 Dajiao Cheng
Dajiao Cheng Ke Hu
Ke Hu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 19 July 2022
Sec. Photocatalysis and Photochemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.958626
This article is part of the Research Topic Catalysis Rising Stars in China View all 10 articles
Atomically precise gold nanoclusters (Au NCs) have high specific surface area and abundant unsaturated active sites. Traditionally, Au NCs are employed as thermocatalysts for multielectron transfer redox catalysis. Meanwhile, Au NCs also exhibit discrete energy levels, tunable photophysical and electrochemical properties, including visible to near infrared absorption, microsecond long-lived excited-state lifetime, and redox chemistry. In recent years, Au NCs are increasingly employed as visible to near infrared photocatalysts for their high photocatalytic activity and unique selectivity. This review focuses on the photophysical properties of a variety of Au NCs and their employment as photocatalysts in photocatalytic reactions and related applications including solar energy conversion and photodynamic therapies.
Metal nanoclusters (NCs) are a new type of nanomaterials with core-shell structures. The core comprises a few to hundreds of metal atoms and the outer shell consists of ligands protecting the core from aggregation (Figure 1A) (Chen and Li, 2020). In recent decades, noble metal nanoclusters especially gold have attracted extensive attention. The size of gold nanoclusters (Au NCs) is typically less than 2 nm, which is between that of small molecules and metal nanoparticles (NPs). Because the size is close to the de Broglie wavelength of the electron at the Fermi level of Au atoms (about 0.5 nm) (Xu and Suslick, 2010), Au NCs exhibit discrete energy levels and a variety of molecular-like properties (Aikens, 2011), such as the atomically precise molecular formula, multiple visible absorption peaks, tunable luminescence, and molecular-like excited-state dynamics (Figure 1B) (Song et al., 2016; Maity et al., 2019; Zhou et al., 2021).
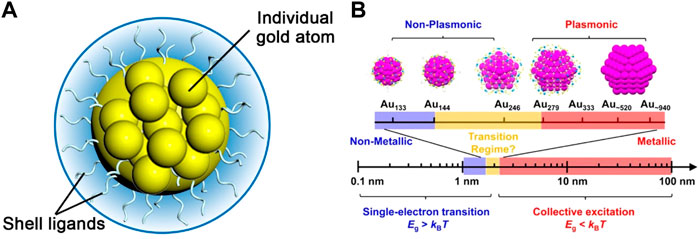
FIGURE 1. (A) Schematic illustration of the core-shell structure of Au NCs (B) Changes in the band gap as the number of atoms in a material decreases. Reprinted with permission from (Zhou et al., 2021). Copyright 2021 American Chemical Society.
Unlike small molecules, Au NCs possess high specific surface areas and a large number of unsaturated reaction sites (Du et al., 2020), which lead to high catalytic activities and unique selectivity in catalytic reactions such as oxidation, hydrogenation, and C-C coupling (Li and Jin, 2013; Fang et al., 2016; Jin et al., 2016). It is worth mentioning that monodisperse Au NCs with well-defined structures can be used as model catalysts to reveal the correlation between catalyst performance and structure at the atomic scale (Du et al., 2020). The focus of this review will be on the optical properties from Au NC’s unique electronic transitions as well as the practical applications of photocatalysis, solar energy conversion, and phototherapies that make use of Au NC’s electronically excited states.
The photophysical properties of atomically precise gold nanoclusters are closely related to the number of gold atoms. Researchers found that Au NCs had a specific number of atoms, known as the “magic number” (Negishi et al., 2004; Negishi et al., 2007). The magic-number gold nanoclusters exhibit high stability and similar photophysical properties. The “superatom electronic theory” proposed by Häkkinen et al. explained the stability of magic-number gold nanoclusters (Hakkinen, 2008; Walter et al., 2008). The core of Au NCs has delocalized ‘‘superatomic orbitals’’ including 1S, 1P, 1D, 2S, 1F, 2P, 1G, 2D, 1H, 3S, 1I.... The effective valence electrons of the gold core are filled into superatomic orbitals in turn according to the aufbau rule. Closed electron shells appear for the gold cores similar to the chemically inert noble gases, so Au NCs are chemically stable. For example, Au102(SR)44 protected by organic thiolate ligands (-SR), the first Au NCs to be comprehensively characterized by X-ray crystal structure determination, has the exact formula of Au79 [Au (p-MBA)2]19 [Au (p-MBA)3]2 (Jadzinsky et al., 2007). The effective valence electron number of the Au79 core is 58 (= 79-19-2), which just meets the closed-electron-shell configuration of 1S21P61D102S21F142P61G18, so the Au79 core exhibits high stability (Walter et al., 2008).
The correlation between the photophysical properties and the number of gold atoms is attributed to the discrete energy levels, which are affected by the quantum size effect. The quantum size effect was explained in detail by the Kubo criterion and the Jellium model (Kubo, 1962; Wood and Ashcroft, 1982).
Kubo proposed that the gap between the highest occupied state and the lowest unoccupied state (called the Kubo gap δ) of a particle consisting of N atoms was proportional to Ef/N, where Ef is the Fermi potential of the bulk metal. Comparing d and thermal energy (kT), it is possible to judge whether a material has metallic or non-metallic properties (Kubo, 1962). When d is less than kT, electrons are excited thermally to generate free electron-hole pairs. However, when d is much larger than kT, free electrons are confined to the discrete energy levels (Roduner, 2006). Metal nanoclusters are usually non-metallic with molecular-like photophysical properties.
Later, the Jellium model clarified the quantitative relationship between the electronic structure of the cores in the metal nanoclusters and the number of metal atoms. In the Jellium model, the valence electrons of the metal core are confined in orbitals that have the same symmetry as the atomic orbitals. The valence electrons are also filled according to the aufbau rule. D. M. Wood and N. W. Ashcroft proved that the energy gap of metal nanoclusters was about Ef/N1/3 through the Jellium model, where N was the number of metal atoms in the cluster and Ef was the Fermi potential energy of the bulk metal (Wood and Ashcroft, 1982).
Ultrasmall-sized gold nanoclusters with discrete energy levels exhibit unique optical absorption, radiative transitions, and excited-state dynamics (Aikens, 2011). Researchers delved into the origin and influencing factors of these photophysical properties, including the cluster geometry such as size, crystal structure and atomic packing, the charge state of the cluster, and the nature of the ligands.
The optical absorption spectra of gold nanoparticles and gold nanoclusters have obvious differences. Gold nanoparticles such as Au2406 NPs have the surface plasmonic resonance peak, while Au NCs do not have (Figure 2A) (Yau et al., 2010). The single-electron transitions between discrete energy levels of Au NCs constitute the molecular absorption (Aikens, 2011).
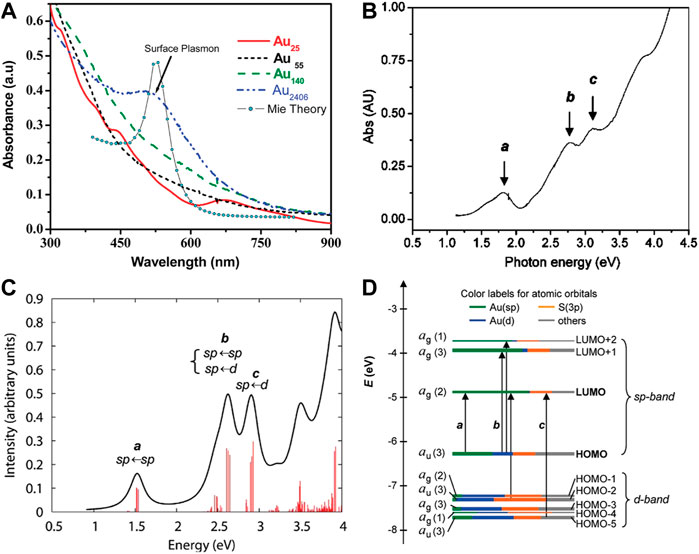
FIGURE 2. (A) UV-vis-NIR absorption spectra of gold nanoparticles and gold nanoclusters. Reprinted with permission from (Yau et al., 2010). Copyright 2010 American Chemical Society (B) The experimental absorption spectrum and (C) the theoretical absorption spectrum of Au25(SH)18- (D) Kohn–Sham orbital energy level diagram for a model compound Au25(SH)18−. Reprinted with permission from (Zhu et al., 2008a). Copyright 2008 American Chemical Society.
In 2008, Jin and co-workers first reported the crystal structure of Au25(SR)18 and studied the correlation between the electronic structure and optical properties by time-dependent density functional theory (TDDFT) calculations (Zhu et al., 2008a). The theoretical absorption spectrum of Au25(SH)18- is in good agreement with the experimental absorption spectrum (Figures 2B,C). Peak a is attributed to the HOMO-LUMO intraband transition (sp←sp). Peak c corresponds to the interband transition (sp←d) from HOMO-n to LUMO. Peak b belongs to the mixed transition (Figure 2D).
Crystal structure analysis shows that the center of Au25(SR)18 is an icosahedral Au13 core and the outer layer is composed of twelve Au atoms and eighteen thiolate ligands. Each outer Au-Au pair (six pairs in total) is bridged by one-SR ligand and with two other-SR ligands bridging between the outer Au atoms and the icosahedral Au13 core. HOMO, LUMO, LUMO+1, LUMO+2 and other orbitals are almost entirely composed of the atomic orbitals of thirteen Au atoms in the icosahedral core. Therefore, the absorption peak a at 1.52 eV (Figures 2B,C) is regarded as a transition caused by the electronic structure and geometry of the Au13 core (Zhu et al., 2008a). Biicosahedral [Au25(PPh3)10(SCnH2n+1)5Cl2]2+ clusters have the completely different crystal structure (Shichibu et al., 2007). The optical absorption peak at 1.76 eV originates from the interaction between two icosahedral Au13 units sharing vertices rather than the electronic transitions within a single Au13 unit. The above two findings indicate that the crystal structure of Au NCs also affects the optical absorption.
Although the Au25(SR)18 cluster is divided into two parts including the Au13 core and the ligand layer, the optical absorption spectrum of Au25(SH)18- cluster is not a simple linear combination of the spectra from the metal core and the ligand, which is demonstrated with more detailed TDDFT calculations. The complex absorption spectrum of the Au25(SR)18 cluster arises from the geometrical and electronic interactions between the metal core and ligands (Aikens, 2008).
Optical absorption of Au NCs is affected by the quantum size effect. As the size of the Au NCs decreases, the gap of discrete energy levels increases and the optical absorption of the clusters shows blue shifts (Figure 3A). For example, the absorption onsets of Au10-12(SG)10-12 cluster, Au15(SG)13 cluster, Au18(SG)14 cluster, and Au25(SG)18 cluster are 450, 650, 700, and 900 nm, respectively (Yu Y. et al., 2013; Stamplecoskie and Kamat, 2014). The absorption onset of the Au144(SR)60 cluster is located at 700 nm (Qian and Jin, 2011). Meanwhile, when the cluster size is reduced to Au25(SR)18, the absorption onset shows the blue shift to 670 nm. Jin summarizes the stable sizes of thiolate-protected gold nanoclusters and the size dependence of the optical absorption in detail (Jin, 2015).
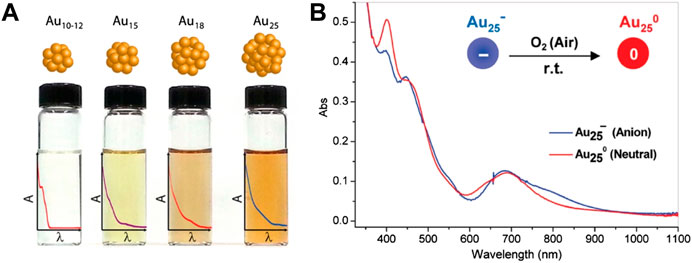
FIGURE 3. (A) The structural diagram and the optical absorption spectra changes of different sizes of Au NCs. Reprinted with permission from (Stamplecoskie and Kamat, 2014). Copyright 2014 American Chemical Society (B) Optical absorption spectra of anionic Au25− cluster (blue) and charge neutral Au25 cluster (red) in solution. Reprinted with permission from (Zhu et al., 2008b). Copyright 2008 American Chemical Society.
However, there are exceptions to the size dependence of the optical absorption. For example, the size of the Au22(SR)18 cluster is larger than that of the Au18(SR)14 cluster, but its absorption onset shows a blue shift of 20 nm (Yu et al., 2014; Pyo et al., 2015; Yu et al., 2015). This phenomenon suggests that the optical absorption of Au NCs is not only related to the size but also to the ratio between the number of ligands and the number of metal atoms.
The electron-withdrawing properties of surface-protecting ligands in Au NCs also affect the optical absorption of the Au NCs. For example, the introduction of electron-withdrawing groups (-X) into p-thiophenol (p-SPhX) ligands induces distortion to the Au25(SPhX)18 framework and the degree of distortion increases with enhancement of the electron-withdrawing properties. As the distorted degree of the cluster framework increases, the HOMO-LUMO gap becomes smaller and the optical absorption exhibits red shifts (Tlahuice-Flores et al., 2013). A similar phenomenon is also observed in the Au38(SPhX)24 cluster, where the optical absorption exhibits a slight red shift with the enhancement of the electron-withdrawing properties of the -X substituent. However, Murray and co-workers believe that the ligand layer affects the electron energy of the gold core in the cluster but does not change the HOMO-LUMO energy gap (Guo and Murray, 2005). The HOMO-LUMO electronic absorption peak of the Au25(SNap)18- cluster (SNap = 1-naphthalenethiolate) shows a red shift of about 10 nm compared with that of the Au25(SC2H4Ph)18- cluster, which is attributed to the expansion of the Au13 core induced by the aromatic thiolate ligands (Li et al., 2016).
Au NC surface charges from surface-protecting ligands also affect the optical absorption of Au NCs. Xie et al. used multiple ligands with different charges, such as −COO-, −NH3+, and −OH, to induce the surface charge anisotropy of the Au25 clusters. This results in structural distortion, which in turn leads to the anomalous optical absorptions at about 780 and 980 nm (Yuan et al., 2016).
In addition to the charges of the ligands, the surface charge states of the clusters also have an effect on the optical absorption spectra (Lee et al., 2004; Negishi et al., 2007; Zhu et al., 2008b). For example, anionic Au25− cluster loses one electron and counterion tetraoctylammonium TOA+ when it is oxidized to charge neutral Au25 cluster in air, resulting in the structural distortion of the gold core which affects the optical absorption of the cluster. The optical absorption spectra of the anionic cluster and the charge neutral cluster are generally similar in shape. However, the absorption around 800 nm in the anionic cluster disappears in the charge neutral cluster, while the weak absorption around 400 nm in the anionic cluster is significantly enhanced in the charge neutral cluster (Figure 3B) (Zhu et al., 2008b). When the Au25(SC2H4Ph)18q cluster is oxidized, the surface charge q gradually changes from negative charge to positive charge, resulting in an enlarged HOMO-LUMO gap and a red shift in the optical absorption (Venzo et al., 2011). Density functional theory (DFT) calculations further verify the correlation between the surface charge states of the cluster and the optical absorption (Antonello et al., 2013).
Au NCs exhibit visible to near-infrared (NIR) emission (Bigioni et al., 2000; Huang and Murray, 2001; Link et al., 2002). The origin of Au NCs photoluminescence remains debatable. Ramakrishna et al. employing time-resolved luminescence spectroscopy of monolayer thiolate-protected Au25 clusters have revealed that the NIR luminescence originates from the S-Au-S-Au-S semi-ring states and the visible luminescence originates from the Au13 core states (Devadas et al., 2010). However, Aikens et al. calculated that photoluminescence of the Au25 cluster were all ascribed to core-based orbitals. Variation of the ligand lengths only influenced photoluminescence spectra from molecular orbital energy shifts. (Weerawardene and Aikens, 2016). A more recent study of the Au25 clusters photoluminescence asserted that NIR emission around 1,100 nm should be the dominant gap emission of the Au13 core state rather than the weak emission around 700 nm of the core-shell charge transfer state because the photoluminescent excitation spectra with emission monitored at ∼700 nm showed a different profile in comparison with the absorption spectra of Au13 clusters. In addition, the lifetime of the Au25 clusters excited state obtained from transient absorption spectroscopy is highly consistent with the lifetime of that obtained from the NIR photoluminescence. (Figure 4) (Zhou and Song, 2021). The detailed photoluminescence origin of the Au NCs is subject to data interpretation possibly due to different charge-donating capability or hydrophilicity of ligands. The changes under different experimental conditions may also vary significantly.
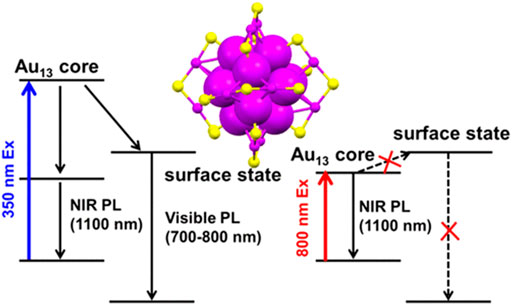
FIGURE 4. Schematic illustration of the photoluminescent origin of Au25 clusters. Reprinted with permission from (Zhou and Song, 2021). Copyright 2021 American Chemical Society.
In earlier studies, the quantum yields (QY) of most thiolate-protected gold nanoclusters are less than 1% and the utilization of incident light is low (Link et al., 2002; Negishi et al., 2004; Negishi et al., 2005). A few Au NCs exhibit higher quantum yields including the Au18(SG)14 cluster with a QY of 5.3% and the Au22(SG)18 cluster with a QY of about 8% (Ghosh et al., 2012; Yu et al., 2014). Researchers find that the size and structure of Au NCs determine the photoluminescence emission wavelength and QY. Meanwhile, both different surface-protecting ligands and aggregation-induced emission (AIE) can enhance QY.
Bulk metals with no band gap and non-radiative decay usually have weak photoluminescent emission. As the size shrinks to the nanometer scale and approaches the Fermi wavelength of electrons, the bulk metal transforms into the cluster. The discrete energy levels of clusters cause strong luminescence in addition to the molecular-like optical absorption. According to the Jellium model (Wood and Ashcroft, 1982), as the atom numbers in the metal core of the cluster increase, there is a red shift in the photoluminescence. For example, pepsin-protected Au8 cluster, Au13 cluster, and Au25 cluster synthesized by changing the pH emit blue, green, and red photoluminescent emission, respectively (Kawasaki et al., 2011). Adjusting the molar ratios among the dendrimer ligands, Au3+, and the amount of NaBH4, a series of Au5 clusters, Au8 clusters, Au13 clusters, Au23 clusters, and Au31 clusters of the same type were synthesized and their photoluminescent emission bands shift from UV to NIR (Figure 5A) (Zheng et al., 2004).
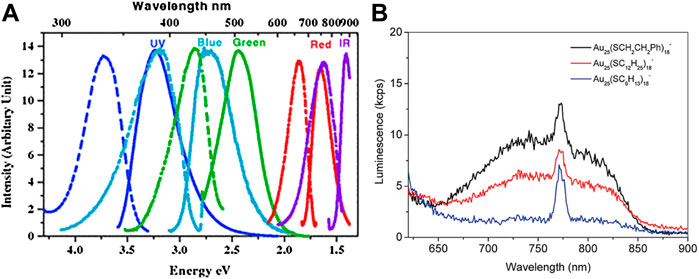
FIGURE 5. (A) Excitation spectra (dashed) and emission spectra (solid) of dendrimer-protected gold nanoclusters different sizes. Reprinted with permission from (Zheng et al., 2004). Copyright 2004 by the American Physical Society (B) Photoluminescence spectra of Au25(SR)18− with different −R groups (−C2H4Ph, −C12H25, −C6H13). Reprinted with permission from (Wu and Jin, 2010). Copyright 2010 American Chemical Society.
Optical properties of Au NCs are not only related to the size but also to the crystal structure. Biicosahedral [Au25(PPh3)10(SCnH2n+1)5Cl2]2+ clusters and the icosahedral Au25(SC2H4Ph)18 have completely different crystal structures (Zhu et al., 2008a). Although both clusters have 25 gold atoms, they have different UV-vis absorption and photoluminescent emission.
The photoluminescence of Au NCs is also affected by the surface ligands, which have little effect on the photoluminescence wavelength but significantly affect QY. Ligands with electron-rich atoms (such as N, O) or groups (such as −COOH, NH2) can interact with the surface of Au NCs and transfer delocalized electrons directly to the Au core to effectively enhance QY. For example, water-soluble thiolate GSH-protected Au25(SG)18− clusters show stronger photoluminescence emission than hydrophobic organic thiolate-protected Au25(SR)18− clusters (Wu and Jin, 2010).
Electron donating ligands can enhance ligand-to-metal charge transfer (LMCT) and ligand-to-metal-metal charge transfer (LMMCT) to strengthen the photoluminescent emission intensity. Jin and co-workers find that the photoluminescence intensity of hydrophobic thiolate-protected gold nanoclusters is proportional to the electron-donating properties of the ligands (Figure 5B) (Wu and Jin, 2010). In the follow-up work, Jin et al. verify the conclusion again that the lower electronegativity or the stronger electron-donating property of the ligands have, the stronger the photoluminescence of the Au36(SR)24 clusters are (Kim et al., 2017). The decreased electronegativity of the cluster can also strengthen the electron transfer between the ligand and the gold core to enhance photoluminescent emission. The higher valence of the surface charge q the Au25(SC2H4Ph)18q cluster has, the stronger photoluminescence intensity the cluster shows (Wu and Jin, 2010).
Polar ligands can also affect the electron transfer between thiolate ligands and the gold core to change the photoluminescence intensity of the cluster. For example, an increase in the polarization of the Au-S bond can enhance the photoluminescence intensity. Murray et al. find that when the non-polar ligands on the surface of Au38 clusters and Au140 clusters are replaced by polar ligands, the NIR photoluminescence intensity of the clusters increases and the intensity is related to the number of polar ligands linearly (Wang et al., 2006).
The photoluminescence intensity of non-photoluminescent polydentate polymer-protected gold nanoclusters is proportional to the electron-donating ability of the ligands and is also affected by the steric hindrance of the polymer (Li et al., 2013). Au NCs protected by sterically hindered polymer PTMP-PtBMA have the highest QY of about 20.1%. The rigid Au(I)-thiolate shell can also improve the QY of the clusters (Pyo et al., 2015). The QY of the Au22(SG)18 cluster is increased to 60% after the gold shell was rigidified by TOA+ (Figure 6).
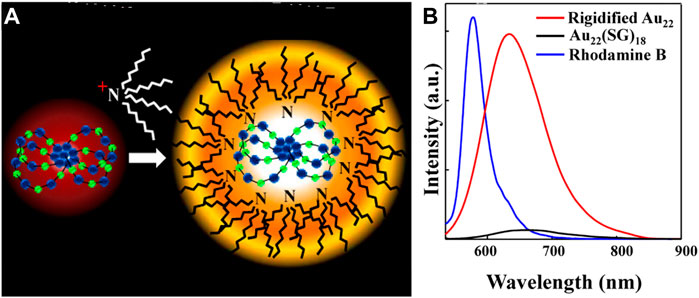
FIGURE 6. (A) Schematic representation of TOA+ binding to the Au22(SG)18 cluster (Au in blue and S in green) (B) Photoluminescence spectra of Au22(SG)18 clusters in aqueous solution and TOA−Au22 clusters in toluene. Reprinted with permission from (Pyo et al., 2015). Copyright 2015 American Chemical Society.
Aggregation-induced emission can significantly enhance the luminescence intensity of Au NCs. Xie and co-workers have synthesized ultrabright Au (0)@Au(I)-thiolate core-shell nanoclusters (Luo et al., 2012). The addition of a weakly polar solvent disrupts the hydration shell and neutralizes the surface charge so that the short Au(I)-thiolate motifs aggregates on the Au (0) core. Conventional Au(I)-thiolate NCs, which originally do not emit light in aqueous solution, emit strong photoluminescence after aggregation with a QY of about 15%.
The excited state dynamics of Au NCs are so unique that they differ significantly from gold nanoparticles. Firstly, the excited-state lifetime of Au NCs (∼ns) is much longer than that of gold nanoparticles (∼70 fs) (Varnavski et al., 2001; Zhou et al., 2017b). Typically, Jin et al. reported the excited state lifetime of body-centered cubic Au38 cluster is about 5 microseconds (Zhou et al., 2019). Furthermore, high pump power prolongs the electron-phonon coupling time of gold nanoparticles. (Zhou et al., 2016). However, the excited state dynamics of Au NCs is independent on the pump laser intensity, which is similar to the properties of molecules (Yau et al., 2010; Stamplecoskie and Kamat, 2014; Zhou et al., 2016). Au55 NCs exhibit a molecular-like single-electron relaxation process. The transient absorption at 640 nm of the Au55 clusters decays from the initial state to the intermediate state quickly and then decay back to the ground state slowly (Varnavski et al., 2010; Yau et al., 2010).
At present, it is generally accepted that the core-shell relaxation model is used to analyze the excited state dynamics of Au NCs. When the Au25(SR)18− cluster is excited, the ultrafast charge relaxation at the Au13 core is less than 200 fs and the relaxation from the gold core state to the semi-ring state is about 1 ps (Miller et al., 2009; Devadas et al., 2010). Goodson and Kamat et al. find that the excited state dynamics of Au NCs are mainly affected by the properties of the S-Au-S-Au-S semi-ring state through transient absorption spectroscopy (Devadas et al., 2010; Yau et al., 2010; Stamplecoskie et al., 2014).
As mentioned previously, the photophysical properties of Au NCs are highly dependent on the size. Lee and Ramakrishna et al. observe that the excited state lifetimes of Au NCs also show the distinct size dependence. As the size of Au NCs decreases or the number of Au atoms decreases, the enlarged HOMO-LUMO gap leads to the reduced nonradiative decay transition rate and the increased exciton lifetime (Figures 7A,B) (Kwak et al., 2017). This phenomenon indicates that the excited state dynamics of Au NCs can be explained by the energy gap law, which states that the nonradiative decay dynamics are inversely proportional to the energy gap (Englman and Jortner, 1970). However, Kamat et al. find that Au18(SR)14 cluster has the longest excited state lifetime among Au10-12(SR)10-12 cluster, Au15(SR)13 cluster, Au18(SR)14 cluster, and Au25(SR)18 cluster (Stamplecoskie and Kamat, 2014). This result deviates from the expectation of the energy gap law. The explanation is that Au18(SG)14 cluster and Au25(SG)18 cluster with the gold core exhibit fast relaxation of less than 1 ps and long-lived LMCT of about 200 ns, while small-sized Au NCs only show long-lived LMCT without fast relaxation processes in the gold core.
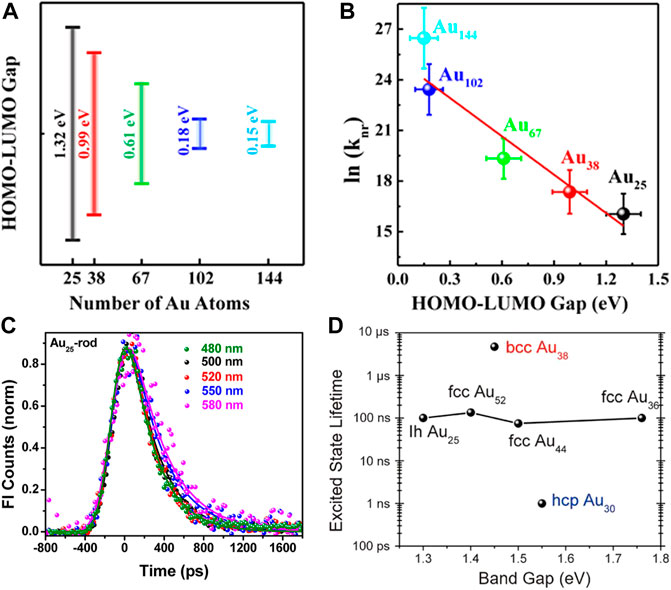
FIGURE 7. (A) Plot of the HOMO-LUMO energy gap versus the number of Au atoms for Au NCs (B) Plot of the logarithmic form of the nonradiative decay transition rate (referred to as ln (knr)) versus HOMO-LUMO gap. Reprinted with permission from (Kwak et al., 2017). Copyright 2017 American Chemical Society (C) Luminescence kinetic decay traces of Au25-rod at different excitation wavelengths. Reprinted with permission from (Devadas et al., 2013). Copyright 2013 American Chemical Society (D) Plot of the Excited state lifetime versus band gap of bcc Au38, hcp Au38, and fcc Au36/Au44/Au52. Reprinted with permission from (Zhou et al., 2019). Copyright 2019 American Association for the Advancement of Science.
The structure of how the gold atoms arrange inside the cluster also affects its excited state dynamics. Ramakrishna et al. compared the femtosecond luminescence dynamics of Au25-rod clusters, Au25-sphere clusters, and Au38-rod clusters. They find that the luminescence kinetic decay traces of Au25-rod are independent of the excitation wavelength (Figure 7C), while the kinetic growth traces and the kinetic decay traces of Au25-spheres and Au38-rods show the specific excitation wavelength dependence, which is attributed to the cascade relaxation from core-gold to shell-gold (Devadas et al., 2013). Jin et al. studied two structural isomers of Au38(SC2H4Ph)24 in which the Au23 core of the Au38Q cluster was the bi-icosahedron and that of the Au38T cluster wass made up of a mono-icosahedral Au13 capped by a Au12 tri-tetrahedron by sharing two atoms (Zhou et al., 2017a). The Au38Q cluster shows a rapid decay of 1.5 ps followed by nanosecond relaxation back to the ground state. The excited state dynamics of the Au38T cluster is similar to that of the Au38Q cluster except that the rapid decay process is accelerated by about 50%. The picosecond decay originates from the core-shell charge transfer or the electron rearrangement in the gold core, so it is speculated that the accelerated picosecond decay in the Au38T cluster is attributed to its unique core structure.
Jin et al. also find that the atomic packing of Au NCs also greatly affects its excited state dynamics (Zhou et al., 2019). As shown in Figure 7D, the HOMO-LUMO gaps of face-centered cubic (fcc) Au NCs with different sizes vary greatly, but the excited state lifetimes hardly change. The excited state lifetime of hexagonal close-packed (hcp) Au30(SR)18 cluster is reduced by about two orders of magnitude and that of body-centered cubic (bcc) Au38S2(SR)20 cluster is increased by nearly two orders of magnitude. This significant excited state lifetime difference is related to the distance and their connection modes between the Au4 tetrahedral units.
The surface charge state of the cluster also affects its excited state dynamics in addition to the geometry and size. Taking the ultrafast electron relaxation dynamics of Au25(SR)18q (q = 0, −1) as an example, the core excitation lifetime of the anionic cluster is about 1,000 times longer than that of the neutral cluster (Qian et al., 2010). The excited state relaxation of Au25(SC2H4Ph)18q (q = 0, −1) mainly originates from the nonradiative energy transfer from the gold core to the ligands. The energy is transferred from the LUMO orbital to the ligands in the neutral cluster, while the energy is transferred from the LUMO+1 orbital above the LUMO orbital for the anionic cluster and the excited state lifetime is extended by about a hundred times (Green and Knappenberger, 2012).
Besides the excited state lifetime, the electron transfer ability of the excited state is also crucial for the material to become a potential photosensitizer. Methyl viologen [(MV2+)] is often used as an electron acceptor to examine the photoinduced electron transfer kinetics in Au NCs and its correlation with photocatalytic activity. Kamat et al. find that only long-lived semi-ring states (or LMCT states) of Au NCs are involved in electron transfer processes (Stamplecoskie et al., 2014). Therefore, the effect of different ligands on the LMCT states can tune the electron transfer efficiency of Au NCs. Meanwhile, Marcus theory suggests that the electron transfer rate decreases rapidly as the distance between the donor and acceptor becomes longer (Silverstein, 2012). Theoretically, the larger the shell ligands are, the smaller the electron transfer rates of Au NCs become (Chen and Li, 2020). Kamat et al. also demonstrate that the electron transfer efficiency is highly correlated with the size of Au NCs. As the size of clusters decreases, the electron transfer efficiency increases (Figure 8) (Stamplecoskie and Kamat, 2014). Au NCs with high electron transfer efficiencies and good visible light absorption have the potential to be excellent photosensitizers (Abbas et al., 2016).
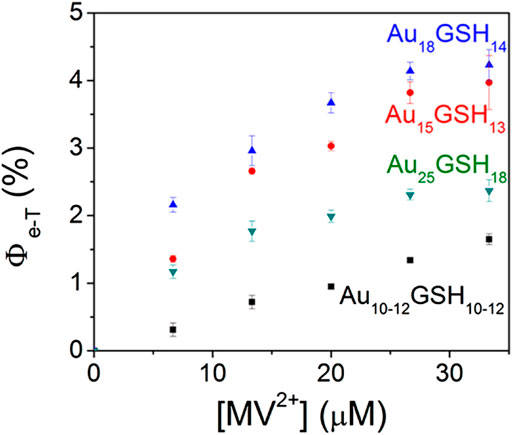
FIGURE 8. Quantum efficiency of electron transfer, Φe-T, following the 355 nm laser pulse excitation of aqueous AuxGSHy solution containing different concentrations of (MV2+). Reprinted with permission from (Stamplecoskie and Kamat, 2014). Copyright 2014 American Association for the Advancement of Science.
The current society is facing the dual pressure of economic development and environmental protection. Photocatalysis is an environmentally friendly technology that can realize solar energy conversion and has broad development prospects. Suitable photocatalysts enable efficient and selective photocatalytic reactions and their optical properties have a great influence on the catalytic efficiency and selectivity (Du et al., 2020). Good photocatalysts have the following characteristics: 1) broad visible light absorption to maximize sunlight utilization and gain selectivity; 2) long-lived photoinduced charge separated states to react with substrate molecules before relaxation to the ground states; 3) high reduction or oxidation potentials to react with a range of substrates; 4) the ability to transfer photogenerated electrons and holes to the reactive interface efficiently (Chai et al., 2019; Witzel et al., 2021).
As mentioned above, Au NCs with the ultra-small size and atomically precise composition exhibit high specific surface area, abundant unsaturated active sites, discrete energy levels, and controllable electronic properties. Therefore, Au NCs have received extensive attention in the fields of thermocatalysis and electrocatalysis (Fang et al., 2016; Chai et al., 2019; Du et al., 2020). Au NCs serving as promising photocatalysts have also raised increasing interest. With the in-depth study of the synthetic methods and optical properties of Au NCs, the energy level gap and the band position of Au NCs can be tuned by adjusting the size, crystal structure, atomic packing, surface charge state or surface ligands. The change of the energy level gap and the band position affect the optical absorption, the separation lifetime of photogenerated electron-hole pairs and the redox ability. At present, Au NCs have been used in photocatalytic conversion (oxidation and reduction) of the organics, photocatalytic degradation of the organic dyes, photocatalytic water splitting, photocatalytic CO2 reduction and phototherapy.
Reactive oxygen species (ROS) are a class of chemically active molecules containing oxygen including singlet oxygen (1O2), superoxide, hydrogen peroxide (H2O2), and hydroxyl radicals (•OH). Photosensitizers can generate ROS through the photochemical reactions, which are mainly divided into two reaction pathways (Foote, 1991). In the type I reaction pathway, various oxygen radicals, such as superoxide and hydroxyl radicals, are generated by the electron transfer from the excited state photosensitizers to the substrates. In the type II reaction pathway, the photosensitizer is excited to form the singlet excited state (S1) and then transformed into the triplet excited state (T1) through the intersystem crossing. The triplet photosensitizer generates 1O2 by the energy transfer with triplet oxygen (3O2).
The photosensitizers are usually organic dye molecules with strong absorption or nanomaterials such as quantum dots (QDs) (Samia et al., 2003; Ma et al., 2008), silicon nanocrystals (Kovalev and Fujii, 2005), and metal nanoparticles (Vankayala et al., 2011; Long et al., 2013; Vankayala et al., 2013). Au NCs have been demonstrated to act as photosensitizers and generate 1O2 efficiently by tuning the electronic structure of the clusters (Kawasaki et al., 2014; Zhang et al., 2018; Du et al., 2020). Au NCs can undergo thermal catalytic oxidation reaction using the organic oxidant PhIO (Li et al., 2012; Liu et al., 2015) or photocatalytic oxidation reaction using 1O2 generated from energy transfer, the latter being more environmentally friendly.
Jin et al. demonstrate that the ability of 1O2 generation is related to the size of Au NCs (Kawasaki et al., 2014). Regardless of the surface charge state and the solubility, Au25 clusters can generate 1O2 under the visible or NIR light irradiation (Figure 9A). Because of the large HOMO-LUMO gap (1.3 eV), Au25 clusters have a high T1 yield. In contrast, the HOMO-LUMO gap of Au38 clusters is small (0.9 eV) and the energy of T1 is lower than that required to excite 3O2 to form 1O2 (0.97 eV). Therefore, Au38 clusters cannot generate 1O2. 1O2 produced by photoexciting Au25 clusters can catalyze the oxidation of organic sulfide to sulfoxide with selectivity close to 100%. Besides, Zhu et al. also find that the arylthiolated Au25(F-Ph)18− nanocluster with NIR emission can photosensitize 3O2 to produce 1O2 for photocatalytic oxidative functionalization of sulfide, β-ketoesters, and 2-aryl-1,2,3,4-tetrahydroisoquinoline (Wang et al., 2022).
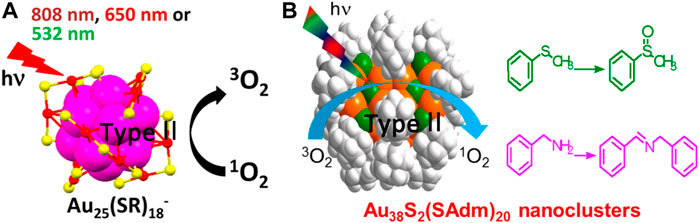
FIGURE 9. (A) Schematic diagram and the characterizations of 1O2 generation from Au25 clusters under the visible or NIR light irradiation. Reprinted with permission from (Kawasaki et al., 2014). Copyright 2014 American Chemical Society (B) Au38 clusters generate 1O2 under light and catalyze the oxidation of sulfides to sulfoxides and that of benzylamines to imines selectively. Reprinted with permission from (Li et al., 2017). Copyright 2017 American Chemical Society.
Maran et al. further investigate the effect of the surface charge state of Au25(SC3)18q clusters on the photocatalytic performance of 1O2 generation by time-resolved electron paramagnetic resonance (TR-EPR) (Agrachev et al., 2019). The anion clusters generate 1O2 under photoexcitation through the type II photoreaction pathway and then the EPR signal is observed. Under the same experimental conditions, neutral clusters cannot generate 1O2 and no EPR signal is observed.
The above study by Jin et al. shows that Au38(SC2H4Ph)24 cannot photo-generate 1O2 due to the small HOMO-LUMO gap (0.9 eV) (Kawasaki et al., 2014). Li et al. have shown that the energy gap of clusters can be adjusted by changing the atomic packing to improve the photocatalytic performance (Li et al., 2017). The Au38S2(SAdm)20 clusters (−SAdm = 1-adamantanethiolate) are body-centered cubic atomic packing, which is different from the face-centered cubic atomic packing of common Au NCs. The Au38S2(SAdm)20 clusters with the HOMO-LUMO gap of 1.57 eV can photo-generate 1O2 and the photocatalytic efficiency is higher than that of Au25(SR)18− described above. In this work, 1O2 can oxidize not only sulfides to sulfoxides but also benzylamines to imines selectively (Figure 9B).
In summary, Au NCs can be used as photosensitizers to photo-generate 1O2 for the selective oxidation of organics. In addition, Au NCs can also be combined with semiconductor materials to improve the efficiency of photocatalytic organic conversion under visible light excitation by broadening the optical absorption range or separating photogenerated electron-hole pairs effectively.
Wang and Li et al. synthesized the Au25(PPh3)10(SC3H6SiO3)5Cl2/TiO2 composite for the photocatalytic oxidation of amines to imines efficiently and selectively under mild reaction conditions (Chen et al., 2017). Au25(PPh3)10(SR)5Cl2 clusters can separate the photogenerated electron-hole pairs effectively leading to the high catalytic activity. The photocatalytic reaction involves an Au-H intermediate and a carbocation intermediate derived from benzylamine (Figure 10), and the reactive sites are the bare Au atoms due to the removal of part of the PPh3 ligands.
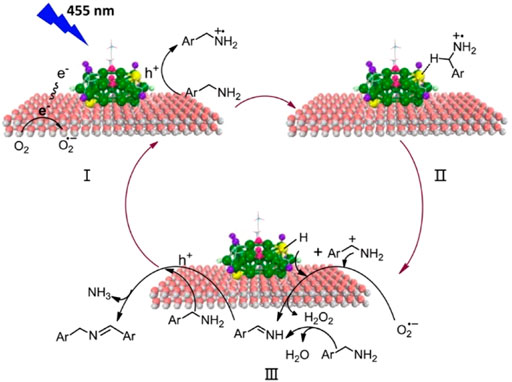
FIGURE 10. (A) The mechanism diagram of the photocatalytic oxidation of benzylamine by Au25/TiO2. Reprinted with permission from (Chen et al., 2017). Copyright 2017 American Chemical Society.
Liu and co-workers construct Aux/NP-TNTA heterostructure by depositing Aux clusters onto highly ordered nanoporous layer-covered TiO2 nanotube arrays (NP-TNA) via a facile electrostatic self-assembly strategy (Xiao et al., 2015). Aux/NP-TNTA heterostructures can be used as photocatalysts for the reduction of nitroaromatics. Aux clusters photo-generate electron-hole pairs under sunlight illumination. Because the potential of LUMO in Aux NCs is more negative than the conduction band (CB) edge of TiO2 and the interaction between them is tight, the photogenerated electrons of Aux clusters are easily injected into the CB of TiO2 to reduce the nitroaromatics adsorbed on the surface of the materials, while the photogenerated holes oxidize ammonium formate as the electron donor. It should be noted that the above reactions occurred in N2 to avoid the participation of O2. The condition ensured that the nitroaromatics were all reduced by the photogenerated electrons. Aux/NP-TNTA heterostructures enhance the performance of photocatalytic reduction of organics through the efficient separation of photogenerated electron-hole pairs.
The composites composed of semiconductors and Au NCs as photosensitizers can also degrade organic dyes through photocatalysis. Jin et al. investigated the performance and mechanism of Au25(SC2H4Ph)18/TiO2 composites to generate ROS for the degradation of methyl orange (MO) under the visible light irradiation (Figure 11A) (Yu C. et al., 2013). Au25(SR)18 clusters expand the optical absorption range to NIR region which results in a 1.6-fold increase in the activity of photocatalytic degradation of MO under the visible light irradiation. The photogenerated electrons of Au25(SR)18 clusters are injected into the conduction band of TiO2 to inhibit the recombination of photogenerated electrons and holes effectively, which also contributes to the improvement of the photocatalytic degradation of MO.
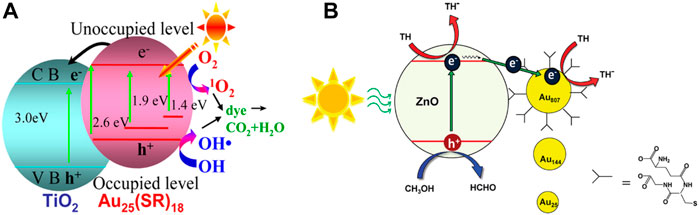
FIGURE 11. (A) Mechanism of the photocatalytic degradation of organic dyes by Au25(SC2H4Ph)18/TiO2 composites. Reprinted with permission from (Yu C. et al., 2013). Copyright 2013 American Chemical Society (B) The photocatalytic degradation of TH of ZnO-Au composites under visible light irradiation. Reprinted with permission from (Lee et al., 2011). Copyright 2011 American Chemical Society.
Aux/NP-TNTA heterostructures constructed by Liu and co-workers are also used for the photocatalytic degradation of MO (Xiao et al., 2015). The photogenerated electrons transferred to the conduction band of TiO2 and the photogenerated holes of Aux clusters can react with H2O to generate ROS for the degradation of MO. The efficient separation of the photogenerated electron-hole pairs enhances the performance of the photocatalytic degradation of organic dyes.
How to improve the photostability of Au NCs at the interface of composites is a long-standing challenge. Xu and Chen et al. report the SiO2-Au GSH clusters-BPEI@TiO2 (SABT) composites assembled by a simple method (Weng et al., 2018). Branched polyethyleneimine (BPEI) is not only the surface modifier but also the stabilizer and the reducing agent, which can prevent the oxidation of the ligands on the surface of the clusters. Therefore, the size and structure of Au GSH clusters can be maintained for more than 10 h under the continuous visible light irradiation. The thickness-controllable TiO2 coating stabilizes the ultra-small Au GSH clusters supported on the SiO2 sphere. As a result, the SABT composite has good photostability and its photocatalytic activity for the degradation of rhodamine B has little change in 10 cycles.
In composites composed of the semiconductor and Au NCs as photocatalysts, Au NCs generally act as photosensitizers to capture the visible light. Au NCs can also accept the photogenerated electrons to catalyze reactions. Both the size and surface ligands of Au NCs affect the efficiency of the photocatalytic degradation of organic dyes.
Three different sizes of Aux (SG)y clusters synthesized by Lee and Song et al. were deposited on the surface of ZnO through the carboxyl groups in their ligands (Lee et al., 2011). It is speculated that the photogenerated electrons of ZnO under visible light irradiation are injected into the Au NCs to reduce thionine (TH) on the surface of the Au NCs. The absorption spectrum of ZnO hardly changes after the cluster deposition indicating that the light-harvesting ability is not enhanced, so the photocatalytic performance is only related to the efficiency of the charge separation. The increased sizes of Au NCs accelerate the electron transfer rate to improve the efficiency of the charge separation (Figure 11B). Therefore, the ZnO-Au composite composed of Au NCs with the large size has higher photocatalytic performance for the degradation of TH.
Xie et al. synthesized thio-β-cyclodextrin (SH-β-CD) protected gold nanoclusters to decorate TiO2 (Zhu et al., 2018). TiO2-Au NCs@β-CD composites can photocatalyze the degradation of MO efficiently. The mechanism indicates that photogenerated electrons from TiO2 under UV light are injected into the gold core to form the catalytic center, which reduces O2 to superoxide anion (O2−•) for the degradation of MO. In addition, the photogenerated holes of TiO2 can also oxidize H2O to •OH for the degradation of MO. The SH-β-CD ligands on the surface of the clusters can capture MO efficiently through the host-guest interaction. Therefore, the enhanced photocatalytic degradation of MO originates from the synergistic effect of the gold core, SH-β-CD ligands and TiO2 in the TiO2-Au NCs@β-CD composite.
Wang et al. synthesized the silane-stabilized Au NCs by a simple photoreduction method. The Au NCs act as stand-alone photocatalysts for the photocatalytic degradation of methylene blue (MB) under visible light irradiation (Zhou S. et al., 2017). The silane-stabilized Au NCs degrade about 96% of MB after exposure to the visible light for 1 h. It has been confirmed that the mechanism of the photocatalytic degradation of organic dyes of the silane-stabilized Au NCs is similar to that of semiconductors.
Au NCs can be used as the cocatalysts in photocatalytic water splitting for the conversion of solar energy to chemical energy. Monodisperse Au25(SG)18 clusters with unchanged sizes are supported on the BaLa4Ti4O15 photocatalysts and the prepared composites can be used for photocatalytic water splitting (Figure 12A) (Negishi et al., 2013). The experimental results show that the catalytic performance of the Au25 cluster cocatalyst is 2.6 times higher than that of the gold nanoparticle cocatalyst. This may be due to the fact that ultra-small Au25 clusters can introduce the same number of active sites as the gold nanoparticles at lower loadings to reduce the interference of cocatalysts on the optical absorption of catalysts. It has also been found that the stability of Au NCs precursors is crucial to achieve the controllable loading. Meanwhile, the photocatalytic activity increases as the size of the cocatalyst decreases (Negishi et al., 2015).
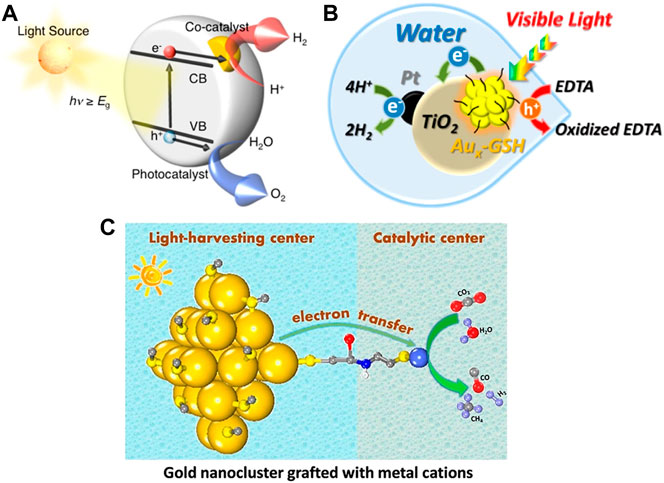
FIGURE 12. (A) Schematic of photocatalytic water splitting by composites via a one-step method: conduction band (CB), valence band (VB), band gap (Eg). Reprinted with permission from (Negishi et al., 2015). Copyright 2015 American Chemical Society (B) Illustration of Aux-GSH NCs sensitized Pt/TiO2 NPs for photocatalytic water splitting reaction. Reprinted with permission from (Chen and Kamat, 2014). Copyright 2014 American Chemical Society (C) Schematic of covalently bridged Au-GSH clusters and metal cations for the visible-light-driven CO2 reduction. Reprinted with permission from (Cui et al., 2018). Copyright 2018 American Chemical Society.
Besides acting as the cocatalysts, Au NCs can also be the photosensitizers for H2 generation in the photocatalytic water splitting. Kamat and coworkers find that the reversible reduction (E0 = −0.63 V vs. RHE) and oxidation (E0 = 0.97 and 1.51 V vs. RHE) potentials of glutathione-capped metal nanoclusters (Aux-GSH NCs) are suitable for driving water splitting (Chen and Kamat, 2014). Aux-GSH NCs sensitized Pt/TiO2 NPs in aqueous slurry system can generate H2 with sacrificial electron donors, such as EDTA, under the visible light irradiation (Figure 12B). The Aux-GSH NCs expand the photoresponse range of the large band gap semiconductor to efficiently realize the conversion of light energy to chemical energy. However, the Aux-GSH NCs aggregate into large NPs upon continuous illumination, which limits the photoconversion efficiency.
Au NCs as photosensitizers can also photocatalyze CO2 reduction. Xiong and co-workers attach M2+ metal cations (Fe2+, Co2+, Ni2+, and Cu2+) to Au-GSH NCs covalently through l-cysteine as a bridging ligand to achieve the stable connection between the light-harvesting center and the catalytic site (Cui et al., 2018). The photogenerated electrons are transferred from the Au-GSH NC to M2+ via the bridge bond and are used to reduce CO2 at the catalytic site M2+ (Figure 12C). It is noteworthy to overcome the instability of Au-GSH NCs in this reaction system to prevent aggregation.
It has been confirmed that the protection of Au NCs by a metal-organic framework (MOF) can improve the stability of Au NCs in photocatalytic CO2 reduction effectively. Recently, Fei et al. prepare N-heterocyclic carbene (NHC)-stabilized ultrasmall Au NCs in a MOF by the heterogeneous nucleation method (Jiang et al., 2021). Au-NC@MOF composites exhibit the stable and excellent catalytic activity for photocatalytic CO2 reduction. It has been demonstrated that the photogenerated electrons are transferred from Au NCs to MOFs via the MOF-NHC-Au covalent bond bridge to separate photogenerated electron-hole pairs rapidly, which enables the synergistic catalysis of Au NCs and MOFs.
Phototherapy is a promising cancer treatment technology with minimal trauma, few side effects and high efficacy (Lin et al., 2021). Photodynamic therapy (PDT) is a clinically licensed non-invasive phototherapy (Dolmans et al., 2003). In the aerobic environment, photosensitizers excited by light with appropriate wavelengths generate ROS through either type I or type II reaction pathways to oxidize adjacent biological macromolecules such as DNA. The photochemical reactions causes the local oxidative damage to kill cancer cells (Li and Grant, 2016).
It has been demonstrated that Au NCs exhibit excellent biocompatibility, pharmacokinetics, renal clearance and biodegradability (Zhang et al., 2012; Zheng et al., 2021). Therefore, Au NCs can be widely used in biomedical fields such as biosensing, photoluminescence imaging, and cancer therapy (Yang et al., 2019; Zheng et al., 2021). At the same time, ultra-small Au NCs exhibit unique optical properties including high two-photon absorption cross section, long-lived triplet state, and high photoluminescence quantum yields. Combining the above two points, Au NCs can be used as photosensitizers in PDT.
Au NCs with high two-photon absorption cross section can improve the efficacy of PDT significantly. Dihydrolipoic acid coated gold nanoclusters (AuNC@DHLA) as photosensitizers can take full advantage of high two-photon absorption and mediate the single electron transfer to generate O2−• for the efficient photodynamic therapy (type I) (Han et al., 2020). Meanwhile, two-photon excitation can increase the penetration depth in tissue.
The combination of Au NCs and semiconductors can improve the charge separation efficiency, which also improves the efficacy of PDT significantly. Li and Zhang et al. prepare TiO2 NPs-Au NC-graphene composites (TAG) for the efficient PDT (Cheng et al., 2017). Au NCs with narrow band gaps have the cut-off absorption wavelength at 617 nm for the efficient utilization of simulated sunlight. Meanwhile, Au NCs, TiO2 NPs, and graphene have staggered energy levels which can separate photogenerated electron-hole pairs effectively. Based on the above two advantages, TAG composites generate a large amount of •OH and O2−• via the water oxidation and oxygen reduction under simulated sunlight irradiation to induce severe cancer cell death. It demonstrates the significant PDT (type I) efficacy of TAG.
The functional molecules can be bound to the surface of Au NCs covalently through the terminal functional groups, which can also enhance the PDT efficacy of the clusters and expand the applications. A common covalent conjugation scheme is the coupling of carboxyl groups to primary amines via 1-ethyl-(3-dimethylaminopropyl)carbodiimide (EDC). For example, a tumor-targeting agent (folic acid) and a photosensitizer (protoporphyrin IX) can be covalently bound to lipoic acid protected Au18 clusters to form a multifunctional PFL-AuC nanocomposites, which can be used as a photosensitizer for photoluminescence imaging assisted PDT (type II) with tumor targeting (Nair et al., 2015). PFL-AuC nanocomposite is more efficient for the photo-generation of 1O2 compared with protoporphyrin IX alone. The tumor-targeting agents cause the local aggregation of PFL-AuC nanocomposites. Therefore, the PFL-AuC nanocomposites can induce PDT to cause the cancer cell death with a low-energy laser. In addition, the NIR emission of the PFL-AuC nanocomposite can track the PDT process in real time. This method of constructing nanocomposites through covalent binding also enables organelle-targeting of Au NCs. For example, the multifunctional nuclear-targeting TAT peptide-Au NCs (peptide sequence: N-GRKKRRQRRR-C) can simultaneously perform photoluminescence imaging, gene delivery, and NIR-excited PDT (type II) to kill cancer cells effectively (Vankayala et al., 2015).
The severely hypoxic environment in solid tumors limits the efficacy of traditional PDT (Li et al., 2018; Liu et al., 2018). Nitroaromatic compounds are bioreductive prodrugs that are activated by nitroreductase (NTR) overexpressed in hypoxic tissues to generate DNA-reactive cytotoxic arylamines, which enables hypoxia-targeting tumor therapy (Searle et al., 2004; Wilson and Hay, 2011). However, the activation of nitroaromatic prodrugs by NTR shows slow reaction kinetics and the regulation of the reaction kinetics is complicated (Searle et al., 2004). In addition, native NTR is difficult to obtain in large quantities and loses its activity easily when pH or temperature changes (Yanto et al., 2010). At present, it has been reported that nanomaterials with catalytic activity can mimic natural enzymes to convert prodrugs into bioactive molecules for cancer treatment (Soldevila-Barreda and Metzler-Nolte, 2019; Oliveira et al., 2020; Vong et al., 2020). Visible light that excites nanomaterials to activate prodrugs is spatially and temporally controllable (Alonso-de Castro et al., 2018; Gurruchaga-Pereda et al., 2019; Zhang and Liu, 2020).
Common photosensitizers are difficult to mimic NTR because the reduction of nitroaromatics to arylamines requires a challenging six-electron/six-proton process. Meanwhile, the selective catalytic reduction of nitroaromatics is also a long-standing challenge. Our group used the glutathione protected Au NCs as photosensitizers to mimic NTRs for the selective photocatalytic reduction of nitrobenzene to aniline (AN) in a hypoxic environment (Figure 13A) (Liu et al., 2021). Notably, the catalytic activity of Au NCs can be regulated by the temporally and spatially controllable light (Figure 13B). Finally, Au NCs photocatalyze the activation of nitroaromatic prodrugs in tumor cells to achieve the photochemotherapy against the hypoxic condition of solid tumors, which overcomes the oxygen dependence of the traditional PDT.
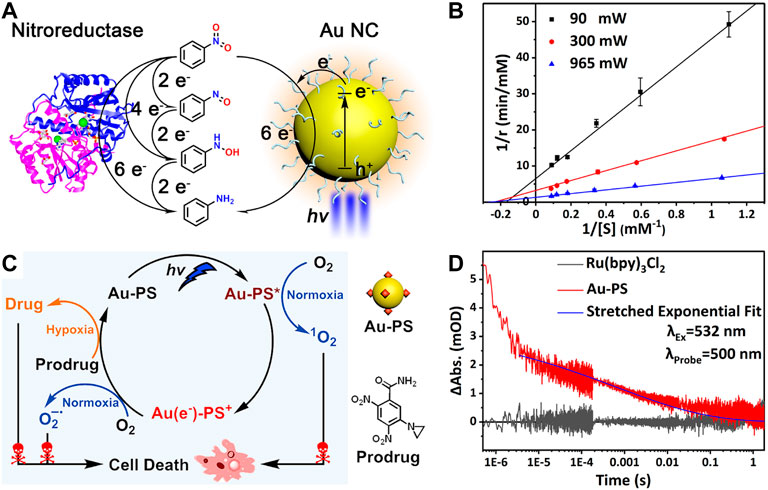
FIGURE 13. (A) Gold nanoclusters perform enzyme-like photocatalysis for nitrobenzene (B) Enzyme-like reaction kinetics of gold nanoclusters tuned by the light intensity. Reprinted with permission from (Liu et al., 2021). Copyright 2021 American Chemical Society (C) Proposed mechanism of the functionalized gold nanoclusters for photodynamic and photocatalytic double killing of cancer cells (D) Time-dependent decay of the transient absorption of charge-separated states in Au-PS. Reprinted with permission from (Cheng et al., 2021). Copyright 2021 American Chemical Society.
However, Au NCs only absorb little blue light and have low utilization of visible light, so the photocatalytic reduction of nitrobenzene is less efficient. In an effort to eliminate this drawback, our group prepared the functionalized gold nanoclusters (Au-PS) as the photocatalyst by covalently linking ruthenium coordination compounds as the photosensitizers to Au NCs (Cheng et al., 2021). In the hypoxic conditions, Au-PS still exhibits the selective photocatalytic reduction of nitrobenzene to aniline under the visible light excitation. At the same time, this functionalization strategy broadens the optical absorption range of the material and prolongs the separation lifetime of photogenerated electron-hole pairs by utilizing the charge transfer from the excited PS to the Au NC core (Figure 13C), so the photocatalytic efficiency is improved significantly. In the normoxic conditions, ROS are generated after the photoexcitation of Au-PS. Therefore, Au-PS can be used both as the photosensitizer for photodynamic therapy in the normoxic conditions as well as being the photocatalyst to activate the nitroaromatic prodrug CB1954 in the hypoxic conditions (Figure 13D). This constitutes a dual kill mechanism to induce the cancer cell death in both normoxic and hypoxic conditions.
The art of synthesizing monodisperse and atomically precise Au NCs makes it possible to finely tune the geometric structure and composition. Au NCs with discrete energy levels exhibit controllable photophysical properties, including optical absorption, redox capacity, and the excited-state lifetime. These tunable photophysical properties give Au NCs great potential for the photocatalytic applications. Although Au NCs can be used for the solar energy conversion, including the degradation of organic pollutants, the conversion of organics, water splitting, and CO2 reduction, the photocatalytic reactions of Au NCs are less reported than their applications in thermal catalysis.
In future endeavors of expanding photocatalytic potentials of Au NCs particularly in solar energy conversion, we think a good research direction would be making the best use of the versatile surface chemistry of Au NCs while combining the excellent visible light absorption and charge separation properties. Molecular light harvesters typically do not possess the capability of both light harvesting and multi-electron transfer catalysis whereas Au NCs do. Some organic transformations such as benzylamine oxidation or nitroaromatics reduction discussed in this review have proven Au NCs their potential. With targeted design of the gold core as well as smart choice of Au NC surface-protecting ligands, Au NCs have a bright future as the photocatalysts to utilize solar energy for value added chemical reactions.
In addition to solar energy conversion, Au NCs with good biocompatibility also have great potential as photocatalysts for biomedical application. They can be delivered into mammalian cells for the photocatalytic generation of bioactive molecules. The functionalization of surface ligands can transform Au NCs into targeted multifunctional nanocomposites and enhance the photocatalytic performance in living organisms. So far, reports on Au NC in vivo photocatalysis are still very rare. More research is waiting ahead.
KH designed the content of the review. DC drafted the review. DC, RL, and KH are all involved in revising the manuscript.
This work was sponsored by the National Natural Science Foundation of China (21872037) and Natural Science Foundation of Shanghai (21ZR1404400).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbas, M. A., Kim, T.-Y., Lee, S. U., Kang, Y. S., and Bang, J. H. (2016). Exploring interfacial events in gold-nanocluster-sensitized solar cells: Insights into the effects of the cluster size and electrolyte on solar cell performance. J. Am. Chem. Soc. 138 (1), 390–401. doi:10.1021/jacs.5b11174
Agrachev, M., Ruzzi, M., Venzo, A., and Maran, F. (2019). Nuclear and electron magnetic resonance spectroscopies of atomically precise gold nanoclusters. Acc. Chem. Res. 52 (1), 44–52. doi:10.1021/acs.accounts.8b00495
Aikens, C. M. (2011). Electronic structure of ligand-passivated gold and silver nanoclusters. J. Phys. Chem. Lett. 2 (2), 99–104. doi:10.1021/jz101499g
Aikens, C. M. (2008). Origin of discrete optical absorption spectra of M25(SH)18− nanoparticles (M = Au, Ag). J. Phys. Chem. C 112 (50), 19797–19800. doi:10.1021/jp8090914
Alonso-de Castro, S., Cortajarena, A. L., Lopez-Gallego, F., and Salassa, L. (2018). Bioorthogonal catalytic activation of platinum and ruthenium anticancer complexes by FAD and flavoproteins. Angew. Chem. Int. Ed. 57 (12), 3143–3147. doi:10.1002/anie.201800288
Antonello, S., Perera, N. V., Ruzzi, M., Gascón, J. A., and Maran, F. (2013). Interplay of charge state, lability, and magnetism in the molecule-like Au25(SR)18 cluster. J. Am. Chem. Soc. 135 (41), 15585–15594. doi:10.1021/ja407887d
Bigioni, T. P., Whetten, R. L., and Dag, Ö. (2000). Near-infrared luminescence from small gold nanocrystals. J. Phys. Chem. B 104 (30), 6983–6986. doi:10.1021/jp993867w
Chai, O. J. H., Liu, Z., Chen, T., and Xie, J. (2019). Engineering ultrasmall metal nanoclusters for photocatalytic and electrocatalytic applications. Nanoscale 11 (43), 20437–20448. doi:10.1039/c9nr07272a
Chen, D., and Li, J. (2020). Ultrasmall Au nanoclusters for bioanalytical and biomedical applications: The undisclosed and neglected roles of ligands in determining the nanoclusters’ catalytic activities. Nanoscale Horiz. 5 (10), 1355–1367. doi:10.1039/d0nh00207k
Chen, H., Liu, C., Wang, M., Zhang, C., Luo, N., Wang, Y., et al. (2017). Visible light gold nanocluster photocatalyst: Selective aerobic oxidation of amines to imines. ACS Catal. 7 (5), 3632–3638. doi:10.1021/acscatal.6b03509
Chen, Y. S., and Kamat, P. V. (2014). Glutathione-capped gold nanoclusters as photosensitizers. Visible light-induced hydrogen generation in neutral water. J. Am. Chem. Soc. 136 (16), 6075–6082. doi:10.1021/ja5017365
Cheng, D., Liu, R., Tian, L., Zhou, Q., Niu, F., Yue, Y., et al. (2021). Photocatalytic nitroaromatic prodrug activation by functionalized gold nanoclusters. ACS Appl. Nano Mat. 4 (12), 13413–13424. doi:10.1021/acsanm.1c02926
Cheng, Y., Chang, Y., Feng, Y., Liu, N., Sun, X., Feng, Y., et al. (2017). Simulated sunlight-mediated photodynamic therapy for melanoma skin cancer by titanium-dioxide-nanoparticle-gold-nanocluster-graphene heterogeneous nanocomposites. Small 13 (20), 1603935–1603946. doi:10.1002/smll.201603935
Cui, X., Wang, J., Liu, B., Ling, S., Long, R., Xiong, Y., et al. (2018). Turning Au nanoclusters catalytically active for visible-light-driven CO2 reduction through bridging ligands. J. Am. Chem. Soc. 140 (48), 16514–16520. doi:10.1021/jacs.8b06723
Devadas, M. S., Kim, J., Sinn, E., Lee, D., Goodson, T., Ramakrishna, G., et al. (2010). Unique ultrafast visible luminescence in monolayer-protected Au25 clusters. J. Phys. Chem. C 114 (51), 22417–22423. doi:10.1021/jp107033n
Devadas, M. S., Thanthirige, V. D., Bairu, S., Sinn, E., and Ramakrishna, G. (2013). Temperature-dependent absorption and ultrafast luminescence dynamics of bi-icosahedral Au25 clusters. J. Phys. Chem. C 117 (44), 23155–23161. doi:10.1021/jp408333h
Dolmans, D. E. J. G. J., Fukumura, D., and Jain, R. K. (2003). Photodynamic therapy for cancer. Nat. Rev. Cancer 3 (5), 380–387. doi:10.1038/nrc1071
Du, Y., Sheng, H., Astruc, D., and Zhu, M. (2020). Atomically precise noble metal nanoclusters as efficient catalysts: A bridge between structure and properties. Chem. Rev. 120 (2), 526–622. doi:10.1021/acs.chemrev.8b00726
Englman, R., and Jortner, J. (1970). The energy gap law for radiationless transitions in large molecules. Mol. Phys. 18 (2), 145–164. doi:10.1080/00268977000100171
Fang, J., Zhang, B., Yao, Q., Yang, Y., Xie, J., Yan, N., et al. (2016). Recent advances in the synthesis and catalytic applications of ligand-protected, atomically precise metal nanoclusters. Coord. Chem. Rev. 322, 1–29. doi:10.1016/j.ccr.2016.05.003
Foote, C. S. (1991). Definition of type I and type II photosensitized oxidation. Photochem. Photobiol. 54 (5), 659. doi:10.1111/j.1751-1097.1991.tb02071.x
Ghosh, A., Udayabhaskararao, T., and Pradeep, T. (2012). One-step route to luminescent Au18SG14 in the condensed phase and its closed shell molecular ions in the gas phase. J. Phys. Chem. Lett. 3 (15), 1997–2002. doi:10.1021/jz3007436
Green, T. D., and Knappenberger, K. L. (2012). Relaxation dynamics of Au25L18 nanoclusters studied by femtosecond time-resolved near infrared transient absorption spectroscopy. Nanoscale 4 (14), 4111–4118. doi:10.1039/c2nr31080e
Guo, R., and Murray, R. W. (2005). Substituent effects on redox potentials and optical gap energies of molecule-like Au38(SPhX)24 nanoparticles. J. Am. Chem. Soc. 127 (34), 12140–12143. doi:10.1021/ja053119m
Gurruchaga-Pereda, J., Martínez-Martínez, V., Rezabal, E., Lopez, X., Garino, C., Mancin, F., et al. (2019). Flavin bioorthogonal photocatalysis toward platinum substrates. ACS Catal. 10 (1), 187–196. doi:10.1021/acscatal.9b02863
Hakkinen, H. (2008). Atomic and electronic structure of gold clusters: Understanding flakes, cages and superatoms from simple concepts. Chem. Soc. Rev. 37 (9), 1847–1859. doi:10.1039/b717686b
Han, R., Zhao, M., Wang, Z., Liu, H., Zhu, S., Huang, L., et al. (2020). Super-efficient in vivo two-photon photodynamic therapy with a gold nanocluster as a type I photosensitizer. ACS Nano 14 (8), 9532–9544. doi:10.1021/acsnano.9b05169
Huang, T., and Murray, R. W. (2001). Visible luminescence of water-soluble monolayer-protected gold clusters. J. Phys. Chem. B 105 (50), 12498–12502. doi:10.1021/jp0041151
Jadzinsky, P. D., Calero, G., Ackerson, C. J., Bushnell, D. A., and Kornberg, R. D. (2007). Structure of a thiol monolayer-protected gold nanoparticle at 1.1 Å resolution. Science 318 (5849), 430–433. doi:10.1126/science.1148624
Jiang, Y., Yu, Y., Zhang, X., Weinert, M., Song, X., Ai, J., et al. (2021). N-heterocyclic carbene-stabilized ultrasmall gold nanoclusters in a metal-organic framework for photocatalytic CO2 reduction. Angew. Chem. Int. Ed. 60 (32), 17388–17393. doi:10.1002/anie.202105420
Jin, R. (2015). Atomically precise metal nanoclusters: Stable sizes and optical properties. Nanoscale 7 (5), 1549–1565. doi:10.1039/c4nr05794e
Jin, R., Zeng, C., Zhou, M., and Chen, Y. (2016). Atomically precise colloidal metal nanoclusters and nanoparticles: Fundamentals and opportunities. Chem. Rev. 116 (18), 10346–10413. doi:10.1021/acs.chemrev.5b00703
Kawasaki, H., Hamaguchi, K., Osaka, I., and Arakawa, R. (2011). ph-Dependent synthesis of pepsin-mediated gold nanoclusters with blue green and red fluorescent emission. Adv. Funct. Mat. 21 (18), 3508–3515. doi:10.1002/adfm.201100886
Kawasaki, H., Kumar, S., Li, G., Zeng, C., Kauffman, D. R., Yoshimoto, J., et al. (2014). Generation of singlet oxygen by photoexcited Au25(SR)18 clusters. Chem. Mat. 26 (9), 2777–2788. doi:10.1021/cm500260z
Kim, A., Zeng, C., Zhou, M., and Jin, R. (2017). Surface engineering of Au36(SR)24 nanoclusters for photoluminescence enhancement. Part. Part. Syst. Charact. 34 (8), 1600388–1600393. doi:10.1002/ppsc.201600388
Kovalev, D., and Fujii, M. (2005). Silicon nanocrystals: Photosensitizers for oxygen molecules. Adv. Mat. 17 (21), 2531–2544. doi:10.1002/adma.200500328
Kubo, R. (1962). Electronic properties of metallic fine particles. I. J. Phys. Soc. Jpn. 17 (6), 975–986. doi:10.1143/JPSJ.17.975
Kwak, K., Thanthirige, V. D., Pyo, K., Lee, D., and Ramakrishna, G. (2017). Energy gap law for exciton dynamics in gold cluster molecules. J. Phys. Chem. Lett. 8 (19), 4898–4905. doi:10.1021/acs.jpclett.7b01892
Lee, D., Donkers, R. L., Wang, G., Harper, A. S., and Murray, R. W. (2004). Electrochemistry and optical absorbance and luminescence of molecule-like Au38 nanoparticles. J. Am. Chem. Soc. 126 (19), 6193–6199. doi:10.1021/ja049605b
Lee, J., Shim, H. S., Lee, M., Song, J. K., and Lee, D. (2011). Size-controlled electron transfer and photocatalytic activity of ZnO–Au nanoparticle composites. J. Phys. Chem. Lett. 2 (22), 2840–2845. doi:10.1021/jz2013352
Li, G., Abroshan, H., Liu, C., Zhuo, S., Li, Z., Xie, Y., et al. (2016). Tailoring the electronic and catalytic properties of Au25 nanoclusters via ligand engineering. ACS Nano 10 (8), 7998–8005. doi:10.1021/acsnano.6b03964
Li, G., and Jin, R. (2013). Atomically precise gold nanoclusters as new model catalysts. Acc. Chem. Res. 46 (8), 1749–1758. doi:10.1021/ar300213z
Li, G., Qian, H., and Jin, R. (2012). Gold nanocluster-catalyzed selective oxidation of sulfide to sulfoxide. Nanoscale 4 (21), 6714–6717. doi:10.1039/c2nr32171h
Li, L., Li, Z., Zhang, H., Zhang, S., Majeed, I., Tan, B., et al. (2013). Effect of polymer ligand structures on fluorescence of gold clusters prepared by photoreduction. Nanoscale 5 (5), 1986–1992. doi:10.1039/c2nr33693f
Li, X., Kwon, N., Guo, T., Liu, Z., and Yoon, J. (2018). Innovative strategies for hypoxic-tumor photodynamic therapy. Angew. Chem. Int. Ed. 57 (36), 11522–11531. doi:10.1002/anie.201805138
Li, Z., and Grant, K. B. (2016). DNA photo-cleaving agents in the far-red to near-infrared range – A review. RSC Adv. 6 (29), 24617–24634. doi:10.1039/c5ra28102d
Li, Z., Liu, C., Abroshan, H., Kauffman, D. R., and Li, G. (2017). Au38S2(SAdm)20 photocatalyst for one-step selective aerobic oxidations. ACS Catal. 7 (5), 3368–3374. doi:10.1021/acscatal.7b00239
Lin, Z., Goswami, N., Xue, T., Chai, O. J. H., Xu, H., Liu, Y., et al. (2021). Engineering metal nanoclusters for targeted therapeutics: From targeting strategies to therapeutic applications. Adv. Funct. Mat. 31 (51), 2105662–2105680. doi:10.1002/adfm.202105662
Link, S., Beeby, A., FitzGerald, S., El-Sayed, M. A., Schaaff, T. G., Whetten, R. L., et al. (2002). Visible to infrared luminescence from a 28-atom gold cluster. J. Phys. Chem. B 106 (13), 3410–3415. doi:10.1021/jp014259v
Liu, C., Yan, C., Lin, J., Yu, C., Huang, J., Li, G., et al. (2015). One-pot synthesis of Au144(SCH2Ph)60 nanoclusters and their catalytic application. J. Mat. Chem. A Mat. 3 (40), 20167–20173. doi:10.1039/c5ta05747g
Liu, R., Cheng, D., Zhou, Q., Niu, F., and Hu, K. (2021). Gold nanoclusters perform enzyme-like photocatalysis for prodrug activation. ACS Appl. Nano Mat. 4 (2), 990–994. doi:10.1021/acsanm.1c00014
Liu, Y., Jiang, Y., Zhang, M., Tang, Z., He, M., Bu, W., et al. (2018). Modulating hypoxia via nanomaterials chemistry for efficient treatment of solid tumors. Acc. Chem. Res. 51 (10), 2502–2511. doi:10.1021/acs.accounts.8b00214
Long, R., Mao, K., Ye, X., Yan, W., Huang, Y., Wang, J., et al. (2013). Surface facet of palladium nanocrystals: A key parameter to the activation of molecular oxygen for organic catalysis and cancer treatment. J. Am. Chem. Soc. 135 (8), 3200–3207. doi:10.1021/ja311739v
Luo, Z., Yuan, X., Yu, Y., Zhang, Q., Leong, D. T., Lee, J. Y., et al. (2012). From aggregation-induced emission of Au(I)-thiolate complexes to ultrabright Au(0)@Au(I)-thiolate core-shell nanoclusters. J. Am. Chem. Soc. 134 (40), 16662–16670. doi:10.1021/ja306199p
Ma, J., Chen, J.-Y., Idowu, M., and Nyokong, T. (2008). Generation of singlet oxygen via the composites of water-soluble thiol-capped CdTe quantum dots-sulfonated aluminum phthalocyanines. J. Phys. Chem. B 112 (15), 4465–4469. doi:10.1021/jp711537j
Maity, S., Bain, D., and Patra, A. (2019). An overview on the current understanding of the photophysical properties of metal nanoclusters and their potential applications. Nanoscale 11 (47), 22685–22723. doi:10.1039/c9nr07963g
Miller, S. A., Womick, J. M., Parker, J. F., Murray, R. W., and Moran, A. M. (2009). Femtosecond relaxation dynamics of Au25L18− monolayer-protected clusters. J. Phys. Chem. C 113 (22), 9440–9444. doi:10.1021/jp9025046
Nair, L. V., Nazeer, S. S., Jayasree, R. S., and Ajayaghosh, A. (2015). Fluorescence imaging assisted photodynamic therapy using photosensitizer-linked gold quantum clusters. ACS Nano 9 (6), 5825–5832. doi:10.1021/acsnano.5b00406
Negishi, Y., Chaki, N. K., Shichibu, Y., Whetten, R. L., and Tsukuda, T. (2007). Origin of magic stability of thiolated gold clusters: A case study on Au25(SC6H13)18. J. Am. Chem. Soc. 129 (37), 11322–11323. doi:10.1021/ja073580+
Negishi, Y., Matsuura, Y., Tomizawa, R., Kurashige, W., Niihori, Y., Takayama, T., et al. (2015). Controlled loading of small aun clusters (n = 10–39) onto BaLa4Ti4O15 photocatalysts: Toward an understanding of size effect of cocatalyst on water-splitting photocatalytic activity. J. Phys. Chem. C 119 (20), 11224–11232. doi:10.1021/jp5122432
Negishi, Y., Mizuno, M., Hirayama, M., Omatoi, M., Takayama, T., Iwase, A., et al. (2013). Enhanced photocatalytic water splitting by BaLa4Ti4O15 loaded with ∼1 nm gold nanoclusters using glutathione-protected Au25 clusters. Nanoscale 5 (16), 7188–7192. doi:10.1039/c3nr01888a
Negishi, Y., Nobusada, K., and Tsukuda, T. (2005). Glutathione-protected gold clusters revisited: Bridging the gap between gold(I)−thiolate complexes and thiolate-protected gold nanocrystals. J. Am. Chem. Soc. 127 (14), 5261–5270. doi:10.1021/ja042218h
Negishi, Y., Takasugi, Y., Sato, S., Yao, H., Kimura, K., Tsukuda, T., et al. (2004). Magic-numbered aun clusters protected by glutathione monolayers (n = 18, 21, 25, 28, 32, 39): isolation and spectroscopic characterization. J. Am. Chem. Soc. 126 (21), 6518–6519. doi:10.1021/ja0483589
Oliveira, B. L., Stenton, B. J., Unnikrishnan, V. B., de Almeida, C. R., Conde, J., Negrao, M., et al. (2020). Platinum-triggered bond-cleavage of pentynoyl amide and N-propargyl handles for drug-activation. J. Am. Chem. Soc. 142 (24), 10869–10880. doi:10.1021/jacs.0c01622
Pyo, K., Thanthirige, V. D., Kwak, K., Pandurangan, P., Ramakrishna, G., Lee, D., et al. (2015). Ultrabright luminescence from gold nanoclusters: Rigidifying the Au(I)-thiolate shell. J. Am. Chem. Soc. 137 (25), 8244–8250. doi:10.1021/jacs.5b04210
Qian, H., and Jin, R. (2011). Ambient synthesis of Au144(SR)60 nanoclusters in methanol. Chem. Mat. 23 (8), 2209–2217. doi:10.1021/cm200143s
Qian, H., Sfeir, M. Y., and Jin, R. (2010). Ultrafast relaxation dynamics of [Au25(SR)18]q nanoclusters: Effects of charge state. J. Phys. Chem. C 114 (47), 19935–19940. doi:10.1021/jp107915x
Roduner, E. (2006). Size matters: Why nanomaterials are different. Chem. Soc. Rev. 35 (7), 583–592. doi:10.1039/b502142c
Samia, A. C. S., Chen, X., and Burda, C. (2003). Semiconductor quantum dots for photodynamic therapy. J. Am. Chem. Soc. 125 (51), 15736–15737. doi:10.1021/ja0386905
Searle, P. F., Chen, M.-J., Hu, L., Race, P. R., Lovering, A. L., Grove, J. I., et al. (2004). Nitroreductase: A prodrug-activating enzyme for cancer gene therapy. Clin. Exp. Pharmacol. Physiol. 31 (11), 811–816. doi:10.1111/j.1440-1681.2004.04085.x
Shichibu, Y., Negishi, Y., Watanabe, T., Chaki, N. K., Kawaguchi, H., Tsukuda, T., et al. (2007). Biicosahedral gold clusters [Au25(PPh3)10(SCnH2n+1)5Cl2]2+ (n = 2−18): A stepping stone to cluster-assembled materials. J. Phys. Chem. C 111 (22), 7845–7847. doi:10.1021/jp073101t
Silverstein, T. P. (2012). Marcus theory: Thermodynamics can control the kinetics of electron transfer reactions. J. Chem. Educ. 89 (9), 1159–1167. doi:10.1021/ed1007712
Soldevila-Barreda, J. J., and Metzler-Nolte, N. (2019). Intracellular catalysis with selected metal complexes and metallic nanoparticles: Advances toward the development of catalytic metallodrugs. Chem. Rev. 119 (2), 829–869. doi:10.1021/acs.chemrev.8b00493
Song, X. R., Goswami, N., Yang, H. H., and Xie, J. (2016). Functionalization of metal nanoclusters for biomedical applications. Analyst 141 (11), 3126–3140. doi:10.1039/c6an00773b
Stamplecoskie, K. G., Chen, Y.-S., and Kamat, P. V. (2014). Excited-state behavior of luminescent glutathione-protected gold clusters. J. Phys. Chem. C 118 (2), 1370–1376. doi:10.1021/jp410856h
Stamplecoskie, K. G., and Kamat, P. V. (2014). Size-dependent excited state behavior of glutathione-capped gold clusters and their light-harvesting capacity. J. Am. Chem. Soc. 136 (31), 11093–11099. doi:10.1021/ja505361n
Tlahuice-Flores, A., Whetten, R. L., and Jose-Yacaman, M. (2013). Ligand effects on the structure and the electronic optical properties of anionic Au25(SR)18 clusters. J. Phys. Chem. C 117 (40), 20867–20875. doi:10.1021/jp407150t
Vankayala, R., Kuo, C.-L., Nuthalapati, K., Chiang, C.-S., and Hwang, K. C. (2015). Nucleus-targeting gold nanoclusters for simultaneous in vivo fluorescence imaging, gene delivery, and NIR-light activated photodynamic therapy. Adv. Funct. Mat. 25 (37), 5934–5945. doi:10.1002/adfm.201502650
Vankayala, R., Kuo, C. L., Sagadevan, A., Chen, P. H., Chiang, C. S., Hwang, K. C., et al. (2013). Morphology dependent photosensitization and formation of singlet oxygen (1Δg) by gold and silver nanoparticles and its application in cancer treatment. J. Mat. Chem. B 1 (35), 4379–4387. doi:10.1039/c3tb20806k
Vankayala, R., Sagadevan, A., Vijayaraghavan, P., Kuo, C. L., and Hwang, K. C. (2011). Metal nanoparticles sensitize the formation of singlet oxygen. Angew. Chem. Int. Ed. 50 (45), 10640–10644. doi:10.1002/anie.201105236
Varnavski, O., Ispasoiu, R. G., Balogh, L., Tomalia, D., and Goodson, T. (2001). Ultrafast time-resolved photoluminescence from novel metal–dendrimer nanocomposites. J. Chem. Phys. 114 (5), 1962–1965. doi:10.1063/1.1344231
Varnavski, O., Ramakrishna, G., Kim, J., Lee, D., and Goodson, T. (2010). Optically excited acoustic vibrations in quantum-sized monolayer-protected gold clusters. ACS Nano 4 (6), 3406–3412. doi:10.1021/nn1003524
Venzo, A., Antonello, S., Gascón, J. A., Guryanov, I., Leapman, R. D., Perera, N. V., et al. (2011). Effect of the charge state (z = −1, 0, +1) on the nuclear magnetic resonance of monodisperse Au25[S(CH2)2Ph]18z clusters. Anal. Chem. 83 (16), 6355–6362. doi:10.1021/ac2012653
Vong, K., Yamamoto, T., Chang, T. C., and Tanaka, K. (2020). Bioorthogonal release of anticancer drugs via gold-triggered 2-alkynylbenzamide cyclization. Chem. Sci. 11 (40), 10928–10933. doi:10.1039/d0sc04329j
Walter, M., Akola, J., Lopez-Acevedo, O., Jadzinsky, P. D., Calero, G., Ackerson, C. J., et al. (2008). A unified view of ligand-protected gold clusters as superatom complexes. Proc. Natl. Acad. Sci. U. S. A. 105 (27), 9157–9162. doi:10.1073/pnas.0801001105
Wang, G., Guo, R., Kalyuzhny, G., Choi, J.-P., and Murray, R. W. (2006). NIR luminescence intensities increase linearly with proportion of polar thiolate ligands in protecting monolayers of Au38 and Au140 quantum dots. J. Phys. Chem. B 110 (41), 20282–20289. doi:10.1021/jp0640528
Wang, S., Tang, L., Cai, B., Yin, Z., Li, Y., Xiong, L., et al. (2022). Ligand modification of Au25 nanoclusters for near-infrared photocatalytic oxidative functionalization. J. Am. Chem. Soc. 144 (9), 3787–3792. doi:10.1021/jacs.2c01570
Weerawardene, K. L. D. M., and Aikens, C. M. (2016). Theoretical insights into the origin of photoluminescence of Au25(SR)18– nanoparticles. J. Am. Chem. Soc. 138 (35), 11202–11210. doi:10.1021/jacs.6b05293
Weng, B., Lu, K. Q., Tang, Z., Chen, H. M., and Xu, Y. J. (2018). Stabilizing ultrasmall Au clusters for enhanced photoredox catalysis. Nat. Commun. 9, 1543–1553. doi:10.1038/s41467-018-04020-2
Wilson, W. R., and Hay, M. P. (2011). Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11 (6), 393–410. doi:10.1038/nrc3064
Witzel, S., Hashmi, A. S. K., and Xie, J. (2021). Light in gold catalysis. Chem. Rev. 121 (14), 8868–8925. doi:10.1021/acs.chemrev.0c00841
Wood, D. M., and Ashcroft, N. W. (1982). Quantum size effects in the optical properties of small metallic particles. Phys. Rev. B 25 (10), 6255–6274. doi:10.1103/PhysRevB.25.6255
Wu, Z., and Jin, R. (2010). On the ligand’s role in the fluorescence of gold nanoclusters. Nano Lett. 10 (7), 2568–2573. doi:10.1021/nl101225f
Xiao, F. X., Hung, S. F., Miao, J., Wang, H. Y., Yang, H., Liu, B., et al. (2015). Metal-cluster-decorated TiO2 nanotube arrays: A composite heterostructure toward versatile photocatalytic and photoelectrochemical applications. Small 11 (5), 554–567. doi:10.1002/smll.201401919
Xu, H., and Suslick, K. S. (2010). Water-soluble fluorescent silver nanoclusters. Adv. Mat. 22 (10), 1078–1082. doi:10.1002/adma.200904199
Yang, J., Wang, F., Yuan, H., Zhang, L., Jiang, Y., Zhang, X., et al. (2019). Recent advances in ultra-small fluorescent Au nanoclusters toward oncological research. Nanoscale 11 (39), 17967–17980. doi:10.1039/c9nr04301b
Yanto, Y., Hall, M., and Bommarius, A. S. (2010). Nitroreductase from Salmonella typhimurium: Characterization and catalytic activity. Org. Biomol. Chem. 8 (8), 1826. doi:10.1039/B926274A
Yau, S. H., Varnavski, O., Gilbertson, J. D., Chandler, B., Ramakrishna, G., Goodson, T., et al. (2010). Ultrafast optical study of small gold monolayer protected clusters: A closer look at emission. J. Phys. Chem. C 114 (38), 15979–15985. doi:10.1021/jp101420g
Yu, C., Li, G., Kumar, S., Kawasaki, H., and Jin, R. (2013). Stable Au25(SR)18/TiO2 composite nanostructure with enhanced visible light photocatalytic activity. J. Phys. Chem. Lett. 4 (17), 2847–2852. doi:10.1021/jz401447w
Yu, Y., Chen, X., Yao, Q., Yu, Y., Yan, N., Xie, J., et al. (2013). Scalable and precise synthesis of thiolated Au10–12, Au15, Au18, and Au25 nanoclusters via pH controlled CO reduction. Chem. Mat. 25 (6), 946–952. doi:10.1021/cm304098x
Yu, Y., Li, J., Chen, T., Tan, Y. N., and Xie, J. (2015). Decoupling the CO-reduction protocol to generate luminescent Au22(SR)18 nanocluster. J. Phys. Chem. C 119 (20), 10910–10918. doi:10.1021/jp510829d
Yu, Y., Luo, Z., Chevrier, D. M., Leong, D. T., Zhang, P., Jiang, D.-e., et al. (2014). Identification of a highly luminescent Au22(SG)18 nanocluster. J. Am. Chem. Soc. 136 (4), 1246–1249. doi:10.1021/ja411643u
Yuan, X., Goswami, N., Chen, W., Yao, Q., and Xie, J. (2016). Insights into the effect of surface ligands on the optical properties of thiolated Au25 nanoclusters. Chem. Commun. 52 (30), 5234–5237. doi:10.1039/c6cc00857g
Zhang, J., and Liu, J. (2020). Light-activated nanozymes: Catalytic mechanisms and applications. Nanoscale 12 (5), 2914–2923. doi:10.1039/c9nr10822j
Zhang, J., Zhou, Y., Zheng, K., Abroshan, H., Kauffman, D. R., Sun, J., et al. (2018). Diphosphine-induced chiral propeller arrangement of gold nanoclusters for singlet oxygen photogeneration. Nano Res. 11 (11), 5787–5798. doi:10.1007/s12274-017-1935-2
Zhang, X.-D., Wu, D., Shen, X., Liu, P.-X., Fan, F.-Y., Fan, S.-J., et al. (2012). In vivo renal clearance, biodistribution, toxicity of gold nanoclusters. Biomaterials 33 (18), 4628–4638. doi:10.1016/j.biomaterials.2012.03.020
Zheng, J., Zhang, C., and Dickson, R. M. (2004). Highly fluorescent, water-soluble, size-tunable gold quantum dots. Phys. Rev. Lett. 93 (7), 077402–077405. doi:10.1103/PhysRevLett.93.077402
Zheng, Y., Wu, J., Jiang, H., and Wang, X. (2021). Gold nanoclusters for theranostic applications. Coord. Chem. Rev. 431, 213689–213715. doi:10.1016/j.ccr.2020.213689
Zhou, M., Du, X., Wang, H., and Jin, R. (2021). The critical number of gold atoms for a metallic state nanocluster: Resolving a decades-long question. ACS Nano 15 (9), 13980–13992. doi:10.1021/acsnano.1c04705
Zhou, M., Higaki, T., Hu, G., Sfeir Matthew, Y., Chen, Y., Jiang, D.-e., et al. (2019). Three-orders-of-magnitude variation of carrier lifetimes with crystal phase of gold nanoclusters. Science 364 (6437), 279–282. doi:10.1126/science.aaw8007
Zhou, M., and Song, Y. (2021). Origins of visible and near-infrared emissions in [Au25(SR)18]− nanoclusters. J. Phys. Chem. Lett. 12 (5), 1514–1519. doi:10.1021/acs.jpclett.1c00120
Zhou, M., Tian, S., Zeng, C., Sfeir, M. Y., Wu, Z., Jin, R., et al. (2017a). Ultrafast relaxation dynamics of Au38(SC2H4Ph)24 nanoclusters and effects of structural isomerism. J. Phys. Chem. C 121 (20), 10686–10693. doi:10.1021/acs.jpcc.6b10360
Zhou, M., Zeng, C., Chen, Y., Zhao, S., Sfeir, M. Y., Zhu, M., et al. (2016). Evolution from the plasmon to exciton state in ligand-protected atomically precise gold nanoparticles. Nat. Commun. 7, 13240–13246. doi:10.1038/ncomms13240
Zhou, M., Zeng, C., Sfeir, M. Y., Cotlet, M., Iida, K., Nobusada, K., et al. (2017b). Evolution of excited-state dynamics in periodic Au28, Au36, Au44, and Au52 nanoclusters. J. Phys. Chem. Lett. 8 (17), 4023–4030. doi:10.1021/acs.jpclett.7b01597
Zhou, S., Duan, Y., Wang, F., and Wang, C. (2017). Fluorescent Au nanoclusters stabilized by silane: Facile synthesis, color-tunability and photocatalytic properties. Nanoscale 9 (15), 4981–4988. doi:10.1039/c7nr01052d
Zhu, H., Goswami, N., Yao, Q., Chen, T., Liu, Y., Xu, Q., et al. (2018). Cyclodextrin–gold nanocluster decorated TiO2 enhances photocatalytic decomposition of organic pollutants. J. Mat. Chem. A Mat. 6 (3), 1102–1108. doi:10.1039/c7ta09443d
Zhu, M., Aikens, C. M., Hollander, F. J., Schatz, G. C., and Jin, R. (2008a). Correlating the crystal structure of a thiol-protected Au25 cluster and optical properties. J. Am. Chem. Soc. 130 (18), 5883–5885. doi:10.1021/ja801173r
Keywords: gold nanoclusters, photophysics, photocatalysis, solar energy conversion, phototherapy
Citation: Cheng D, Liu R and Hu K (2022) Gold nanoclusters: Photophysical properties and photocatalytic applications. Front. Chem. 10:958626. doi: 10.3389/fchem.2022.958626
Received: 31 May 2022; Accepted: 28 June 2022;
Published: 19 July 2022.
Edited by:
Yulian He, Shanghai Jiao Tong University, ChinaReviewed by:
Meng Zhou, University of Science and Technology of China, ChinaCopyright © 2022 Cheng, Liu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Hu, a2h1QGZ1ZGFuLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.