 Ara Cho
Ara Cho Byoung Joon Park
Byoung Joon Park Jeong Woo Han
Jeong Woo Han- Department of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, South Korea
Metal-N-doped carbon is a promising replacement for non-precious-metal catalysts such as Pt for the oxygen reduction reaction (ORR) in polymer electrolyte membrane fuel cells (PEMFCs). Although these materials have relatively good catalytic activity and are cost-effective, they still have lower ORR activity than Pt, and so improving their performances is greatly required. In this study, high-throughput screening was employed based on density functional theory (DFT) calculations to search for good candidate catalysts with a transition metal atom coordinated by heteroatoms (B, N, S, O, and P) embedded in a graphene structure. In addition, coordinating a transition metal with two types of heteroatom dopants in a graphene structure was also considered. We calculated the binding energies of ORR intermediates on metal-heteroatom-based graphene structures because they are known to play a key role in ORR. Based on our results, the new group of electrocatalysts imparts excellent ORR activity for PEMFCs, and we suggest that our approach provides useful insight into exploring other promising candidate catalysts.
Introduction
Developing high-performance oxygen reduction reaction (ORR) catalysts is a key for clean energy technologies such as fuel cells and metal-air batteries. ORR activity can be determined using the adsorption energy of oxygen intermediates as a descriptor. Although Pt invokes a high reaction rate due to its appropriate adsorption strength of oxygen intermediates (Nørskov et al., 2004), its high price and scarcity are impediments to its practical use in industry. Therefore, numerous efforts have been made to develop catalysts composed of cheap and abundant elements to replace Pt and Pt-based catalysts.
Metal single atom materials coordinated to nitrogen (M-N-Cs), especially the Fe-N-C catalyst (Lefèvre et al., 2009; Chen et al., 2019; He et al., 2020; Lu et al., 2020), have attracted research interest as the most promising alternative to Pt. Numerous studies of M-N-Cs have focused on modifying the electronic structure of the active site by controlling the coordination environment or changing the d-block element at the metal center. For example, Xu et al. computationally screened M-N-Cs by tuning the metal species and number of nitrogen atoms to be coordinated with the metal atom (Xu et al., 2018). They found that Fe-pyridine/pyrrole-N4 catalysts lying near the top of a volcano plot showed the highest ORR activity, which indicates that it would be challenging for M-N-C catalysts with only nitrogen ligands to outperform Fe-N-C catalysts. Therefore, a new strategy is required to improve ORR activity beyond the Fe-N-C catalyst.
Recently, a new approach of introducing a p-block element (B, N, O, P, or S) into M-N-Cs that can adjust the adsorption strength of intermediates by changing the electronic and geometric structures has been suggested. For example, Jung et al. found that hydrogen peroxide (H2O2) production rate and selectivity can be regulated by adding electron-rich or electron-poor species into Co-N-C (Jung et al., 2020). Their theoretical and experimental results demonstrated that the adsorption energy of *OOH was increased with the addition of electron-rich species such as oxygen, but decreased with the addition of electron-poor species such as protons, thereby affecting the charge amount of the metal center. Similarly, Mun et al. modulated the adsorption strength of Fe atoms in an Fe-N-C catalyst by controlling the charge density of the carbon plane via the electron withdrawing and donating properties (Mun et al., 2019). In addition, Shang et al. showed that Cu coordinated with N and S (Cu-N/S-C) formed an unsymmetrical structure with a higher ORR activity than either Cu-N-C or Fe-N-C (Shang et al., 2020). Due to their asymmetric structure, additional π-bonds between the Cu atom and the oxygen intermediates were formed, thereby enhancing the adsorption strength. Although the evidence from many studies suggests that M-N-C catalysts in which a single metal atom is coordinated with two other elements can enhance ORR activity, no one has yet examined all possible combinations.
In this work, we computationally screened all possible atomic combinations in MX4 and MYX3 structures, where M is a transition metal atom (3d, 4d, or 5d) and X and Y are heteroatoms (B, N, O, S, or P). First, we obtained structural stability based on the formation energy, and then the stable structures were filtered out and their limiting potentials for ORR were calculated. We found a linear scaling relationship to establish an ORR activity volcano plot from which we were able to derive several potential candidates.
Methods
Spin-polarized density functional theory (DFT) calculations were performed by using the Vienna Ab Initio Simulation Package (VASP) (Kresse and Hafner, 1993; Kresse and Hafner, 1994; Kresse and Furthmüller, 1996a; Kresse and Furthmüller, 1996b) with projector augmented wave (PAW) pseudopotentials (Blöchl, 1994; Kresse and Joubert, 1999). Electron-exchange correlation energy values were treated with the Perdew-Burke-Enrnzerhof (PBE) functional of the generalized gradient approximation (Perdew et al., 1996). In the expansion of the plane wave, the cutoff energy was set as 400 eV. Geometry relaxation was stopped when the difference in the total force was less than 0.03 eV/Å. To avoid an artificial electrostatic field, dipole corrections were used to compute all of the energy values reported here (Neugebauer and Scheffler, 1992). In our calculations, we used semi-empirical dispersion correction of the DFT-D3(BJ) method (Grimme et al., 2011).
The MX4 and MYX3 structures were constructed based on a (5 × 5) supercell graphene with a vacuum layer of 20 Å by using Monkhorst-Pack 3 × 3 × 1 k-point meshes (Monkhorst and Pack, 1976). We adopted a computational hydrogen electrode (CHE) model including proton-coupled electron transfer (Nørskov et al., 2004) for the ORR free energy pathway. Based on this model, the reference electrode potential was a standard hydrogen electrode (SHE) where the free energy of protons can be related to that of 1/2 H2(g) under conditions of pH = 0 and 1 bar of H2 gas at 298 K. The Gibbs free energy of the proton-electron transfer step was given by
where ∆E is the reaction energy obtained from the DFT calculations, ∆ZPE is the change in zero-point energy, T is the temperature (298 K), ∆S is the change in entropy, U is the applied potential to the SHE (when a potential is applied, the free energy is shifted by ∆GU = -eU, where e is the elementary charge of an electron), and ∆Gsol is a solvation correction of 0.3 eV for ∗OH and ∗OOH (Xu et al., 2018).
Results and Discussion
Computational Screening Procedure for Metal-Heteroatom-Doped Carbon
As shown in Figure 1, we only focused on the porphyrin-like active sites where the metal center atom is coordinated with four heteroatoms in a carbon plane with two carbon vacancies due to the high computational cost. In the case of dual heteroatom doping, one of the four heteroatoms was changed, resulting in a structure with a 3:1 ratio. From the results, there were 700 possible combinations (= 28 transition metals × 5 host heteroatoms × 5 dopant heteroatoms). The variation of the d-orbital for transition metals ultimately increases the catalytic performance by controlling the binding strength of electrocatalytic reaction intermediates in the hydrogen evolution reaction (HER), ORR, the oxygen evolution reaction (OER), and the carbon dioxide reduction reaction (CO2RR) (Xu et al., 2018; Zhu et al., 2019; Park et al., 2021). Therefore, we considered 28 d-block transition metal elements, including 3d (from Sc to Zn), 4d (from Y to Cd), and 5d (from Hf to Au). Heteroatoms including B, N, O, P, and S acted as glue atoms for the transition metal in the heteroatom-doped carbon structures by changing the covalent bonding in the coordinating environment when their p-orbital positions were dissimilar. Moreover, local strain in the carbon lattice plane was introduced at different levels because of the different atomic sizes between the host and dopant heteroatoms.

FIGURE 1. A schematic illustration of the possible combinations of metal-heteroatom-doped carbon with 28 transition metals and five heteroatoms when the coordinated heteroatoms are either the same or different at a ratio of 3:1. The gray, orange, blue, and yellow balls represent carbon, metal, host heteroatom, and dopant heteroatom, respectively.
Various forms of metal-heteroatom-doped carbon (M-YX3, M = transition metal, Y = dopant heteroatom, and X = host heteroatom) were considered. For example, the Co-ON3 structure comprises a Co atom fourfold coordinated with one O atom and three N atoms in the presence of two C vacancies within the carbon matrix. In the computational screening process, we first estimated the stability of a metal-heteroatom-doped carbon structure by considering the formation energy and single atom binding energy. Second, the free energy changes for ORR on the stable metal-heteroatom-doped carbon structures were calculated to construct a 2-D volcano plot with ΔGOOH and ΔGOH as descriptors to measure ORR activity.
Structural Stability Analysis of the Metal-Heteroatom-Doped Carbon Structures
First, we examined the stability of 700 M-YX3 structures by using the formation energy (Eform) calculated as follows:
where
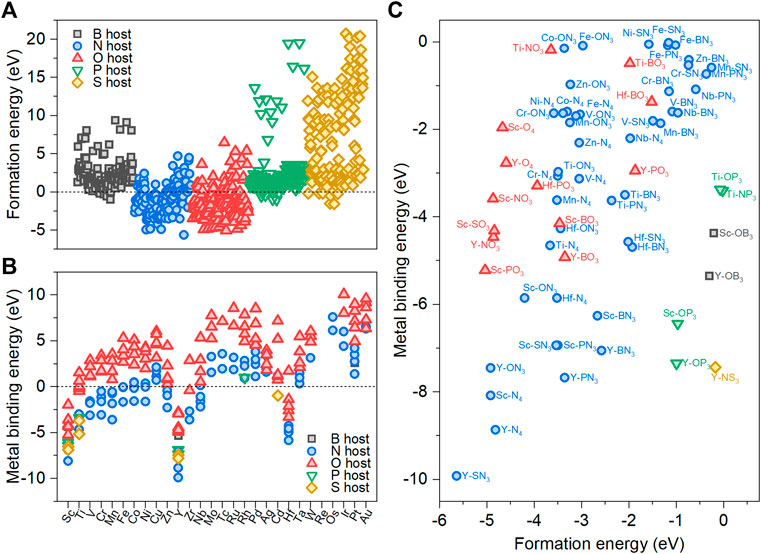
FIGURE 2. (A) Formation energies (Eform) of 700 M-YX3. (B) Metal-binding energies (Eb) of 222 M-YX3 with negative Eform as a function of transition metals. (C) Stable 66 M-YX3 structures with both negative Eform and negative Eb.
Next, we calculated the metal-binding energy (Eb) for the 222 thermodynamically stable M-YX3 structures based on the formation energy. Under the applied potential for ORR, metal atoms embedded in a carbon matrix can dissolve into the electrolyte, thereby causing the matrix to lose its active sites as follows:
Thus, the corresponding dissolution reaction energy at applied potential U was calculated by using the followed equation:
where
Therefore, we define the metal-binding energy using the dissolution reaction energy at operating ORR condition U = 0.8 VRHE as follows:
where
Considering the adsorption characteristics of key reaction intermediates OOH, O, and OH during ORR is highly important for understanding the difference in electrocatalytic performance between different metal-heteroatom-doped carbon structures, and thus offers guidelines for identifying the electrocatalyst with the best ORR activity. Supplementary Table S1 summarizes all of the free energy levels of the ORR intermediates in structurally stable metal-heteroatom-doped carbon. To reduce the computational cost, we only considered the metal atom center as the active site and assumed that the associative mechanism of ORR occurs on the M-YX3 structure. The four-electron transfer pathway for ORR under acidic conditions, which has been well studied previously (Kulkarni et al., 2018), proceeds as follows:
Thus, the overall four-electron pathway for ORR can be summarized as
The elementary reaction step of the ORR with the maximum free energy change (ΔGmax) value is defined as the potential-determining step (PDS). Moreover, a PDS with the positive ΔG value is thermodynamically unfavorable for the ORR reaction, and results in a large overpotential. The relationship between the limiting potential (UL) and a negative ΔGmax can be expressed as
The free energy diagrams for metal-heteroatom-doped carbon structures having a positive UL value are shown in Figure 3A. The UL means the maximum output potential at which the ORR elementary reaction steps are still exothermic (Kulkarni et al., 2018). Therefore, we selected the metal-heteroatom-doped carbon with a positive UL value because the electrocatalyst with a larger UL value has a higher ORR activity. The free energy changes of various metal-heteroatom-doped carbon catalysts were analyzed, for which the PDS is either OOH∗ formation or proton-electron transfer of OH∗ to form H2O, which is in agreement with the findings of previous research (Liang et al., 2014). Optimal adsorption strengths of the ORR intermediates are required for efficient ORR catalysis. ORR intermediates with adsorption strengths that are too weak result in insufficient O2 activation, and ones with adsorption strengths that are too strong result in unfavorable conditions for reducing and removing the ORR intermediates.
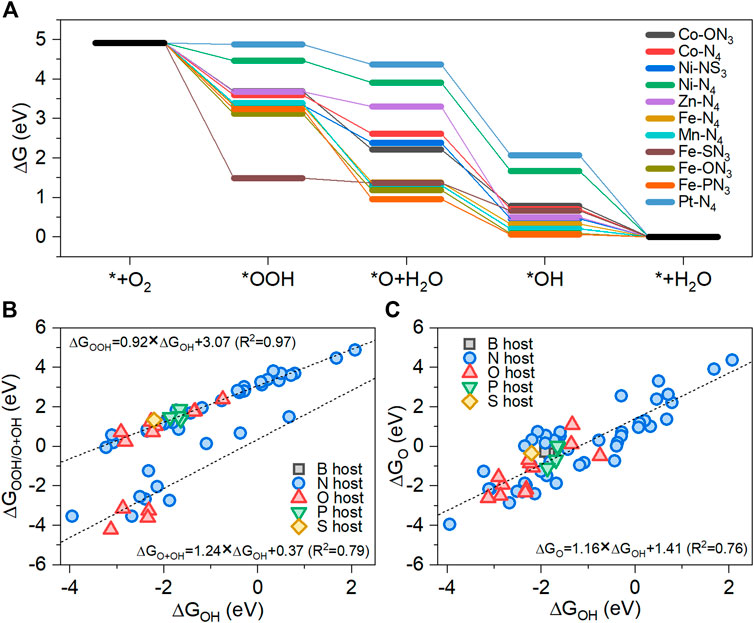
FIGURE 3. (A) Free energy changes during the ORR on 11 M-YX3 with positive limiting potential. The linear adsorption free energy scaling relationships of the ORR intermediates on the stable M-YX3 structures: (B) OOH*/O*+OH* vs. OH* and (C) O* vs. OH*.
For M-N4, the UL values of Co-N4, Fe-N4, Ni-N4, Zn-N4, Mn-N4, and Pt-N4 were found to be positive. The PDSs of Co-N4, Fe-N4, and Mn-N4 consist of the OH∗ removal step (UL = 0.71, 0.33, and 0.21 VRHE, respectively). The OOH∗ formation step is the PDS for Ni-N4 and Pt-N4 (UL = 0.45 and 0.04 VRHE). Thus, M-N4 structures with a strong OH∗ adsorption strength have the OH∗ removal step as the PDS, whereas those with a weak OH∗ adsorption strength have the OOH∗ formation step as the PDS because O2 activation is more difficult than in the counterparts with strong OH∗ adsorption. The PDS of the Zn-N4 structures was the ∗O formation step due to relatively weak O∗ adsorption strength compared to the other M-N4 structures. From the M-YN3 group, Co-ON3, Ni-SN3, Fe-ON3, Fe-SN3, and Fe-PN3 all showed a positive UL; the PDSs of Co-ON3, Ni-SN3, Fe-ON3, and Fe-PN3 were the OH∗ removal step with UL = 0.79, 0.46, 0.09, and 0.07 VRHE, respectively, while the PDS of Fe-SN3 was the ∗O formation step due to relatively strong OOH∗ adsorption strength compared to the other M-N4 structures.
Linear scaling correlations among the ORR intermediates on the structurally stable metal-heteroatom-doped carbon structures were observed for ΔGOOH or ΔGO vs. ΔGOH. The slope and intercept for the correlation between OH and OOH were 0.92 and 3.07, respectively, with a correlation coefficient (R2) value of 0.97, which is consistent with previous research (Calle-Vallejo et al., 2017; Kulkarni et al., 2018; Wan et al., 2019) (Figure 3B). Meanwhile, the scaling relationship between OH and O+OH was highly linear with a slope value of 1.24, an intercept value of 0.37, and an R2 value of 0.79. Figure 3C shows the scaling relationship between O and OH. The slope and the intercept for the correlation between OH and O were 1.16 and 1.41, respectively, with an R2 of 0.76. The O intermediates caused more oxidation of metal than OOH and OH because O species must form a double bond with the metal atom, resulting in more structural changes compared to the initial structure before the optimization. Therefore, the scaling relationship between O and OH has a larger variance than that between OOH and OH. Remarkably, for the structurally stable metal-heteroatom-doped carbon structures, the PDS is either the OOH∗ formation step or the OH∗ removal step. As shown in Supplementary Table S1, only four of the metal-heteroatom-doped carbon structures have either the O∗ formation step or the OH∗ formation step as the PDS. It is well known that the free energy changes in the OOH∗ formation step and the OH∗ removal step are determined by the relationship between ΔGOOH and ΔGOH, and thus the UL can be predicted based on this (Kulkarni et al., 2018).
General Activity Volcano Mapping for ORR on the Metal-Heteroatom-Doped Carbon Structures
We first investigated the ORR selectivity of 11 M-YX3 structures with a positive limiting potential. The equilibrium potential for the four and two electron pathways are shown in the dashed line at 1.23 and 0.7 eV, respectively (Figure 4A). According to the results, Mn-N4, Zn-N4, Ni-N4, and Pt-N4 had a lower overpotential of H2O2 production than H2O production. These results were consistent with previous theoretical findings (Jung et al., 2020). Figure 4B shows a 2-D volcano map for the theoretical limiting potential of ORR on various metal-heteroatom-doped carbon structures favoring the four-electron pathway for the H2O production by using ΔGOOH and ΔGOH as descriptors. Because UL is more positive in the red area, its optimum is at ΔGOOH = 3.69 eV and ΔGOH = 1.23 eV. The dashed line represents the scaling relationship between ΔGOOH and ΔGOH (ΔGOOH = 0.92 × ΔGOH + 3.07). Interestingly, the O-dopant heteroatom shifted Co-N4 closer to the ORR optimal value (ΔGOOH increased from 3.60 to 3.70 eV and ΔGOH from 0.71 to 0.79 eV). When an electron-rich heteroatom species is substituted for an N atom, this positively changes the charge state of the Co atom. Thus, changing the coordinating heteroatom from -N4 to -ON3 weakens the adsorption strength of the ORR intermediates on the structure. This is because the electronegativity of O (3.44) is higher than that of N (3.04), so the metal coordinated by O is more oxidative than the one by N, resulting in the adsorption strength being weakened.
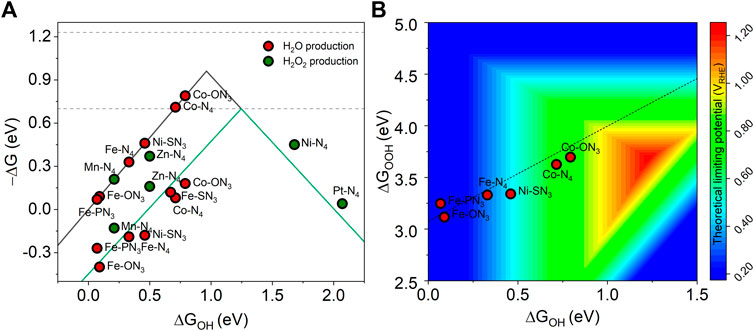
FIGURE 4. (A) Catalytic activity toward the H2O production (black solid line) and H2O2 production (green solid line), (B) A 2-D volcano map with ΔGOOH and ΔGOH as descriptors for the theoretical limiting potential of ORR on M-YX3 with a positive limiting potential.
It has already been reported that Co-N-C is a good catalyst for ORR through previous DFT computational screening studies and experimental validation (Zheng et al., 2016). Using O-dopant as a heteroatom has been reported for an O-C(Al) catalyst synthesized by using isomorphic metal-organic framework MIL-53(Al, Ga) (Yang et al., 2020) or a multiwalled carbon nanotube catalyst (Jiang et al., 2019). Moreover, the role of O-dopant heteroatoms has been elucidated in Co-NG(O) catalysts (Jung et al., 2020). The Co-ON3 structure derived from our screening results is stable and shows the highest ORR efficiency. Based on our results and those from previous studies, we expect that it can be successfully implemented experimentally.
In this study, to simplify the procedure, we calculated the adsorption energies of the oxygen intermediates using a metal atom as the active site. However, for the materials doped with low-electronegativity heteroatoms, such as P, the oxygen may adsorb on the heteroatom rather than on the metal atom, thereby stabilizing the structure (Dipojono et al., 2019). In addition, as the applied potential increases, the oxygen intermediates can be poisoned at the active site, resulting in different adsorption configurations as well as different ORR mechanisms (Li et al., 2016). These limitations will be assessed further in the future research. Nonetheless, our work is worth noting that a large-scale screening of metal-heteroatom-doped carbon materials that include many unknown combinations was conducted and the synthesis feasibility was determined based on the set of stability criteria. Finally, we provided the ORR activity data of those stable catalysts and demonstrated an unreported promising catalyst.
Conclusion
We have developed a computational screening process for the efficient metal-heteroatom-doped carbon ORR catalysts. M-N4 structures comprising 28 transition metals in conjunction with five dopant heteroatoms (B, N, O, P, and S) were constructed. Structural stability was evaluated by using the formation energy and the metal binding energy on heteroatom-doped carbon. We were able to identify 66 metal-heteroatom-doped carbon structures that were stable through rigorous stability testing. To compare the ORR activities of the 66 structurally stable metal-heteroatom-doped carbon structures, we calculated the free energy changes for ORR and selected 11 metal-heteroatom-doped carbon structures with a positive limiting potential. Finally, a 2-D volcano map constructed using ΔGOOH and ΔGOH as descriptors revealed that Co-ON3 was located nearest to the optimal point for ORR catalysis. This high-throughput screening process provides a promising path for the rational design of heterogenous electrocatalysts for energy conversion as well as scientific insight into the effect of heteroatom doping on carbon structures.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation. The coordinates of 66 structures and corresponding DFT energies used in this study are available on Catalysis-hub.org (Winther et al., 2019) under following link: https://www.catalysis-hub.org/publications/AraComputational2022.
Author Contributions
JWH directed and supervised this research work. AC and BJP designed the research and performed the theoretical calculations. AC, BJP and JWH co-wrote the manuscript. All of the authors discussed the results and revised the manuscript.
Funding
The authors acknowledge financial support from the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (NRF-2019M3D1A1079303 and NRF-2016R1A5A1009592). The authors also acknowledge the supercomputing resource including technical support from the Supercomputing Center, the Korean Institute of Science and Technology Information (KSC-2019-CRE-0016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors JH.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.873609/full#supplementary-material
References
Blöchl, P. E. (1994). Projector Augmented-Wave Method. Phys. Rev. B 50, 17953–17979. doi:10.1103/physrevb.50.17953
Bondi, A. (1964). van der Waals Volumes and Radii. J. Phys. Chem. 68, 441–451. doi:10.1021/j100785a001
Calle-Vallejo, F., Krabbe, A., and García-Lastra, J. M. (2017). How Covalence Breaks Adsorption-Energy Scaling Relations and Solvation Restores Them. Chem. Sci. 8, 124–130. doi:10.1039/c6sc02123a
Chen, M., He, Y., Spendelow, J. S., and Wu, G. (2019). Atomically Dispersed Metal Catalysts for Oxygen Reduction. ACS Energ. Lett. 4, 1619–1633. doi:10.1021/acsenergylett.9b00804
Dipojono, H. K., Saputro, A. G., Fajrial, A. K., Agusta, M. K., Akbar, F. T., Rusydi, F., et al. (2019). Oxygen Reduction Reaction Mechanism on a Phosporus-Doped Pyrolyzed Graphitic Fe/N/C Catalyst. New J. Chem. 43, 11408–11418. doi:10.1039/c9nj02118c
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 32, 1456–1465. doi:10.1002/jcc.21759
He, Y., Liu, S., Priest, C., Shi, Q., and Wu, G. (2020). Atomically Dispersed Metal-Nitrogen-Carbon Catalysts for Fuel Cells: Advances in Catalyst Design, Electrode Performance, and Durability Improvement. Chem. Soc. Rev. 49, 3484–3524. doi:10.1039/c9cs00903e
Jiang, K., Back, S., Akey, A. J., Xia, C., Hu, Y., Liang, W., et al. (2019). Highly Selective Oxygen Reduction to Hydrogen Peroxide on Transition Metal Single Atom Coordination. Nat. Commun. 10, 3997. doi:10.1038/s41467-019-11992-2
Jung, E., Shin, H., Lee, B.-H., Efremov, V., Lee, S., Lee, H. S., et al. (2020). Atomic-level Tuning of Co-N-C Catalyst for High-Performance Electrochemical H2O2 Production. Nat. Mater. 19, 436–442. doi:10.1038/s41563-019-0571-5
Kresse, G., and Furthmüller, J. (1996a). Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 6, 15–50. doi:10.1016/0927-0256(96)00008-0
Kresse, G., and Furthmüller, J. (1996b). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186. doi:10.1103/physrevb.54.11169
Kresse, G., and Hafner, J. (1993). Ab Initiomolecular Dynamics for Liquid Metals. Phys. Rev. B 47, 558–561. doi:10.1103/physrevb.47.558
Kresse, G., and Hafner, J. (1994). Norm-conserving and Ultrasoft Pseudopotentials for First-Row and Transition Elements. J. Phys. Condens. Matter 6, 8245–8257. doi:10.1088/0953-8984/6/40/015
Kresse, G., and Joubert, D. (1999). From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 59, 1758–1775. doi:10.1103/physrevb.59.1758
Kulkarni, A., Siahrostami, S., Patel, A., and Nørskov, J. K. (2018). Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 118, 2302–2312. doi:10.1021/acs.chemrev.7b00488
Lefèvre, M., Proietti, E., Jaouen, F., and Dodelet, J. P. (2009). Iron-Based Catalysts with Improved Oxygen Reduction Activity in Polymer Electrolyte Fuel Cells. Science 324, 71–74. doi:10.1126/science.1170051
Li, J., Ghoshal, S., Liang, W., Sougrati, M.-T., Jaouen, F., Halevi, B., et al. (2016). Structural and Mechanistic Basis for the High Activity of Fe-N-C Catalysts toward Oxygen Reduction. Energy Environ. Sci. 9, 2418–2432. doi:10.1039/c6ee01160h
Liang, W., Chen, J., Liu, Y., and Chen, S. (2014). Density-Functional-Theory Calculation Analysis of Active Sites for Four-Electron Reduction of O2 on Fe/N-Doped Graphene. ACS Catal. 4, 4170–4177. doi:10.1021/cs501170a
Lu, B., Liu, Q., and Chen, S. (2020). Electrocatalysis of Single-Atom Sites: Impacts of Atomic Coordination. ACS Catal. 10, 7584–7618. doi:10.1021/acscatal.0c01950
Mantina, M., Chamberlin, A. C., Valero, R., Cramer, C. J., and Truhlar, D. G. (2009). Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A. 113, 5806–5812. doi:10.1021/jp8111556
Monkhorst, H. J., and Pack, J. D. (1976). Special Points for Brillouin-Zone Integrations. Phys. Rev. B 13, 5188–5192. doi:10.1103/physrevb.13.5188
Mun, Y., Lee, S., Kim, K., Kim, S., Lee, S., Han, J. W., et al. (2019). Versatile Strategy for Tuning ORR Activity of a Single Fe-N4 Site by Controlling Electron-Withdrawing/Donating Properties of a Carbon Plane. J. Am. Chem. Soc. 141, 6254–6262. doi:10.1021/jacs.8b13543
Neugebauer, J., and Scheffler, M. (1992). Adsorbate-substrate and Adsorbate-Adsorbate Interactions of Na and K Adlayers on Al(111). Phys. Rev. B 46, 16067–16080. doi:10.1103/physrevb.46.16067
Nørskov, J. K., Rossmeisl, J., Logadottir, A., Lindqvist, L., Kitchin, J. R., Bligaard, T., et al. (2004). Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. The J. Phys. Chem. B 108, 17886–17892.
Park, B. J., Wang, Y., Lee, Y., Noh, K. J., Cho, A., Jang, M. G., et al. (2021). Effective Screening Route for Highly Active and Selective Metal−Nitrogen‐Doped Carbon Catalysts in CO2 Electrochemical Reduction. Small 17, 2103705. doi:10.1002/smll.202103705
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Shang, H., Zhou, X., Dong, J., Li, A., Zhao, X., Liu, Q., et al. (2020). Engineering Unsymmetrically Coordinated Cu-S1N3 Single Atom Sites with Enhanced Oxygen Reduction Activity. Nat. Commun. 11, 3049. doi:10.1038/s41467-020-16848-8
Wan, H., Østergaard, T. M., Arnarson, L., and Rossmeisl, J. (2019). Climbing the 3D Volcano for the Oxygen Reduction Reaction Using Porphyrin Motifs. ACS Sustain. Chem. Eng. 7, 611–617. doi:10.1021/acssuschemeng.8b04173
Winther, K. T., Hoffmann, M. J., Boes, J. R., Mamun, O., Bajdich, M., Bligaard, T., et al. (2019). Catalysis-Hub.org, An Open Electronic Structure Database for Surface Reactions. Scientific Data 6, 75.
Xu, H., Cheng, D., Cao, D., and Zeng, X. C. (2018). A Universal Principle for a Rational Design of Single-Atom Electrocatalysts. Nat. Catal. 1, 339–348. doi:10.1038/s41929-018-0063-z
Yang, Q., Xu, W., Gong, S., Zheng, G., Tian, Z., Wen, Y., et al. (2020). Atomically Dispersed Lewis Acid Sites Boost 2-electron Oxygen Reduction Activity of Carbon-Based Catalysts. Nat. Commun. 11, 5478. doi:10.1038/s41467-020-19309-4
Zheng, Y., Yang, D.-S., Kweun, J. M., Li, C., Tan, K., Kong, F., et al. (2016). Rational Design of Common Transition Metal-Nitrogen-Carbon Catalysts for Oxygen Reduction Reaction in Fuel Cells. Nano Energy 30, 443–449. doi:10.1016/j.nanoen.2016.10.037
Keywords: heteroatom doping, metal-nitrogen-doped carbon, single-metal-atom catalysts, oxygen reduction reaction, electrocatalysts, computational screening
Citation: Cho A, Park BJ and Han JW (2022) Computational Screening of Single-Metal-Atom Embedded Graphene-Based Electrocatalysts Stabilized by Heteroatoms. Front. Chem. 10:873609. doi: 10.3389/fchem.2022.873609
Received: 11 February 2022; Accepted: 11 March 2022;
Published: 06 April 2022.
Edited by:
Di Chen, Tsinghua University, ChinaReviewed by:
WooChul Jung, Korea Advanced Institute of Science and Technology, South KoreaShijun Zhao, City University of Hong Kong, Hong Kong SAR, China
Copyright © 2022 Cho, Park and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeong Woo Han, andoYW5AcG9zdGVjaC5hYy5rcg==
†These authors have contributed equally to this work.