 Jie Hu
Jie Hu Panpan Wang2,3†
Panpan Wang2,3†
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 13 June 2022
Sec. Inorganic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.914699
This article is part of the Research Topic Non-precious Metal-based Materials: Design, Fundamental and Functionality View all 8 articles
The organic small molecule fuel battery has attracted wild attention in recent years. Unfortunately, the inherent catalyst poisoning phenomenon hinders its commercialization. Exploring the anodic catalytic reaction mechanism is urgent. This article investigates the nucleation mechanism of HCOOH on the catalyst electrode surface. The electrochemical results indicate that the HCOOH oxidation conforms to the two-dimensional instantaneous nucleation process. The corresponding adsorption model of CO on the catalyst surface was finally established.
Developing clean energy is the key to solving environmental pollution and energy crisis problems (Xu et al., 2020a; Shuai et al., 2020; Xu et al., 2021). The organic small molecule fuel battery has attracted wild attention in recent years due to its high efficiency, friendly environment and so on (Xu et al., 2020b). However, the electrochemical oxidation process and the catalyst poisoning phenomenon of organic molecules are unclear, severely restricting its development. Thus, it is necessary to develop research on the anodic reaction mechanism of organic small molecules on the catalyst electrode surface, which can provide a new idea to improve the fuel battery efficiency.
The electrocatalytic oxidation reaction of formic acid (HCOOH) on the metal surface is often used as a reaction model to study the structure-performance relationship of other complex catalytic systems due to its simple molecular structure and favorable sensitivity on the electrocatalyst surface (Chen et al., 2006a). In recent years, various electrochemical techniques have been used to study the effect of different factors on the electrocatalytic oxidation mechanism of HCOOH (Samjeské et al., 2005; Chen et al., 2006b; Mukouyama et al., 2006; Osawa et al., 2011). In addition, the calculation of HCOOH oxidation based on DFT theory has also been developed rapidly (Wang and Liu, 2015). Parson proposed that the oxidation of formic acid on the catalyst electrode surface may follow a dual pathway mechanism, namely, the indirect pathway and the direct pathway (Capon and Parsons, 1973). In the direct pathway, formic acid removes H atomic to produce CO2. In the indirect pathway, formic acid first dehydrates to produce CO toxic intermediate, closely adsorbed on the electrode surface, and CO is oxidized to CO2 with the increase in the electric potential (Watanabe and Motoo, 1975).
Although there are many studies on the electrochemical oxidation mechanism of HCOOH on the catalyst electrode surface, they mainly focus on detecting the dissociation of adsorbed species of organic small molecules on the electrode surface. The reports of nucleation/growth dynamics are very few, which can provide abundant information about the molecular mechanism with the electrode surface. The phenomenon of the solid-phase transition is widespread in the electrode surface, such as metal electrodeposition, Ag2O and AgO transformation, and adsorption-desorption process (Gómez et al., 1992). Sun et al. obtained the i-t transient current curves of HCOOH oxidation on catalyst electrode surface at different adsorption times using the electrical step experiment. However, they did not discuss the nucleation and growth kinetics of adsorptive species on the catalyst electrode surface (Sun et al., 1992). Therefore, it is necessary and significant to explore the nucleation mechanism and kinetic process of HCOOH on the catalyst electrode surface. The dimensionless processed transient curves confirmed that the nucleation/growth process of HCOOH on the catalyst electrode surface followed the diffusion-controlled two-dimensional (2D) instantaneous nucleation model. This groundbreaking research can help us understand the redox process mechanism of HCOOH and provide a theoretical basis for choosing high-performance fuel battery catalysts.
All chemicals were of analytical grade. The testing solution is 0.5 mmol/L HCOOH + 0.1 mmol/L HClO4. The electrochemical measurements were conducted using CHI 660E electrochemical workstation at room temperature. A conventional three-electrode system was conducted with catalyst as the counter electrode, Ag/AgCl as the reference electrode, and catalyst electrode as the working electrode. The electric step parameters are as shown in Scheme 1.
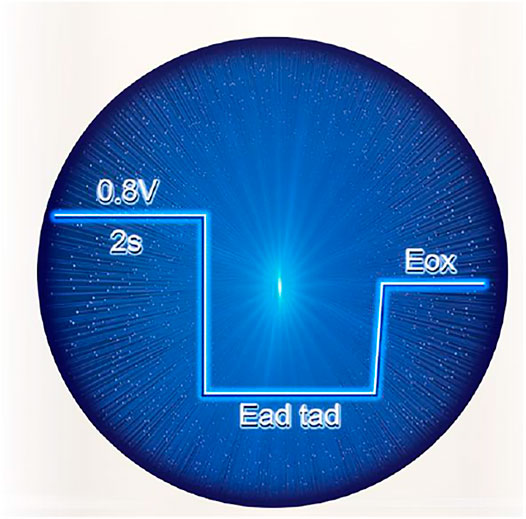
SCHEME 1. Multi-potential step parameter.
Figure 1 shows the i-t curves of HCOOH on the catalyze electrode surface, which can reflect the dissociation and adsorption process of the intermediate HCOOH reaction. The net current curves were obtained by subtracting the background current; namely, the adsorption time was 0 ms, which can reflect the oxidation current of the adsorbed CO. It can be found that the oxidation peak became more obvious and the peak type changed from high and thin to low and fat with the increase in the adsorption time. In addition, all oxidation processes were completed within 100 ms, which was related to the low HCOOH concentration.
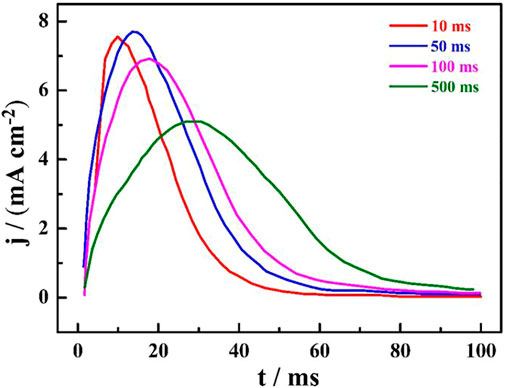
FIGURE 1. The i-t net current curves of HCOOH on a single crystal catalyst electrode surface at different adsorption times.
Figure 2 shows that the i-t curves display a typical electrocrystallization nucleation characteristic, which is the appearance of the maximum peak current in the transient curves. Although the mathematical expression models of the metal nucleation are abundant, the most widely used is the dimensionless expression with undetermined parameters in the research process of electrocrystallization mechanism using i-t transient curves, which can be expressed as follows (Fleiu et al., 1994):
where Im and tm denote the maximum current density (A.cm−2) and its corresponding time, respectively. Comparing the obtained I/Im-t/tm curves with the theoretical nucleation curves obtained in Eqs 1, 2 and confirming the solid phase nucleation mode of CO on the catalyst electrode surface, the experimental results show that the nucleation process of the oxidation intermediates of HCOOH (CO) on the single crystal catalyst electrode surface conforms to the 2D instantaneous nucleation model at any testing time. It is worth noting that the coincidence degree of the obtained curves with the 2D instantaneous nucleation curve is higher with the extension of testing time.
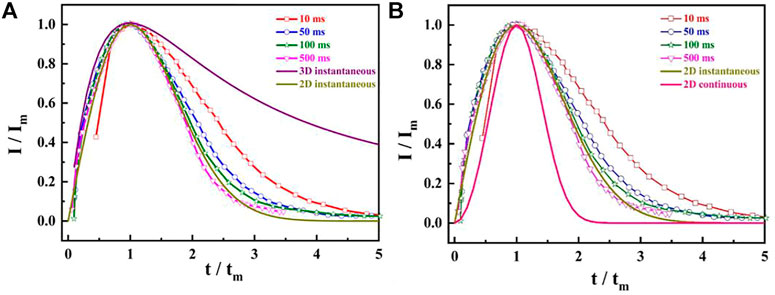
FIGURE 2. The dimensionless (I/Im ∼ t/tm) curves at different adsorption times: (A) 2D and 3D instantaneous model; (B) 2D instantaneous and continuous model.
The dimensionless expressions of 2D instantaneous nucleation can be further arranged as follows (Bewick et al., 1962):
To further verify the above experimental conclusion, the experimental data were further processed according to Formula 3, and the results are shown in Figure 3. It can be seen that the In (I/t) ∼t2 curves under different adsorption times displayed a good linear relationship with the obtained theoretical curves.
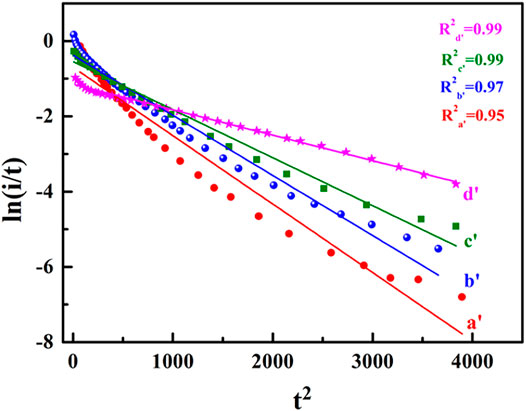
FIGURE 3. The In (I/t) ∼t2 curves of HCOOH on a single crystal catalyst electrode.
The results further confirmed that the CO oxidation process of HCOOH hydrolysis and adsorption on the surface of the single crystal catalyst electrode was a 2D instantaneous nucleation process, and the curve fitting degree increased with the increase of adsorption time.
According to the above experimental results, the adsorption model of the HCOOH oxidation intermediate CO at the catalyst electrode surface is shown in Figure 4. Based on the definition of 2D instantaneous nucleation, assuming that the adsorbed CO on the catalyst electrode surface can be seen as the independent island, the small islands could collide fusion and gradually occupy the electrode surface with the extension of adsorption time. Finally, the catalyst electrode surface was completely dominated by CO molecules and lost the catalytic activity, namely, the catalyst poisoning. CO was oxidized to CO2 when the applied signal reached oxidation potential, and the catalyst electrode was exposed again to restore the catalytic activity. Moreover, atoms can promote the activation of H2O molecules to form OHads and reaction with CO to form CO2 and further promote the HCOOH oxidation on the electrode surface.
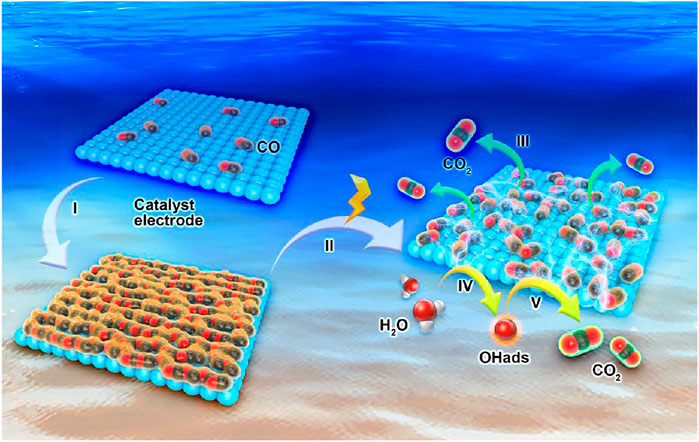
FIGURE 4. Scheme of the CO adsorption model.
In conclusion, the experimental result indicated that the adsorption of HCOOH oxidation intermediates (CO) on the catalyst electrode surface conformed to the 2D instantaneous nucleation mechanism at any testing adsorption time and the corresponding nucleation model was established. This research can help us understand the redox process mechanism of HCOOH and provide a theoretical basis for choose of high-performance fuel battery catalysts.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
JH: conceptualization, methodology, writing—original draft. PW: data curation, formal analysis, investigation. CC: methodology, resources, writing—reviewing and editing.
Author PW was employed by the China Merchants Testing Vehicle Technology Research Institute Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bewick, A., Fleischmann, M., and Thirsk, H. R. (1962). Kinetics of the Electrocrystallization of Thin Films of Calomel. Trans. Faraday Soc. 58, 2200–2216. doi:10.1039/tf9625802200
Capon, A., and Parsons, R. (1973). The Oxidation of Formic Acid on Noble Metal Electrodes. J. Electroanal. Chem. Interfacial Electrochem. 44, 239–254. doi:10.1016/s0022-0728(73)80250-5
Chen, Y. X., Heinen, M., Jusys, Z., and Behm, R. J. (2006a). Bridge-Bonded Formate: Active Intermediate or Spectator Species in Formic Acid Oxidation on a Pt Film Electrode? Langmuir 22, 10399–10408. doi:10.1021/la060928q
Chen, Y. X., Heinen, M., Jusys, Z., and Behm, R. J. (2006b). Kinetics and Mechanism of the Electrooxidation of Formic Acid-Spectroelectrochemical Studies in a Flow Cell. Angew. Chem. Int. Ed. 45, 981–985. doi:10.1002/anie.200502172
Fleiu, J. M., Orts, J. M., Gomez, R., and Aldaz, A. (1994). New Information on the Unusual Adsorption States of Pt(111) in Sulphuric Acid Solutions from Potentiostatic Adsorbate Replacement by CO. J. Electroanal. Chem. 372, 265–268. doi:10.1016/0022-0728(93)03259-R
Gómez, E., Muller, C., and Proud, W. G. (1992). Electrodeposition of Nickel on Vitreous Carbon: Influence of Potential on Deposit Morphology. J. Apple Electrochem 22, 872–876. doi:10.1007/BF01023732
Mukouyama, Y., Kikuchi, M., Samjeské, G., Osawa, M., and Okamoto, H. (2006). Potential Oscillations in Galvanostatic Electrooxidation of Formic Acid on Platinum: A Mathematical Modeling and Simulation. J. Phys. Chem. B 110, 11912–11917. doi:10.1021/jp061129j
Osawa, M., Komatsu, K.-i., Samjeské, G., Uchida, T., Ikeshoji, T., Cuesta, A., et al. (2011). The Role of Bridge-Bonded Adsorbed Formate in the Electrocatalytic Oxidation of Formic Acid on Platinum. Angew. Chem. Int. Ed. 50, 1159–1163. doi:10.1002/anie.201004782
Samjeské, G., Miki, A., Ye, S., Yamakata, A., Mukouyama, Y., Okamoto, H., et al. (2005). Potential Oscillations in Galvanostatic Electrooxidation of Formic Acid on Platinum: A Time-Resolved Surface-Enhanced Infrared Study. J. Phys. Chem. B 109, 23509–23516. doi:10.1021/jp055220j
Shuai, H., Xu, J., and Huang, K. (2020). Progress in Retrospect of Electrolytes for Secondary Magnesium Batteries. Coord. Chem. Rev. 422, 213478. doi:10.1016/j.ccr.2020.213478
Sun, S. G., and Yang, Y. (1999). Studies of Kinetics of HCOOH Oxidation on Pt(100), Pt(110), Pt(111), Pt(510) and Pt(911) Single Crystal Electrodes. J. Electroanal. Chem. 467, 121–131. doi:10.1016/S0022-0728(99)00032-7
Wang, H. F., and Liu, Z. P. (2015). Formic Acid Oxidation at Pt/H2O Interface from Periodic DFT Calculations Integrated with a Continuum Solvation Model. J. Phys. Chem. C 113, 17502–17508. doi:10.1021/jp9059888
Watanabe, M., and Motoo, S. (1975). Electrocatalysis by Ad-Atoms. J. Electroanal. Chem. Interfacial Electrochem. 60, 267–273. doi:10.1016/s0022-0728(75)80261-0
Xu, J., Wei, Z., Tang, L., Wang, A., Zhang, Y., Qiao, Y., et al. (2020). Effects of Short Pulse Current on the Voltage Delay Behavior of Magnesium Battery. J. Power Sources 454, 227869. doi:10.1016/j.jpowsour.2020.227869
Xu, J., Wang, P., Yu, R., Zheng, Z., Shoaib Ahmad Shah, S., and Chen, C. (2020b). A New Insight into the Effect of Scan Rate and Mass Transport from Pt Rotating Disk Electrode on the Electrochemical Oxidation Process of Methanol. Mater. Lett. 260, 126950. doi:10.1016/j.matlet.2019.126950
Keywords: adhesion, crystal structure, interfaces, catalyst, nucleation process
Citation: Hu J, Wang P and Chen C (2022) The Electrochemical Oxidation and Mass Transfer Mechanism of Formic Acid on the Catalyst Electrode Surface. Front. Chem. 10:914699. doi: 10.3389/fchem.2022.914699
Received: 07 April 2022; Accepted: 02 May 2022;
Published: 13 June 2022.
Edited by:
Xiaohang Zheng, Harbin Institute of Technology, ChinaCopyright © 2022 Hu, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Hu, amllaHU2Njg4OEAxNjMuY29t; Changguo Chen, Y2djaGVuQGNxdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.