 Chaoyue Chen
Chaoyue Chen Xin Wang
Xin Wang Tinghai Yang
Tinghai Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Chem. , 13 April 2022
Sec. Organic Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fchem.2022.883474
This article is part of the Research Topic New Hypervalent Iodine Reagents for Oxidative Coupling - Volume II View all 8 articles
With the renaissance of chemical electrosynthesis in the last decade, the electrochemistry of hypervalent iodine compounds has picked up the pace and achieved significant improvements. By employing traceless electrons instead of stoichiometric oxidants as the alternative clean “reagents”, many hypervalent iodine compounds were efficiently electro-synthesized via anodic oxidation methods and utilized as powerful redox mediators triggering valuable oxidative coupling reactions in a more sustainable way. This minireview gives an up-to-date overview of the recent advances during the past 3 years, encompassing enhanced electrosynthesis technologies, novel synthetic applications, and ideas for improving reaction sustainability.
Hypervalent iodine reagents (HIRs) have recently attracted considerable attention in organic synthesis because of their valuable oxidizing abilities, superior leaving group abilities, unique electrophilic properties, and environment-friendly features (Yoshimura and Zhdankin, 2016; Wang and Studer, 2017). To date, their synthetic applications have expanded from ligand transfer reactions, oxidative coupling processes, halogenations, oxidative rearrangements, heterocycle synthesis, and numerous other reactions (Chen et al., 2020; Takenaga et al., 2020). Furthermore, the applications of chiral HIRs and asymmetric precursors have resulted in many valuable contributions to modern asymmetric synthesis (Yang et al., 2021).
HIRs are often considered as environmentally benign alternatives to various chemical oxidants, especially the highly toxic heavy-metal oxidizers, i.e., Pb(IV), Hg(II), and Tl(III) reagents (Yoshimura and Zhdankin, 2016; Takenaga et al., 2020). Nevertheless, the current common preparation methods of HIRs pose environmental concerns to their widespread application and upscaling (Liu et al., 2018; Elsherbini and Wirth, 2018; Frey et al., 2022). Typically, these preparation methods often require (over)stoichiometric amounts of hazardous or costly oxidants, such as m-chloroperbenzoic acid (m-CPBA), CH3CO3H, NaBO3, NaIO4, oxone (2KHSO5·KHSO4·K2SO4), H2O2, and Selectfluor, thus inevitably leading to an excess of waste products (Francke et al., 2018; Francke, 2019; Elsherbini and Wirth, 2018). Hence, more straightforward and sustainable methods for HIRs preparation and derivatization are highly desired.
Nowadays, organic electrosynthesis has witnessed a considerable renaissance for the excellent redox efficacy and tunable cell conditions (Francke and Little, 2014; Yan et al., 2017; Moeller, 2018). Because this methodology utilizes electrons instead of stoichiometric oxidants/reductants as alternative clean redox reagents and can produce important intermediates or reagents in situ, so, organic electrosynthesis is recognized as a reliable and sustainable alternative to conventional approaches (Pollok and Waldvogel, 2020; Novaes et al., 2021).
With the renaissance of organic electrosynthesis, many synthetic chemists have switched to the electrochemical preparation of HIRs via the anodic oxidation of iodine (I) precursors, and attempted to use in situ electrogenerated HIRs as important mediators for various iodine (III)-mediated transformations (Francke et al., 2018; Elsherbini and Wirth, 2018; Francke, 2019; Francke, 2021). And this research front has undergone a significant expansion and currently become an increasingly active research area of hypervalent iodine chemistry. Given the importance and relevance envisaged in future transformations, herein, we intend to summarize recent applications of electrogenerated HIRs and their applications as redox mediators in various chemical transformations. Due to the limited space, this minireview will only highlight the advances over the past 3 years and serves as an update to the earlier reviews conducted by Francke et al. (Francke et al., 2018; Francke, 2019) and Elsherbini and Wirth (2018), respectively.
The first example of the electrochemical synthesis of hypervalent iodine derivatives can be traced back to 1960 by the work of Schmidt and Meinert (1960). In a brief report, they described a procedure for electrochemical generation of well-known (difluoroiodo)benzene (PhIF2) by anodic oxidation of iodobenzene in the presence of silver fluoride. Shortly after that, the first electrochemical preparation of diaryliodonium salts was disclosed by Miller and Hoffmann in 1967 (Miller and Hoffmann, 1967). (Dialkoxyiodo)arenes, another traditional iodine (III) reagents, were first electro-synthesized by Nishiyama and Amano in 2006 (Amano and Nishiyama, 2006). Apart from these pioneering researches, only a few scattered reports on electro-generation of aryl iodide(III) species are available.
During the past decade, with the development of equipment and technologies in electrochemistry, the efforts on electrochemical synthesis of hypervalent iodine reagents have picked up the pace and achieved significant improvements (Scheme 1).
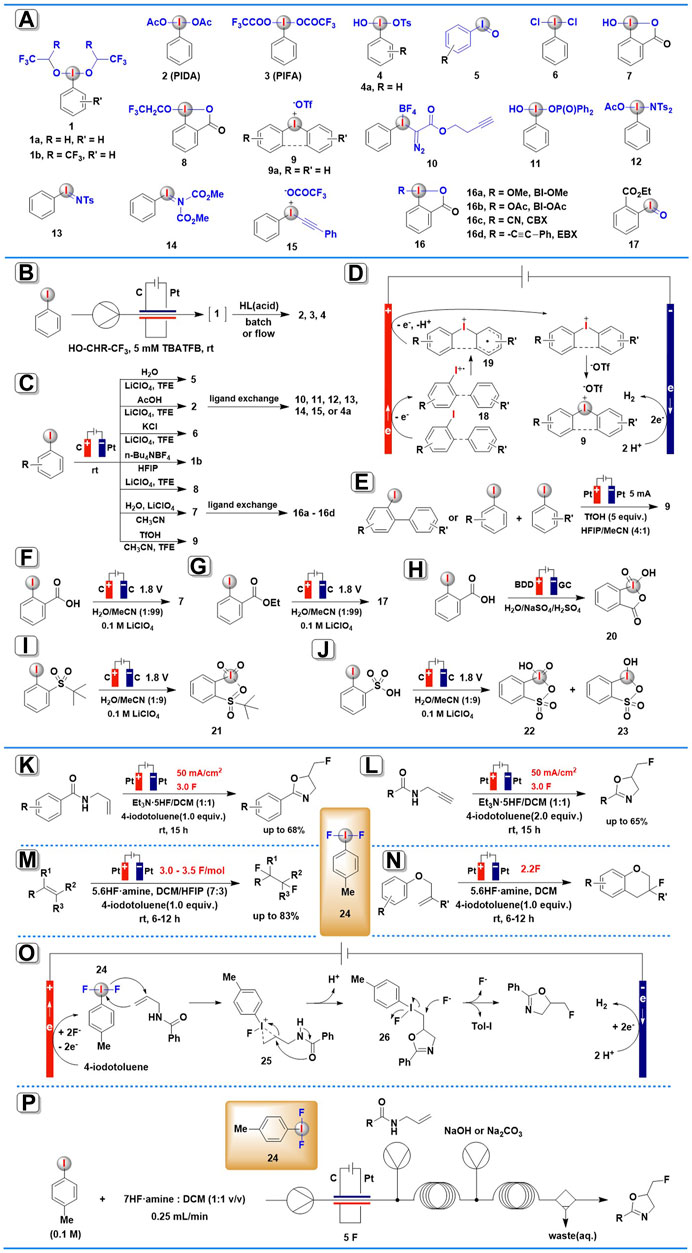
SCHEME 1. Electrochemical synthesis of HIRs and organic transformations mediated by anodically generated (difluoroiodo)arene. (A) Electrogenerated hypervalent iodine derivatives. (B) The continuous-flow electrochemical generation of HIRs. (C) Anodic oxidation for the synthesis of aryliodine(III) reagents. (D) The possible mechanism for electrochemical synthesis of diaryliodonium salts. (E) Scalable electrochemical synthesis of diaryliodonium salts. (F,G) Electrochemical synthesis of HIRs using water as the O-atom source. (H–J) Electrochemical synthesis of organic iodine (V) compounds. (K–N) Organic transformations mediated by anodically generated (difluoroiodo)arene. (O) The possible mechanism for electrochemical fluorocyclization of N-allylcarboxamides mediated by ArIF2. (P) Fluorinations using flow electrolysis.
In 2019, the first generic method for synthesizing bench-stable classic HIRs via anodic oxidation-ligand exchange in an electrochemical flow reactor was described by Wirth and co-workers (Elsherbini et al., 2019). By using a glassy carbon anode and a platinum cathode, iodoarene precursors were anodically oxidized to the corresponding active (dialkoxyiodo)arenes 1 (Scheme 1A) either in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) or in 2,2,2-trifluoroethanol (TFE) under continuous-flow conditions (Scheme 1B). Nevertheless, the electrogenerated (dialkoxyiodo)arenes 1 are only stable in solution, and they disintegrate as soon as the solvent is removed, so, in situ electrochemically generated 1 commonly needs to be immediately used in the next transformation. When unstable 1a or 1b been further treated with the appropriate acids, many useful stable HIRs, such as (diacetoxyiodo)benzene (PIDA, 2), [(bistrifluoroacetoxy)iodo]benzene (PIFA, 3), and Koser’s reagent and its derivatives 4 were achieved via facile ligand exchange reactions. Furthermore, this protocol allowed for the simple synthesis of electron-deficient HIRs that were previously difficult to prepare using conventional methods. In addition, the productivity of this procedure (electrochemical oxidation-ligand exchange) is higher than the similar transformations under batch electrolysis conditions.
Very recently (2021), He and others (Zu et al., 2021) have presented a comprehensive investigation on the usage of anodic oxidation of aryl iodides in the synthesis of diverse hypervalent iodine compounds (Scheme 1C). They provided a sustainable and broadly applicable approach for accessing four types of aryl iodide(III) reagents: iodosylarenes, (difunctionaliodo)arenes, benziodoxoles, and diaryliodonium salts, among others. After a careful examination of the anodic oxidation conditions, electrochemical synthesis of iodosylarenes 5 was carried out in an undivided cell equipped with a graphite anode and a Pt plate cathode and using LiClO4 as the electrolyte and TFE as the solvent under a constant current of 15 mA. Three traditional (difunctionaliodo)arenes, PIDA 2, IBD (PhICl2, 6), and PhI[OCH(CF3)2]2 1b, could also be produced in good to outstanding yields with great selectivity by simply substituting H2O with AcOH, KCl, or replacing the solvent to HFIP in the absence of H2O. 2-Iodobenzoic acids precursors could be efficiently converted into benziodoxoles 7 and 8 in high yields under slightly modified electrochemical conditions. Furthermore, a variety of diaryliodonium salts 9 were obtained in satisfying yields employing TfOH as both the electrolyte and counteranion, and CH3CN and TFE as the mixed solvent. When subjected to additional ligand-exchange, the electrogenerated PIDA 2 or hydroxy benziodoxolone 7 could readily convert into many useful HIRs, including Koser’s reagent 4, Weiss’ reagent 10, [hydroxy(phosphoryloxy)iodo]benzene 11, amidoiodane 12, iodonium imide 13, alkynyliodonium salt 14, aryl iodonium ylide 15, and benziodoxole reagents 16 (BI-OAc, BI-OMe, CBX, and EBX).
The authors suggested two mechanisms to explain the electro-generation of HIRs, one for three types of iodosylarenes, (difunctionaliodo)arenes, and benziodoxoles, and the other for diaryliodonium salts. In the first case, the aryl iodide(III) reagents are proposed to proceed via two-electron anodic oxidation of iodoarene in the presence of a suitable ligand. The cathodic half-reaction is the reduction of protons, which produces H2 as a by-product. In the latter case (Scheme 1D), one electron anodic oxidation of iodoarene(I) affords radical cation intermediate 18, which is then converted into intermediate 19 by radical addition with arene species. Subsequently, the resulting intermediate 19 performs a second one-electron anodic oxidation and further combines with –OTf ion to yield the desired diaryliodonium salts 9.
Almost at the same time, Moran and co-workers also established an electrochemical approach to synthesis cyclic and acyclic diaryliodonium salts 9 (Elsherbini and Moran, 2021). In a simple undivided electrolysis cell in MeCN-HFIP-TfOH without any added electrolyte salts, more than 30 diaryliodonium salts 9 were achieved in high to excellent yields via anodic oxidation of iodobiaryls and iodoarene/arene (Scheme 1E). It was worth noting that this protocol has high scalability, in less than 3 h, the reaction was scaled up to a 10 mmol scale, yielding more than 4 g of 9a (>95%). The large-scale experiment’s solvent mixture was easily recovered (>97%) and recycled several times without a notable drop in yield.
Also in 2021, Folkman and others have demonstrated a novel electrochemical synthetic route to HIRs using water as an abundant, green chemistry O-atom source (Folkman et al., 2021). The bulk electrolysis was conducted in a U-cell equipped with a glassy carbon electrode at 1.8 V vs. Ag/AgNO3, using 0.1 M LiClO4 as the supporting electrolyte and 1% H2O in MeCN as the O-atom source (Schemes 1F,G). IBA (7) and its derivative (IBA-ester, 17) were obtained with Faradaic efficiency of 89 and 78%, respectively. According to isotopic (18O) labeling studies using H218O, the authors suggest that the O-atom in the hypervalent product is initially coming from the oxidized, O-atom-containing surface of the glassy carbon electrode, and then the surface oxide of the glassy carbon is regenerated by the O-atoms from water solution. These results provide evidence that H2O serves as the oxygen source for the newly formed functional groups in hypervalent iodine products in this electrosynthesis process.
Whereas several methods for electrochemical synthesis of iodine (III) compounds have been developed during the past few decades, the methods for anodic electrosynthesis of iodine (V) species were less explored and have just recently been established.
In 2018 Bystron’s group reported the first electrochemical synthesis of organic iodine (V) compound, 2-iodoxybenzoic acid (IBX, 20), through anodic oxidation of 2-iodobenzoic acid (IBA) in a divided cell using a boron-doped diamond (BDD) electrode in 0.2 M H2SO4 aqueous solution (Bystron et al., 2018; Devadas et al., 2020) (Scheme 1H). This anodic oxidation process exhibited excellent selectivity and high current efficiency (>50%). No undesired organic products were generated with O2 as the only by-product. The aqueous environment was recognized as a crucial aspect of this anodic oxidation process because iodine (V) compounds could be electrochemically generated only in aqueous electrolyte solutions (Devadas et al., 2020).
Apart from iodine (III) compounds, the aforementioned method by Folkman and others (Folkman et al., 2021) also enables the electrochemical synthesis of iodine (V) species (Schemes 1I,J). In the same paper, the widely used IBX-sulfone 21 and a mixture of IBA-sulfonic acid 22 with IBX-sulfonic acid 23 were obtained via bulk anodic oxidation electrolysis of PhI-sulfone and PhI-sulfonic acid using water as the O-atom source. However, attempts to isolate these iodine (V) derivatives were unsuccessful due to decomposition under experimental conditions.
In many organic electrosynthesis practices, HIRs which in situ generated at the anode, often immediately used as in-cell or ex-cell or in-flow mediators in subsequent redox reactions, thus efficiently triggering many valuable organic transformations, including fluorination, oxidative cyclization, trifluoroethoxy-lactonization, aziridination, and enantioselective cyclizations, etc. (Schemes 1K–P, 2). Since these reactions are not suitable to be classified according to the type of reaction or the type of chemical bond formed, they are classified and described herein according to the iodine (III) redox mediators employed.
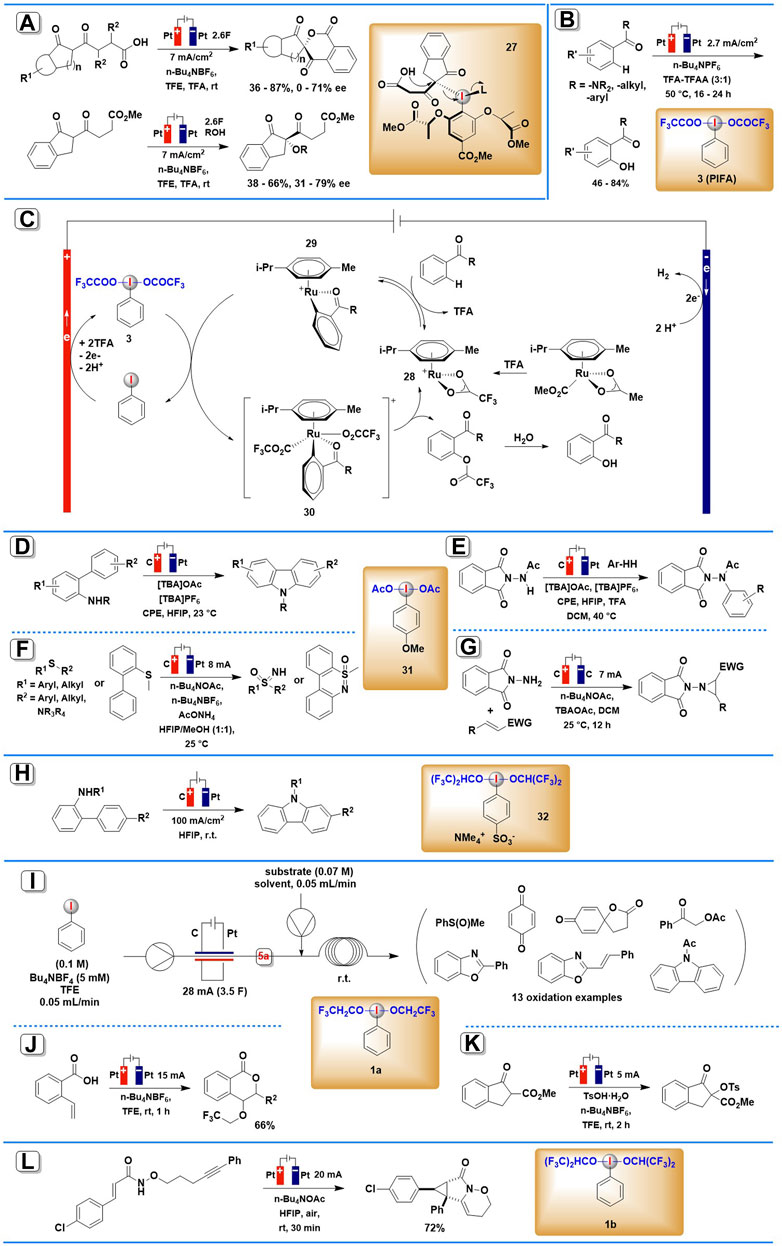
SCHEME 2. Organic transformations mediated by anodically generated (diacyloxyiodo)arenes or (dialkoxyiodo)arenes. (A) The asymmetric spirolactonization mediated by anodically generated chiral iodine (III) intermediate. (B) The reaction and the possible mechanism (C) of iodine(III)/Ruthenium complexes electrocatalyzed C–H oxygenation. (D,E) The electrocatalytic C-N coupling mediated by anodically generated ArI(RCOO)2. (F) The electrochemical synthesis of NH-sulfoximines and NH-sulfonimidamides mediated by anodically generated ArI(RCOO)2. (G) The electrochemical aziridination mediated by anodically generated ArI(RCOO)2. (H) The intramolecular oxidative C-N bond formation mediated by anodically generated (dialkoxyiodo)arene. (I–K)Organic transformations mediated by anodically generated PhI(OCH2CF3)2. (L) The electrochemical synthesis of cyclopropane mediated by anodically generated (dialkoxyiodo)arene.
A novel electrochemical fluorocyclization of N-allylcarboxamides for the construction of 5-fluoromethyl-2-oxazolines was developed in 2019 by Waldvogel and co-workers (Haupt et al., 2019). The reaction was carried out under constant current in an undivided cell charged with two Pt electrodes and anodically generated ArIF2 24 was employed as the redox mediator in the presence of Et3N·5HF (Scheme 1K). It was noteworthy that the iodoarene precursors could be partly or even quantitatively isolated after electrolysis.
The proposed mechanism (Scheme 1O) showed that 4-iodotoluene is oxidated at the anode to generate the activated ArIF2 24, which is then transformed to a cyclic iodonium species 25 by addition with an allyl moiety. The intermediate formed undergoes intramolecular ring-opening leading to 5-(λ3-iodanyl)methyl oxazoline 26, which then suffers an SN2 nucleophilic attack from fluorine, resulting in the formation of the halogenated product along with the regeneration of ArI.
Encouraged by these findings, the same group continued to explore more organic transformations mediated by electrochemically generated ArIF2 24, a similar electrochemical protocol (Scheme 1L) has been successful in the conversion of a wide range of N-propargylamides into their corresponding 5-fluoromethyl-2-oxazoles (Herszman et al., 2019). It was noted by the authors that the stoichiometry of iodobenzene ArI proved to be a crucial factor in determining product selectivity. In addition, this electrochemical approach allows the fluorocyclization of electron-rich N-propargylamides that are difficult or impossible in conventional reagent-based methods. The proposed mechanism showed that the fluorocyclization proceeds through ArIF2 24, which is generated by direct electrochemical oxidation of iodoarene at the anode.
In 2020, Lennox and co-workers report a method for electrochemical vicinal 1,2-difluorination of alkenes using Tol-IF2 24 as a mediator (Doobary et al., 2020). In this case, fluorinated products yielded a range from moderate to outstanding across a wide range of alkene substrates (Scheme 1M). Superior to conventional methods, this ex-cell approach permits the difluorination of electron-rich moieties, anilines, and substituted internal alkenes. The authors noted that the electrochemical synthesis of a hypervalent iodine mediator via an ex-cell approach, which avoids oxidative substrate degradation, is the key factor for success.
Based on this result, Lennox’s group continued to utilize this electrochemical approach for intramolecular fluoroarylation of phenolic ethers mediated by electrochemically generated ArIF2 24 (Doobary et al., 2021) (Scheme 1N). The use of an ex-cell procedure has proven to be critical to the success of this process. Notably, this electrochemical approach exhibited broad functional group compatibility, even electron-withdrawing groups on the aromatic ring and gem substituents on the alkene were well tolerated in the reaction system. Furthermore, this reaction could be easily scaled up with good efficiency, which is important for future applications.
Unlike the batch approaches outlined above, Wirth and co-workers described a continuous flow method for electrochemical generation of ArIF2 24 and its synthetic applications in 2021 (Winterson et al., 2021). Several fluorination transformations, including fluorocyclizations, difluorinations, monofluorinations, iodofluorinations, and ring contractions, are conveniently implemented in continuous flow (Scheme 1P). Because (difluoroiodo)arene mediators are not bench-stable and degrade as soon as the solvents are removed, their continuous generation and immediate application in flow are particularly useful. Under high flow rates, the continuous-flow procedure achieved higher yields and up to 834 mg h−1 productivities with drastically decreased reaction times.
The first application of electrogenerated chiral hypervalent iodine compound in enantioselective electrosynthesis (Scheme 2A) was developed by Wirth and co-workers in 2019 (Gao et al., 2019). The asymmetric spirolactonization process utilized an undivided cell under constant current reaction conditions (J = 7 mA/cm2) with platinum electrodes and the addition of 1.2 equiv. of mediator and Bu4NBF4 electrolyte. Fluorinated alcohol TFE was chosen as the solvent for stabilizing iodine (III) reagents generated by the anodic oxidation of iodoarenes. Under these conditions, a range of diketo carboxylic acids were smoothly converted into chiral six-membered lactones in 36–87% yield and 0–71% ee mediated by in situ generated chiral iodine (III) intermediate 27. Intermolecular α-alkoxylation of diketo ester derivatives can also be achieved using the same protocol, and the corresponding coupling products were obtained in 38–66% yield and 31–79% ee. Furthermore, this batch-type process could be adapted to an electrochemical flow microreactor to reduce or minimize the use of supporting electrolytes.
In 2020, Ackermann and co-workers demonstrated how iodoarene mediators can be integrated into metallic-electrocatalytic processes (Massignan et al., 2020). By a combination of the catalytic electro-regeneration of iodine (III) intermediate 3 with ruthenium-(II) catalyzed C-H activation, a wide array of aromatic amides and ketones were selectively converted to their corresponding oxygenated products (Scheme 2B). The reaction was carried out in an undivided cell equipped with Pt plate electrodes under a relatively low current density (2.7 mA/cm2), using a mixture consisting of TFA, TFAA, and n-Bu4PF6 served as electrolyte. Chemical oxidants such as m-CPBA or ozone, which are commonly employed to synthesize the HIRs, failed to give the desired products in adequate yields. Mechanistic studies by experiment, computation, cyclic voltammetry, and in operando NMR spectroscopy showed that an in situ formed HIR served the dual role of electron-shuttle and a transfer reagent of carboxylate anions. The proposed mechanism is shown in Scheme 2C. The catalytic cycle starts with ruthenium-(II)-catalyzed C-H activation of 28, and then the resulting complex 29 suffer indirect oxidation by anodically generated PIFA, leading to the formation of ruthenium(IV) intermediate 30. The ruthenium(IV) intermediate undergoes reductive elimination, releasing the product and closing the catalytic cycle.
Also in 2020, Powers and others described electrochemical C–N coupling methodology to achieve N-acetylcarbazole mediated by anodically generated iodine (III) intermediate 31 (Maity et al., 2020). An undivided cell was equipped with a glassy carbon electrode at the anode and a platinum electrode at the cathode using HFIP as solvent at room temperature (Schemes 2D,E). This electrosynthesis process can apply to both intra- and inter-molecular C–H/N–H coupling. The crucial significance of acetate ions in stabilizing initially produced iodanyl radical intermediates has been demonstrated in mechanistic investigations.
An efficient synthesis of NH-sulfoximines, NH-sulfonimidamides, and dibenzothiazines via anodically generated iodine (III) intermediate 31 was reported by Wang, Xu, and co-workers in 2021 (Kong et al., 2021). The reaction was conducted in an undivided cell equipped with graphite (C) anode and Pt plate cathode, at a constant current of 8 mA using n-Bu4NPF6 as the electrolyte (Scheme 2F). It is noteworthy that this in-cell method employs catalytic amounts of iodoarene and simple ammonia sources, thus avoiding the use of an excess of HIRs.
In 2021, Dai, Xu, and co-workers reported a protocol for electrochemical aziridination of electron-deficient alkenes mediated by electro-generated iodine (III) species 31 (Liu et al., 2021). By using graphene sheet as an anode, graphite felt as cathode, and n-Bu4NBF4 as supporting electrolyte, a number of aziridines were smoothly achieved in DCM under a current of 7 mA (Scheme 2G). A broad substrate scope was achieved in this protocol, including various cinnamate, cinnamide, α,β-unsaturated nitrile, and ketones.
In 2019, Francke’s group developed an electrochemical protocol to achieve intramolecular oxidative C–N bond formation (Roesel et al., 2019). In this protocol, a novel ionically tagged iodophenylsulfonate was employed as both mediator and supporting electrolyte, thus allowing electrolysis without supporting electrolyte additives and facilitating the recovery from the reaction mixture (Scheme 2H). Anodic oxidation of 4-iodobenzenesulfonate led to the formation of redox-active iodine (III) species 32, which could trigger the oxidative cyclization of 2-acetamidobiphenyls.
Wirth and co-workers demonstrated that the electrochemical generator of iodine (III) 1a exhibits excellent efficiency and reliability in organic electrosynthesis (Elsherbini et al., 2019). By coupling the flow of the in situ electrochemically generated PhI(OCH2CF3)2 with the second flow of thioanisoles, hydroquinone, 3-(4-hydroxyphenyl)propanoic acid, ortho-hydroxyimines, acetophenones, propiophenones, or acetamide in TFE solvent, a series of oxidative transformations including mono-oxidation, dearomatization, cyclization, α-acetoxylation, tosyloxylation, etc. were successfully achieved using simple reaction setup (Scheme 2I). The yields of the end products are normally high, and they can be easily enhanced by adjusting the second reaction’s residence time and temperature. Furthermore, all these reactions can be scaled up to 0.4–1.0 mmol.
Based on their method for the electrochemical synthesis of aryl iodide(III) reagents mentioned above, He and his co-workers further developed two oxidative transformations, trifluoro-ethoxylactonization, and tosyloxylation, enabled by anodically generated iodine (III) intermediate 1a (Zu et al., 2021). The two reactions were both conducted in an undivided cell equipped with platinum electrodes under a constant current of 5 mA, using n-Bu4NBF4 as the electrolyte and TFE as the solvent (Schemes 2J,K). The corresponding products were achieved in moderate yields.
In 2022, Wright, Wen, Zhang, and co-workers reported one example of cation-induced [2 + 1] cyclization of alkenyl amide mediated by electrochemically generated iodine (III) intermediate (Yuan et al., 2022). The iodine (III) species 1b, which in situ generated from iodobenzene in the presentence of n-Bu4NOAc in HFIP, smoothly promoted the annulation process and constructed highly strained cyclopropane-fused lactam in 72% yield (Scheme 2L).
As shown herein, the growing interest in hypervalent iodine chemistry together with the “renaissance” of organic electrosynthesis opened new possibilities for developing synthetically attractive transformations in a more sustainable way. By means of anodic oxidation of iodine (I) precursors, HIRs can be electrochemically generated using electric current as an inexpensive and traceless oxidant under environmentally benign conditions. The in situ generated HIRs can utilize as powerful batch or flow mediators and trigger valuable oxidative coupling reactions, including fluorocyclizations, trifluoro-ethoxylactonisation, aziridination, asymmetric spirolactonization, etc.
Despite considerable progress and achievements, there are major challenges still yet to be overcome. Firstly, the number and types of electrochemically accessible HIRs, especially iodine (V) species, still need to be expanded. Moreover, the development of low-cost, non-toxic, and more sustainable electrolytes as a replacement of commonly used NEt3·nHF electrolytes or fluorinated alcohols is highly desirable. Finally, the exploration of more challenging electro-synthetic applications, especially asymmetric electrocatalysis, mediated by electro-generated iodine (III) or iodine (V) is also in urgent demand. Although electrochemistry of hypervalent iodine compounds is still in its infancy, the authors believe that this research front may enjoy imminent prosperity in the near future.
TY designed this proposal and determined the contents. CC collected the literature data related and wrote the minireview. XW prepared the schemes. All authors contributed to the final version of the manuscript.
This work was partially supported by the Industry-University-Research Cooperation Project of Jiangsu Province, China (No. BY2021335) and the Changzhou Sci and Tech Program (CZ20210029).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Amano, Y., and Nishiyama, S. (2006). Oxidative Synthesis of Azacyclic Derivatives through the Nitrenium Ion: Application of a Hypervalent Iodine Species Electrochemically Generated from Iodobenzene. Tetrahedron Lett. 47, 6505–6507. doi:10.1016/j.tetlet.2006.07.050
Bystron, T., Horbenko, A., Syslova, K., Hii, K. K. M., Hellgardt, K., and Kelsall, G. (2018). 2-Iodoxybenzoic Acid Synthesis by Oxidation of 2-iodobenzoic Acid at a boron-doped diamond Anode. ChemElectroChem 5, 1002–1005. doi:10.1002/celc.201800027
Chen, C., Wang, X., and Yang, T. (2020). Recent Synthetic Applications of the Hypervalent Iodine(III) Reagents in Visible-Light-Induced Photoredox Catalysis. Front. Chem. 8, 551159. doi:10.3389/fchem.2020.551159
Devadas, B., Svoboda, J., Krupička, M., and Bystron, T. (2020). Electrochemical and Spectroscopic Study of 2-iodobenzoic Acid and 2-iodosobenzoic Acid Anodic Oxidation in Aqueous Environment. Electrochimica Acta 342, 136080. doi:10.1016/j.electacta.2020.136080
Doobary, S., Poole, D. L., and Lennox, A. J. J. (2021). Intramolecular Alkene Fluoroarylation of Phenolic Ethers Enabled by Electrochemically Generated Iodane. J. Org. Chem. 86, 16095–16103. doi:10.1021/acs.joc.1c01946
Doobary, S., Sedikides, A. T., Caldora, H. P., Poole, D. L., and Lennox, A. J. J. (2020). Electrochemical Vicinal Difluorination of Alkenes: Scalable and Amenable to Electron‐Rich Substrates. Angew. Chem. Intl Edit 59, 1155–1160. doi:10.1002/anie.201912119
Elsherbini, M., and Moran, W. J. (2021). Scalable Electrochemical Synthesis of Diaryliodonium Salts. Org. Biomol. Chem. 19, 4706–4711. doi:10.1039/D1OB00457C
Elsherbini, M., Winterson, B., Alharbi, H., Folgueiras‐Amador, A. A., Génot, C., and Wirth, T. (2019). Continuous‐Flow Electrochemical Generator of Hypervalent Iodine Reagents: Synthetic Applications. Angew. Chem. Int. Ed. 58, 9811–9815. doi:10.1002/anie.201904379
Elsherbini, M., and Wirth, T. (2018). Hypervalent Iodine Reagents by Anodic Oxidation: a Powerful green Synthesis. Chem. Eur. J. 24, 13399–13407. doi:10.1002/chem.201801232
Folkman, S. J., Finke, R. G., Galán-Mascarós, J. R., and Miyake, G. M. (2021). Carbon-electrode-mediated Electrochemical Synthesis of Hypervalent Iodine Reagents Using Water as the O-Atom Source. ACS Sust. Chem. Eng. 9, 10453–10467. doi:10.1021/acssuschemeng.1c01315
Francke, R., Broese, T., and Roesel, A. F. (2018). “Electrochemistry of Hypervalent Iodine Compounds,” in Hypervalent Halogen Compounds. Editor I. Marek (John Wiley & Sons), 1–22. doi:10.1002/9780470682531.pat0960
Francke, R. (2019). Electrogenerated Hypervalent Iodine Compounds as Mediators in Organic Synthesis. Curr. Opin. Electrochemistry 15, 83–88. doi:10.1016/j.coelec.2019.03.012
Francke, R., and Little, R. D. (2014). Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev. 43, 2492–2521. doi:10.1039/C3CS60464K
Francke, R. (2021). Recent Progress in the Electrochemistry of Hypervalent Iodine Compounds. Curr. Opin. Electrochemistry 28, 100719. doi:10.1016/j.coelec.2021.100719
Frey, B., Maity, A., Tan, H., Roychowdhury, P., and Powers, D. C. (2022). “Sustainable Methods in Hypervalent Iodine Chemistry,” in Iodine Catalysis in Organic Synthesis. Editors K. Ishihara, and K. Muñiz (Wiley-VCH), 335–386. doi:10.1002/9783527829569.ch12
Gao, W.-C., Xiong, Z.-Y., Pirhaghani, S., and Wirth, T. (2019). Enantioselective Electrochemical Lactonization Using Chiral Iodoarenes as Mediators. Synthesis 51, 276–284. doi:10.1055/s-0037-1610373
Haupt, J. D., Berger, M., and Waldvogel, S. R. (2019). Electrochemical Fluorocyclization of N-Allylcarboxamides to 2-oxazolines by Hypervalent Iodine Mediator. Org. Lett. 21, 242–245. doi:10.1021/acs.orglett.8b03682
Herszman, J. D., Berger, M., and Waldvogel, S. R. (2019). Fluorocyclization of N-Propargylamides to Oxazoles by Electrochemically Generated ArIF2. Org. Lett. 21, 7893–7896. doi:10.1021/acs.orglett.9b02884
Kong, X., Lin, L., Chen, X., Chen, Y., Wang, W., and Xu, B. (2021). Electrochemical Oxidative Syntheses of NH-Sulfoximines, NH-Sulfonimidamides and Dibenzothiazines via Anodically Generated Hypervalent Iodine Intermediates. ChemSusChem. 14, 3277–3282. doi:10.1002/cssc.202101002
Liu, K., Song, C., and Lei, A. (2018). Recent Advances in Iodine Mediated Electrochemical Oxidative Cross-Coupling. Org. Biomol. Chem. 16, 2375–2387. doi:10.1039/C8OB00063H
Liu, F., Dai, J., and Cheng, X. (2021). Aryl-iodide-mediated Electrochemical Aziridination of Electron-Deficient Alkenes. Chin. J. Org. Chem. 41, 4014–4020. doi:10.6023/cjoc202105046
Maity, A., Frey, B. L., Hoskinson, N. D., and Powers, D. C. (2020). Electrocatalytic C-N Coupling via Anodically Generated Hypervalent Iodine Intermediates. J. Am. Chem. Soc. 142, 4990–4995. doi:10.1021/jacs.9b13918
Massignan, L., Tan, X., Meyer, T. H., Kuniyil, R., Messinis, A. M., and Ackermann, L. (2020). C−H Oxygenation Reactions Enabled by Dual Catalysis with Electrogenerated Hypervalent Iodine Species and Ruthenium Complexes. Angew. Chem. Int. Ed. 59, 3184–3189. doi:10.1002/anie.201914226
Miller, L. L., and Hoffmann, A. K. (1967). The Electrochemical Formation of Carbonium and Iodonium Ions from Alkyl and Aryl Iodides. J. Am. Chem. Soc. 89, 593–597. doi:10.1021/ja00979a022
Moeller, K. D. (2018). Using Physical Organic Chemistry to Shape the Course of Electrochemical Reactions. Chem. Rev. 118, 4817–4833. doi:10.1021/acs.chemrev.7b00656
Novaes, L. F. T., Liu, J., Shen, Y., Lu, L., Meinhardt, J. M., and Lin, S. (2021). Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 50, 7941–8002. doi:10.1039/D1CS00223F
Pollok, D., and Waldvogel, S. R. (2020). Electro-organic Synthesis - a 21st century Technique. Chem. Sci. 11, 12386–12400. doi:10.1039/D0SC01848A
Roesel, A. F., Broese, T., Májek, M., and Francke, R. (2019). Iodophenylsulfonates and Iodobenzoates as Redox‐Active Supporting Electrolytes for Electrosynthesis. ChemElectroChem 6, 4229–4237. doi:10.1002/celc.201900540
Schmidt, H., and Meinert, H. (1960). Zum Mechanismus der elektrochemischen Fluorierung und über die Bildung von Jod-monofluorid. Angew. Chem. 72, 109–110. doi:10.1002/ange.19600720305
Takenaga, N., Kumar, R., and Dohi, T. (2020). Heteroaryliodonium(III) Salts as Highly Reactive Electrophiles. Front. Chem. 8, 599026. doi:10.3389/fchem.2020.599026
Wang, X., and Studer, A. (2017). Iodine(III) Reagents in Radical Chemistry. Acc. Chem. Res. 50, 1712–1724. doi:10.1021/acs.accounts.7b00148
Winterson, B., Rennigholtz, T., and Wirth, T. (2021). Flow Electrochemistry: a Safe Tool for Fluorine Chemistry. Chem. Sci. 12, 9053–9059. doi:10.1039/D1SC02123K
Yan, M., Kawamata, Y., and Baran, P. S. (2017). Synthetic Organic Electrochemical Methods since 2000: on the Verge of a Renaissance. Chem. Rev. 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Yoshimura, A., and Zhdankin, V. V. (2016). Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Yuan, D.-F., Wang, Z.-C., Geng, R.-S., Ren, G.-Y., Wright, J. S., Ni, S.-F., et al. (2022). Hypervalent Iodine Promoted the Synthesis of Cycloheptatrienes and Cyclopropanes. Chem. Sci. 13, 478–485. doi:10.1039/D1SC05429E
Yang, X.-G., Hu, Z.-N., Jia, M.-C., Du, F.-H., and Zhang, C. (2021). Recent Advances and the prospect of Hypervalent Iodine Chemistry. Synlett 32, 1289–1296. doi:10.1055/a-1492-4943
Keywords: hypervalent iodine reagent, organic electrosynthesis, anodic oxidation, redox mediator, synthetic method
Citation: Chen C, Wang X and Yang T (2022) Recent Updates on Electrogenerated Hypervalent Iodine Derivatives and Their Applications as Mediators in Organic Electrosynthesis. Front. Chem. 10:883474. doi: 10.3389/fchem.2022.883474
Received: 25 February 2022; Accepted: 25 March 2022;
Published: 13 April 2022.
Edited by:
Jian-Wei Han, East China University of Science and Technology, ChinaCopyright © 2022 Chen, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaoyue Chen, bmp1Y2hhb3l1ZWNoZW5AMTYzLmNvbQ==; Tinghai Yang, dGluZ2hhaV95YW5nQGhvdG1haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.