 Zhenhua Liu
Zhenhua Liu Yang Fang1†
Yang Fang1† Bo Tang
Bo Tang- 1Key Laboratory of Molecular and Nano Probes, Shandong Provincial Key Laboratory of Clean Production of Fine Chemicals, College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Ministry of Education, Shandong Normal University, Jinan, China
- 2Department of Pharmacy, Zibo Central Hospital, Zibo, China
A novel difunctionalization of aryldiazonium salts was realized for the one-step generation of symmetric and asymmetric p-azophenols. This approach is proceeded by the sequentially regioselective aromatic C-O and C-N bond construction under mild reaction conditions, unlocking a new reaction strategy to facilitate the synthesis of p-azophenols.
Introduction
Azoaromatic compounds are significant core motifs and found the prevalence in a variety of applications due to their unique properties of the azo unit (Pang et al., 2020). They are universally applied in synthetic chemistry (Nguyen et al., 2018), sensing probes (Dong et al., 2015), organic dyes (Bafana et al., 2011), smart materials (Mohamed et al., 2015; Gerkman et al., 2020), and therapeutic agents (Lizard et al., 2020; Babamale et al., 2021). Considering the widespread utility of aromatic azo scaffolds, a handful of methods for their formation were established by adding an external oxidant or noble metal catalyst (Grirrane et al., 2008; Dai et al., 2018; Han et al., 2021; Sitter and Vannucci, 2021). Therefore, the synthesis route to achieve azobenzene derivatives in an efficient and handy manner continues to be keenly pursued.
Aryldiazonium salts (Griess, 1858), abundant and inexpensive building blocks, have stimulated a series of remarkable name reactions as useful and efficient synthons over the next hundred years (Sandmeyer, 1884; Hodgson, 1947; Gomberg and Bachmann, 1924; Balzand Schiemann, 1927; Meerwein et al., 1939; Kikukawa and Matsuda, 1977; Kikukawa et al., 1979). Even to this day, the aryldiazonium compounds still receive much attention and constantly result in plenty of new functionalization conversions. For example, the diazo-oriented substitutions of aryldiazonium salts with various coupling partners via the denitrogenation step have been successfully explored (Figure 1A) (Mo et al., 2018; Felpin and Sengupta, 2019; Yuan et al., 2021). Aryldiazonium salts by the nucleophilic addition-type reaction have also been extensively used for inserting the azo group into the target molecules (Figure 1A) (Liu F et al., 2017; Liu Z et al., 2017; Abrams et al., 2018; Liu et al., 2018). Recently, cyclization by retaining the privileged N2 fragment has been a thriving pattern in heterocyclic chemistry (Figure 1B) (Mo et al., 2013; Zhang et al., 2020). Apart from the familiar aryl or/and aryl azo precursors described above, the aryl-based difunctionalization pattern of aryldiazonium salts has seriously remained in the shadow of underdevelopment. Herein, we exposed a first example utilizing this aryl-based difunctionalization tactics of aryldiazonium salts with H2O to directly assemble versatile p-azophenols in good yields under mild reaction conditions (Figure 1C).
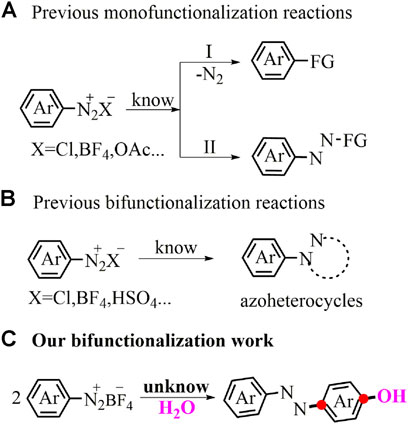
FIGURE 1. Functionalizations of aryldiazonium salts.
Results and Discussion
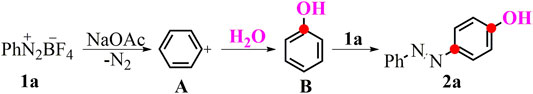
To optimize our aryl difunctionalization scenario, we first employed phenyldiazonium salt 1a as the standard substrate to evaluate reaction conditions with the high-yielding product 4-(phenyldiazenyl)phenol 2a in mind. When MeOH was conducted as a solvent in the difunctionalization reaction of the phenyldiazonium salt 1a, an unprecedented preference was smoothly delivered with the yielding product 2a in 46% yield (Table 1, entry 1). The absolute structure of 2a was also confirmed by X-ray diffraction analysis (CCDC No. 2070994, see details in SI). In light of this initial experimental result, we further investigated other solvents for this aryl-based cascade reaction, while keeping the moderate yield permanent (Table 1, entries 2–5). With the consideration of a hydroxyl source in the target product, we next screened the amount of H2O in the reaction (Table 1, entries 6–8). The optimal ratio of MeOH/H2O was found to be 3:1 with a superior increase in the yield to 89%. Then, other bases were probed in the reaction conditions and gave somewhat unsatisfied yields of the desired product (Supplementary Table S1, entries 1-8 in SI). Similarly, adjusting the amount of the base or reaction temperature was proved to impede the desired conversion (Supplementary Table S1, entries 9–11 in SI).
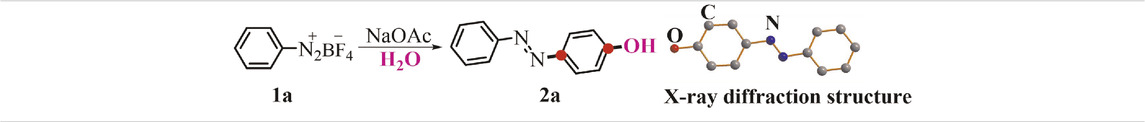
TABLE 1. Optimization of the reaction conditions.a
TABLE 1. Continued.
Having the optimized reaction conditions in hand, we began examining the reactivity of various aryldiazonium salts to give the corresponding p-azophenol derivatives (Scheme 1). Aryldiazonium salts with ortho- and meta-substituents including electron-donating or electron-withdrawing groups (1b-1g) smoothly reacted with H2O to produce the symmetrical products (2b-2g) in good yields. In addition, poly-substituents with different positions on the aryldiazonium salts were fully compatible in the standard conditions, delivering azo-containing phenolic compounds (2h-2l) in 73–82% yields. Regrettably, when the substrate bearing the meta-trifluoromethyl group was performed in the standard tandem reaction, no desired difunctionalized product 2m was observed.
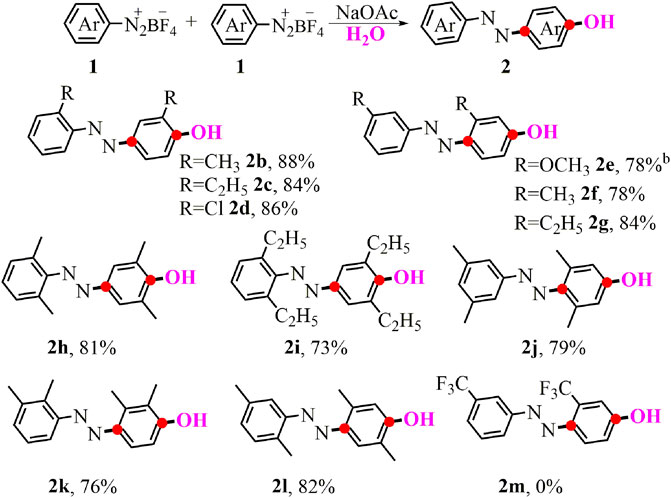
SCHEME 1. Synthesis of symmetrical p-azophenol. aConditions: 1 (0.5 mmol), NaOAc (0.5 mmol, 1 eq.), in 2 ml of the mixed solvent (VMeOH/VH2O = 3:1) at 0°C-r.t. for 3 h; yields of isolated products. bWithout 1.0 eq. NaOAc.
A cross reaction between two different diazonium salts by using this efficient difunctionalization procedure is undoubtedly an attractive extension (Scheme 2). Just as we speculated, the reaction with the phenyldiazonium salt 1a and 2-chlorobenzene diazonium salt 1dwas viable in this cascade conversion and provided the unsymmetrical p-azophenol 3a in 86% yield. Then, we further examined other cross-coupling partners with the 2-chlorobenzene diazo salt as one model reactant. To our delight, aryldiazonium salts attached to the ortho- or meta-group were smoothly subjected into the reaction, yielding the aimed unsymmetrical products (3b-3e) in 82–87% yields. Similarly, the substrate with multiple substituents at different positions was also found to successfully deliver the target products (3f-3g). The X-ray structure of products 3f and 3g (CCDC No. 2070995; 2070996, see details in SI) also validated the asymmetrical structure of the assumed product. Note that the 3-chlorobenzene diazonium salt by combining with electron-neutral or electron-donating ones was well suitable in the reaction, producing the unsymmetrical p-azophenol (3i-3l) in 63–80% yields.
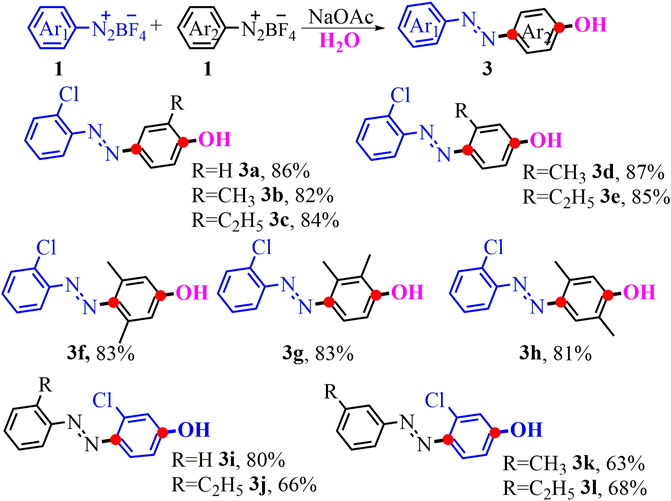
SCHEME 2. Synthesis of unsymmetrical p-azophenol. aConditions: 1 (0.5 mmol), NaOAc (0.5 mmol, 1 eq.), in 2 ml of the mixed solvent (VMeOH/VH2O = 3:1) at 0°C-r.t. for 3 h; yields of isolated products.
To improve the scalability and practicality of this difunctionalization approach, large-scale synthesis was applied with the substrate 1b in the standard one-pot reaction, leading to the p-azophenol derivative 2b with a satisfied yield (Scheme 3). Regarding the possibilities for further decoration of the hydroxyl group, this difunctionalized product 2b was readily introduced into allylation (4a), propargylation (4b), and acetylation (4c) with the corresponding modification reagents.
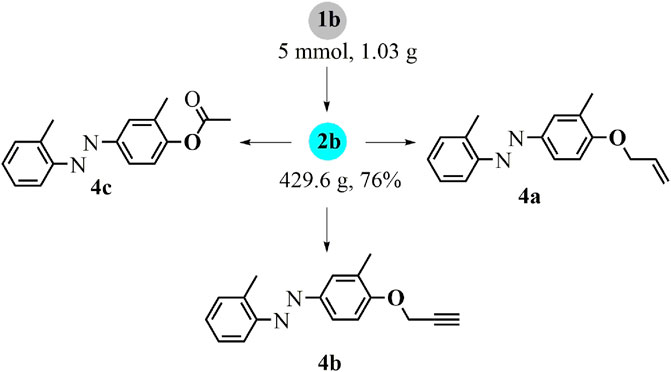
SCHEME 3. Large-scale reaction and derivatizations.
To obtain the information of this difunctionalization pathway, free radical trap experiments (Liu et al., 2019; Liu et al., 2020) with using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) did not give the target product 2a or the corresponding radical coupling byproduct with the assistance of HRMS (Supplementary Scheme S1A in SI). Those inhibited results might ascribe to the acidity of BHT (Huang and Lumb, 2019) or oxidability of TEMPO (Ryan et al., 2019) influencing on the stability of diazonium salts in the standard reaction. So an ionic process was still heavily weighted in the optimal process. To examine the sequence of hydroxylation and azo-modification, a hypothesized PhOH intermediate led to the para-functionalization product 2b’ rather than the target product 2b (Supplementary Scheme S1B in SI). Diphenylazo 5a (Okumura et al., 2013) replacing the diazonium salt was used as the reactant whereas the desired formation of 2a was completely shut down (Supplementary Scheme S1C in SI). Those observations were more likely to highlight that the diazo-localized hydroxylation of the aryldiazonium salt preceded over the para-selective azo reaction in the standard reaction. In order to further help understand the reaction mechanism, the charge densities of the carbon adjacent to the azo group in the aryldiazonium cation were calculated by the natural population analysis (NPA) charge analysis (see Supplementary Scheme S2 in SI).
In view of the above experimental observations and the results of calculated NPA charges for selective carbon in aryl azo ions (see Supplementary Scheme S2 in SI), a proposed pathway is displayed in Scheme 4. Initially, the releasing nitrogen of substrate 1a produced the active phenyl cation A (Canning et al., 1999; Canning et al., 2002), which quickly underwent the electrophilic substitution reaction with H2O by forming the phenol intermediate B (Ando, 1978). If different diazonium salts were employed, the substrate bearing more positive charges at the carbon adjacent to the azo group could be preferentially converted to phenol species. Sequentially, the in situ para-selective bias of species B with another diazonium salt through electrophilic aromatic substitution (EArS) occurred (Fiege et al., 2000; Xu and Zeng, 2010; Huang and Lumb, 2019), offering the final product 2/3 in one-pot.
SCHEME 4. Possible mechanism for the reaction.
Conclusion
In summary, we have successfully demonstrated a novel and practical aryl-based difunctionalization strategy of aryldiazonium salts with H2O, providing a convenient avenue to valuable symmetric/asymmetric p-azophenols in a one-pot operation. Obviously, this tandem method directly forming aromatic C-O and C-N bonds results in tolerance of readily available substrates, step efficiency, and practical reaction operations. Mechanistically, an ionic mechanism was suggested by relying on the preliminary findings of our designed experiments and the corresponding calculated NPA charges. As far as we know, this is the first example by achieving the aryl-based difunctionalization strategy of aryldiazonium salts with H2O to straightforwardly construct versatile p-azophenol derivatives in one-pot. Our difunctionalization model of diazosalts undoubtedly presents a seductive blueprint to assess the significant skeleton in organic synthetic chemistry.
Experiment
General Information
All reagents were purchased from commercial sources and used without treatment, unless otherwise indicated. The products were purified by column chromatography over silica gel. 1H NMR and 13C NMR spectra were recorded at 25°C on a Varian 400 and 100 MHz, respectively, and TMS was used as the internal standard. Mass spectra were recorded on a BRUKER AutoflexIII Smartbeam MS-spectrometer. High-resolution mass spectra (HRMS) were recorded on Bruck microTof by using the ESI method.
General procedure for the synthesis of 2/3: Phenyldiazonium tetrafluoroborate 1 (0.5 mmol, 1.0 eq.) and sodium acetate (0.5 mmol, 1.0 eq.) were added in an oven-dried 15-ml Schlenk tube with 2 ml of mixed solvent (MeOH/H2O = 3/1) at 0°C; the reaction mixture was then stirred at r.t. for 3 h. The resulting reaction mixture was taken up by dichloromethane (3 × 15 ml). The organic layer was washed with brine (3 × 40 ml), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by a silica gel column chromatography (petroleum ether/ethyl acetate = 5:2), and the target product two or three was afforded.
(E)-4-(phenyldiazenyl)phenol (2a). Compound 2a was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 89%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.6 Hz, 4H), 7.50–7.33 (m, 3H), 6.86 (d, J = 8.6 Hz, 2H), and 5.21 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 157.3, 151.6, 146.0, 129.4, 128.0, 123.9, 121.5, and 114.7; HRMS (ESI-TOF)m/z calculated for C12H9N2O [M-H]-: 197.0715, found: 197.0704.
(E)-2-methyl-4-(o-tolyldiazenyl)phenol (2b). Compound 2b was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 88%, yellow solid. 1H NMR (400 MHz, CDCl3) δ7.78–7.74 (m, 1H), 7.73–7.70 (m, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.35–7.29 (m, 2H), 7.28–7.22 (m, 1H), 6.87 (d, J = 8.4 Hz, 1H), 5.41 (s, 1H), 2.70 (s, 3H), and 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.5, 150.8, 147.4, 137.4, 131.1, 130.2, 126.3, 125.6, 124.4, 123.0, 115.4, 115.1, 17.5, and 15.8; HRMS (ESI-TOF) m/z calculated for C14H13N2O [M-H]-: 225.1028, found: 225.1018.
(E)-2-ethyl-4-((2-ethylphenyl)diazenyl)phenol (2c). Compound 2b was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 84%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.76–7.70 (m, 1H), 7.65–7.59 (m, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.33–7.23 (m, 2H), 7.20–7.14 (m, 1H), 6.73 (d, J = 8.4 Hz, 1H), 5.43 (s, 1H), 3.14–2.99 (m, 2H), 2.70–2.56 (m, 2H), and 1.27–1.14 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 155.0, 149.2, 146.4, 142.3, 129.5, 129.4, 128.5, 125.4, 123.6, 121.0, 23.6, 21.9, 15.3, and 12.6; HRMS (ESI-TOF) m/z calculated for C16H17N2O [M-H]-: 253.1341, found: 253.1320.
(E)-2-chloro-4-((2-chlorophenyl)diazenyl)phenol (2d). Compound 2d was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 86%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.93–7.87 (m, 1H), 7.76–7.69 (m, 1H), 7.61–7.56 (m, 1H), 7.49–7.4 (m, 1H), 7.30–7.22 (m, 2H), 6.77 (d, J = 8.6 Hz, 1H), and 4.43 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 147.7, 145.0, 144.6, 133.5, 129.8, 129.5, 126.2, 123.7, 123.4, 118.3, 116.5, and 113.8; HRMS (ESI-TOF) m/z calculated for C12H7Cl2N2O [M-H]-: 264.9935, found: 264.9907.
(E)-3-methoxy-4-((3-methoxyphenyl)diazenyl)phenol (2e). Compound 2e was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 78%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.68–7.58 (m, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.33–7.25 (m, 2H), 6.90–6.83 (m, 1H), 6.32–6.12 (m, 2H), 4.07 (s, 1H), 3.89 (s, 3H), and 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.1, 158.1, 153.6, 150.4, 133.9, 128.5, 117.5, 115.2, 115.1, 106.4, 104.7, 96.7, 55.0, and 54.4; HRMS (ESI-TOF) m/z calculated for C14H13N2O3 [M-H]-: 257.0926, found: 257.0911.
(E)-3-methyl-4-(m-tolyldiazenyl)phenol (2f). Compound 2f was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 78%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.69–7.63 (m, 3H), 7.40–7.35 (m, 1H), 7.24 (d, J = 7.6 Hz, 1H), 6.79–6.75 (m, 1H), 6.72–6.68 (m, 1H), 5.30 (s, 1H), 2.70 (s, 3H), and 2.45 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.1, 153.1, 145.1, 141.0, 138.8, 130.9, 128.8, 123.0, 120.0, 117.3, 117.0, 113.5, 21.4, and 17.6; HRMS (ESI-TOF) m/z calculated for C14H13N2O [M-H]-: 225.1028, found: 225.1003.
(E)-3-ethyl-4-((3-ethylphenyl)diazenyl)phenol (2g). Compound 2g was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 84%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.66–7.57 (m, 2H), 7.53–7.46 (m, 1H), 7.34–7.21 (m, 1H), 7.19–7.08 (m, 1H), 6.70–6.62 (m, 1H), 6.59–6.50 (m, 1H), 4.98 (s, 1H), 3.12–2.86 (m, 2H), 2.63 (q, J = 7.6 Hz, 2H), and 1.23–1.10 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 157.6, 152.0, 146.0, 144.2, 143.3, 128.8, 127.9, 121.4, 118.6, 116.1, 114.8, 112.7, 27.7, 23.6, 15.0, and 14.4; HRMS (ESI-TOF) m/z calculated for C16H17N2O [M-H]-: 253.1341, found: 253.1311.
(E)-4-((2,6-dimethylphenyl)diazenyl)-2,6-dimethylphenol (2h). Compound 2h was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 81%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.52 (s, 2H), 7.01 (s, 3H), 5.00 (s, 1H), 2.25 (s, 6H), and 2.21 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 154.1, 150.9, 145.4, 128.9, 127.8, 126.3, 122.5, 122.2, 17.4, and 14.9; HRMS (ESI-TOF) m/z calculated for C16H17N2O [M-H]-: 253.1341, found: 253.1306.
(E)-4-((2,6-diethylphenyl)diazenyl)-2,6-diethylphenol (2i). Compound 2i was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 73%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 2H), 7.13–7.04 (m, 3H), 5.00 (s, 1H), 2.68–2.61 (m, 4H), 2.59–2.51 (m, 4H), 1.28–1.23 (m, 6H), and 1.10–1.03 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 153.2, 150.7, 145.8, 134.6, 128.6, 126.3, 126.1, 120.4, 23.8, 22.0, 14.3, and 12.6; HRMS (ESI-TOF) m/z calculated for C20H25N2O [M-H]-: 309.1967, found: 309.1942.
(E)-4-((3,5-dimethylphenyl)diazenyl)-3,5-dimethylphenol (2j). Compound 2j was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 79%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.39 (s, 2H), 7.02 (s, 1H), 6.49 (s, 2H), 5.21 (s, 1H), and 2.38–2.30 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 155.5, 153.2, 144.9, 138.7, 134.4, 132.0, 120.0, 115.8, 21.3, and 19.7; HRMS (ESI-TOF) m/z calculated for C16H17N2O [M-H]-: 253.1341, found: 253.1333.
(E)-4-((2,3-dimethylphenyl)diazenyl)-2,3-dimethylphenol (2k). Compound 2k was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 76%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.48–7.42 (m, 1H), 7.40–7.31 (m, 1H), 7.14 (d, J = 7.2 Hz, 1H), 7.10–7.03 (m, 1H), 6.63–6.55 (m, 1H), 5.04 (s, 1H), 2.62 (s, 3H), 2.55 (s, 3H), 2.29 (s, 3H), and 2.18 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.8, 150.3, 144.8, 138.6, 137.0, 135.2, 130.2, 124.6, 122.0, 113.4, 112.5, 111.9, 18.9, 12.7, 12.2, and 10.6; HRMS (ESI-TOF) m/z calculated for C16H17N2O [M-H]-: 253.1341, found: 253.1336.
(E)-4-((2,5-dimethylphenyl)diazenyl)-2,5-dimethylphenol (2l). Compound 2l was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 82%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.45 (s, 1H), 7.31 (s, 1H), 7.15–7.02 (m, 2H), 6.63 (s, 1H), 5.03 (s, 1H), 2.60 (d, J = 3.0 Hz, 6H), 2.29 (s, 3H), and 2.19 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.2, 149.9, 144.3, 137.2, 134.8, 133.3, 129.9, 129.7, 120.9, 117.5, 115.6, 115.1, 20.0, 16.2, and 14.5; HRMS (ESI-TOF) m/z calculated for C16H17N2O [M-H]-: 253.1341, found: 253.1313.
(E)-4-((2-chlorophenyl)diazenyl)phenol (3a). Compound 3a was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 86%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.94–7.80 (m, 2H), 7.62–7.54 (m, 1H), 7.51–7.43 (m, 1H), 7.34–7.20 (m, 2H), 6.97–6.78 (m, 2H), and 5.44 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 157.7, 147.7, 146.3, 133.6, 130.0, 129.5, 126.2, 124.5, 116.5, and 114.8; HRMS (ESI-TOF) m/z calculated for C12H8ClN2O [M-H]-: 231.0325, found: 231.0315.
(E)-4-((2-chlorophenyl)diazenyl)-2-methylphenol (3b). Compound 3b was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 82%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.76–7.66 (m, 2H), 7.63–7.53 (m, 1H), 7.49–7.42 (m, 1H), 7.30–7.21 (m, 2H), 6.80 (d, J = 8.4 Hz, 1H), 5.49 (s, 1H), and 2.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.2, 147.8, 146.2, 133.5, 129.8, 129.5, 126.2, 124.8, 123.6, 122.8, 116.5, 114.2, and 14.8; HRMS (ESI-TOF) m/z calculated for C13H10ClN2O [M-H]-: 245.0482, found: 245.0445.
(E)-4-((2-chlorophenyl)diazenyl)-2-ethylphenol (3c). Compound 3c was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 84%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.62–7.51 (m, 1H), 7.50–7.40 (m, 1H), 7.32–7.18 (m, 2H), 6.79–6.53 (m, 1H), 5.46 (s, 1H), 2.63 (q, J = 7.4 Hz, 2H), and 1.27–1.10 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 155.8, 147.8, 146.3, 133.4, 129.8, 129.7, 129.5, 126.2, 123.8, 122.0, 116.6, 114.5, 21.9, and 12.6; HRMS (ESI-TOF) m/z calculated for C14H12ClN2O [M-H]-: 259.0638, found: 259.0630.
(E)-4-((2-chlorophenyl)diazenyl)-3-methylphenol (3d). Compound 3d was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 87%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.71–7.63 (m, 1H), 7.61–7.51 (m, 1H), 7.49–7.41 (m, 1H), 7.32–7.21 (m, 2H), 6.79–6.57 (m, 2H), 5.22 (s, 1H), and 2.62 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 157.7, 148.0, 144.4, 140.6, 133.6, 129.7, 129.5, 126.1, 117.2, 116.8, 116.2, 112.8, and 16.7; HRMS (ESI-TOF) m/z calculated for C13H10ClN2O [M-H]-: 245.0482, found: 245.0471.
(E)-4-((2-chlorophenyl)diazenyl)-3-ethylphenol (3e). Compound 3e was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 85%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.75–7.62 (m, 1H), 7.60–7.52 (m, 1H), 7.49–7.39 (m, 1H), 7.36–7.17 (m, 2H), 6.79–6.53 (m, 2H), 5.19 (s, 1H), 3.05 (q, J = 7.4 Hz, 2H), and 1.27–1.10 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 157.9, 148.0, 146.6, 143.7, 133.6, 129.7, 129.5, 126.1, 117.0, 116.7, 114.7, 112.8, 23.6, and 15.1; HRMS (ESI-TOF) m/z calculated for C14H12ClN2O [M-H]-: 259.0638, found: 259.0621.
(E)-4-((2-chlorophenyl)diazenyl)-3,5-dimethylphenol (3f). Compound 3f was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 83%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.60–7.54 (m, 1H), 7.52–7.45 (m, 1H), 7.31–7.22 (m, 2H), 6.55 (s, 2H), 5.27 (s, 1H), and 2.49 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 155.5, 148.4, 143.1, 135.7, 133.9, 129.7, 129.4, 126.1, 115.9, 115.0, and 19.8; HRMS (ESI-TOF) m/z calculated for C14H12ClN2O [M-H]-: 259.0638, found: 259.0616.
(E)-4-((2-chlorophenyl)diazenyl)-2,3-dimethylphenol (3g). Compound 3g was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 83%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.62–7.51 (m, 2H), 7.49–7.40 (m, 1H), 7.31–7.20 (m, 2H), 6.60 (d, J = 8.8 Hz, 1H), 5.26 (s, 1H), 2.61 (s, 3H), and 2.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.7, 148.2, 144.6, 139.5, 133.5, 129.59, 129.50, 126.1, 122.2, 116.9, 113.9, 112.2, 12.7, and 10.6; HRMS (ESI-TOF) m/z calculated for C14H12ClN2O [M-H]-: 259.0638, found: 259.0608.
(E)-4-((2-chlorophenyl)diazenyl)-2,5-dimethylphenol (3h). Compound 3h was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 81%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.61–7.51 (m, 2H), 7.48–7.42 (m, 1H), 7.29–7.21 (m, 2H), 6.65 (s, 1H), 5.22 (s, 1H), 2.59 (s, 3H), and 2.19 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.3, 148.2, 144.1, 138.2, 133.4, 129.57, 129.50, 126.1, 121.3, 117.9, 116.8, 115.6, 16.2, and 14.4; HRMS (ESI-TOF) m/z calculated for C14H12ClN2O [M-H]-: 259.0638, found: 259.0619.
(E)-3-chloro-4-(phenyldiazenyl)phenol (3i). Compound 3i was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 80%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.86–7.74 (m, 3H), 7.72–7.65 (m, 1H), 7.40–7.27 (m, 2H), 6.86 (d, J = 8.8 Hz, 2H), and 4.43 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 157.7, 152.5, 145.8, 134.0, 129.1, 129.0, 124.2, 121.0, 120.5, and 114.9; HRMS (ESI-TOF) m/z calculated for C12H8ClN2O [M-H]-: 231.0325, found: 231.0320.
(E)-3-chloro-4-((2-ethylphenyl)diazenyl)phenol (3j). Compound 3j was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 66%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.83–7.62 (m, 4H), 7.42–7.29 (m, 2H), 6.84–6.74 (m, 1H), 5.28 (s, 1H), 2.71–2.57 (m, 2H), and 1.26–1.21 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 155.6, 152.6, 145.9, 134.0, 129.6, 129.0, 128.9, 123.1, 122.0, 121.0, 120.4, 114.5, 21.8, and 12.6; HRMS (ESI-TOF) m/z calculated for C14H12ClN2O [M-H]-: 259.0638, found: 259.0628.
(E)-3-chloro-4-(m-tolyldiazenyl)phenol (3k). Compound 3k was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 63%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.85–7.74 (m, 2H), 7.67 (d, J = 8.8 Hz, 1H), 7.48–7.35 (m, 2H), 6.83–6.67 (m, 2H), 3.63 (s, 1H), and 2.69 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.7, 153.9, 144.9, 141.8, 135.0, 130.0, 129.8, 122.2, 121.5, 117.4, 117.2, 113.8, and 17.6; HRMS (ESI-TOF) m/z calculated for C13H10ClN2O [M-H]-: 245.0482, found: 245.0473.
(E)-3-chloro-4-((3-ethylphenyl)diazenyl)phenol (3l). Compound 3l was prepared in a manner of the general procedure for the synthesis of 2/3. Yield 68%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.78–7.66 (m, 2H), 7.60 (d, J = 8.8 Hz, 1H), 7.39–7.26 (m, 2H), 6.76–6.70 (m, 1H), 6.67–6.59 (m, 1H), 3.79 (s, 1H), 3.11–2.98 (m, 2H), and 1.25–1.18 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 158.0, 152.9, 146.7, 143.2, 133.9, 129.0, 128.8, 121.0, 120.6, 116.3, 114.7, 112.8, 23.6, and 15.1; HRMS (ESI-TOF) m/z calculated for C14H12ClN2O [M-H]-: 259.0638, found: 259.0627.
The dry sealed tube was charged with 2b (113.1 mg, 0.5 mmol, 1.0 eq.), allyl bromide (52.0 μL, 0.6 mmol, 1.2 eq.), and K2CO3 (138.0 mg, 1.0 mmol, 2.0 eq.) in anhydrous acetone (4 ml). Then, the reaction was heated at 50°C for 5 h. The mixture was extracted with DCM (3 × 30 ml), and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative TLC (PE/EA = 5/1, v/v) to give 4a.
(E) -1-(4-(allyloxy)-3-methylphenyl)-2-(o-tolyl)diazene (4a). Compound 4a was prepared in a manner of the above procedure. Yield 93%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.84–7.72 (m, 2H), 7.64–7.53 (m, 1H), 7.34–7.19 (m, 3H), 6.91–6.83 (m, 1H), 6.15–5.95 (m, 1H), 5.51–5.38 (m, 1H), 5.34–5.23 (m, 1H), 4.67–4.53 (m, 2H), 2.69 (s, 3H), and 2.32 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.2, 151.0, 147.1, 137.4, 133.1, 131.1, 130.1, 127.7, 126.4, 124.2, 123.6, 117.3, 115.5, 110.9, 68.9, 17.6, and 16.5; HRMS (ESI-TOF) m/z calculated for C17H18N2ONa [M + Na]+: 289.1317, found: 289.1297.
The dry sealed tube was charged with 2b (113.1 mg, 0.5 mmol, 1.0 eq.), 3-bromopropyne (51.7 μL, 0.6 mmol, 1.2 eq.), and K2CO3 (138.0 mg, 1.0 mmol, 2.0 eq.), in anhydrous acetone (4 ml). Then, the reaction was heated at 50°C for 4.5 h. The mixture was extracted with DCM (3 × 30 ml), and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative TLC (PE/EA = 5/1, v/v) to give 4b.
(F) -1-(3-methyl-4-(prop-2-yn-1-yloxy)phenyl)-2-(o-tolyl)diazene (4b). Compound 4b was prepared in a manner of the above procedure. Yield 92%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.77–7.65 (m, 2H), 7.50 (d, J = 7.8 Hz, 1H), 7.25–7.21 (m, 2H), 7.19–7.12 (m, 1H), 6.94 (d, J = 8.6 Hz, 1H), 4.68 (d, J = 2.4 Hz, 2H), 2.61 (s, 3H), 2.44 (t, J = 2.4 Hz, 1H), and 2.23 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 156.9, 149.8, 146.4, 136.4, 130.0, 129.2, 126.9, 125.3, 123.4, 122.2, 114.4, 110.3, 77.3, 74.6, 55.0, 16.4, and 15.3; HRMS (ESI-TOF) m/z calculated for C17H16N2ONa [M + Na]+: 287.1160, found: 287.1132.
The dry sealed tube was charged with 2b (113.1 mg, 0.5 mmol, 1.0 eq.), acetyl chloride (71.2 μL, 1.0 mmol, 2.0 eq.), and triethylamine (Et3N) (139.0 μL, 1.0 mmol, 2.0 eq.) in anhydrous DCM (4 ml). Then, the reaction was stirred at r.t. for 3 h. The mixture was extracted with DCM (3 × 30 ml), and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative TLC (PE/EA = 5/1, v/v) to give 4c.
(E) -2-methyl-4-(o-tolyldiazenyl)phenyl acetate (4c). Compound 4c was prepared in a manner of the above procedure. Yield 95%, yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.74–7.66 (m, 2H), 7.53–7.47 (m, 1H), 7.28–7.20 (m, 2H), 7.18–7.11 (m, 1H), 7.06 (d, J = 8.4 Hz, 1H), 2.61 (s, 3H), 2.24 (s, 3H), and 2.18 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.8, 150.1, 149.7, 149.6, 136.9, 130.1, 129.9, 129.8, 125.3, 124.5, 121.5, 120.5, 114.3, 19.7, 16.4, and 15.3; HRMS (ESI-TOF) m/z calculated for C16H16N2O2Na [M + Na]+: 291.1109, found: 291.1084.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
ZL and YF designed the study, performed the experiments, and collected the data. WF performed theoretical calculations and edited the manuscript. YL and XG prepared some substrates (40%). ZL, WG, and BT conceived this project, gave financial support, wrote, and revised the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank for the financial support provided by the National Natural Science Foundation of China (21976112, 21775092), the National Key R and D Program of China (2019YFA0210100), Major Science and Technology Innovation Project of Shandong Province, 2021ZDSYS09, the Natural Science Foundation of Shandong Province of China (ZR2021MB031, ZR2020JQ10), and the China Postdoctoral Science Foundation (2020M682204).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.818627/full#supplementary-material
References
Abrams, R., Lefebvre, Q., and Clayden, J. (2018). Transition Metal Free Cycloamination of Prenyl Carbamates and Ureas Promoted by Aryldiazonium Salts. Angew. Chem. Int. Ed. 57, 13587–13591. doi:10.1002/anie.201809323
Ando, W. (1978). “Photochemistry of the Diazonium and Diazo Groups,” in The Chemistry of Diazonium and Diazo Groups. Editor S. Patai (Chichester: Wiley), 341–487. Part 1. doi:10.1002/9780470771549.ch9
Babamale, H. F., Sangeetha, T., Tan, J. S., and Yam, W. (2021). Synthesis and Characterization of Azobenzene Derivatives and Azobenzene-Imidazolium Conjugates with Selective Antimicrobial Potential. J. Mol. Struct. 1232, 130049–130058. doi:10.1016/j.molstruc.2021.130049
Bafana, A., Devi, S. S., and Chakrabarti, T. (2011). Azo Dyes: Past, Present and the Future. Environ. Rev. 19, 350–371. doi:10.1139/a11-018
Balz, G., and Schiemann, G. (1927). Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung. Ber. Dtsch. Chem. Ges. A/b 60, 1186–1190. doi:10.1002/cber.19270600539
Canning, P. S. J., Maskill, H., McCrudden, K., and Sexton, B. (2002). A Product Analytical Study of the Thermal and Photolytic Decomposition of Some Arenediazonium Salts in Solution. Bcsj 75, 789–800. doi:10.1246/bcsj.75.789
Canning, P. S. J., McCrudden, K., Maskill, H., and Sexton, B. (1999). Rates and Mechanisms of the Thermal Solvolytic Decomposition of Arenediazonium Ions. J. Chem. Soc. Perkin Trans. 2 2 (12), 2735–2740. doi:10.1039/a905567c
Dai, Y., Li, C., Shen, Y., Lim, T., Xu, J., Li, Y., et al. (2018). Light-tuned Selective Photosynthesis of Azo- and Azoxy-Aromatics Using Graphitic C3N4. Nat. Commun. 9, 60–67. doi:10.1038/s41467-017-02527-8
Dong, M., Babalhavaeji, A., Samanta, S., Beharry, A. A., and Woolley, G. A. (2015). Red-Shifting Azobenzene Photoswitches for In Vivo Use. Acc. Chem. Res. 48, 2662–2670. doi:10.1021/acs.accounts.5b00270
Felpin, F.-X., and Sengupta, S. (2019). Biaryl Synthesis with Arenediazonium Salts: Cross-Coupling, CH-Arylation and Annulation Reactions. Chem. Soc. Rev. 48, 1150–1193. doi:10.1039/c8cs00453f
Fiege, H., Voges, H. W., and Paulus, W. (2000). “Phenol Derivatives,” in Ullmann’s Encyclopedia of Industrial Chemistry. Editor F. Ullmann (Weinheim, Germany: Wiley VCH), 521–582. doi:10.1002/14356007.a19_313
Gerkman, M. A., Gibson, R. S. L., Calbo, J., Shi, Y., Fuchter, M. J., and Han, G. G. D. (2020). Arylazopyrazoles for Long-Term Thermal Energy Storage and Optically Triggered Heat Release below 0 °C. J. Am. Chem. Soc. 142, 8688–8695. doi:10.1021/jacs.0c00374
Gomberg, M., and Bachmann, W. E. (1924). The Synthesis of Biaryl Compounds by Means of the Diazo Reaction. J. Am. Chem. Soc. 46, 2339–2343. doi:10.1021/ja01675a026
Griess, J. P. (1858). Vorläufige Notizüber die Einwirkung von salpetrigerSäure auf Amidinitro-und Aminitrophenylsäure. Ann. Chem. Justus Liebigs 106, 123–126. doi:10.1002/jlac.18581060114
Grirrane, A., Corma, A., and García, H. (2008). Gold-Catalyzed Synthesis of Aromatic Azo Compounds from Anilines and Nitroaromatics. Science 322, 1661–1664. doi:10.1126/science.1166401
Han, S., Cheng, Y., Liu, S., Tao, C., Wang, A., Wei, W., et al. (2021). Selective Oxidation of Anilines to Azobenzenes and Azoxybenzenes by a Molecular Mo Oxide Catalyst. Angew. Chem. Int. Ed. 60, 6382–6385. doi:10.1002/anie.202013940
Huang, Z., and Lumb, J.-P. (2019). Phenol-Directed C-H Functionalization. ACS Catal. 9, 521–555. doi:10.1021/acscatal.8b04098
Kikukawa, K., and Matsuda, T. (1977). Reaction of Diazonium Salts with Transition Metals. I. Arylation of Olefins with Arenediazonium Salts Catalyzed by Zero Valent Palladium. Chem. Lett. 6, 159–162. doi:10.1246/cl.1977.159
Kikukawa, K., Nagira, K., Terao, N., Wada, F., and Matsuda, T. (1979). Reaction of Diazonium Salts with Transition Metals. II. Palladiumcatalyzed Arylation of Ethylene with Arenediazonium Salts. Bcsj 52, 2609–2610. doi:10.1246/bcsj.52.2609
Liu, F., Wang, J.-Y., Zhou, P., Li, G., Hao, W.-J., Tu, S.-J., et al. (2017). Merging [2+2] Cycloaddition with Radical 1,4-Addition: Metal-free Access to Functionalized Cyclobuta[a ]naphthalen-4-Ols. Angew. Chem. Int. Ed. 56, 15570–15574. doi:10.1002/anie.201707615
Liu, J.-Q., Shen, X., Wang, Y., Wang, X.-S., and Bi, X. (2018). [3 + 2] Cycloaddition of Isocyanides with Aryl Diazonium Salts: Catalyst-dependent Regioselective Synthesis of 1,3- and 1,5-Disubstituted 1,2,4-Triazoles. Org. Lett. 20, 6930–6933. doi:10.1021/acs.orglett.8b03069
Liu, Z., Hao, W., Gao, W., Zhu, G., Li, X., Tong, L., et al. (2019). Silver-catalyzed Three-Component Reaction: Synthesis of N2-Substituted 1,2,3-triazoles via Direct Benzylic Amination. Sci. China Chem. 62, 1001–1006. doi:10.1007/s11426-019-9455-0
Liu, Z., Ji, H., Gao, W., Zhu, G., Tong, L., Lei, F., et al. (2017). Copper(i)-mediated Carboamination of Vinyl Azides by Aryldiazonium Salts: Synthesis of N2-Substituted 1,2,3-triazoles. Chem. Commun. 53, 6259–6262. doi:10.1039/c7cc02391j
Liu, Z., Wang, L., Yu, T., Sun, Y., Chen, H., Gao, W., et al. (2020). A Bench-Stable Low-Molecular-Weight Vinyl Azide Surrogate for a Cascade Reaction: Facile Access to Novel N-Vinyl-1,2,3-Triazoles. Org. Chem. Front. 7, 2628–2633. doi:10.1039/d0qo00714e
Lizard, G., Latruffe, N., and Vervandier-Fasseur, D. (2020). Aza- and Azo-Stilbenes: Bio-Isosteric Analogs of Resveratrol. Molecules 25, 605–618. doi:10.3390/molecules25030605
Meerwein, H., Büchner, E., and van Emster, K. (1939). Über die Einwirkung aromatischer Diazoverbindungen auf α,β-ungesättigte Carbonylverbindungen. J. Prakt. Chem. 152, 237–266. doi:10.1002/prac.19391520705
Mo, F., Dong, G., Zhang, Y., and Wang, J. (2013). Recent Applications of Arene Diazonium Salts in Organic Synthesis. Org. Biomol. Chem. 11, 1582–1593. doi:10.1039/c3ob27366k
Mo, F., Qiu, D., Zhang, Y., and Wang, J. (2018). Renaissance of Sandmeyer-type Reactions: Conversion of Aromatic C-N Bonds into C-X Bonds (X = B, Sn, P, or CF3). Acc. Chem. Res. 51, 496–506. doi:10.1021/acs.accounts.7b00566
Mohamed, A. A., Salmi, Z., Dahoumane, S. A., Mekki, A., Carbonnier, B., and Chehimi, M. M. (2015). Functionalization of Nanomaterials with Aryldiazonium Salts. Adv. Colloid Interf. Sci. 225, 16–36. doi:10.1016/j.cis.2015.07.011
Nguyen, T. H. L., Gigant, N., and Joseph, D. (2018). Advances in Direct Metal-Catalyzed Functionalization of Azobenzenes. ACS Catal. 8, 1546–1579. doi:10.1021/acscatal.7b03583
Okumura, S., Lin, C.-H., Takeda, Y., and Minakata, S. (2013). Oxidative Dimerization of (Hetero)aromatic Amines Utilizing T-BuOI Leading to (Hetero)aromatic Azo Compounds: Scope and Mechanistic Studies. J. Org. Chem. 78, 12090–12105. doi:10.1021/jo402120w
Pang, J., Gao, C., Shu, L., Hu, X., and Li, M. (2020). DFT Calculations: Bridged-Azo Working with Visible Light. Comput. Theor. Chem. 1191, 113041–113047. doi:10.1016/j.comptc.2020.113041
Ryan, M. C., Whitmire, L. D., McCann, S. D., and Stahl, S. S. (2019). Copper/TEMPO Redox Redux: Analysis of PCET Oxidation of TEMPOH by Copper(II) and the Reaction of TEMPO with Copper(I). Inorg. Chem. 58, 10194–10200. doi:10.1021/acs.inorgchem.9b01326
Sandmeyer, T. (1884). Ueber die Ersetzung der Amidgruppe durch Chlor in den aromatischen Substanzen. Ber. Dtsch. Chem. Ges. 17, 1633–1635. doi:10.1002/cber.18840170219
Sitter, J. D., and Vannucci, A. K. (2021). Photocatalytic Oxidative Coupling of Arylamines for the Synthesis of Azoaromatics and the Role of O2 in the Mechanism. J. Am. Chem. Soc. 143, 2938–2943. doi:10.1021/jacs.0c13101
Xu, H., and Zeng, X. (2010). Synthesis of Diaryl-Azo Derivatives as Potential Antifungal Agents. Bioorg. Med. Chem. Lett. 20, 4193–4195. doi:10.1016/j.bmcl.2010.05.048
Yuan, T., Tang, Q., Shan, C., Ye, X., Wang, J., Zhao, P., et al. (2021). Alkyne Trifunctionalization via Divergent Gold Catalysis: Combining π-Acid Activation, Vinyl-Gold Addition, and Redox Catalysis. J. Am. Chem. Soc. 143, 4074–4082. doi:10.1021/jacs.1c01811
Keywords: aryldiazonium salts, difunctionalization, p-azophenols, one-step, metal-free
Citation: Liu Z, Fang Y, Liu Y, Fu W, Gan X, Gao W and Tang B (2022) One-Pot Difunctionalization of Aryldiazonium Salts for Synthesis of para-Azophenols. Front. Chem. 10:818627. doi: 10.3389/fchem.2022.818627
Received: 19 November 2021; Accepted: 03 January 2022;
Published: 26 January 2022.
Edited by:
Zhendong Jin, The University of Iowa, United StatesReviewed by:
Balaraman Ekambaram, National Chemical Laboratory (CSIR), IndiaNan Zheng, University of Arkansas, United States
Copyright © 2022 Liu, Fang, Liu, Fu, Gan, Gao and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Gao, Z2Fvd2VuQHNkbnUuZWR1LmNu; Bo Tang, dGFuZ2JAc2RudS5lZHUuY24=
†These authors have contributed equally to this work