and Summers, M. F. (2013). Database proton NMR chemical shifts for RNA signal assignment and validation. J. Biomol. NMR 55, 33–46. doi:10.1007/s10858-012-9683-9
Bass, T. M., Carr, C. R., Sherbow, T. J., Fettinger, J. C., and Berben, L. A. (2020). Syntheses of square planar gallium complexes and a proton NMR correlation probing metalloaromaticity. Inorg. Chem. 59 (18), 13517–13523. doi:10.1021/acs.inorgchem.0c01908
Belić, D., Shawrav, M. M., Bertagnolli, E., and Wanzenboeck, H. D. (2017). Direct writing of gold nanostructures with an electron beam: On the way to pure nanostructures by combining optimized deposition with oxygen-plasma treatment. Beilstein J. Nanotechnol. 8, 2530–2543. doi:10.3762/bjnano.8.253
Benitez, D., Shapiro, N. D., Tkatchouk, E., Wang, Y., Goddard, W. A., and Toste, F. D. (2009). A bonding model for gold(I) carbene complexes. Nat. Chem. 1 (6), 482–486. doi:10.1038/nchem.331
Blaya, M., Bautista, D., and Gil-Rubio, J. (2014a). Synthesis of Au(I) trifluoromethyl complexes. Oxidation to Au(III) and reductive elimination of halotrifluoromethanes. J. Organometallics 33, 6358–6368. doi:10.1021/om500669j
Blaya, M., Bautista, D., Gil-Rubio, J., and Vicente, J. (2014b). Synthesis of Au(I) trifluoromethyl complexes. Oxidation to Au(III) and reductive elimination of halotrifluoromethanes. Organometallics 33 (22), 6358–6368. doi:10.1021/om500669j
Botman, A., Mulders, J. J. L., and Hagen, C. W. (2009). Creating pure nanostructures from electron beam-induced deposition using purification techniques: A technology perspective. Nanotechnology 20, 372001. doi:10.1088/0957-4484/20/37/372001
Brintlinger, T., Fuhrer, M. S., Melngailis, J., Utke, I., Bret, T., Perentes, A., et al. (2005). Electrodes for carbon nanotube devices by focused electron beam induced deposition of gold. J. Vac. Sci. Technol. B 23, 3174. doi:10.1116/1.2130355
Bull, J. N., Lee, J. W. L., Gardiner, S. H., and Vallance, C. (2014). Account: An introduction to velocity-map imaging mass spectrometry (VMImMS). Eur. J. Mass Spectrom. 20 (2), 117–129. doi:10.1255/ejms.1264
Cano-Higuita, D. M., Villa-Vélez, H. A., Telis-Romero, J., Váquiro, H. A., and Nicoletti Telis, V. R. (2015). Influence of alternative drying aids on water sorption of spray dried mango mix powders: A thermodynamic approach. Food Bioprod. Process. 93, 19–28. doi:10.1016/j.fbp.2013.10.005
Carden, W. G., Lu, H., Spencer, J. A., Fairbrother, D. H., and McElwee-White, L. (2018). Mechanism-based design of precursors for focused electron beam-induced deposition. MRS Commun. 8, 343–357. doi:10.1557/mrc.2018.77
Carden, W. G., Thorman, R. M., Unlu, I., Abboud, K. A., Fairbrother, H., and McElwee-White, L. (2019). Design, synthesis, and evaluation of CF3AuCNR precursors for focused electron beam-induced deposition of gold. ACS Appl. Mat. Interfaces 11, 11976–11987. doi:10.1021/acsami.8b18368
Chang, S.-Y., Uehara, A., Booth, S. G., Ignatyev, K., Frederick, J., Mosselmans, W., et al. (2015). Structure and bonding in Au(I) chloride species: A critical examination of X-ray absorption spectroscopy (XAS) data. RSC Adv. 5, 6912–6918. doi:10.1039/c4ra13087a
Cheng, B., and Ceriotti, M. (2018). Computing the absolute Gibbs free energy in atomistic simulations: Applications to defects in solids. Phys. Rev. B 97, 054102. doi:10.1103/physrevb.97.054102
Chien, M.-H., Shawrav, M. M., Hingerl, K., Taus, P., Schinnerl, M., Wanzenboeck, H. D., et al. (2021). Analysis of carbon content in direct-write plasmonic Au structures by nanomechanical scanning absorption microscopy. J. Appl. Phys. 129, 063105. doi:10.1063/5.0035234
Doumeng, M., Makhlouf, L., Berthet, B., Marsan, O., Denape, J., Chabert, F., et al. (2021). A comparative study of the crystallinity of polyetheretherketone by using density, DSC, XRD, and Raman spectroscopy techniques. Polym. Test. Vol. 93, 106878. doi:10.1016/j.polymertesting.2020.106878
Fernández-Moreira, V., Marzo, I., and Concepción Gimeno, M. (2014). Luminescent Re(i) and Re(i)/Au(i) complexes as cooperative partners in cell imaging and cancer therapy. Chem. Sci. 5, 4434–4446. doi:10.1039/c4sc01684j
Fowlkes, J. D., Winkler, R., Lewis, B. B., Fernandez-Pacheco, A., Skoric, L., Sanz-Hernandez, D., et al. (2018). High-fidelity 3D-nanoprinting via focused electron beams: Computer-aided design (3BID). ACS Appl. Nano Mat. 1, 1028–1041. doi:10.1021/acsanm.7b00342
Fukaya, H., Ono, T., and Abe, T. (2001). Bond dissociation energies of CF3-X bonds (X = C, O, N, S, Br): Ab initio molecular orbital calculation and application to evaluation of fire suppression ability. J. Phys. Chem. A 105 (31), 7401–7404. doi:10.1021/jp011641z
Furst, M. R. L., and Cazin C.S., (2010). Copper N-heterocyclic carbene (NHC) complexes as carbene transfer reagents. J. Chem. Commun. 46, 6924–6925. doi:10.1039/c0cc02308f
Galassi, R., Oumarou, C. S., Burini, A., Dolmella, A., Micozzi, D., Vincenzettic, S., et al. (2015). A study on the inhibition of dihydrofolate reductase (DHFR) from Escherichia coli by gold(I) phosphane compounds. X-ray crystal structures of (4,5 – dichloro – 1H – imidazolate – 1 – yl) triphenylphosphane - gold(I) and (4,5 – dicyano – 1Himidazolate – 1 – yl) – triphenylphosphane - gold(I). Dalton Trans. 44, 3043–3056. doi:10.1039/c4dt01542h
Gao, Xiaoyu, and Lowry, Gregory V. (2018). Progress towards standardized and validated characterizations for measuring physicochemical properties of manufactured nanomaterials relevant to nano health and safety risks. Nano Impact 9, 14–30. doi:10.1016/j.impact.2017.09.002
Gil-Rubio, J., and Vicente, J. (2015). Gold trifluoromethyl complexes. Dalton Trans. 44, 19432–19442. doi:10.1039/c5dt02023a
Glessi, C., Mahgoub, A., Hagen, C. W., and Tilset, M. (2021). Gold(I) N-heterocyclic carbene precursors for focused electron beam-induced deposition. Beilstein J. Nanotechnol. 12, 257–269. doi:10.3762/bjnano.12.21
González-Rubio, S., Salgado, C., Manzaneda-González, V., Muñoz-Úbeda, M., Ahijado-Guzmán, R., Natale, P., et al. (2022). Tunable gold nanorod/NAO conjugates for selective drug delivery in mitochondria-targeted cancer therapy. Nanoscale 14, 8028–8040. doi:10.1039/d2nr02353a
Gope, K., Prabhudesai, V. S., Mason, N. J., and Krishnakumar, E. (2016). Probing the resonant states of Cl2 using velocity slice imaging. J. Phys. B At. Mol. Opt. Phys. 49, 015201. doi:10.1088/0953-4075/49/1/015201
Hagen, C. W., van Dorp, W. F., Crozier, P. A., and Kruit, P. (2008). Electronic pathways in nanostructure fabrication. Surf. Sci. 602, 3212–3219. doi:10.1016/j.susc.2007.11.034
Hasan, M., Kozhevnikov, I. V., Siddiqui, M. R. H., Steiner, A., and Winterton, N. (1999). Gold compounds as ionic liquids. Synthesis, structures, and thermal properties of N,N‘-Dialkylimidazolium tetrachloroaurate salts. Inorg. Chem. 38 (25), 5637–5641. doi:10.1021/ic990657p
Holder, C. F., and Schaak, R. E. (2019). Tutorial on powder X-ray diffraction for characterizing nanoscale materials. ACS Nano 13 (7), 7359–7365. doi:10.1021/acsnano.9b05157
Hopkinson, M. N., Richter, C., Schedler, M., and Glorius, F. (2014). Glorius, an overview of N-heterocyclic carbenes, F. Nature 510, 485–496. doi:10.1038/nature13384
Hosseini, M., and Mohammadi, A. H. (2020). A Gibbs free energy minimization based model for liquid–liquid equilibrium calculation of a system containing oil, brine, and surfactant. . IFP Energies Nouv. 75, 17. doi:10.2516/ogst/2020012
Jäger, A., and Span, R. (2012). Equation of state for solid carbon dioxide based on the Gibbs free energy. J. Chem. Eng. Data 57, 590–597. doi:10.1021/je2011677
Jiao, J., Xiao, D., Zhao, X., and Deng, Y. (2016). Analysis of the molecules structure and vertical electron affinity of organic gas impact on electric strength. Plasma Sci. Technol. 18, 554–559. doi:10.1088/1009-0630/18/5/19
Johnson, A. (2016). An efficient and sustainable synthesis of NHC gold complexes. Chem. Commun. 52, 9664–9667. doi:10.1039/c6cc05190a
Kaminskya, J., Mataby, R. A., Wernerb, H.-J., and Jensen, F. (2008). The accuracy of local MP2 methods for conformational energies. Mol. Phys. 106 (No. 15), 1899–1906. doi:10.1080/00268970802360355
Khan, H., Yerramilli, A. S., D'Oliveira, A., Alford, T. L., Boffitov, D. C., and Patience, G. S. (2020). Experimental methods in chemical engineering: X-Ray diffraction spectroscopy—XRD. Can. J. Chem. Eng. 98, 1255–1266. doi:10.1002/cjce.23747
Kuhness, D., Gruber, A., Winkler, R., Sattelkow, J., Fitzek, H., Letofsky-Papst, I., et al. (2021). High-fidelity 3D nanoprinting of plasmonic gold nanoantennas. ACS Appl. Mat. Interfaces 13, 1178–1191. doi:10.1021/acsami.0c17030
Lee, J.-C., Chai, J.-D., and Lin, S.-T. (2015). Assessment of density functional methods for exciton binding energies and related optoelectronic properties. RSC Adv. 5, 101370–101376. doi:10.1039/c5ra20085g
Levchenko, V., Glessi, C., Øien-Ø⁰aard, S., and Tilset, C. (2020). Organometallic chemistry in aqua regia: Metal and ligand based oxidations of (NHC)AuCl complexes. M. Dalton Trans. 49, 3473–3479. doi:10.1039/c9dt04472h
Li, X., Cai, Z., and Sevilla, M. D. (2002). DFT calculations of the electron affinities of nucleic acid bases: Dealing with negative electron affinities. J. Phys. Chem. A 106 (8), 1596–1603. doi:10.1021/jp013337b
Liu, H. -T., Xiong, X. -G., Dau, P. D., Wang, Y. -L., Huang, D. -L., Li, J., et al. (2013). Probing the nature of gold–carbon bonding in gold–alkynyl complexes. Nat. Commun. 4, 2223. doi:10.1038/ncomms3223
Lomzov, A. A., Vorobjev, Y. N., and Pyshnyi, D. V. (2015). Evaluation of the Gibbs free energy changes and melting temperatures of DNA/DNA duplexes using hybridization enthalpy calculated by molecular dynamics simulation. J. Phys. Chem. B 119 (49), 15221–15234. doi:10.1021/acs.jpcb.5b09645
Longevial, J.-F., Langlois, A., Buisson, A., Devillers, C. H., Clément, S., van der Lee, A., et al. (2016). Synthesis, characterization, and electronic properties of porphyrins conjugated with N-heterocyclic carbene (NHC)–Gold(I) complexes. Organometallics 35, 663–672. doi:10.1021/acs.organomet.5b00966
López-Vidaña, E. C., Castillo Téllez, M., Pilatowsky Figueroa, I., Santis Espinosa, L. F., and Castillo-Téllez, B. (2021). Moisture sorption isotherms, isosteric heat, and Gibbs free energy of stevia leaves. J. Food Process. Preserv 45, 15016. doi:10.1111/jfpp.15016
Luo, Y.-R. (2007). Comprehensive handbook of chemical bond energies. Boca Raton, FL: CRC Press.Bond dissociation energies
Lüttge, A. (2006). Crystal dissolution kinetics and Gibbs free energy. J. Electron Spectrosc. Relat. Phenom. 150 (Issues 2–3), 248–259. doi:10.1016/j.elspec.2005.06.007
Magén, C., Pablo-Navarro, J., and María De Teresa, J. (2021). Focused-electron-beam engineering of 3D magnetic nanowires. Nanomaterials 11, 402.
Malet-Martino, M., and Martino, R. (2002). Clinical studies of three oral prodrugs of 5-fluorouracil (capecitabine, UFT, S-1): A review. Oncologist 7 (4), 288–323. doi:10.1634/theoncologist.7-4-288
Manaa, M. R. (2017). Determination of adiabatic ionization potentials and electron affinities of energetic molecules with the Gaussian-4 method. Chem. Phys. Lett. 678, 102–106. doi:10.1016/j.cplett.2017.04.038
Marashdeh, A., Tiesma, T., van Velzen, N. J. C., Harder, S., Havenith, R. W. A., De Hosson, J. T. M., et al. (2017). The rational design of a Au(I) precursor for focused electron beam induced deposition. Beilstein J. Nanotechnol. 8, 2753–2765. doi:10.3762/bjnano.8.274
Marell, D. J., Emond, S. J., Kulshrestha, A., and Hoye, T. R. (2014). Analysis of seven-membered lactones by computational NMR methods: Proton NMR chemical shift data are more discriminating than carbon. J. Org. Chem. 79, 752–758. doi:10.1021/jo402627s
Marion, N., and Nolan, S. P. (2008). N-Heterocyclic carbenes in gold catalysis. Chem. Soc. Rev. 37, 1776–1782. doi:10.1039/b711132k
Mármol, I., Quero, J., Rodríguez-Yoldi, M. J., and Cerrada, E. (2019). Gold as a possible alternative to platinum-based chemotherapy for colon cancer treatment. Cancers 11 (6), 780. doi:10.3390/cancers11060780
Martínez-Salvador, S., Forniés, J., Menjón, Martín A., and Menjón, B. (2011). [Au(CF3)(CO)]: A gold carbonyl compound stabilized by a trifluoromethyl group. B. Angew. Chem. Int. Ed. 50, 6571–6574. doi:10.1002/anie.201101231
Martins, G. F., and Cabral, B. J. C. (2019). Electron propagator theory approach to the electron binding energies of a prototypical photo-switch molecular system: Azobenzene. J. Phys. Chem. A 123, 2091–2099. doi:10.1021/acs.jpca.9b00532
Mirzadeh, N., Srinivasa Reddy, T., and Bhargava, S. K. (2019). Advances in diphosphine ligand-containing gold complexes as anticancer agents. Coord. Chem. Rev. 388, 343–359. doi:10.1016/j.ccr.2019.02.027
Moroz, E. M. (2011). X-Ray diffraction structure diagnostics of nanomaterials. Russ. Chem. Rev. 80, 293–312. doi:10.1070/rc2011v080n04abeh004163
Nag, P., Polášek, M., and Fedor, J. (2019). Dissociative electron attachment in NCCN: Absolute cross sections and velocity-map imaging. Phys. Rev. A 99, 052705. doi:10.1103/physreva.99.052705
Nobili, S., Mini, E., Landini, I., Gabbiani, C., Casini, A., and Messori, L. (2010). Gold compounds as anticancer agents: Chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 30 (3), 550–580. doi:10.1002/med.20168
Ochterski, J. W. (2000). Thermochemistry in Gaussian. Available at https://gaussian.com/thermo/.
Olivotos, S., and Economou-Eliopoulos, M. (2016). Gibbs free energy of formation for selected platinum group minerals (PGM). Geosciences 6, 2. doi:10.3390/geosciences6010002
Parr, R. G., and Pearson, R. G. (1983). Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516. doi:10.1021/ja00364a005
Péres-Britrián, A., Baya, M., Casas, J. M., Falvello, L. R., Martín, A., and Menjón, B. (2017). (CF3)3Au as a highly acidic organogold(iii) fragment. Chem. Eur. J. 23, 14918–14930. doi:10.1002/chem.201703352
Porchia, M., Pellei, M., Marinelli, M., Tisato, F., Del Bello, F., and Santini, C. (2018). New insights in Au-NHCs complexes as anticancer agents. Eur. J. Med. Chem. 146, 709–746. doi:10.1016/j.ejmech.2018.01.065
Prabhudesai, V. S., Tadsare, V., Ghosh, S., Gope, K., Davis, D., and Krishnakumar, E. (2014). Dissociative electron attachment studies on acetone. J. Chem. Phys. 141, 164320. doi:10.1063/1.4898144
Puydinger dos Santos, M. V., Szkudlarek, A., Rydosz, A., Guerra-Nuñez, C., Béron, F., Pirota, K. R., et al. (2018). Comparative study of post-growth annealing of Cu(hfac)2, Co2(CO)8 and Me2Au(acac) metal precursors deposited by FEBID. Beilstein J. Nanotechnol. 9, 91–101. doi:10.3762/bjnano.9.11
Richardson, J. H., Stephenson, L. M., and Brauman, J. I. (1975). Photodetachment of electrons from trifluoromethyl and trifluorosilyl ions; the electron affinities of CF3− and SiF3−. Phys. Lett. 30, 17–20. doi:10.1016/0009-2614(75)85487-x
Salike, S., and Bhatt, N. (2020). Thermodynamically consistent estimation of Gibbs free energy from data: Data reconciliation approach. Bioinformatics 36 (Issue 4), 1219–1225. doi:10.1093/bioinformatics/btz741
Sauer, W., Drexel, H., Grill, V., Pelc, A., Gstir, B., Hanl, G., et al. (2002). Electron impact ionization studies for SF5CF3. J. Phys. B Atomic Mol. Opt. Phys. 35 (11), 2567–2574. doi:10.1088/0953-4075/35/11/314
Scheunemann, H. U., Illenberger, E., and Baumgärtel, H. (1980). Dissociative electron attachment to CCl4, CHCl3, CH2Cl2 and CH3Cl. Ber. Bunsenges. Phys. Chem. 14, 580–585. doi:10.1002/bbpc.19800840612
Schuh, E., Pflüger, C., Citta, A., Folda, A., Rigobello, M. P., Bindoli, A., et al. (2012). Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 55 (11), 5518–5528. doi:10.1021/jm300428v
Shawrav, M. M., Taus, P., Wanzenboeck, H. D., SchinnerlStöger-Pollach, M. M., Schwarz, S., Steiger-Thirsfeld, A., et al. (2016). Highly conductive and pure gold nanostructures grown by electron beam induced deposition. Sci. Rep. 6, 34003. doi:10.1038/srep34003
Shuman, N. S., Miller, T. M., Friedman, J. F., Viggiano, A. A., Maergoiz, A. I., and Troe, J. (2011). Pressure and temperature dependence of dissociative and non-dissociative electron attachment to CF3: Experiments and kinetic modelling. J. Chem. Phys. 135, 054306. doi:10.1063/1.3614471
Solovyev, A., Ueng, S.-H., Monot, J., Fensterbank, L., Malacria, M., Lacôte, E., et al. (2010). Estimated rate constants for hydrogen abstraction from N-heterocyclic Carbene−Borane complexes by an alkyl radical. D. P. Org. Lett. 12, 2998–3001. doi:10.1021/ol101014q
Tahir, K. M., Sajid, A., Tariq, Z. M., Chandra, K. A., Iqbal, M. S., and Dong-Qing, W. (2020). Gibbs free energy calculation of mutation in PncA and RpsA associated with pyrazinamide resistance. Front. Mol. Biosci. 7, 52. doi:10.3389/fmolb.2020.00052
Tan, S. J., Yan, Y. K., Lee, P. P. F., and Lim, K. H. (2010). Copper, gold and silver compounds as potential new anti-tumor metallodrugs. Future Med. Chem. 2, 1591–1608. doi:10.4155/fmc.10.234, No
Taraban, M. B., DePaz, R. A., Lobo, B., and Yu, Y. B. (2017). Water proton NMR: A tool for protein aggregation characterization. Anal. Chem. 89, 5494–5502. doi:10.1021/acs.analchem.7b00464
Thorman, R. M., Ragesh Kumar, T. P., Howard Fairbrother, D., and Ingólfsson, O. (2015). The role of low-energy electrons in focused electron beam induced deposition: Four case studies of representative precursors. Beilstein J. Nanotechnol. 6, 1904–1926. doi:10.3762/bjnano.6.194
Utke, I., Hoffmann, P., and Melngailis, J. (2008). Gas-assisted focused electron beam and ion beam processing and fabrication. J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 26, 1197. doi:10.1116/1.2955728
Wang, H. M. J., and Lin, I. J. B. (1998). Facile synthesis of silver(I)−Carbene complexes. Useful carbene transfer agents. Organometallics 17, 972–975. doi:10.1021/om9709704
Winkler, R., Schmidt, F.-P., Haselmann, U., Fowlkes, J. D., Lewis, B. B., Kothleitner, G., et al. (2017). Direct-Write 3D nanoprinting of plasmonic structures. ACS Appl. Mat. Interfaces 9, 8233–8240. doi:10.1021/acsami.6b13062
Woldu, A. S., and Mai, J. (2012). Computation of the bond dissociation enthalpies and free energies of hydroxylic antioxidants using the ab initio Hartree–Fock method. Redox Rep. 17 (6), 252–274. doi:10.1179/1351000212y.0000000030
Wozniak, D., Hicks, A., Sabbers, W. A., and Dobereiner, G. (2019). Imidazolyl-phenyl (imp) anions: A modular structure for tuning solubility and coordinating ability. Dalton Trans. 48, 14138–14155. doi:10.1039/c9dt03511g
Xu, X., and Truhlar, D. G. (2011). Accuracy of effective core potentials and basis sets for density functional calculations, including relativistic effects, as illustrated by calculations on arsenic compounds. J. Chem. Theory Comput. 7, 2766–2779. doi:10.1021/ct200234r
Xuan, C.-j., Wang, X.-d., Xia, L., Wu, B., Li, H., and Tian, S.-x. (2014). Dissociative electron attachment to 1,2-dichlorobenzene using mass spectrometry with phosphor screen. Chin. J. Chem. Phys. 27, 628–630. No. 6. doi:10.1063/1674-0068/27/06/628-630
Ye, H., Trippel, S., Di Fraia, M., Fallahi, A., Mücke, O. D., Kärtner, F. X., et al. (2018). Velocity-map imaging for emittance characterization of multiphoton electron emission from a gold surface. Phys. Rev. Appl. 9, 044018. doi:10.1103/physrevapplied.9.044018
Zhang, D., Luo, R., and Zeng, Z. (2019). Characterization of surface free energy of mineral filler by spreading pressure approach. Constr. Build. Mater. 218, 126–134. doi:10.1016/j.conbuildmat.2019.05.128
Zhao, D., Han, A., and Qiu, M. (2019). Ice lithography for 3D nanofabrication. Sci. Bull. 64, 865–871. doi:10.1016/j.scib.2019.06.001
Zhong, Y., Ping, D., Song, X., and Yin, F. (2009). Determination of grain size by XRD profile analysis and TEM counting in nano-structured Cu. J. Alloys Compd. 476 (Issues 1–2), 113–117. doi:10.1016/j.jallcom.2008.08.075
Zhou, X., Liu, D., Bu, H., Deng, L., Liu, H., Yuan, P., et al. (2018). XRD-based quantitative analysis of clay minerals using reference intensity ratios, mineral intensity factors, rietveld, and full pattern summation methods: A critical review. Solid Earth Sci. 3 (Issue 1), 16–29. doi:10.1016/j.sesci.2017.12.002
Keywords: dissociative electron attachment, gold imidazolyl compounds, focused electron beam deposition, XRD, gold precursors
Citation: Pintea M, Mason N, Peiró-Franch A, Clark E, Samanta K, Glessi C, Schmidtke IL and Luxford T (2023) Dissociative electron attachment to gold(I)-based compounds: 4,5-dichloro-1,3-diethyl-imidazolylidene trifluoromethyl gold(I). Front. Chem. 11:1028008. doi: 10.3389/fchem.2023.1028008
Received: 26 August 2022; Accepted: 01 June 2023;
Published: 19 June 2023.
Edited by:
Malgorzata Biczysko, Shanghai University, ChinaReviewed by:
Vincenzo Laporta, National Research Council (CNR), ItalyCopyright © 2023 Pintea, Mason, Peiró-Franch, Clark, Samanta, Glessi, Schmidtke and Luxford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Pintea, bWFyeWlhcGludGVhQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.