
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 14 July 2022
Sec. Theoretical and Computational Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.920661
This article is part of the Research TopicRecent Advances, New Perspectives and Applications of Conceptual Density Functional TheoryView all 7 articles
 Maricruz Rangel-Galván1
Maricruz Rangel-Galván1 María Eugenia Castro2*
María Eugenia Castro2* Jose Manuel Perez-Aguilar1
Jose Manuel Perez-Aguilar1 Norma A. Caballero3
Norma A. Caballero3 Francisco J. Melendez1*
Francisco J. Melendez1*The anandamide is a relevant ligand due to its capacity of interacting with several proteins, including the T-type calcium channels, which play an important role in neuropathic pain and depression disorders. Hence, a detailed characterization of the chemical properties and conformational stability of anandamide may provide valuable information to understand its behavior in a biological context. Herein, conceptual DFT and QTAIM analyses were performed to theoretically characterize the chemical reactivity properties and the structural stability of conformations of anandamide, using the BP86/cc-pVTZ level of theory. Global reactivity description, based on conceptual DFT, indicates that the hardness increases and the electrophilicity index decreases for both, the hairpin and U-shape conformers relative to the extended conformers. Also, an increase in the chemical potential value and a decrease in the electronegativity and the electrophilicity index is observed in the ethanolamide open ring conformers in comparison with the corresponding closed ring structures. In addition, regarding the characterization of local reactivity descriptors, the maximum values of the Fukui and Parr functions indicate that the most probable location for a nucleophilic attack is either the hydroxyl oxygen located in the ethanolamide closed ring conformers or the carbonyl oxygen present in the open ring conformers. The most probable location for an electrophilic attack is in the alkyl double bond region in all anandamide conformers. According to the QTAIM results, the intramolecular hydrogen bond formation stabilizing the structure of anandamide has interaction energy values for the closed ring conformations of 12.33–12.46 kcal mol−1, indicating a strong interaction. Lastly, molecular docking calculations determined that a region in the pore, denominate as pore-blocking, is a probable site for the interaction of anandamide with the human Cav3.2 isoform of the T-type calcium channel family. The pore-blocking site contains hydrophobic residues where the non-polar part in the final alkyl region of anandamide established mainly alkyl-alkyl interactions, while the polar part (the ethanolamide group) interacts with the polar residue S900. The information based on conceptual DFT presented may aid in the design of drugs with similar chemical characteristics as those identified in anandamide so as to bind anandamide-interacting proteins, including the T-type calcium channels.
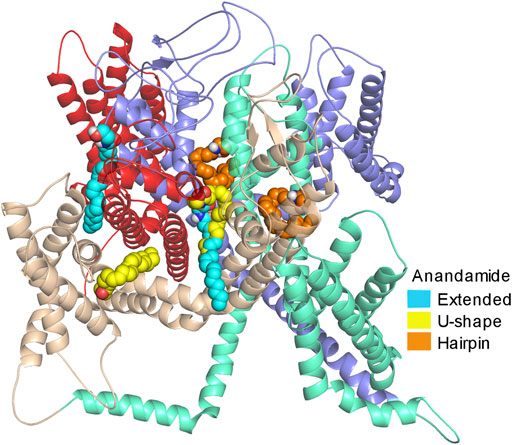
GRAPHICAL ABSTRACT
Anandamide is a fatty acid neurotransmitter that is synthesized enzymatically in the brain from arachidonic acid (AA) and ethanolamide substrates (Devane and Axelrod, 1994); however, other synthetic pathways involving the N-arachidonoyl phosphatidylethanolamine (PE) precursor and the phosphodiesterase enzyme, have been also proposed (Sugiura et al., 2002). Although anandamide is an endogenous endocannabinoid ligand known to bind mainly to the cannabinoid receptors, it is also capable of directly modulating a variety of ion channels, such as potassium channels, sodium channels, TRPV1 (Transient Receptor Potential Vanilloid type 1), and the T-type calcium channels (Van Der Stelt and Di Marzo, 2005; Oz, 2006). Along these lines, it has been proved that blocking both, the T-type calcium channel and the cannabinoid receptors is an important therapeutic strategy for treating neuropathic and inflammatory pain (You et al., 2011; Gadotti et al., 2013; Bladen et al., 2015). Moreover, the T-type calcium channels and the CB1/CB2 cannabinoid receptors are involved in disorders associated with neuropathic pain, mental pain, suicidal behavior, and depression (Pacher et al., 2006; Todorovic and Jevtovic-Todorovic, 2013; Tibbs et al., 2016; Colino et al., 2018; Snutch and Zamponi, 2018; Yang et al., 2018). Among the three isoforms that constitute the T-type calcium channels (Cav3.1, Cav3.2, and Cav3.3), the Cav3.2 contributes significantly to the nociceptive pathway which is responsible for the physiological and pathological mechanism of pain transmission. It was demonstrated by electrophysiological experiments that anandamide directly inhibits the T-type calcium channels during neural activities with a preference for the Cav3.2 isoform displaying IC50 values for the Cav3.1, Cav3.2, and Cav3.3 channels of 4.15 µM, 330 nM, and 1.10 µM, respectively (Chemin et al., 2001). Using electrophysiology recordings, it was found that the hydroxyl group and the alkyl chain from anandamide play important roles in the inhibition mechanism of the T-type calcium channel by anandamide. In detail, it was found that the chain length and the presence, position, and all-cis isomerism of the alkyl chain double bonds, are all important factors for the degree of anandamide inhibition. Interestingly, the inhibition can occur even though the alkyl chain contains either 18 or 22 carbons instead of 20 carbons. Also, it was observed that a decrease in the degree of unsaturation in anandamide reduces its blocking effect, with a fully saturated alkyl chain displaying negligible inhibitory effect. The inhibition increases when double bonds are near the carboxyl group and have no effect when a trans-configuration and triple bonds are considered (Chemin et al., 2007).
Experimental X-ray structures found the alternated form of anandamide (cis-trans-cis-trans) and all-trans anandamide in interaction with its intracellular transporter, the fatty acid binding protein FABP5 in mouse (mFABP5) and human (hFABP5), respectively (Sanson et al., 2014). Also, both isomer molecules the synthesized all-trans anandamide and the natural cis-anandamide are equally good substrates for fatty acid amide hydrolase FAAH enzyme (Ferreri et al., 2008). Nevertheless, it has been observed that endogenous trans and cis-trans of AA isomerization originate from diet and by the action of the nitrogen dioxide radicals (NO2) (Zghibeh et al., 2004). This could indicate that the alternated (cis-trans-cis-trans) and all-trans anandamide forms may be present in certain conditions but the most relevant form during receptor interaction is the all-cis anandamide.
Regarding the interaction of anandamide with the cannabinoid receptors, a mutagenesis study demonstrated the importance of the amino acid F3.25 (following B&W nomenclature) for binding anandamide in the binding site of the CB1 receptor while a computational docking study showed interactions between anandamide, in a hairpin conformation, with the same receptor (McAllister et al., 2003). In a crystallographic study where the structure of the CB1 receptor was solved, the authors used the X-ray structure to dock various agonist molecules, including anandamide and found that the molecule prefers a hairpin conformation in the binding site of the receptor (Hua et al., 2016). Although there is significant information regarding the interaction of anandamide and various proteins, there is no information regarding the interaction of this important compound with the T-type calcium channels. Herein we used computational methods to characterize the reactivity of anandamide in the ligand-receptor context, evaluate its structural stability, and study the interaction of anandamide with the structure of the Cav3.2 channel. Conceptual DFT is used for the chemical reactivity interpretation by global and local reactivity descriptors, which are used to identify the possible interacting regions of anandamide at the Cav3.2 channel. QTAIM analyses were performed to theoretically characterize the structural stability of the preferred conformations of anandamide (Rangel-Galván et al., 2022a). These calculations were made using the BP86/cc-pVTZ level of theory. Finally, molecular docking calculations are used to characterize the main amino acids interacting in the Cav3.2/anandamide complex.
The conceptual DFT approach (Domingo et al., 2016; Frau and Glossman-Mitnik, 2017; Frau and Glossman-Mitnik, 2018) was used to evaluate the global reactivity descriptors, such as chemical potential (μ), electronegativity (χ), hardness (η), softness (s), and electrophilicity index (ω), from the Frontier molecular orbitals energies, EHOMO and ELUMO, using the following equations: μ = (EHOMO + ELUMO) ⁄ 2; χ = −(EHOMO + ELUMO) ⁄ 2; η = ELUMO−EHOMO; s = 1 ⁄ η, ω = μ2 ⁄ 2η. In addition, the local reactivity descriptors, such as the Fukui functions
Topological parameters, based on the QTAIM approach (Matta and Boyd, 2007; Hernández-Paredes et al., 2017; Bayat and Fattahi, 2019), were calculated, such as electronic density,
Molecular docking calculations were carried out for the Cav3.2 calcium channel and the conformers of anandamide. The rigid blind docking was performed using the AutoDockVina (version 1.1.2) program (Trott and Olson, 2010). AutoDockVina uses a genetic algorithm as a searching method and the binding free energy parameter (−ΔG) as the scoring function. The binding free energy is calculated through the equation:
Six minimum energy structures for anandamide were selected for the conceptual DFT analysis: extended with closed EA ring (Eclosed), extended with open EA ring (Eopen), U-shape with closed EA ring (Uclosed), U-shape with open EA ring (Uopen), hairpin with closed EA ring (Hclosed), and hairpin with open EA ring (Hopen). Global reactivity descriptors for the six conformers of anandamide Eclosed, Eopen, Uclosed, Uopen, Hclosed, and Hopen, were evaluated: chemical potential (μ), electronegativity (χ), hardness (η), softness (s), and electrophilicity index (ω), according to conceptual DFT approach (Domingo et al., 2016; Frau and Glossman-Mitnik, 2017). Table 1 summarized the HOMO and LUMO energies and the global reactivity descriptors for the conformers of anandamide.
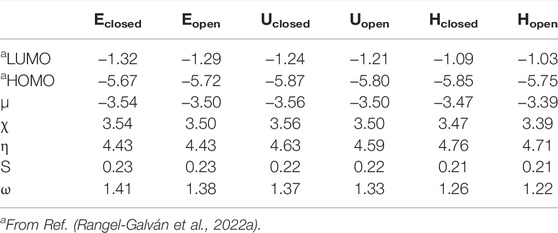
TABLE 1. HOMO and LUMO energies and global reactivity descriptors (eV) of the conformers of anandamide obtained at the BP86/cc-pVTZ level of theory in chloroform.
For the Eclosed/Eopen, Uclosed/Uopen, and Hclosed/Hopen shapes of anandamide, in Table 1 it is observed that there are no significant changes when anandamide changes the curvature of this structure. Some slight differences are found in hardness, softness, and the electrophilicity index. The hardness increases a 4.6/3.5 and 7.4/6.3% for Uclosed/Uopen and Hclosed/Hopen shapes, respectively, relative to the Eclosed/Eopen conformers. The softness suffers a decrement keeping the same proportion. Finally, a decrease of the electrophilicity index of 3.3/3.6 and 10.6/12.0% is observed for Uclosed/Uopen and Hclosed/Hopen shapes, respectively, with respect to the Eclosed/Eopen conformers. The effect of the ethanolamide (EA) open ring conformers in comparison with the corresponding closed shapes is seen in slight differences, an increase in chemical potential, and a decrease in electronegativity and electrophilicity index. For example, for the electrophilicity index, the decrease represents a 2, 2.3, and 3.5% for Eopen, Uopen, and Hopen with respect to their corresponding closed shapes.
In addition to anandamide as a dual ligand for the T-type calcium channels and the cannabinoid receptors, the NMP compounds have been shown to be capable of binding both proteins (You et al., 2011; Gadotti et al., 2013; Bladen et al., 2015), which is relevant in the treatment of pain disorders.
Taking into account the conceptual DFT analysis previously executed for the NMP compounds, the global reactivity descriptor that changes the most when anandamide and NMP compounds are compared (NMP-7 and Hclosed shape for representation), is the hardness of 51%, from 4.76 eV in the Hclosed conformation of anandamide and 3.14 eV in the NMP-7 ligand (Rangel-Galván et al., 2022b). Anandamide has a higher resistance to changing electronic distribution compared to the NMP compounds (Rangel-Galván et al., 2022b). It is worth mentioning that significant changes in the hardness index have been observed when a ligand-receptor interaction process is modeled (Aliste, 2000). The electrophilicity index is another uneven descriptor; a higher value corresponds to the NMP compounds with respect to anandamide conformers with a difference of 37.6%. The electrophilicity index results are well correlated to the receptor affinity properties and biological activity (Parthasarathi et al., 2004).
Respect the local reactivity descriptors, Table 2 shows the condensed Fukui functions,
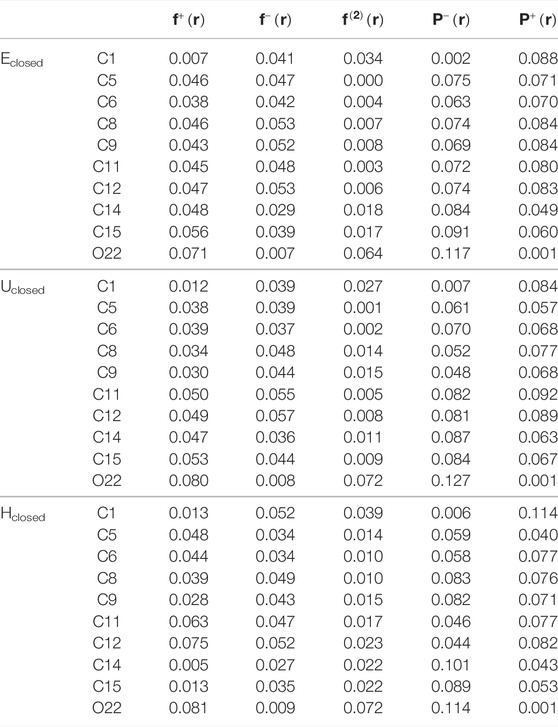
TABLE 2. Fukui functions,
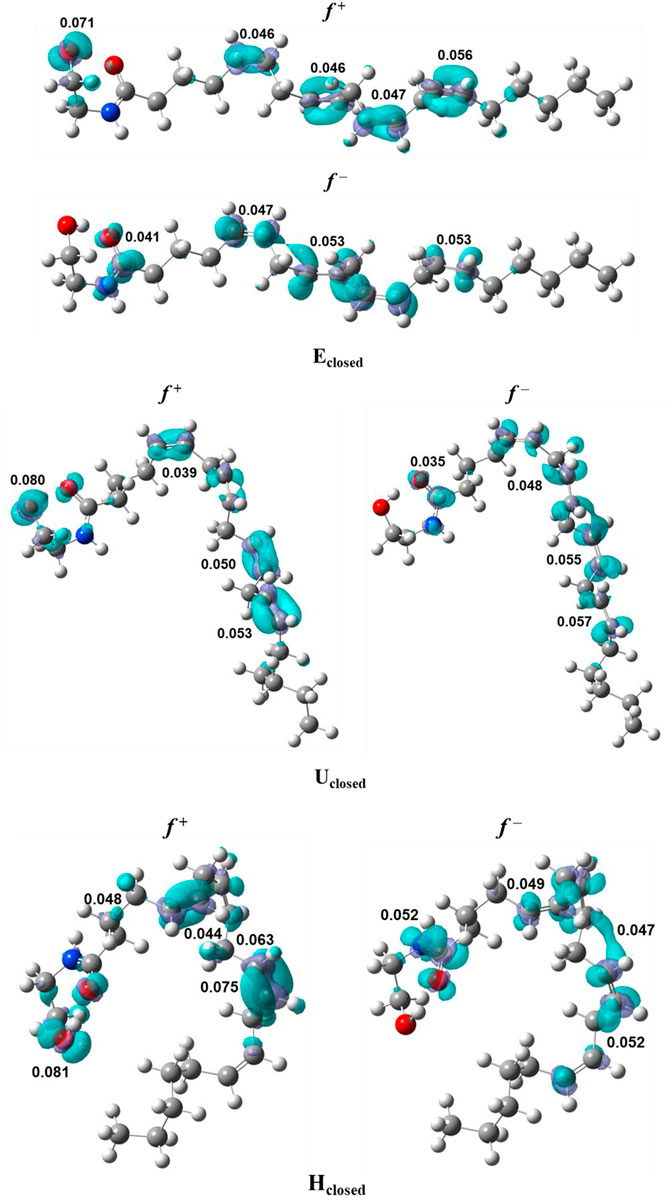
FIGURE 1. Fukui functions, f + and f –, of the conformers of anandamide: Eclosed, Uclosed, and Hclosed, obtained at the BP86/cc-pVTZ level of theory in chloroform.
The QTAIM analysis was focused to characterize the intramolecular hydrogen bond of the closed ring formed in the ethanolamide group in the conformers Eclosed, Uclosed, and Hclosed of anandamide. This interaction of the hydrogen bond generates a ring structure of seven atoms in the EA group. The stability of the conformers with a closed ring of anandamide by the hydrogen bond formation effect is analyzed by means of interaction energy for each closed ring anandamide conformer from electronic density obtained from the BP86/cc-pVTZ level of theoretical calculations in chloroform. Table 3 and Supplementary Table S2 show the values of electronic density,
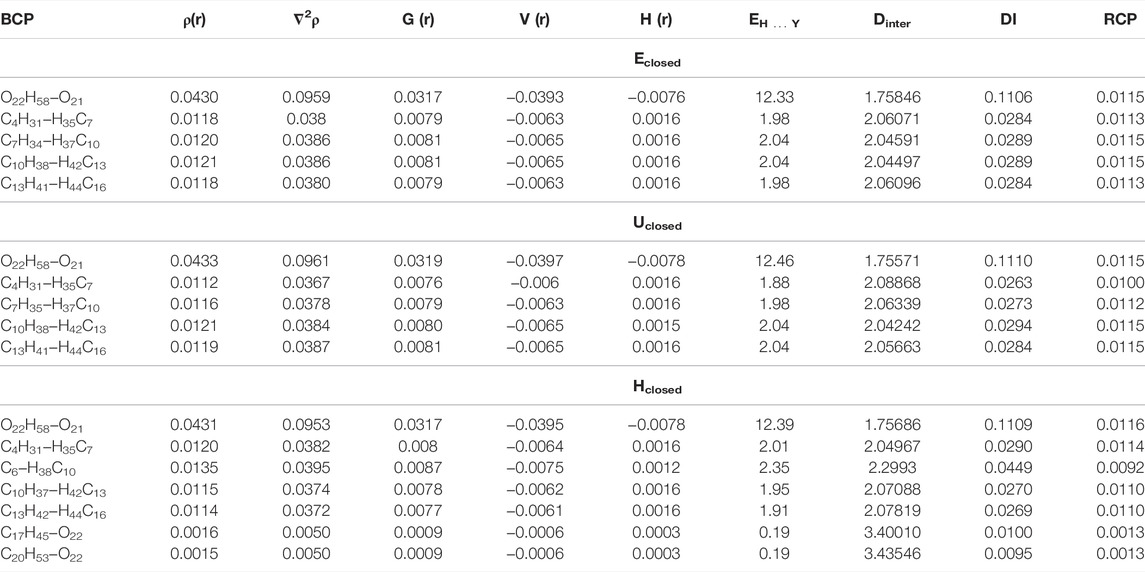
TABLE 3. Topological parameters (a.u.), EH … Y (kcal mol−1), and interatomic distances (Dinter, Å) of the conformers of anandamide: Eclosed, Uclosed, and Hclosed, obtained at the BP86/cc-pVTZ level of theory in chloroform.
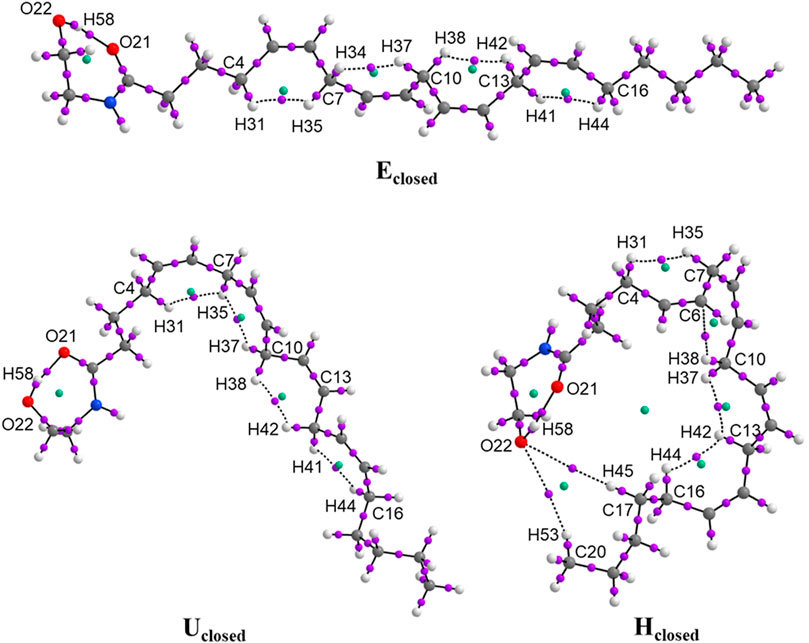
FIGURE 2. Molecular graphs of the conformers of anandamide: Eclosed, Uclosed, and Hclosed, obtained at the BP86/cc-pVTZ level of theory in chloroform.
From the results, it is observed the non-covalent interaction corresponds to the hydrogen bond in the EA group in O21⋯H58-O22 with ρ(r) = 0.043 and ∇2ρ(r) = ∼ 0.096 a.u. for conformers Eclosed, Uclosed, and Hclosed. These values are in agreement with those reported in the literature of 0.002–0.035 and 0.024–0.139 a.u., respectively (Popelier, 2000). On the other hand, the sign of H(r) parameter indicates, in the three closed conformers, that the accumulation of charge density on the hydrogen bond is a stabilizing bond (H(r) < 0) (Cremer and Kraka, 1984). In addition, the DI parameters have values in the range of 0.1106–0.1110 a.u. for the intramolecular hydrogen bond O21⋯H58−O22 in the three closed conformers, indicating that is an interaction <1.0 corresponding to a non-covalent interaction, while the DI for BCP between H58−O22 have values in the range of 0.5371–0.5402 a.u., indicating that this bond has a minor strength than a single bond (value close to 1.0 a.u.), due to the H58 is also contributing to the hydrogen bond formation with the atom O21. The interaction energy of the intramolecular hydrogen bond O21⋯H58−O22 corresponds to 12.33–12.46 kcal mol−1 and it forms an RCP with ρ(r) = 0.0115–0.0116 a.u. for the three conformers, being slightly major the EH … Y value in the conformer Uclosed. It indicates that this hydrogen bond has a stabilizing effect larger than in the other conformers with the closed ring. The value of ρ(r) in the RCP of ethanolamide indicates that the ring formation stabilizes in the same order as the structures in the three conformers of the closed ring.
The non-covalent interactions H⋯H in the Alkyl-M region with a value of ρ(r) in the range of 0.011–0.020 a.u. generate the formation of four ring structures with ρ(r) in the RCP of 0.010–0.015 a.u. for the three closed conformers. The interaction energies between H⋯H are very small indicating weak interactions with values of DI of 0.03 a.u. approximately. Only in the conformer Hclosed, is observed the non-covalent interaction C6−H37C10 with values of ρ(r) in the BCP and RCP of 0.0135 and 0.0092 a.u., respectively, and a DI value of 0.045 a.u. In addition, the conformer Hclosed forms an additional ring structure for the interactions between C17H45−O24 and C20H53−O24 with the value of ρ(r) = 0.0013 a.u. in the RCP. On the other hand, the DI values between atoms C5 = C6, C8 = C9, C11 = C12, and C14 = C15 in the Alkyl-M region are 1.77 a.u., corroborating the cis double bonds formation in the three closed conformers. Figure 2 and Supplementary Figure S2 show the molecular graphs of the anandamide conformers. The purple dots represent the bond critical points (BCP) and the green points represent the ring critical points (RCP).
Given the importance of mixed T-type/cannabinoid blockage in the pain diseases context, in this molecular docking analysis, and as a comparison, we consider the five proposed sites of the complex formed by the Cav3.2 channel and other dual blockers, the NMP compounds. The sites named S6DI, S6DII, S6DIII, S5DIV, and pore-blocking (Figure 3), are located in the transmembrane region, where aromatic and hydrophobic amino acids are prevalent (Rangel-Galván et al., 2022b). In this work, were analyzed the conformers Eclosed, Uclosed, and Hclosed in interaction with the Cav3.2 calcium channel. In addition, the conformers Eopen, Uopen, and Hopen were included to confirm our previous observations that the closed ring structures are more stable than their open counterparts (Rangel-Galván et al., 2022a) and probably more like to participate in the interaction with biological macromolecules, specifically with the Cav3.2 calcium channel. Figure 3 shows the anandamide conformers as ligands and the Cav3.2 channel as the receptor, showing the main interacting segments.
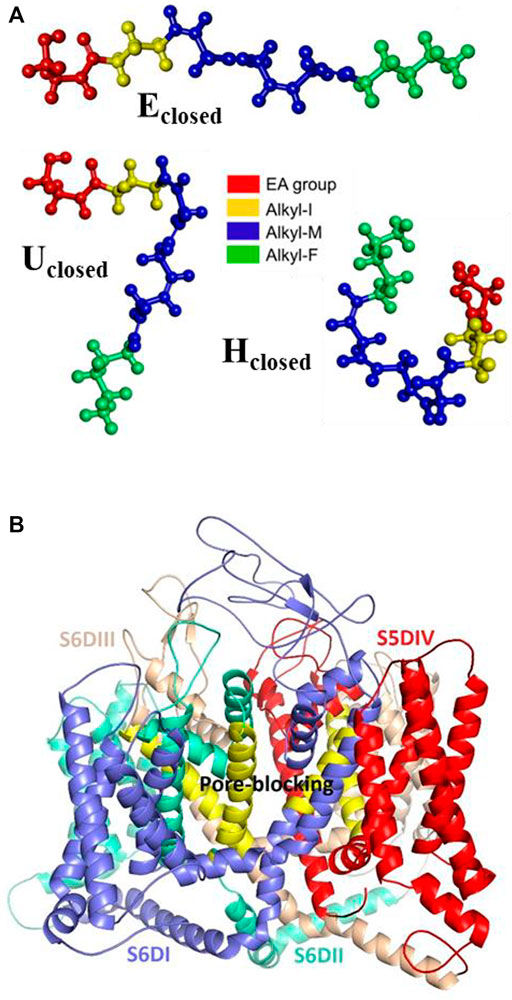
FIGURE 3. Molecular structures. (A) Anandamide conformers Eclosed, Uclosed, and Hclosed, and (B) Cav3.2 channel showing the main interacting segments.
In the S5DIV site (Figure 4), the Uclosed and Uopen conformers place their polar head groups into the S4DIII segment, where the polar interactions are formed with the hydrogen N23−H⋯O of Uclosed and the carbonyl oxygen of L806 (backbone), and the hydrogen O22−H⋯O of Uopen and the carbonyl oxygen of R807 (both residues are located at 2.6 Å from the ligands). Also, is observed an unfavorable acceptor-acceptor interaction with O21 of Uclosed and Uopen conformers and the side chain carbonyl oxygen of D767 residue (Figure 4). The Uclosed conformer forms an additional hydrogen bond, formed by hydrogen O22−H⋯O and the side chain carbonyl oxygen of acidic residue D767, however, this interaction does not contribute to the affinity of the ligand for the calcium channel with interaction energy of −9.2 kcal mol−1 for both conformers. The non-polar portion of both conformers (Uclosed and Uopen) is placed in the S5DIV segment where the hydrophobic residues are located forming alkyl–alkyl interactions with the L815, L1151, L1154, and V1249 residues. In the X-ray structures of the TRPV5, TRPV2, and TRPV1 channels, which are close members of the T-type calcium channels, in complex with econazole (Hughes et al., 2018), resiniferatoxin (Zubcevic et al., 2018), and capsazepine (Gao et al., 2016), respectively, we observed ligand binding sites located in equivalent positions to that defined as the segment S5DIV in the Cav3.2.
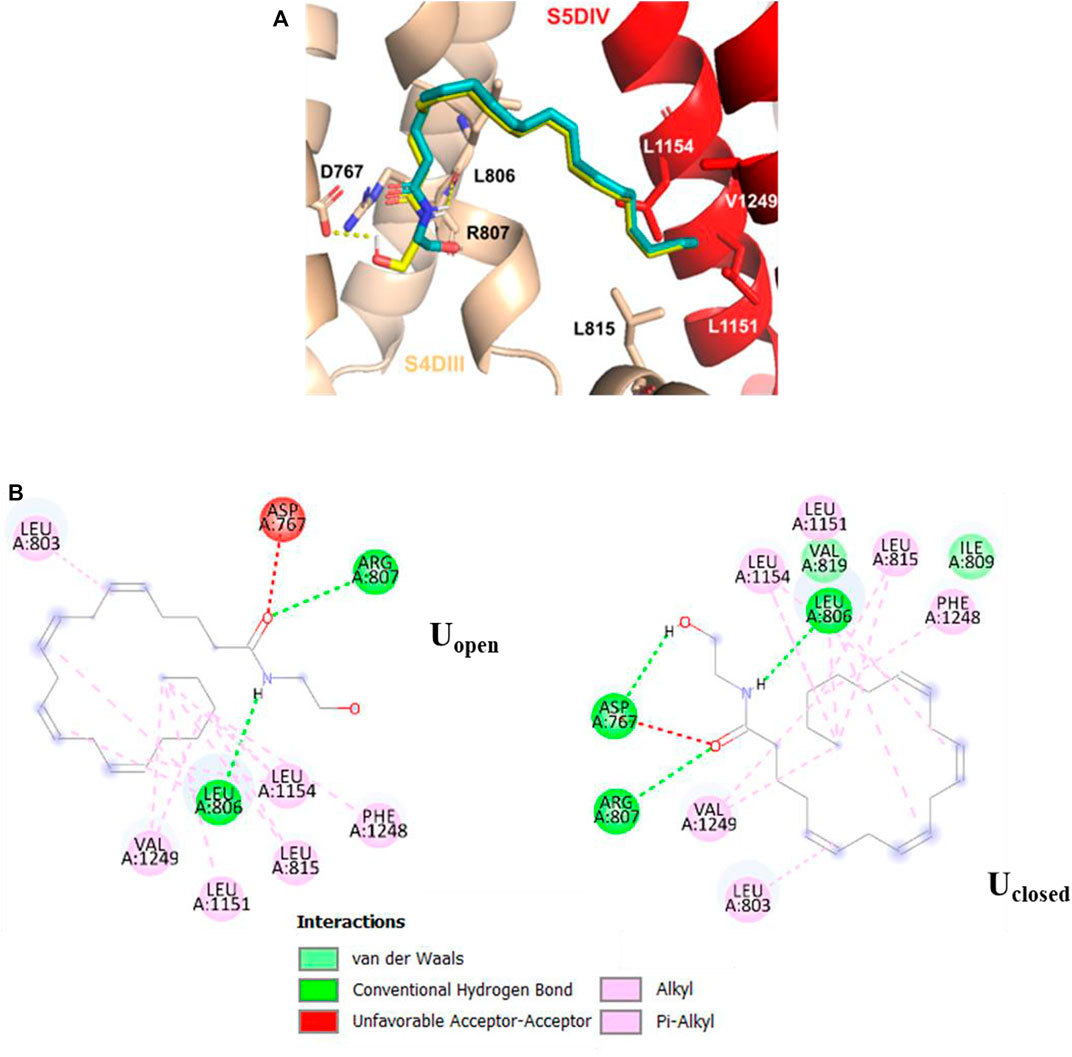
FIGURE 4. Molecular interactions of Uopen (cyan) and Uclosed (yellow) anandamide conformers with Cav3.2. (A) 3D perspective of S5DIV site, Uclosed and Uopen conformers forming two hydrogen bonds with residues L806 and R807, Uclosed additionally forms a hydrogen bond with D767 (B) 2D perspective interaction for each anandamide conformer in the S5DIV site.
As for the pore-blocking site, we identified a common hydrophobic region formed by the residues V1251, V1254, M948, F949, V945, L946, I331, and M330, where the non-polar tail (Alkyl-F) of the anandamide conformer Uopen and the pentyl group of the NMP-181 compound established alkyl-alkyl interactions. In these molecular poses, the carbonyl oxygen of S900 and T568 residues in Cav3.2, form a hydrogen bond with the polar head (C2’−H⋯O) of the conformer Uopen of anandamide and the amine group (C7−H⋯O) of the NMP-181, respectively. The conformer Uclosed, in the pore-blocking site, is located very similar to the conformer Uopen (Figure 5) but the EA group is oriented towards the segment P of the DI forming a polar interaction (C−H⋯O22) with O22 of anandamide and the carbon of G1206. In this context, it can be observed an equivalence of the interaction modes of the functional groups of anandamide and the NMP ligands (Rangel-Galván et al., 2022b) with the channel Cav3.2. Electrophysiological experimental registers show a blocking preference of the inactivation phase of the Cav3.2 channel by both anandamide (Chemin et al., 2001) and the NMP compounds (You et al., 2011; Gadotti et al., 2013; Bladen et al., 2015) was the MFV residues, located in the site S6-DIII, contribute to the inactivation phase of the Cav3 channels (Marksteiner et al., 2001). In the case of the Cav3.2, the corresponding residues are M948, F949, and V950 which are located in the pore-blocking site. Also, in a recent study, the Cav3.1/Z944 complex was solved by cryo-EM (Zhao et al., 2019), where common hydrophobic interacting residues stabilize the ligand binding pose, including the residue T921 (which is equivalent to T586 of the Cav3.2 located in the pore-blocking site). This ligand binding site was proposed also for the genistein/Cav3.3 complex using a combination of in vitro and in silico technics for the study (Rangel-Galván et al., 2021). Another recent study mapping the binding site of the Cav3.1 channel with a small cyclic peptide PnCs1 using docking and molecular dynamics showed a pocket located mainly in the pore region and including some fenestration (Depuydt et al., 2021). Based on the aforementioned experimental evidence, it is possible that the most probable pose for this type of ligand is located in the pore-blocking site.
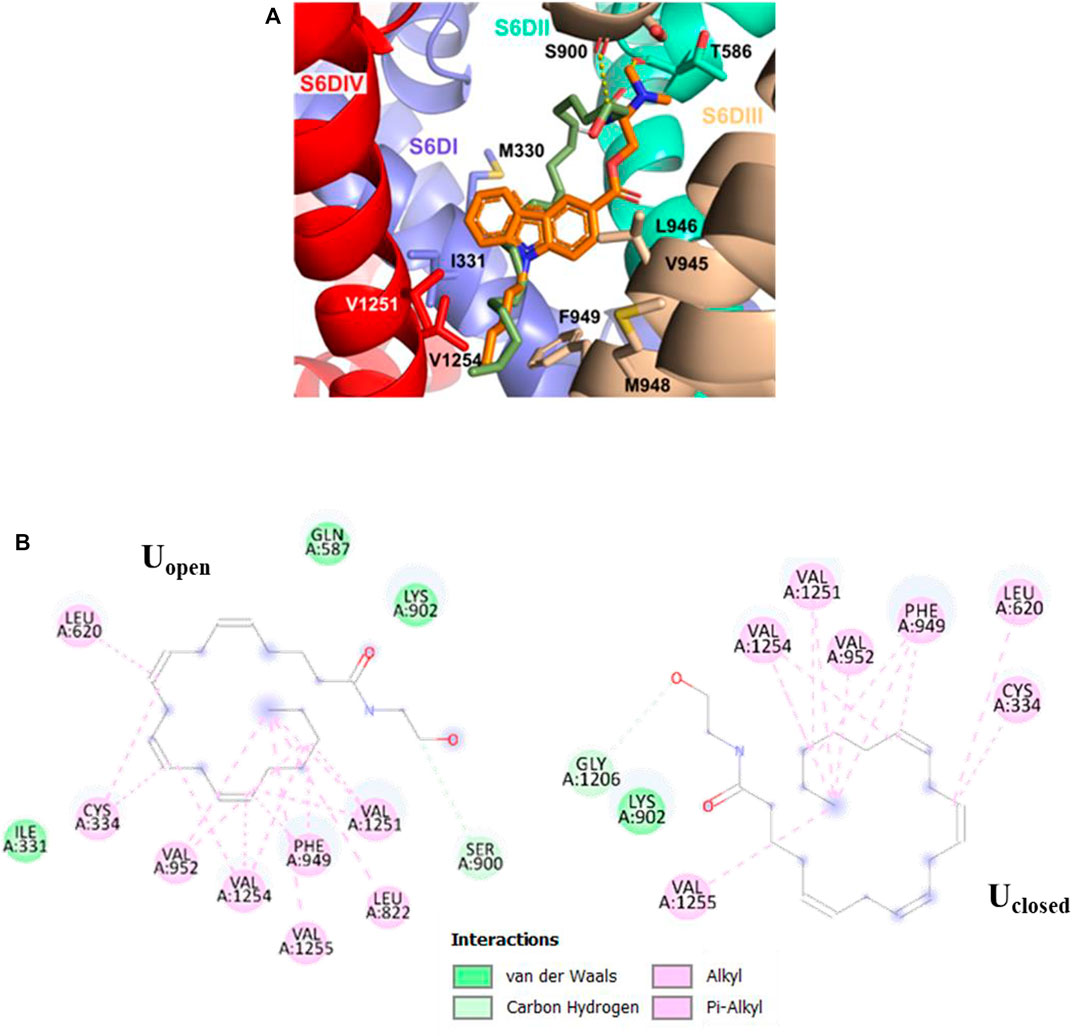
FIGURE 5. Molecular interactions of Uclosed, Uopen (green), and NMP-181 (orange) with Cav3.2. (A) 3D perspective of Uopen and NMP-181 at the pore-blocking site in the Cav3.2 channel; (B) 2D perspective interaction for Uclosed and Uopen anandamide conformer in the pore-blocking site.
In the pose S6DII, the Eclosed and Eopen anandamide conformers (Figure 6A) form a π-σ interaction with residue F161 while the Alkyl-F region forms an alkyl interaction with residues M330 and L333. The Hclosed and Hopen conformers form a π−σ interaction with the residues F316 and F1197 in the S6DI site (Figure 6B) and with the F943 in the S6DIII site (Figure 6C).
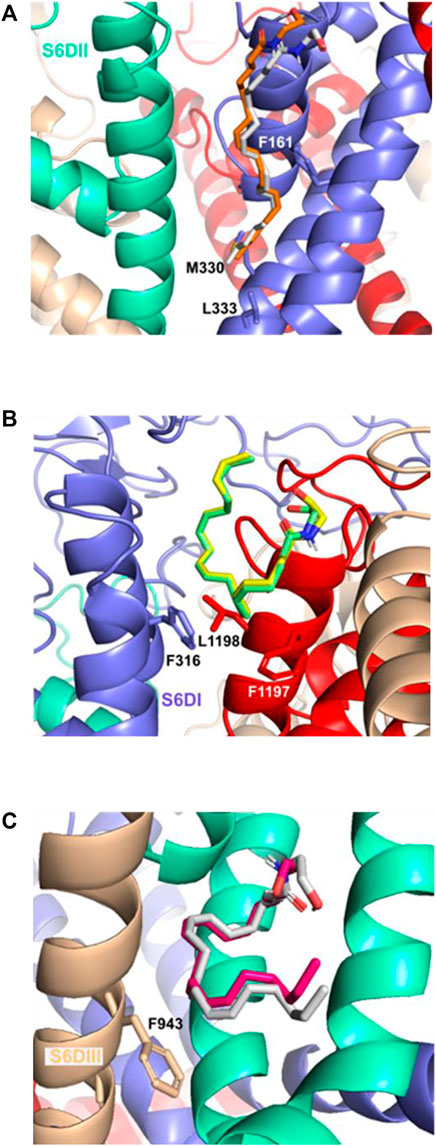
FIGURE 6. Important residues interaction of the Cav3.2 channel and the anandamide conformers: (A) S6DII site and the Eclosed and Eopen conformer, (B) S6DI site and the Hclosed and Hopen conformers, (C) S6DIII site and the Hclosed and Hopen anandamide conformers with π interactions mainly with the F943 residue.
The energy values are summarized in Table 4. According to this analysis, the higher interaction energy in the Cav3.2/anandamide complex corresponds to the Eclosed conformer in the S6DI site. Nevertheless, due to anandamide flexibility is more probable for anandamide to prefer folded conformers. Indeed, the X-ray data of anandamide interacting with its protein transporter (FABP5) show a hairpin shape conformer (Sanson et al., 2014) and according to MD simulations, anandamide prefers curved shapes in the pocket binding FAAH interaction (Palermo et al., 2013).
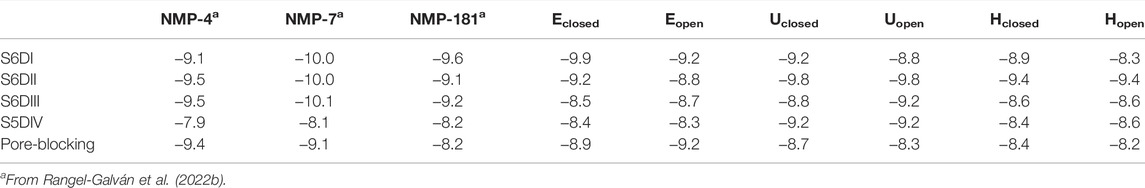
TABLE 4. Interaction energies ΔG (kcal mol−1) of the conformers of anandamide with the channel Cav3.2.
It is observed that there is a negligible difference in the values for the interaction energy related to the formation of a seven-atom ring in the EA group (open versus closed conformations). Only in the S6DI site, a preference for the closed ring conformers is obtained with a difference not greater than 0.7 kcal mol−1. In addition, our results indicate that better interaction energy for the U-shape conformers (Uclosed/Uopen) with respect to the hairpin shape conformers (Hclosed/Hopen) is observed. Along these lines, the drug Z944 is placed in a U-shape form in the binding pocket of the T-type calcium channel (Zhao et al., 2019).
Using conceptual DFT and QTAIM approaches with the BP86/cc-pVTZ level of theory, the structural stability and chemical reactivity properties of six preferred conformers of the endocannabinoid anandamide were analyzed. Our results indicate slight differences for U and H shapes relative to extended forms, in the global reactivity descriptors, concluding an increase in hardness, and a decrease in softness and the electrophilicity index. From these parameters, a decrease in the electrophilicity index was observed in the case of the open ring conformers with respect to their corresponding closed ring counterparts. Also, the hardness and the electrophilicity indexes change when the values for anandamide are compared with those of the NMP compounds. According to the local reactivity descriptors (Fukui and Parr functions), the most probable locations for electrophilic and nucleophilic attacks are located in the ethanolamide group and in the Alkyl-M region for all the anandamide conformers considered in this study. Molecular docking analysis showed important π-σ interactions of anandamide with the side chain of phenylalanine residues. From our results, we observed that, despite their marked structural differences, the NMP compounds (NMP-4, NMP-7, and NMP-181) and anandamide may share a similar physiological activity profile regarding ligand/receptor interactions. The carbazole group is an important region of electronic density accumulation for the NMP compounds in the same way that the Alkyl-M region is for anandamide. According to the literature and the results obtained, the pore-blocking site of the Cav3.2 calcium channel could be a probable binding site for the anandamide molecule.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Conceptualization and design, MC and FM; methodology, MR-G, MC, FM, and JP-A; software, MC, JP-A, NC, and FM; validation, MR-G, and NC; writing the original draft preparation, MR-G, MC, and FM; writing, reviewing and editing, JP-A and NC. All authors have read and agreed to the published version of the manuscript.
Vicerrectoría de Investigación y Estudios de Posgrado (VIEP-BUAP, Mexico) (VIEP2022 project), and the PRODEP Academic Group BUAP-CA-263 (SEP, Mexico).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MR-G thanks CONACYT-México for financial support [Ph.D. fellowship No. 286497]. Authors thank the Laboratorio Nacional de Supercómputo del Sureste de México (LNS-BUAP) of the CONACYT network of national laboratories, for the computer resources and support provided and the PRODEP Academic Group BUAP-CA-263 (SEP, Mexico).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.920661/full#supplementary-material
Aliste, M. P. (2000). Theoretical Study of Dopamine. Application of the HSAB Principle to the Study of Drug-Receptor Interactions. J. Mol. Struct. THEOCHEM 507, 1–10. doi:10.1016/S0166-1280(99)00253-5
Bayat, A., and Fattahi, A. (2019). Influence of Remote Intramolecular Hydrogen Bonding on the Acidity of Hydroxy‐1,4‐Benzoquinonederivatives: A DFT Study. J. Phys. Org. Chem. 32, e3919–10. doi:10.1002/poc.3919
Becke, A. D. (1988). Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Bladen, C., McDaniel, S. W., Gadotti, V. M., Petrov, R. R., Berger, N. D., Diaz, P., et al. (2015). Characterization of Novel Cannabinoid Based T-type Calcium Channel Blockers with Analgesic Effects. ACS Chem. Neurosci. 6, 277–287. doi:10.1021/cn500206a
Chamorro, E., Pérez, P., and Domingo, L. R. (2013). On the Nature of Parr Functions to Predict the Most Reactive Sites along Organic Polar Reactions. Chem. Phys. Lett. 582, 141–143. doi:10.1016/j.cplett.2013.07.020
Chemin, J., Monteil, A., Perez-Reyes, E., Nargeot, J., and Lory, P. (2001). Direct Inhibition of T-type Calcium Channels by the Endogenous Cannabinoid Anandamide. EMBO J. 20, 7033–7040. doi:10.1093/emboj/20.24.7033
Chemin, J., Nargeot, J., and Lory, P. (2007). Chemical Determinants Involved in Anandamide-Induced Inhibition of T-type Calcium Channels. J. Biol. Chem. 282, 2314–2323. doi:10.1074/jbc.M610033200
Chemin, J., Cazade, M., and Lory, P. (2014). Modulation of T-type Calcium Channels by Bioactive Lipids. Pflugers Arch. - Eur. J. Physiol. 466, 689–700. doi:10.1007/s00424-014-1467-5
Colino, L., Herranz-Herrer, J., Gil-Benito, E., Ponte-Lopez, T., del Sol-Calderon, P., Rodrigo-Yanguas, M., et al. (2018). Cannabinoid Receptors, Mental Pain and Suicidal Behavior: a Systematic Review. Curr. Psychiatry Rep. 20, 1–9. doi:10.1007/s11920-018-0880-4
Cremer, D., and Kraka, E. (1984). Chemical Bonds without Bonding Electron Density ? Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. Engl. 23, 627–628. doi:10.1002/anie.198406271
De Proft, F., Vivas‐Reyes, R., Biesemans, M., Willem, R., Martin, J. M. L., and Geerlings, P. (2003). Density Functional Study of the Complexation Reaction of Sn(CH 3 ) 3 X (X = F, Cl, Br and I) with Halide Anions. Eur. J. Inorg. Chem. 2003, 3803–3810. doi:10.1002/ejic.200300044
Depuydt, A.-S., Rihon, J., Cheneval, O., Vanmeert, M., Schroeder, C. I., Craik, D. J., et al. (2021). Cyclic Peptides as T-type Calcium Channel Blockers: Characterization and Molecular Mapping of the Binding Site. ACS Pharmacol. Transl. Sci. 4, 1379–1389. doi:10.1021/acsptsci.1c00079
Devane, W. A., and Axelrod, J. (1994). Enzymatic Synthesis of Anandamide, an Endogenous Ligand for the Cannabinoid Receptor, by Brain Membranes. Proc. Natl. Acad. Sci. U.S.A. 91, 6698–6701. doi:10.1073/pnas.91.14.6698
Domingo, L. R., Pérez, P., and Sáez, J. A. (2013). Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 3, 1486–1494. doi:10.1039/c2ra22886f
Domingo, L., Ríos-Gutiérrez, M., and Pérez, P. (2016). Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 748, 1–22. doi:10.3390/molecules21060748
Dunning, T. H. (1989). Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 90, 1007–1023. doi:10.1063/1.456153
Ferreri, C., Anagnostopoulos, D., Lykakis, I. N., Chatgilialoglu, C., and Siafaka-Kapadai, A. (2008). Synthesis of All-Trans Anandamide: A Substrate for Fatty Acid Amide Hydrolase with Dual Effects on Rabbit Platelet Activation. Bioorg. Med. Chem. 16, 8359–8365. doi:10.1016/j.bmc.2008.08.054
Frau, J., and Glossman-Mitnik, D. (2017). Conceptual DFT Descriptors of Amino Acids with Potential Corrosion Inhibition Properties Calculated with the Latest Minnesota Density Functionals. Front. Chem. 5, 1–8. doi:10.3389/fchem.2017.00016
Frau, J., and Glossman-Mitnik, D. (2018). Conceptual DFT Study of the Local Chemical Reactivity of the Colored BISARG Melanoidin and its Protonated Derivative. Front. Chem. 6, 1–9. doi:10.3389/fchem.2018.00136
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., CheesemanScalmani, J. R. G., et al. (2016). Gaussian 16, Revision B.O1. Wallingford: Gaussian, Inc.
Gadotti, V. M., You, H., Petrov, R. R., Berger, N. D., Diaz, P., and Zamponi, G. W. (2013). Analgesic Effect of a Mixed T-type Channel inhibitor/CB2 Receptor Agonist. Mol. Pain 9, 1744–8069. doi:10.1186/1744-8069-9-32
Gao, Y., Cao, E., Julius, D., and Cheng, Y. (2016). TRPV1 Structures in Nanodiscs Reveal Mechanisms of Ligand and Lipid Action. Nature 534, 347–351. doi:10.1038/nature17964
Hernández-Paredes, J., Carrillo-Torres, R. C., Hernández-Negrete, O., Sotelo-Mundo, R. R., Glossman-Mitnik, D., Esparza-Ponce, H. E., et al. (2017). Experimental and Theoretical Study on the Molecular Structure, Covalent and Non-covalent Interactions of 2,4-dinitrodiphenylamine: X-Ray Diffraction and QTAIM Approach. J. Mol. Struct. 1141, 53–63. doi:10.1016/j.molstruc.2017.03.087
Hua, T., Vemuri, K., Pu, M., Qu, L., Han, G. W., Wu, Y., et al. (2016). Crystal Structure of the Human Cannabinoid Receptor CB1. Cell. 167, 750–762. doi:10.1016/j.cell.2016.10.004
Hughes, T. E. T., Pumroy, R. A., Yazici, A. T., Kasimova, M. A., Fluck, E. C., Huynh, K. W., et al. (2018). Structural Insights on TRPV5 Gating by Endogenous Modulators. Nat. Commun. 9, 1–11. doi:10.1038/s41467-018-06753-6
Lu, T., and Chen, F. (2012). Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 33, 580–592. doi:10.1002/jcc.22885
Marksteiner, R., Schurr, P., Berjukow, S., Margreiter, E., Perez‐Reyes, E., and Hering, S. (2001). Inactivation Determinants in Segment IIIS6 of Ca V 3.1. J. Physiol. 537, 27–34. doi:10.1111/j.1469-7793.2001.0027k.x
Matta, C. F., and Boyd, R. J. (2007). An Introduction to the Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design. 1st ed. Bonn, Germany, Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA. doi:10.1002/9783527610709.ch1
McAllister, S. D., Rizvi, G., Anavi-Goffer, S., Hurst, D. P., Barnett-Norris, J., Lynch, D. L., et al. (2003). An Aromatic Microdomain at the Cannabinoid CB1 Receptor Constitutes an Agonist/Inverse Agonist Binding Region. J. Med. Chem. 46, 5139–5152. doi:10.1021/jm0302647
Morell, C., Grand, A., and Toro-Labbé, A. (2005). New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 109, 205–212. doi:10.1021/jp046577a
Oz, M. (2006). Receptor-independent Actions of Cannabinoids on Cell Membranes: Focus on Endocannabinoids. Pharmacol. Ther. 111, 114–144. doi:10.1016/j.pharmthera.2005.09.009
Pacher, P., Bátkai, S., and Kunos, G. (2006). The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 58, 389–462. doi:10.1124/pr.58.3.2
Palermo, G., Campomanes, P., Neri, M., Piomelli, D., Cavalli, A., Rothlisberger, U., et al. (2013). Wagging the Tail: Essential Role of Substrate Flexibility in FAAH Catalysis. J. Chem. Theory Comput. 9, 1202–1213. doi:10.1021/ct300611q
Parthasarathi, R., Subramanian, V., Roy, D. R., and Chattaraj, P. K. (2004). Electrophilicity Index as a Possible Descriptor of Biological Activity. Bioorg. Med. Chem. 12, 5533–5543. doi:10.1016/j.bmc.2004.08.013
Popelier, P. L. (2000). Atoms in Molecules: An Introduction. 1st ed. Harlow, Great Britain: Prentice Hall College Div.
Rangel-Galván, M., Rangel, A., Romero-Méndez, C., Dávila, E. M., Castro, M. E., Caballero, N. A., et al. (2021). Inhibitory Mechanism of the Isoflavone Derivative Genistein in the Human Cav3.3 Channel. ACS Chem. Neurosci. 12, 651–659. doi:10.1021/acschemneuro.0c00684
Rangel-Galván, M., Castro, M. E., Caballero, N. A., Perez-Aguilar, J. M., and Melendez, F. J. (2022a). NMR and IR Spectroscopic Analysis of the Preferred Conformations of Neurotransmitter Anandamide. [Non-Published Work].
Rangel-Galván, M., Castro, M. E., Perez-Aguilar, J. M., Caballero, N. A., Rangel-Huerta, A., and Melendez, F. J. (2022b). Theoretical Study of the Structural Stability, Chemical Reactivity, and Protein Interaction for NMP Compounds as Modulators of the Endocannabinoid System. Molecules 27, 414–416. doi:10.3390/molecules27020414
Sanner, M. F. (1999). Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 17, 57–61. Available at: https://www.academia.edu/download/25505223/10.1.1.35.6459.pdf.
Sanson, B., Wang, T., Sun, J., Wang, L., Kaczocha, M., Ojima, I., et al. (2014). Crystallographic Study of FABP5 as an Intracellular Endocannabinoid Transporter. Acta Cryst. D. Biol. Crystallogr. 70, 290–298. doi:10.1107/S1399004713026795
Schrödinger, L. (2017). The PyMOL Molecular Graphics System; Version 2.0. Palo Alto, CA, USA: DeLano Sci. LCC.
Snutch, T. P., and Zamponi, G. W. (2018). Recent Advances in the Development of T-type Calcium Channel Blockers for Pain Intervention. Br. J. Pharmacol. 175, 2375–2383. doi:10.1111/bph.13906
Sugiura, T., Kobayashi, Y., Oka, S., and Waku, K. (2002). Biosynthesis and Degradation of Anandamide and 2-arachidonoylglycerol and Their Possible Physiological Significance. Prostagl. Leukot. Essent. Fat. Acids (PLEFA) 66, 173–192. doi:10.1054/plef.2001.0356
Tibbs, G. R., Posson, D. J., and Goldstein, P. A. (2016). Voltage-Gated Ion Channels in the PNS: Novel Therapies for Neuropathic Pain? Trends Pharmacol. Sci. 37, 522–542. doi:10.1016/j.tips.2016.05.002
Todorovic, S. M., and Jevtovic-Todorovic, V. (2013). Neuropathic Pain: Role for Presynaptic T-type Channels in Nociceptive Signaling. Pflugers Arch. - Eur. J. Physiol. 465, 921–927. doi:10.1007/s00424-012-1211-y
Tood, A., K. (2017). AIMAll (Version 17.11.14). TK Gristmill Software, Overland Park KS, USA (aim.tkgristmill.com). Available at: aim.tkgristmill.com (Accessed April 10, 2022).
Trott, O., and Olson, A. J. (2010). AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31, 455–461. doi:10.1002/jcc.21334.AutoDock
Van Der Stelt, M., and Di Marzo, V. (2005). Anandamide as an Intracellular Messenger Regulating Ion Channel Activity. Prostagl. Other Lipid Mediat. 77, 111–122. doi:10.1016/j.prostaglandins.2004.09.007
Vivas-Reyes, R., De Proft, F., Biesemans, M., Willem, R., and Geerlings, P. (2003). A DFT Study of Tin‐ and Crown‐Ether‐Based Host Molecules Capable of Binding Anions and Cations Simultaneously. Eur. J. Inorg. Chem. 2003, 1315–1324. doi:10.1002/ejic.200390171
Vivas-Reyes, R., Mercado, L. D., Anaya-Gil, J., Marrugo, A. G., and Martinez, E. (2008). Theoretical Study to Evaluate Polyfuran Electrical Conductivity and Methylamine, Methoxy Substituent Effects. J. Mol. Struct. THEOCHEM 861, 137–141. doi:10.1016/j.theochem.2008.04.019
Yang, W., and Mortier, W. J. (1986). The Use of Global and Local Molecular Parameters for the Analysis of the Gas-Phase Basicity of Amines. J. Am. Chem. Soc. 108, 5708–5711. doi:10.1021/ja00279a008
Yang, Y., Cui, Y., Sang, K., Dong, Y., Ni, Z., Ma, S., et al. (2018). Ketamine Blocks Bursting in the Lateral Habenula to Rapidly Relieve Depression. Nature 554, 317–322. doi:10.1038/nature25509
You, H., Gadotti, V. M., Petrov, R. R., Zamponi, G. W., and Diaz, P. (2011). Functional Characterization and Analgesic Effects of Mixed Cannabinoid receptor/T-type Channel Ligands. Mol. Pain 7, 1744–8069. doi:10.1186/1744-8069-7-89
Zghibeh, C. M., Raj Gopal, V., Poff, C. D., Falck, J. R., and Balazy, M. (2004). Determination of Trans-arachidonic Acid Isomers in Human Blood Plasma. Anal. Biochem. 332, 137–144. doi:10.1016/j.ab.2004.04.030
Zhao, Y., Huang, G., Wu, Q., Wu, K., Li, R., Lei, J., et al. (2019). Cryo-EM Structures of Apo and Antagonist-Bound Human Cav3.1. Nature 576, 492–497. doi:10.1038/s41586-019-1801-3
Keywords: anandamide, conceptual DFT, QTAIM, molecular docking, chemical reactivity, pain and depression
Citation: Rangel-Galván M, Castro ME, Perez-Aguilar JM, Caballero NA and Melendez FJ (2022) Conceptual DFT, QTAIM, and Molecular Docking Approaches to Characterize the T-Type Calcium Channel Blocker Anandamide. Front. Chem. 10:920661. doi: 10.3389/fchem.2022.920661
Received: 14 April 2022; Accepted: 26 May 2022;
Published: 14 July 2022.
Edited by:
Daniel Glossman-Mitnik, Centro de Investigación de Materiales Avanzados (CIMAV), MexicoReviewed by:
Ricardo Vivas-Reyes, Universidad de Cartagena, ColombiaCopyright © 2022 Rangel-Galván, Castro, Perez-Aguilar, Caballero and Melendez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Eugenia Castro, bWFyZXVnLmNhc3Ryb0Bjb3JyZW8uYnVhcC5teA==; Francisco J. Melendez, ZnJhbmNpc2NvLm1lbGVuZGV6QGNvcnJlby5idWFwLm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.