 Yulia H. Budnikova
Yulia H. Budnikova Egor L. Dolengovsky
Egor L. Dolengovsky Maxim V. Tarasov
Maxim V. Tarasov Tatyana V. Gryaznova
Tatyana V. Gryaznova- 1FRC Kazan Scientific Center of RAS, Arbuzov Institute of Organic and Physical Chemistry, Kazan, Russia
- 2Organic Chemistry Department, Kazan National Research Technological University, Kazan, Russia
The activation of C–H bond, and its direct one-step functionalization, is one of the key synthetic methodologies that provides direct access to a variety of practically significant compounds. Particular attention is focused on modifications obtained at the final stages of the synthesis of complicated molecules, which requires high tolerance to the presence of existing functional groups. Phosphorus is an indispensable element of life, and phosphorus chemistry is now experiencing a renaissance due to new emerging applications in medicinal chemistry, materials chemistry (polymers, flame retardants, organic electronics, and photonics), agricultural chemistry (herbicides, insecticides), catalysis (ligands) and other important areas of science and technology. In this regard, the search for new, more selective, low-waste synthetic routes become relevant. In this context, electrosynthesis has proven to be an eco-efficient and convenient approach in many respects, where the reagents are replaced by electrodes, where the reactants are replaced by electrodes, and the applied potential the applied potential determines their “oxidizing or reducing ability”. An electrochemical approach to such processes is being developed rapidly and demonstrates some advantages over traditional classical methods of C-H phosphorylation. The main reasons for success are the exclusion of excess reagents from the reaction system: such as oxidants, reducing agents, and sometimes metal and/or other improvers, which challenge isolation, increase the wastes and reduce the yield due to frequent incompatibility with these functional groups. Ideal conditions include electron as a reactant (regulated by applied potential) and the by-products as hydrogen or hydrocarbon. The review summarizes and analyzes the achievements of electrochemical methods for the preparation of various phosphorus derivatives with carbon-phosphorus bonds, and collects data on the redox properties of the most commonly used phosphorus precursors. Electrochemically induced reactions both with and without catalyst metals, where competitive oxidation of precursors leads to either the activation of C-H bond or to the generation of phosphorus-centered radicals (radical cations) or metal high oxidation states will be examined. The review focuses on publications from the past 5 years.
Introduction
The development of a new methodology for creating highly selective, low-waste, low-stage and atom-economical reactions, including the formation of the phosphorus-carbon bond, is one of the topical fundamental problems of synthetic and applied chemistry. At present, there is again a revival of interest in the chemistry of organophosphorus compounds in connection with their growing importance in many areas of materials science, medicinal chemistry, organic synthesis, and catalysis (Corbridge, 2013; Carroll and Guiry, 2014; Duffy et al., 2016; Jones et al., 2020; Strasser and Teasdale, 2020; Higham et al., 2021). The appending of phosphorus-carrying functional groups into substrates can be used for the effective altering of their healing properties and biological reactions. Phosphonic derivatives are involved in many biological functions in nature. Some molecules selected as leaders of potential drug candidates, e.g., as anti-cancer, antibacterial, and antiviral agents (Dang et al., 2008; De Clercq, 2011) also contain phosphorus groups. Phosphorylation often greatly improves the hydrophilicity and/or bioavailability of the precursor compounds. Phosphorus ligands with suspended amine relays inside metal complexes imitate hydrogenases in hydrogen reactions, and some organometallic frameworks with bridged arene-phosphorus fragments also work as biomimetic catalysts (Hu et al., 2017; Khrizanforova et al., 2017; Budnikova, 2020; Budnikova and Khrizanforova, 2020), which reveals their nature-like functioning.
Taking into account all the above-mentioned features of organic phosphonates and phosphines, interest in developing methods for constructing phosphorus-carbon bonds continues unabated, and some results partially described in reviews (Budnikova, 2002; Schwan, 2004; Milyukov et al., 2005; Klein et al., 2007; Budnikova and Sinyashin, 2015; Budnikova et al., 2017; Yang et al., 2018; Budnikova and Dudkina, 2019; Rossi-Ashton et al., 2020; Sbei et al., 2020; Liu et al., 2021a). The search for new selective reactions for the preparation of compounds bearing phosphorus groups requires a deeper understanding of the properties and reactivity of key intermediates, which include phosphorus-centered radicals. Methods for the construction of phosphorus-carbon bonds have been developed for over 40 years. Earlier methods and reactions were mainly based on substituted organic derivatives (halides, triflates, borates, etc.) (Hirao et al., 1981; Petrakis and Nagabhushan, 1987; Gelman et al., 2003; Vaillard et al., 2007; Andaloussi et al., 2009; Luo and Wu, 2009; Berrino et al., 2010; Bruch et al., 2010; Zhuang et al., 2011; Zhao et al., 2012; Hu et al., 2013). Recently, new variants of C–H bond phosphorylation have been proposed, for example, transition metal catalysis (Klein et al., 2007; Budnikova and Sinyashin, 2015; Budnikova et al., 2017; Shen et al., 2019; Liu et al., 2021b; Guo et al., 2021; Ma et al., 2021; Zhao et al., 2021; Li et al., 2022a; Li et al., 2022b; Chen et al., 2022; Ji et al., 2022) or radical aromatic phosphorylation (Gao et al., 2017; Gao et al., 2018; Cai et al., 2019; Mai et al., 2019; Chen et al., 2020; Liu et al., 2020; Rawat et al., 2020) (photoinitiated or catalyzed by the same metals or other activators), which provide a direct atom-economical road to C-P functionalized products of different nature (alkenes, alkanes, arenes).
Electrocatalytic or electrochemical reactions of organophosphorus compounds are important and promising tools for the development of environmentally friendly methods of organic transformation. If earlier work in this field demonstrated successful eco-efficient conversions of white phosphorus and organic halides into practically significant products (Budnikova et al., 1999; Budnikova et al., 2001; Kargin et al., 2001; Budnikova et al., 2005; Mikhaylov et al., 2013), the achievements of recent years are mainly associated with the implementation of a one-stage C-H/P-H cross-coupling or electrophilic substitution in arenes under conditions of anodic generation of cation radical precursors; and the establishment of reaction mechanisms using rich capabilities of electrochemical research methods, which will be analysed and summarized in detail hereafter.
Electrochemical metal-free C–H phosphorylation of aromatic hydrocarbons
The non-metal and non-catalytic reactions of direct coupling between aromatic C (sp2)-H and organophosphorus partners seem to be more advantageous than ones involving catalysts, provided that high selectivity and yields are maintained. However, more than 40 years have passed between the first examples of such anodic C-H phosphorylation reactions [the late 70s—early 80s (Ohmori et al., 1979; Nikitin et al., 1983)] and current multi-dimensional and scalable reactions.
Phosphorus precursors used in electrochemical phosphorylation reactions are redox active in many cases, and electron transfer to or from these molecules may be the trigger or primary step of the entire process. The electron transfer products are radical ions or radicals and can be detected by electron paramagnetic resonance (EPR) methods. Table 1 lists some of the recently established potentials of organophosphorus precursor molecules and data on their magnetic resonance parameters, if known. EPR, as it turned out, is a sensitive method for detecting certain phosphorus radical (radical ion) particles, and both the shape of the signal and its parameters depend on the nature of the particles and can be further used to interpret the intermediates and mechanisms of new phosphorylation reactions.
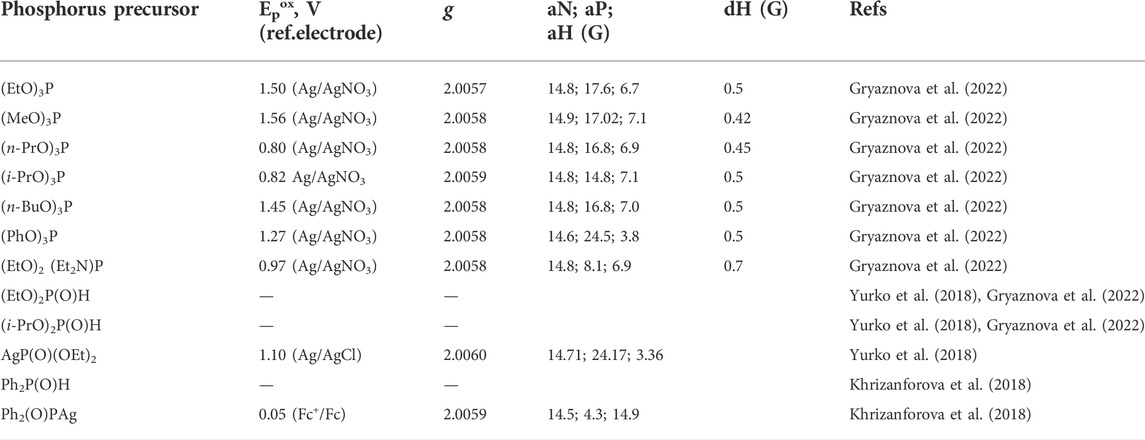
TABLE 1. Potentials of electrooxidation of phosphorus precursors and EPR parameters of phosphorus-centered radicalstrapped by the spin-radical trap N-tert-butyl-α-phenylnitrone (PBN). CH3CN.
But the first ideas were pitched long ago, so, Matsui (Ohmori et al., 1979) and Kargin (Nikitin et al., 1983) hypothesized the initial oxidation of tri- or diorganyl phosphites to radical cations, which can react with benzene, and results in the formation of phosphonium salt (Scheme 1). The latter turns into aryl phosphonate under the influence of the base (Na3PO4, etc.). It should be noted that an electrolyzer with separated anode and cathode spaces was used.
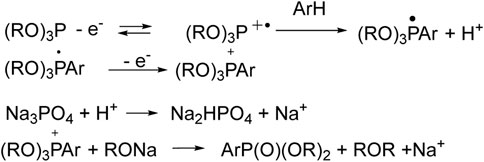
SCHEME 1. Metal-free arene phosphorylation by Matsui and Kargin (Ohmori et al., 1979; Nikitin et al., 1983).
The Gallardo approach implements electrophosphonation of nitro derivatives of benzene in the presence of dialkyl phosphite (Cruz et al., 2011) under oxidative conditions. Unsubstituted benzene does not enter into this reaction, since the key step of the proposed mechanism involves the nucleophilic addition of H-phosphonate to the aromatic nucleus. This step is not possible here and does not produce key σ-Н-adducts, the oxidation of which should lead to the desired C-P product (Scheme 2).
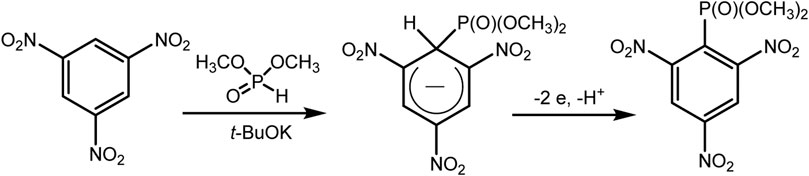
SCHEME 2. Gallardo electrosynthesis (second reaction) (Cruz et al., 2011).
This reaction of a direct anodic coupling of arenes and triorganyl phosphites (RO)3P was thoroughly studied in 2021: with a wider range of aromatic substrates, and even on a large scale in undivided continuous flow cells (Yurko et al., 2018). The authors suggested that the key stage of the process is the formation of phosphorus-centered radical cations on the anode. These electrophilic intermediates react with arenes and form C–P bonds under mild conditions (Scheme 3) with high yields. The high reactivity of P-cation radicals in combination with electrosynthesis ensures not only effective interaction of arenes with various electronic properties (both donor and acceptor substituents in the ring) but also mild functionalization of natural products and bioactive compounds.
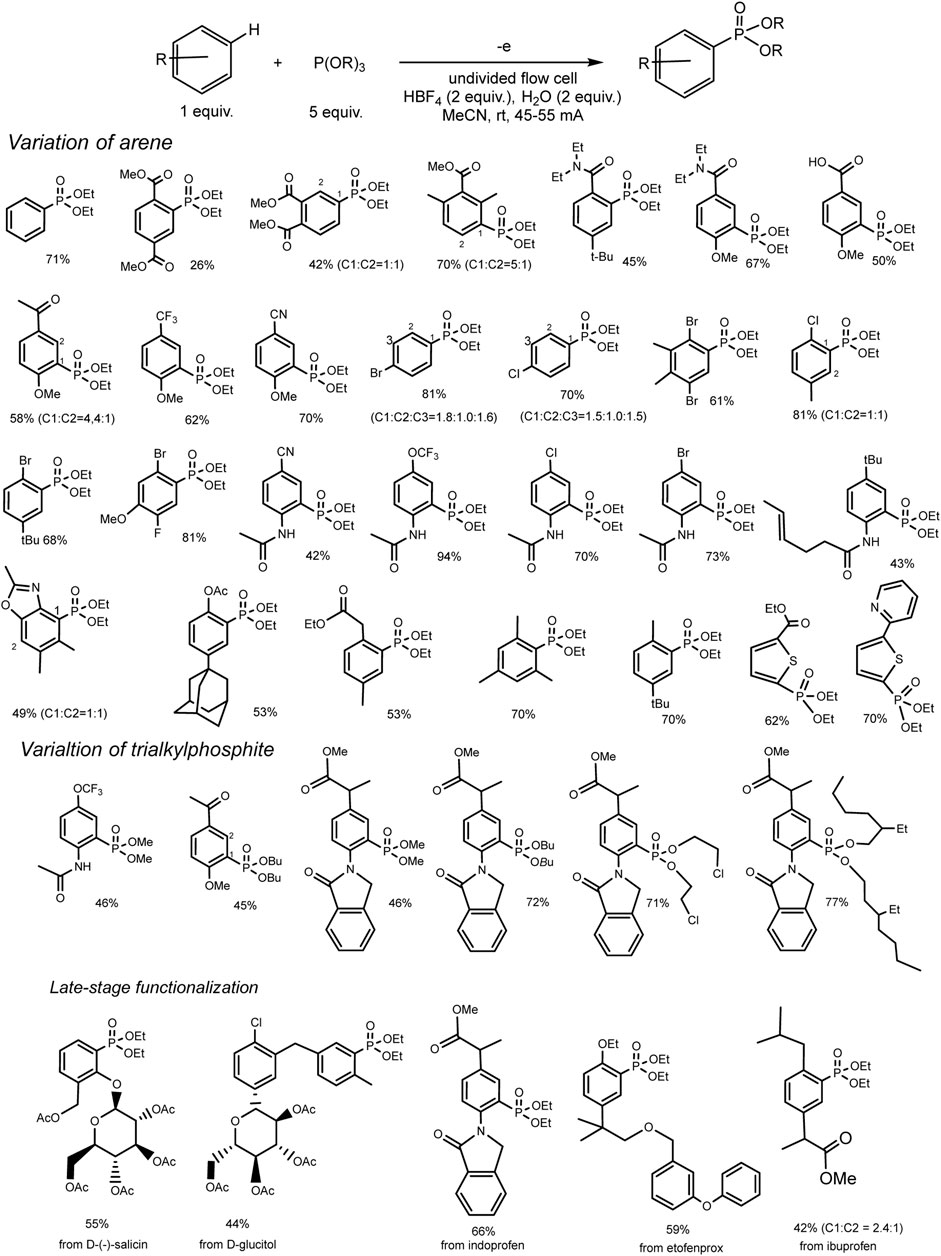
SCHEME 3. Reaction scope, isolated yields. The ratio of regio-isomers is given according to the NMR data of the reactor feed (Long et al., 2021).
A rather unexpected key to success is the additional H2O (2 eqv.) or (RO)2P(O)H (2 eqv.) and electrolysis in a flow cell (Long et al., 2021). Conditions of electrosynthesis in a continuous flow have proved favourable for the conversion of electron-rich and electron-deficient arenes into various arylphosphonates with high yields. All participants in this reaction are easily accessible materials, and the process itself is easily scalable. Scaling was demonstrated by obtaining 55 g of phosphonate product in a single electrolysis.
Although there is no proof of the mechanism in this publication, a logical path has been suggested, as shown in Scheme 4. A possible mechanism of electrochemical C–H phosphorylation involves oxidation of trialkyl phosphite at the anode, resulting in the formation of a phosphorus-centred cation radical, which further reacts with arene and forms a distal cation radical. The next steps are oxidation at the anode and deprotonation to phosphonium. Loss of the alkyl group gives the final phosphonate product. The released protons are reduced at Pt cathode forming H2. The addition of acid HBF4 serves as a background electrolyte and a source of protons for the isolation of H2, which makes it possible to avoid an unwanted cathode reduction of electron-deficient aryl phosphonates. HPO (OR)2 produced through hydrolysis of P (OR)3 in situ probably reacts reversibly with cation radical (RO)3P+ with the formation of the dimeric adduct. The role of the latter in the mechanism is not clear.
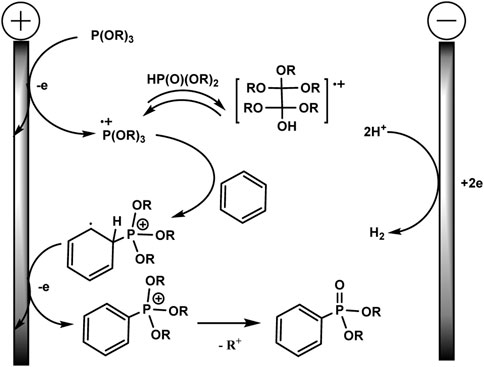
SCHEME 4. Proposed mechanism (Long et al., 2021).
However, the role of water or diethyl phosphite additives is unclear and a relatively large excess of the phosphorus partner in the electrolyte leads to phosphorus-containing wastes in amounts significantly higher than those in Matsui and Kargin reaction (Ohmori et al., 1979; Nikitin et al., 1983). Aromatic compounds exist in excess in this reaction and are usually easy disposable.
Since dialkyl phosphites do not have redox activity in traditional solvents with large windows of available potentials (Yurko et al., 2018; Gryaznova et al., 2022) [or they are oxidized at high anode potentials according to some inconclusive data (Li et al., 2019)], it is impossible to oxidize them to cation radicals and start C-H phosphorylation, as in case of trialkyl phosphites. However, successful examples of electrochemical phosphorylation involving (RO)2P(O)H have been described, but apparently through a different mechanism. Thus, Cheng-Chu Zeng et al. (Li et al., 2019) developed an effective electrochemical protocol for phosphorylation of quinoxaline-2(1H)-ones and xanthenes, C (sp2)–H or C (sp3)–H bonds respectively. This reaction was implemented in an undivided cell according to galvanostatic, transition metals and external oxidants-free protocol. A wide range of substrates is subjected to dehydrogenative C–H/P–H cross-combination with a yield up to 99% (Scheme 5).
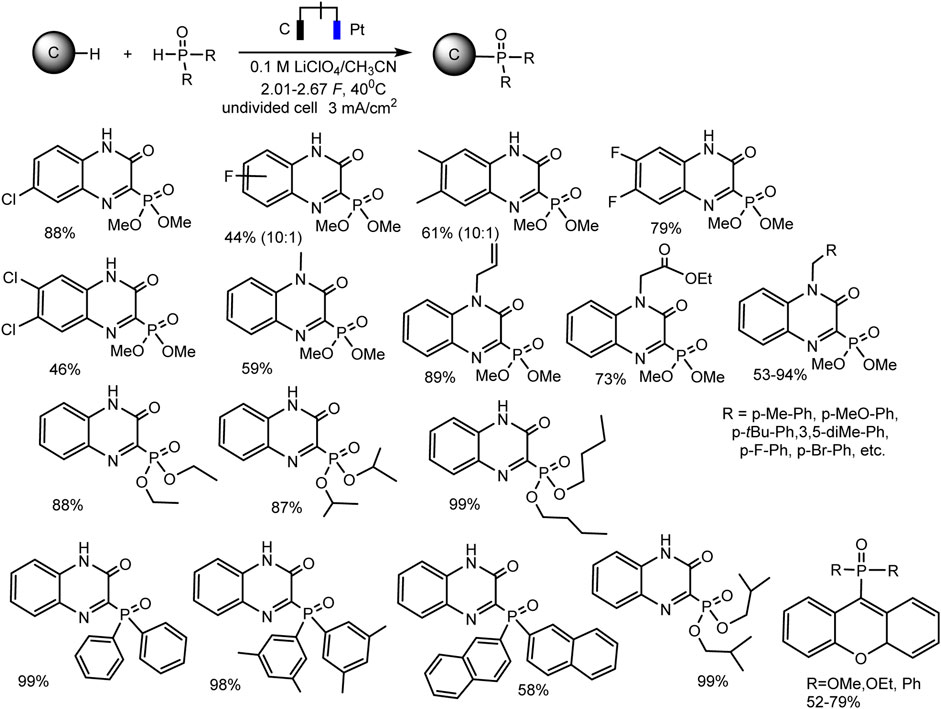
SCHEME 5. Substrate scope for phosphonation of quinoxalin-2(1H)-ones and xanthenes (Li et al., 2019).
Optimal reaction conditions are the following: constant current 3 mA·cm−2, an undivided cell, graphite anode and platinum cathode, 0.1 M LiClO4 background salt, CH3CN, 40°C.
It is assumed that the mechanism of C-H phosphorylation begins with the oxidation of heteroarene, since its oxidative potential is much lower (1.55 V for quinoxaline-2(1H)-one and 1.29 V rel. Ag/Ag + for xanthene) than for phosphite. A diagram for quinoxalinone (Scheme 6) is shown below. The authors believe that quinoxalinone acts as a mediator, forms a cation radical that oxidizes dialkyl phosphite to radical, and the latter finally phosphorylates the C-H bond. In the case of xanthene, its two-electronic oxidation results in the formation of benzyl carbocation, which then reacts with nucleophilic (RO)2P(O)H yielding a desired phosphorylated product. The cathode release of hydrogen is observed. But these are hypothetical mechanisms (Li et al., 2019).
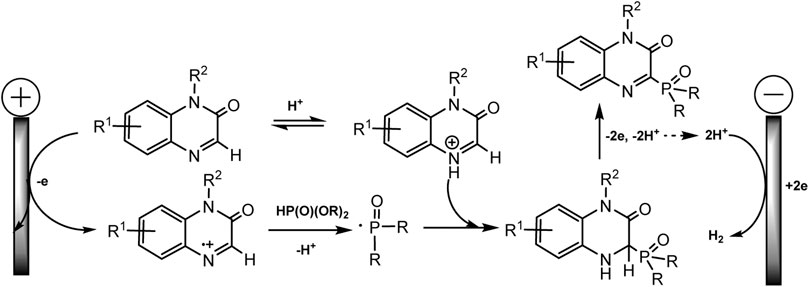
SCHEME 6. Oxidative electrosynthesis of quinoxalin-2(1H)-one phosphonates (Li et al., 2019).
Almost at the same time, the Li-Min Wang group repgraphiorted a metal- and additional oxidant-free protocol for cross-coupling of diaryl phosphine oxides with quinoxalin-2(1H)-ones by electro-dehydrogenation reaction (Hu et al., 2019). Manifold of C3-phosphorylated products was obtained in up to excellent yields, in optimal conditions: undivided cell, platinum as anode and graphite as cathode, acetonitrile as the best solvent, 50°C, constant current 10 mA (Scheme 7).
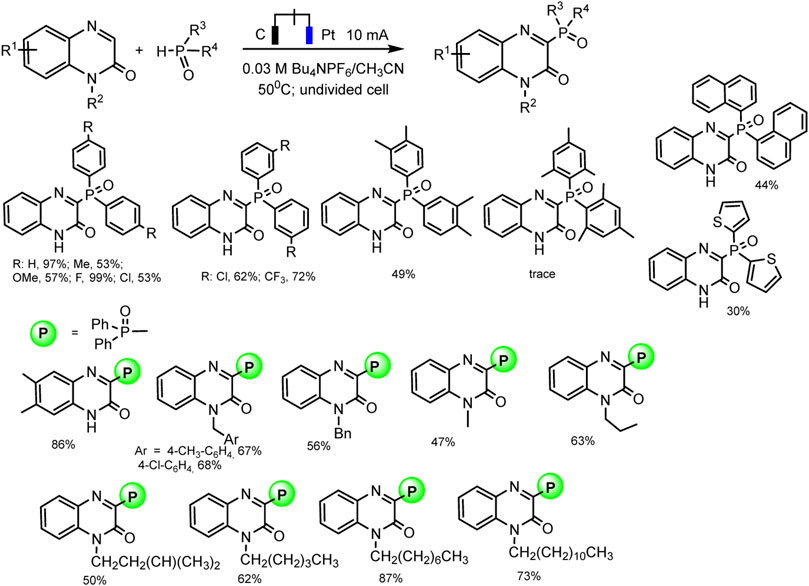
SCHEME 7. Results of quinoxalin-2(1H)-ones and diaryl phosphine oxides coupling (Hu et al., 2019).
The possible mechanism of the process is approximately the same as that proposed in the Zeng’s work (Li et al., 2019).
Electrochemical direct oxidative C–H phosphorylation of thiazole derivatives is the new example of dehydrogenative C–H phosphorylation with H2 evolution (Zhu et al., 2022) (Scheme 8).
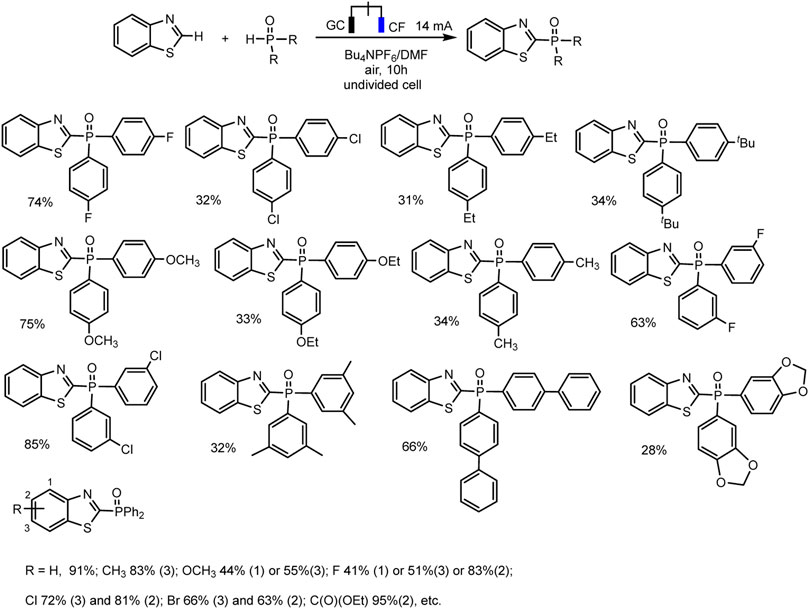
SCHEME 8. Substrate scope for C (sp2)–H phosphonation of thiazole derivatives (Zhu et al., 2022).
The optimal conditions include galvanostatic mode at 14 mA, single-compartment cell, DMF, Bu4NPF6 electrolyte, glassy carbon anode, and copper foam as cathode. The phosphorylation product was obtained in up to 91% yield at room temperature under air conditions (Zhu et al., 2022). Also, as in all electrochemical syntheses discussed here, no external metal or oxidizing agent is required.
R2P(O)H (R = alkyl), as well as (EtO)2P(O)H, exhibited no reactivity toward this cross-coupling. The control experiments have been conducted to gain some mechanistic insights into the process. A radical pathway was suggested based on the reaction inhibition of 2,4- di-t- butyl 4-methylphenol (BHT) as a usual trapping agent, and the observation of Ph2P(O)-adduct in the mass spectrum. A plausible mechanism is depicted in Scheme 6, only with a thiazole as С-H substrate. The oxidation of thiazole derivatives at the anode to form an intermediate radical cation initiates the process. Simultaneously, protons reduction gives hydrogen evolution at the cathode (Zhu et al., 2022).
The electrochemical regioselective N1/C2 phosphorylation of nitrogen-containing heterocycles, and related derivatives, is an efficient protocol representing an affordable “green” strategy for the production of phosphorylated indoles (Dong et al., 2020; Deng et al., 2021). Both N1 and C2 phosphoindoles are biologically active molecules but also widely used in materials chemistry and catalytic processes. It was shown that the selectivity of the joint electrochemical oxidation of indoles and a phosphorus partner depends on the reaction conditions, primarily on the composition of the electrolyte and can lead to various products, both C-H and N-H substitution (Dong et al., 2020; Deng et al., 2021).
Recently, Jin and Liu (Dong et al., 2020) reported on the dehydrogenative N-H/P-H coupling of N-heterocycles with R2P(O)-H phosphorus precursors, in electrochemical oxidative conditions (Scheme 9A. The imidazolium salt, Cs2CO3 were selected as the best electrolyte and base. Gao et al. discovered, that iodide-ion mediates the N-P coupling of indoles and (RO)3P during joint oxidation in undivided cells (Scheme 9B). The EPR method captures the spectra of intermediate iodine radical of and nitrogen-centered radical, which may lie in the N1 pathway of indole phosphorylation.
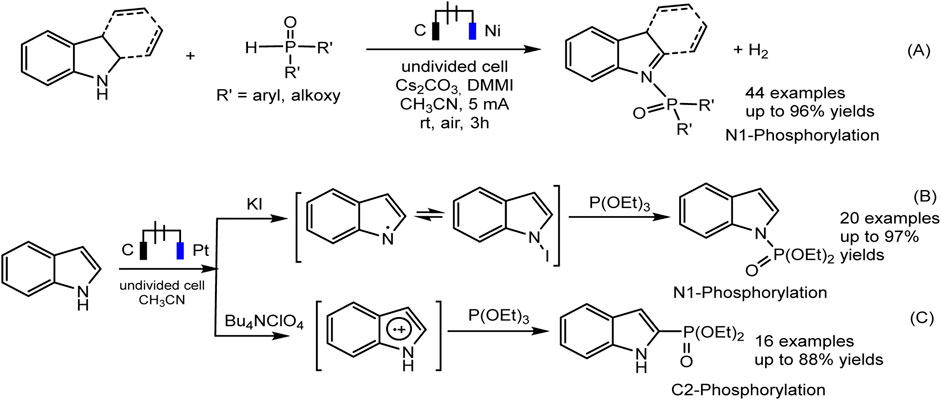
SCHEME 9. The electrochemical oxidative N1 or C2 phosphorylation of indoles (Dong et al., 2020; Deng et al., 2021).
With respect to C2 phosphorylation in the presence of Bu4NClO4 electrolyte, the process is started by the oxidation of indole at the anode, with the formation of an intermediate radical cation, which then reacts with phosphite. The last adduct undergoes further anodic oxidation, and the dehydrogenation and dealkylation steps give the target C2-phosphorylation product (Scheme 9C). (EtO)2P(O)H does not enter into this reaction. The scope of the C2 phosphorylation was subsequently explored. A lot of 1H-indoles (Scheme 10) bearing either electron-withdrawing or electron-donating substituents, tertiary phosphites with i-Pr and n-Bu substituents are also suitable for the reaction and give the desired products in moderate yields. 1Н-pyrrolo [2,3-b] pyridine and melatonin also give phosphonation products in 56% and 43% yields, respectively (Deng et al., 2021).
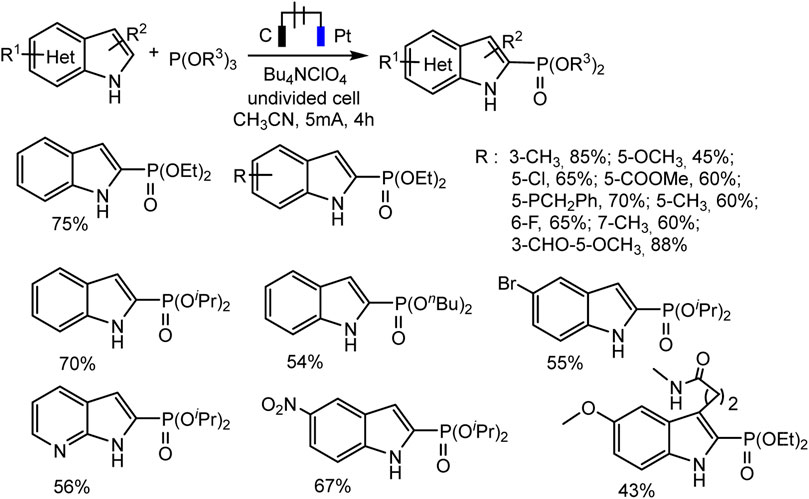
SCHEME 10. C(sp2)–H phosphonation of indoles (Deng et al., 2021).
Léonel and coworkers (Ollivier et al., 2020) used the protected amine for the synthesis of α-aminophosphonates in moderate to good yields by direct phosphorylation of N-carbamate-tetrahydroisoquinoline in an undivided cell (Scheme 11).
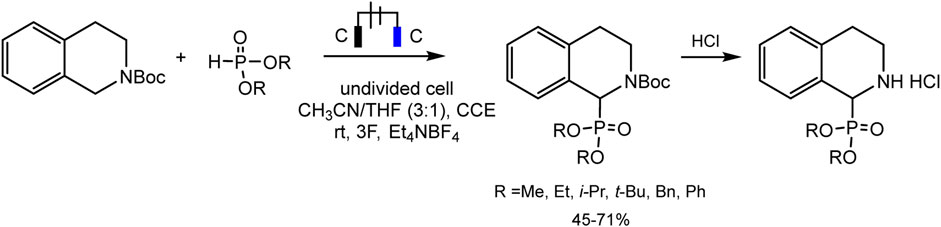
SCHEME 11. Electrophosphorylation of N-carbamate-tetrahydroisoquinoline (Ollivier et al., 2020).
The mechanism is described as a convergent pair electrochemical process (Scheme 12). THIQ-N-Boc oxidizes at the anode by donating two electrons to form an iminium intermediate with proton elimination. The proton is apparently reduced at the cathode with the evolution of hydrogen gas. The phosphorus nucleophile, phosphite anion, is thought to be formed either by one-electron direct reduction of the dialkyl phosphite or by electrogenerated base deprotonation from acetonitrile at the cathode. These intermediate electrophile and nucleophile of iminium and phosphite anion will then react in solution to form the target C-P product.
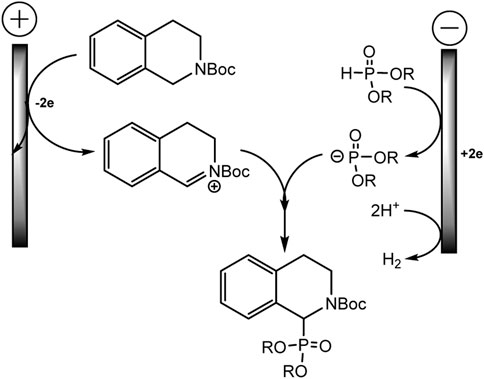
SCHEME 12. Electrooxidative phosphorylation of THIQ-N-Boc (Ollivier et al., 2020).
A relatively simple C–H phosphorylation protocol demonstrating the advantages of electrosynthesis as an environmentally friendly synthesis method was proposed by Aiwen Lei’s group (Yuan et al., 2019). An important aspect is the involvement of substrates of different nature, both with aromatic C (sp2)–H bonds, and O- and N-heterocycles with C (sp3)–H bonds in oxidant- and metal-free conditions (Schemes 13, 14).
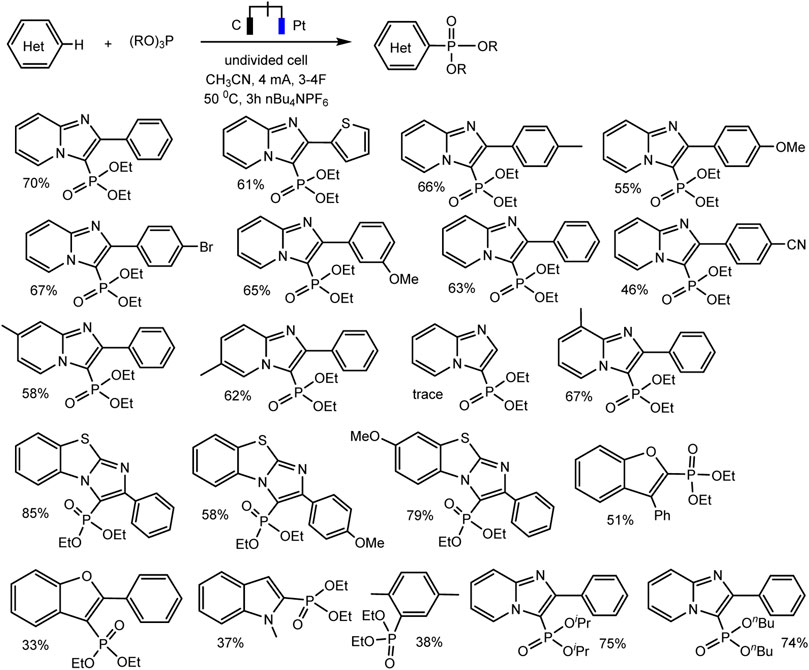
SCHEME 13. Substrate scope for electrochemical oxidative C (sp2)–H phosphorylation of arenes. Yields were determined by 31P NMR (Yuan et al., 2019).
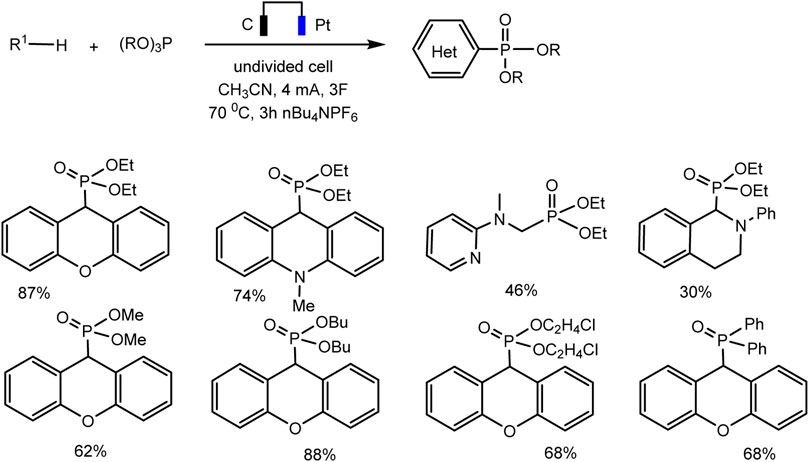
SCHEME 14. Substrate scope for electrochemical oxidative C (sp3)–H phosphorylation of heterocycles. Yields were determined by 31P NMR (Yuan et al., 2019).
Surprisingly, the authors were unable to obtain a voltammogram of the oxidation of trialkyl phosphites from 0 to 2.5 V ref. Ag/AgCl in acetonitrile containing nBu4NPF6, although they are known to oxidize at moderate potentials, which has been shown by other authors (Ohmori et al., 1979; Nikitin et al., 1983; Long et al., 2021; Gryaznova et al., 2022). Thus, the proposed mechanism raises certain doubts, since the substrates used are oxidized relatively far. A tentative mechanism for this C–H phosphorylation was proposed to start from the arene (or heterocycle) oxidation at the electrode to form the radical cation, which is then captured by P(OR)3 [for C (sp2)–H phosphorylation] or stepwise loses proton and electron to turn into another intermediate particle, which is then captured by phosphite [for C (sp3)–H phosphorylation], etc., (Yuan et al., 2019). However, the errors in the determination of potentials do not allow us to confirm these mechanisms with certainty.
Recently, the process of electrochemical phosphorylation with the participation of tertiary phosphites (RO)3P has been studied in detail using the example of acridines, and the redox properties of not only all participants in the reaction, but also key intermediates have been established (Gryaznova et al., 2022). Consequently a mild, efficient acridine C (sp2)–H electro-phosphonation has been developed (Gryaznova et al., 2022). Voltammetry and EPR spectroscopy were used to study the pathways of selective C9 phosphorylation. Some key intermediates such as dihydroacridine dialkyl phosphonates have been obtained and characterized using voltammetry, X-ray diffraction analysis, etc., but triorganyl phosphite cation radicals were identified by EPR (Scheme 15).
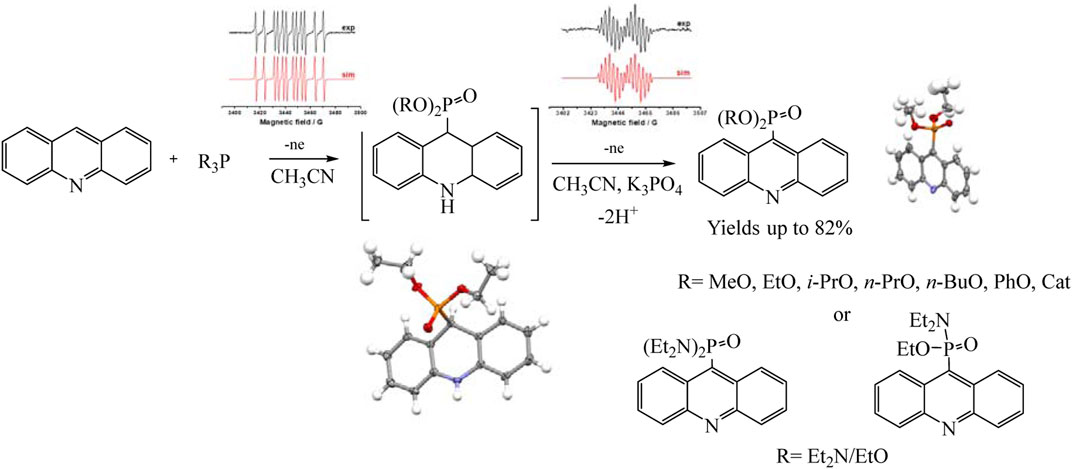
SCHEME 15. Synthetic routes to phosphorylated acridines (Gryaznova et al., 2022).
Knowledge of the redox properties and the nature of intermediates made it possible to suggest two competing pathways. The oxidation potentials of phosphorus and acridine precursors determine the predominance of one or another synthesis route (Scheme 16).
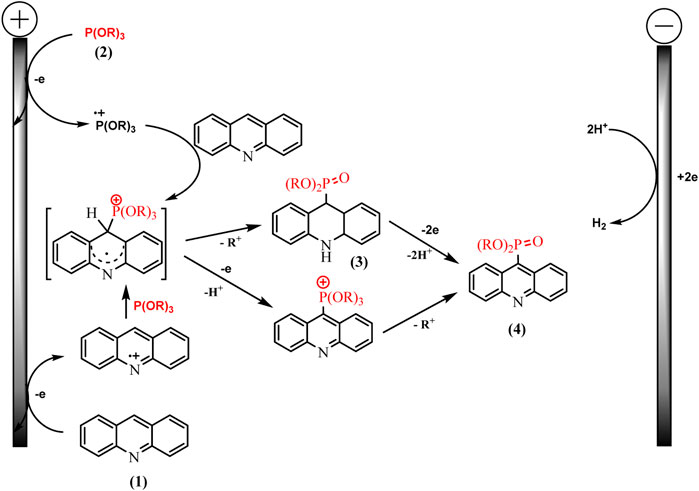
SCHEME 16. Proposed mechanism for acridine phosphonation by trialkylphosphites (Gryaznova et al., 2022).
An example of autocatalysis was recently described in regioselective electrochemically driven C−H phosphorylation of metallocenes (Zheng et al., 2022). The authors suggest that oxidized metallocene, an electrophile, is generated at the anode and reacts with the phosphoryl radical, the formation of the latter being promoted by the metallocene itself (Scheme 17):
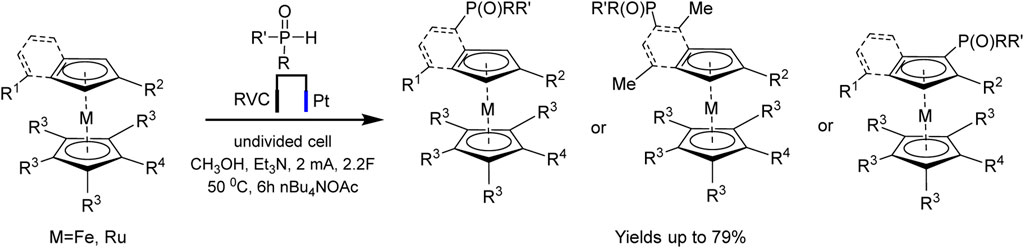
SCHEME 17. Synthesis of metallocene phosphine ligands (Zheng et al., 2022).
The proposed method is applicable to a large number of substrates, so more than 60 phosphorylated (benzo) ferrocenes and rutenocenes have been obtained. No special directing groups or pyrophoric alkyl lithium reagent were used. C−P bond is formed through a radical substitution between phosphoryl radical and metallocene. Moreover, the metallocene functions as a SET (single-electron transfer) reagent triggering the phosphorus-centered radical generation (Scheme 18). It was assumed that the unequal distribution of electron density on the indenyl part should be responsible for the regioselectivity of benzoferrocene phosphorylation.
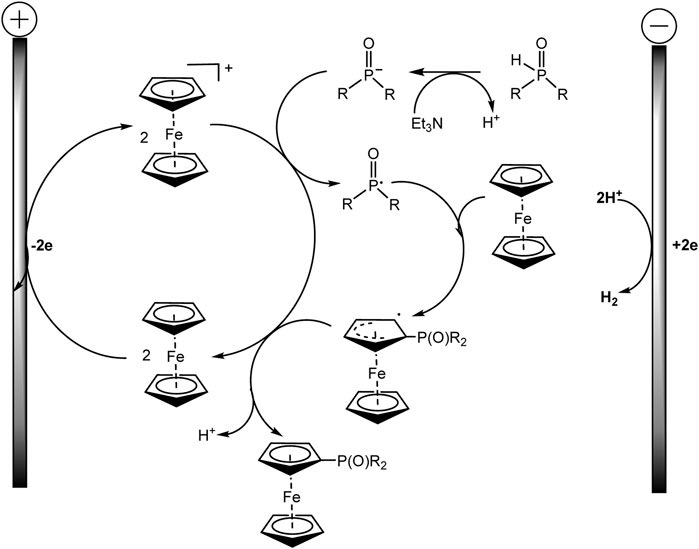
SCHEME 18. Proposed mechanism for ferrocene phosphorylation (Zheng et al., 2022).
Electrochemical metal-catalyzed C–H phosphorylation
Despite recent advances in the anodic phosphorylation of arenes and other derivatives without the participation of metals, metal-catalyzed processes for the introduction of a phosphorus residue into various substrates have been actively worked out over the last years. The coordination of the directing groups with the metal provides high positional selectivity, which is difficult to achieve by other methods. Also, the metal can contribute to the activation of phosphorus partners, if they are not redox-active in the available potential range, for example, dialkyl phosphites. Although the role of metals is not always understood.
The joint oxidation of an aromatic molecule and a dialkyl phosphite catalyzed by a metal proceeds under comparatively mild conditions of a single-stage electrosynthesis of arylphosphonates (Khrizanforov et al., 2017; Khrizanforov et al., 2018). Aryl phosphonates were obtained in 402%–92% yields (Schemes 14A,B) from benzene and coumarin derivatives having both electron-withdrawing and donating substituents in the ring. A bimetallic catalyst system MnIIL/NiIIL (1%, L = bpy) in the anode process (Scheme 19A) (Khrizanforov et al., 2017) or bpyCoCl2 catalyst (5%) in the presence of in EtOH-H2O solution in the cathodic process (Khrizanforov et al., 2018) (Scheme 19B) were applied.
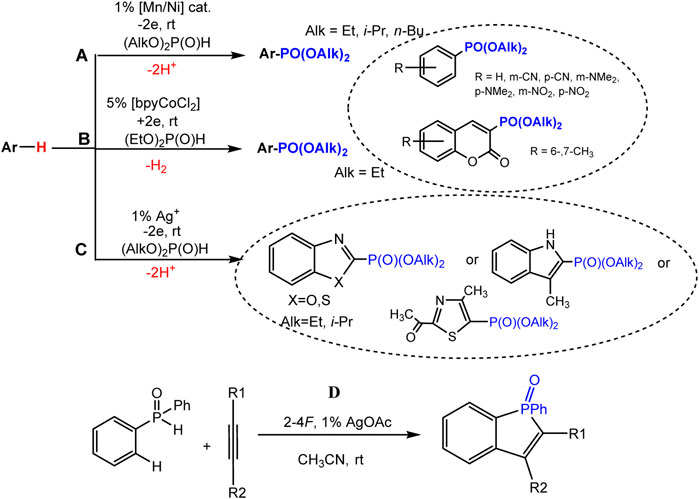
SCHEME 19. Electrochemical metal-catalyzed arene C(sp2)-H phosphonation. (A) reaction with benzene derivatives and coumarins, (B) with azoles, (C) C-H/P-H cyclization-phosphonation of acetylenes.
Mild C-H phosphonation of various azole derivatives (3-methylindole, benzo-1,3-azoles, 4-methyl-2-acetylthiazole) using dialkyl H-phosphonates and silver salts or oxide (1%) as catalysts in anode compartment of electrolyzer (Scheme 19C) was described (Yurko et al., 2018).
It has been shown that the anodic generation of phosphorus-centered radicals makes it possible to realize the cyclization-addition reaction to the triple bond. Such formation of P-radicals was proved under the conditions of joint electrolysis of diphenylphosphine oxide and substituted acetylene at the oxidation potentials of the intermediate silver salt of diphenylphosphine oxide (10% of silver acetate was used as the catalyst), and this reaction affords benzo [b] phosphole derivatives (Scheme 19D) (Khrizanforova et al., 2018). Based on studies of the redox properties of the intermediates of the catalytic cycle by EPR spectroscopy and voltammetry, a radical mechanism of C-H/P-H coupling was proposed.
Different mechanisms of cross-coupling have been proposed, both radical (Khrizanforova et al., 2018; Yurko et al., 2018) and with the participation of higher oxidation states of metal catalysts, for example, AgII (Budnikova and Sinyashin, 2015; Gao et al., 2017).
To increase the selectivity of the C-H functionalization reaction, so-called directing groups are often used, which are coordinated with the metal, sometimes contributing to the formation of a metal-carbon bond, usually in the ortho position. Further substitution of a metal for a functional group makes it possible to obtain a product with high positional selectivity. For the С-H/P-Р phosphonation reaction, there is a complicating factor—the competitive formation of a metal-catalyst phosphonate, which are often low soluble and poorly reactive. Electrochemical C-H functionalization directed by metal-nitrogen coordination was performed in the presence of Pd(II) acetate (Scheme 20). Arenes with functional groups capable of bonding to the palladium atom of the catalyst guide phosphonation to the nearest CH bond.
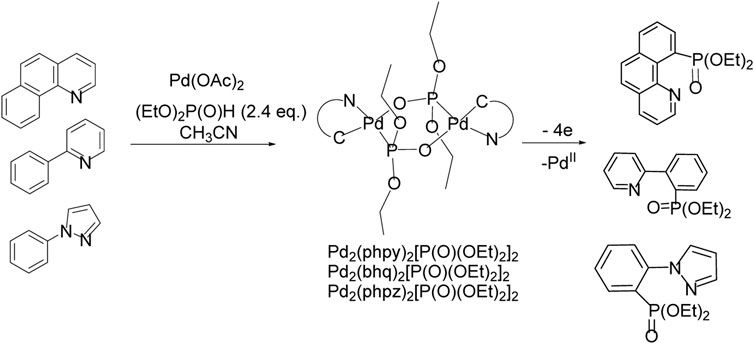
SCHEME 20. Pd-catalyzed ligand-directed ortho-phosphonation of arenes by electrooxidation in divided electrolyzer.
Palladacycles with a changeable oxidation state during electrolysis Pd(II)/Pd(III)/Pd(IV) were suggested to be crucial intermediates in oxidative process (Scheme 20) (Dudkina et al., 2012; Dudkina et al., 2013; Dudkina et al., 2014; Gryaznova et al., 2015a; Gryaznova et al., 2015b; Dudkina et al., 2015; Dudkina et al., 2017). Palladacycles were isolated, their crystal structure and redox properties were studied. It was shown that preparative electrolysis of diphosphonate-bridged Pd-cycle leads to the formation of a phosphonate of the arene, which is a ligand in this complex, and since 2 electrons are spent for each palladium atom, the reductive elimination induced by oxidation proceeds through the Pd(IV) state.
Phosphorylation mechanisms, both radical and with the participation of high-valent metal catalysts, as well as metal-radical ones, are actively proposed and discussed in the literature; it is especially useful to involve voltammetry and EPR studies (Kargin and Budnikova, 2001; Khrizanforov et al., 2016; Dudkina et al., 2017; Khrizanforov et al., 2017; Khrizanforov et al., 2018; Budnikova, 2021; Gryaznova et al., 2022).
Ruan’s group developed the phosphorylation of hydrazones of aldehydes, which proceeds successfully during the electrocatalysis using MnBr2 in an electrolyzer with separation of electrode spaces (Xu et al., 2020) (Scheme 21). The synthesis protocol makes it possible to obtain a number of highly functionalized α-iminophosphine oxides. It is important that the technique is tolerant to different functional groups and can be easily implemented on a gram scale. A likely mechanistic scenario is shown in Scheme 22 without taking into account the influence of MnBr2. The role of MnBr2 remained unclear. Since the oxidation potential of the aldehyde hydrazone is much lower than Eox of Ph2P(O)H, this hydrazone is oxidized first to the carbocation intermediate. The next step involves reaction with a phosphorus partner to form a C-N bond in an intermediate aminyl radical. Further anodic oxidation and subsequent deprotonation give the desired P-C product. Molecular hydrogen is released at the cathode (Xu et al., 2020).
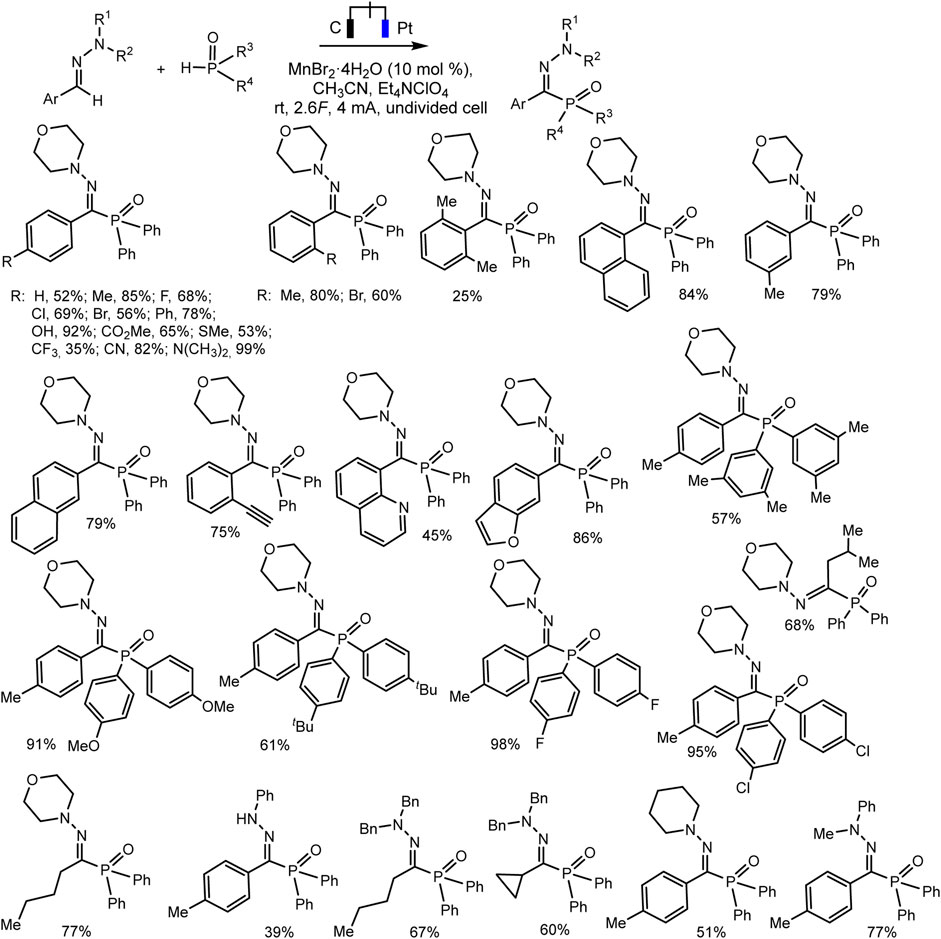
SCHEME 21. Electrochemical phosphorylation of aldehyde hydrazones with diarylphosphine oxide (Xu et al., 2020).
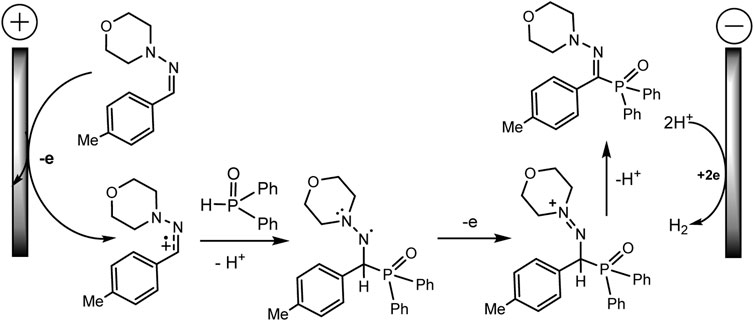
SCHEME 22. Proposed mechanism of phosphorylation of aldehyde hydrazones with diarylphosphine oxide (Xu et al., 2020).
The Lei’ group documented Mn(II)/(III) induced electrochemical C-H/P-H dehydrogenative cross-coupling between aromatic C (sp2)−H of thiophene and furan derivatives and diphenyl phosphine oxides (Wang et al., 2021) (Scheme 23). It was possible to isolate a number of products of both phosphorylation and diphosphorylation by varying the proportions of the substrate in undivided cells. It has been established that C-H activation is not rate-determining based on the data of the experiment with the kinetic isotope effect.
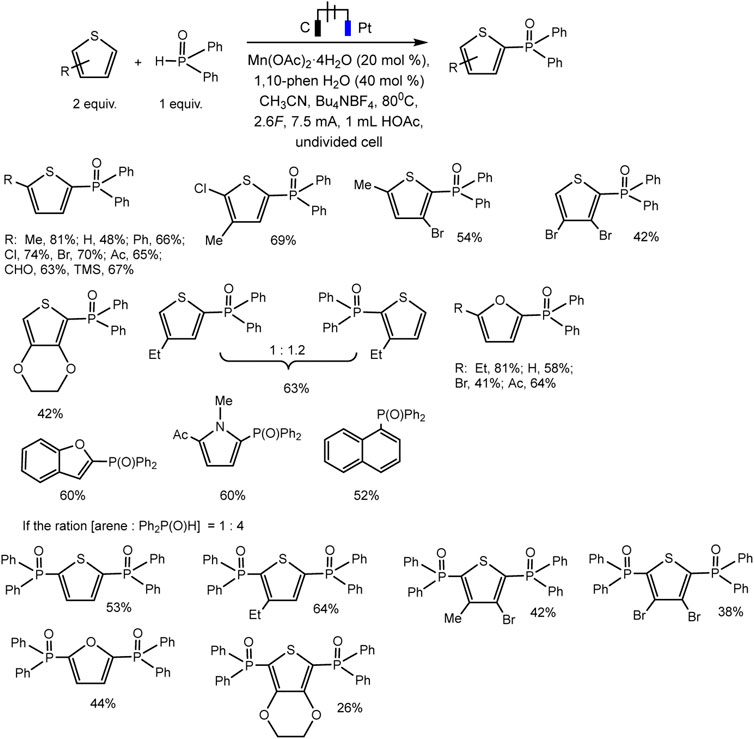
SCHEME 23. Electrochemical C(sp2)–H phosphorylation of thiophene and furan derivatives (Wang et al., 2021).
Mn (II) catalyst is oxidized prior to the aromatic substrate and phosphine oxide, and 1,10-phenanthroline ligand favors the oxidation of Mn(II) to Mn(III) at the anode, which is confirmed by cyclic voltammetry. Afterward, the Mn(III)–P(O)Ph2 is formed with proton elimination by the acetate anion (Scheme 24). The Mn-P intermediate then reacts with arene to form a phosphorylated arene radical, and Mn(III) is regenerated to Mn(II). In a last step, the intermediate allyl radical is oxidized by Mn(III) and subsequent deprotonation gives the target product. As usual, the hydrogen H2 is released at the cathode (Wang et al., 2021).
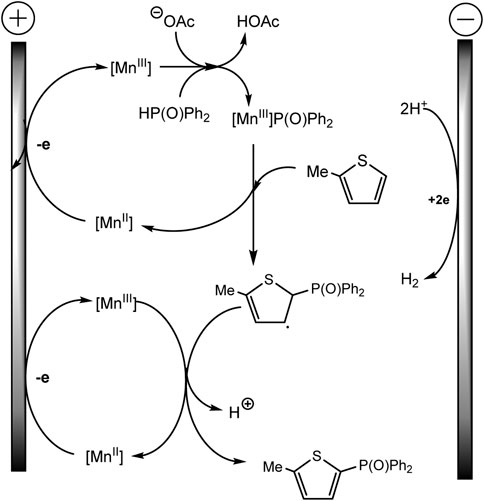
SCHEME 24. The mechanism of phosphorylation of thiophene derivatives (Wang et al., 2021).
Xu et al. have developed RhIII−catalyzed aryl C-H phosphorylation reaction in electrochemical conditions (Wu et al., 2019). The protocol is befitting to a wide range of aromatic substrates and phosphorus precursors and makes it possible to obtain various triarylphosphine oxide easily with low heating (65°C). The best results were obtained in an undivided electrolyzer under galvanostatic conditions at reflux and using Cp*Rh(OAc)2 catalyst, KPF6 background electrolyte, and MeOH solvent (Scheme 25). Oxidation at the anode of the ruthenium (III)-phosphorus complex apparently provides reductive elimination of the final triarylphosphine oxide product and catalyst regeneration.
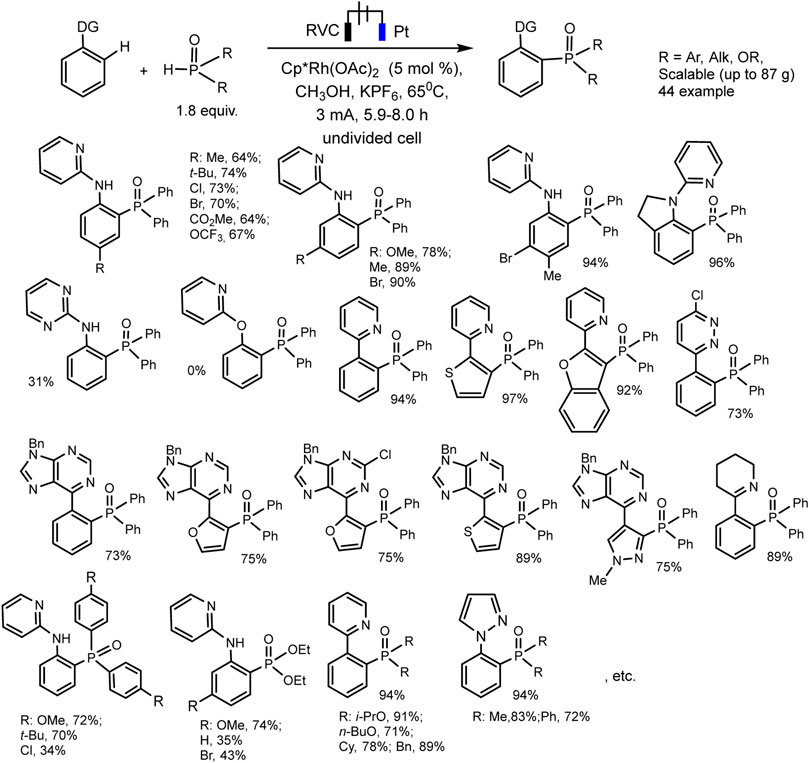
SCHEME 25. Substrate scope for Rh (III)-catalyzed aryl C-H phosphorylation (Wu et al., 2019).
An expectative mechanism is shown in Scheme 26. 2-Phenylpyridine coordinates with the rhodium complex to form a rhodacycle as a result of activation of the ortho-CH bond. Ligand exchange with Ph2P(O)H gives a complex that is more easily oxidized. The reductive elimination induced by anodic oxidation yields the desired C-H phosphorylation product and ends with catalyst regeneration. At the cathode, protons are reduced to H2. These electrochemical reactions are flexibly scalable.
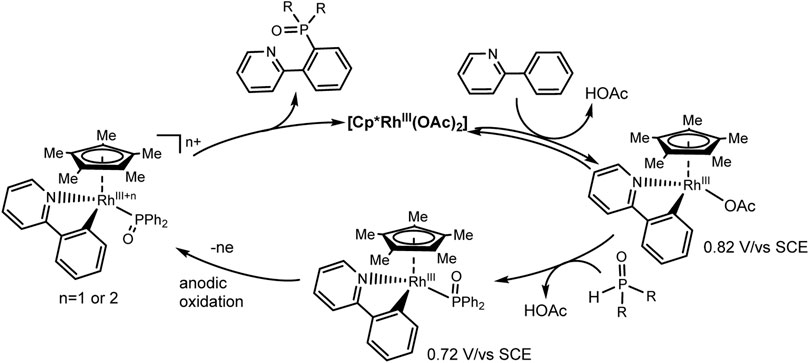
SCHEME 26. Proposed mechanism of Rh(III)-catalyzed aryl C-H phosphorylation (Wu et al., 2019).
A new electrocatalytic protocol for the C-H chlorophosphinoylation of simple alkenes or alkynes was reported by Lin et al. (Lu et al., 2019). The hetero difunctionalization reaction takes place with high efficiency and regioselectivity catalyzed by Mn(II). The optimal conditions include Mn(OTf)2 catalyst, bipyridine ligand, LiClO4 electrolyte, and MeCN solvent (Scheme 27). The reaction is tolerant to a variety of C-H precursors, both derivatives of unsaturated hydrocarbons and P-H partners. The substituted styrenes, vinylpyridine, electronically unactivated alkenes with mono-, di-, and trisubstitution reacted smoothly to form desired products bearing a variety of functional groups. The cyclic alkenes yield the product with excellent diastereoselectivity. The authors suggested that such a successful result is due to the steric loading of the phosphine oxide group (Scheme 27). The catalytic cycle is expected to involve the formation of radicals. This fact is supported by a radical cyclization experiment using a diene, which yields pyrrolidine as a pair of diastereomers. The data of cyclic voltammetry confirm the proposed mechanism involving two different radical intermediates with the participation of the mediator system Mn(II)/Mn(III).
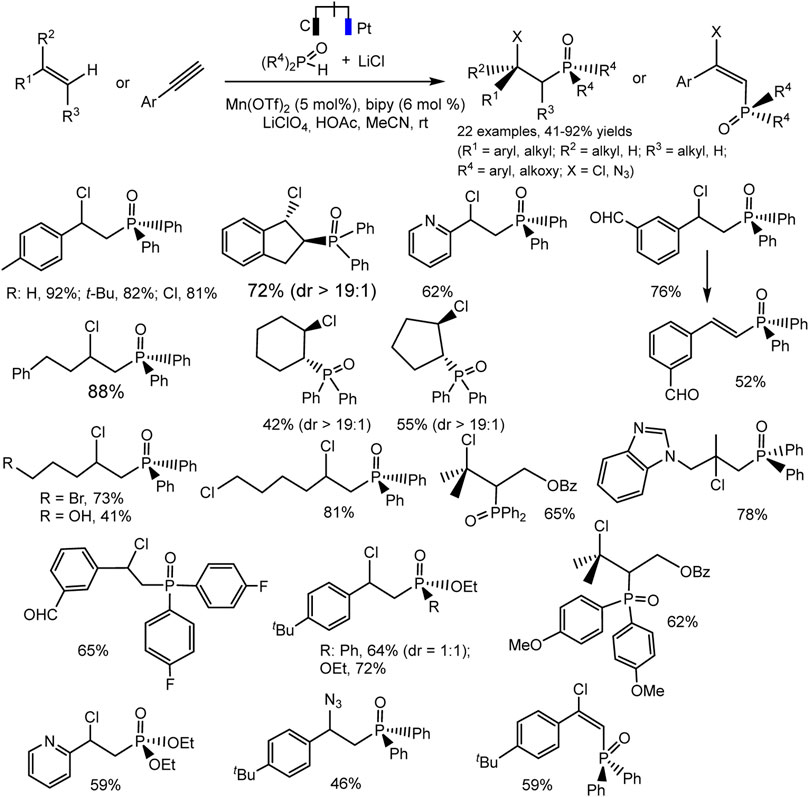
SCHEME 27. Electrocatalytic chlorophosphinoylation of alkenes, substrate scope (Lu et al., 2019).
Recently, the Ackermann group described electrocatalytic ligand-directed C–H phosphorylation of hetarenes and olefins through nickel (III/IV/II) catalysis in an undivided cell (Schemes 28, 29) (Zhang et al., 2021). It can be stated that various benzamides and phosphorus reagents can be involved in the electrosynthesis of arylphosphonates. Moreover, this is a rare example of the use of nickel catalysts in the synthesis of compounds with phosphorus-carbon bonds. The synthesis scheme is very complex and ambiguous, since the reaction system contains many nontrivial additives, the role of which is not clear, for example, iodide ions, triarylphosphine, and guanidine, which themselves are easily oxidized. However, Ni(II) and Ni(III) cyclometallic structures have been proven to be important intermediates. The work uses a whole range of physicochemical data to confirm the conclusions and outcomes, including quantum chemical calculations, HR-ESI-MS and cyclovoltammetric analysis, and, of course, NMR analysis, including on phosphorus nuclei.
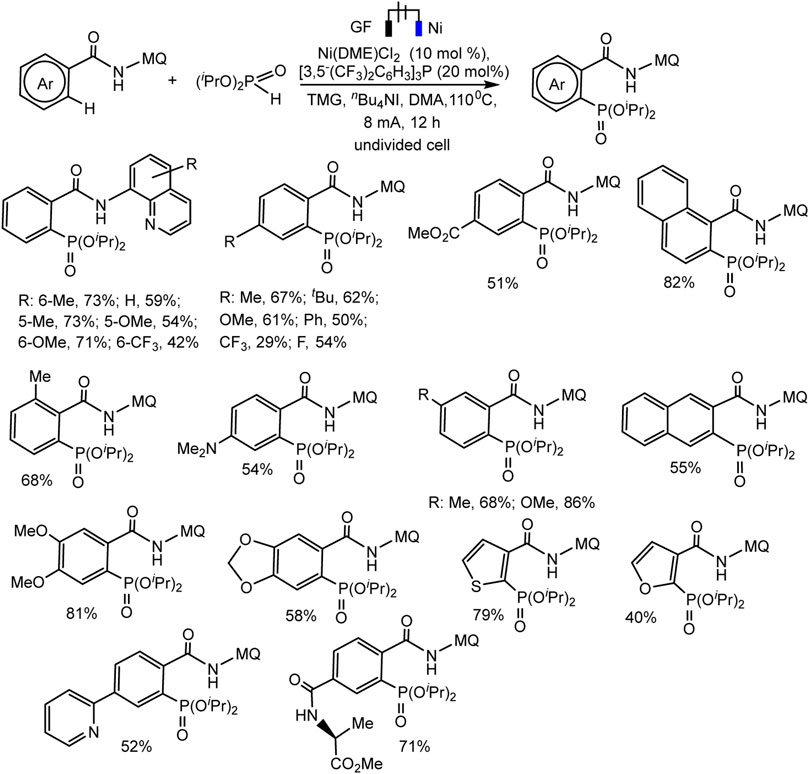
SCHEME 28. Ni-catalysed heteroarene phosphorylation using (iPrO)2P(O)H as phosphorylating reagent (Zhang et al., 2021).
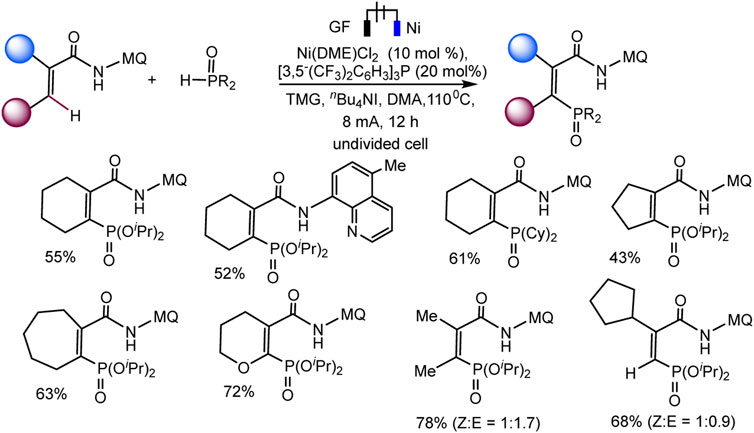
SCHEME 29. Nickela-electrooxidative C (sp2)–H phosphorylation of olefins with (iPrO)2P(O)H or (Cy)2P(O)H as phosphorylating reagents (Zhang et al., 2021).
The range of possibilities for C–H phosphorylation was demonstrated by the successful synthesis of organylphosphonates based on a wide variety of starting phosphorus derivatives -phosphonates, phosphine oxides, diazaphospholidine oxide, mono- or dialkylphosphine oxides, diarylphosphine oxides, with substituents of different electronic nature and steric parameters (Zhang et al., 2021).
Scheme 30 illustrates the mechanism of the catalytic cycle. The deprotonated substrate-ligand binds to Ni(II) into a complex, which is oxidized to Ni(III) with activation of the ortho-C–H bond of the phenyl moiety. At the next stage, the dialkylphosphonate is coordinated to the nickel center, which is oxidized to Ni(IV). Oxidation-induced reductive elimination requires the organic base 1,1,3,3-tetramethylguanidine (TMG) to be successful. The protons released in this way are converted into molecular hydrogen at the cathode (Zhang et al., 2021). However, the scheme does not take into account the fact that the iodide ion is oxidized first, and the role of the substituted triarylphosphine, which is also relatively easily oxidized, is not clear. Possibly, steps of iodination of partners or mediator oxidation of participants also take place.
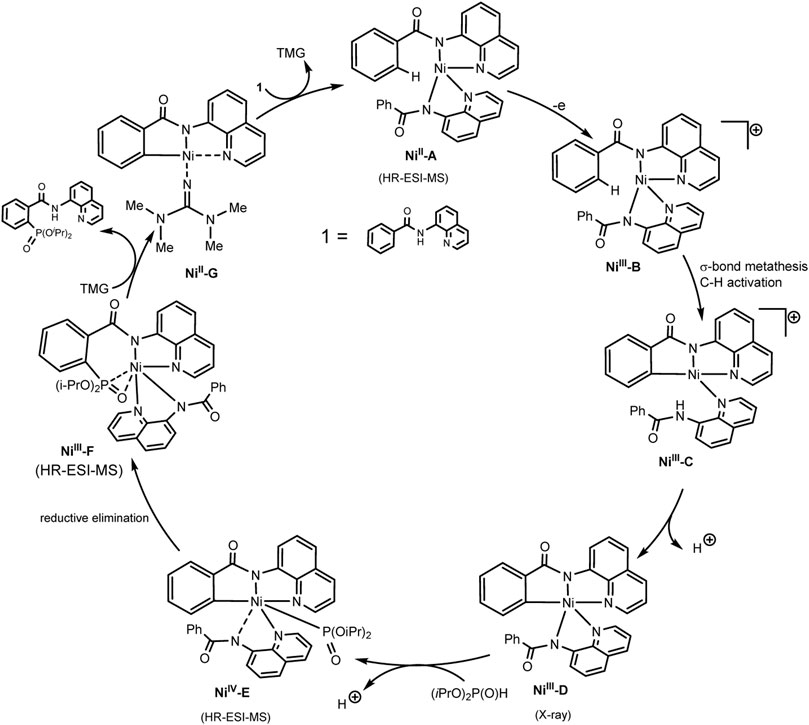
SCHEME 30. Proposal mechanism of Ni(III)/Ni(IV)/Ni(II) catalytic cycle of C-H phosphorylation (Zhang et al., 2021).
Conclusion
The electrolysis method can be successfully used to synthesize numerous P-C-bonded derivatives from available and stable phosphorus precursors through C-H/P-H coupling or electrophilic substitution - phosphorylation of C-H bond, and most importantly, electrochemical engineering enables control of this complex process, achieving high regional, chemo- and Faraday selectivity in some cases. The advantages of electrochemical synthesis in comparison with traditional chemical methods are the absence of additional reagents, a reaction in an almost closed system with a minimal number of mediators-catalysts, which in some cases can be cyclically regenerated to achieve high selectivity and yield. Equally important is the possibility of reducing or eliminating waste, and regenerating potential environmental pollutants, while reducing capital and labour costs for processes, and of course relatively mild reaction conditions. Given the reliability and versatility of functionalization-phosphorylation of C-H bond at the late stages of the synthesis of complicated organic molecules, further progress is expected in the future in obtaining known and new compounds in medicinal chemistry, agrochemistry, and chemical biology. Today, electrochemical functionalization reactions of both aromatic C (sp2)-H and aliphatic C (sp3)-H and some other (N-H, etc.) bonds have been implemented on a huge number of substrates of different nature, and often the advantage is the tolerance of the process to different functional groups in molecules. Scaling electrosynthesis does not usually cause any particular problems, and is done successfully in the cases described if tried. Excellent diastereoselectivity is achieved with a good choice of substituents on the phosphorus precursor and certain synthesis conditions. Mild functionalization of natural products and bioactive compounds has been successfully implemented, and a number of important biologically active molecules and promising compounds for materials chemistry and catalytic processes have been obtained. Reaction mechanisms have been extensively studied, but they are often difficult to prove. Reactions proceed both in the absence of a catalyst metal and in the absence of it; screening of the conditions for each pair of partners is necessary, and there is not always an explanation for the need for the presence of one or another reagent in the reaction mixture. This indicates the importance of continuing research into the mechanisms and application of already made developments for large-scale syntheses and industrial installations. Further efforts should be aimed at clarifying and confirming the mechanisms of these reactions, and implementing a wider range of phosphorus precursors, both inorganic and organic, with yet unstudied phosphorus-element bonds.
Author contributions
Coordination of manuscript—YB. Preparation of the manuscript—ED. MT and TG contributed to the literature review and the writing of the manuscript.
Funding
This work was supported by the Russian Science Foundation Grant No. 22-13-00017.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andaloussi, M., Lindh, J., Sävmarker, J., Sjöberg, R., and Larhed, M. (2009). Microwave-promoted palladium(II)-Catalyzed CP bond formation by using arylboronic acids or aryltrifluoroborates. Chem. Eur. J. 15, 13069–13074. doi:10.1002/chem.200901473
Berrino, R., Cacchi, S., Fabrizi, G., Goggiamani, A., and Stabile, P. (2010). Arenediazonium tetrafluoroborates in palladium-catalyzed C–P bond-forming reactions. Synthesis of arylphosphonates, -phosphine oxides, and -phosphines. Org. Biomol. Chem. 8, 4518–4520. doi:10.1039/C0OB00243G
Bruch, A., Ambrosius, A., Fröhlich, R., Studer, A., Guthrie, D. B., Zhang, H., et al. (2010). Memory of axial chirality in aryl radical phosphanylations. J. Am. Chem. Soc. 132, 11452–11454. doi:10.1021/ja105070k
Budnikova, Y. H., and Dudkina, Y. B. (2019). Progress of electrochemical С(sp2)-H phosphonation. Phosphorus Sulfur Silicon Relat. Elem. 194, 415–419. doi:10.1080/10426507.2018.1540480
Budnikova, Y. H., and Khrizanforova, V. V. (2020). Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials. Pure Appl. Chem 92, 1305–1320. doi:10.1515/pac-2019-1207
Budnikova, Yu. H., and Sinyashin, O. G. (2015). Phosphorylation of C–H bonds of aromatic compounds using metals and metal complexes. Russ. Chem. Rev. 84, 917–951. doi:10.1070/RCR4525
Budnikova, Y. H. (2002). Metal complex catalysis in organic electrosynthesis. Russ. Chem. Rev. 71, 111–139. doi:10.1070/RC2002v071n02ABEH000697
Budnikova, Y. H. (2020). Recent advances in metal–organic frameworks for electrocatalytic hydrogen evolution and overall water splitting reactions. Dalton Trans. 49, 12483–12502. doi:10.1039/d0dt01741h
Budnikova, Y. H. (2021). Electrochemical insight into mechanisms and metallocyclic intermediates of C−H functionalization. Chem. Rec. 21, 2148–2163. doi:10.1002/tcr.202100009
Budnikova, Y. H., Kargin, Y. M., Périchon, J., and Nedelec, J.-Y. (1999). Nickel-catalysed electrochemical coupling between mono- or di-chlorophenylphosphines and aryl or heteroaryl halides. J. Organomet. Chem. 575, 63–66. doi:10.1016/S0022-328X(98)00963-2
Budnikova, Yu. H., Périchon, J., Yakhvarov, D. G., Kargin, Yu.M., and Sinyashin, O. G. (2001). Highly reactive σ-organonickel complexes in electrocatalytic processes. J. Organomet. Chem. 630, 185–192. doi:10.1016/S0022-328X(01)00813-0
Budnikova, Y. H., Yakhvarov, D., and Sinyashin, O. G. (2005). Electrocatalytic eco-efficient functionalization of white phosphorus. J. Organomet. Chem. 690, 2416–2425. doi:10.1016/j.jorganchem.2004.11.008
Budnikova, Y. H., Gryaznova, T. V., Grinenko, V. V., Dudkina, Y. B., and Khrizanforov, M. N. (2017). Eco-efficient electrocatalytic C-P bond formation. Pure Appl. Chem. 89, 311–330. doi:10.1515/pac-2016-1001
Cai, B., Xuan, J., and Xiao, W. (2019). Visible light-mediated C-P bond formation reactions. Sci. Bull. 64, 337–350. doi:10.1016/j.scib.2019.02.002
Carroll, M. P., and Guiry, P. J. (2014). P, N ligands in asymmetric catalysis. Chem. Soc. Rev. 43, 819–833. doi:10.1039/C3CS60302D
Chen, L., Liu, X., and Zou, Y. (2020). Recent advances in the construction of phosphorus-substituted heterocycles, 2009–2019. Adv. Synth. Catal. 362, 1724–1818. doi:10.1002/adsc.201901540
Chen, Y.-Z., Bao, G.-Y., Zhan, X.-C., Fu, J.-G., Ji, X.-M., Zhang, S.-S., et al. (2022). Highly stereoselective synthesis of 2, 2-disubstituted vinylphosphonates via aryl to vinyl 1, 4-palladium migration. Chin. J. Chem. 40, 2188–2192. doi:10.1002/cjoc.202200270
Corbridge, D. E. C. (2013). Phosphorus: Chemistry, biochemistry and technology, sixth edition. Boca Raton: CRC Press.
Cruz, H., Gallardo, I., and Guirado, G. (2011). Electrochemical synthesis of organophosphorus compounds through nucleophilic aromatic substitution: Mechanistic investigations and synthetic scope. Eur. J. Org. Chem. 36, 7378–7389. doi:10.1002/ejoc.201101357
Dang, Q., Kasibhatla, S. R., Jiang, T., Fan, K., Liu, Y., Taplin, F., et al. (2008). Discovery of phosphonic diamide prodrugs and their use for the oral delivery of a series of fructose 1, 6-bisphosphatase inhibitors. J. Med. Chem. 51 (14), 4331–4339. doi:10.1021/jm8001235
De Clercq, E. (2011). The next ten stories on antiviral drug discovery (part E): Advents, advances, and adventures. Med. Res. Rev. 31, 118–160. doi:10.1002/med.20179
Deng, Y., You, S., Ruan, M., Wang, Y., Chen, Z., Yang, G., et al. (2021). Electrochemical regioselective phosphorylation of nitrogen-containing heterocycles and related derivatives. Adv. Synth. Catal. 363 (2), 464–469. doi:10.1002/adsc.202000997
Dong, X., Wang, R., Jin, W., and Liu, C. (2020). Electrochemical oxidative dehydrogenative phosphorylation of N-heterocycles with P(O)–H compounds in imidazolium-based ionic liquid. Org. Lett. 22, 3062–3066. doi:10.1021/acs.orglett.0c00814
Dudkina, Y. B., Mikhaylov, D. Y., Gryaznova, T. V., Sinyashin, O. G., Vicic, D. A., and Budnikova, Y. H. (2012). MII/MIII-Catalyzed ortho-Fluoroalkylation of 2-Phenylpyridine. Eur. J. Org. Chem. 2012 (11), 2114–2117. doi:10.1002/ejoc.201200050
Dudkina, Y. B., Mikhaylov, D. Y., Gryaznova, T. V., Tufatullin, A. I., Kataeva, O. N., Vicic, D. A., et al. (2013). Electrochemical ortho functionalization of 2-phenylpyridine with perfluorocarboxylic acids catalyzed by palladium in higher oxidation states. Organometallics 32, 4785–4792. doi:10.1021/om400492g
Dudkina, Y. B., Gryaznova, T. V., Kataeva, O. N., Budnikova, Y. G., and Sinyashin, O. G. (2014). Electrochemical C-H phosphorylation of 2-phenylpyridine in the presence of palladium salts. Russ. Chem. Bull. 63, 2641–2646. doi:10.1007/s11172-014-0792-3
Dudkina, Y. B., Gryaznova, T. V., Sinyashin, O. G., and Budnikova, Y. H. (2015). Ligand-directed electrochemical functionalization of C(sp2)—H bonds in the presence of the palladium and nickel compounds. Russ. Chem. Bull. 64, 1713–1725. doi:10.1007/s11172-015-1067-3
Dudkina, Y. B., Kholin, K. V., Gryaznova, T. V., Islamov, D. R., Kataeva, O. N., Rizvanov, I. K., et al. (2017). Redox trends in cyclometalated palladium(ii) complexes. Dalton Trans. 46, 165–177. doi:10.1039/C6DT03786K
Duffy, M. P., Delaunay, W., Bouit, P. A., and Hissler, M. (2016). π-Conjugated phospholes and their incorporation into devices: Components with a great deal of potential. Chem. Soc. Rev. 45, 5296–5310. doi:10.1039/C6CS00257A
Gao, Y., Tang, G., and Zhao, Y. (2017). Recent progress toward organophosphorus compounds based on phosphorus-centered radical difunctionalizations. Phosphorus Sulfur Silicon Relat. Elem. 192, 589–596. doi:10.1080/10426507.2017.1295965
Gao, Y., Tang, G., and Zhao, Y. (2018). Recent advances of phosphorus-centered radical promoted difunctionalization of unsaturated carbon-carbon bonds. Chin. J. Org. Chem. 38, 62–74. doi:10.6023/cjoc201708023
Gelman, D., Jiang, L., and Buchwald, S. L. (2003). Copper-catalyzed C−P bond construction via direct coupling of secondary phosphines and phosphites with aryl and vinyl halides. Org. Lett. 5, 2315–2318. doi:10.1021/ol0346640
Gryaznova, T. V., Dudkina, Y. B., Islamov, D. R., Kataeva, O. N., Sinyashin, O. G., Vicic, D. A., et al. (2015). Pyridine-directed palladium-catalyzed electrochemical phosphonation of C(sp2)–H bond. J. Organomet. Chem. 785, 68–71. doi:10.1016/j.jorganchem.2015.03.001
Gryaznova, T., Dudkina, Y., Khrizanforov, M., Sinyashin, O., Kataeva, O., and Budnikova, Y. (2015). Electrochemical properties of diphosphonate-bridged palladacycles and their reactivity in arene phosphonation. J. Solid State Electrochem. 19, 2665–2672. doi:10.1007/s10008-015-2875-y
Gryaznova, T. V., Nikanshina, E. O., Fayzullin, R. R., Islamov, D. R., Tarasov, M. V., Kholin, K. V., et al. (2022). EPR-electrochemical monitoring of P–C coupling: Towards one-step electrochemical phosphorylation of acridine. Electrochim. Acta 428, 140946. doi:10.1016/j.electacta.2022.140946
Guo, L., Su, M., Zhan, H., Liu, W., and Wang, S. (2021). Silver-catalyzed direct regioselective C3 phosphonation of 4hpyrido[1, 2-a]pyrimidin-4-ones with H-phosphites. Asian J. Org. Chem. 10 (7), 1660–1664. doi:10.1002/ajoc.202100235
Higham, L. J., Allen, D. W., and Tebby, J. C. (2021). Spr – organophosphorus chemistry, 50. Cambridge, UK: RSC. doi:10.1039/9781839163814
Hirao, T., Masunaga, T., Ohshiro, Y., and Agawa, T. (1981). A novel synthesis of dialkyl arenephosphonates. Synthesis 1981 (1), 56–57. doi:10.1055/s-1981-29335
Hu, G., Chen, W., Fu, T., Peng, Z., Qiao, H., Cao, Y., et al. (2013). Nickel-catalyzed C–P cross-coupling of arylboronic acids with P(O)H compounds. Org. Lett. 15, 5362–5365. doi:10.1021/ol402672e
Hu, Z., Shen, Z., and Yu, J. C. (2017). Phosphorus containing materials for photocatalytic hydrogen evolution. Green Chem. 19, 588–613. doi:10.1039/C6GC02825J
Hu, C., Hong, G., Zhou, C., Tang, Z.-C., Han, J.-W., and Wang, L.-M. (2019). Electrochemically facilitated oxidative coupling of quinoxalin‐2(1 H )‐Ones with diarylphosphine oxides and pyrroles: A green protocol for C−P, C−C( sp2 ) bond formation. Asian J. Org. Chem. 8, 2092–2096. doi:10.1002/ajoc.201900484
Ji, X.-M., Chen, Y.-Z., Fu, J.-G., Zhang, S.-S., and Feng, C.-G. (2022). Phosphorylation of C(sp3)−H bonds via 1, 4-palladium migration. Org. Lett. 24, 3781–3785. doi:10.1021/acs.orglett.2c01303
Jones, J. L., Yingling, Y. G., Reaney, I. M., and Westerhoff, P. (2020). Materials matter in phosphorus sustainability. MRS Bull. 45, 7–10. doi:10.1557/mrs.2020.4
Kargin, Yu.M., and Budnikova, Yu.G. (2001). Electrochemistry of organophosphorus compounds. Russ. J. Gen. Chem. 71, 1393–1421. doi:10.1023/A:1013906019685
Kargin, Yu.M., Budnikova, Yu.H., Martynov, B. I., Turygin, V. V., and Tomilov, A. P. (2001). Electrochemical synthesis of organophosphorus compounds with P-O, P-N and P-C bonds from white phosphorus. J. Electroanal. Chem. (Lausanne). 507, 157–169. doi:10.1016/S0022-0728(01)00435-1
Khrizanforov, M. N., Strekalova, S. O., Kholin, K. V., Khrizanforova, V. V., Grinenko, V. V., Gryaznova, T. V., et al. (2016). One-stage synthesis of FcP(O)(OC2H5)2 from ferrocene and α-hydroxyethylphosphonate. RSC Adv. 6, 42701–42707. doi:10.1039/C6RA04480H
Khrizanforov, M. N., Strekalova, S. O., Kholin, K. V., Khrizanforova, V. V., Kadirov, M. K., Gryaznova, T. V., et al. (2017). Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 279, 133–141. doi:10.1016/j.cattod.2016.06.001
Khrizanforova, V. V., Karasik, A. A., and Budnikova, Y. H. (2017). Synthetic organometallic models of iron-containing hydrogenases as molecular electrocatalysts for hydrogen evolution or oxidation. Russ. Chem. Rev. 86, 298–317. doi:10.1070/RCR4676
Khrizanforov, M., Strekalova, S., Khrizanforova, V., Dobrynin, A., Kholin, K., Gryaznova, T., et al. (2018). Cobalt-catalyzed green cross-dehydrogenative C(sp2)-H/P-H coupling reactions. Top. Catal. 61, 1949–1956. doi:10.1007/s11244-018-1014-2
Khrizanforova, V. V., Kholin, K. V., Khrizanforov, M. N., Kadirov, M. K., and Budnikova, Yu. H. (2018). Electrooxidative CH/PH functionalization as a novel way to synthesize benzo[b]phosphole oxides mediated by catalytic amounts of silver acetate. New J. Chem. 42, 930–935. doi:10.1039/C7NJ03717A
Klein, A., Budnikova, Y. H., and Sinyashin, O. G. (2007). Electron transfer in organonickel complexes of α-diimines: Versatile redox catalysts for C-C or C-P coupling reactions - a review. J. Organomet. Chem. 692, 3156–3166. doi:10.1016/j.jorganchem.2007.01.021
Li, K.-J., Jiang, Y.-Y., Xu, K., Zeng, C.-C., and Suna, B.-G. (2019). Electrochemically dehydrogenative C–H/P–H cross-coupling: Effective synthesis of phosphonated quinoxalin-2(1H)-ones and xanthenes. Green Chem. 21, 4412–4421. doi:10.1039/c9gc01474h
Li, C.-K., Tao, Z.-K., Shoberu, A., Zhang, W., and Zou, J.-P. (2022). Copper-catalyzed cross-coupling of alkyl and phosphorus radicals for C(sp3)−P bond formation. Org. Lett. 24 (32), 6083–6087. doi:10.1021/acs.orglett.2c02454
Li, Q., Zhao, C.-Q., Chen, T., and Han, L.-B. (2022). Direct phosphorylation of benzylic C–H bonds under transition metal-free conditions forming sp3C–P bonds. RSC Adv. 12, 18441–18444. doi:10.1039/d2ra02812c
Liu, X.-C., Chen, X.-L., Liu, Y., Sun, K., Peng, Y.-Y., Qu, L.-B., et al. (2020). Visible-Light-induced metal-free synthesis of 2-phosphorylated thioflavones in water. ChemSusChem 13, 298–303. doi:10.1002/cssc.201902817
Liu, J., Xiao, H.-Z., Fu, Q., and Yu, D.-G. (2021). Advances in radical phosphorylation from 2016 to 2021. Chem. Synth. 1, 9. doi:10.20517/cs.2021.07
Liu, T., Wei, L., Zhao, B., Liu, Y., and Wan, J.-P. (2021). Copper-catalyzed enaminone C(sp2)–N bond phosphonation for stereoselective synthesis of alkenylphosphonates. J. Org. Chem. 86 (14), 9861–9868. doi:10.1021/acs.joc.1c00862
Long, H., Huang, C., Zheng, Y.-T., Li, Z.-Y., Jie, L.-H., Song, J., et al. (2021). Electrochemical C–H phosphorylation of arenes in continuous flow suitable for late-stage functionalization. Nat. Commun. 12, 6629. doi:10.1038/s41467-021-26960-y
Lu, L., Fu, N., and Lin, S. (2019). Three-component chlorophosphinoylation of alkenes via anodically coupled electrolysis. Synlett 30, 1199–1203. doi:10.1055/s-0039-1689934
Luo, Y., and Wu, J. (2009). Synthesis of arylphosphonates via palladium-catalyzed coupling reactions of aryl imidazolylsulfonates with H-phosphonate diesters. Organometallics 28, 6823–6826. doi:10.1021/om900771v
Ma, L., Shang, S., Yuan, H., Zhang, Y., Zeng, Z., and Chen, Y. (2021). Ag(I)-catalyzed synthesis of (E)-alkenyl phosphonates by oxidative coupling of H-phosphites with β-nitroolefins. Tetrahedron Lett. 87, 153530. doi:10.1016/j.tetlet.2021.153530
Mai, W.-P., Yuan, J.-W., Zhu, J.-L., Li, Q.-Q., Yang, L.-R., Xiao, Y.-M., et al. (2019). Selectfluor-Mediated direct C-H phosphonation of quinoxalin-2(1H)-ones under base and transition-metal free conditions. ChemistrySelect 4, 11066–11070. doi:10.1002/slct.201903478
Mikhaylov, D. Y., Gryaznova, T. V., Dudkina, Y. B., Polyancev, F. M., Latypov, S. K., Sinyashin, O. G., et al. (2013). Novel electrochemical pathway to fluoroalkyl phosphines and phosphine oxides. J. Fluor. Chem. 153, 178–182. doi:10.1016/j.jfluchem.2013.04.009
Milyukov, V. A., Budnikova, Yu. H., and Sinyashin, O. G. (2005). Organic chemistry of elemental phosphorus. Russ. Chem. Rev. 74, 781–805. doi:10.1070/RC2005v074n09ABEH001182
Nikitin, E. V., Romakhin, A. S., Parakin, O. V., Romanov, G. V., Kargin, Y. M., and Pudovik, A. N. (1983). Electrochemical synthesis of aryl phosphonates. Russ. Chem. Bull. 32, 566–569. doi:10.1007/BF00953100
Ociepa, M., Knouse, K. W., He, D., Vantourout, J. C., Flood, D. T., Padial, N. M., et al. (2021). Mild and chemoselective phosphorylation of alcohols using a Ψ-reagent. Org. Lett. 23 (24), 9337–9342. doi:10.1021/acs.orglett.1c02736
Ohmori, H., Nakai, S., and Masui, M. (1979). Anodic oxidation of organophosphorus compounds. Part 2. Formation of dialkyl arylphosphonates via arylation of trialkyl phosphites. J. Chem. Soc. Perkin 1, 2023–2026. doi:10.1039/P19790002023
Ollivier, A., Sengmany, S., Rey, M., Martens, T., and Léonel, E. (2020). Direct phosphonylation of N-Carbamate-tetrahydroisoquinoline by convergent paired electrolysis. Synlett 31, 1191–1196. doi:10.1055/s-0039-1690899
Petrakis, K. S., and Nagabhushan, T. L. (1987). Palladium-catalyzed substitutions of triflates derived from tyrosine-containing peptides and simpler hydroxyarenes forming 4-(diethoxyphosphinyl)phenylalanines and diethyl arylphosphonates. J. Am. Chem. Soc. 109, 2831–2833. doi:10.1021/ja00243a050
Rawat, D., Kumar, R., and Subbarayappa, A. (2020). Visible-light induced phosphonation of quinoxalines and quinoxalin-2(1H)-ones under aerobic metal-free conditions. Green Chem. 22, 6170–6175. doi:10.1039/d0gc02168g
Rossi-Ashton, J. A., Clarke, A. K., Unsworth, W. P., and Taylor, R. J. K. (2020). Phosphoranyl radical fragmentation reactions driven by photoredox catalysis. ACS Catal. 10, 7250–7261. doi:10.1021/acscatal.0c01923
Sbei, N., Martins, G. M., Shirinfar, B., and Ahmed, N. (2020). Electrochemical phosphorylation of organic molecules. Chem. Rec. 20, 1530–1552. doi:10.1002/tcr.202000096
Schwan, A. L. (2004). Palladium catalyzed cross-coupling reactions for phosphorus–carbon bond formation. Chem. Soc. Rev. 33, 218–224. doi:10.1039/B307538A
Shen, J., Xiao, B., Hou, Y., Wang, X., Li, G-Z., Chen, J.-C., et al. (2019). Cobalt(II)-Catalyzed bisfunctionalization of alkenes with diarylphosphine oxide and peroxide. Adv. Synth. Catal. 361 (22), 5198–5209. doi:10.1002/adsc.201900873
Strasser, P., and Teasdale, I. (2020). Main-chain phosphorus-containing polymers for therapeutic applications. Molecules 25, 1716. doi:10.3390/molecules25071716
Vaillard, S. E., Mück-Lichtenfeld, C., Grimme, S., and Studer, A. (2007). Homolytic substitution at phosphorus for the synthesis of alkyl and aryl phosphanes. Angew. Chem. Int. Ed. 46 (34), 6533–6536. doi:10.1002/anie.200701650
Wang, S., Xue, Q., Guan, Z., Ye, Y., and Lei, A. (2021). Mn-catalyzed electrooxidative undirected C–H/P–H cross-coupling between aromatics and diphenyl phosphine oxides. ACS Catal. 11, 4295–4300. doi:10.1021/acscatal.1c00549
Wu, Z.-J., Su, F., Lin, W., Song, J., Wen, T.-B., Zhang, H.-J., et al. (2019). Scalable rhodium(III)-Catalyzed aryl C−H phosphorylation enabled by anodic oxidation induced reductive elimination. Angew. Chem. Int. Ed. 58, 16770–16774. doi:10.1002/anie.201909951
Xu, Z., Li, Y., Mo, G., Zheng, Y., Zeng, S., Sun, P.-H., et al. (2020). Electrochemical oxidative phosphorylation of aldehyde hydrazones. Org. Lett. 22, 4016–4020. doi:10.1021/acs.orglett.0c01343
Xu, Z., Fu, L., Fang, X., Huang, B., Zhou, L., and Wan, J.-P. (2021). Tunable trifunctionalization of tertiary enaminones for the regioselective and metal-free synthesis of discrete and proximal phosphoryl nitriles. Org. Lett. 23 (13), 5049–5053. doi:10.1021/acs.orglett.1c01581
Yang, Q.‐L., Fang, P., and Mei, T.‐S. (2018). Recent advances in organic electrochemical C-H functionalization. Chin. J. Chem. 36, 338–352. doi:10.1002/cjoc.201700740
Yuan, Y., Qiao, J., Cao, Y., Tang, J., Wang, M., Ke, G., et al. (2019). Exogenous-oxidant-free electrochemical oxidative C–H phosphonylation with hydrogen evolution. Chem. Commun. 55, 4230–4233. doi:10.1039/C9CC00975B
Yurko, E. O., Gryaznova, T. V., Kholin, K. V., Khrizanforova, V. V., and Budnikova, Y. H. (2018). External oxidant-free cross-coupling: Electrochemically induced aromatic C–H phosphonation of azoles with dialkyl-H-phosphonates under silver catalysis. Dalton Trans. 47, 190–196. doi:10.1039/C7DT03650G
Zhang, S.-K., Del Vecchio, A., Kuniyil, R., Messinis, A. M., Lin, Z., and Ackermann, L. (2021). Electrocatalytic C–H phosphorylation through nickel(III/IV/II) catalysis. Chem 7, 1379–1392. doi:10.1016/j.chempr.2021.04.009
Zhao, Y.-L., Wu, G.-J., Li, Y., Gao, L.-X., and Han, F.-S. (2012). [NiCl2(dppp)]-Catalyzed cross-coupling of aryl halides with dialkyl phosphite, diphenylphosphine oxide, and diphenylphosphine. Chem. Eur. J. 18, 9622–9627. doi:10.1002/chem.201103723
Zhao, L., Sun, M., Yang, F., and Wu, Y. (2021). Silver(I) promoted the C4−H bond phosphonation of 1-naphthylamine derivatives with H-phosphonates. J. Org. Chem. 86 (17), 11519–11530. doi:10.1021/acs.joc.1c00971
Zheng, H., Liu, C.-H., Guo, S.-Y., He, G.-C., Min, X.-T., Zhou, B.-C., et al. (2022). Electrochemically driven regioselective C−H phosphorylation of group 8 metallocenes. Nat. Commun. 13 (1), 3496. doi:10.1038/s41467-022-31178-7
Zhu, P.-W., Yang, Y.-T., Li, Y., Zhu, J., and Wu, L. (2022). Electrochemical Oxidative C-H Phosphonylation of thiazole derivatives in ambient conditions. Mol. Catal. 517, 112022. doi:10.1016/j.mcat.2021.112022
Keywords: electrochemistry, C-H bond, phosphonates, phosphine oxides, catalysis
Citation: Budnikova YH, Dolengovsky EL, Tarasov MV and Gryaznova TV (2022) Recent advances in electrochemical C—H phosphorylation. Front. Chem. 10:1054116. doi: 10.3389/fchem.2022.1054116
Received: 26 September 2022; Accepted: 17 October 2022;
Published: 03 November 2022.
Edited by:
Bing Yu, Zhengzhou University, ChinaCopyright © 2022 Budnikova, Dolengovsky, Tarasov and Gryaznova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yulia H. Budnikova, eXVsaWFAaW9wYy5ydQ==