 Jie Wu
Jie Wu Tairen Long
Tairen Long Haiyan Wang
Haiyan Wang Jin-Xia Liang
Jin-Xia Liang Chun Zhu
Chun Zhu- School of Chemistry and Chemical Engineering, Guizhou University, Guiyang, China
Methane is the simplest alkane and can be used as an alternative energy source for oil and coal, but the greenhouse effect caused by its leakage into the air is not negligible, and its conversion into liquid methanol not only facilitates transportation, but also contributes to carbon neutrality. In order to find an efficient method for converting methane to methanol, CH4 oxidation catalyzed by Fe(IV)-Oxo-corrolazine (Fe(IV)-Oxo-Cz) and its reaction mechanism regulation by oriented external electric fields (OEEFs) are systematically studied by density functional calculations. The calculations show that Fe(IV)-Oxo-Cz can abstract one H atom from CH4 to form the intermediate with OH group connecting on the corrolazine ring, with the energy barrier of 25.44 kcal mol−1. And then the product methanol is formed through the following rebound reaction. Moreover, the energy barrier can be reduced to 20.72 kcal mol−1 through a two-state reaction pathway. Furthermore, the effect of OEEFs on the reaction is investigated. We found that OEEFs can effectively regulate the reaction by adjusting the stability of the reactant and the transition state through the interaction of electric field-molecular dipole moment. When the electric field is negative, the energy barrier of the reaction decreases with the increase of electric intensity. Moreover, the OEEF aligned along the intrinsic Fe‒O reaction axis can effectively regulate the ability of forming the OH on the corrolazine ring by adjusting the charges of O and H atoms. When the electric field intensity is −0.010 a.u., the OH can be directly rebounded to the CH3· before it is connecting on the corrolazine ring, thus forming the product directly from the transition state without passing through the intermediate with only an energy barrier of 17.34 kcal mol−1, which greatly improves the selectivity of the reaction.
1 Introduction
Compared with oil or coal, methane is an environment-friendly energy, but it is also a greenhouse gas, and its greenhouse effect is much larger than that of carbon dioxide (Chong et al., 2016; Huang et al., 2018; Denning et al., 2021; Huang et al., 2021; Sánchez-López et al., 2021). A quarter of the greenhouse effect caused by man-made greenhouse gases is caused by the leakage of methane into the atmosphere (Kemp et al., 2016). In 2021, the United Nations called for a reduction in methane emission in the atmosphere, aiming to reduce global methane emission by 30% by the end of the century (Brenneis et al., 2022). Therefore, if an efficient method can be found to convert methane into methanol efficiently and economically, it can not only solve the difficulty of methane transportation, but also provide a large number of cheap raw materials for industrial production and reduce methane pipelines leakage (Park et al., 2019; Yan et al., 2020; Januario et al., 2021), thus providing feasible methods for methane emission reduction.
The high valent metal-oxygen systems have been characterized as key intermediates of heme and non heme enzymes (Solomon et al., 2000; Ogliaro et al., 2001; Meunier et al., 2004; Shaik et al., 2005; Rittle and Green, 2010; Visser et al., 2013; Adam et al., 2018; Huang and Groves, 2018; Dubey and Shaik, 2019; Ehudin et al., 2019; Cummins et al., 2020; Shaik and Dubey, 2021), which can effectively hydroxylate aliphatic hydrocarbons (Altun et al., 2007; Hazan et al., 2007), epoxidation (Niwa and Nakada, 2012; Nayak et al., 2020), halogenation (Liu and Groves, 2015), N-demethylation (Yang et al., 2018), and dehydrogenation reactions (Kumar et al., 2009). In particular, Fe(IV)-oxo porphyrin π-cation radical species, known as Cpd-I in heme proteins such as cytochrome P450, can mediate many key oxidative processes (Meunier et al., 2004; Shaik et al., 2005; Cho et al., 2012; Zhang et al., 2017; Caddell Haatveit et al., 2019; Caulfield et al., 2019). Corrolazines, formed by replacing the meso-position carbon atoms of corroles with N atoms, are very similar in structure to porphyrins, but have more π electrons than porphyrins, and can better stabilize high-valent metals (Ramdhanie et al., 2001; Goldberg et al., 2003; Fox et al., 2004; Lansky et al., 2005; Lansky and Goldberg, 2006; Goldberg 2007; McGown et al., 2009; Prokop et al., 2011; Pierloot et al., 2012; Baglia et al., 2015; Joslin et al., 2016; Jung et al., 2016; Zaragoza et al., 2016; Ghosh 2017; Zhu et al., 2018; Dedić et al., 2021; Wang et al., 2021; Zhu et al., 2021). As Fe is the active center metal of methane monooxygenase (Shteinman 2020; Freakley et al., 2021), which can selectively convert methane to methanol under natural environmental conditions. Fe-corrolazine is very likely to catalyze the oxidation of methane to methanol under very mild conditions. Therefore, it is necessary to study the oxidation of methane catalyzed by Fe-Oxo-corrolazine.
Recently, Sason Shaik et al. (Hirao et al., 2008; Gorin et al., 2012; Fried and Boxer, 2015; Li et al., 2015; Akamatsu et al., 2017; Che et al., 2018; Ciampi et al., 2018; He et al., 2018; Yu and Coote, 2019; Shaik et al., 2020; Shaik et al., 2004b; Kraskov et al., 2021; Yu et al., 2021; Zhang et al., 2021; de Visser et al., 2022) found that oriented external electric fields (OEEFs) can be used as a new type of catalyst to catalyze reactions by stabilizing transition states through the interactions between OEEFs and the molecular dipole moments, and even can increase the selectivity of reactions through adjusting the direction of OEEFs. Through theoretical calculations, our group also found that the OEEFs can modulate the reaction process through the interactions with the dipole moment of the reaction molecules (Wang et al., 2019).
Herein, we systematically study the reaction process of the oxidation of methane to methanol catalyzed by Fe(IV)-Oxo-corrolazine, and discuss the regulation of its reaction mechanism catalyzed by OEEFs, which provides a theoretical basis for the direct and efficient conversion of methane to methanol.
2 Computational Details
All the calculations were performed in Gaussian16 package (Frisch et al., 2016), using the B3LYP-D3(BJ) (Stephens et al., 1994; Grimme et al., 2010; Grimme et al., 2011) hybrid functional with the LANL2TZ (Roy et al., 2008) basis set coupled with the effective core potential for Fe atom and the all-electron 6–31++G (d,p) (Hariharan and Pople, 1973) basis set for other atoms. The structures of reactant (RC), transition state (TS), intermediate (INT) and product (P) were fully optimized without any symmetry constraints. Then the natures of these optimized structures were assessed by frequencies calculation, for RC, INT, and P with only real frequencies, and for TS with only one imaginary frequency. Moreover, all TS species were further verified by intrinsic reaction coordinate (IRC) calculations. The calculated output file was analyzed by Multiwfn to obtain the spin density (Lu and Chen, 2012).
As shown in Figure 1, using the keyword “Field = M ± N″, the two OEEFs, Fz1 and Fz2 along the Fe‒O axis and O‒H axis respectively, were applied to regulate the CH4 oxidation reaction catalyzed by Fe(IV)-Oxo-Cz. The positive direction of the electric field vector follows the Gaussian 16 convention, i.e., the direction from negative charge to positive charge is Fz > 0. The electric field intensity ranges from −0.010 a.u to +0.010 a.u. for Fz1 and Fz2 (1 a.u. = 51.4 V Å−1).
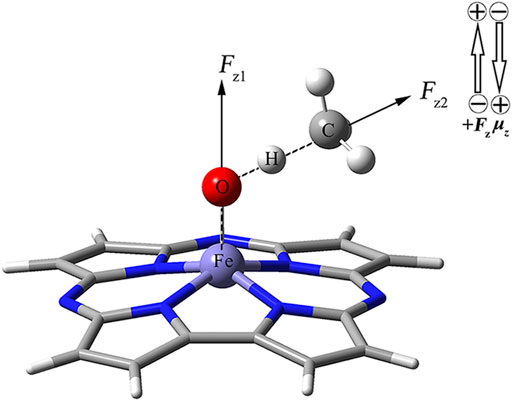
FIGURE 1. Definitions of two OEEFs Fz1 is along the Fe‒O axis perpendicular to the corrolazine ring, and Fz2 is along the O‒H axis.
3 Results and Discussion
3.1 CH4 Oxidation Catalyzed by Fe(IV)-Oxo-Corrolazine Under the Field-Free Condition
3.1.1 Structure and Electronic Properties of Fe(IV)-Oxo-Cz
The geometry structures of Fe(IV)-Oxo-Cz in doublet, quartet and sextet states were optimized and their relative energies and selective structure parameters are collected in Table 1. As seen from Table 1, the quartet state is lower in free energies than the double and sextet states by 10.25 and 40.05 kcal mol−1, respectively (absolute energies in Supplementary Table S1). Moreover, the calculated Fe‒O bond length in quartet state is 1.615 Å, which is close to the experimentally determined value of 1.640 Å (Cho et al., 2012). Therefore, the quartet state is the ground state, and due to the stronger interaction between the Fe atom and the O atom, the Fe atom deviates upward from the corrolazine ring, as shown in Figure 2. Furthermore, the spin densities and NPA charges for the different spin states of Fe(IV)-Oxo-Cz were calculated (see Supplementary Tables S2, S3). For the quartet state, the Fe‒O moiety only occupied two single electrons, and the remaining one single electron was occupied by the corrolazine ring as shown in Figure 2. And the single electron occupying molecular orbitals (SOMO) of the quartet state Fe(IV)-Oxo-Cz is shown in Figure 3. Two single electrons are in the orthogonal π orbitals of the Fe‒O moiety and the other mainly distributes on the corrolazine ring, which is exactly the same as the electronic configuration of Cpd-I (Huang and Groves, 2017; Zaragoza et al., 2017), reflecting the potential enzymatic catalytic activity of Fe(IV)-Oxo-Cz.
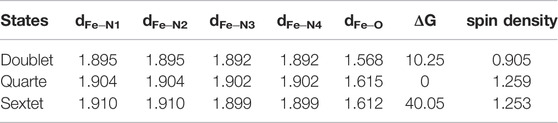
TABLE 1. Selected Bond Lengths (Å), Mulliken spin density of Fe, and relative energies (ΔG, kcal·mol−1) of Fe(IV)-Oxo-Cz, in doublet, quartet and sextet states.
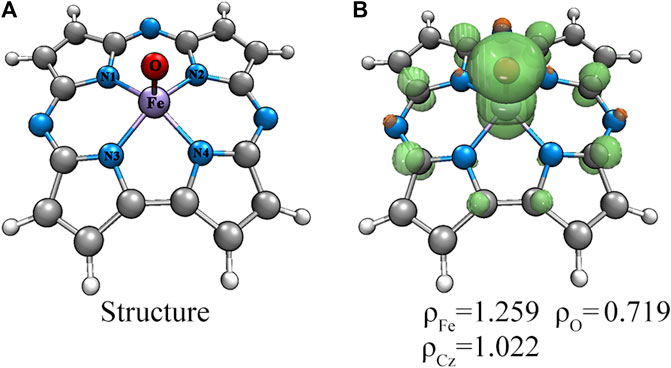
FIGURE 2. Structure (A) and spin densities (B) of the quartet Fe(IV)-Oxo-Cz.

FIGURE 3. The single electron occupying molecular orbitals of the quartet Fe(IV)-Oxo-Cz.
3.1.2 CH4 Oxidation Catalyzed by Fe(IV)-Oxo-Corrolazine Under the Field-free Condition
Based on the quartet ground state structure of Fe(IV)-Oxo-Cz, the reaction between Fe(IV)-Oxo-Cz and CH4 in quartet state in the absence of OEEF was studied. As illustrated in Figure 4; Table 2, the terminal O of Fe‒O first abstracts a hydrogen atom of CH4, and the bond length of Fe‒O bond increases from 1.615 Å of the reactant complex (RC) to 1.725 Å of the transition state (TS1), while the distance between O atom and H atom decreases significantly from 2.393 Å of RC to 1.187 Å of TS1 to yield O‒H bond, with the energy barrier of 25.44 kcal mol−1. With the progress of the reaction, the Fe‒O bond length grows gradually and the O‒H bond length further decreases, yielding the intermediates (INT) of Fe(IV)‒OH and CH3·. Then the product (P) CH3OH is produced through the rebound reaction where the newly formed OH is rapidly rebounded to the CH3· from Fe-corrolazine. (Cummins et al., 2020; Huang and Groves, 2017; Shaik et al., 2004a). And in this step, the reaction barrier is only 1.98 kcal mol−1, and the reaction energy of 40.93 kcal mol−1 will promote the reaction to the right.
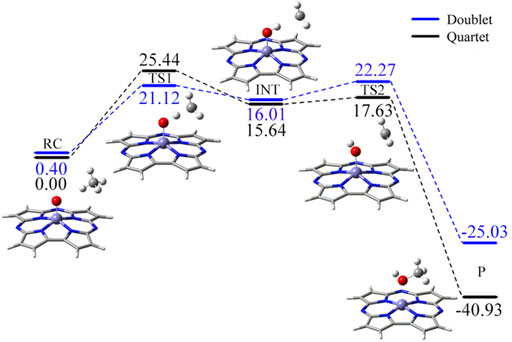
FIGURE 4. The predicted reaction pathway of CH4 oxidation catalyzed Fe(IV)-Oxo-Cz in the electric field free.
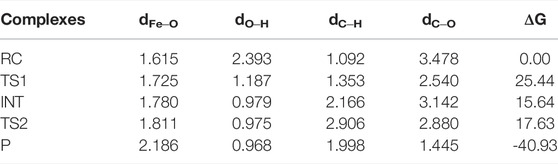
TABLE 2. Selected Bond Lengths (Å) and the relative electronic energies (ΔG, kcal·mol−1) of species involved in the reaction of CH4 oxidation catalyzed for the quartet of Fe(IV)-Oxo-Cz.
Moreover, considering that the reaction may proceed at different potential energy surfaces, we further calculated the double state potential energy surface. As shown in Figure 4 and Supplementary Table S4, the TS1 of the double state is lower than that of the quartet state, indicating that the reaction is a two-state reaction and the reaction is easier to carry out (Schröder et al., 2000; Stuyver et al., 2020). However, considering that the quartet state of RC is the ground state, and the quartet state of P is much more stable than the double state, and that when the OEEF with Fz1 = −0.010 a.u. is applied, the energy order of the quartet and double states of reactant dose not changed and there is no energy crossing points along the reaction pathway, so we further study the effect of OEEFs on the reaction in quartet state in detail. For comparison, we also selected the representative external electric fields −0.010, −0.004, −0.002, +0.002, +0.004, and +0.010 a.u. for the calculations of the double state reaction, as shown in Supplementary Tables S5, S6.” As shown in Supplementary Table S7, the OEEF does not change the rate-determining step of the reaction.
3.2 OEEFs Regulating the Reaction Mechanism
3.2.1 The Effect of OEEF on the Stabilities of the RC and TS1
In order to explore the regulation mechanism of OEEFs in the reaction, we first systematically studied the effect of OEEFs on the TS1 of the reaction. As shown in Figure 5A, due to the application of OEEFs, the relative energies of the reaction TS1 change significantly. For Fz > 0, the structure of TS1 is stabilized by OEEFs, and its relative energy decreases with the increase of the electric field intensity. While for Fz < 0, OEEFs in different directions have different effects on the TS1. For Fz1, when it is in the range of 0 ∼ −0.002 a.u., the TS1 is destabilized by the OEEF, and its relative energy increases with the increase of the electric field intensity. However, as the electric field intensity further increases more than -0.002 a.u. the TS1 is stabilized by the OEEF again, and its relative energy decreases with the increase of the electric field intensity. For Fz2, the TS1 is always stabilized by the OEEF, and its relative energy decreases with the increase of the electric field intensity.
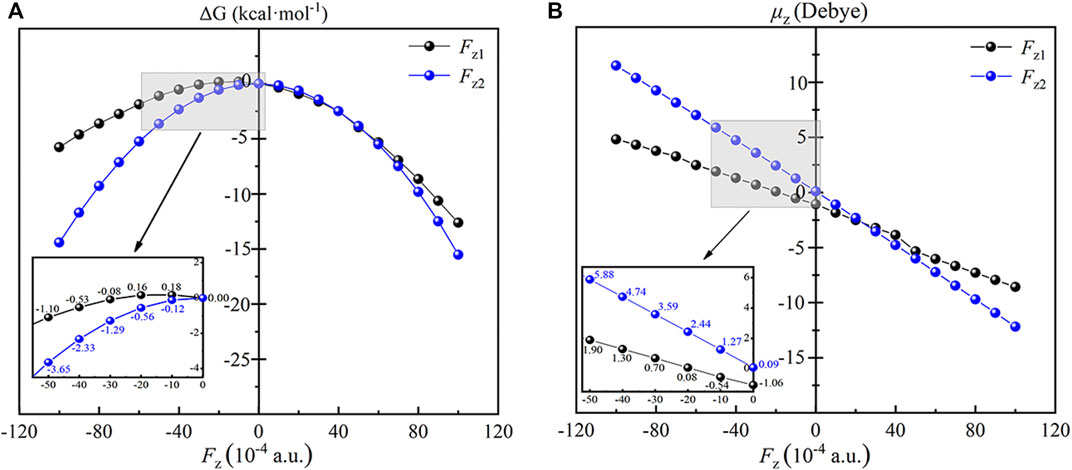
FIGURE 5. Plots of the relative energies (A) and the dipole moments (B) of the TS1 as a function of the applied OEEFs. The inset is the enlarged view at Fz1 and Fz2 = −0.005 a.u. to 0 a.u. black curve for Fz1 and blue curve for Fz2.
To explore the essential reason for the stability change of the TS1 effected by OEEFs, the dipole moments in z orientation of the TS1 of Fe(IV)-Oxo-Cz and CH4 at different electric field intensities are analyzed. As shown in Figure 5B, for Fz1 > 0, the dipole moment in z1 direction of the TS1 increases from -1.06 D in the electric field free to -8.58 D in Fz1 = +0.010 a.u. Therefore, the TS1 is stabilized by the applied OEEFs originating from the attractive interaction between the increased dipole moment in z1 direction and the OEEF. When the OEEF is reversed to Fz1 < 0, the interaction between the dipole moment and the OEEF becomes complex. For −0.002 a.u. < Fz1 < 0, the OEEF decreases the dipole moment of in z1 direction of the TS1, and the repulsion between Fz1 and the dipole moment of the TS1 destabilizes the TS1. However, when Fz1 becomes more negative, it flips the orientation of the molecular dipole of the TS1. Therefore, the increasing OEEF increases the dipole moment in z1 orientation of the TS1, thus again stabilizing the TS1 originating from the attraction between the increased dipole moment and Fz1. For Fz2, the orientation of molecular dipole moment in z2 direction is opposite to the direction of the OEEF, and the dipole moment always increases with the increase of electric field intensity, thus always stabilizing the TS1 of the retion originating from the attractive interaction between the increased dipole moment in z2 direction and the OEEF.
Similar to the TS1, the RC of the reaction of Fe(IV)-Oxo-Cz and CH4 are also effected by the OEEF remarkably. As shown in Figure 6, for Fz1 and Fz2 > 0, the dipole moment in z orientation of the RC increases with the increase of OEEFs. Therefore, the RC is stabilized by OEEFs, originating from the attraction between OEEFs and the increased dipole moment in z orientation. For Fz1 and Fz2 < 0, the dipole moment in z orientation first decreases with the increase of the electric field intensity in the initial part of Fz < 0, and then increases with the increase of the electric field intensity, which is more than -0.006 a.u. for Fz1 and -0.002 a.u. for Fz2 resulting from the reverse of the molecular dipole in z direction of the RC. Thus, the RC is first destabilized and then stabilized by OEEFs resulting from the repulsion and attraction between OEEFs and the increased dipole moment in z orientation, respectively.
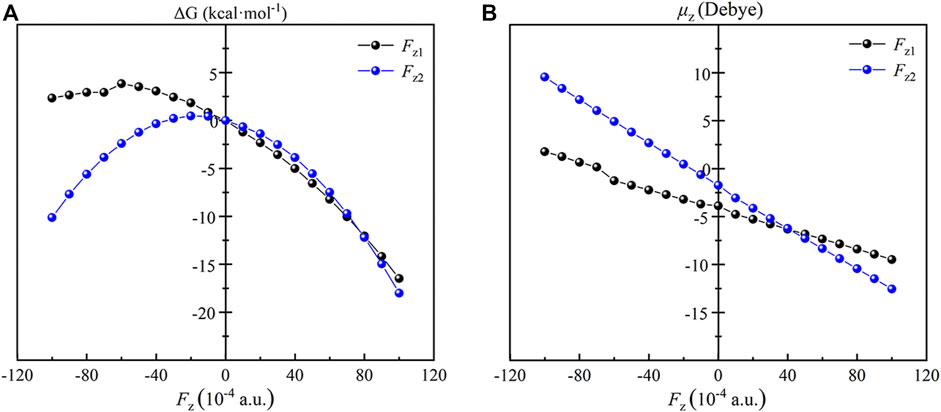
FIGURE 6. Plots of the relative energies (A) and the dipole moments (B) of the RC as a function of the applied OEEFs.
3.2.2 The Effect of OEEFs on the Energy Barrier of CH4 Oxidation Catalyzed by Fe(IV)-Oxo-Corrolazine
The effect of OEEFs on the energy barrier of the reaction of CH4 oxidation by Fe(IV)-Oxo-Cz is further investigated. As shown in Table 3; Figure 7, for Fz1 and Fz2 > 0, the energy barriers of the reaction increase with the increase of electric field intensity, because the dipole moments in z direction of the RC and TS1 increase with the increase of electric field intensity, and the dipole moments of the RC is always larger than the TS1 in the electric intensity range, thus resulting in its stronger stabilization by OEEFs. However, for Fz1 and Fz2 < 0, the stabilization of the TS1 by OEEFs is always stronger than that of the RC, originating from the stronger dipole moment of the TS1, so the energy barriers of the reaction decrease with the increase of electric field intensity, in which the energy barriers decrease to 17.34 and 21.16 kcal mol−1 for Fz1 = −0.010 and Fz2 = −0.010 a.u., respectively, thus greatly promoting the reaction. Especially for Fz1, it can more effectively regulate the reaction than Fz2, resulting from its greater slope of energy barrier curve in Figure 7. Moreover, as its direction is nearly perpendicular to the corrolazine ring, Fz1 can be more easily aligned, thus making it easier to apply in practice.
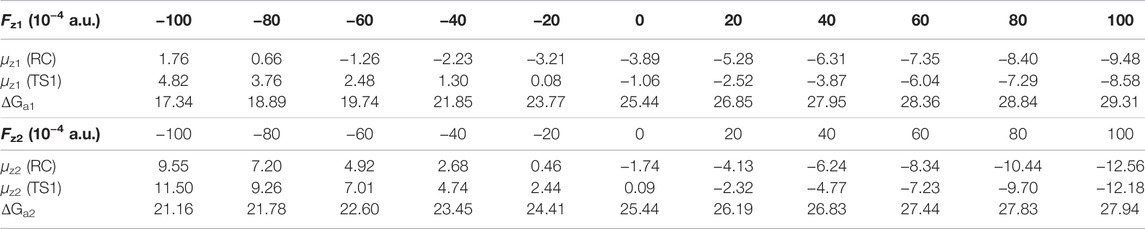
TABLE 3. The dipole moments of the RC, TS1 and the energy barrier (ΔG, kcal·mol−1) of CH4 oxidation catalyzed by Fe(IV)-Oxo-Cz under different electric field intensities.
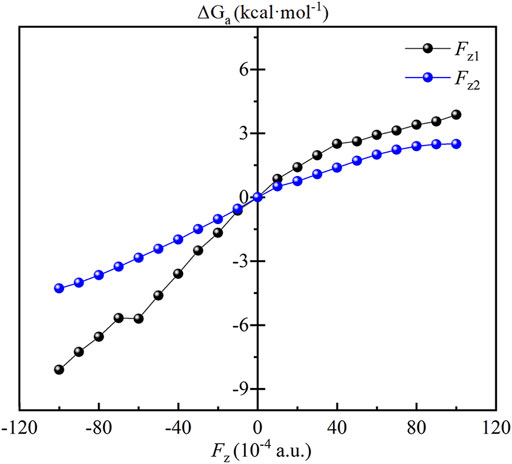
FIGURE 7. Plots of the relative energy barrier (ΔGa, kcal·mol−1) of the CH4 oxidation catalyzed Fe(IV)-Oxo-Cz.
3.2.3 OEEFs Optimizing the Process of CH4 Oxidation Catalyzed by Fe(IV)-Oxo-Corrolazine
As shown in Table 4, for Fz1 < 0, with the increase of electric field intensity, the negative charge of O atom and the positive charge of H atom in the TS1 decrease, while the O‒H distance increases. Therefore, the ability of H to transfer to O to form stable the OH groups on the corrolazine ring decreases, which is the key process of forming the INT. When the intensity of the electric field reaches -0.010 a.u., both the negative charge of the O atom and the positive charge of the H atom reach the smallest, and the OH distance is the largest. Therefore, before the OH group forming on the corrolazine ring, it directly returns to the C atom through the rebound reaction from the P, as shown in Supplementary Figure S1, thereby simplifying the process of the reaction without passing through the INT to the product, thus avoiding the coupling between the intermediates to generate other products (Cho et al., 2012), greatly improving the selectivity of the reaction, and being beneficial to industrial applications.
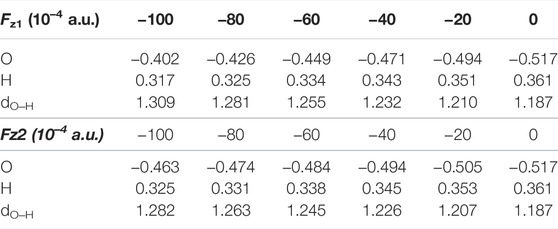
TABLE 4. The O‒H Lengths (Å), the NPA charges (|e|) of the O and H atoms of the TS1 under different electric field intensities.
4 Conclusion
Extensive density functional calculations have been carried out to explore the CH4 oxidation reaction catalyzed by Fe(IV)-Oxo-Cz and its regulatory mechanism by OEEFs. The calculations show one H atom of CH4 is captured by Fe(IV)-Oxo-Cz to form INT, in which OH group is connecting on the corrolazine ring, and then the product methanol is formed through the following rebound reaction. And the energy barrier of the reaction is 25.44 kcal mol−1. Moreover, the energy barrier can be reduced to 20.72 kcal mol−1 through a two-state reaction pathway. To facilitate the reaction, we applied OEEFs Fz1 and Fz2 along the Fe‒O axis and the O‒H axis to modulate the reaction, respectively. When the positive OEEFs are applied, the energy barrier of the reaction increases with the increase of the electric field intensity. However, while flipping OEEFs to the negative direction, the energy barrier of the reaction decreases with the increase of the electric field intensity originating from the interaction of electric field-molecular dipole moment, which can facilitate the reaction. Especially, the Fz1 is easier be applied in practice because its direction is along the intrinsic Fe‒O reaction axis approximately perpendicular to the corrolazine ring, and it can effectively modulate the ability of forming the OH on the corrolazine ring by adjusting the charge of O and H atoms. When its intensity is −0.010 a.u., Fz1 can simplify the reaction path to directly form the reaction product from the transition state without passing through the intermediate, with only an energy barrier of 17.34 kcal mol−1, in which the OH is directly rebounded to CH3· before it is connecting on the corrolazine ring, thus greatly improving the selectivity of the reaction.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
CZ, J-XL, and HW directed the research. JW conducted DFT calculations. JW and TL analyzed the data. All the authors discussed the results and co-write the manuscript.
Funding
This work was supported by the National Science Foundation of China (Nos. 21963005, 21763006), Natural Science Foundation of Guizhou University (No. [2021]40 and [2020]32).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.896944/full#supplementary-material
References
Adam, S. M., Wijeratne, G. B., Rogler, P. J., Diaz, D. E., Quist, D. A., Liu, J. J., et al. (2018). Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function. Chem. Rev. 118, 10840–11022. doi:10.1021/acs.chemrev.8b00074
Akamatsu, M., Sakai, N., and Matile, S. (2017). Electric-Field-Assisted Anion-π Catalysis. J. Am. Chem. Soc. 139, 6558–6561. doi:10.1021/jacs.7b02421
Altun, A., Shaik, S., and Thiel, W. (2007). What Is the Active Species of Cytochrome P450 during Camphor Hydroxylation? QM/MM Studies of Different Electronic States of Compound I and of Reduced and Oxidized Iron-Oxo Intermediates. J. Am. Chem. Soc. 129, 8978–8987. doi:10.1021/ja066847y
Baglia, R. A., Prokop-Prigge, K. A., Neu, H. M., Siegler, M. A., and Goldberg, D. P. (2015). Mn(V)(O) versus Cr(V)(O) Porphyrinoid Complexes: Structural Characterization and Implications for Basicity Controlling H-Atom Abstraction. J. Am. Chem. Soc. 137, 10874–10877. doi:10.1021/jacs.5b05142
Brenneis, R. J., Johnson, E. P., Shi, W., and Plata, D. L. (2022). Atmospheric- and Low-Level Methane Abatement via an Earth-Abundant Catalyst. ACS Environ. Au. 2, 223–231. doi:10.1021/acsenvironau.1c00034
Caddell Haatveit, K., Garcia-Borràs, M., and Houk, K. N. (2019). Computational Protocol to Understand P450 Mechanisms and Design of Efficient and Selective Biocatalysts. Front. Chem. 6, 663. doi:10.3389/fchem.2018.00663
Caulfield, K. P., Conradie, J., Arman, H. D., Ghosh, A., and Tonzetich, Z. J. (2019). Iron(II) Corrole Anions. Inorg. Chem. 58, 15225–15235. doi:10.1021/acs.inorgchem.9b02209
Che, F., Gray, J. T., Ha, S., Kruse, N., and ScottMcEwen, S. L. J.-S. (2018). Elucidating the Roles of Electric Fields in Catalysis: A Perspective. ACS Catal. 8, 5153–5174. doi:10.1021/acscatal.7b02899
Cho, K., Leeladee, P., McGown, A. J., DeBeer, S., and Goldberg, D. P. (2012). A High-Valent Iron-Oxo Corrolazine Activates C‒H Bonds via Hydrogen-Atom Transfer. J. Am. Chem. Soc. 134, 7392–7399. doi:10.1021/ja3018658
Chong, Z. R., Yang, S. H. B., Babu, P., Linga, P., and Li, X. S. (2016). Review of Natural Gas Hydrates as an Energy Resource: Prospects and Challenges. Appl. Energy 162, 1633–1652. doi:10.1016/j.apenergy.2014.12.061
Ciampi, S., Darwish, N., Aitken, H. M., Diez-Perez, I., and Coote, M. L. (2018). Harnessing Electrostatic Catalysis in Single Molecule, Electrochemical and Chemical Systems: a Rapidly Growing Experimental Tool Box. Chem. Soc. Rev. 47, 5146–5164. doi:10.1039/c8cs00352a
Cummins, D. C., Alvarado, J. G., Zaragoza, J. P. T., Effendy Mubarak, M. Q., Lin, Y.-T., de Visser, S. P., et al. (2020). Hydroxyl Transfer to Carbon Radicals by Mn(OH) vs Fe(OH) Corrole Complexes. Inorg. Chem. 59, 16053–16064. doi:10.1021/acs.inorgchem.0c02640
de Visser, S. P., Mukherjee, G., Ali, H. S., and Sastri, C. V. (2022). Local Charge Distributions, Electric Dipole Moments, and Local Electric Fields Influence Reactivity Patterns and Guide Regioselectivities in α-Ketoglutarate-Dependent Non-heme Iron Dioxygenases. Acc. Chem. Res. 55, 65–74. doi:10.1021/acs.accounts.1c00538
Dedić, D., Dorniak, A., Rinner, U., and Schöfberger, W. (2021). Recent Progress in (Photo-)-Electrochemical Conversion of CO2 with Metal Porphyrinoid-Systems. Front. Chem. 9, 685619. doi:10.3389/fchem.2021.685619
Denning, S., Majid, A. A. A., Lucero, J. M., Crawford, J. M., and CarreonKoh, M. A. C. A. (2021). Methane Hydrate Growth Promoted by Microporous Zeolitic Imidazolate Frameworks ZIF-8 and ZIF-67 for Enhanced Methane Storage. ACS Sustain. Chem. Eng. 9, 9001–9010. doi:10.1021/acssuschemeng.1c01488
Dubey, K., and Shaik, S. (2019). Cytochrome P450 -The Wonderful Nanomachine Revealed through Dynamic Simulations of the Catalytic Cycle. Acc. Chem. Res. 52, 389–399. doi:10.1021/acs.accounts.8b00467
Ehudin, M. A., Gee, L. B., Sabuncu, S., Braun, A., Moënne-Loccoz, P., Hedman, B., et al. (2019). Tuning the Geometric and Electronic Structure of Synthetic High-Valent Heme Iron(IV)-Oxo Models in the Presence of a Lewis Acid and Various Axial Ligands. J. Am. Chem. Soc. 141, 5942–5960. doi:10.1021/jacs.9b00795
Fox, J. P., Ramdhanie, B., Zareba, A. A., Czernuszewicz, R. S., and Goldberg, D. P. (2004). Copper(III) and Vanadium(IV)-Oxo Corrolazines. Inorg. Chem. 43, 6600–6608. doi:10.1021/ic049384a
Freakley, S. J., Dimitratos, N., Willock, D. J., Taylor, S. H., and KielyHutchings, C. J. G. J. (2021). Methane Oxidation to Methanol in Water. Acc. Chem. Res. 54, 2614–2623. doi:10.1021/acs.accounts.1c00129
Fried, S. D., and Boxer, S. G. (2015). Measuring Electric Fields and Noncovalent Interactions Using the Vibrational Stark Effect. Acc. Chem. Res. 48, 998–1006. doi:10.1021/ar500464j
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, Revision A.03. Wallingford, CT: Gaussian, Inc.
Ghosh, A. (2017). Electronic Structure of Corrole Derivatives: Insights from Molecular Structures, Spectroscopy, Electrochemistry, and Quantum Chemical Calculations. Chem. Rev. 117, 3798–3881. doi:10.1021/acs.chemrev.6b00590
Goldberg, D. P. (2007). Corrolazines: New Frontiers in High-Valent Metalloporphyrinoid Stability and Reactivity. Acc. Chem. Res. 40, 626–634. doi:10.1021/ar700039y
Goldberg, D. P., Ramdhanie, B., Mandimutsira, B. S., Wang, H., and Fox, J. P. (2003). Corrolazines: Novel Porphyrinoid Compounds Capable of Oxygen Binding, Stabilization of High-Valent Metal-Oxo Species, and More. J. Inorg. Biochem. 96, 21. doi:10.1016/S0162-0134(03)80440-3
Gorin, C. F., Beh, E. S., and Kanan, M. W. (2012). An Electric Field-Induced Change in the Selectivity of a Metal Oxide-Catalyzed Epoxide Rearrangement. J. Am. Chem. Soc. 134, 186–189. doi:10.1021/ja210365j
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 132, 154104. doi:10.1063/1.3382344
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 32, 1456–1465. doi:10.1002/jcc.21759
Hariharan, P. C., and Pople, J. A. (1973). The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta (Berl.) 28, 213–222. doi:10.1007/BF00533485
Hazan, C., Kumar, D., de Visser, S. P., and Shaik, S. (2007). A Density Functional Study of the Factors that Influence the Regioselectivity of Toluene Hydroxylation by Cytochrome P450 Enzymes. Eur. J. Inorg. Chem. 2007, 2966–2974. doi:10.1002/ejic.200700117
He, Z., Cui, H., Hao, S., Wang, L., and Zhou, J. (2018). Electric-Field Effects on Ionic Hydration: A Molecular Dynamics Study. J. Phys. Chem. B 122, 5991–5998. doi:10.1021/acs.jpcb.8b02773
Hirao, H., Chen, H., Carvajal, M. A., Wang, Y., and Shaik, S. (2008). Effect of External Electric Fields on the C‒H Bond Activation Reactivity of Nonheme Iron-Oxo Reagents. J. Am. Chem. Soc. 130, 3319–3327. doi:10.1021/ja070903t
Huang, K., Miller, J. B., Huber, G. W., Dumesic, J. A., and Maravelias, C. T. (2018). A General Framework for the Evaluation of Direct Nonoxidative Methane Conversion Strategies. Joule 2, 349–365. doi:10.1016/j.joule.2018.01.001
Huang, X. Y., and Groves, J. T. (2017). Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C‒H Activation. J. Biol. Inorg. Chem. 22, 185–207. doi:10.1007/s00775-016-1414-3
Huang, X. Y., and Groves, J. T. (2018). Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 118, 2491–2553. doi:10.1021/acs.chemrev.7b00373
Huang, Y. F., Shao, Y. M., Bai, Y., Yuan, Q. C., Ming, T. Z., Davies, P., et al. (2021). Feasibility of Solar Updraft Towers as Photocatalytic Reactors for Removal of Atmospheric Methane-The Role of Catalysts and Rate Limiting Steps. Front. Chem. 9, 745347. doi:10.3389/fchem.2021.745347
Januario, E. R., Silvaino, P. F., Machado, A. P., Moreira Vaz, J., and Spinace, E. V. (2021). Methane Conversion under Mild Conditions Using Semiconductors and Metal-Semiconductors as Heterogeneous Photocatalysts: State of the Art and Challenges. Front. Chem. 9, 685073. doi:10.3389/fchem.2021.685073
Joslin, E. E., Zaragoza, J. P. T., Baglia, R. A., Siegler, M. A., and Goldberg, D. P. (2016). The Influence of Peripheral Substituent Modification on PV, MnIII, and MnV(O) Corrolazines: X-Ray Crystallography, Electrochemical and Spectroscopic Properties, and HAT and OAT Reactivities. Inorg. Chem. 55, 8646–8660. doi:10.1021/acs.inorgchem.6b01219
Jung, J., Neu, H. M., Leeladee, P., Siegler, M. A., Ohkubo, K., Goldberg, D. P., et al. (2016). Photocatalytic Oxygenation of Substrates by Dioxygen with Protonated Manganese(III) Corrolazine. Inorg. Chem. 55, 3218–3228. doi:10.1021/acs.inorgchem.5b02019
Kemp, C. E., Ravikumar, A. P., and Brandt, A. R. (2016). Comparing Natural Gas Leakage Detection Technologies Using an Open-Source “Virtual Gas Field” Simulator. Environ. Sci. Technol. 50, 4546–4553. doi:10.1021/acs.est.5b06068
Kraskov, A., von Sass, J., Nguyen, A. D., Hoang, T. O., Buhrke, D., Katz, S., et al. (2021). Local Electric Field Changes during the Photoconversion of the Bathy Phytochrome Agp2. Biochemistry 60, 2967–2977. doi:10.1021/acs.biochem.1c00426
Kumar, D., Tahsini, L., de Visser, S. P., Kang, H. Y., and KimNam, S. J. W. (2009). Effect of Porphyrin Ligands on the Regioselective Dehydrogenation versus Epoxidation of Olefins by Oxoiron(IV) Mimics of Cytochrome P450. J. Phys. Chem. A 113, 11713–11722. doi:10.1021/jp9028694
Lansky, D. E., and Goldberg, D. (2006). Hydrogen Atom Abstraction by a High-Valent Manganese(V)-Oxo Corrolazine. Inorg. Chem. 45, 5119–5125. doi:10.1021/ic060491+
Lansky, D. E., Mandimutsira, B., Ramdhanie, B., Clausén, M., Penner-Hahn, J., Zvyagin, S. A., et al. (2005). Synthesis, Characterization, and Physicochemical Properties of Manganese(III) and Manganese(V)-Oxo Corrolazines. Inorg. Chem. 44, 4485–4498. doi:10.1021/ic0503636
Li, H. X., Su, T. A., Zhang, V., Steigerwald, M. L., and NuckollsVenkataraman, C. L. (2015). Electric Field Breakdown in Single Molecule Junctions. J. Am. Chem. Soc. 137, 5028–5033. doi:10.1021/ja512523r
Liu, W., and Groves, J. T. (2015). Manganese Catalyzed C‒H Halogenation. Acc. Chem. Res. 48, 1727–1735. doi:10.1021/acs.accounts.5b00062
Lu, T., and Chen, F. W. (2012). Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 33, 580–592. doi:10.1002/jcc.22885
McGown, A. J., Kerber, W. D., Fujii, H., and Goldberg, D. P. (2009). Catalytic Reactivity of a Meso-N-Substituted Corrole and Evidence for a High-Valent Iron-Oxo Species. J. Am. Chem. Soc. 131, 8040–8048. doi:10.1021/ja809183z
Meunier, B., de Visser, S. P., and Shaik, S. (2004). Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 104, 3947–3980. doi:10.1021/cr020443g
Nayak, M., Nayak, P., Sahu, K., and Kar, S. (2020). Synthesis, Characterization, and Application of Oxo-Molybdenum(V)-Corrolato Complexes in Epoxidation Reactions. J. Org. Chem. 85, 11654–11662. doi:10.1021/acs.joc.0c01146
Niwa, T., and Nakada, M. (2012). A Non-heme Iron(III) Complex with Porphyrin-like Properties that Catalyzes Asymmetric Epoxidation. J. Am. Chem. Soc. 134, 13538–13541. doi:10.1021/ja304219s
Ogliaro, F., Visser, S., Cohen, S., Kaneti, J., and Shaik, S. (2001). The Experimentally Elusive Oxidant of Cytochrome P450: A Theoretical “Trapping” Defining More Closely the “Real” Species. Chembiochem 2, 848–851. doi:10.1002/1439-7633(20011105)2:11<848::AID-CBIC848>3.0.CO;2-0
Park, M. B., Park, E. D., and Ahn, W.-S. (2019). Recent Progress in Direct Conversion of Methane to Methanol over Copper-Exchanged Zeolites. Front. Chem. 7, 514. doi:10.3389/fchem.2019.00514
Pierloot, K., Langner, E., Swarts, J., Conradie, J., and Ghosh, A. (2012). Low-Energy States of Manganese-Oxo Corrole and Corrolazine: Multiconfiguration Reference Ab Initio Calculations. Inorg. Chem. 51, 4002–4006. doi:10.1021/ic201972f
Prokop, K. A., Neu, H. M., de Visser, S. P., and Goldberg, D. P. (2011). A Manganese(V)-Oxo π-Cation Radical Complex: Influence of One-Electron Oxidation on Oxygen-Atom Transfer. J. Am. Chem. Soc. 133, 15874–15877. doi:10.1021/ja2066237
Ramdhanie, B., Stern, C. L., and Goldberg, D. P. (2001). Synthesis of the First Corrolazine: A New Member of the Porphyrinoid Family. J. Am. Chem. Soc. 123, 9447–9448. doi:10.1021/ja011229x
Rittle, J., and Green, M. T. (2010). Cytochrome P450 Compound I: Capture, Characterization, and C‒H Bond Activation Kinetics. Science 330, 933–937. doi:10.1126/science.1193478
Roy, L. E., Hay, P. J., and Martin, R. L. (2008). Revised Basis Sets for the LANL Effective Core Potentials. J. Chem. Theory Comput. 4, 1029–1031. doi:10.1021/ct8000409
Sánchez-López, P., Kotolevich, Y., Yocupicio-Gaxiola, R. I., Antúnez-García, J., Chowdari, R. K., Petranovskii, V., et al. (2021). Recent Advances in Catalysis Based on Transition Metals Supported on Zeolites. Front. Chem. 9, 716745. doi:10.3389/fchem.2021.716745
Schröder, D., Shaik, S., and Schwarz, H. (2000). Two-State Reactivity as a New Concept in Organometallic Chemistry. Acc. Chem. Res. 33, 139–145. doi:10.1021/ar990028j
Shaik, S., Cohen, S., Visser, S., Sharma, P., Kumar, D., Kozuch, S., et al. (2004a). The “Rebound Controversy”: An Overview and Theoretical Modeling of the Rebound Step in C‒H Hydroxylation by Cytochrome P450. Eur. J. Inorg. Chem. 2004, 207–226. doi:10.1002/ejic.200300448
Shaik, S., Danovich, D., Joy, J., Wang, Z. F., and Stuyver, T. (2020). Electric-Field Mediated Chemistry: Uncovering and Exploiting the Potential of (Oriented) Electric Fields to Exert Chemical Catalysis and Reaction Control. J. Am. Chem. Soc. 142, 12551–12562. doi:10.1021/jacs.0c05128
Shaik, S., de Visser, S. P., and Kumar, D. (2004b). External Electric Field Will Control the Selectivity of Enzymatic-like Bond Activations. J. Am. Chem. Soc. 126, 11746–11749. doi:10.1021/ja047432k
Shaik, S., and Dubey, K. D. (2021). The Catalytic Cycle of Cytochrome P450: a Fascinating Choreography. Trends Chem. 3, 1027–1044. doi:10.1016/j.trechm.2021.09.004
Shaik, S., Kumar, D., de Visser, S. P., Altun, A., and Thiel, W. (2005). Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 105, 2279–2328. doi:10.1021/cr030722j
Shteinman, A. A. (2020). Bioinspired Oxidation of Methane: From Academic Models of Methane Monooxygenases to Direct Conversion of Methane to Methanol. Kinet. Catal. 61, 339–359. doi:10.1134/s0023158420030180
Solomon, E. I., Brunold, T. C., Davis, M. I., Kemsley, J. N., Lee, S.-K., Lehnert, N., et al. (2000). Geometric and Electronic Structure/Function Correlations in Non-heme Iron Enzymes. Chem. Rev. 100, 235–350. doi:10.1021/cr9900275
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 98, 11623–11627. doi:10.1021/j100096a001
Stuyver, T., Ramanan, R., Mallick, D., and Shaik, S. (2020). Oriented (Local) Electric Fields Drive the Millionfold Enhancement of the H-Abstraction Catalysis Observed for Synthetic Metalloenzyme Analogues. Angew. Chem. Int. Ed. 59, 7915–7920. doi:10.1002/anie.201916592
Visser, S., Porro, C., Quesne, M., Sainna, M., and Munro, A. (2013). Overview on Theoretical Studies Discriminating the Two-Oxidant versus Two-State-Reactivity Models for Substrate Monoxygenation by Cytochrome P450 Enzymes. Curr. Top. Med. Chem. 13, 2218–2232. doi:10.2174/15680266113136660155
Wang, C., Danovich, D., Chen, H., and Shaik, S. (2019). Oriented External Electric Fields: Tweezers and Catalysts for Reactivity in Halogen-Bond Complexes. J. Am. Chem. Soc. 141, 7122–7136. doi:10.1021/jacs.9b02174
Wang, X. Q., Wang, H. Y., Zheng, L. F., Zhu, C., and Liang, J.-X. (2021). Oriented External Electric Fields Regulating the Oxidation Reaction of CH4 Catalyzed by Mn-Corrolazine. Int. J. Quantum Chem. 121, e26443. doi:10.1002/qua.26443
Yan, Y., Chen, C. L., Zou, S. H., Liu, J. J., and XiaoFan, L. P. J. (2020). High H2O2 Utilization Promotes Selective Oxidation of Methane to Methanol at Low Temperature. Front. Chem. 8, 252. doi:10.3389/fchem.2020.00252
Yang, L. L., Chen, X., Qu, Z. X., and Gao, J. L. (2018). Combined Multistate and Kohn-Sham Density Functional Theory Studies of the Elusive Mechanism of N-Dealkylation of N,N-Dimethylanilines Mediated by the Biomimetic Nonheme Oxidant FeIV(O)(N4Py)(ClO4)2. Front. Chem. 6, 406. doi:10.3389/fchem.2018.00406
Yu, L. J., and Coote, M. L. (2019). Electrostatic Switching between SN1 and SN2 Pathways. J. Phys. Chem. A 123, 582–589. doi:10.1021/acs.jpca.8b11579
Yu, S., Vermeeren, P., Hamlin, T. A., and Bickelhaupt, F. M. (2021). How Oriented External Electric Fields Modulate Reactivity. Chem. Eur. J. 27, 5683–5693. doi:10.1002/chem.202004906
Zaragoza, J. P. T., Siegler, M. A., and Goldberg, D. P. (2016). Rhenium(v)-oxo Corrolazines: Isolating Redox-Active Ligand Reactivity. Chem. Commun. 52, 167–170. doi:10.1039/C5CC07956J
Zaragoza, J. P. T., Yosca, T. H., Siegler, M. A., Moenne-Loccoz, P., and GreenGoldberg, M. T. D. P. (2017). Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc. 139, 13640–13643. doi:10.1021/jacs.7b07979
Zhang, L., Yang, X. H., Li, S., and Zhang, J. M. (2021). Functionalized Silicon Electrodes toward Electrostatic Catalysis. Front. Chem. 9, 715647. doi:10.3389/fchem.2021.715647
Zhang, X. Q., Li, X.-X., Liu, Y. F., and Wang, Y. (2017). Suicide Inhibition of Cytochrome P450 Enzymes by Cyclopropylamines via a Ring-Opening Mechanism: Proton-Coupled Electron Transfer Makes a Difference. Front. Chem. 5, 3. doi:10.3389/fchem.2017.00003
Zhu, C., Liang, J.-X., and Cao, Z. X. (2018). Mn-O-O Electron Spin Flip Mechanism Triggered by the Visible-Light Irradiation for the Generation of an Active Mn(V)-Oxo Complex from O2: Insight from Density Functional Calculations. J. Phys. Chem. C 122, 20781–20786. doi:10.1021/acs.jpcc.8b05531
Keywords: density functional calculations, Fe(IV)-Oxo-Corrolazine, CH4 oxidation, oriented external electric fields, catalysis
Citation: Wu J, Long T, Wang H, Liang J-X and Zhu C (2022) Oriented External Electric Fields Regurating the Reaction Mechanism of CH4 Oxidation Catalyzed by Fe(IV)-Oxo-Corrolazine: Insight from Density Functional Calculations. Front. Chem. 10:896944. doi: 10.3389/fchem.2022.896944
Received: 16 March 2022; Accepted: 08 June 2022;
Published: 29 June 2022.
Edited by:
Miquel Solà, University of Girona, SpainReviewed by:
Sam P. De Visser, The University of Manchester, United KingdomMiquel Torrent Sucarrat, University of the Basque Country, Spain
Thijs Stuyver, Massachusetts Institute of Technology, United States
Copyright © 2022 Wu, Long, Wang, Liang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-Xia Liang, bGlhbmdqeDIwMDlAMTYzLmNvbQ==; Chun Zhu, Y3podTIwMTRAMTYzLmNvbQ==