 Hui-Min He
Hui-Min He Hui Yang
Hui Yang Ying Li3
Ying Li3- 1 Department of Physics, Institute of Computational and Applied Physics, Taiyuan Normal University, Jinzhong, China
- 2 School of Chemistry and Chemical Engineering, Shanxi Datong University, Datong, China
- 3 Laboratory of Theoretical and Computational Chemistry, Jilin University, Changchun, China
By replacing one Al or N atom of aluminum nitride nanocage Al12N12 with an alkaline-earth metal atom, two series of compounds, namely, M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca), were constructed and investigated in theory. The substituted effect of alkaline-earth metal on the geometric structure and electronic properties of Al12N12 is studied in detail by density functional theory (DFT) methods. The calculated binding energies, HOMO–LUMO gaps, and VIE values of these compounds reveal that they possess high stability, though the NBO and HOMO analyses show that they are also excess electron compounds. Due to the existence of diffuse excess electrons, these alkaline-earth metal-substituted compounds exhibit larger first hyperpolarizabilities (β 0) than pure Al12N12 nanocage. In particular, these considered compounds exhibit satisfactory infrared (IR) (>1800 nm) and ultraviolet (UV) (˂ 250 nm) transparency. Therefore, these proposed excess electron compounds with high stability may be regarded as potential candidates for new UV and IR NLO molecules.
Introduction
Over the past few decades, the design and synthesis of nonlinear optical (NLO) materials with excellent performance have exerted a tremendous fascination on researchers in consideration of their widespread applications in optics and optoelectronics (Marder Seth et al., 1994; Priyadarshy et al., 1996; Nakano et al., 2002; Zhang et al., 2005; Xiao et al., 2008; Xu et al., 2013). Up to now, abundant strategies have been proposed to acquire high-performance NLO materials of the new type, for instance, designing donor–π-conjugated-bridge-acceptor (D–π-A) models (Kanis et al., 1994), decorating or modifying sp2-hybridized carbon nanomaterials (Bai et al., 2013; Muhammad et al., 2013; Yu et al., 2013; Karamanis et al., 2014; Zhou et al., 2014), synthesizing octupolar molecules (Ja Lee et al., 2003), utilizing a multideck sandwich cluster as the building block (Wang et al., 2015), applying the bond length alternation (BLA) theory (Meyers et al., 1994), enhancing push–pull effects (Liu et al., 2010), and doping metal atoms (Di Bella, 2001; Zhong et al., 2012b; Wu et al., 2014), etc. In particular, Chen et al. (2004) and Li et al. (2004) have revealed that the introduction of loosely bound excess electrons into a molecule is an effective measure to dramatically enhance its NLO response. Therein, the diffuse excess electron is responsible for improved NLO response. Subsequently, a lot of compounds with dispersed excess electrons, namely, excess electron compounds, are designed in theory, and as expected, they exhibit considerable large NLO responses (Zhong et al., 2015).
In previous studies, it has been found that alkali-metal-doped organic complexants exhibit large first hyperpolarizabilities (Chen et al., 2005; Xu et al., 2007; Wang et al., 2012). In these systems, excess electrons are formed when organic complexants push/pull valence electrons of alkali metal atoms. Therefore, such systems were classified as excess electron compounds, where the alkali metal atom acts as the excess electron source. However, the introduction of active alkali metal atoms reduces stability of these compounds. Therefore, selecting the proper excess electron source to achieve new excess electron compounds will be an efficient way to obtain NLO materials with high stability. To achieve this aim, we have used alkaline-earth metal atoms as an electron source to design several types of excess electron compounds (He et al., 2017a; He et al., 2017b; He et al., 2019) with large first hyperpolarizabilities and satisfying stability.
On the other word, fullerene-like hollow nanocages with noncarbon have attracted great attention owing to their special optoelectronic properties in recent years (Golberg et al., 1998; Strout, 2000; Fu et al., 2001; Stafström et al., 2001; Bertolus et al., 2004; Wang et al., 2005; Beheshtian et al., 2011; Ahmadi Peyghan et al., 2012; Beheshtian et al., 2012c). In particular, Group III–V nitrides are the most promising nanoscale materials in various fields. Therefore, a lot of theoretical and experimental studies have been carried out on the Group III-V fullerene-like nanostructures, especially the most significant AlN nanocages because of their high thermal conductivity and chemical stability (Silaghi-Dumitrescu et al., 1996; Oku et al., 2004; Koi et al., 2005; Yang et al., 2005; Zhang and Zhang, 2005; Zope and Dunlap, 2005; Li et al., 2007; Beheshtian et al., 2012a; Liu et al., 2013; Saeedi et al., 2013). Ab initio calculations revealed that the Al12N12 nanocage is the most stable AlN nanostructure and thereby can be regarded as an ideal inorganic fullerene-like candidate (Wu et al., 2003). Considering the unique structural and electronic structure of this cage, it has been used as an excellent starting material to design NLO molecules. For instance, two inorganic electrides, M@Al12N12(M = Li, Na, and K) (Niu et al., 2014) and M3O@Al12N12 (M = Li, Na, and K) (Sun et al., 2016), were achieved by doping alkali metal atoms and superalkali clusters on the Al12N12 nanocage, while the M@Al12N11 and M@Al11N12 compounds were designed by substituting one atom of Al12N12 with an alkali-metal atom (Maria et al., 2016). All these Al12N12-based excess electron compounds exhibit considerably high NLO responses.
In order to enhance the NLO responses and stability of such Al12N12-based compounds, two series of inorganic compounds, M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca), were obtained by substituting Al or N atom in the Al12N12 with alkaline-earth metal in the current work. We mainly focus on the following issues: 1) Can loosely bound excess electrons that can dramatically enhance molecular NLO response be generated in these proposed inorganic compounds with alkaline-earth substituted? 2) Do these novel compounds possess larger stability and first hyperpolarizability (β 0) than those of previously reported alkali-metal-substituted systems M@Al12N11 and M@Al11N12 (M = Li, Na, and K)? Our results revealed that these inorganic compounds M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca) indeed contain diffuse excess electrons and thus exhibit larger β 0 values than alkali-metal-substituted systems. In particular, These excess electron compounds exhibit excellent infrared (IR) (>1800 nm) and ultraviolet (UV) (˂ 250 nm) transparency for their potential applications with modern laser frequency conversion technology and optical parameter oscillator processes (Zhang et al., 2009; Luo et al., 2014).
Computational Details
The geometric structures with all real frequencies are obtained by using a combination of Becke’s hybrid 3-parameter exchange functional (Becke, 1993a) and Lee–Yang–Parr’s correlation function (Lee et al., 1988) (B3LYP). In a previous study, the Pople-type (Bilbrey et al., 2013) basis set 6-31+G(d) was selected because it has been proved to be reliable for the geometry optimization of similar systems (Niu et al., 2014). Natural bond orbital (NBO) analyses, vertical ionization energies (VIE), and binding energy (E b) calculations were also performed at the B3LYP/6-31+G(d) level. The binding energy of the alkaline-earth metal atom M was calculated using the counterpoise procedure (Alkorta and Elguero, 1999) and is defined as follows: For M@Al12N11,
For M@Al12N11,
The VIE is the energy difference between neutral molecule and cation systems at the neutral optimization geometry (Wang et al., 2012; Bai et al., 2013; Sun et al., 2015; He et al., 2019).
Polarizability and first hyperpolarizability calculations are carried out on long-range correlated methods CAM-B3LYP (Tawada et al., 2004; Yanai et al., 2004) and BHandHLYP (Becke, 1993b) in conjunction with the 6-31+G(d) basis set. As all the systems are in the doublet states, values of spin eigenvalue ⟨S2⟩ involved in the structural optimization and NLO computations are in the range of 0.760–0.789, which shows an error range of 1.3%–4.9%, indicating that the spin contamination is negligible in the current calculations.
The static polarizability (α 0) and the first hyperpolarizability (β 0) are defined as follows:
where
For electronic transition properties, the transition energy ΔE, oscillator strength f 0, and the difference of dipole moment Δμ between the ground and the crucial excited state are estimated by the time-dependent density functional theory, TD-CAM-B3LYP, with the 6-31+G(d) basis set. Simultaneously, ultraviolet-visible–infrared (UV-VIS-IR) absorption spectra of all the systems were also obtained at the same level. All the UV-VIS-IR spectra were reflected with Gaussian curves under a full-width at half-maximum (FWHM) of 0.10 eV.
All of the calculations were carried out by using the Gaussian 16 program package (Frisch et al., 2016). Molecular configurations and molecular orbital (MO) plots were generated with the GaussView program (Dennington et al., 2016).
Results and Discussion
First, the isolated aluminum nitride nanocage Al12N12 was optimized, and its structure is shown in Figure 1. In this study, the pure Al12N12 nanocage is found to be a T h-symmetric fullerene-like cage consisting of six 4-membered rings and eight 6-membered rings, in which the Al–N bond lengths are 1.794 and 1.858 Å, respectively, in consistent with earlier reports (Beheshtian et al., 2012a; Beheshtian et al., 2012b; Niu et al., 2014). Then, the initial geometry structures of M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca) were constructed by replacing one atom (Al or N) in the Al12N12 nanocage with an alkaline-earth metal atom M. In the M@Al12N11 series, one alkaline-earth metal atom is substituted for one nitrogen atom, whereas one aluminum atom is replaced with one alkaline-earth metal atom in M@Al11N12. After optimization, six equilibrium conformations of M@Al12N11 and M@Al11N12 have been obtained and are shown in Figure 1. The selected geometrical parameters, VIE values, and HOMO–LUMO gap values of these resulting compounds as well as the binding energies and NBO charges of alkaline-earth metal atoms in them are listed in Table 1.
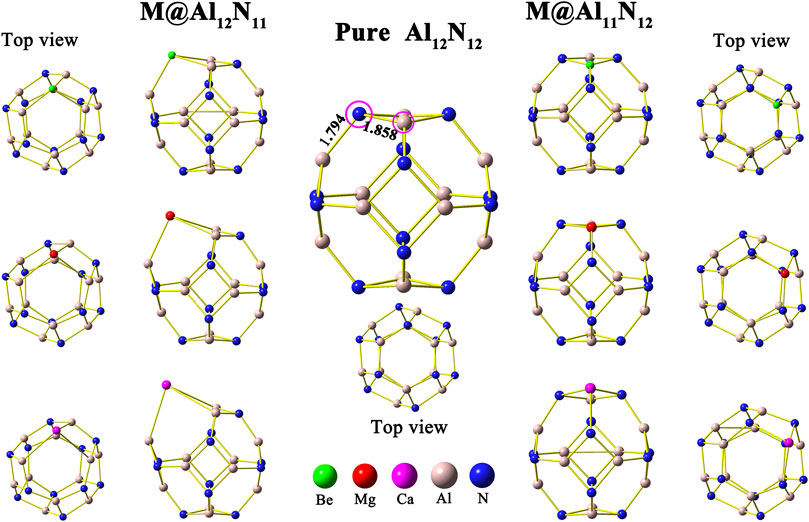
FIGURE 1. Optimized geometric structure of undoped Al12N12 and alkaline-earth metal-doped M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca).
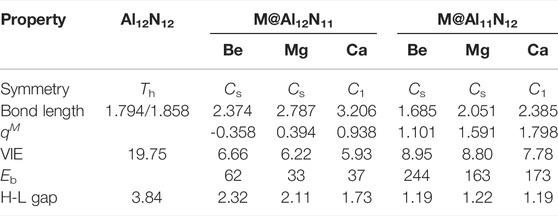
TABLE 1. Symmetry, average M–Al bond length (in Å), average M–N bond length (in Å), HOMO–LUMO gap (H-L Gap, in eV), and vertical ionization energies (VIE, in eV) as well as the binding energy (E b, in kcal/mol) and NBO charge of alkaline-earth metal atom for M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca).
As shown in Table 1, when one nitrogen or aluminum atom is replaced, the original Al-N bond is elongated. That is to say, the bond lengths of M–Al and M–N in the displaced systems are longer than those of the original Al-N in the pure Al12N12 nanocage. Nevertheless, the Be-N bond is a special case in Be@Al11N12. The Be-N bond in the Be@Al11N12 decreases by ca. 0.175 Å as compared with the Al-N bond in the isolated Al12N12 nanocage, which may be attributed to the fact that the nitrogen atom possesses larger electronegativity and the Be atom has smaller atomic radii. It is also found that the bond lengths of M–Al and M–N show a monotonous increase with the increasing atomic number of M. In addition, the M–Al bond length of M@Al12N11 series exhibits a larger increment than the M-N bond length of the M@Al11N12 series as compared with the Al-N bond length of pure Al12N12 nanocage.
Figure 2 displays HOMOs of Al12N12 nanocage, M@Al12N11, and M@Al11N12 (M = Be, Mg, and Ca). It can be seen that the HOMO of a pure Al12N12 nanocage consists of p atomic orbitals of N atoms. Differently, all the HOMOs of M@Al12N11 and M@Al11N12 systems possess diffuse excess electrons, reflecting the unique electric characteristics of these studied compounds. Therefore, these proposed M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca) systems can be regarded as a new type of inorganic excess electron compounds. Interestingly, it is found that these systems exhibit almost the same HOMOs.
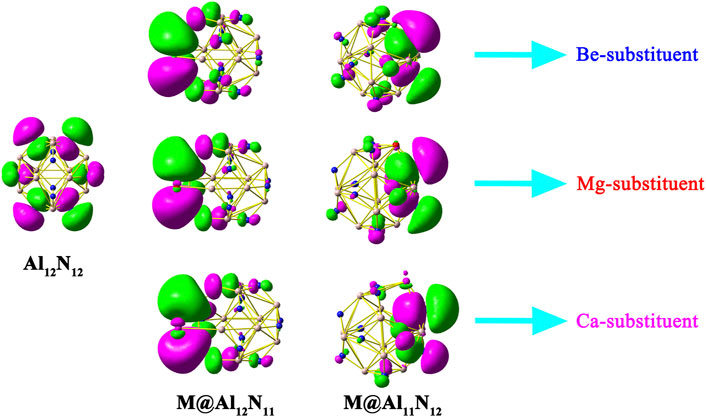
FIGURE 2. HOMOs of Al12N12, M@Al12N11, and M@Al11N12 (M = Be, Mg, and Ca) systems.
NBO analysis was carried out to analyze charge transfer in nanocages and electronic structures of these systems. The computed NBO charges on alkaline-earth metal atoms in these studied compounds are given in Table 1. From Table 1, it can be seen that the NBO charges of alkaline-earth metal atoms in M@Al11N12 (M = Be, Mg, and Ca) are in the range of (1.101–1.798) |e|, which is much larger than the charges (0.496–0.788) |e| (Maria et al., 2016) on alkali metal atoms in M@Al11N12 (M = Li, Na, and K) and the charges of (0.32-0.60) |e| (Ullah et al., 2020) on alkaline-earth metal atoms in AEM@Al12N12 (M = Be, Mg, and Ca), indicating that more charges are transferred from alkaline-earth metal atoms to the nanocages in these compounds. In addition, it is also found that the charges on M atoms in the M@Al11N12 series are more electropositive than those of the M@Al12N11 series, which may be attributed to the fact that the electronegativity of the nitrogen atom is larger than that of the aluminum atom. When an alkaline-earth metal atom is substituted for an Al atom or N atom, the nitrogen atom has a stronger ability to absorb electrons than the Al atom. Therefore, introducing M atoms in the M@Al11N12 series loses more electrons and displays more electropositivity. Additionally, it is found that the charges on M atoms increase along with the increasing M atomic number in both series because of the increasing electropositivity of M. Moreover, the NBO analysis also reveals that the alkaline-earth metal atoms can serve as the source of diffuse excess electrons for these excess electron compounds.
It is known that the stability of molecules is of great significance for their synthesis in the laboratory and further application in practice. Moreover, the kinetic stability, chemical reactivity, and optical polarizability of any molecule can be described from the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). HOMO–LUMO gaps of all the studied excess electron compounds are calculated and are summarized in Table 1. As can be seen from Table 1, pure Al12N12 has a large band gap of 3.84 eV, which is a barrier in the way of its applications in electronic devices. As compared with Al12N12, a crucial decrease in the HOMO–LUMO gap was noticed for all replaced systems. That is to say, the HOMO–LUMO gaps of M@Al12N11 and M@Al11N12 are reduced to (1.19–2.32) eV. Even so, the gap values of M@Al12N11 (M = Be, Mg, and Ca) compounds are much larger than those (1.39–1.78) eV (Maria et al., 2016) of M@Al12N11 (M = Li, Na, and K) compounds, comparable to that (1.57 eV) (Wang et al., 1999) of the kinetically stable C60, and those (1.59–3.79) eV (Ullah et al., 2020) of AEM@Al12N12 (M = Be, Mg, and Ca), suggesting the large chemical stability of the studied M@Al12N11 (M = Be, Mg, and Ca) species. From Figure 3, it is also found that the gap values decrease along with the decreasing M atomic number for the M@Al12N11 series, whereas they are hardly equal for the M@Al11N12 series.
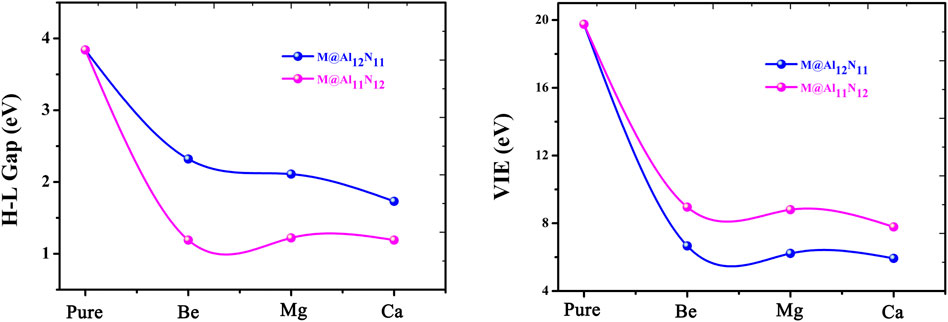
FIGURE 3. Relationship between H-L Gaps and VIE values with the atomic number of alkaline-earth metals.
Furthermore, the large electronic stability of these excess electron compounds can also be characterized by their higher vertical ionized energies (VIEs) of (5.93–8.95) eV, which are slightly higher than the reported values of inorganic and organic excess electron compounds (Chen et al., 2005; Zhong et al., 2012a; He et al., 2017a; He et al., 2017b; Ullah et al., 2020), indicating that these proposed nanocage compounds exhibit high electronic stability. Figure 3 demonstrates the alkaline-earth metal atomic number effect on VIE values, namely, the larger the atomic number, the smaller the VIE value.
Additionally, the binding energies (E b) of these designed M@Al12N11 and M@Al11N12 compounds are also calculated and given in Table 1. The E b values are defined as the negative of the intramolecular interaction energies between the alkaline-earth metal M and the remaining Al12N11 or Al11N12 moieties. Thus, the larger the E b value is, the stronger the interaction between two subunits. From Table 1, it can be seen that all the proposed excess electron compounds exhibit much larger E b values of (33.0-244.0) kcal/mol than those (31-89 kcal/mol) (Maria et al., 2016) of previously reported alkali-metal substituted compounds, indicating that alkaline-earth metal atoms are more tightly bound to the remaining Al12N11 or Al11N12 units than the alkali metal atoms in M@Al12N11 and M@Al11N12 (M = Li, Na, and K). More importantly, the M@Al11N12 (M = Be, Mg, and Ca) series exhibit extremely large E b values up to (173-244) kcal/mol, which are far more than those of (64-89) kcal/mol for M@Al11N12 (M = Li, Na, and K) (Maria et al., 2016). Thus, as compared with alkali metal atoms, the introduction of alkaline-earth metal atoms into the Al12N12 nanocage can produce more stable species with excess electrons. In addition, the E b values of proposed alkaline-earth metal substituted excess electron compounds are also comparable to those (59-106) kcal/mol of small clusters with alkaline-earth metal atoms (Srivastava et al., 2018).
To evaluate nonlinear optical response, the dipole moments (µ 0), polarizabilities (α 0), and first hyperpolarizabilities (β 0) of pure Al12N12 and the proposed M@Al12N11 and M@Al11N12 compounds were calculated and are summarized in Table 2. To better visualize the results, the dependences of the polarizability (α 0) and first hyperpolarizability (β 0) values on the alkaline-earth metal atomic number are exhibited in Figure 4. Our results revealed that the α 0 (286 au) of the Al12N12 nanocage is increased to 319-410 au for replaced compounds, indicating that the substitution of alkaline-earth metal atoms virtually affects the α 0 value of the Al12N12 molecule. As shown in Table 2 and Figure 4, the α 0 changes in the order of 331 au (Be) < 369 au (Mg) < 410 au (Ca) in the M@Al12N11 series and similarly, varies in the order of 312 au (Be) < 326au (Mg) < 345au (Ca) in the M@Al11N12 series. In short, α 0 increases along with the increasing M atomic number. Also, it is observed that the M@Al12N11 exhibits a relatively larger α 0 value than the corresponding M@Al11N12, indicating that the excess electrons in the HOMOs of the former are more diffuse than the latter because static polarizability is sensitive to electronic delocalization.
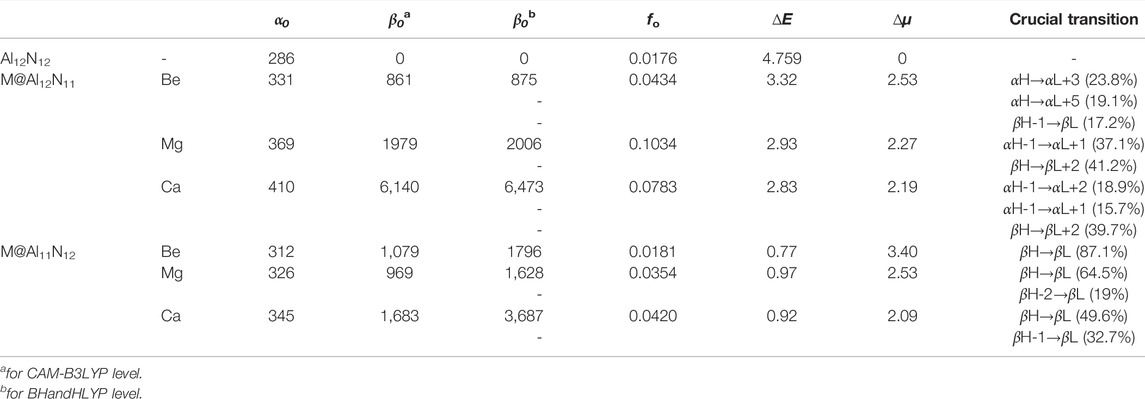
TABLE 2. Static polarizability (α 0 ), static first hyperpolarizability β 0 , oscillator strength (f o), transition energy (∆E, in eV), and difference of dipole moment (∆μ in D) between the ground and the crucial excited states and crucial transition for M@Al12N11 and M@Al11N12 (M = Be, Mg and Ca).
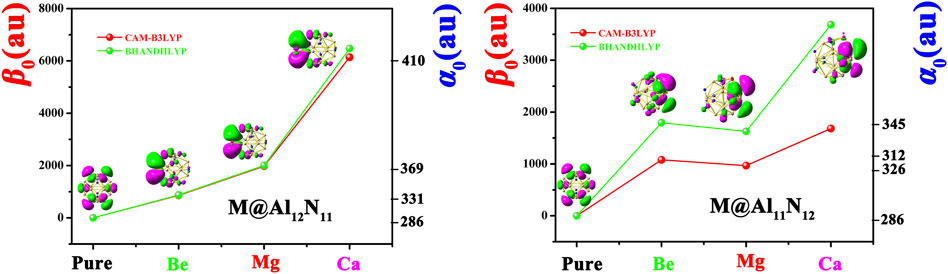
FIGURE 4. Dependences of α 0 and β 0 values on the M atomic number for Al12N12, M@Al12N11, and M@Al11N12 and the first hyperpolarizability comparison of two different methods.
Due to the pure Al12N12 nanocage being centrosymmetric, its β 0 values are zero. Thus, different from α 0, the substitution of M for Al or N atom in Al12N12 brings a prominent improvement of first hyperpolarizability (β 0) of the Al12N12 nanocage, which is because that M-substitution not only destroys the centrosymmetry of Al12N12 but also makes two kinds of replaced systems possessing the dispersed excess electrons. Two different long-range corrected methods were used to calculate the hyperpolarizability β 0 values, which have been listed in Table 2. Figure 4 manifests the first hyperpolarizability at CAM-B3LYP for M@Al12N11 and M@Al11N12 follows the same trend as for BHandHLYP. However, the calculated values at CAM-B3LYP are slightly lower than BHandHLYP.
Additionally, it can be distinctly seen from Table 2 and Figure 4 that the β 0 values of Ca-substitution compounds are greatly larger than those of Be-substitution and Mg-substitution compounds, signifying an evident effect of the M atomic number on the NLO responses of M@Al12N11 and M@Al11N12. To be specific, the varying order for β 0 is 861au (Be) < 1979au (Mg) < 6140au (Ca) in the M@Al12N11 series, which is consistent with the change sequence of α 0 values. That is, the compounds with higher polarizabilities also present relatively higher NLO responses because of the more diffuse excess electrons in them.
The comparison of β 0 values between our proposed excess electron compounds and previously reported alkali-metal substituted compounds is meaningful. In the M@Al12N11 series, it can be seen that reported alkali-metal substituted compounds exhibit much larger β 0 values of (2,500-9,100) au (in CAM-B3LYP/6-311 + g* level) (Maria et al., 2016) than those (861-6,140) au of our proposed alkaline-earth metal substituted excess electron compounds. However, the resultant comparison of β 0 values is inverse in the M@Al11N12 series, namely, β 0 values (1,628–3,687) au of alkaline-earth metal-substituted compounds are larger than those (420–770) kcal/mol of alkali-metal-substituted compounds. Thus, when a N atom is substituted by alkaline-earth metal atoms in an Al12N12 nanocage, excess electron compounds M@Al11N12 (M = Be, Mg, and Ca) can exhibit larger NLO responses than M@Al11N12 (M = Li, Na, and K).
For intensive discernment of this significant increase in first hyperpolarizability due to substitution of alkaline-earth metal in the Al12N12 nanocage, let us consider the simplest two-level model (Maroulis, 1996; Xu et al., 2009), which is derived from the sum-over states method:
where involved ΔE, f 0, and Δμ are the transition energy, oscillator strength, and difference of the dipole moment between the ground state and the crucial excited state, respectively. The expression distinctly displays that β 0 is inversely proportional to the third power of ∆E. Therefore, crucial transition energy plays an important role in the evaluation of β 0. In the current work, the TD-CAM-B3LYP calculations are performed to obtain the dominated excited states of these studied compounds. The crucial transitions and the corresponding ΔE, f 0, and Δμ values are presented in Table 2 and Figure 5, respectively. It is noted that the electrons involved in the crucial excited states of these considered species primarily come from their HOMO, HOMO-1, and HOMO-2 orbitals. Meanwhile, from Figure 5, it can be seen that crucial transitions make the electrons more diffuse, which may lead to the large β 0.H
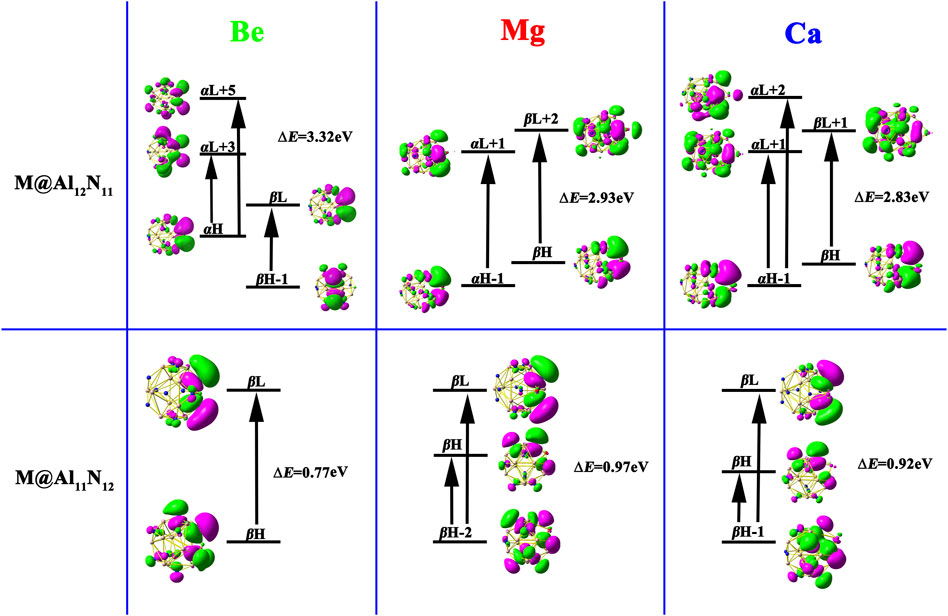
FIGURE 5. Crucial transition states of M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca) compounds.
Moreover, compared to the large ΔE value (4.759 eV) in pure Al12N12, the M@Al12N11 and M@Al11N12 compounds exhibit much smaller ΔE values of (0.77-3.32) eV, which are comparable to those of (1.295–1.982) eV for the alkali metal-based electrides, including Li@(calix [4]pyrrole) (Chen et al., 2005), Li n -H-(CF2-CH2)3-H (n = 1, 2) (Xu et al., 2007), H4C4N2⋯Na2 (Ma et al., 2008), and Li@B10H14 (Muhammad et al., 2009), and far less than (4.6–6.7) eV of M@Al12N11 and M@Al11N12(M = Li, Na, and K) (Maria et al., 2016), as well as (1.80–4.76) eV of AEM@Al12N12 (Ullah et al., 2020). Hence, these small ΔE values bring forth the large β 0 values of these proposed alkaline-earth metal-based excess electron compounds.
It is well-known that the main applications for NLO materials are in doubling frequency and second harmonic generation (SHG). Accordingly, the superior NLO materials not only need large NLO response but also must be transparent under the applied laser region. Therefore, the ultraviolet-visible–infrared (UV-VIS-NIR) absorption spectra of these M@Al12N11 and M@Al11N12 compounds are gained and shown in Figure 6. From Figure 6A, it can be seen that the main absorption region of M@Al12N11 compounds is from 300 to 500 nm. The absorption of these compounds in the infrared spectral region is weak, especially for Be@Al12N11, there is no absorption in the visible region of (510–780) nm and the infrared spectral region, which suggests that Be@Al12N11 has satisfying transparency in both the visible region of (510–780) nm and infrared spectral region. In addition, Figure 6B shows that the M@Al11N12 series have an infrared (IR) transparent region at wavelength >1800 nm. Thus, it is hoped that these six excess electron compounds could be used as new IR NLO materials. Simultaneously, it is observed that they also have an ultraviolet (UV) transparent region at wavelength ˂ 250 nm. Then, they may be taken as a new candidate for UV NLO materials.
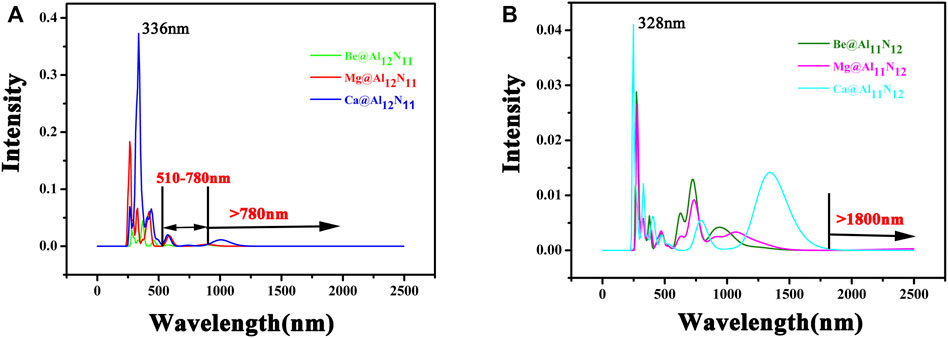
FIGURE 6. Electronic spectra of M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca) compounds.
Conclusions
Using the density functional theory (DFT), two new series of excess electron compounds, i.e., M@Al12N11 and M@Al11N12 (M = Be, Mg, and Ca), have been obtained and studied theoretically in this work. The substituted effect of alkaline-earth metal on the geometric structures and electronic properties of aluminum nitride (Al12N12) nanocage has been investigated in detail. Binding energy calculations display that these proposed compounds, particularly the Al-replaced nanocages have high structural stability. In addition, the substitution of alkaline-earth metal for Al and N in Al12N12 significantly reduces its HOMO–LUMO gap and VIE value, which may bring forth large optical polarizability. More importantly, these studied compounds contain diffuse excess electrons and thus show high NLO responses. Particularly, our results reveal that all these considered compounds show satisfying infrared (IR) transparent region (>1800 nm) and ultraviolet (UV) region (< 250 nm). Thus, we hope that this study could not only provide new candidates of potential NLO molecules but also promote future applications of Al-N fullerene-like nanocages in the field of nonlinear optics.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the Science and Technology Innovation Project of Higher Education in Shanxi Province (2020L0507, 2020L0516, and 2021L370).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the National Supercomputing Center in Shenzhen for providing computational resources.
References
Ahmadi Peyghan, A., Pashangpour, M., Bagheri, Z., and Kamfiroozi, M. (2012). Energetic, Structural, and Electronic Properties of Hydrogenated Al12P12 Nanocluster. Phys. E Low-dimensional Syst. Nanostructures 44 (7), 1436–1440. doi:10.1016/j.physe.2012.03.007
Alkorta, I., and Elguero, J. (1999). Theoretical Study of Strong Hydrogen Bonds between Neutral Molecules: The Case of Amine Oxides and Phosphine Oxides as Hydrogen Bond Acceptors. J. Phys. Chem. A 103 (2), 272–279. doi:10.1021/jp982644n
Bai, Y., Zhou, Z.-J., Wang, J.-J., Li, Y., Wu, D., Chen, W., et al. (2013). New Acceptor-Bridge-Donor Strategy for Enhancing NLO Response with Long-Range Excess Electron Transfer from the NH2.M/M3O Donor (M = Li, Na, K) to inside the Electron Hole Cage C20F19 Acceptor through the Unusual σ Chain Bridge (CH2)4. J. Phys. Chem. A 117 (13), 2835–2843. doi:10.1021/jp3120594
Becke, A. D. (1993b). A New Mixing of Hartree-Fock and Local Density‐functional Theories. J. Chem. Phys. 98 (2), 1372–1377. doi:10.1063/1.464304
Becke, A. D. (1993a). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98 (7), 5648–5652. doi:10.1063/1.464913
Beheshtian, J., Bagheri, Z., Kamfiroozi, M., and Ahmadi, A. (2012a). A Comparative Study on the B12N12, Al12N12, B12P12 and Al12P12 Fullerene-like Cages. J. Mol. Model. 18 (6), 2653–2658. doi:10.1007/s00894-011-1286-y
Beheshtian, J., Bagheri, Z., Kamfiroozi, M., and Ahmadi, A. (2011). Toxic CO Detection by B12N12 Nanocluster. Microelectron. J. 42 (12), 1400–1403. doi:10.1016/j.mejo.2011.10.010
Beheshtian, J., Peyghan, A. A., Bagheri, Z., and Kamfiroozi, M. (2012c). Interaction of Small Molecules (NO, H2, N2, and CH4) with BN Nanocluster Surface. Struct. Chem. 23 (5), 1567–1572. doi:10.1007/s11224-012-9970-9
Beheshtian, J., Peyghan, A. A., and Bagheri, Z. (2012b). Selective Function of Al12N12 Nano-Cage towards NO and CO Molecules. Comput. Mater. Sci. 62, 71–74. doi:10.1016/j.commatsci.2012.05.041
Bertolus, M., Finocchi, F., and Millié, P. (2004). Investigating Bonding in Small Silicon-Carbon Clusters: Exploration of the Potential Energy Surfaces of Si3C4, Si4C3, and Si4C4 Using Ab Initio Molecular Dynamics. J. Chem. Phys. 120 (9), 4333–4343. doi:10.1063/1.1636717
Bilbrey, J. A., Kazez, A. H., Locklin, J., and Allen, W. D. (2013). Exact Ligand Cone Angles. J. Comput. Chem. 34 (14), 1189–1197. doi:10.1002/jcc.23217
Chen, W., Li, Z.-R., Wu, D., Gu, F.-L., Hao, X.-Y., Wang, B.-Q., et al. (2004). The Static Polarizability and First Hyperpolarizability of the Water Trimer anion:Ab Initiostudy. J. Chem. Phys. 121 (21), 10489–10494. doi:10.1063/1.1811609
Chen, W., Li, Z.-R., Wu, D., Li, Y., Sun, C.-C., and Gu, F. L. (2005). The Structure and the Large Nonlinear Optical Properties of Li@Calix[4]pyrrole. J. Am. Chem. Soc. 127 (31), 10977–10981. doi:10.1021/ja050601w
Dennington, R. D., Keith, T. A., and Millam, J. M. (2016). GaussView, Version6. Shawnee Mission. KS: Semichem Inc.
Di Bella, S. (2001). Second-order Nonlinear Optical Properties of Transition Metal Complexes. Chem. Soc. Rev. 30 (6), 355–366. doi:10.1039/B100820J
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, Revision A.03. Wallingford CT, USA: Gaussian, Inc.
Fu, C.-C., Weissmann, M., Machado, M., and Ordejón, P. (2001). Ab Initiostudy of Silicon-Multisubstituted Neutral and Charged Fullerenes. Phys. Rev. B 63 (8), 085411. doi:10.1103/PhysRevB.63.085411
Golberg, D., Bando, Y., Stéphan, O., and Kurashima, K. (1998). Octahedral Boron Nitride Fullerenes Formed by Electron Beam Irradiation. Appl. Phys. Lett. 73 (17), 2441–2443. doi:10.1063/1.122475
He, H.-M., Li, Y., Yang, H., Yu, D., Li, S.-Y., Wu, D., et al. (2017a). Efficient External Electric Field Manipulated Nonlinear Optical Switches of All-Metal Electride Molecules with Infrared Transparency: Nonbonding Electron Transfer Forms an Excess Electron Lone Pair. J. Phys. Chem. C 121 (1), 958–968. doi:10.1021/acs.jpcc.6b11919
He, H.-M., Li, Y., Yang, H., Yu, D., Wu, D., Zhong, R.-L., et al. (2017b). Effects of the Cage Number and Excess Electron Number on the Second Order Nonlinear Optical Response in Molecular All-Metal Electride Multicage Chains. J. Phys. Chem. C 121 (45), 25531–25540. doi:10.1021/acs.jpcc.7b06464
He, H.-M., Luis, J. M., Chen, W.-H., Yu, D., Li, Y., Wu, D., et al. (2019). Nonlinear optical response of endohedral all-metal electride cages 2e−Mg2+(M@E12)2−Ca2+ (M = Ni, Pd, and Pt; E = Ge, Sn, and Pb). J. Mat. Chem. C 7 (3), 645–653. doi:10.1039/C8TC05647A
Ja Lee, M., Piao, M., Jeong, M.-Y., Hae Lee, S., Min Kang, K., Jeon, S.-J., et al. (2003). Novel azo octupoles with large first hyperpolarizabilities. J. Mat. Chem. 13 (5), 1030–1037. Electronic supplementary information (ESI) is available: molar absorptivity spectra of 1a,d,e, P1 and P2 in THF and plots of I2ω vs. number density for 1a,d,e and 2a,c,e in THF at 1064 nm. See http://www.rsc.org/suppdata/jm/b3/b300777d/. doi:10.1039/b300777d
Kanis, D. R., Ratner, M. A., and Marks, T. J. (1994). Design and Construction of Molecular Assemblies with Large Second-Order Optical Nonlinearities. Quantum Chemical Aspects. Chem. Rev. 94 (1), 195–242. doi:10.1021/cr00025a007
Karamanis, P., Otero, N., and Pouchan, C. (2014). Unleashing the Quadratic Nonlinear Optical Responses of Graphene by Confining White-Graphene (H-BN) Sections in its Framework. J. Am. Chem. Soc. 136 (20), 7464–7473. doi:10.1021/ja502631w
Koi, N., Oku, T., Narita, I., and Suganuma, K. (2005). Synthesis of Huge Boron Nitride Cages. Diam. Relat. Mater. 14 (3), 1190–1192. doi:10.1016/j.diamond.2004.12.018
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 37 (2), 785–789. doi:10.1103/PhysRevB.37.785
Li, J., Xia, Y., Zhao, M., Liu, X., Song, C., Li, L., et al. (2007). Theoretical Prediction for the (AlN)12 Fullerene-like Cage-Based Nanomaterials. J. Phys. Condens. Matter 19 (34), 346228. doi:10.1088/0953-8984/19/34/346228
Li, Y., Li, Z.-R., Wu, D., Li, R.-Y., Hao, X.-Y., and Sun, C.-C. (2004). An Ab Initio Prediction of the Extraordinary Static First Hyperpolarizability for the Electron-Solvated Cluster (FH)2%7BBe%7D(HF). J. Phys. Chem. B 108 (10), 3145–3148. doi:10.1021/jp036808y
Liu, Z.-B., Zhou, Z.-J., Li, Y., Li, Z.-R., Wang, R., Li, Q.-Z., et al. (2010). Push-pull Electron Effects of the Complexant in a Li Atom Doped Molecule with Electride Character: a New Strategy to Enhance the First Hyperpolarizability. Phys. Chem. Chem. Phys. 12 (35), 10562–10568. doi:10.1039/c004262e
Liu, Z., Wang, X., Liu, G., Zhou, P., Sui, J., Wang, X., et al. (2013). Low-density Nanoporous Phases of Group-III Nitrides Built from Sodalite Cage Clusters. Phys. Chem. Chem. Phys. 15 (21), 8186–8198. doi:10.1039/C3CP50814E
Luo, Z.-Z., Lin, C.-S., Zhang, W.-L., Zhang, H., He, Z.-Z., and Cheng, W.-D. (2014). Ba8Sn4S15: A Strong Second Harmonic Generation Sulfide with Zero-Dimensional Crystal Structure. Chem. Mat. 26 (2), 1093–1099. doi:10.1021/cm403369m
Ma, F., Li, Z.-R., Xu, H.-L., Li, Z.-J., Li, Z.-S., Aoki, Y., et al. (2008). Lithium Salt Electride with an Excess Electron Pair-A Class of Nonlinear Optical Molecules for Extraordinary First Hyperpolarizability. J. Phys. Chem. A 112 (45), 11462–11467. doi:10.1021/jp8040023
Marder, S. R., Gorman, C. B., Meyers, F., Perry, J. W., Bourhill, G., Brédas, J.-L., et al. (1994). A Unified Description of Linear and Nonlinear Polarization in Organic Polymethine Dyes. Science 265 (5172), 632–635. doi:10.1126/science.265.5172.632
Maria, M., Iqbal, J., and Ayub, K. (2016). Theoretical Study of the Non Linear Optical Properties of Alkali Metal (Li, Na, K) Doped Aluminum Nitride Nanocages. RSC Adv. 6 (96), 94228–94235. doi:10.1039/C6RA21797D
Maroulis, G. (1996). Electric Properties of Carbon Tetrafluoride. Chem. Phys. Lett. 259 (5), 654–660. doi:10.1016/0009-2614(96)00728-2
Meyers, F., Marder, S. R., Pierce, B. M., and Bredas, J. L. (1994). Electric Field Modulated Nonlinear Optical Properties of Donor-Acceptor Polyenes: Sum-Over-States Investigation of the Relationship between Molecular Polarizabilities (.alpha., .beta., and .gamma.) and Bond Length Alternation. J. Am. Chem. Soc. 116 (23), 10703–10714. doi:10.1021/ja00102a040
Muhammad, S., Xu, H.-L., Zhong, R.-L., Su, Z.-M., Al-Sehemi, A. G., and Irfan, A. (2013). Quantum Chemical Design of Nonlinear Optical Materials by Sp2-Hybridized Carbon Nanomaterials: Issues and Opportunities. J. Mat. Chem. C 1 (35), 5439–5449. doi:10.1039/c3tc31183j
Muhammad, S., Xu, H., Liao, Y., Kan, Y., and Su, Z. (2009). Quantum Mechanical Design and Structure of the Li@B10H14 Basket with a Remarkably Enhanced Electro-Optical Response. J. Am. Chem. Soc. 131 (33), 11833–11840. doi:10.1021/ja9032023
Nakano, M., Fujita, H., Takahata, M., and Yamaguchi, K. (2002). Theoretical Study on Second Hyperpolarizabilities of Phenylacetylene Dendrimer: Toward an Understanding of Structure−Property Relation in NLO Responses of Fractal Antenna Dendrimers. J. Am. Chem. Soc. 124 (32), 9648–9655. doi:10.1021/ja0115969
Niu, M., Yu, G., Yang, G., Chen, W., Zhao, X., and Huang, X. (2014). Doping the Alkali Atom: An Effective Strategy to Improve the Electronic and Nonlinear Optical Properties of the Inorganic Al12N12 Nanocage. Inorg. Chem. 53 (1), 349–358. doi:10.1021/ic4022917
Oku, T., Nishiwaki, A., and Narita, I. (2004). Formation and Atomic Structure of B12N12 Nanocage Clusters Studied by Mass Spectrometry and Cluster Calculation. Sci. Technol. Adv. Mater. 5 (5), 635–638. doi:10.1016/j.stam.2004.03.017
Priyadarshy, S., Therien, M. J., and Beratan, D. N. (1996). Acetylenyl-Linked, Porphyrin-Bridged, Donor−Acceptor Molecules: A Theoretical Analysis of the Molecular First Hyperpolarizability in Highly Conjugated Push−Pull Chromophore Structures. J. Am. Chem. Soc. 118 (6), 1504–1510. doi:10.1021/ja952690q
Saeedi, M., Anafcheh, M., Ghafouri, R., and Hadipour, N. L. (2013). A Computational Investigation of the Electronic Properties of Octahedral Al n N n and Al n P N Cages (n = 12, 16, 28, 36, and 48). Struct. Chem. 24 (2), 681–689. doi:10.1007/s11224-012-0119-7
Silaghi-Dumitrescu, I., Lara-Ochoa, F., and Haiduc, I. (1996). A12B12 (A = B,Al; B = N,P) Fullerene-like Cages and Their Hydrogenated Forms Stabilized by Exohedral Bonds. An AM1 Molecular Orbital Study. J. Mol. Struct. THEOCHEM 370 (1), 17–23. doi:10.1016/s0166-1280(96)04677-5
Srivastava, A. K., Misra, N., and Pandey, S. K. (2018). DFT Study on Planar (CaO)n Rings (N = 1-5) and Their Hydrogen Storage Behavior: Ca-O versus Mg-O Clusters. J. Clust. Sci. 29 (1), 57–65. doi:10.1007/s10876-017-1306-x
Stafström, S., Hultman, L., and Hellgren, N. (2001). Predicted Stability of A New aza[60]Fullerene Molecule, C48N12. Chem. Phys. Lett. 340 (3), 227–231. doi:10.1016/S0009-2614(01)00418-3
Strout, D. L. (2000). Structure and Stability of Boron Nitrides: Isomers of B12N12. J. Phys. Chem. A 104 (15), 3364–3366. doi:10.1021/jp994129a
Sun, W.-M., Li, X.-H., Wu, D., Li, Y., He, H.-M., Li, Z.-R., et al. (2016). A Theoretical Study on Superalkali-Doped Nanocages: Unique Inorganic Electrides with High Stability, Deep-Ultraviolet Transparency, and a Considerable Nonlinear Optical Response. Dalton Trans. 45 (17), 7500–7509. doi:10.1039/C6DT00342G
Sun, W.-M., Wu, D., Li, Y., Liu, J.-Y., He, H.-M., and Li, Z.-R. (2015). A Theoretical Study on Novel Alkaline Earth-Based Excess Electron Compounds: Unique Alkalides with Considerable Nonlinear Optical Responses. Phys. Chem. Chem. Phys. 17 (6), 4524–4532. doi:10.1039/C4CP04951A
Tawada, Y., Tsuneda, T., Yanagisawa, S., Yanai, T., and Hirao, K. (2004). A Long-Range-Corrected Time-dependent Density Functional Theory. J. Chem. Phys. 120 (18), 8425–8433. doi:10.1063/1.1688752
Ullah, F., Kosar, N., Ali, A., MariaMahmood, T., Mahmood, T., and Ayub, K. (2020). Design of Novel Inorganic Alkaline Earth Metal Doped Aluminum Nitride Complexes (AEM@Al12N12) with High Chemical Stability, Improved Electronic Properties and Large Nonlinear Optical Response. Optik 207, 163792. doi:10.1016/j.ijleo.2019.163792
Wang, J.-J., Zhou, Z.-J., Bai, Y., Liu, Z.-B., Li, Y., Wu, D., et al. (2012). The Interaction between Superalkalis (M3O, M = Na, K) and a C20F20 Cage Forming Superalkali Electride Salt Molecules with Excess Electrons inside the C20F20 Cage: Dramatic Superalkali Effect on the Nonlinear Optical Property. J. Mat. Chem. 22 (19), 9652–9657. doi:10.1039/C2JM15405F
Wang, R., Zhang, D., and Liu, C. (2005). Theoretical Prediction of a Novel Inorganic Fullerene-like Family of Silicon-Carbon Materials. Chem. Phys. Lett. 411 (4), 333–338. doi:10.1016/j.cplett.2005.06.055
Wang, S.-J., Wang, Y.-F., and Cai, C. (2015). Multidecker Sandwich Cluster VnBenn+1(n= 1, 2, 3, 4) as a Polarizable Bridge for Designing 1D Second-Order NLO Chromophore: Metal−π Sandwich Multilayer Structure as a Particular Charge-Transfer Axis for Constructing Multidimensional NLO Molecules. J. Phys. Chem. C 119 (28), 16256–16262. doi:10.1021/acs.jpcc.5b04656
Wang, X.-B., Ding, C.-F., and Wang, L.-S. (1999). High Resolution Photoelectron Spectroscopy of C60 −Anions and Accurate Determination of the Electron Affinity of C60. J. Chem. Phys. 110 (17), 8217–8220. doi:10.1063/1.478732
Wu, H.-Q., Xu, H.-L., Sun, S.-L., and Su, Z.-M. (2014). Li Doped Effect of through Novel Noncovalent Charge Transfer on Nonlinear Optical Properties. Dyes Pigments 106, 7–13. doi:10.1016/j.dyepig.2014.01.031
Wu, H.-S., Zhang, F.-Q., Xu, X.-H., Zhang, C.-J., and Jiao, H. (2003). Geometric and Energetic Aspects of Aluminum Nitride Cages. J. Phys. Chem. A 107 (1), 204–209. doi:10.1021/jp027300i
Xiao, D., Bulat, F. A., Yang, W., and Beratan, D. N. (2008). A Donor−Nanotube Paradigm for Nonlinear Optical Materials. Nano Lett. 8 (9), 2814–2818. doi:10.1021/nl801388z
Xu, H.-L., Li, Z.-R., Wu, D., Ma, F., Li, Z.-J., and Gu, F. L. (2009). Lithiation and Li-Doped Effects of [5]Cyclacene on the Static First Hyperpolarizability. J. Phys. Chem. C 113 (12), 4984–4986. doi:10.1021/jp806864w
Xu, H.-L., Li, Z.-R., Wu, D., Wang, B.-Q., Li, Y., Gu, F. L., et al. (2007). Structures and Large NLO Responses of New Electrides: Li-Doped Fluorocarbon Chain. J. Am. Chem. Soc. 129 (10), 2967–2970. doi:10.1021/ja068038k
Xu, X., Hu, C.-L., Kong, F., Zhang, J.-H., Mao, J.-G., and Sun, J. (2013). Cs2GeB4O9: a New Second-Order Nonlinear-Optical Crystal. Inorg. Chem. 52 (10), 5831–5837. doi:10.1021/ic302774h
Yanai, T., Tew, D. P., and Handy, N. C. (2004). A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3lyp). Chem. Phys. Lett. 393 (1), 51–57. doi:10.1016/j.cplett.2004.06.011
Yang, Z., Shi, L., Chen, L., Gu, Y., Cai, P., Zhao, A., et al. (2005). Synthesis, Characterization and Properties of Novel BN Nanocages from a Single-Source Precursor. Chem. Phys. Lett. 405 (1), 229–233. doi:10.1016/j.cplett.2005.02.041
Yu, G., Zhao, X., Niu, M., Huang, X., Zhang, H., and Chen, W. (2013). Constructing a Mixed π-conjugated Bridge: a Simple and Effective Approach to Realize a Large First Hyperpolarizability in Carbon Nanotube-Based Systems. J. Mat. Chem. C 1 (24), 3833–3841. doi:10.1039/c3tc00022b
Zhang, D., and Zhang, R. Q. (2005). Geometrical Structures and Electronic Properties of AlN Fullerenes: A Comparative Theoretical Study of AlN Fullerenes with BN and C Fullerenes. J. Mat. Chem. 15 (29), 3034–3038. doi:10.1039/B503724G
Zhang, Q., Chung, I., Jang, J. I., Ketterson, J. B., and Kanatzidis, M. G. (2009). Chalcogenide Chemistry in Ionic Liquids: Nonlinear Optical Wave-Mixing Properties of the Double-Cubane Compound [Sb7S8Br2](AlCl4)3. J. Am. Chem. Soc. 131 (29), 9896–9897. doi:10.1021/ja903881m
Zhang, T.-G., Zhao, Y., Asselberghs, I., Persoons, A., Clays, K., and Therien, M. J. (2005). Design, Synthesis, Linear, and Nonlinear Optical Properties of Conjugated (Porphinato)Zinc(II)-Based Donor−Acceptor Chromophores Featuring Nitrothiophenyl and Nitrooligothiophenyl Electron-Accepting Moieties. J. Am. Chem. Soc. 127 (27), 9710–9720. doi:10.1021/ja0402553
Zhong, R.-L., Xu, H.-L., Li, Z.-R., and Su, Z.-M. (2015). Role of Excess Electrons in Nonlinear Optical Response. J. Phys. Chem. Lett. 6 (4), 612–619. doi:10.1021/jz502588x
Zhong, R.-L., Xu, H.-L., Muhammad, S., Zhang, J., and Su, Z.-M. (2012a). The Stability and Nonlinear Optical Properties: Encapsulation of an Excess Electron Compound LiCN⋯Li within Boron Nitride Nanotubes. J. Mat. Chem. 22 (5), 2196–2202. doi:10.1039/C1JM14358A
Zhong, R.-L., Xu, H.-L., Sun, S.-L., Qiu, Y.-Q., and Su, Z.-M. (2012b). The Excess Electron in a Boron Nitride Nanotube: Pyramidal NBO Charge Distribution and Remarkable First Hyperpolarizability. Chem. Eur. J. 18 (36), 11350–11355. doi:10.1002/chem.201201570
Zhou, Z.-J., Yu, G.-T., Ma, F., Huang, X.-R., Wu, Z.-J., and Li, Z.-R. (2014). Theoretical Investigation on Nonlinear Optical Properties of Carbon Nanotubes with Stone-Wales Defect Rings. J. Mat. Chem. C 2 (2), 306–311. doi:10.1039/c3tc31904k
Keywords: nonlinear optical response (NLO), excess electron compound, aluminum nitride nanocage, first hyperpolarizabilities, density functional theory
Citation: He H-M, Yang H, Li Y and Li Z-R (2022) Theoretical Study of Alkaline-Earth Metal (Be, Mg, and Ca)-Substituted Aluminum Nitride Nanocages With High Stability and Large Nonlinear Optical Responses. Front. Chem. 10:918704. doi: 10.3389/fchem.2022.918704
Received: 12 April 2022; Accepted: 16 May 2022;
Published: 21 June 2022.
Edited by:
Ambrish Kumar Srivastava, Deen Dayal Upadhyay Gorakhpur University, IndiaReviewed by:
Sarvesh Kumar Pandey, Indian Institute of Science (IISc), IndiaRuby Srivastava, Centre for Cellular and Molecular Biology (CCMB), India
Copyright © 2022 He, Yang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Yang, eWFuZ2h1aTA5MTkwMEAxMjYuY29t