 Majid Khan1,2†
Majid Khan1,2† Satya Kumar Avula1†
Satya Kumar Avula1† Sobia Ahsan Halim1
Sobia Ahsan Halim1 Muhammad Waqas1
Muhammad Waqas1 Mufarreh Asmari3
Mufarreh Asmari3 Ajmal Khan1*
Ajmal Khan1* Ahmed Al-Harrasi1*
Ahmed Al-Harrasi1*- 1Natural and Medical Sciences Research Center, University of Nizwa, Birkat Al Mauz, Nizwa, Oman
- 2H.E.J Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan
- 3Department of Pharmaceutical Chemistry, College of Pharmacy, King Khalid University, Abha, Saudi Arabia
A series of 1H-1,2,3-triazole analogs (7a–7d and 9a–9s) were synthesized via “click” chemistry and evaluated for in vitro carbonic anhydrase-II (bovine and human) inhibitory activity. The synthesis of intermediates, 7a and 7c, was achieved by using (S)-(-)ethyl lactate as a starting material. These compounds (7a and 7c) underwent Suzuki–Miyaura cross-coupling reaction with different arylboronic acids in 1,4-dioxane, reflux at 90–120°C for 8 h using Pd(PPh3)4 as a catalyst (5 mol%), and K2CO3 (3.0 equiv)/K2PO4 (3.0 equiv) as a base to produce target 1H-1,2,3-triazole molecules (9a–9s) for a good yield of 67–86%. All the synthesized compounds were characterized through NMR spectroscopic techniques. Furthermore, all those compounds have shown significant inhibitory potential for both sources of carbonic anhydrase-II (CA-II). In the case of bCA-II, compounds 9i, 7d, 9h, 9o, 9g, and 9e showed potent activity with IC50 values in the range of 11.1–17.8 µM. Whereas for hCA-II, compounds 9i, 9c, 9o, and 9j showed great potential with IC50 values in the range of 10.9–18.5 µM. The preliminary structure–activity relationship indicates that the presence of the 1H-1,2,3-triazole moiety in those synthesized 1H-1,2,3-triazole analogs (7a–7d and 9a–9s) significantly contributes to the overall activity. However, several substitutions on this scaffold affect the activity to several folds. The selectivity index showed that compounds 9c, 9k, and 9p are selective inhibitors of hCA-II. Kinetics studies showed that these compounds inhibited both enzymes (bCA-II and hCA-II) in a competitive manner. Molecular docking indicates that all the active compounds fit well in the active site of CA-II. This study has explored the role of 1H-1,2,3-triazole-containing compounds in the inhibition of CA-II to combat CA-II-related disorders.
1 Introduction
Carbonic anhydrases (EC 4.2.1.1) are zinc metalloenzymes, which serve as therapeutic targets for numerous health issues such as glaucoma, cerebral edema, epilepsy, and various types of cancers (Nocentini and Supuran, 2019). These enzymes mainly catalyze a key physiological reaction, i.e., interconversion of carbon dioxide and bicarbonate to acutely lower the level of carbon dioxide in the blood and other cells of the body (Smith and Ferry, 2000; Khan et al., 2022a). Several other important physiological processes are also regulated by these enzymes, such as trafficking of carbon dioxide between the lungs and other body tissues; maintaining the body homeostasis; biosynthesis reactions such as gluconeogenesis, lipogenesis, bone resorption, calcification, and tumor formation; and other vital pathological or physiological reactions (Ghiasi et al., 2012; Čapkauskaitė et al., 2012; Aslan et al., 2019; Khan et al., 2020; Khan et al., 2021; Aspatwar et al., 2022; Rasool et al., 2022). So far, 16 isozymes of CA with specific functions have been discovered in mammals, such as cytosolic carbonic anhydrases (e.g., CA I-III and CAVII), membrane-bound isozymes (e.g., CA-IV, -IX, -XII, -XIV, and -XV), mitochondrial isozyme (e.g., CA-V), and secreted CA (e.g., CA-VI). Recently, two CA isozymes (viz., CA-IX and -XII) gained attention due to their association with tumors. CA-IX is one of the key enzymes which are controlled by the hypoxia-induced transcription factor (HIF-1α). This membrane-bound enzyme catalyzes the reversible hydration of carbon dioxide to bicarbonate in the extracellular space and plays an important role in the progression of tumor. CA-XII is also implicated in extracellular acidification in many types of tumor (Nocentini and Supuran. 2018). Consequently, CA-IX and -XII are driving factors for tumor growth, invasiveness, proliferation, metastasis, and resistance to radio- and chemotherapy (Supuran et al., 2018; Daunys and Petrikaitė, 2020; Rafiq et al., 2021a; Khan et al., 2022b; Rehman et al., 2022).
Inhibition of CA isoenzyme is important in the treatment of several diseases such as edema, glaucoma, obesity, cancer, epilepsy, and osteoporosis. It makes these enzymes a valuable therapeutic target in the treatment of primary tumor growth, metastasis, and the reduction of cancer stem cell population (Beyza Öztürk Sarıkaya et al., 2010; Nocentini et al., 2019; Hashmi et al., 2021; Rehman et al., 2022).
Compounds with the 1H-1,2,3-triazole moiety are crucially important in pharmaceuticals, agrochemicals, and medicinal chemistry (Abdel-Wahab et al., 2012; Avula et al., 2021a). The 1H-1,2,3-triazole group is significant in organic chemistry due to their wide range of applications in bio-medicinal, biochemical, material sciences, and pharmaceuticals (Singh et al., 2010). 1H-1,2,3-triazole-bearing molecules have undergone substantial growth over the past few decades (Thirumurugan et al., 2013). Those compounds have wide industrial applications such as dyes, photostabilizers, photographic materials, corrosion inhibitors, and agrochemicals (Green et al., 1995). Therefore, the synthesis of 1H-1,2,3-triazole derivatives could be a topic of special interest for synthetic chemists because of their biological, medicinal, and industrial applications. Due to this reason, great effort has been directed toward developing new synthetic methodologies for 1H-1,2,3-triazole moieties.
Recent literature studies (Kumar et al., 2018; Sharma et al., 2019) have reported the superior carbonic anhydrase inhibitory potential of 1H-1,2,3-triazole ring-containing heterocycles (Figure 1). In continuation of our research work on 1H-1,2,3-triazole analogues (Avula et al., 2018; Avula et al., 2019; Avula et al., 2021a; Avula et al., 2021b), we, herein, report the synthesis of a new series of novel 1H-1,2,3-triazole analogues (7a–7d and 9a–9s) via “click” chemistry and their in vitro CA-II inhibitory activities for the first time.
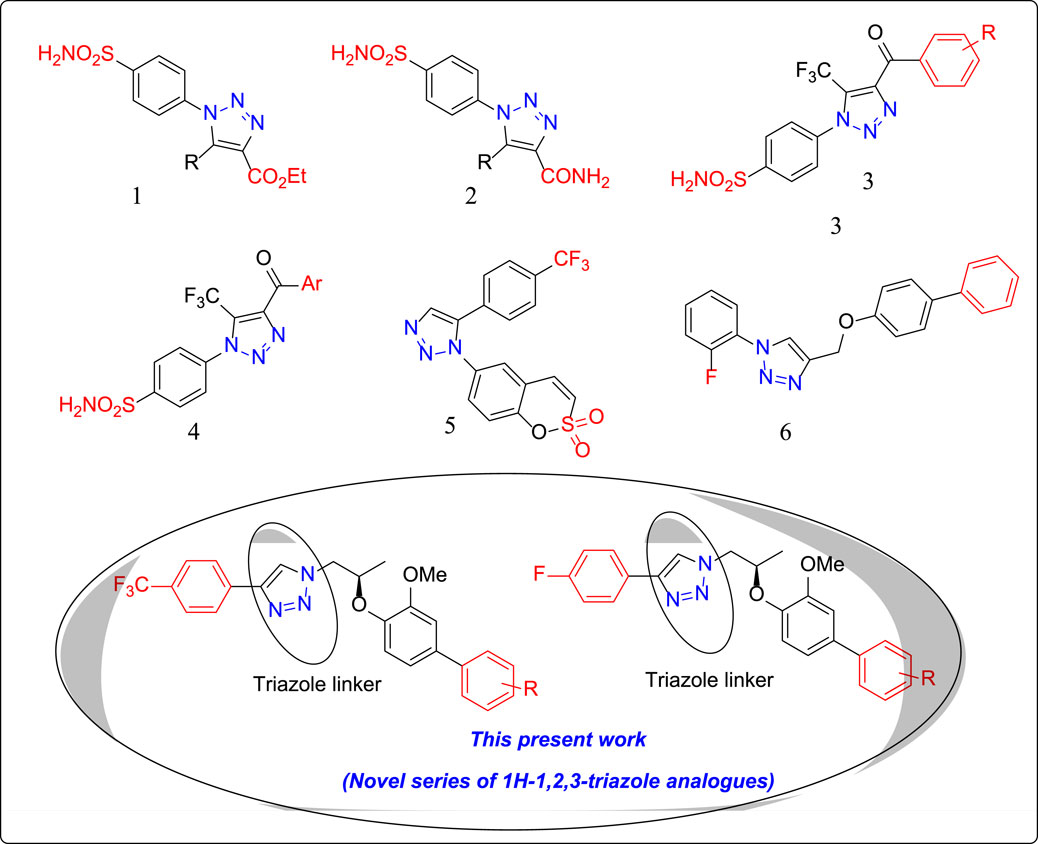
FIGURE 1. Structures of few clinically used 1H-1,2,3-triazole analogues as potential CA-II inhibitors. 1: 1H-1,2,3-triazol-1-yl) benzenesulfonamide 2: 1H-1,2,3-triazole-4-carboxamide 3: 1H-1,2,3-triazol-1-yl) benzenesulfonamide 4: 1H-1,2,3-triazol-1-yl) benzenesulfonamide 5: 1H-1,2,3-triazol-1-yl) benzo [e][1,2] oxathiine 2,2-dioxide 6: 4-{[(1,1′-biphenyl)-4-yloxy] methyl}-1-(2-fluorophenyl)-1H-1,2,3-triazole.
“Click” chemistry refers to a group of reactions that are fast, simple to use, easy to purify, versatile, region specific, and give a high product yield. Among several reactions that fulfill the criteria, Huisgen 1,3-dipolar cycloaddition of azides and terminal alkynes has emerged as a frontrunner. It has applications in a wide variety of research areas, including material sciences, polymer chemistry, and pharmaceutical sciences (Thi et al., 2016; Nabih et al., 2020; Kaur et al., 2021).
The current study aimed to thoroughly investigate the biological activities of novel synthesized compounds (7a–7d and 9a–9s) as effective CA inhibitors. Their inhibitory effect on bovine (bCA-II) and human (hCA-II) enzymes was examined, and molecular modeling was performed to predict their possible mode of binding inside the active sites of bCA and hCA.
2 Materials and methods
2.1 Chemistry
Reagents were obtained from Sigma-Aldrich, Germany. Silica gel was used for column chromatography with 100–200 mesh. All solvents were purified by following the standard procedure. Thin-layer chromatography (TLC) was performed on silica gel F254 pre-coated plates. UV-light and I2 stain were used to visualize the spots. The 1H and 13C NMR spectra were recorded on an NMR spectrometer (Bruker: 600 MHz for 1H, 150 MHz for 13C, and 564 MHz for 19F) using CDCl3 as a solvent. High-resolution electrospray ionization mass spectra (HR-ESI-MS) were recorded on the Agilent 6530 LC Q-TOF instrument. Organic extracts and solutions of pure compounds were dried over anhydrous MgSO4.
2.2 Biological enzyme inhibition
2.2.1 Carbonic anhydrase assay
CA inhibitory activity was determined according to the spectrophotometric method described in the literature (Rafiq et al., 2021b). Total assay volume comprises 140 µl of HEPES-Tris buffer, 20 µl of bCA-II solution (0.1 mg/ml HEPES-Tris buffer), 20 µl of the test compound (prepared in DMSO), and 20 µl p-NPA (0.7 mM, methanol). First, the test compound and enzyme were incubated for 15 min at 25°C; after incubation, in quick sequence, substrate was added to each experiment in 96-well plates, which were placed in the xMARK microplate spectrophotometer (Bio-Rad, United States), and the absorbance was set to 400 nm with 1 min interval time. The activity of the controlled compound was taken as 100%. All experiments were carried out thrice for each used concentration, and the results were presented as a mean of the triplicate, and % inhibition of each compound was calculated using the following formula:
2.2.2 Kinetics protocol
For mechanistic studies, both human and bovine isozymes (0.1 mg/ml/well) were incubated with different concentrations of test compounds (with respect to their determined IC50 values; two concentrations higher and two concentrations lower than their respective IC50) for 15 min at 25°C. Soon after the incubation, various concentrations of substrate were added to the reaction medium (01–0.8 mM). The enzyme activity was measured under steady-state conditions by observing changes in absorbance for 30 min at 400 nm on the microtiter plate reader (Bio-Rad X-MarkTm molecular spectrometer, United States). The results were analyzed and processed by Grafit 7 software (Erithacus Software Limited, United Kingdom). Different kinetic parameters of enzymes (e.g., Vmax, Km, Vmaxapp, and Kmapp) and values of correlation coefficients, intercepts, slopes, and their standard error mean were calculated by the linear regression graph by Grafit 7. Linear regression analysis was used to calculate the “best fit” line or curve through a dataset by minimizing the deviation of the data from the curve.
2.2.3 Docking protocol
In silico docking is widely used to explore the binding mechanism of biologically active molecules. The X-ray structures of bCA-II (PDB ID: 1V9E, resolution = 1.95 Å) (Saito et al., 2004) and hCA-II (PDB code: 1BN1, resolution = 2.1 Å) (Boriack-Sjodin et al., 1998) were downloaded from the Protein Data Bank (www.rcsb.org) for docking studies. Chain A was retained if more than one chain was present in the structure. The Molecular Operating Environment (MOE) (MOE version 2020.0901) was used in docking. Initially, protein structures were treated using the Protein Preparation Wizard of MOE by adding hydrogen atoms and making partial charges on residues with the AMBER12:EHT forcefield. Subsequently, the protein structures were subjected to short energy minimization and geometric optimization with the AMBER12:EHT forcefield with an RMSD gradient of 0.1 kcal mol−1Å−1. The structures of ligands were prepared by ChemDraw and minimized by the MOE with an RMSD gradient of 0.1 kcal∙mol−1Å−1 and MMFF94x forcefield. For docking, the Triangle Matcher placement method and London dG scoring function were applied. After docking, 30 docked possess of each compound were saved, and the best scoring docked pose was visualized.
3 Results and discussion
3.1 Synthesis of 1H-1,2,3-triazole analogues (7a–7d)
The synthetic scheme of a new series of 1H-1,2,3-triazole analogues via “click” chemistry (7a-7d) is depicted in Scheme 1. In an initial step, Mitsunobu reaction was carried out between compound 1 and 4-bromo-2-methoxyphenol in the dry THF solvent using diisopropylazodicarboxylate (DIAD), which produced desired compound 2 (86%). In the next step, compound 2 underwent reduction by using DIBAL-H from 0°C to room temperature and in dry DCM, to produce desired compound 3 (90%). In compound 3, a free –OH group was protected with p-toluenesulfonyl chloride in the presence of triethylamine (Et3N) and in dry DCM at 0°C to room temperature to give compound 4, which was treated with NaN3 in DMF at 70°C to generate desired compound 5.
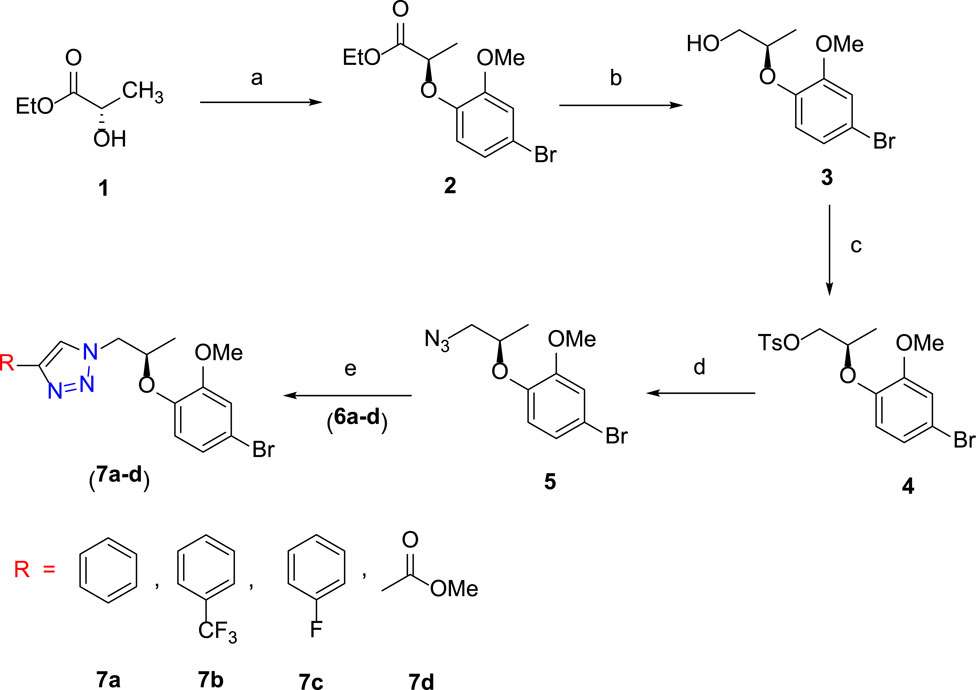
SCHEME 1. Reagents and conditions: (A) 4-bromo-2-methoxyphenol, PPh3, DIAD, dry THF, 0 °C to room temperature, 3 h, 86%; (B) DIBAL-H, dry DCM, 0°C to room temperature, 3 h, 90%; (C) TsCl, Et3N, dry DCM, DMAP, 0°C to room temperature, 5 h, 95%; (D) NaN3, DMF, 70°C, 3 h, 78%; (E) alkyne derivative (6a–6d), CuI, Et3N, MeCN, room temperature, 3 h, 7a–7d (74–80%).
The final step was carried out using “click” chemistry (Avula et al., 2018; Avula et al., 2019) where a 1,3-dipolar cycloaddition reaction occurred between alkyne derivatives 6a–6d and compound 5 in the presence of Hunig’s base and CuI in the acetonitrile solvent to produce target 1H-1,2,3-triazole analogues (7a–7d).
3.2 Synthesis of cross-coupled 1H-1,2,3-triazole analogs (9a–9s)
The synthetic scheme of cross-coupled 1H-1,2,3-triazole analogues (9a–9s) is depicted in Scheme 2. The final step was the functionalization of the aryl bromide moiety by the Suzuki–Miyaura cross-coupling reaction (Hassan et al., 2015; Hassan et al., 2017) of 7b and 7c with different arylboronic acids (8a–8o) in 1,4-dioxane solvent, refluxed at 90–120°C for 8 h to afford a novel series of 1H-1,2,3-triazole analogues (9a–9s) for a good yield (67–86%) (Table 1). The best yield was obtained in 1,4-dioxane solvent, refluxed at 90–120°C for 8 h using Pd(PPh3)4 as a catalyst (5 mol%) and K2CO3 (3.0 equiv)/K2PO4 (3.0 equiv) as a base. The structures of newly synthesized compounds (9a–9s) were confirmed by NMR spectroscopic techniques.
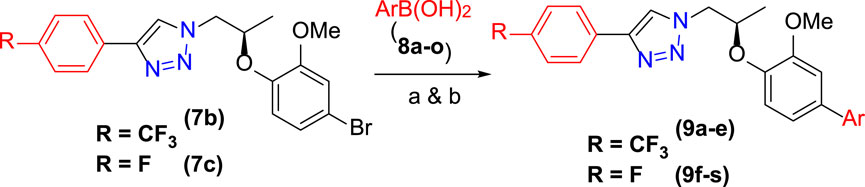
SCHEME 2. Synthesis of 9a–9s: reagents and conditions: (A) 7b (1.0 equiv), 8a–8e (1.2–1.5 equiv), Pd(PPh3)4 (5 mol%), K2PO4 (3.0 equiv), 1,4-dioxane, 90–110°C, 8 h. (B) 7c (1.0 equiv), 8f–8s (1.5 equiv), Pd(PPh3)4 (5 mol%), K2CO3 (3.0 equiv), 1,4-dioxane, 120°C, 8 h.
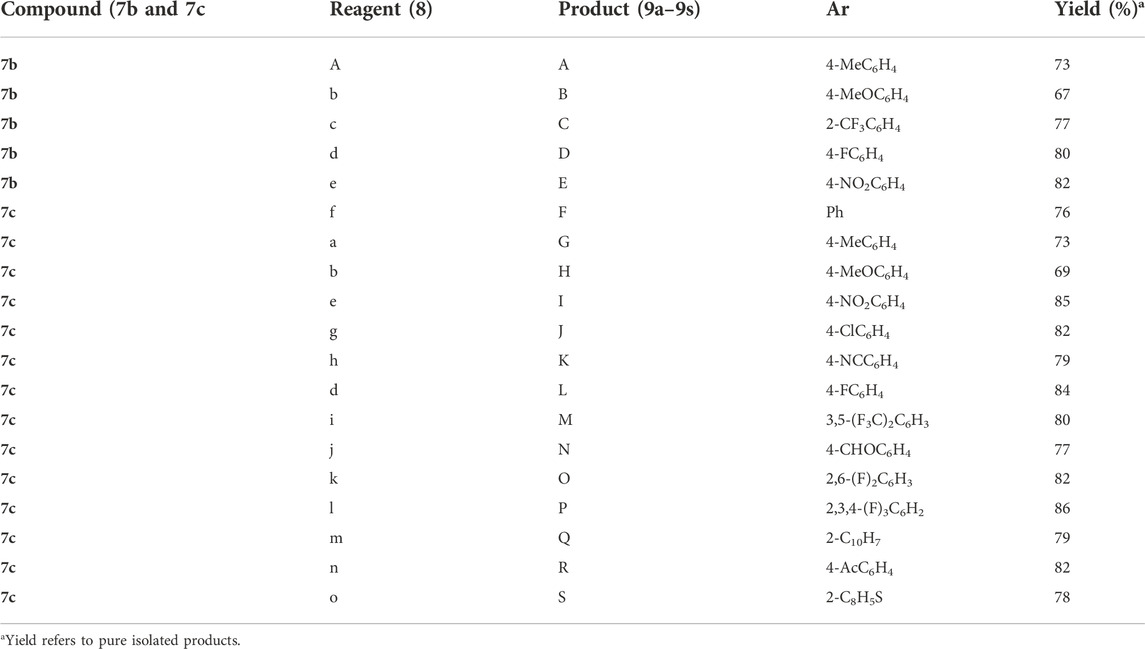
TABLE 1. Synthesis of cross-coupled 1H-1,2,3-triazole analogues (9a–9s).
3.2.1 General reaction procedure for cross-coupled 1H-1,2,3-triazole analogs (9a–9s)
1,4-dioxane (5 ml per 1 mmol) solution of 7b or 7c (1.0 equiv), K2CO3 (3.0 equiv)/K2PO4 (3.0 equiv), Pd (PPh3)4 (5 mol%), and arylboronic acid 8a–8s (1.2 or 1.5 equiv) were stirred at 90–120°C for 8 h. After cooling down to 20°C, H2O was added. The organic and the aqueous layers were separated, and the latter was extracted with CH2Cl2 (15 × 3 ml). The combined organic layer was dried over anhydrous MgSO4 and filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (EtOAc/Hexane 9:1) to give pure cross-coupled products 9a–9s.
3.3 In vitro inhibition of bCA-II by 1H-1,2,3-triazole analogues (7a–7d and 9a–9s)
The present study focused on the in vitro inhibition of bovine and human CA-II (bCA-II and hCA-II) by triazole derivatives (7a–7d, and 9a–9s) and their binding interactions with the enzyme active site. As the efficient and safer CA inhibitors have been urgently required to expand therapeutic regimes for glaucoma and epilepsy. Compounds 7a–7d and 9a–9s showed IC50 values in the range of 11.1–88.4 µM. Compound 9i (IC50 = 11.1 ± 0.2 µM) is the most potent inhibitor of bCA-II in the current series. Similarly, 7d (IC50 = 12.3 ± 0.4 µM), 9h (IC50 = 12.9 ± 1.0 µM), 9o (IC50 = 14.1 ± 0.2 µM), 9g (IC50 = 16.4 ± 0.4 µM), 9e (IC50 = 17.8 ± 0.8 µM), 7c (IC50 = 20.4 ± 0.3 µM), 7b (IC50 = 20.6 ± 0.3 µM), and 7a (IC50 = 21.4 ± 0.1 µM) showed potent activity as compared to the standard drug, acetazolamide (IC50 = 18.6 ± 0.4 µM). Whereas 9a (IC50 = 25.7 ± 0.2 µM), 9d (IC50 = 28.8 ± 0.4 µM), and 9r (IC50 = 29.0 ± 1.0 µM) are moderately active against bCA-II, and 9b (IC50 = 39.1 ± 0.3 µM), 9m (IC50 = 41.0 ± 0.4 µM), and 9f (IC50 = 88.4 ± 0.3 µM) are the least active against bCA-II (Table 2).

TABLE 2. In vitro inhibition of bCA-II and hCA-II by 7a–7d and 9a–9s.
3.3.1 Kinetics studies of most potent compounds against bCA-II and hCA-II
The mechanism of action of potent compounds was studied in a concentration-dependent manner against both the targeted enzymes. Therefore, compounds 9i and 9o were selected for kinetics studies (Table 3). These compounds (9i and 9o) showed competitive inhibition of both enzymes. 9i and 9o inhibited bCA-II with a dissociation constant (Ki) of 9.4 ± 0.3 and 11.3 ± 0.17 µM, respectively. Similarly, for hCA-II, the dissociation constant (Ki) of 9i and 9o is 8.1 ± 0.04, and 6.5 ± 0.19 µM, respectively. The Lineweaver–Burk plot of 9o for hCA-II and bCA-II is, respectively, presented in Figure 2 and Figure 3.
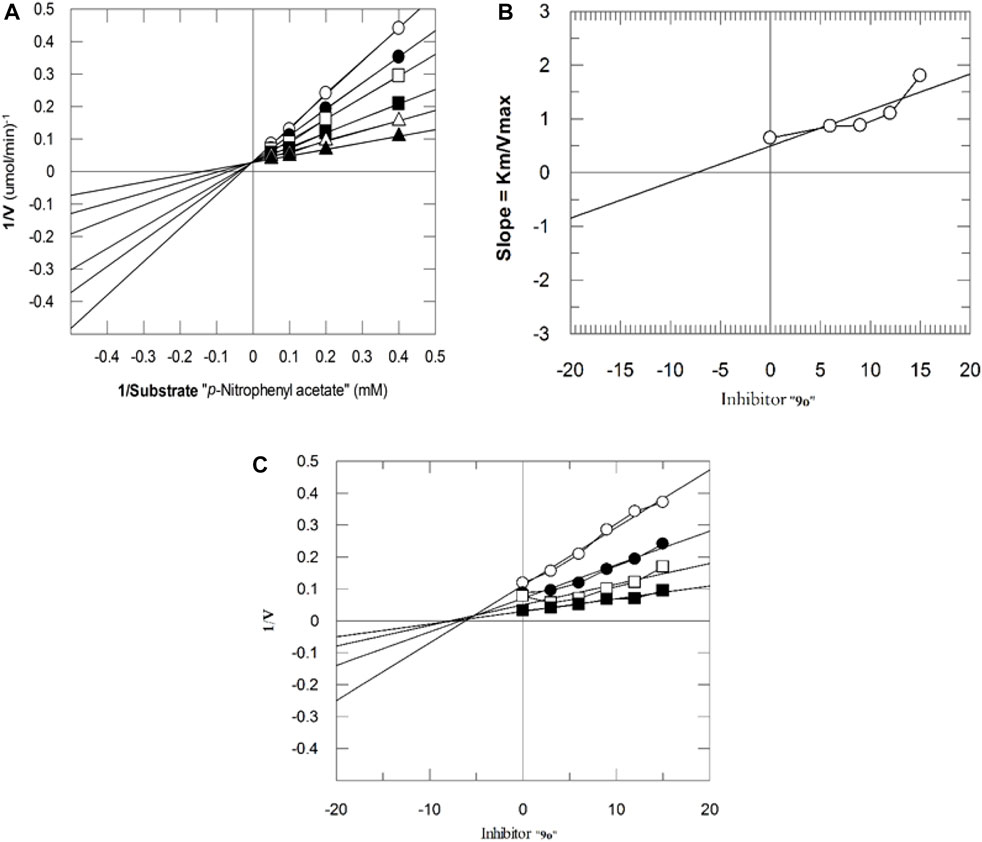
FIGURE 2. Inhibition of hCA-II by 9o. (A) Lineweaver–Burk plot of the reciprocal of the rate of reaction (velocities) vs. reciprocal of the substrate (p-nitrophenyl acetate) in the absence (▲), 6 µM (∆), 9 µM (■), 12 µM (□), 15 µM (●), and 18 µM (○) of 9o. (B) Secondary replot of the Lineweaver–Burk plot between the slopes of each line on the Lineweaver–Burk plot vs. different concentrations of 9o. (C) Dixon plot of the reciprocal of the rate of reaction (velocities) vs. different concentrations of 9o.
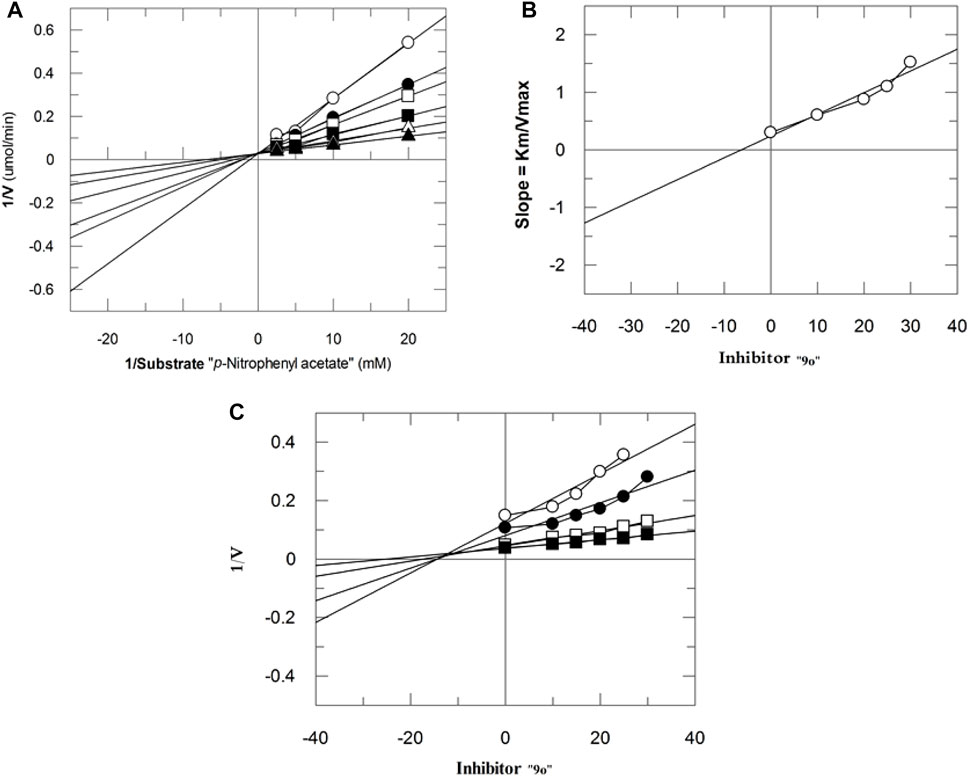
FIGURE 3. Inhibition of bCA-II by 9o. (A) Lineweaver–Burk plot of the reciprocal of the rate of reaction (velocities) vs. reciprocal of the substrate (p-nitrophenyl acetate) in the absence (▲), 7 µM (∆), 10 µM (■), 13 µM (□), 16 µM (●), and 19 µM (○) of 9o. (B) Secondary replot of Lineweaver–Burk plot between the slopes of each line on the Lineweaver–Burk plot vs. different concentrations of 9o. (C) Dixon plot of the reciprocal of the rate of reaction (velocities) vs. different concentrations of 9o.
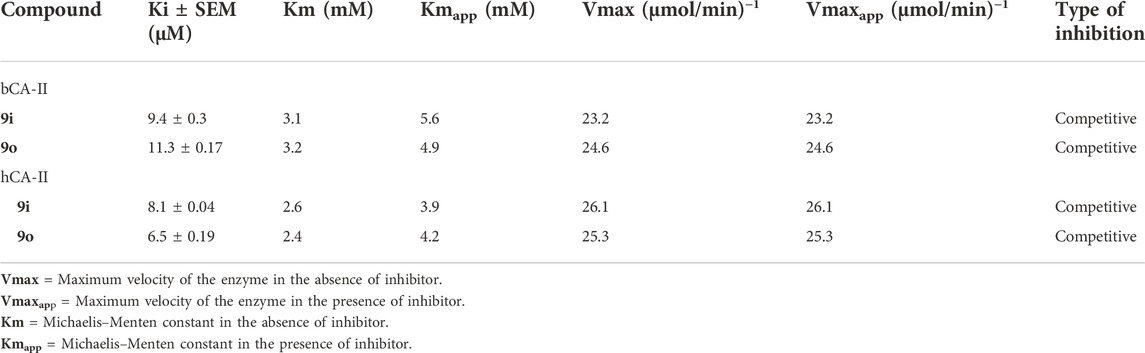
TABLE 3. Kinetic results of compounds 9i and 9o for enzymes bCA-II and hCA-II.
3.3.2 Analysis of binding interactions and the structure–activity relationship (SAR) against bCA-II
All the active compounds were docked at the active site of bCA-II (Figure 4), where compounds showed significant binding interactions. An interesting correlation was obtained from the detailed analysis. The compounds bearing nitro oxygen, benzaldehyde, and thiophenone groups significantly interacted with the metallo center, “Zn” ion, in the active site. Whereas other derivatives showed contact with active site residues through their triazole moiety. The most active compound 9i was chelated with a Zn ion in the active site via its nitro oxygen; in addition, 9i mediated hydrogen bonds (H-bond) with two water molecules (WAT462 and WAT435) and side chain of Gln91. The bond distances are given in Table 4. Similarly, 7d interacted with the Zn atom, a water molecule (WAT488), and with the side chain of Gln91. However, compounds 9h, 9o, and 9g lost interactions with Zn, while retained their interactions with the active site residues. Compound 9h mediated H-bonds with the side chain of Asn66, Gln91, and Thr199, and two water molecules (WAT279 and WAT435). Whereas 9o formed a halogen bond with the side chain of His93 through its fluorine atom and mediated H-bonds with the side chains of Thr198 and Gln91, and WAT435. Similarly, 9g also formed H-bonds with the side chain Gln91, WAT279 and WAT493; furthermore, 9g was stabilized by the π–π stacking interaction by the phenyl ring of Phe129. Compound 9e was chelated with the Zn ion through its nitro oxygen and formed H-bonds with the side chain of Gln91 and WAT279. The binding mode of 7c shows that the compound mediated H-bonds with the side chains of Thr198 and Gln91, and WAT279. In contrast, 7b interacted with the Zn atom through its acetophenone-substituted carbonyl moiety and made H-bonds with the side chain of Thr198 and WAT466. The docked orientation of 7a revealed that 7a interacted with the side chains of Gln91 and Asn66 and WAT466 through H-bonds. Compound 9r showed contact with the side chain of Gln91 and WAT310 (2.32 and 2.27 Å), respectively.
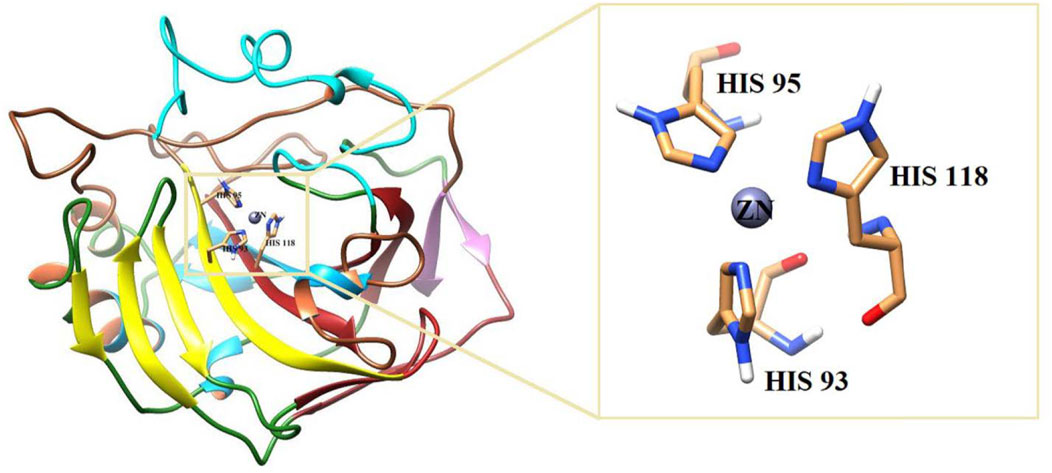
FIGURE 4. 3D-structure of bCA is shown. The active site is highlighted in the box.
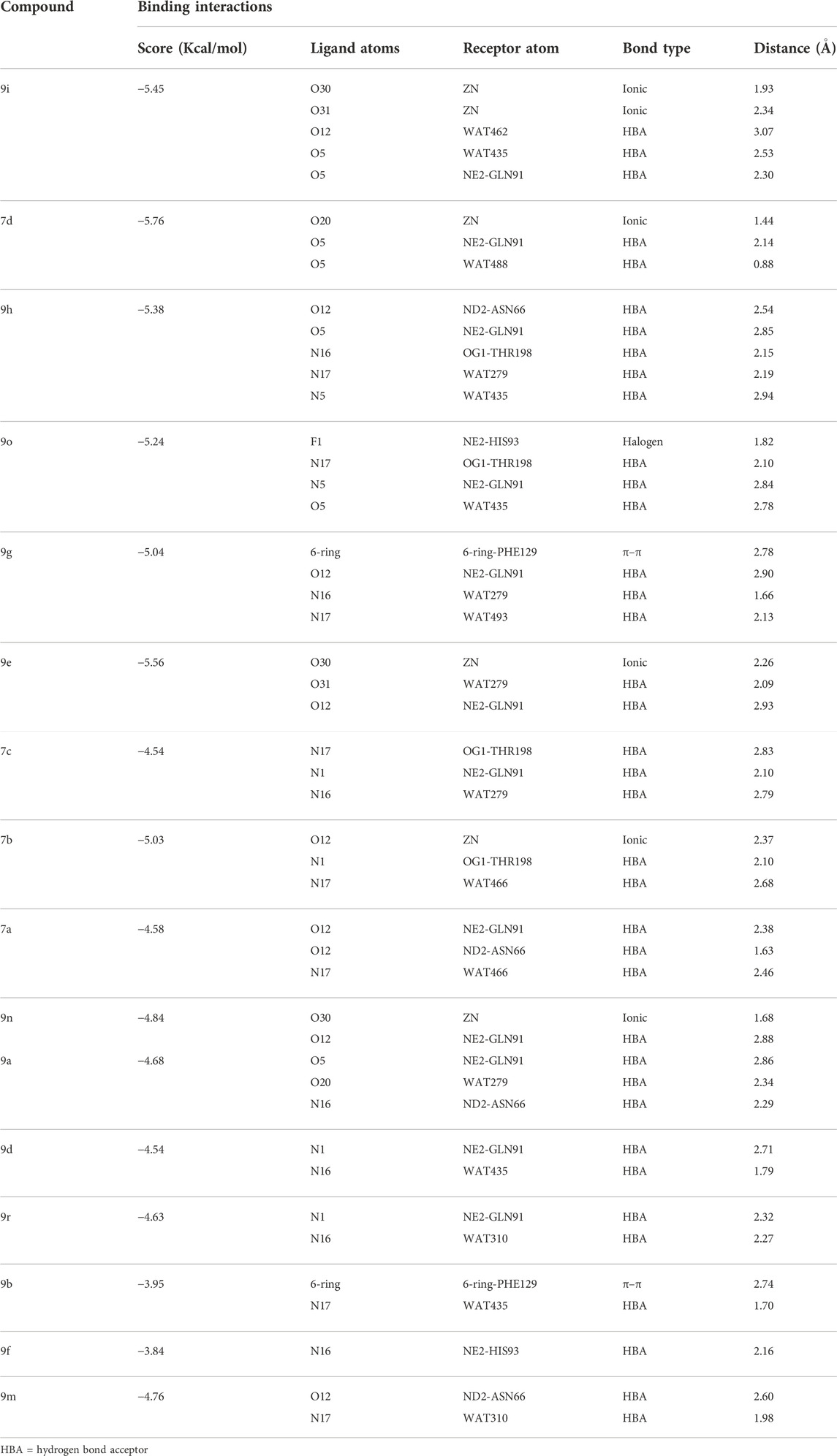
TABLE 4. Binding analysis of active 1H-1,2,3-triazole analogues in the active site of bCA-II.
The binding modes of moderate active compounds 9a, 9d, and 9r demonstrated that all those molecules mediated H-bonds with the side chain of Gln91; however, 9a also interacted with the side chain of Asn66 and WAT279, whereas 9d and 9r formed H-bonds with WAT435 and WAT310. On the other hand, the least active compounds, 9b, 9f, and 9m formed less H-bonds and hydrophobic interactions within the active site. The phenyl ring of Phe129 provided π–π stacking interactions to 9b which also formed an H-bond with WAT435. Similarly, 9f formed an H-bond with the side chain of His93, and 9m made H-bonds with the side chain of Asn66 and WAT310. The detailed binding interactions of the active compounds are tabulated in Table 4, and the binding modes are depicted in Figure 5.
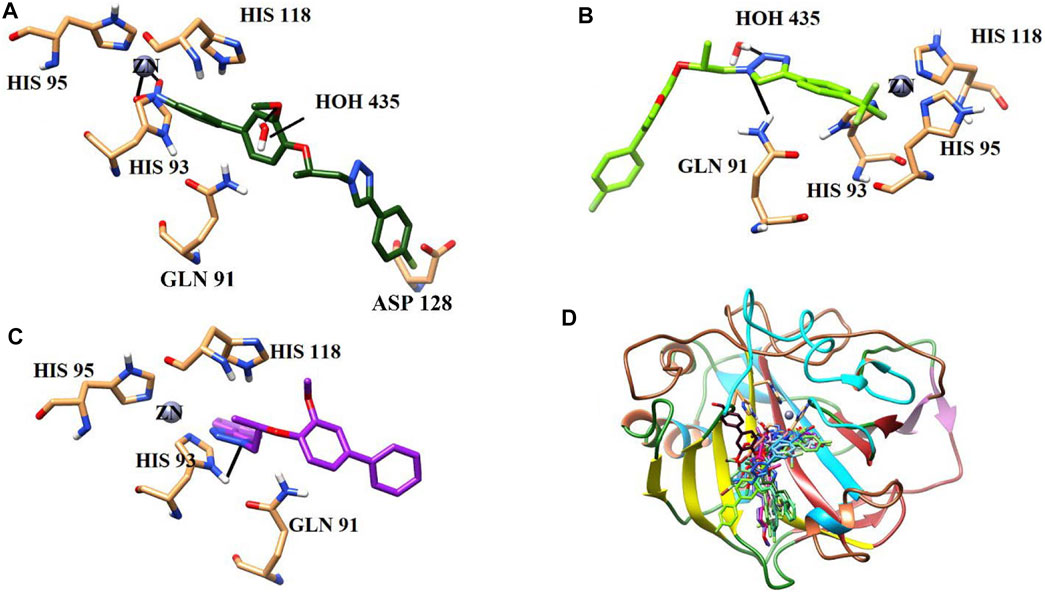
FIGURE 5. Binding orientations of (A) most active compound 9i (dark green stick model), (B) moderate active 9d (light green stick model), (C) least active 9f (purple stick model), and (D) all the active compounds are shown in the active site of bCA. The active site residues are shown in coral sticks, and H-bonds are depicted in black lines.
3.4 In vitro inhibition of hCA by 7a–7d and 9a–9s
In the in vitro testing, compound 9i (IC50 = 10.9 ± 1.0 µM) was retrieved as the most potent inhibitor of hCA-II, followed by 9c (IC50 = 12.5 ± 0.6 µM), 9o (IC50 = 13.6 ± 1.0 µM), 9j (IC50 = 18.5 ± 0.4 µM), 9p (IC50 = 19.3 ± 0.2 µM), 9a (IC50 = 21.0 ± 1.0 µM), 9k (IC50 = 21.3 ± 0.8 µM), 9b (IC50 = 22.4 ± 0.4 µM), 7d (IC50 = 25.1 ± 0.4 µM), and 9n (IC50 = 23.0 ± 0.3 µM). Whereas compounds 7c (IC50 = 22.8 ± 0.2 µM) and 9m (IC50 = 27.4 ± 0.9 µM) demonstrated moderate inhibition, while 7a (IC50 = 35.3 ± 0.6 µM) and 7b (IC50 = 45.1 ± 0.5 µM) showed weak inhibitory activity against hCA as compared to acetazolamide (IC50 = 19.5 ± 1.0 µM). The results are given in Table 2.
3.4.1 Analysis of binding interactions and the structure–activity relationship (SAR) against hCA-II
The current library of compounds was also evaluated against hCA-II to understand the specificity of these compounds. Among all the compounds, 9i (IC50 = 10.9 ± 1.10 µM) showed the highest inhibitory potential for hCA-II. The binding analysis of 9i revealed that it mediates significant interaction with a Zn ion and active site residues. The nitro group of 9i formed an ionic interaction with the Zn atom and bidentate interactions with the side chain of Asn62. Whereas compounds 9c and 9o interacted with the side chains of Asn62, Gln92, Thr199, His96, and Thr200. 9c mediated H-bonds with the side chains of Asn62 and Gln92 and further formed a halogen bond with the side chain of Thr199. Similarly, 9o formed H-bonds with the side chains of His96 and Thr200 through its methoxybenzene and triazole moieties, respectively. The triazole nitrogen of 9j formed an ionic bond with Zn and was further stabilized by the side chain of Gln92 through H-bonds; in addition, this compound formed a halogen bond with the side chain of Thr199. Compounds 9p, 9a, 9k, and 9b were stabilized by the side chains of Thr200, Asn62, Gln92, Thr199, and His96. The methoxyethane moiety of 9p mediated a H-bond with a side chain of Thr200 and a π–cation interaction with the side chain of Thr199. Whereas 9a mediated multiple H-bonds with the side chains of Asn62 and Gln92 and a halogen bond with the side chain of Thr199 through its fluoro group. Similarly, 9k displayed a halogen bond with the side chain of His96 and stabilized by the side chains of Thr199 and Thr200 through H-bonds. Similarly, compound 9b was stabilized by the side chains of Asn62 and His64 by H-bonds and mediated a π–cation interaction with the side chain of Thr199. Compounds 7d and 9n formed ionic interactions with Zn ions and H-bonds with the side chains of Thr199, Gln92, and Thr200. Additionally, 9n also demonstrated a π–cation bond with the side chain of Gln92.
Compounds 7c and 9m exhibited moderate inhibition of hCA. The docked view of 7c depicted that it was stabilized by the side chains of Gln92, Thr200, and Asn62 through H-bonds; moreover, the bromo group of 7c mediated a halogen bond with the side chain of His64. Similarly, 9m formed a halogen bond with Thr199, a H-bond with the side chain of Gln91, and a hydrophobic interaction with the phenyl ring of Phe131. Although 7a and 7b exhibited weak inhibition of hCA, those compounds displayed good interactions with the side chains of Thr200 and Gln92 via their triazole moieties. Additionally, 7b also formed a halogen bond with Thr200. The binding interactions of compounds (Figure 6) and their docking scores are given in Table 5.
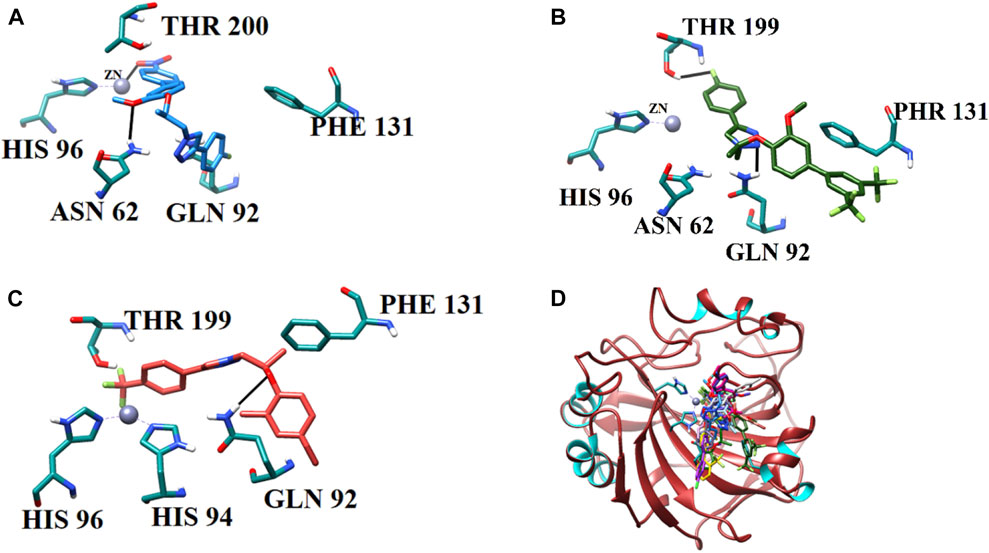
FIGURE 6. Binding orientations of (A) most active compound 9i (sky blue stick model), (B) moderate active 9m (green stick model), (C) least active 7b (salmon stick model), and (D) all the active compounds (stick models) are shown in the active site of hCA-II. The active site residues are shown in cyan sticks, and H-bonds are depicted in black lines.
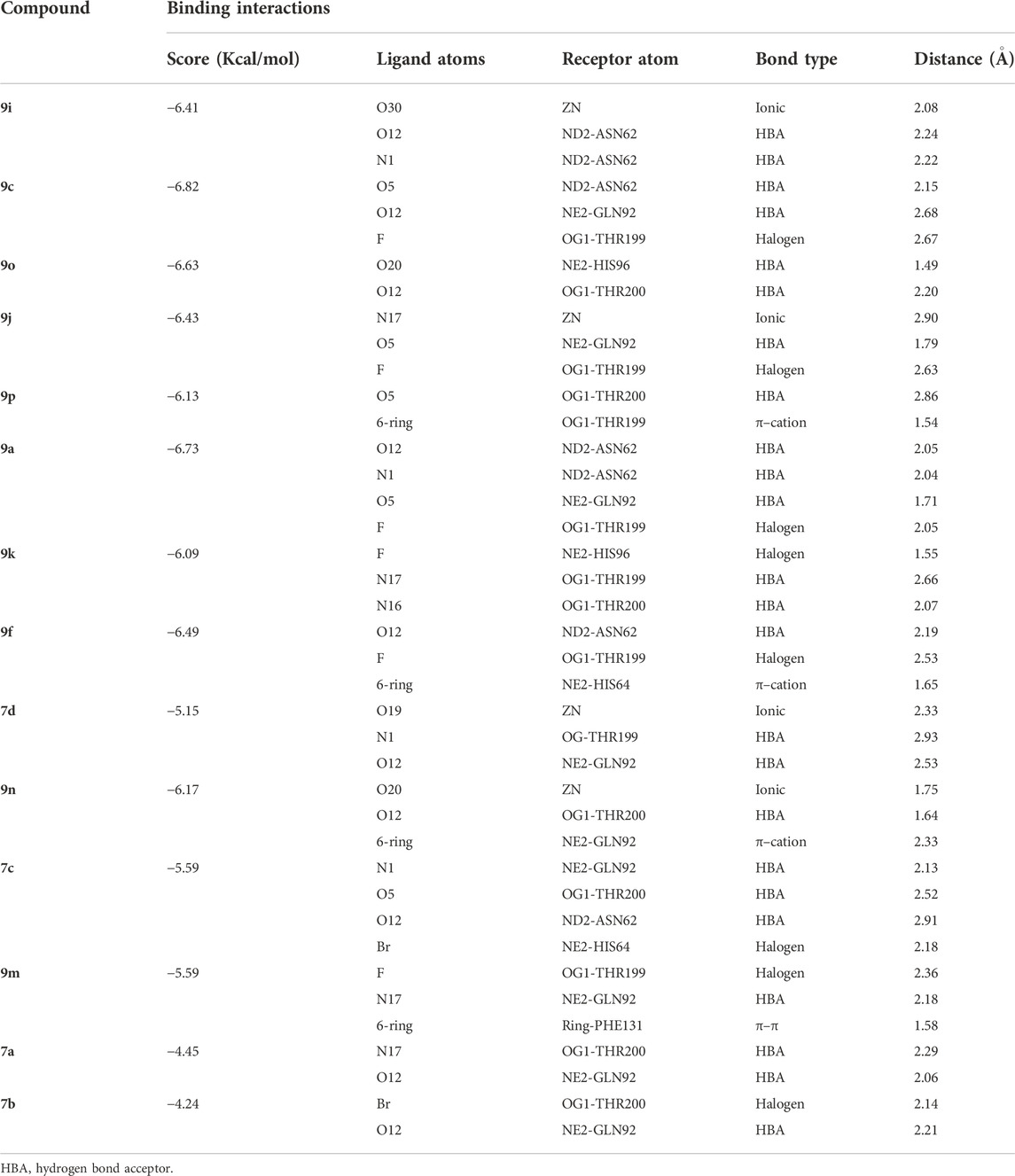
TABLE 5. Binding analysis of 1H-1,2,3-triazole analogues in the active site of hCA-II.
3.5 Selectivity of 1H-1,2,3 triazole analogs against CA-II enzymes
As per selectivity of the evaluated analogues, it was found that compounds 9c, 9k, and 9p are selective inhibitors of the hCA-II, whereas compounds 9d–9h and 9r are selective inhibitors of bCA-II.
4 Conclusion
A series of novel 1H-1,2,3-triazole analogues were synthesized (7a–7d, and 9a–9s) and evaluated for their carbonic anhydrase-II inhibitory activity in vitro. (S)-(-)ethyl lactate was used as the starting material to introduce the chirality of the target molecules. The triazole moiety was prepared via “click” chemistry and the aryl derivatives through the Suzuki–Miyaura cross-coupling reaction. The compounds were scrutinized for their possibility to inhibit bovine and human carbonic anhydrase-II (CA-II) for the first time. All the compounds demonstrated moderate inhibitory potential against CA-II. Herein, compound 9i was identified as a common inhibitor of both bovine and human CA-II. Furthermore, in silico docking analyses revealed that all the active compounds are well-accommodated in the active site of the CA-II enzyme.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author contributions
AA-H and AK conceived and designed the study. SA synthesized all the compounds. AK and MK performed the in vitro assay. MW and SH performed the computational studies and analyzed the data. AA-H and AK wrote the manuscript with inputs and comments from all co-authors. All authors have read and approved the final version of the manuscript.
Funding
The project was funded by The Research Council (TRC), Oman through the funded project (BFP/RGP/CBS/21/006).
Acknowledgments
The authors would like to thank University of Nizwa and The Research Council (TRC), Oman, for their generous support for this project. We thank technical staff for assistance. The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Small Groups under grant number (RGP.1/200/43).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdel-Wahab, B. F., Abdel-Latif, E., Mohamed, H. A., and Awad, G. E. (2012). Design and synthesis of new 4-pyrazolin-3-yl-1, 2, 3-triazoles and 1, 2, 3-triazol-4-yl-pyrazolin-1-ylthiazoles as potential antimicrobial agents. Eur. J. Med. Chem. 52, 263–268. doi:10.1016/j.ejmech.2012.03.023
Aslan, H. E., Demir, Y., Özaslan, M. S., Türkan, F., Beydemir, Ş., and Küfrevioğlu, Ö. I. (2019). The behavior of some chalcones on acetylcholinesterase and carbonic anhydrase activity. Drug Chem. Toxicol. 42 (6), 634–640. doi:10.1080/01480545.2018.1463242
Aspatwar, A., Tolvanen, M. E., Barker, H., Syrjänen, L., Valanne, S., Purmonen, S., et al. (2022). Carbonic anhydrases in metazoan model organisms: Molecules, mechanisms, and physiology. Physiol. Rev. 102 (3), 1327–1383. doi:10.1152/physrev.00018.2021
Avula, S. K., Khan, A., Halim, S. A., Al-Abri, Z., Anwar, M. U., Al-Rawahi, A., et al. (2019). Synthesis of novel (R)-4-fluorophenyl-1H-1, 2, 3-triazoles: A new class of α-glucosidase inhibitors. Bioorg. Chem. 91, 103182. doi:10.1016/j.bioorg.2019.103182
Avula, S. K., Khan, A., Rehman, N. U., Anwar, M. U., Al-Abri, Z., Wadood, A., et al. (2018). Synthesis of 1H-1, 2, 3-triazole derivatives as new α-glucosidase inhibitors and their molecular docking studies. Bioorg. Chem. 81, 98–106. doi:10.1016/j.bioorg.2018.08.008
Avula, S. K., Khan, M., Halim, S. A., Khan, A., Al-Riyami, S. A., Csuk, R., et al. (2021a). Synthesis of new 1H-1, 2, 3-triazole analogs in aqueous medium via “click” chemistry: A novel class of potential carbonic anhydrase-II inhibitors. Front. Chem. 9, 642614. doi:10.3389/fchem.2021.642614
Avula, S. K., Rehman, N. U., Khan, M., Halim, S. A., Khan, A., Rafiq, K., et al. (2021b). New synthetic 1H-1, 2, 3-triazole derivatives of 3-O-acetyl-β-boswellic acid and 3-O-acetyl-11-keto-β-boswellic acid from Boswellia sacra inhibit carbonic anhydrase II in vitro. Med. Chem. Res. 30 (6), 1185–1198. doi:10.1007/s00044-021-02723-8
Beyza Öztürk Sarıkaya, S., Gülçin, İ., Supuran, C. T., and design, d. (2010). Carbonic anhydrase inhibitors: Inhibition of human erythrocyte isozymes I and II with a series of phenolic acids. Chem. Biol. Drug Des. 75 (5), 515–520. doi:10.1111/j.1747-0285.2010.00965.x
Boriack-Sjodin, P. A., Zeitlin, S., Christianson, D. W., Chen, H. H., Crenshaw, L., Gross, S., et al. (1998). Structural analysis of inhibitor binding to human carbonic anhydrase II. Protein Sci. 7 (12), 2483–2489. doi:10.1002/pro.5560071201
Čapkauskaitė, E., Zubrienė, A., Baranauskienė, L., Tamulaitienė, G., Manakova, E., Kairys, V., et al. (2012). Design of [(2-pyrimidinylthio) acetyl] benzenesulfonamides as inhibitors of human carbonic anhydrases. Eur. J. Med. Chem. 51, 259–270. doi:10.1016/j.ejmech.2012.02.050
Daunys, S., and Petrikaitė, V. (2020). The roles of carbonic anhydrases IX and XII in cancer cell adhesion, migration, invasion and metastasis. Biol. Cell 112 (12), 383–397. doi:10.1111/boc.201900099
Ghiasi, M., Kamalinahad, S., Arabieh, M., and Zahedi, M. (2012). Carbonic anhydrase inhibitors: A quantum mechanical study of interaction between some antiepileptic drugs with active center of carbonic anhydrase enzyme. Comput. Theor. Chem. 992, 59–69. doi:10.1016/j.comptc.2012.05.005
Green, G., Evans, J., Vong, A., Katritzky, A., Rees, C., and Scriven, E. (1995). Comprehensive heterocyclic chemistry II. Oxford, New York: Pergamon Press.
Hashmi, S., Khan, S., Shafiq, Z., Taslimi, P., Ishaq, M., Sadeghian, N., et al. (2021). Probing 4-(diethylamino)-salicylaldehyde-based thiosemicarbazones as multi-target directed ligands against cholinesterases, carbonic anhydrases and α-glycosidase enzymes. Bioorg. Chem. 107, 104554. doi:10.1016/j.bioorg.2020.104554
Hassan, Z., Al-Harrasi, A., Rizvi, T., Hussain, J., and Langer, P. (2017). Selective synthesis, characterization of isomerically pure arylated benzo [1, 2-b: 6, 5-b′] dithiophenes by regioselective suzuki–miyaura reaction and evaluation of the catalytic properties of nickel versus palladium complexes. Synthesis 49 (03), 557–564. doi:10.1055/s-0035-1562538
Hassan, Z., Al-Shidhani, S., Al-Ghafri, A., Al-Harrasi, A., Hussain, J., and Csuk, R. (2015). Palladium-catalyzed chemo-and regioselective cross-coupling reactions of 2, 3-dichloronaphthalene-1, 4-bistriflate. Tetrahedron Lett. 56 (52), 7141–7144. doi:10.1016/j.tetlet.2015.11.013
Kaur, J., Saxena, M., and Rishi, N. (2021). An overview of recent advances in biomedical applications of click chemistry. Bioconjug. Chem. 32 (8), 1455–1471. doi:10.1021/acs.bioconjchem.1c00247
Khan, A., Khan, M., Halim, S. A., Khan, Z. A., Shafiq, Z., and Al-Harrasi, A. (2020). Quinazolinones as competitive inhibitors of carbonic anhydrase-II (human and bovine): Synthesis, in-vitro, in-silico, selectivity, and kinetics studies. Front. Chem. 8, 598095. doi:10.3389/fchem.2020.598095
Khan, I., Khan, A., Halim, S. A., Khan, M., Zaib, S., Al-Yahyaei, B. E. M., et al. (2021). Utilization of the common functional groups in bioactive molecules: Exploring dual inhibitory potential and computational analysis of keto esters against α-glucosidase and carbonic anhydrase-II enzymes. Int. J. Biol. Macromol. 167, 233–244. doi:10.1016/j.ijbiomac.2020.11.170
Khan, M., Halim, S. A., Shafiq, Z., Islam, M., Shehzad, M. T., Ibrar, A., et al. (2022a). Inhibitory efficacy of thiosemicarbazones for carbonic Anhydrase II (bovine and human) as a target of calcification and tumorigenicity. Curr. Pharm. Des. 28 (36), 3010–3022. doi:10.2174/1381612828666220729105849
Khan, M., Shah, S. R., Khan, F., Halim, S. A., Rahman, S. M., Khalid, M., et al. (2022b). Efficient synthesis with green chemistry approach of novel pharmacophores of imidazole-based hybrids for tumor treatment: Mechanistic insights from in situ to in silico. Cancers 14 (20), 5079. doi:10.3390/cancers14205079
Kumar, R., Vats, L., Bua, S., Supuran, C. T., and Sharma, P. K. (2018). Design and synthesis of novel benzenesulfonamide containing 1, 2, 3-triazoles as potent human carbonic anhydrase isoforms I, II, IV and IX inhibitors. Eur. J. Med. Chem. 155, 545–551. doi:10.1016/j.ejmech.2018.06.021
Molecular Operating Environment (MOE) (2020). Chemical computing group ULC, 1010 sherbooke st. West, suite 910, Montreal, QC.
Nabih, K., Maatallah, M., Baouid, A., and Jarid, A. (2020). Theoretical analysis of the mechanism of the 1, 3-dipolar cycloaddition of benzodiazepine with N-aryl-C-ethoxycarbonylnitrilimine. J. Chem. 2020, 1–8. doi:10.1155/2020/8695405
Nocentini, A., Lucidi, A., Perut, F., Massa, A., Tomaselli, D., Gratteri, P., et al. (2019). Α, γ-diketocarboxylic acids and their esters act as carbonic anhydrase IX and XII selective inhibitors. ACS Med. Chem. Lett. 10 (4), 661–665. doi:10.1021/acsmedchemlett.9b00023
Nocentini, A., and Supuran, C. T. (2019). Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discov. 14 (11), 1175–1197. doi:10.1080/17460441.2019.1651289
Nocentini, A., and Supuran, C. T. (2018). Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: A patent review (2008–2018). Expert Opin. Ther. Pat. 28 (10), 729–740. doi:10.1080/13543776.2018.1508453
Rafiq, K., Khan, A., Ur Rehman, N., Halim, S. A., Khan, M., Ali, L., et al. (2021a). New carbonic anhydrase-II inhibitors from marine macro Brown alga dictyopteris hoytii supported by in silico studies. Molecules 26 (23), 7074. doi:10.3390/molecules26237074
Rafiq, K., Khan, M., Muhammed, N., Khan, A., Ur Rehman, N., Al-Yahyaei, B. E. M., et al. (2021b). New amino acid clubbed schiff bases inhibit carbonic anhydrase II, α-glucosidase, and urease enzymes: In silico and in vitro. Med. Chem. Res. 30 (3), 712–728. doi:10.1007/s00044-020-02696-0
Rasool, A., Batool, Z., Khan, M., Halim, S. A., Shafiq, Z., Temirak, A., et al. (2022). Bis-pharmacophore of cinnamaldehyde-clubbed thiosemicarbazones as potent carbonic anhydrase-II inhibitors. Sci. Rep. 12 (1), 16095–16111. doi:10.1038/s41598-022-19975-y
Rehman, N. U., Halim, S. A., Khan, A., Khan, M., Al-Hatmi, S., and Al-Harrasi, A. (2022). Commikuanoids AC: New cycloartane triterpenoids with exploration of carbonic anhydrase-II inhibition from the resins of Commiphora kua by in vitro and in silico molecular docking. Fitoterapia 157, 105125. doi:10.1016/j.fitote.2022.105125
Saito, R., Sato, T., Ikai, A., and Tanaka, N. (2004). Structure of bovine carbonic anhydrase II at 1.95 Å resolution. Acta Crystallogr. D. Biol. Crystallogr. 60 (4), 792–795. doi:10.1107/s0907444904003166
Sharma, V., Kumar, R., Bua, S., Supuran, C. T., and Sharma, P. K. (2019). Synthesis of novel benzenesulfonamide bearing 1, 2, 3-triazole linked hydroxy-trifluoromethylpyrazolines and hydrazones as selective carbonic anhydrase isoforms IX and XII inhibitors. Bioorg. Chem. 85, 198–208. doi:10.1016/j.bioorg.2019.01.002
Singh, N., Pandey, S., and Tripathi, R. P. (2010). Regioselective [3+ 2] cycloaddition of chalcones with a sugar azide: Easy access to 1-(5-deoxy-D-xylofuranos-5-yl)-4, 5-disubstituted-1H-1, 2, 3-triazoles. Carbohydr. Res. 345 (12), 1641–1648. doi:10.1016/j.carres.2010.04.019
Smith, K. S., and Ferry, J. G. (2000). Prokaryotic carbonic anhydrases. FEMS Microbiol. Rev. 24 (4), 335–366. doi:10.1111/j.1574-6976.2000.tb00546.x
Supuran, C. T., Alterio, V., Di Fiore, A., D’Ambrosio, K., Carta, F., Monti, S. M., et al. (2018). Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med. Res. Rev. 38 (6), 1799–1836. doi:10.1002/med.21497
Thi, T. P., Le Nhat, T. G., Hanh, T. N., Quang, T. L., The, C. P., Thi, T. A. D., et al. (2016). Synthesis and cytotoxic evaluation of novel indenoisoquinoline-substituted triazole hybrids. Bioorg. Med. Chem. Lett. 26 (15), 3652–3657. doi:10.1016/j.bmcl.2016.05.092
Keywords: synthesis, Suzuki–Miyaura cross-coupling, “click” chemistry, 1H-1,2,3-triazole analogs, carbonic anhydrase-II inhibitory activity, molecular docking studies
Citation: Khan M, Avula SK, Halim SA, Waqas M, Asmari M, Khan A and Al-Harrasi A (2022) Biochemical and in silico inhibition of bovine and human carbonic anhydrase-II by 1H-1,2,3-triazole analogs. Front. Chem. 10:1072337. doi: 10.3389/fchem.2022.1072337
Received: 17 October 2022; Accepted: 09 November 2022;
Published: 25 November 2022.
Edited by:
Mahmood Ahmed, University of Education Lahore, PakistanReviewed by:
Parham Taslimi, Bartin University, TurkeyYilei Xiao, Liaocheng People’s Hospital, China
Copyright © 2022 Khan, Avula, Halim, Waqas, Asmari, Khan and Al-Harrasi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ajmal Khan, YWptYWxraGFuQHVuaXp3YS5lZHUub20=; Ahmed Al-Harrasi, YWhhcnJhc2lAdW5pendhLmVkdS5vbQ==
†These authors have contributed equally to this work