
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 02 June 2022
Sec. Chemical Physics and Physical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.899461
This article is part of the Research Topic Electronic and Nuclear Quantum Dynamics of Molecules in Intense Laser Fields View all 13 articles
 Tim Bayer
Tim Bayer  Matthias Wollenhaupt*
Matthias Wollenhaupt*Atomic and molecular free electron vortices (FEVs), characterized by their spiral-shaped momentum distribution, have recently attracted a great deal of attention due to their varied shapes and their unusual topological properties. Shortly after their theoretical prediction by the single-photon ionization (SPI) of He atoms using pairs of counterrotating circularly polarized attosecond pulses, FEVs have been demonstrated experimentally by the multiphoton ionization (MPI) of alkali atoms using single-color and bichromatic circularly polarized femtosecond pulse sequences. Recently, we reported on the analysis of the experimental results employing a numerical model based on the ab initio solution of the time-dependent Schrödinger equation (TDSE) for a two-dimensional (2D) atom interacting with a polarization-shaped ultrashort laser field. Here, we apply the 2D TDSE model to study molecular FEVs created by SPI and MPI of a diatomic molecule using polarization-tailored single-color and bichromatic femtosecond pulse sequences. We investigate the influence of the coupled electron-nuclear dynamics on the vortex formation dynamics and discuss the effect of CEP- and rotational averaging on the photoelectron momentum distribution. By analyzing how the molecular structure and dynamics is imprinted in the photoelectron spirals, we explore the potential of molecular FEVs for ultrafast spectroscopy.
Vortex beams, such as optical vortices (Babiker et al., 2002; Shen et al., 2019; Eickhoff et al. 2020b) and electron vortex beams (Verbeeck et al., 2010; Lloyd et al., 2017), which are characterized by their helical wave fronts, are currently attracting much attention both theoretically and experimentally. Recently, the analogies between free electron vortices (FEVs) in transmission electron microscopy and multiphoton ionization (MPI) have been pointed out (Eickhoff et al., 2020c). In photoionization, FEVs are created by ionizing a quantum system with tailored ultrashort circularly polarized laser pulse sequences. Introducing a time-delay τ between two counterrotating circularly polarized subpulses results in an “unusual kind of Ramsey interference” Ngoko Djiokap et al. (2015), which gives rise to an azimuthal interference pattern in the photoelectron momentum distribution (PMD) forming a multi-armed Archimedean spiral. Atomic FEVs have been studied theoretically employing different numerical methods, including the ab initio solution of the time-dependent Schrödinger equation (TDSE) (Ngoko Djiokap et al. 2015; Ngoko Djiokap et al., 2016; Djiokap and Starace., 2017; Ngoko Djiokap et al., 2017; Ivanov et al., 2017; Jia et al., 2019; Xiao et al., 2019; Bayer et al., 2020; Qin et al., 2020; Wang et al., 2020; Zhen et al., 2020; Geng et al., 2020; Maxwell et al., 2020; Djiokap et al., 2021; Geng et al., 2021), calculations based on the strong-field approximation (Hasović et al., 2016; Ivanov et al., 2017; Li M. et al., 2017; Li et al., 2018a; Li et al., 2018b; Gazibegović-Busuladžić et al., 2018; Kong et al., 2018; Li et al., 2019b; Xiao et al., 2019; Geng et al., 2020; Maxwell et al. 2020; Qin et al., 2020; Geng et al., 2021; Becker and Milošević, 2022), perturbative analytical approaches (Ngoko Djiokap et al., 2015; Ngoko Djiokap et al., 2016; Djiokap and Starace, 2017; Ngoko Djiokap et al., 2017; Djiokap et al., 2021), emerging techniques such as the
The first experimental demonstration of atomic FEVs was reported in (Pengel et al., 2017b). By MPI of potassium atoms with single-color time-delayed counterrotating circularly polarized pulse sequences, we observed FEVs with four-, six and eight-fold rotational symmetry (Pengel et al., 2017a; Pengel et al., 2017b). Subsequently, we demonstrated the creation of seven-fold rotationally symmetric FEVs by MPI of sodium atoms (Kerbstadt et al., 2019b) using time-delayed bichromatic sequences. In the same work, FEVs with odd rotational symmetry have been created using bichromatic fields in the limit τ → 0. In addition, we reported on crescent-shaped FEVs (Kerbstadt et al., 2019b; Eickhoff et al., 2020a; Eickhoff et al., 2021c) and a five-fold symmetric FEV (Eickhoff et al., 2021c) created by bichromatic MPI. In the strong-field ionization regime, Mancuso et al. observed crescent-shaped and three-fold symmetric FEVs using co- and counterrotating bichromatic fields, respectively (Mancuso et al., 2015, Mancuso et al., 2016). Similarly, Eckart et al. created three-fold symmetric FEVs (Eckart et al., 2016; Eckart et al., 2018) and measured the PMD using cold target recoil-ion momentum spectroscopy (Dörner et al., 2000; Ullrich et al., 2003). In our experiments, we applied photoelectron tomography (Wollenhaupt et al., 2009b; Wollenhaupt et al., 2013) to reconstruct the 3D PMD of the atomic FEVs (Pengel et al., 2017a; Pengel et al., 2017b; Kerbstadt et al., 2019b; Eickhoff et al., 2021c). Recently, this method has been developed further into a holographic scheme to reconstruct the photoelectron wave function by measuring interferograms in the PMD (Eickhoff et al., 2020a; Eickhoff et al., 2021b; Eickhoff et al., 2021c).
Meanwhile, the theoretical research focus has evolved from atomic FEVs towards the investigation of FEVs created on molecular systems. Bandrauk and coworkers were the first to recognize the potential of molecular FEVs as spectroscopic tools sensitive to the molecular geometry and ultrafast electron dynamics by investigating the MPI of
Building on our recent numerical study on atomic FEVs (Bayer et al., 2020), here we apply our 2D TDSE model to numerically investigate molecular FEVs created by photoionization of a diatomic molecular ion with polarization-tailored laser pulses. Using polarization-shaped pulses, at least 2D numerical simulations are required to describe the generally asymmetric polarization profiles. In many cases, the 2D approach is sufficient to capture the essential features of the PMD. Compared to a full 3D calculation, 2D simulations benefit from a greatly reduced computational cost. Recently, we employed the 2D TDSE model to accurately reproduce atomic FEVs created experimentally by MPI of alkali atoms (Bayer et al., 2020) to validate our numerical approach. Considering a molecule aligned in the laser polarization plane, the 2D model is expected to yield similarly realistic results for molecular FEVs. The central inset to Figure 1A illustrates the 2D laser-molecule interaction by showing a counterrotating circularly polarized pulse sequence impinging on a diatomic molecule aligned in the laser polarization-plane along with the created 2D PMD. Compared to a full 3D calculation, the polar information of the PMD is lost. However, in the case of FEVs, this information is generally less relevant, since the characteristic spiral pattern is an azimuthal feature of the PMD which is fully contained in a central section parallel to the polarization plane. We note that, although the description in 2D “flatland” is considered an approximation of real space, 2D approaches are relevant for confined condensed matter systems such as surfaces, interfaces or monolayers of e.g., graphene (Novoselov et al., 2012) or transition-metal dichalcogenides (Li et al., 2022). In this contribution, we focus on three aspects of molecular FEVs which are relevant for the design of future experiments and provide insights into the rich variety of physical mechanisms behind the generation of molecular FEVs by polarization-shaped pulses. First, we compare the PMD from different ionization regimes, i.e., SPI vs. MPI, at a range of fixed internuclear separations. Besides the influence of the increased angular momentum transfer in MPI, we investigate to which extend intermediate resonances are involved in the formation of the FEVs. Second, we identify the fingerprints of the coupled electron-nuclear dynamics in the interference structures of the FEVs. To this end, we compare the PMDs from photoionization of a rigid and a vibrating molecule and study the interaction between nuclear motion and electronic ionization dynamics. Specifically, we discuss the role of transient electronic resonances arising during the vibration for FEVs created by MPI and examine signatures of non-adiabatic bound state dynamics in the PMD. Third, we investigate molecular FEVs in the molecular frame vs. laboratory frame by considering molecular rotation and optical phase averaging. We analyze the sensitivity of the PMD to the experimental parameters and define optimal conditions to observe the molecular FEVs in the experiment.
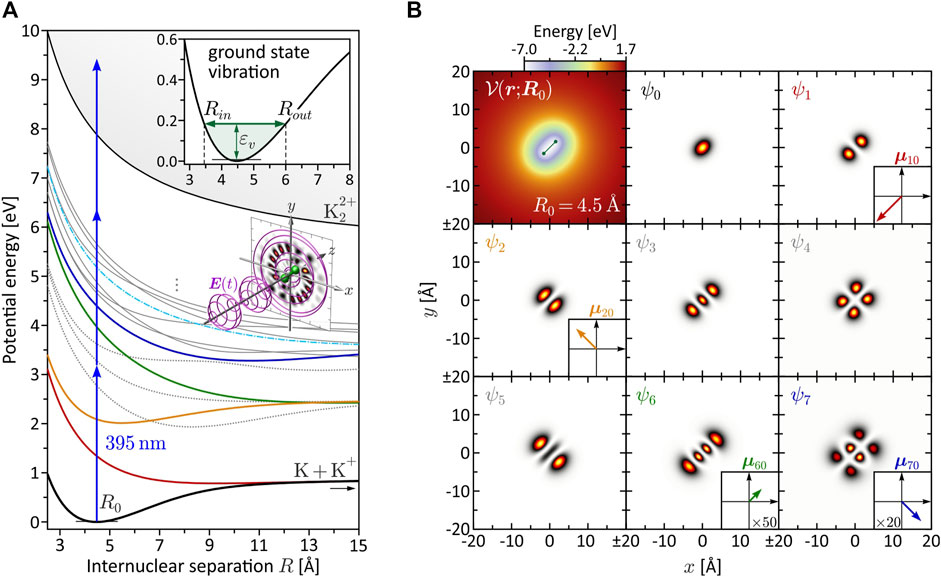
FIGURE 1. (Color online.) Characterization of the bound molecular system. (A) BO potential energy curves Vn(R). The equilibrium distance in the electronic ground state is found at R0 = 4.48 Å. Among the eight lowest states, only V1(R) (red), V2(R) (yellow), V6(R) (green) and V7(R) (blue) are dipole-coupled to the ground state. These states are relevant for the investigated MPI schemes. Especially state V6(R) plays a prominent role in the dynamic three-photon ionization scenario (see Section 3.2.2), because it becomes near-resonant with a pulse of central wavelength λ0 = 395 nm at the outer turning point Rout = 6.0 Å of the vibration (see inset). (B) Molecular potential
The paper is organized as follows. After the introduction of the numerical model and the characterization of the molecular system in Section 2, we start in Section 3.1 by investigating a rigid molecule interacting with laser pulses of different polarization in the SPI and MPI regime. In Section 3.2, we consider a vibrating molecule and focus on the creation of molecular FEVs via SPI and MPI in the presence of coupled electron-nuclear dynamics. Section 3.3 addresses different experimental averaging mechanisms which we discuss on a molecular FEV from bichromatic two- vs. three-photon ionization. In Section 4, we conclude the paper and give a brief outlook.
In this section, we provide the theoretical background to describe the interaction of a diatomic molecule with a polarization-shaped laser field in two dimensions, and present the numerical methods used to solve the 2D TDSE. The 2D TDSE model is based on established numerical techniques (Tannor, 2007; Bauer, 2017) and has been described in more detail in Bayer et al. (2020) for the generation of atomic FEVs. Here we focus on the extension of the model from an atomic to a molecular system. In particular, we describe our semi-classical approach to incorporate the vibrational nuclear dynamics into the 2D TDSE model, in order to investigate the interplay between the nuclear motion and the electronic excitation, i.e., the coupled electron-nuclear dynamics.
We consider the interaction of a diatomic molecular ion with a polarization-shaped ultrashort laser pulse propagating in the z-direction. The two atoms are arranged in the x-y-plane which coincides with the laser polarization plane. Their nuclei are separated by R = R eR, with eR being a unit vector parallel to the internuclear axis. Throughout this paper, we consider a homonuclear molecule. The x-y-coordinate frame is chosen such that
with r = (x, y) and erf denoting the error function. Motivated by our recent experiments on potassium atoms (Pengel et al., 2017b) and dimers (Bayer et al., 2013), we choose an effective nuclear charge of z = 0.9085 and a softcore parameter of a = 2.3065 Å (Eickhoff et al., 2021c) to mimic the singly charged potassium dimer
with me being the mass of the electron, for different values of R. The higher-lying Rydberg states are mainly characterized by Coulomb repulsion between the two nuclei and converge to the repulsive double-ionization potential Vi(R) (thin black line). The BO potentials aid us in the design of specific excitation scenarios and, in particular, serve to identify intermediate resonances encountered along the MPI pathways. The color coding among the lowest eight states ψn indicates their dipole-coupling to the electronic ground state ψ0. States plotted as gray dotted lines are not coupled to the ground state. The transition dipole moments μn0 = −e⟨ψn|r|ψ0⟩, with the electron charge −e, are determined using the electronic eigenfunctions ψn (r; R). The eigenfunctions ψn (r; R0) at the equilibrium distance R0 = 4.48 Å are shown in Figure 1B, along with the molecular potential
The temporal laser electric field is described in the spherical basis (Wollenhaupt et al., 2009a; Bayer et al., 2019; Kerbstadt et al., 2019b). We consider a bichromatic double pulse sequence of the general form (Eickhoff et al., 2021a; Eickhoff et al., 2021c).
Each subpulse n = 1, 2 is characterized by an individual peak amplitude
The laser-molecule interaction is described in the dipole approximation. Initially, we consider a rigid molecule with fixed internuclear separation R. As a consequence, the nuclear kinetic energy vanishes. The corresponding TDSE for the wave function ψ(r; R, t) of the valence electron in the length gauge reads
The TDSE is solved numerically using established methods described in Section 2.2. The ab initio TDSE calculation inherently includes all intermediate states, which are relevant for the MPI schemes discussed in Sections 3.2.2, 3.3. Therefore, no further approximations concerning the electronic structure of the molecule are required, as confirmed in Bayer et al. (2020). In our model, the electron dynamics is treated quantum mechanically, whereas the slower dynamics of the nuclei is treated classically, which is an established strategy to reduce the dimensionality of the calculation (Carrasco et al., 2022). Specifically, we model the vibrational nuclear dynamics by inserting a time-dependent function R = R(t) into the electronic potential
The TDSE in Eq. 4 is solved numerically on a discrete 2D spatial grid. The wave function of the active electron is propagated in time according to
where
where
Motivated by recent attosecond studies on molecular FEVs (Yuan et al., 2016; Djiokap et al., 2018; Guo et al., 2021), we start in Section 3.1 by considering a rigid diatomic molecule interacting with various standard pulse shapes, including simple linearly and circularly polarized pulses as well as counterrotating circularly polarized double pulse sequences (Pengel et al., 2017b). For a selection of fixed internuclear separations, we analyze the PMD resulting from SPI and MPI to identify the basic physical mechanisms in the creation of molecular FEVs. Molecular vibration is introduced in Section 3.2, where we study the influence of the nuclear motion on the electronic excitation for SPI and MPI. In Section 3.3, we address different types of spatial averaging in view of the experimental implementation of molecular FEV scenarios. For this purpose, we consider a CEP-sensitive bichromatic two- vs. three-photon ionization scenario to compare the calculated molecular frame PMD with the PMD measured in the laboratory frame. In particular, we investigate the influence of molecular rotation averaging and CEP-averaging, both of which play a crucial role in molecular photoionization experiments.
To get a first idea of the ionization dynamics of diatomic molecules, especially when ionized with polarization shaped pulses, we start with a rigid diatomic molecule whose nuclei are fixed in space. This so-called static nuclear frame represents a particularly good approximation for the interaction of molecules with attosecond pulses (Yuan et al., 2016; Djiokap et al., 2018; Guo et al., 2021), because the nuclei are virtually frozen on the attosecond timescale. For our purpose, freezing the nuclei serves as a simplification to provide a clear physical picture of the various interference mechanisms involved in molecular photoionization. The influence of the nuclear motion is investigated next in Section 3.2. In the following, two types of perturbative ionization processes are compared. First, we study SPI by XUV/UV pulses with central wavelengths in the range of λ0 = 113−210 nm and a fixed pulse duration of Δt = 2 fs. Second, we study MPI by UV/VIS pulses with central wavelengths of λ0 = 353−680 nm and a duration of Δt = 5 fs. The pulse duration in the MPI scheme is chosen slightly larger than in the SPI scheme, because the photoelectron signal from (non-resonant) perturbative N-photon ionization is determined by the N-th order optical spectrum (Meshulach and Silberberg, 1999). For a Gaussian-shaped pulse, the signal is spectrally broadened by a factor of
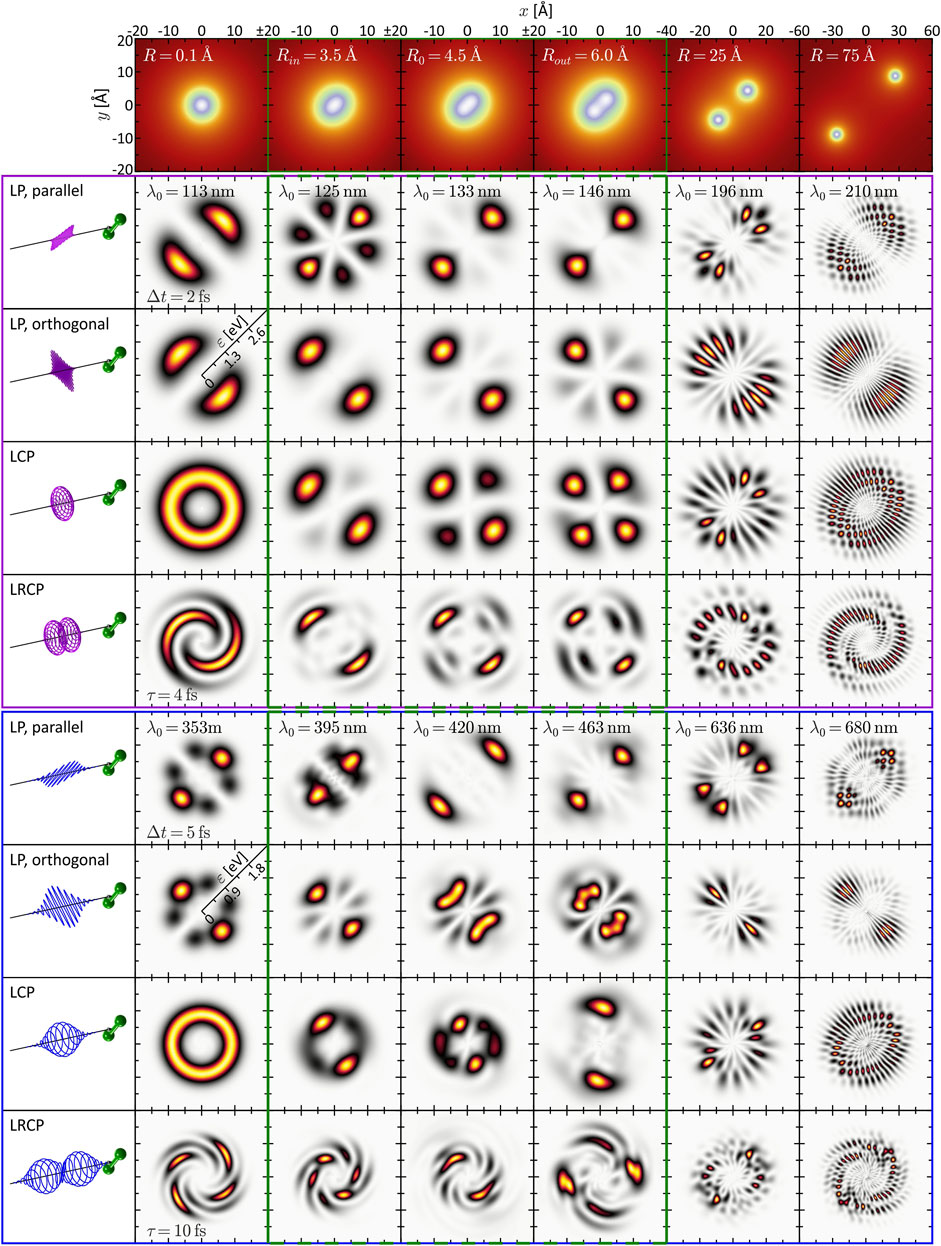
FIGURE 2. (Color online.) Gallery of calculated and energy-calibrated PMDs created by photoionization of the rigid molecule at different internuclear separations (columns) with different types of laser pulses (rows). The corresponding molecular potentials are shown in the top row. The laser pulses are illustrated on the left-hand side. The upper part (magenta frame) shows the PMDs resulting from one-photon ionization, the PMDs in the lower part (blue frame) result from three-photon ionization. The green frames (second to fourth column) indicate the vibration window.
We start by inspecting the results from the SPI scheme. In the limit R → 0 (first column of Figure 2), the PMDs are straightforwardly described in an atomic picture. The potential becomes approximately spherically symmetric entailing an |s, m = 0⟩-type ground state wave function. In accordance with the dipole selection rules, SPI by an LP laser pulse therefore creates a |p, 0⟩-type, dumbbell-shaped photoelectron wave packet aligned along the polarization direction. Similarly, an LCP pulse produces a |p, 1⟩-type, torus-shaped wave packet. In general, the superposition principle applies to perturbative SPI, i.e., the free electron wave packet from photoionization with a superposition of laser fields is given by the coherent superposition of the partial waves from the individual fields (Wollenhaupt et al., 2002). For example, the photoelectron wave packets created by an LCP pulse are a coherent superposition of the wave packets created by the corresponding LP pulses parallel and orthogonal to the internuclear axis. SPI by a time-delayed LRCP sequence yields an FEV in the shape of a two-armed Archimedean spiral with a counterclockwise sense of rotation (Ngoko Djiokap et al., 2015). This spiral-shaped PMD is understood in the framework of the perturbative ionization model presented in Pengel et al. (2017a), Pengel et al. (2017b) applied to one-photon ionization. According to the model, the two-armed FEV arises from the superposition of the
Next, we consider the PMD from SPI in the dissociation limit R ≫ R0. The PMDs shown in the last column of Figure 2 exhibit the same coarse (average) structure as in the atomic limit but are modulated by an intricate interference pattern. Some of the PMDs resemble the trilobite states currently discussed in the context of long-range Rydberg molecules (Shaffer et al., 2018; Eiles, 2019) and initially reported in (Greene et al., 2000). The observed pattern results from the interplay between two interference mechanisms already discussed by Cohen and Fano (1966) and reviewed recently in Yuan and Bandrauk (2015). The first mechanism is two-center interference, i.e., the superposition of partial waves emitted from the individual nuclei—hence both being of similar shape as in the atomic limit—spatially displaced by R. The second mechanism is Coulomb diffraction of each partial wave at the potential of the neighboring nucleus. Both mechanisms are illustrated in Figure 3 on the example of the two-armed FEV from ionization with an LRCP sequence. Panel (a) shows the spiral-shaped PMD
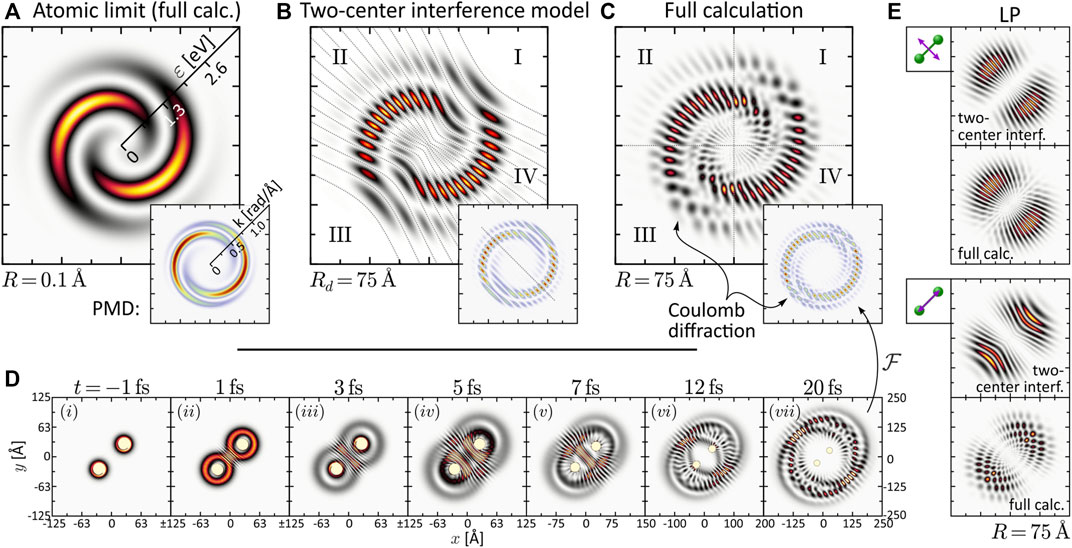
FIGURE 3. (Color online.) Discussion of interference mechanisms on the example of the two-armed FEV created by one-photon ionization with an LRCP pulse sequence (cf. Panel 2). The two pulses are centered at τ1 = −τ2 = −2 fs. (A) Energy-calibrated PMD of the FEV in the atomic limit R = 0.1 Å. The inset shows the actual PMD in momentum representation. (B) Two-center interference model based on the superposition of two atomic partial wave packets with origins displaced by Rd = 75 Å. (C) Full calculation of the two-armed FEV in the dissociation limit R = 75 Å. (D) Time series of the electron density |ψ(r; R, t)|2 illustrating the diffraction of the partial wave packets emitted from one nucleus at the Coulomb potential of the neighboring nucleus. (E) Comparison between two-center interference model and full calculation for the LP single-pulse scenarios orthogonal and parallel to the internuclear axis.
While the interpretation of the PMDs in the atomic and the dissociation limit is quite transparent, the analysis of the PMDs in the intermediate regime, presented in the central columns of Figure 2, is more demanding. The penultimate column, with R = 25 Å, represents the transition between the two limits. Here the PMDs are already characterized by the two interference mechanisms, the main difference being the lower modulation frequency of the two-center interference fringes in comparison to the last column. Around the equilibrium distance R0 = 4.48 Å (third column; see also Figure 1A) of the molecule, however, the two mechanisms are less meaningful for the interpretation of the PMDs. In this regime, the internuclear separation is difficult to extract directly from the PMD but could be retrieved by comparison to an analytical model of the photoelectron wave function (Fernández et al., 2009) or by application of a machine learning procedure (Shvetsov-Shilovski and Lein, 2022). Some general features of the PMDs, which we confirmed numerically, can be identified. For example, the PMDs created by LCP and RCP pulses (the latter are not shown) are mirror images of one another, when reflected at the internuclear axis or orthogonal to this axis. Mirroring the field at one of these axes inverts its rotational sense and transforms an LCP into an RCP pulse and vice versa. Currently, the mirror symmetry of LCP and RCP pulses is also referred to when discussing chiro-optical effects, such as photoelectron circular dichroism (Hergenhahn et al., 2004; Powis, 2008; Lux et al., 2012). Altogether, the second (R = 3.45 Å), third and fourth (R = 6.0 Å) column correspond to the nuclear vibration window which will be investigated in the following Section 3.2. The PMDs shown here build the basis for the discussion of the vibrational dynamics.
Unlike SPI, MPI depends not only on the initial state and the final state in the continuum, but is also highly sensitive to intermediate resonances. Moreover, the absorption of multiple photons opens up numerous ionization pathways leading to target continuum states characterized by superpositions of angular momenta that are generally larger than in SPI. As a consequence, PMDs created by MPI have a richer structure than those created by SPI. The simulation results shown in the bottom part of Figure 2 (blue frame) are based on three-photon ionization of the potassium molecular ion, motivated by recent experimental studies on potassium atoms (Wollenhaupt et al., 2009a; Pengel et al., 2017a; Pengel et al., 2017b) and molecules (Bayer et al., 2013; Braun et al., 2014). The PMDs in the first column are again well understood in an atomic picture invoking the dipole selection rules. Accordingly, three-photon ionization of the |s, 0⟩-type ground state by an LP pulse creates an |f, 0⟩-type photoelectron wave packet aligned in laser polarization direction (Wollenhaupt et al., 2009a). Analogously, the LCP pulse creates an |f, 3⟩-type, torus-shaped wave packet, slightly more confined in radial direction compared to the SPI scheme. The radial width of the PMD in the MPI scheme is determined by the bandwidth of the third-order spectrum of the 5 fs pulse, which is actually smaller than the fundamental bandwidth of the 2 fs pulse. The LRCP sequence creates a six-armed FEV with counterclockwise sense of rotation, as observed and discussed in detail in Pengel et al. (2017a), Pengel et al. (2017b).
In the dissociation limit (last column), we recognize the same interference mechanisms at play as in the SPI scheme (cf. Section 3.1.1). Again, the average structure of the PMDs is the same as in the atomic limit (first column). Along the internuclear axis, however, the PMDs are modulated by speckle-shaped interference patterns due to the combined effect of Coulomb diffraction and two-center interference, whereas in lateral direction, we observe the fringe-type patterns characteristic for almost pure two-center interference.
The interpretation of the intermediate R-regime is more involved. The PMDs shown in the central columns depend strongly on the central wavelength λ0, which indicates the presence of intermediate resonances. Also, a comparison of the different PMDs within each row shows that the shape of the PMDs changes drastically with the internuclear separation. In contrast, in the SPI scheme, where resonances of the bound system play no role, the PMDs in the vibration window (green frame) are qualitatively similar and change smoothly with R. Another indication of intermediate resonances is the rotation of the PMDs in the circularly polarized scenarios, discernible e.g., by the angular alignment of the main lobes in the 2D plane. Resonances introduce additional ionization phases in the photoelectron wave packet (Eickhoff et al., 2022) which, in the circularly polarized case, translate into an azimuthal rotation of the PMD (Eickhoff et al., 2020a). The observed rotation is particularly pronounced in the PMDs from LCP and LRCP ionization in the fourth column, i.e., for R = Rout = 6.0 Å. The excitation scheme in Figure 1A suggests that at this internuclear separation the transition from the ground state to state ψ6 (green line in Figure 1A) becomes near-resonant with a pulse of central wavelength λ0 = 463 nm (see also Figure 6B).
The quantitative analysis of the bound state population dynamics pn(t) for the lowest eight electronic states is shown in Figure 4. At the equilibrium distance R0 and λ0 = 420 nm (third column), shown in panel (a), we see the transient population of states ψ1 and ψ2 by a few percent, followed by a population return to the ground state. This result confirms the non-resonant character of the interaction and verifies the perturbative interaction conditions. Panel (b) shows the dynamics at Rout and the same central wavelength. In this case, we observe an efficient population transfer to state ψ6 of about 10%. The induced electronic coherence results in a pronounced charge oscillation in the bound molecular system along the internuclear axis. This dynamics is illustrated in the insets to (b) which show the time-dependent electron density |ψ(r; R, t)|2 over one half-cycle of the charge oscillation after the interaction. These findings confirm the above assumption of a resonance-enhanced multiphoton ionization (REMPI) process at Rout via state ψ6. Our simulations show that the exact resonance at Rout is found around λ0 = 413 nm. At λ0 = 463 nm, as in the fourth column, we still register about 1% population transfer due to the broad bandwidth of the pulses. Additional high-lying resonances are expected at the two-photon level, due to the high density of Rydberg states, but their analysis is beyond the scope of this paper. The resonance ψ0 → ψ6 at Rout, however, will also be relevant in the discussion of coupled vibrational and electronic (vibronic) MPI dynamics in Sections 3.2.2, 3.3.
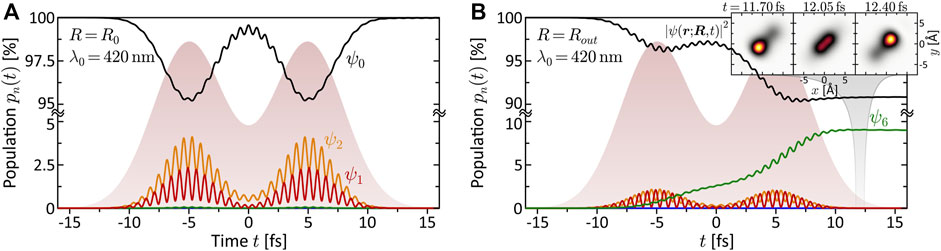
FIGURE 4. (Color online.) Bound state population dynamics of the rigid molecule excited by the LRCP sequences (shaded background) from the MPI scheme. Shown are the time-dependent populations of the eight lowest electronic states ψn (cf. Panel 1B). (A) Non-resonant excitation at the equilibrium distance R0 = 4.48 Å using a central wavelength of λ0 = 420 nm. Only states ψ1 and ψ2 are transiently populated, mediating the MPI process. Finally, almost the entire population returns to the ground state. (B) Resonant excitation of state ψ6 at the outer turning point Rout = 6.0 Å, using the same central wavelength λ0 = 420 nm as in (A). The coherent superposition of states ψ0 and ψ6 corresponds to an efficient ultrafast charge oscillation in the bound molecular system, illustrated in the inset by a time series of the electron density |ψ(r; R, t)|2.
The coupled electron-nuclear dynamics in molecules has been studied extensively on the femtosecond timescale, as reviewed for example in Gatti (2014), Bircher et al. (2017), and very recently on the attosecond timescale (Nisoli et al., 2017; Cattaneo et al., 2018). Experimentally, the coupled electron-nuclear dynamics of potassium dimers has been investigated in Bayer et al. (2013), Braun et al. (2014). In this section, we account for the vibration of the nuclei along the internuclear axis and study the coupling between the nuclear motion and the electronic ionization dynamics. To this end, we initialize the molecule in a vibrationally excited state of the electronic ground state potential V0(R), which we can think of as being prepared, e.g., by stimulated Raman excitation. We use LRCP sequences to study the creation of FEVs by ionization of the vibrating molecule. Femtosecond double pulse sequences are well-established in femtochemistry and the time-resolved investigation of molecular dynamics (Zewail, 1995), being the basis of pump-probe techniques (Zewail, 2000). Here, however, the focus is not primarily on time-resolved imaging of the dynamics, but on exploring their signatures imprinted in the photoelectron spirals. In the following, we design specific vibration-scenarios via the timing of the pulse sequence. Specifically, we lock the two pulses to distinct stages of the nuclear oscillation, i.e., the inner and the outer turning point. The resulting PMD is analyzed for both the SPI and the MPI scheme (cf. Section 3.1).
The timescale of the vibrational dynamics depends, amongst others, on the reduced mass μ of the nuclear system. For the purpose of this study, μ is chosen such that the nuclear timescale is adapted to the laser pulses employed in Section 3.1.1, which yields the most transparent results. Specifically, μ is adapted such that the period of the vibration (cf. top inset to Figure 1) is Tv = 13.7 fs. This value is compatible with the laser pulse duration of Δt = 2 fs. The laser central wavelength is fixed to λ0 = 125 nm as in the second column of Figure 2 (magenta frame). The variation parameter is the time-delay τ between the two subpulses of the LRCP sequence.
Two distinct scenarios are investigated. First, the pulses are separated by half a vibrational cycle, τ = Tv/2 = 6.84 fs, and locked to different turning points. Second, the pulses are separated by τ = Tv and locked to the same turning point of two consecutive vibration cycles. The numerical results are presented in Figure 5. The top frames show the nuclear motion R(t) (green curve) together with the x-component of the circularly polarized laser field E(t) (magenta curve). During the most intense part of the pulses, i.e. within Δt, the nuclei move by about 0.2 Å at the inner and 0.1 Å at the outer turning point. The calculated photoelectron spectra are shown below in different representations, including the 2D PMD
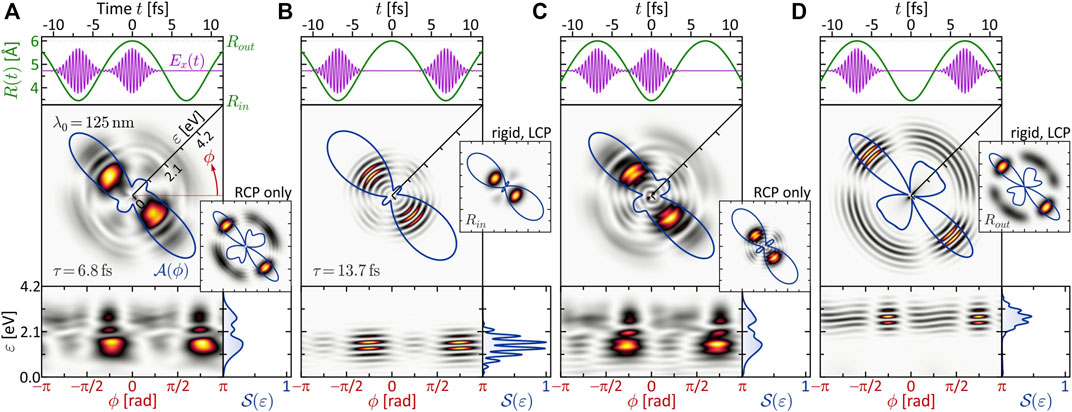
FIGURE 5. One-photon ionization of the vibrating molecule by single-color LRCP sequences locked to the two turning points of the nuclear vibration R(t). The 2D PMD in energy representation
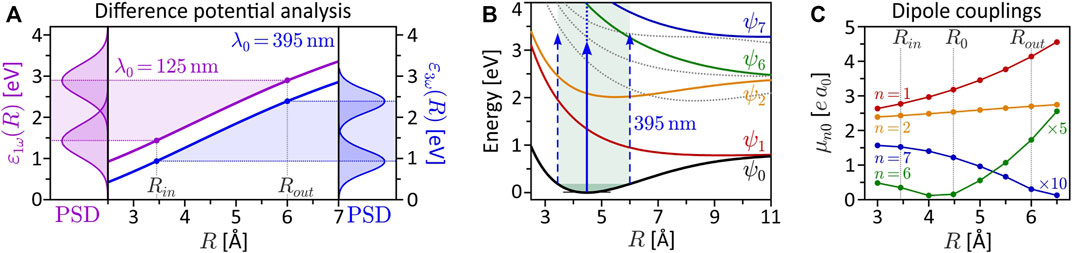
FIGURE 6. (Color online.) Derived quantities to characterize the interdependence of electronic and nuclear dynamics. (A) Difference potential analysis for the SPI scheme (magenta curve) and the MPI scheme (blue curve). The left frame depicts the power spectral density (PSD) of the 125 nm, 2 fs pulse mapped at the inner and outer turning point of the vibration. The right frame shows the PSD of the third order spectrum of the 395 nm, 5 fs pulse. (B) Excitation scheme of the vibrating molecule interacting with the 395 nm pulse employed in the MPI scheme. At the outer turning point, state ψ6 is excited resonantly from the ground state. (C) R-dependent transition dipole moments μn0(R) = |e⟨ψn|r|ψ0⟩| for ground state transitions to the seven lowest bound electronic states (vanishing dipole couplings are not shown). At the outer turning point Rout, the coupling of the ground state to the resonant state ψ6 is one order of magnitude larger than at the equilibrium distance R0.
Specifically, the inner turning point is mapped to an energy of ɛ1ω(Rin) = 1.4 eV, while the outer turning point is mapped to ɛ1ω(Rout) = 2.9 eV. This mapping gives rise to the localized rings observed in Figures 5B,D. In these SPI scenarios, the laser pulses probe the nuclear dynamics at two consecutive inner and outer turning points, respectively. The enhancement of the photoelectron amplitude in these regions indicates the increased probability of finding the nuclei at one of the two turning points, in analogy to a classical oscillator. The corresponding angular distributions
In panels (a) and (c) of Figure 5, we observe no such regular interference pattern as in (b) and (d). Here, the two partial wave packets are mapped into different energy windows and interfere only in a narrow overlap region around ɛ = 2.1 eV (see left frame of Figure 6A), where indeed some interference fringes are observed. The energetic disentanglement of the free electron wave packets is also indicated by the slight asymmetry of the side lobes in the angular distribution
The results obtained in the dynamic MPI scheme are especially rich in information. Not only is MPI sensitive to intermediate resonances, but unlike the static case, in the presence of nuclear dynamics, electronic resonances may occur at certain internuclear distances and be absent in others. To study the signatures of such transient resonances in the PMD, we again lock two pulses, which are short compared to the vibrational period of the molecule, to different stages of the nuclear dynamics. The vibrational dynamics are adapted to the longer pulse duration of Δt = 5 fs used in Section 3.1.2 by choosing the reduced mass of the nuclei such that the vibration period is increased to Tv = 30.6 fs. We employed the LRCP sequences from Section 3.1.2 with a central wavelength of λ0 = 395 nm (second column of Figure 2). This central wavelength is very close to the one-photon resonance ψ0 → ψ6 at the outer turning point Rout of the vibration, as illustrated in the excitation scheme shown in Figure 6B (see also Section 3.1.2). At the inner turning point Rin and at the equilibrium distance R0, however, the field is far detuned from any one-photon resonance, since the states ψ3, ψ4 and ψ5 (gray dashed curves) are not dipole-coupled to the ground state. Consequently, we expect the transient resonance ψ0 → ψ6 to play a prominent role in the scenarios involving MPI at the outer turning point. The R-dependent transition dipole moments plotted in Figure 6C show that the coupling between the ground state and the ψ6-state (green curve) at the outer turning point is one order of magnitude larger than at the equilibrium distance (cf. Figure 1B). In general, the variation of a transition dipole moment along a nuclear coordinate can be indicative of a change of the electronic structure in this direction (Wollenhaupt et al., 2003). However, in this case, the variation of the dipole coupling is attributed to the R-dependent contributions of the inner and outer lobes of ψ6 to the matrix element (cf. Figure 1B). In the vicinity of R0, at R ≈ 4.2 Å, these contributions cancel each other and the matrix element vanishes. The numerical results are presented in Figure 7, with the same organization as in Figure 5 to facilitate the comparison. We start with the discussion of panel (b), where both pulses ionize the vibrating molecule at the inner turning point. This is the only scenario, where the above-mentioned resonance is not crucial. According to the difference potential analysis in Figure 6A (blue curve), the inner turning point is mapped to an energy of ɛ3ω(Rin) = 3ℏω0 − ΔV (Rin) = 0.9 eV. This energy is marked by a blue arrow in the bottom right frame of Figure 7B. In fact, the calculated PMD is red-shifted towards a kinetic energy of ɛ = 0.6 eV, best seen in the energy spectrum
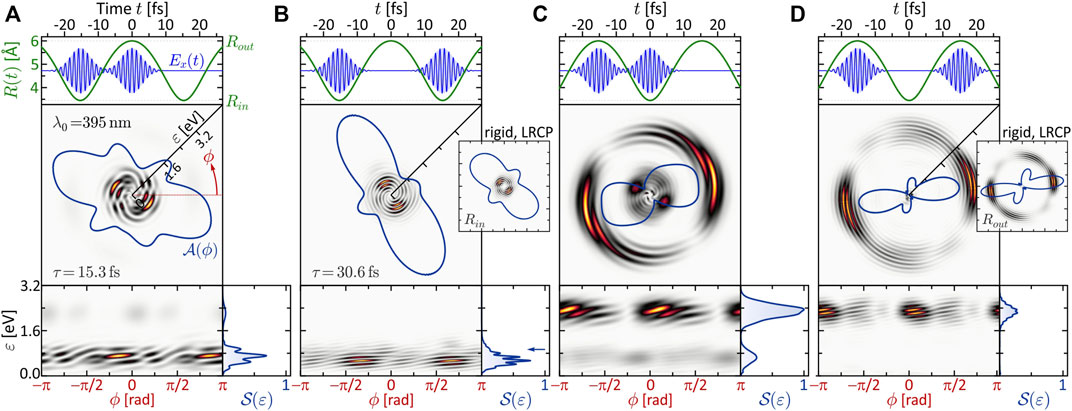
FIGURE 7. (Color online.) Three-photon ionization of the vibrating molecule by single-color LRCP sequences locked to the two turning points of the nuclear vibration R(t). The organization of panels (A–D) is analogous to Figure 5. Striking differences to the SPI scheme are observed, especially in panels (A,C), which are attributed to the occurrence of transient intermediate resonances.
A similar beating pattern is observed in Figure 7D, where both pulses ionize the molecule at the outer turning point. The created photoelectron wave packets are localized energetically around ɛ3ω(Rout) = 2.4 eV, in accordance with the difference potential analysis in Figure 6A. Both pulses excite the ψ6-state near-resonantly driving a (1 + 2) REMPI process, which we verified numerically by evaluation of the population dynamics (not shown) similar to Figure 4. Our results show that the first subpulse transfers about 5% of population from the ground state to the resonant ψ6-state. Hence, the second pulse finds the molecule in a coherent superposition of states ψ0 and ψ6 and maps both into the continuum, the former by three- and the latter by two-photon ionization. One might suspect that the additional photoelectron contribution from state ψ6 is responsible for the observed beating pattern in the PMD. However, since the beating—unlike the ψ0 → ψ6 resonance—is absent in the static LRCP scenario shown in the inset, we conclude that the beating is indeed a signature of the non-adiabatic ground state dynamics. The same alignment of the angular distributions in the static and the dynamic LRCP scenario confirms the absence of additional resonances in the course of the nuclear vibration.
In the scenarios of panels (a) and (c), we expect again a broader energy distribution of the photoelectrons, because the laser field ionizes the molecule at both turning points. In fact, the PMD in panel (c) displays a bimodal energy spectrum
Eventually, in panel (a), the role of the two subpulses is exchanged. The first subpulse ionizes the molecule at the inner turning point via direct (2 + 1) REMPI, creating a low-energy wave packet around ɛ = 0.9 eV and transferring some population to ψ11. At the outer turning point, the second subpulse therefore ionizes the molecule by direct (1 + 2) REMPI via the resonant ψ6-state, creating a high-energy wave packet around ɛ = 2.4 eV and, in addition, maps the ψ11-state into the continuum, creating another wave packet centered around ɛ = 0.5 eV. The interference of the two low-energy wave packets gives rise to the pronounced vortex structure observed in the low-energy region. We note that, although the different intensities of slow and fast electrons in Figures 7A,C suggest a ground state depletion by the first pulse of each scenario, the ionization is indeed perturbative. In both cases, the first pulse depopulates the ground state only by several %, comparable to Figure 4B.
The results presented in this section demonstrate that the coupled electron-nuclear dynamics leaves distinct fingerprints in the spiral-shaped PMDs of molecular FEVs. For example, the striking difference between Figures 7A,C, as well as the comparison between the PMD from the vibrating and the rigid molecule, clearly reveal that, in molecular MPI, the interplay between nuclear dynamics and electronic resonances is of great importance. Therefore, model calculations that predict the fully differential PMDs are invaluable tools to decipher the sophisticated structures of the PMD and identify the multiple interference phenomena underlying the creation of molecular FEVs. However, the results also show, that more refined models including the quantum mechanical description of nuclear dynamics are needed to accurately predict the fine details of the coupled electron-nuclear dynamics. Given the high degree of complexity of the interference patterns, further processing and analysis of the PMDs will in practice be necessary to extract detailed information on the underlying molecular dynamics. Experimentally, the creation and differential detection of molecular FEVs promises to be a highly-sensitive spectroscopic technique to probe molecular structure and dynamics, provided the delicate interference patterns are not averaged out by the experimental conditions—an issue which we will address in the following section.
So far, we have considered the laser-molecule interaction in the molecular frame, i.e., for a fixed alignment of the molecule in space. In addition, we have assumed a phase-stable wave form and polarization profile of the laser field with a fixed orientation relative to the molecular axis in the experiment. Any deviation from these idealizations will, in general, wash out the intricate interference patterns observed in the molecular frame PMD. For example, measurements are typically performed on isotropic ensembles of molecules. The PMD measured in the laboratory frame is then a rotational average of all molecular alignments relative to the laser propagation direction. In addition, the measured PMD is typically accumulated over multiple laser shots and locations within the laser focus introducing wave form averaging via phase fluctuations of the laser and the Gouy phase (Hoff et al., 2017; Bayer et al., 2020). While these phase fluctuations might be less significant in the case of a single multi-cycle pulse, they become crucial in the case of few-cycle pulses, multi-pulse sequences and combinations thereof. Specifically, a variation of the relative phase between the subpulses in the LRCP sequences used here for the creation of molecular FEVs rotates the spiral-shaped interference pattern in the molecular frame PMD (Pengel et al., 2017a). As a result, fluctuating relative phases blur the interference pattern in the angular direction. Since bichromatic LRCP sequences, consisting of two subpulses with commensurable central frequencies, control the rotation of the PMD via the relative phase and the CEP (Kerbstadt et al., 2019b; Eickhoff et al., 2020a), even CEP fluctuations wash out the observable interference pattern in the bichromatic case.
In this section, we investigate the influence of the different averaging mechanisms on the PMD in a single test scenario. Besides the averaging over the molecular alignment, we examine CEP-averaging. Unlike the relative optical phase, which can be controlled experimentally with zeptosecond precision (Köhler et al., 2011), the CEP is generally more difficult to stabilize and inherently varies over the laser focus due to the Gouy-phase (Hoff et al., 2017). Under perturbative conditions, however, FEVs created by single-color pulses are CEP-insensitive (Pengel et al., 2017b). Therefore, we consider a molecular FEV created by dynamic two- vs. three-photon ionization using a bichromatic (2ω: 3ω) LRCP pulse sequence, which is highly sensitive to the CEP (Kerbstadt et al., 2019b; Eickhoff et al., 2021c). The pulses are separated by a full vibrational cycle, τ = Tv, and locked to consecutive instants of the nuclei traversing the equilibrium distance R0, heading outwards. Thus, both pulses probe the same vibrational stage and a maximum range of internuclear separations. Maintaining a pulse duration of Δt = 5 fs as in Sections 3.1.2, 3.2.2, we adapted the reduced mass such that the vibrational period is Tv = 18.0 fs to fully separate both pulses in time and minimize photoelectron contributions due to frequency mixing between the two colors (Kerbstadt et al., 2017a; Eickhoff et al., 2021c). The initial LCP subpulse has a central wavelength of λ1 = 395 nm as in the MPI scheme discussed in the previous sections. The time-delayed RCP subpulse has a central wavelength of λ2 = 2λ1/3 = 263 nm to ensure the energetic overlap of two- and three-photon ionization pathways at 2ℏω2 = 3ℏω1. The amplitude ratio of the two subpulses is chosen such that in a static MPI scenario with R = R0 both produce the same photoelectron yield. This procedure, which is also followed in the experiments (Kerbstadt et al., 2019b; Eickhoff et al., 2020a), generally optimizes the interference contrast between the two partial wave packets in the full LRCP scenario.
The numerical results are presented in Figure 8. Panel (a) shows the PMD in the molecular frame for a fully stabilized pulse. We observe a structured five-armed FEV consistent with the perturbative ionization model applied to two- vs. three-photon ionization. The FEV is localized in the low-energy region around ɛ = 0.75 eV. The reason for the enhancement of the photoelectron signal near the ionization threshold is the resonant excitation of the ψ6-state by the first subpulse (cf. discussion in Section 3.2.2), which is then mapped into the continuum around ɛ = 0.75 eV by absorption of another photon from the second subpulse. The inset shows the PMD obtained in the atomic limit (rigid molecule with R = 0.1 Å). Compared to the dynamic case, it displays a more regularly shaped five-armed Archimedean spiral, similar to the result obtained recently for the 2D model of a single potassium atom (Eickhoff et al., 2021c). Figures 8B–D show three types of rotational averaging procedures. In panel (b), both the molecule and the laser field are rotated by 360°, in 16 steps of δϕ = 22.5°, keeping their relative orientation locked. In an experiment, this procedure corresponds to a rotation of the detector around an aligned molecule interacting with a phase-stable field. As expected, the average result is an isotropic, torus-shaped PMD which serves as a reference for the other two—experimentally more relevant—averaging studies.
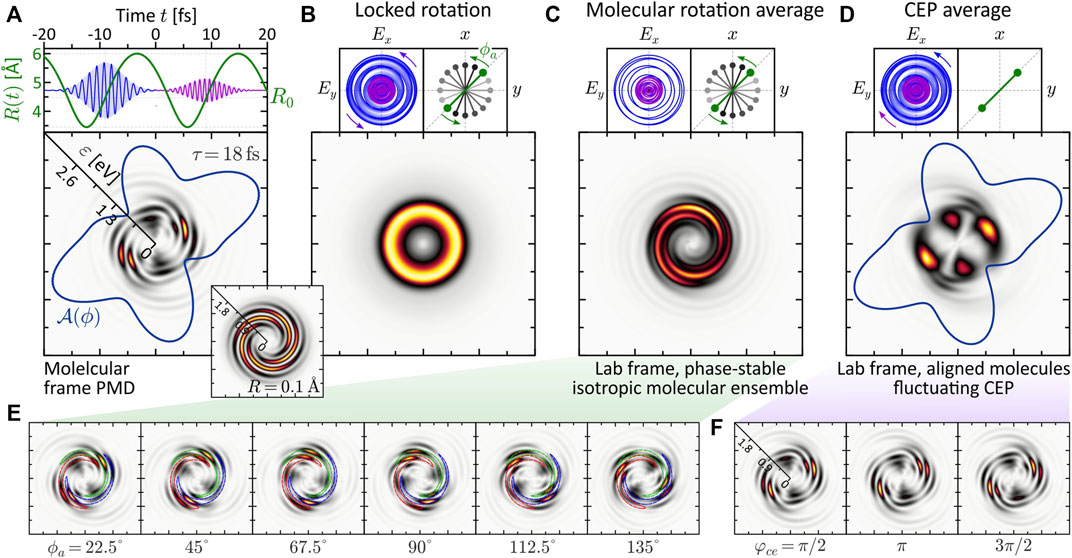
FIGURE 8. (Color online.) Influence of experimental conditions on a molecular FEV created by dynamic two- vs. three-photon ionization using an LRCP bichromatic (2ω: 3ω) pulse sequence. All frames show energy-calibrated 2D PMDs. (A) The molecular frame PMD created by a phase-stable field exhibits an intricate five-armed spiral pattern. (B) Rotating both the molecule and the field about the laser propagation direction washes out the interference pattern completely and yields an isotropic, torus-shaped PMD in the lab frame. (C) Lab frame PMD obtained for an isotropic molecular ensemble and a phase-stable pulse sequence. Surprisingly, the highly structured five-armed FEV transforms into a regularly shaped three-armed Archimedean spiral. (D) Lab frame PMD obtained for an ensemble of aligned molecules and a fluctuating CEP. The CEP controls the rotation of the bichromatic field. (E,F) show selected subsets of molecular frame PMDs underlying the molecular rotation averaging and the CEP-averaging procedure, respectively.
Panel (c) shows the effect of molecular rotation averaging on the PMD. This type of averaging is relevant when measuring on an ensemble of unaligned molecules. In contrast to the real 3D space, where the molecule has two angular degrees of freedom (polar and azimuthal), in 2D we consider the rotation about the azimuthal angle ϕ. Due to the reflection symmetry of the homonuclear dimer, only rotations up to an angle of ϕa = 180° need to be considered. The alignment angle ϕa is taken relative to the positive grid-diagonal in counterclockwise direction. Keeping the laser field fixed, we calculated 16 molecular frame PMDs in which we successively rotated the molecule in steps of δϕa = 11.25°. A subset of these calculations is shown in panel (e). Surprisingly, the five-armed spiral pattern is not completely washed out by the molecular rotation, but evolves into a three-armed Archimedean spiral. To rationalize the formation of the three-armed spiral in the lab frame PMD, the series of images in panel (e) illustrates how the maxima of the five-armed spiral shift along a contour of the three-armed spiral of the averaged PMD under the molecular rotation. So far, the exact mechanism behind the counterintuitive transformation of the five-armed spiral into a three-armed spiral upon averaging has not been fully analyzed. Clearly, in order to obtain averaged PMDs that can be compared with experimental results, rotation averaging needs to be performed in 3D. It is quite possible that 3D averaging also blurs other features of the PMD. The results from the 2D model presented here are a natural first step in this direction and serve to compare rotational vs. CEP averaging.
Panel (d) shows the influence of CEP-averaging on the PMD. This type of averaging is especially relevant in bichromatic MPI schemes based on the interference of photoelectron wave packets of different parity (Kerbstadt et al., 2018; Kerbstadt et al., 2019b). In the case of MPI with a bichromatic LRCP sequence, the CEP controls the rotation of the field in the polarization plane and therefore determines the relative orientation between field and molecule. Here, we considered an aligned molecule (ϕa = 0) and varied the CEP from φce = 0 to 2π in 16 steps of δφce = π/8. As a result, the fine five-armed spiral pattern observed in the phase-stable scenario (a) is averaged out. The main lobes of the angular distribution
The above results show, that both the molecular alignment and the stability of the optical phase are required for the observation of the fine details characterizing the molecular FEVs. Hence, the retrieval of the rich structural and dynamical information encoded in the PMD from laboratory measurements requires optimal experimental conditions including molecular alignment and phase stability. An alternative to the alignment of molecules is the use of coincidence detection techniques (Ullrich et al., 2003) and post-selection of ionization events corresponding to a specific molecular alignment.
Free electron vortices (FEVs) created by photoionization of atoms with ultrashort polarization-tailored laser pulse sequences already exhibit a great variety of shapes, as recently reviewed in (Kerbstadt et al., 2019a; Kerbstadt et al., 2020, Eickhoff et al., 2021c). The photoelectron momentum distribution (PMD) of atomic FEVs is characterized by a regularly shaped multi-armed Archimedean spiral (in energy representation) (Pengel et al., 2017b), with controllable symmetry, rotational sense and spiral arm pitch (Kerbstadt et al., 2019b). However, molecular FEVs, studied in this contribution, display an even richer structure than their atomic counterparts, resulting from the interplay of three key features. The basic characteristic is the spiral-shaped interference pattern, fully analogous to that of atomic FEVs. This vortex shape is superimposed by a structural feature, due to the broken spherical symmetry of the molecule as opposed to an atom. Finally, a dynamical feature arises due to the nuclear degrees of freedom and the coupling between the electron and nuclear dynamics. The interplay of all these features results in a wide variety of details in the PMD and opens the door to the emerging field of molecular FEVs.
In this numerical study, we have explored the rich structures in the molecular FEVs and studied multiple interference phenomena underlying the ionization dynamics. Our strategy was based on the ab initio solution of the 2D time-dependent Schrödinger equation (TDSE) for a diatomic molecular ion with a single active electron, aligned in the polarization plane of a polarization-shaped ultrashort laser pulse. The molecular system was thoroughly characterized by the calculation of the Born-Oppenheimer potentials, eigenfunctions and transition dipole moments. The 2D TDSE model for the light-induced coupled electron-nuclear dynamics was employed to study the PMD created by a number of established laser pulse shapes (Eickhoff et al., 2021a), focusing on the creation of molecular FEVs by counterrotating circularly polarized double pulse sequences. We compared the results from single-photon ionization (SPI) and multiphoton ionization (MPI). The SPI findings were easier to interpret than the MPI results because the latter give rise to multiple ionization pathways (Eickhoff et al., 2021c) and are highly sensitive to intermediate resonances in the bound system. The physical properties of the molecular FEVs were analyzed taking into account various observables, including the fully differential 2D PMD, the (angle-integrated) energy spectrum and the (energy-integrated) angular distribution. In addition, we followed the time evolution of the electron density |ψ|2 in coordinate space, to reveal the ionization dynamics in the bound molecular system and rationalize the vortex formation dynamics in the continuum. In the investigated MPI schemes, the evaluation of the bound state population dynamics allowed us to unambiguously identify intermediate resonances and unravel their signatures imprinted in the PMD.
Specifically, we presented three interrelated studies. In the first study, we explored the influence of the nuclear configuration on the molecular FEVs. For this purpose, we considered the interaction of a rigid molecule with different standard pulse shapes, already used for experiments on MPI of atoms, and analyzed the created PMDs for different internuclear separations R. In the limit R → 0, the findings reproduced the experimental atomic FEV results and were well-understood in an atomic picture. In the limit R → ∞, complex interference patterns were observed in the PMD, which were created by an interplay of two-center interference and Coulomb diffraction. In the intermediate R-regime, around the equilibrium distance R0, the structure of the PMDs was found to be more intricate and more difficult to explain in a simple picture. This regime was relevant for the second study, in which we considered the interaction of a vibrating molecule with specifically designed counterrotating circularly polarized pulse sequences. By synchronization of the two subpulses to the molecular vibration, we investigated the interplay between the nuclear motion and the electronic excitation dynamics and identified signatures of the coupled electron-nuclear dynamics in the PMD of the molecular FEVs. First, we found that the nuclear dynamics result in a bimodal photoelectron kinetic energy spectrum, rationalized by Mullikan’s difference potential analysis. Second, we observed the fingerprint of non-adiabatic phase dynamics of the ground state wave function, which manifested in a subtle but clearly visible radial modulation of the PMD. The latter finding demonstrates the high sensitivity of the PMD to non-adiabatic electron-nuclear dynamics in molecules and underscores the power of highly differential photoelectron detection techniques to reveal even subtle changes in the bound electronic wave function. Further characteristic features of the FEVs created by MPI could be attributed to the influence of two intermediate resonances, which were excited during different stages of the molecular vibration. All these results highlight the great importance of the interplay between the nuclear dynamics and electronic resonances in molecular MPI. The third study was relevant in view of the experimental implementation of molecular FEV scenarios. On the example of bichromatic two- vs. three-photon ionization, we compared the PMD obtained in the molecular frame to the PMD measured in the laboratory which is, in general, subject to different types of averaging. Specifically, we studied the influence of molecular rotation averaging and CEP-averaging. We found that in both cases, some of the fine details of the molecular FEVs were washed out. In order to retrieve the rich structural and dynamical information encoded in the PMD, it is essential to align the molecules prior to photoionization, or detect the momentum of the photoions in coincidence and post-select the events corresponding to a specific molecular alignment, and to stabilize the phase of the laser pulses.
In perspective, our 2D TDSE model will be refined towards the inclusion of quantum mechanical nuclear wave packet dynamics, i.e., a full quantum treatment of the coupled electron-nuclear dynamics. The refined model will allow more accurate simulation of the molecular FEVs generated by MPI in the presence of intermediate resonances. The numerical study presented here provides first insights into the rich variety of relevant physical mechanisms involved in the generation of molecular FEVs with shaped laser pulses and serves as preparatory work for the experimental implementation in the laboratory. The combination of white-light polarization pulse shaping (Pengel et al., 2017a; Kerbstadt et al., 2017b) with photoelectron tomography (Wollenhaupt et al., 2009b; Kerbstadt et al., 2019a) will enable the use of molecular FEVs as powerful spectroscopic tools to probe molecular structure and dynamics.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Building on previous experimental studies by both authors, TB and MW proposed to study the physics of molecular vortices by direct simulation of the two-dimensional TDSE. They developed the concept and structure of the manuscript and interpreted the results. In close cooperation with MW, TB prepared the figures, performed the numerical calculations, and wrote the manuscript. TB and MW contributed to the revision of the manuscript, read and approved the submitted version.
This research was funded by the Deutsche Forschungsgemeinschaft (DFG) via the priority program SPP1840 QUTIF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Armstrong, G. S. J., Clarke, D. D. A., Benda, J., Wragg, J., Brown, A. C., and van der Hart, H. W. (2019). Modeling Tomographic Measurements of Photoelectron Vortices in Counter-Rotating Circularly Polarized Laser Pulses. Phys. Rev. A 100, 063416. doi:10.1103/physreva.100.063416
Assion, A., Geisler, M., Helbing, J., Seyfried, V., and Baumert, T. (1996). Femtosecond Pump-Probe Photoelectron Spectroscopy: Mapping of Vibrational Wave-Packet Motion. Phys. Rev. A 54, R4605–R4608. doi:10.1103/physreva.54.r4605
Babiker, M., Bennett, C. R., Andrews, D. L., and Dávila Romero, L. C. (2002). Orbital Angular Momentum Exchange in the Interaction of Twisted Light with Molecules. Phys. Rev. Lett. 89, 143601. doi:10.1103/physrevlett.89.143601
Bandrauk, A. D., and Shen, H. (1993). Exponential Split Operator Methods for Solving Coupled Time‐dependent Schrödinger Equations. J. Chem. Phys. 99, 1185–1193. doi:10.1063/1.465362
Bauer, D. (2017). Computational Strong-Field Quantum Dynamics: Intense Light-Matter Interactions, 1. Berlin/Boston: De Gruyter.
Baumert, T., Buehler, B., Grosser, M., Thalweiser, R., Weiss, V., Wiedenmann, E., et al. (1991). Femtosecond Time-Resolved Wave Packet Motion in Molecular Multiphoton Ionization and Fragmentation. J. Phys. Chem. 95, 8103–8110. doi:10.1021/j100174a019
Bayer, T., Braun, H., Sarpe, C., Siemering, R., von den Hoff, P., de Vivie-Riedle, R., et al. (2013). Charge Oscillation Controlled Molecular Excitation. Phys. Rev. Lett. 110, 123003. doi:10.1103/physrevlett.110.123003
Bayer, T., Gräfing, D., Kerbstadt, S., Pengel, D., Eickhoff, K., Englert, L., et al. (2019). Time-resolved 3d Imaging of Ultrafast Spin-Orbit Wave Packet Dynamics. New J. Phys. 21, 033001. doi:10.1088/1367-2630/aafb87
Bayer, T., Philipp, C., Eickhoff, K., and Wollenhaupt, M. (2020). Atomic Photoionization Dynamics in Ultrashort Cycloidal Laser Fields. Phy. Rev. A 102, 013104. doi:10.1103/physreva.102.013104
Becker, W., and Milošević, D. B. (2022). Elliptic Dichroism in Strong-Field Ionization of Atoms Subjected to Tailored Laser Fields. Phys. Chem. Chem. Phys. 24, 7014–7027. doi:10.1039/d1cp05209h
Ben, S., Chen, S., Bi, C.-R., Chen, J., and Liu, X.-S. (2020). Investigation of Electron Vortices in Time-Delayed Circularly Polarized Laser Pulses with a Semiclassical Perspective. Opt. Express 28, 29442–29454. doi:10.1364/oe.400846
Bian, X. B., and Bandrauk, A. D. (2012). Attosecond Time-Resolved Imaging of Molecular Structure by Photoelectron Holography. Phys. Rev. Lett. 108, 263003. doi:10.1103/physrevlett.108.263003
Bircher, M. P., Liberatore, E., Browning, N. J., Brickel, S., Hofmann, C., Patoz, A., et al. (2017). Nonadiabatic Effects in Electronic and Nuclear Dynamics. Struct. Dyn. 4, 061510. doi:10.1063/1.4996816
Bliokh, K. Y., Ivanov, I. P., Guzzinati, G., Clark, L., Van Boxem, R., Béché, A., et al. (2017). Theory and Applications of Free-Electron Vortex States. Phys. Rep. 690, 1–70. doi:10.1016/j.physrep.2017.05.006
Braun, H., Bayer, T., Sarpe, C., Siemering, R., Vivie-Riedle, R., Baumert, T., et al. (2014). Coupled Electron-Nuclear Wavepacket Dynamics in Potassium Dimers. J. Phys. B 47, 124015. doi:10.1088/0953-4075/47/12/124015
Carrasco, S., Rogan, J., Valdivia, J. A., Chang, B. Y., Malinovsky, V. S., and Sola, I. R. (2022). Circularly Polarized Light-Induced Potentials and the Demise of Excited States. Phys. Chem. Chem. Phys. 24, 2966–2973. doi:10.1039/d1cp04523g
Cattaneo, L., Vos, J., Bello, R. Y., Palacios, A., Heuser, S., Pedrelli, L., et al. (2018). Attosecond Coupled Electron and Nuclear Dynamics in Dissociative Ionization of H2. Nat. Phys. 14, 733–738. doi:10.1038/s41567-018-0103-2
Chen, Z., and He, F. (2020). Interference of Nuclear Wave Packets Carrying Different Angular Momenta in the Dissociation of H2+ in Strong Circularly Polarized Laser Pulses. Phys. Rev. A 102, 033107. doi:10.1103/physreva.102.033107
Chen, Z., He, P.-L., and He, F. (2020). Spiral Nuclear Momentum Distribution for the Dissociation of H2+ in a Circularly Polarized Laser Pulse. Phys. Rev. A 101, 033406. doi:10.1103/physreva.101.033406
Clarke, D. D. A., Armstrong, G. S. J., Brown, A. C., and van der Hart, H. W. (2018). R-matrix-with-time-dependence Theory for Ultrafast Atomic Processes in Arbitrary Light Fields. Phys. Rev. A 98, 053442. doi:10.1103/physreva.98.053442
Cohen, H. D., and Fano, U. (1966). Interference in the Photo-Ionization of Molecules. Phys. Rev. 150, 30–33. doi:10.1103/physrev.150.30
Djiokap, J. M. N., and Starace, A. F. (2017). Doubly-excited State Effects on Two-Photon Double Ionization of Helium by Time-Delayed, Oppositely Circularly-Polarized Attosecond Pulses. J. Opt. 19, 124003. doi:10.1088/2040-8986/aa8fc0
Djiokap, J. M. N., and Starace, A. F. (2021). Temporal Coherent Control of Resonant Two-Photon Double Ionization of the Hydrogen Molecule via Doubly Excited States. Phys. Rev. A 103, 053110. doi:10.1103/physreva.103.053110
Djiokap, J. M. N., Meremianin, A. V., Manakov, N. L., Madsen, L. B., Hu, S. X., and Starace, A. F. (2018). Dynamical Electron Vortices in Attosecond Double Photoionization of H2. Phys. Rev. A 98, 063407. doi:10.1103/physreva.98.063407
Djiokap, J. M. N., Meremianin, A. V., and Manakov, N. L. (2021). Electron Interference in Atomic Ionization by Two Crossing Polarized Ultrashort Pulses. Phys. Rev. A 103, 023103. doi:10.1103/physreva.103.023103
Dörner, R., Mergel, V., Jagutzki, O., Spielberger, L., Ullrich, J., Moshammer, R., et al. (2000). Cold Target Recoil Ion Momentum Spectroscopy: a ’momentum Microscope’ to View Atomic Collision Dynamics. Phys. Rep. 330, 95–192. doi:10.1016/s0370-1573(99)00109-x
Eckart, S., Richter, M., Kunitski, M., Hartung, A., Rist, J., Henrichs, K., et al. (2016). Nonsequential Double Ionization by Counterrotating Circularly Polarized Two-Color Laser Fields. Phys. Rev. Lett. 117, 133202. doi:10.1103/physrevlett.117.133202
Eckart, S., Kunitski, M., Ivanov, I., Richter, M., Fehre, K., Hartung, A., et al. (2018). Subcycle Interference upon Tunnel Ionization by Counter-rotating Two-Color Fields. Phys. Rev. A 97, 041402. doi:10.1103/physreva.97.041402
Eickhoff, K., Kerbstadt, S., Bayer, T., and Wollenhaupt, M. (2020a). Dynamic Quantum State Holography. Phys. Rev. A 101, 013430. doi:10.1103/physreva.101.013430
Eickhoff, K., Köhnke, D., Feld, L.-C., Englert, L., Bayer, T., and Wollenhaupt, M. (2020b). Tailored Holograms for Superimposed Vortex States. New J. Phys. 22, 123015. doi:10.1088/1367-2630/abc8b0
Eickhoff, K., Rathje, C., Köhnke, D., Kerbstadt, S., Englert, L., Bayer, T., et al. (2020c). Orbital Angular Momentum Superposition States in Transmission Electron Microscopy and Bichromatic Multiphoton Ionization. New J. Phys. 22, 103045. doi:10.1088/1367-2630/abbe54
Eickhoff, K., Englert, L., Bayer, T., and Wollenhaupt, M. (2021a). Multichromatic Polarization-Controlled Pulse Sequences for Coherent Control of Multiphoton Ionization. Front. Phys. 9, 444. doi:10.3389/fphy.2021.675258
Eickhoff, K., Feld, L., Köhnke, D., Bayer, T., and Wollenhaupt, M. (2021b). Trichromatic Shaper-Based Quantum State Holography. Phys. Rev. A 104, 052805. doi:10.1103/physreva.104.052805
Eickhoff, K., Feld, L., Köhnke, D., Englert, L., Bayer, T., and Wollenhaupt, M. (2021c). Coherent Control Mechanisms in Bichromatic Multiphoton Ionization. J. Phys. B Atomic, Mol. Opt. Phys. 54, 164002. doi:10.1088/1361-6455/ac11a0
Eickhoff, K., Köhnke, D., Feld, L., Bayer, T., and Wollenhaupt, M. (2022). Determination of Atomic Multiphoton Ionization Phases by Trichromatic Multichannel Wave Packet Interferometry. Phys. Rev. A 105, 053113.
Eiles, M. T. (2019). Trilobites, Butterflies, and Other Exotic Specimens of Long-Range Rydberg Molecules. J. Phys. B 52, 113001. doi:10.1088/1361-6455/ab19ca
Erdmann, M., Gross, E. K. U., and Engel, V. (2004). Time-dependent Electron Localization Functions for Coupled Nuclear-Electronic Motion. J. Chem. Phys. 121, 9666–9670. doi:10.1063/1.1806812
Feit, M. D., Fleck, J. A., and Steiger, A. (1982). Solution of the Schrödinger Equation by a Spectral Method. J. Comp. Phys. 47, 412–433. doi:10.1016/0021-9991(82)90091-2
Fernández, J., Yip, F. L., Rescigno, T. N., McCurdy, C. W., and Martín, F. (2009). Two-center Effects in One-Photon Single Ionization of H2+, H2, and Li2+ with Circularly Polarized Light. Phys. Rev. A 79, 043409. doi:10.1103/physreva.79.043409
Gatti, F. (2014). Molecular Quantum Dynamics: From Theory to Applications. Berlin Heidelberg: Springer.
Gazibegović-Busuladžić, A., Becker, W., and Milošević, D. B. (2018). Helicity Asymmetry in Strong-Field Ionization of Atoms by a Bicircular Laser Field. Opt. Express 26, 12684–12697. doi:10.1364/oe.26.012684
Geng, L., Cajiao, V., F., Kamiński, J. Z., Peng, L.-Y., and Krajewska, K. (2020). Vortex Structures in Photodetachment by Few-Cycle Circularly Polarized Pulses. Phys. Rev. A 102, 043117. doi:10.1103/physreva.102.043117
Geng, L., Cajiao Vélez, F., Kamiński, J. Z., Peng, L.-Y., and Krajewska, K. (2021). Structured Photoelectron Distributions in Photodetachment Induced by Trains of Laser Pulses: Vortices versus Spirals. Phys. Rev. A 104, 033111. doi:10.1103/physreva.104.033111
Greene, C. H., Dickinson, A. S., and Sadeghpour, H. R. (2000). Creation of Polar and Nonpolar Ultra-long-range Rydberg Molecules. Phys. Rev. Lett. 85, 2458–2461. doi:10.1103/physrevlett.85.2458
Grossmann, F. (2018). Theoretical Femtosecond Physics: Atoms and Molecules in Strong Laser Fields. Berlin Heidelberg: Springer.
Guo, J., Zhang, S. Q., Zhang, J., Zhou, S. P., and Guan, P. F. (2021). Exploration of Electron Vortices in the Photoionization of Diatomic Molecules in Intense Laser Fields. Laser Phys. 31, 065301. doi:10.1088/1555-6611/abfb73
Hasović, E., Becker, W., and Milošević, D. B. (2016). Electron Rescattering in a Bicircular Laser Field. Opt. Express 24, 6413–6424. doi:10.1364/OE.24.006413
Heather, R., and Metiu, H. (1987). An Efficient Procedure for Calculating the Evolution of the Wave Function by Fast Fourier Transform Methods for Systems with Spatially Extended Wave Function and Localized Potential. J. Chem. Phys. 86, 5009–5017. doi:10.1063/1.452672
Hergenhahn, U., Rennie, E. E., Kugeler, O., Marburger, s., Lischke, T., Powis, I., et al. (2004). Photoelectron Circular Dichroism in Core Level Ionization of Randomly Oriented Pure Enantiomers of the Chiral Molecule Camphor. J. Chem. Phys. 120, 4553–4556. doi:10.1063/1.1651474
Hoff, D., Kruger, M., Maisenbacher, L., Sayler, A. M., Paulus, G. G., and Hommelhoff, P. (2017). Tracing the Phase of Focused Broadband Laser Pulses. Nat. Phys. 13, 947. doi:10.1038/nphys4185
Huismans, Y., Rouzée, A., Gijsbertsen, A., Jungmann, J. H., Smolkowska, A. S., Logman, P., et al. (2011). Time-resolved Holography with Photoelectrons. Science 331, 61–64. doi:10.1126/science.1198450
Ivanov, I. A., Nam, C. H., and Kim, K. T. (2017). Photoionization in the Presence of Circularly Polarized Fundamental and Odd-Order Harmonic Fields. Phys. Rev. A 95, 053401. doi:10.1103/physreva.95.053401
Jia, J., Cui, H., Zhang, C., Shao, J., Ma, J., and Miao, X. (2019). Investigation of the Photoionization Process of Helium Ion in Bichromatic Circularly Xuv Fields with Different Time Delays. Chem. Phys. Lett. 725, 119–123. doi:10.1016/j.cplett.2019.04.008
Ke, Q., Zhou, Y., Tan, J., He, M., Liang, J., Zhao, Y., et al. (2019). Two-dimensional Photoelectron Holography in Strong-Field Tunneling Ionization by Counter Rotating Two-Color Circularly Polarized Laser Pulses. Opt. Express 27, 32193–32209. doi:10.1364/oe.27.032193
Kerbstadt, S., Pengel, D., Johannmeyer, D., Englert, L., Bayer, T., and Wollenhaupt, M. (2017a). Control of Photoelectron Momentum Distributions by Bichromatic Polarization-Shaped Laser Fields. New J. Phys. 19, 103017. doi:10.1088/1367-2630/aa83a4
Kerbstadt, S., Timmer, D., Englert, L., Bayer, T., and Wollenhaupt, M. (2017b). Ultrashort Polarization-Tailored Bichromatic Fields from a CEP-Stable White Light Supercontinuum. Opt. Express 25, 12518. doi:10.1364/oe.25.012518
Kerbstadt, S., Pengel, D., Englert, L., Bayer, T., and Wollenhaupt, M. (2018). Carrier-envelope-phase Control of Asymmetries in the Multiphoton Ionization of Xenon Atoms by Ultrashort Bichromatic Fields. Phys. Rev. A 97, 063402. doi:10.1103/physreva.97.063402
Kerbstadt, S., Eickhoff, K., Bayer, T., and Wollenhaupt, M. (2019a). Control of Free Electron Wave Packets by Polarization-Tailored Ultrashort Bichromatic Laser Fields. Adv. Phys. X 4, 1672583. doi:10.1080/23746149.2019.1672583
Kerbstadt, S., Eickhoff, K., Bayer, T., and Wollenhaupt, M. (2019b). Odd Electron Wave Packets from Cycloidal Ultrashort Laser Fields. Nat. Comm. 10, 658. doi:10.1038/s41467-019-08601-7
Kerbstadt, S., Eickhoff, K., Bayer, T., and Wollenhaupt, M. (2020). Bichromatic Control of Free Electron Wave Packets. Cham, Switzerland: Springer, 43–76. book section 3. doi:10.1007/978-3-030-47098-2_3
Köhler, J., Wollenhaupt, M., Bayer, T., Sarpe, C., and Baumert, T. (2011). Zeptosecond Precision Pulse Shaping. Opt. Express 19, 11638–11653. doi:10.1364/oe.19.011638
Kong, X., Zhang, G., Li, M., Wang, T., Ding, X., and Yao, J. (2018). Odd-fold-symmetric Spiral Momentum Distributions and Their Stark Distortions in Hydrogen. JOSA B 35, 2163–2168. doi:10.1364/josab.35.002163
Kosloff, R., and Kosloff, D. (1986). Absorbing Boundaries for Wave Propagation Problems. J. Comp. Phys. 63, 363–376. doi:10.1016/0021-9991(86)90199-3
Li, M., Zhang, G., Zhao, T., Ding, X., and Yao, J. (2017a). Electron Vortices in Photoionization by a Pair of Elliptically Polarized Attosecond Pulses. Chin. Opt. Lett. 15, 120202. doi:10.3788/col201715.120202
Li, Z. L., Li, Y. J., and Xie, B. S. (2017b). Momentum Vortices on Pairs Production by Two Counter-rotating Fields. Phys. Rev. D. 96, 076010. doi:10.1103/physrevd.96.076010
Li, M., Zhang, G., Ding, X., and Yao, Y. (2018a). Symmetric Electron Vortices of Hydrogen Ionized by Orthogonal Elliptical Fields. IEEE Photonics J. 10, 1–9. doi:10.1109/jphot.2018.2854237
Li, M., Zhang, G., Kong, X., Wang, T., Ding, X., and Yao, J. (2018b). Dynamic Stark Induced Vortex Momentum of Hydrogen in Circular Fields. Opt. Express 26, 878–886. doi:10.1364/oe.26.000878
Li, Z. L., Xie, B. S., and Li, Y. J. (2018c). Vortices in Multiphoton Pair Production by Two-Color Rotating Laser Fields. J. Phys. B 52, 025601. doi:10.1088/1361-6455/aaf3f9
Li, M., Zhang, G., Ding, X., and Yao, J. (2019a). Ac Stark Effect on Vortex Spectra Generated by Circularly Polarized Pulses. IEEE Photonics J. 11, 1–11. doi:10.1109/jphot.2019.2916106
Li, M., Zhang, G. Z., Ding, X., and Yao, J. Q. (2019b). Carrier Envelope Phase Description for an Isolated Attosecond Pulse by Momentum Vortices. Chin. Phys. Lett. 36, 063201. doi:10.1088/0256-307x/36/6/063201
Li, D., Shan, H., Rupprecht, C., Knopf, H., Watanabe, K., Taniguchi, T., et al. (2022). Hybridized exciton-photon-phonon states in a transition metal dichalcogenide van der waals heterostructure microcavity. Phys. Rev. Lett. 128, 087401. doi:10.1103/PhysRevLett.128.087401
Lloyd, S. M., Babiker, M., Thirunavukkarasu, G., and Yuan, J. (2017). Electron Vortices: Beams with Orbital Angular Momentum. Rev. Mod. Phys. 89, 035004. doi:10.1103/revmodphys.89.035004
Lux, C., Wollenhaupt, M., Bolze, T., Liang, Q., Köhler, J., Sarpe, C., et al. (2012). Circular Dichroism in the Photoelectron Angular Distributions of Camphor and Fenchone from Multiphoton Ionization with Femtosecond Laser Pulses. Angew. Chem. Int. Ed. 51, 5001–5005. doi:10.1002/anie.201109035
Madelung, E. (1927). Quantentheorie in Hydrodynamischer Form. Z. Phys. 40, 322–326. doi:10.1007/bf01400372
Mancuso, C. A., Hickstein, D. D., Grychtol, P., Knut, R., Kfir, O., Tong, X. M., et al. (2015). Strong-field Ionization with Two-Color Circularly Polarized Laser Fields. Phys. Rev. A 91, 031402R. doi:10.1103/physreva.91.031402
Mancuso, C. A., Hickstein, D. D., Dorney, K. M., Ellis, J. L., Hasovic, E., Knut, R., et al. (2016). Controlling Electron-Ion Rescattering in Two-Color Circularly Polarized Femtosecond Laser Fields. Phys. Rev. A 93, 053406. doi:10.1103/physreva.93.053406
Marston, C. C., and Balint-Kurti, G. G. (1989). The Fourier Grid Hamiltonian Method for Bound State Eigenvalues and Eigenfunctions. J. Chem. Phys. 91, 3571–3576. doi:10.1063/1.456888
Maxwell, A., Armstrong, G. S. J., Ciappina, M., Pisanty, E., Kang, Y., Brown, A., et al. (2020). Manipulating Twisted Electrons in Strong Field Ionization. Faraday Discuss. 228, 394–412. doi:10.1063/1.1675522
Meshulach, D., and Silberberg, Y. (1999). Coherent Quantum Control of Multiphoton Transitions by Shaped Ultrashort Optical Pulses. Phys. Rev. A 60, 1287–1292. doi:10.1103/physreva.60.1287
Mulliken, R. S. (1971). Role of Kinetic Energy in the Franck-Condon Principle. J. Chem. Phys. 55, 309–314. doi:10.1063/1.1675522
Ngoko Djiokap, J. M., Hu, S. X., Madsen, L. B., Manakov, N. L., Meremianin, A. V., and Starace, A. F. (2015). Electron Vortices in Photoionization by Circularly Polarized Attosecond Pulses. Phys. Rev. Lett. 115, 113004. doi:10.1103/physrevlett.115.113004
Ngoko Djiokap, J. M., Meremianin, A. V., Manakov, N. L., Hu, S. X., Madsen, L. B., and Starace, A. F. (2016). Multistart Spiral Electron Vortices in Ionization by Circularly Polarized Uv Pulses. Phys. Rev. A 94, 013408. doi:10.1103/physreva.94.013408
Ngoko Djiokap, J. M., Meremianin, A. V., Manakov, N. L., Hu, S. X., Madsen, L. B., and Starace, A. F. (2017). Kinematical Vortices in Double Photoionization of Helium by Attosecond Pulses. Phys. Rev. A 96, 013405. doi:10.1103/physreva.96.013405
Nisoli, M., Decleva, P., Calegari, F., Palacios, A., and Martín, F. (2017). Attosecond Electron Dynamics in Molecules. Chem. Rev. 117, 10760–10825. doi:10.1021/acs.chemrev.6b00453
Novoselov, K. S., Fal’ko, V. I., Colombo, L., Gellert, P. R., Schwab, M. G., and Kim, K. (2012). A Roadmap for Graphene. Nature 490, 192–200. doi:10.1038/nature11458
Pengel, D., Kerbstadt, S., Englert, L., Bayer, T., and Wollenhaupt, M. (2017a). Control of Three-Dimensional Electron Vortices from Femtosecond Multiphoton Ionization. Phys. Rev. A 96, 043426. doi:10.1103/physreva.96.043426
Pengel, D., Kerbstadt, S., Johannmeyer, D., Englert, L., Bayer, T., and Wollenhaupt, M. (2017b). Electron Vortices in Femtosecond Multiphoton Ionization. Phys. Rev. Lett. 118, 053003. doi:10.1103/PhysRevLett.118.053003
Powis, I. (2008). Photoelectron Circular Dichroism in Chiral Molecules. Adv. Chem. Phys. 138, 267–329. doi:10.1002/9780470259474.ch5
Qin, Y. N., Li, M., Feng, Y., Luo, S., Zhou, Y., and Lu, P. (2020). Extracting the Phase Distribution of the Electron Wave Packet Ionized by an Elliptically Polarized Laser Pulse. Front. Phys. 16, 32502. doi:10.1007/s11467-020-1017-x
Santra, R. (2006). Why Complex Absorbing Potentials Work: A Discrete-Variable-Representation Perspective. Phys. Rev. A 74, 034701. doi:10.1103/physreva.74.034701
Shaffer, J. P., Rittenhouse, S. T., and Sadeghpour, H. R. (2018). Ultracold Rydberg Molecules. Nat. Comm. 9, 1965. doi:10.1038/s41467-018-04135-6
Shen, Y., Wang, X., Xie, Z., Min, C., Fu, X., Liu, Q., et al. (2019). Optical Vortices 30 Years on: Oam Manipulation from Topological Charge to Multiple Singularities. Light Sci. Appl. 8, 1–29. doi:10.1038/s41377-019-0194-2
Shin, S., and Metiu, H. (1995). Nonadiabatic Effects on the Charge Transfer Rate Constant: A Numerical Study of a Simple Model System. J. Chem. Phys. 102, 9285–9295. doi:10.1063/1.468795
Shin, S., and Metiu, H. (1996). Multiple Time Scale Quantum Wavepacket Propagation: Electron-Nuclear Dynamics. J. Phys. Chem. 100, 7867–7872. doi:10.1021/jp952498a
Shu, C.-C., Guo, Y., Yuan, K.-J., Dong, D., and Bandrauk, A. D. (2020). Attosecond All-Optical Control and Visualization of Quantum Interference between Degenerate Magnetic States by Circularly Polarized Pulses. Opt. Lett. 45, 960–963. doi:10.1364/ol.386879
Shvetsov-Shilovski, N. I., and Lein, M. (2022). Deep Learning for Retrieval of the Internuclear Distance in a Molecule from Interference Patterns in Photoelectron Momentum Distributions. Phys. Rev. A 105, L021102. doi:10.1103/physreva.105.l021102
Sprik, M., and Klein, M. L. (1988). Optimization of a Distributed Gaussian Basis Set Using Simulated Annealing: Application to the Adiabatic Dynamics of the Solvated Electron. J. Chem. Phys. 89, 1592–1607. doi:10.1063/1.455156
Tal-Ezer, H., and Kosloff, R. (1986). A Direct Relaxation Method for Calculating Eigenfunctions and Eigenvalues of the Schr Odinger Equation. Chem. Phys. Lett. 127, 223.
Tannor, D. (2007). Introduction to Quantum Mechanics: A Time-dependent Perspective. Sausalito: University Science Books.
Ullrich, J., Moshammer, R., Dorn, A., Dörner, R., Schmidt, L. P. H., and Schmidt-Böcking, H. (2003). Recoil-ion and Electron Momentum Spectroscopy: Reaction-Microscopes. Rep. Prog. Phys. 66, 1463–1545. doi:10.1088/0034-4885/66/9/203
Verbeeck, J., Tian, H., and Schattschneider, P. (2010). Production and Application of Electron Vortex Beams. Nature 467, 301. doi:10.1038/nature09366
Wang, R., Zhang, Q., Ran, C., Cao, W., and Lu, P. (2020). Proposal for Detecting Ring Current via Electron Vortices. Opt. Lett. 45, 1383–1386. doi:10.1364/ol.388516
Winter, M., Wollenhaupt, M., and Baumert, T. (2006). Coherent Matter Waves for Ultrafast Laser Pulse Characterization. Opt. Comm. 264, 285–292. doi:10.1016/j.optcom.2005.12.079
Wollenhaupt, M., Assion, A., Liese, D., Sarpe-Tudoran, C., Baumert, T., Zamith, S., et al. (2002). Interferences of Ultrashort Free Electron Wave Packets. Phys. Rev. Lett. 89, 173001. doi:10.1103/physrevlett.89.173001
Wollenhaupt, M., Assion, A., Graefe, O., Liese, D., Sarpe-Tudoran, C., Winter, M., et al. (2003). Changes of the Electronic Structure along the Internuclear Coordinate Studied by Ultrafast Photoelectron Spectroscopy: the 21 Σu+ Na2 Double Minimum State. Chem. Phys. Lett. 376, 457–464. doi:10.1016/s0009-2614(03)00994-1
Wollenhaupt, M., Engel, V., and Baumert, T. (2005). Femtosecond Laser Photoelectron Spectroscopy on Atoms and Small Molecules: Prototype Studies in Quantum Control. Annu. Rev. Phys. Chem. 56, 25–56. doi:10.1146/annurev.physchem.56.092503.141315
Wollenhaupt, M., Krug, M., Köhler, J., Bayer, T., Sarpe-Tudoran, C., and Baumert, T. (2009a). Photoelectron Angular Distributions from Strong-Field Coherent Electronic Excitation. Appl. Phys. B 95, 245–259. doi:10.1007/s00340-009-3431-1
Wollenhaupt, M., Krug, M., Köhler, J., Bayer, T., Sarpe-Tudoran, C., and Baumert, T. (2009b). Three-dimensional Tomographic Reconstruction of Ultrashort Free Electron Wave Packets. Appl. Phys. B 95, 647–651. doi:10.1007/s00340-009-3513-0
Wollenhaupt, M., Lux, C., Krug, M., and Baumert, T. (2013). Tomographic Reconstruction of Designer Free-Electron Wave Packets. Chem. Phys. Chem. 14, 1341–1349. doi:10.1002/cphc.201200968
Xiao, X.-R., Wang, M.-X., Liang, H., Gong, Q., and Peng, L.-Y. (2019). Proposal for Measuring Electron Displacement Induced by a Short Laser Pulse. Phys. Rev. Lett. 122, 053201. doi:10.1103/PhysRevLett.122.053201
Yuan, K.-J., and Bandrauk, A. D. (2015). Electron Interference in Molecular Circular Polarization Attosecond Xuv Photoionization. Photonics 2, 71–92. doi:10.3390/photonics2010071
Yuan, K.-J., Chelkowski, S., and Bandrauk, A. D. (2016). Photoelectron Momentum Distributions of Molecules in Bichromatic Circularly Polarized Attosecond UV Laser Fields. Phys. Rev. A 93, 053425. doi:10.1103/physreva.93.053425
Yuan, K. J., Lu, H. Z., and Bandrauk, A. D. (2017). Photoionization of Triatomic Molecular Ions H32+ by Intense Bichromatic Circularly Polarized Attosecond Uv Laser Pulses. J. Phys. B 50, 124004.
Zewail, A. H. (1995). Femtochemistry: Concepts and Applications. Weinheim: VCH Verlagsgesellschaft mbH, 15–130.
Zewail, A. H. (2000). Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond Using Ultrafast Lasers. Angew. Chem. Int. Ed. 39, 2586–2631. doi:10.1002/1521-3773(20000804)39:15<2586::aid-anie2586>3.0.co;2-o
Keywords: free electron vortices, polarization-tailored femtosecond laser pulses, diatomic molecules, multiphoton ionization, coupled electron-nuclear dynamics, polarization-shaped bichromatic fields, photoelectron momentum distribution
Citation: Bayer T and Wollenhaupt M (2022) Molecular Free Electron Vortices in Photoionization by Polarization-Tailored Ultrashort Laser Pulses. Front. Chem. 10:899461. doi: 10.3389/fchem.2022.899461
Received: 18 March 2022; Accepted: 06 May 2022;
Published: 02 June 2022.
Edited by:
Yuichi Fujimura, Tohoku University, JapanReviewed by:
Martin Centurion, University of Nebraska-Lincoln, United StatesCopyright © 2022 Bayer and Wollenhaupt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Wollenhaupt, bWF0dGhpYXMud29sbGVuaGF1cHRAdW9sLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.