 Wanyue Peng
Wanyue Peng Sviatoslav Baranets
Sviatoslav Baranets Svilen Bobev
Svilen Bobev- Department of Chemistry and Biochemistry, University of Delaware, Newark, DE, United States
A new ternary phase has been synthesized and structurally characterized. BaLixCd13–x (x ≈ 2) adopts the cubic NaZn13 structure type (space group Fm
1 Introduction
In recent papers, we described new results from our exploratory work in the Ba–Li–In–Ge (Ovchinnikov and Bobev, 2019) and Ba–Li–Cd–Ge systems (Baranets et al., 2021b). As noted therein, our synthetic approach employed molten In and Cd as metal fluxes, allowing us to grow crystals of the quaternary phases, BaLi2+xIn2–xGe2 (0 ≤ x ≤ 0.66) and BaLi2Cd2Ge2 (CaCu4P2 type, space group R
Cadmium metal is known to have considerable toxicity with a destructive impact on most living systems. Due to its low boiling point, Cd is also far from an ideal solvent for high-temperature reactions. However, the crucial role of Cd in the flux growth of new intermetallic compounds makes the current study fit into this special issue on Crystal Growth Under Extreme Conditions. BaLi2Cd2Ge2 (Baranets et al., 2021) together with Yb2CdSb2 (Xia and Bobev, 2007a), Sr2CdAs2 (Wang et al., 2011), Ba2CdAs2 (Wang et al., 2011), Eu2CdAs2 (Wang et al., 2011), Ba11Cd8Bi14 (Xia and Bobev, 2006a), RE2CdGe2 (RE = Pr, Nd, Sm, Gd–Yb; Y) (Guo et al., 2012), Ba2Cd3Bi4 (Xia and Bobev, 2006b), A14Cd1+xPn11 (0 ≤ x ≤ 0.3; A = Sr, Eu; Pn = As, Sb) (Makongo et al., 2015), Sr3Cd8Ge4 (Suen et al., 2018), Eu3Cd8Ge4 (Suen et al., 2018), and Eu10Cd6Bi12 (Xia and Bobev, 2007b) from our laboratory have come into being by employing molten Cd as a flux for the synthesis. This is also the case with the title compound, BaLixCd13−x (x ≈ 2). As mentioned above, BaLixCd13−x (x ≈ 2) was serendipitously obtained during the exploratory work in the AE–Li–Cd–Si and AE–Li–Cd–Ge systems (AE = alkaline earth metals Ca, Sr, Ba, and the nominally divalent Eu, Yb). As a result of such work, the new cubic BaLixCd13–x (x ≈ 2) phase was discovered. It is the first structurally characterized compound between the respective elements and crystallizes with the common NaZn13 structure type (space group Fm
The crystal growth from Cd flux, the structural characterization, as well as a brief analysis of the chemical bonding of BaLixCd13–x (x ≈ 2) and BaCd11 are the main subject of discussion in this paper.
2 Materials and methods
2.1 Synthesis
The starting materials were purchased from Alfa Aesar, all with stated purity of 99.9 wt% or better. The metals were stored in an argon-filled glovebox and used as received. The surface of the Li rod was cleaned with a scalpel blade prior to cutting and using it.
Single crystals were synthesized via the flux growth method. As stated already, reactions aimed at AELi2Cd2Ge2 (AE = alkaline-earth metal) were the starting point for this research, (Baranets et al., 2021b), and the title compound BaLixCd13–x (x ≈ 2) was identified as a minor side product of such experiment. Subsequently, the samples were prepared without Ge in the elemental mixtures, where excessive stoichiometric amounts of Cd served as the flux for all compositions. A starting composition of Ba:Li:Cd: of 1:12:20 was used for BaLixCd13−x (x ≈ 2). The 6-fold excess Li was chosen due to its high vapor pressure and losses at the reaction temperature. The corresponding amounts of elements (Ba rod, Cd shot, Zn shot, Ca shot, Yb shot, Li rod) were loaded into alumina crucibles. A piece of quartz wool was placed at the bottom of a fused silica ampoule. The alumina crucible was then loaded to the bottom of an ampoule. Another piece of quartz wool was placed on top of the crucible without touching the elements inside. The ampoules were sealed under a vacuum level of ca. 30 millitorr.
All the samples in this study were heated in muffle furnaces with a temperature profile as the following: 100°C → 20°C/h to 200°C → 50°C/h to 700°C → held at 700°C for 12.5 h → 4°C/h to 550°C. At this point of the crystal growth, the sealed tube was taken out from the furnace, flipped, and the molten metallic flux was separated from the grown crystals by centrifugation. After that, the sealed ampoule was brought in the glovebox and break-opened. Inspection of the specimen under an optical microscope revealed the presence of many small, grey crystals with a metallic luster, usually clustered together. Single-crystal X-ray diffraction confirmed them to be the new cubic BaLixCd13−x (x ≈ 2) phase. A portion of the crystallites was then ground into fine powders with a mortar and pestle for powder X-ray diffraction, attesting the presence of the cubic phase in the bulk. The experiment was repeated in a Nb-tube to confirm that the results are repeatable in both alumina and Nb containers. Such reactions also verified that no inadvertent reduction of the Al2O3 can become a source of Al metal, as experienced recently by us in another materials system (Baranets and Bobev, 2021a).
Following the structure elucidation of BaLixCd13–x (x ≈ 2), reactions aimed at AELixCd13–x (x ≈ 2) (AE = Ca, Sr, Eu, Yb) were set up with the same nominal compositions as the ones described in the preceding paragraph, but they were unsuccessful. An attempt to grow crystals from Cd-flux reaction but without Li in the nominal mixture resulted in the growth of the known binary phase BaCd11 (Sanderson and Baenziger, 1953). Since its structure has not been refined from single-crystal X-ray diffraction data and since the quality of the obtained crystals was excellent, herein we supply this information as well.
Crystals of BaLixCd13−x (x ≈ 2) do not appear to degrade in air over a period of 1 week. Powder X-ray diffraction patterns also show the polycrystalline material to be air- and moisture-stable for the same amount of time and possibly even longer.
2.2 Powder X-ray diffraction and single-crystal X-ray diffraction
The X-ray powder diffraction patterns were collected using a Rigaku Miniflex powder diffractometer utilizing Ni-filtered Cu Kα radiation (λ = 1.5418 Å) and were used for phase identification only. All additional structural work was done using single-crystal X-ray diffraction methods.
Single-crystal X-ray diffraction measurements (SC-XRD) were performed using a Bruker APEX II diffractometer with monochromated Mo Kα radiation. Single crystal geometries are typically in block shape, with each side smaller than 60 µm in length. Crystals were selected under a microscope in dry Paratone-N oil. The measurements were conducted at a temperature of 200 K. Data integration and semiempirical absorption correction were performed with the Bruker-supplied software (Bruker AXS Inc, 2014). The crystal structure was solved with the intrinsic phasing method and was refined with full-matrix least-squares minimization on F2 using ShelXL (Sheldrick, 2015). Olex2 software was used as a graphical interface (Dolomanov et al., 2009). Atomic coordinates of all compounds reported in this paper were standardized with the Structure Tidy program (Gelato and Parthé, 1987). All sites were refined with anisotropic displacement parameters. Final difference Fourier map was flat and featureless. Selected crystallographic data are summarized in Table 1.

TABLE 1. Selected crystallographic data and structure refinement parameters for BaLixCd13–x (x ≈ 2).
One specific aspect of the refinements concerning the site occupation factors (SOF) requires a special mention. All SOFs were checked by freeing an individual SOF, while other variables were kept fixed. No statistically significant deviations were observed for the SOF of the Ba site (8a); the Li position (8b) indicated a freely-refined SOF of ca. 105%. The detected “over-occupation” of the Li atom barely had statistical significance (deviations were within ca. 3-4σ). The Cd site (96i) showed approximately 94–95% occupancy (within ca. 8-9σ), which is indicative of the existence of either 1) vacancies, or 2) potential disordering with a lighter element, such as Li in this case. The model with Li/Cd co-occupation was chosen on the basis of expected greater electronic structure stability of BaLixCd13–x (x ≈ 2) vs. BaLiCd12–x (x ≈ 0.6–0.7). We must also note that ignoring the above-mentioned signs for a small disorder on the Cd site (96i) and refining an ordered BaLiCd12 model leads to a converging least-squares minimization, however, with increased R-values (R1 = 0.0182; wR2 = 0.0443) compared to those in presented Table 1 for the BaLixCd13–x (x ≈ 2) structure. Residual difference peak and hole in the Fourier synthesis map were also much higher, therefore, this possibility was excluded from further consideration. Further details of the structural work are discussed later on. The corresponding crystallographic information files (CIF) for BaLixCd13–x (x ≈ 2) and BaCd11 have been deposited with CSD, and the data for this paper can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, United Kingdom; Fax: +44-1223-336033; E-mail: ZGVwb3NpdEBjY2RjLmNhbS5hYy51aw==). Depository numbers are 2184513 (BaLixCd13–x) and 2184514 (BaCd11).
2.3 Electronic structure calculations
To investigate the chemical bonding of all compositions, electronic structure calculations were performed within the local density approximation (density functional theory) using the TB-LMTO-ASA program (Tank et al., 1994). Experimental unit cell parameters and atomic coordinates obtained in this study were used as the input parameters in our calculation. In order to satisfy the atomic sphere approximation (ASA), we employed von-Barth-Hedin functional (Von Barth and Hedin, 1972) and introduced empty spheres during the calculation. The Brillouin zone was sampled by 1,000 k-point grid. Electronic density of states (DOS), atom-projected electronic density of states (PDOS), and crystal orbital Hamilton population (COHP) were calculated with modules in the LMTO program (Steinberg and Dronskowski, 2018).
3 Results and discussion
3.1 Crystal structure
The structures of some AM13 compounds (space group Fm

TABLE 2. Atomic coordinates and equivalent displacement parameters (Å2) for BaLixCd13–x (x ≈ 2).
There are over a hundred compounds reported to crystallize in the NaZn13 structure type (Villars and Calvert, 1991). In the past 2 decades, the NaZn13 family has been expanded to include many ternary counterparts, some of which are cubic alloys, such as EuCuxAl13−x (Phelan et al., 2012) and RECo13−xGax (RE = La, Ce, Pr, Nd, and mischmetal) (Weitzer et al., 1990), while some other show crystallographic ordering. Examples of the latter group are BaLi7Al6 (Häussermann et al., 1998), AMxT13−x (A = Ba, Sr, La, Eu, M = Cu and Ag; T = Al, Ga and In; x = 5–6.5.) (Nordell and Miller, 1999), ACu9Tt4 (A = Ca, Sr, Ba, Eu; Tt = Si, Ge, Sn) (Schäfer et al., 2012), and BaAuxZn13−x (Gupta and Corbett, 2012). One should notice that most structurally characterized phases are known to follow the “1-13” stoichiometry, while other members are shown to be slightly off-stoichiometric, such as SrZn13–x (Wendorff and Röhr, 2006), and EuZn13–x (Saparov and Bobev, 2008), where the Zn-deficiency is necessitated for the sake of attaining more favorable valence electron count. More discussion on that topic will follow later. In some cases, particularly among the ternaries, ordering of the elements leads to cubic to tetragonal distortion (Fm
We also need to draw attention to the fact that while BaZn13 (rather BaZn∼12.8) is known to crystallize with the cubic NaZn13 structure type (Wendorff and Röhr, 2006), a binary compound BaCd13 with the said structure does not exist. The most Cd-rich phase is BaCd11 and it crystallizes in its own structure type (tetragonal space group I41/amd (no. 141); Pearson symbol tI48) (Sanderson and Baenziger, 1953). Despite the compositional difference and despite crystallizing in different space groups, the NaZn13 and BaCd11 structure types share a lot in common in terms of the large polyhedra surrounding the Na and Ba atoms, respectively (Figure 1). In the tetragonal structure, each Ba atom is surrounded by 22 Cd atoms (Cd1), while the Cd2 atoms have 12 nearest neighboring Cd ones. A more comprehensive structural description of the BaCd11 structure type can be found in Ref. (Sanderson and Baenziger, 1953). Besides BaCd11, YbZn11 (Kuzma et al., 1965), SrCd11 (Köster and Meixner, 1965), CeZn11 (Zelinska et al., 2004), and PrZn11 (Iandelli and Palenzona, 1967) are also reported to form with the same structure type.
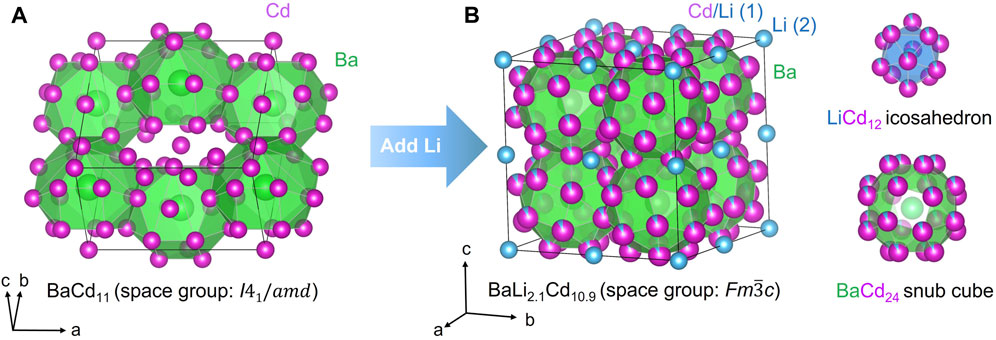
FIGURE 1. (A) BaCd11 crystallizes in a tetragonal structure type. It cannot transition to the cubic NaZn13 structure even with the presence of excessive Cd. (B) BaLi2.1Cd10.9 crystallizes in the cubic NaZn13 structure type).
Apparently, the NaZn13 structure can only be stabilized with Ba and Cd only when a small amount of lithium is present, with BaLi2.1Cd10.9 being the final refined composition (Table 1). One lithium atom takes over the Zn2 site in the NaZn13 prototype structure, while the balance of lithium atoms per formula unit is admixed with Cd at the Zn2 site in a ratio of ca. 1:10 (Table 2). The consequence of the Li atoms being in such positions is that, instead of having the same kind of atoms on both the vertices and the centers of the icosahedra, the center of the 12-membered polyhedron is Li, while the vertices are mostly Cd atoms. We confirmed that the hypothetic cubic BaCd13 phase was inaccessible without Li (vide supra). In such a Cd-rich environment, the crystals that grow are the tetragonal BaCd11. The exact opposite scenario is observed when one moves up in group 12 to Zn—the most Zn-rich phase in the Ba–Zn binary system is the cubic BaZn13 (Wendorff and Röhr, 2006)) while tetragonal BaZn11 does not exist.
Based on the above and given the similarities between the two structure types, one may ask the question as to what is the role Li atoms play to stabilize the cubic BaLixCd13–x (x ≈ 2) phase, and whether or not the homogeneity range can be expanded. Our experimental findings are limited to compositions close to the refined, and an unambiguous answer to the two questions is not possible at present. However, we can speculate that since most known compounds within the NaZn13 structural family show valence electron counts close to 27e− per formula unit (Häussermann et al., 1998; Nordell and Miller, 1999), one should expect, based on electronic arguments alone, that the composition BaLixCd13–x (x ≈ 1) would be preferred. Recognizing that there are geometric factors at play, too, and that they might be in competition with the electronic factors, could lead to different conclusions. In fact, there has been much debate about the favorable conditions of which structure type (NaZn13 or BaCd11) is optimal, yet, no universal conclusion has been reached. The hard-sphere colloidal model shows that the ideal size ratio between A and M is between 0.49 and 0.63 (Hachisu and Yoshimura, 1980; Yoshimura and Hachisu, 1983; Bartlett et al., 1992; Shevchenko et al., 2005). Later, Hudson (Hudson, 2010) approached this problem with the optimal packing fraction analysis. It is shown that the majority of the AM13 compounds yield a close packing fraction between 0.69 and 0.77. However, multiple known compounds break this rule. A typical example is SrBe13 (Matyushenko et al., 1964) yielding a size ratio of 2, which is far from the ideal range mentioned previously. It can be also suggested, again from a purely geometrical perspective, that the size of the atom in the icosahedral environment (recall that the covalent radii of Li and Zn are nearly identical while Li and Cd differ (Pauling, 1960)) is also a contributing factor, and could tip the scales when it comes to the phase-preference between cubic NaZn13 vs. tetragonal BaCd11.
Therefore, we can argue that the composition BaLixCd13–x (x ≈ 2) may be the limiting/preferred one for the stabilization of the NaZn13-type structure for both electronic and geometric reasons.
3.2 Electronic structure
The bonding characteristics of BaLi2.1Cd10.9 and BaCd11 are investigated via electronic structure calculations using the LMTO program (Tank et al., 1994). Accurate modeling of the electronic properties of BaLi2.1Cd10.9 with the LMTO program is possible, yet challenging, due to the mixed occupancy of Li and Cd on the 96i site. The input of the mixed occupancy to the model would only be possible by reducing the cubic symmetry to triclinic P1, while assigning corresponding positions to Li and Cd atoms according to the refined occupancy. To simplify the calculation and to avoid symmetry reduction, we evaluated the electronic properties of BaLi2.1Cd10.9 in its original symmetry without considering the existence of Li on the Cd site. The model employed was the idealized BaLiCd12 compound with an ordered cubic structure, where each Ba, Cd, and Li atom resides on an independent site. Atomic coordinates were taken from Table 2.
The atom-projected electronic density of states (DOS) for BaLiCd12 and BaCd11 are shown in Figures 2A,C, respectively. For BaLiCd12, a dip of the total DOS to ca. Three states eV−1 unit cell−1 can be observed at around E−EF = 0.19 eV. In the rigid-band approximation, shifting the Fermi level ca. 0.2 eV would correspond to seven additional electrons per unit cell, or 0.88e− per formula unit. For BaCd11, a dip to ∼5 eV−1 unit cell−1 can be observed right near the Fermi level. However, neither are deep enough to be identified as pseudogaps.
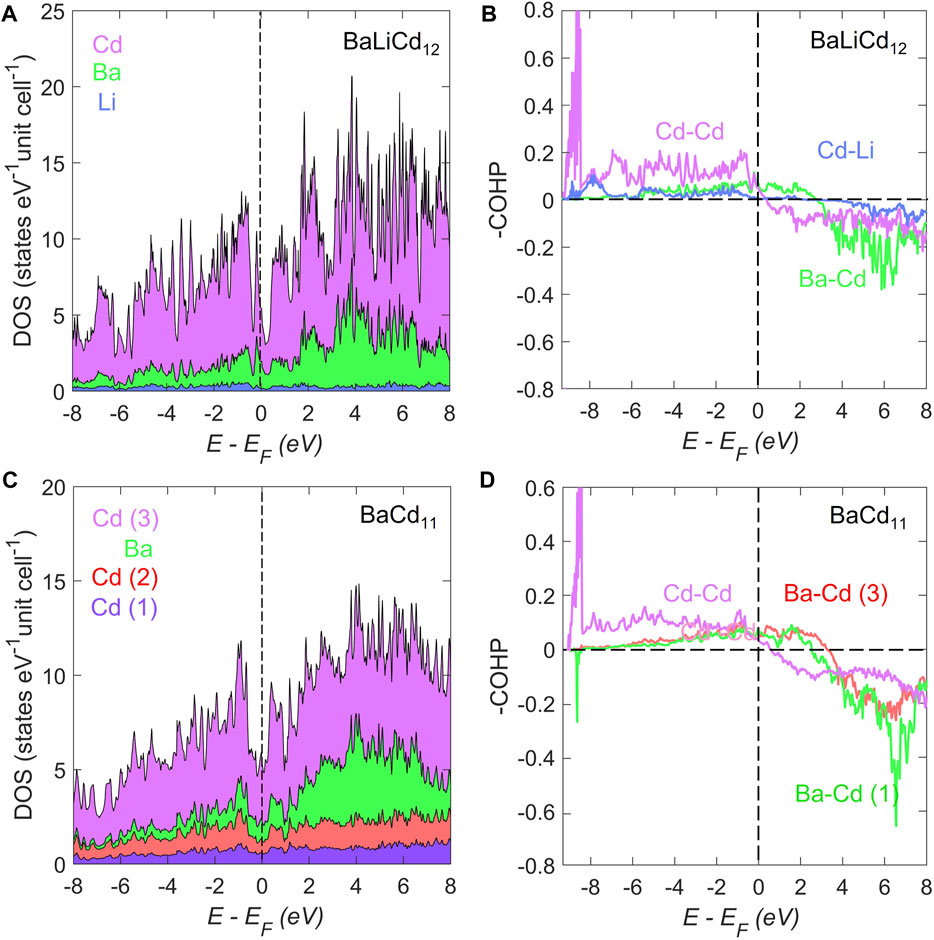
FIGURE 2. The atom-projected electronic density of states (DOS) for (A) BaLiCd12 and (C) BaCd11. The DOS of both compounds exhibit characteristics of metallic bonding. A relatively larger dip can be observed in the BaLiCd12 compound compared to that of BaCd11. The crystal orbital Hamilton population curves (COHP) for the averaged selected interactions of (B) BaLiCd12 and (D) BaCd11. For BaLiCd12, the Ba–Cd interactions are underoptimized at the Fermi level, while the Cd–Li and Cd–Cd interactions are almost optimized at the Fermi level. In contrast, for BaCd11, all interactions are under-optimized at the Fermi level.
In both compounds, the states in the vicinity of the dips are primarily contributed by the s-orbitals of Cd and Ba. Actually, the density of states plots of both compounds exhibit little structuring, with no gaps in the range of −3 eV < E−EF < 9 eV. This is the characteristic of metallic bonding, indicating the absence of localized states or lone pairs. It appears that both systems are stabilized by the interaction between the delocalized electrons.
The crystal orbital Hamilton population curves (COHP) for selected averaged interactions in BaLiCd12 and BaCd11 are plotted in Figures 2B,D. For BaLi2Cd11, the Ba–Cd interactions are under-optimized at the Fermi level, while the Cd–Li and Cd–Cd interactions are almost optimized at the Fermi level. In contrast, for BaCd11, all interactions are under-optimized at the Fermi level. The optimized Cd–Li and Cd–Cd bondings in the cubic BaLiCd12 compared to the under-optimized bonding in the BaCd11 phase could be an indication of higher stability of the former. Given the similarity between the COHP curves of the Ba–Cd contacts in both compounds, the bond strength of Cd–Li and Cd–Cd in the icosahedron might be the driving force of the transition between these two phases. This inference is in line with the geometric explanation in the previous subsection.
4 Summary and outlook
The bonding characteristics of the Cd-rich phases, BaLi2.1Cd10.9 and BaCd11, both made from Cd flux, were studied both experimentally and computationally. In addition to providing insight into the relative stability of the tetragonal BaCd11 and cubic NaZn13 structures, this study reports the discovery of a new phase BaLi2.1Cd10.9, expanding the variety of compounds that crystallize in the NaZn13 structure type. Combined with the first-principles calculations of the electronic structure of both phases, we found that the bond strength of Cd–Li and Cd–Cd within the icosahedron might be one of the driving forces leading to the stabilization of the cubic NaZn13 type structure in this section of the Ba–Li–Cd phase diagram.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: The corresponding crystallographic information files (CIF) for BaLixCd13–x (x ≈ 2) and BaCd11 have been deposited with CSD, and the data for this paper can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, United Kingdom; Fax: +44-1223-336033; E-mail: ZGVwb3NpdEBjY2RjLmNhbS5hYy51aw==). Depository numbers are 2184513 (BaLixCd13–x) and 2184514 (BaCd11).
Author contributions
WP: Investigation, Methodology, Formal analysis, Visualization, Writing—original draft. SB: Conceptualization, Investigation, Formal analysis, Writing—review and editing. SB: Conceptualization, Supervision, Project administration, Writing—review and editing.
Funding
This work received financial support from the National Science Foundation (NSF) through an award number DMR-2004579.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Baranets, S., and Bobev, S. (2021a). Caught in action. The late rare earths thulium and lutetium substituting aluminum atoms in the structure of Ca14AlBi11. J. Am. Chem. Soc. 143, 65–68. doi:10.1021/jacs.0c11026
Baranets, S., Ovchinnikov, A., and Bobev, S. (2021b). Synthesis, crystal and electronic structure of BaLi2Cd2Ge2. Z. fur Naturforsch. B 76, 689–697. doi:10.1515/znb-2021-0114
Bartlett, P., Ottewill, R., and Pusey, P. (1992). Superlattice formation in binary mixtures of hard-sphere colloids. Phys. Rev. Lett. 68, 3801–3804. doi:10.1103/physrevlett.68.3801
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A., and Puschmann, H. (2009). OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 42, 339–341. doi:10.1107/s0021889808042726
Gelato, L., and Parthé, E. (1987). Structure tidy – A computer program to standardize crystal structure data. J. Appl. Crystallogr. 20, 139–143. doi:10.1107/s0021889887086965
Guo, S.-P., Meyers, J. J., Tobash, P. H., and Bobev, S. (2012). Eleven new compounds in the RE–Cd–Ge systems (RE = Pr, Nd, Sm, Gd–Yb; Y): Crystal chemistry of the RE2CdGe2 series. J. Solid State Chem. 192, 16–22. doi:10.1016/j.jssc.2012.03.045
Gupta, S., and Corbett, J. D. (2012). BaAuxZn13−x: Electron-poor cubic NaZn13-type intermetallic and its ordered tetragonal variant. Inorg. Chem. 51, 2247–2253. doi:10.1021/ic2022787
Hachisu, S., and Yoshimura, S. (1980). Optical demonstration of crystalline superstructures in binary mixtures of latex globules. Nature 283, 188–189. doi:10.1038/283188a0
Häussermann, U., Svensson, C., and Lidin, S. (1998). Tetrahedral stars as flexible basis clusters in sp-bonded intermetallic frameworks and the compound BaLi7Al6 with the NaZn13 structure. J. Am. Chem. Soc. 120, 3867–3880. doi:10.1021/ja973335y
Hudson, T. S. (2010). Dense sphere packing in the NaZn13 structure type. J. Phys. Chem. C 114, 14013–14017. doi:10.1021/jp1045639
Iandelli, A., and Palenzona, A. (1967). Zinc-rich phases of the rare Earth-zinc alloys. J. Less Common Metals 12, 333–343. doi:10.1016/0022-5088(67)90001-x
Ketelaar, J. (1937). The crystal structure of alloys of zinc with the alkali and alkaline Earth metals and of cadmium with potassium. J. Chem. Phys. 5, 668. doi:10.1063/1.1750098
Köster, W., and Meixner, J. (1965). Der Aufbau der Systeme des Europiums mit Silber, Kadmium und Indium sowie das System Kadmıum-Strontium. Int. J. Mater. Res. 56, 695–703. doi:10.1515/ijmr-1965-561009
Kuzma, Y. B., Kripyakevich, P., and Frankevich, D. (1965). Compounds of rare-Earth metals with zinc and their crystal structures. Izvestiia Akademii nauk SSSR Neorganicheskie Mater. 1, 1547.
Makongo, J. P., Darone, G. M., Xia, S.-Q., and Bobev, S. (2015). Non-stoichiometric compositions arising from synergistic electronic and size effects. Synthesis, crystal chemistry and electronic properties of A14Cd1+xPn11 compounds (0 ≤ x ≤ 0.3; A = Sr, Eu; Pn = As, Sb). J. Mat. Chem. C 3, 10388–10400. doi:10.1039/c5tc01605c
Matyushenko, N., Verkhorobin, L., and Karev, V. (1964). Strontium beryllide. Sov. Phys. Crystallogr. Engl. Transl. 9, 273.
Nordell, K. J., and Miller, G. J. (1999). Linking intermetallics and Zintl compounds: An investigation of ternary trielides (Al, Ga, In) forming the NaZn13 structure type. Inorg. Chem. 38, 579–590. doi:10.1021/ic980772k
Ovchinnikov, A., and Bobev, S. (2019). Layered quaternary germanides—Synthesis and crystal and electronic structures of AELi2In2Ge2 (AE = Sr, Ba, Eu). Inorg. Chem. 58, 7895–7904. doi:10.1021/acs.inorgchem.9b00588
Phelan, W. A., Kangas, M. J., McCandless, G. T., Drake, B. L., Haldolaarachchige, N., Zhao, L. L., et al. (2012). Synthesis, structure, and physical properties of Ln(Cu, Al, Ga)13–x (Ln = La–Pr, and Eu) and Eu(Cu, Al)13–x. Inorg. Chem. 51, 10193–10202. doi:10.1021/ic301024t
Sanderson, M., and Baenziger, N. (1953). The crystal structure of BaCd11. Acta Crystallogr. 6, 627–631. doi:10.1107/s0365110x53001745
Saparov, B., and Bobev, S. (2008). Zinc-deficiency in intermetallics with the NaZn13 type: Re-Determination of the crystal structure and physical properties of EuZn13−x (x = 0.25(1)). J. Alloys Compd. 461, 119–123. doi:10.1016/j.jallcom.2007.09.040
Schäfer, M. C., Yamasaki, Y., Fritsch, V., and Bobev, S. (2012). Synthesis and structural characterization of ACu9Tt4 (A = Ca, Sr, Ba, Eu; Tt = Si, Ge, Sn) – tetragonally distorted ternary variants of the cubic NaZn13 structure type. Improved structure refinement of SrCu2Ge2. Z. Anorg. Allg. Chem. 638, 1204–1211. doi:10.1002/zaac.201200062
Sheldrick, G. M. (2015). Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 71, 3–8. doi:10.1107/s2053229614024218
Shevchenko, E. V., Talapin, D. V., O’brien, S., and Murray, C. B. (2005). Polymorphism in AB13 nanoparticle superlattices: An example of semiconductor-metal metamaterials. J. Am. Chem. Soc. 127, 8741–8747. doi:10.1021/ja050510z
Steinberg, S., and Dronskowski, R. (2018). The crystal orbital Hamilton population (COHP) method as a tool to visualize and analyze chemical bonding in intermetallic compounds. Crystals 8, 225. doi:10.3390/cryst8050225
Suen, N.-T., Huang, L., Meyers, J. J., and Bobev, S. (2018). An unusual triple-decker variant of the tetragonal BaAl4 structure type: Synthesis, structural characterization, and chemical bonding of Sr3Cd8Ge4 and Eu3Cd8Ge4. Inorg. Chem. 57, 833–842. doi:10.1021/acs.inorgchem.7b02781
Tank, R., Jepsen, O., Burkhardt, A., and Andersen, O. (1994). TB-LMTO-ASA program. Stuttgart, Germany: Max-Planck-Institut für Festkörperforschung.
Villars, P., and Calvert, L. D. (1991). Pearson’s handbook of crystallographic data for intermetallic phases. Material Park, OH: ASM.
Von Barth, U., and Hedin, L. (1972). A local exchange-correlation potential for the spin polarized case. i. J. Phys. C. Solid State Phys. 5, 1629–1642. doi:10.1088/0022-3719/5/13/012
Wang, J., Yang, M., Pan, M.-Y., Xia, S.-Q., Tao, X.-T., He, H., et al. (2011). Synthesis, crystal and electronic structures, and properties of the new pnictide semiconductors A2CdPn2 (A = Ca, Sr, Ba, Eu; Pn = P, As). Inorg. Chem. 50, 8020–8027. doi:10.1021/ic200286t
Weitzer, F., Hiebl, K., Grin, Y. N., Rogl, P., and Nöel, H. (1990). Magnetism and structural chemistry of RECo13−xGax alloys (RE = La, Ce, Pr, Nd, and mischmetal MM). J. Appl. Phys. 68, 3504–3507. doi:10.1063/1.346362
Wendorff, M., and Röhr, C. (2006). Polar binary Zn/Cd-rich intermetallics: Synthesis, crystal and electronic structure of A(Zn/Cd)13 (A = alkali/alkaline Earth) and Cs1.34Zn16. J. Alloys Compd. 421, 24–34. doi:10.1016/j.jallcom.2005.11.016
Xia, S.-Q., and Bobev, S. (2006). Ba11Cd8Bi14: Bismuth zigzag chains in a ternary alkaline-Earth transition-metal Zintl phase. Inorg. Chem. 45, 7126–7132. doi:10.1021/ic060583z
Xia, S.-Q., and Bobev, S. (2007). Cation-anion interactions as structure directing factors: Structure and bonding of Ca2CdSb2 and Yb2CdSb2. J. Am. Chem. Soc. 129, 4049–4057. doi:10.1021/ja069261k
Xia, S.-Q., and Bobev, S. (2007). Synthesis, structural characterization, electronic structure, and magnetic properties of the Zintl phase Eu10Cd6Bi12. Chem. Asian J. 2, 619–624. doi:10.1002/asia.200700016
Xia, S.-Q., and Bobev, S. (2006). Temperature-dependent crystallographic studies and electronic structure of Ba2Cd3Bi4. J. Solid State Chem. 179, 3371–3377. doi:10.1016/j.jssc.2006.07.003
Yoshimura, S., and Hachisu, S. (1983). “Order formation in binary mixtures of monodisperse lattices,” in Front. Colloid. Sci. In memoriam professor dr. Bun-ichi Tamamushi Progress in Colloid and Polymer Science (Berlin, Germany: Springer), Vol. 68, 59–70.
Zelinska, O., Conrad, M., and Harbrecht, B. (2004). Refinement of the crystal structure of cerium zinc (1:11), CeZn11. Z. Kristallogr. - New Cryst. Struct. 219, 389–390. doi:10.1524/ncrs.2004.219.14.389
Keywords: Cd, Li, single-crystal X-ray diffraction, crystal structure, synthesis
Citation: Peng W, Baranets S and Bobev S (2022) Synthesis, crystal and electronic structure of BaLixCd13–x (x ≈ 2). Front. Chem. 10:991625. doi: 10.3389/fchem.2022.991625
Received: 11 July 2022; Accepted: 10 August 2022;
Published: 07 September 2022.
Edited by:
Hans-Conrad zur Loye, University of South Carolina, United StatesReviewed by:
Arthur Mar, University of Alberta, CanadaBoniface Fokwa, University of California, Riverside, United States
Copyright © 2022 Peng, Baranets and Bobev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svilen Bobev, Ym9iZXZAdWRlbC5lZHU=