 Chao Wang1,2*†
Chao Wang1,2*† Jing Chang1,2†
Jing Chang1,2† Shanbo Yang1,2
Shanbo Yang1,2 Lingyu Shi1,2
Lingyu Shi1,2 Yujing Zhang3
Yujing Zhang3 Wenjing Liu1,2
Wenjing Liu1,2 Jingsen Meng1,2
Jingsen Meng1,2 Jun Zeng1,2
Jun Zeng1,2 Renshuai Zhang1,2
Renshuai Zhang1,2 Dongming Xing1,4*
Dongming Xing1,4*- 1Cancer Institute, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
- 2Qingdao Cancer Institute, Qingdao University, Qingdao, China
- 3The Affiliated Cardiovascular Hospital of Qingdao University, Qingdao University, Qingdao, China
- 4School of Life Sciences, Tsinghua University, Beijing, China
Combretastatin A-4 (CA-4) is a potent inhibitor of tubulin polymerization and a colchicine binding site inhibitor (CBSI). The structure-activity relationship study of CA-4 showed that the cis double bond configuration and the 3,4,5-trimethoxy group on the A ring were important factors to maintain the activity of CA-4. Therefore, starting from this condition, chemists modified the double bond and also substituted 3,4,5-trimethoxyphenyl with various heterocycles, resulting in a new generation of CA-4 analogs such as chalcone, Flavonoid derivatives, indole, imidazole, etc. Quinoline derivatives have strong biological activity and have been sought after by major researchers for their antitumor activity in recent years. This article reviews the research progress of novel CA-4 containing quinoline analogs in anti-tumor from 1992 to 2022 and expounds on the pharmacological mechanisms of these effective compounds, including but not limited to apoptosis, cell cycle, tubulin polymerization inhibition, immune Fluorescence experiments, etc., which lay the foundation for the subsequent development of CA-4 containing quinoline analogs for clinical use.
1 Introduction
Microtubules, a key building block of the cytoskeleton, are dynamic polymers of tubulin that form an ordered network of polarized tubules (Honore et al., 2005). Microtubules are formed by combining α and β heterodimers, which are important components of the eukaryotic cytoskeleton, and they have played important roles in mitosis and cytokinesis (Lu et al., 2012). The vast majority of these molecules act by binding to a heterodimer of the protein microtubulin (α, β) that forms the microtubule core (Wang et al., 2021). The microtubulin heterodimer contains six binding sites (Pryor et al., 2002): Pironetin, Taxane, Laulimalide/Peloruside, Vinca, Maytansine, and Colchicine binding sites. Targeted drugs that act on microtubules and exert anti-tumor effects by inhibiting microtubule proteins are called microtubule protein inhibitors. They were classified into two categories according to their mechanism of action on microtubules: microtubule protein depolymerization inhibitors, such as Paclitaxel (1, Figure 1), and microtubule protein polymerization inhibitors, such as Vinblastine (2, Figure 1) and Colchicine (3, Figure 1).
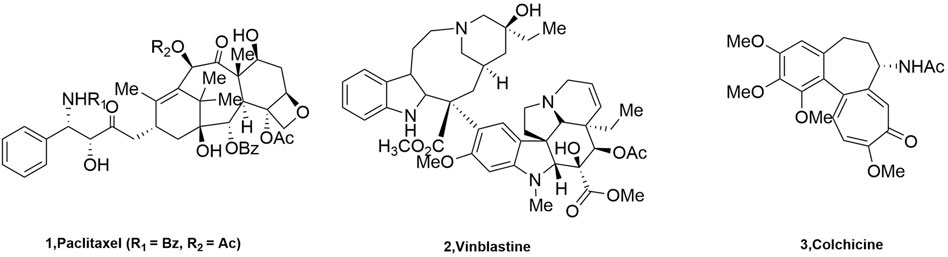
FIGURE 1. The structure of tubulin inhibitors.
Current studies show that there are many drugs used in clinical oncology that act on the paclitaxel and vincristine binding sites. These drugs are very effective but have complex structures due to their large molecular weights, which makes the major researchers discouraged. However, the compounds acting on CBSI have the characteristics of small molecular weight, simple structure, diverse molecules, and easy synthesis and transformation, which are sought after by major researchers (Gigant et al., 2005). A number of representative tubulin inhibitors (currently in clinical trials) targeting the colchicine binding site have emerged (Figure 2).
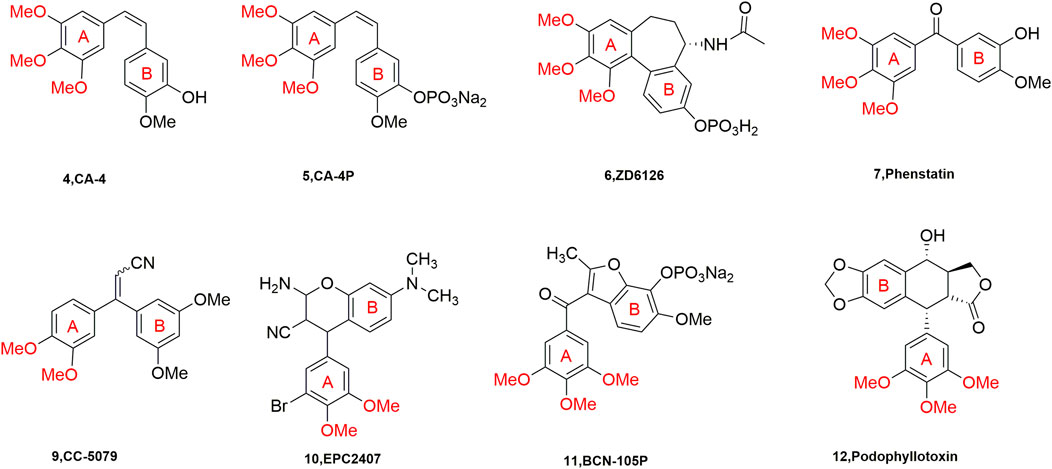
FIGURE 2. Chemical structures of some CBSIs (currently in clinical trials).
It is well known that quinoline was isolated in the 19th century and since then more and more related natural products have been isolated and identified. Derivatives containing quinoline show great advantages due to nitrogen atoms, such as can increase the basicity of molecules, because of its basic properties and the possibility of nitrogen atoms forming strong hydrogen bonds with the target. Another important property is polarity, which can be used as a means to reduce lipophilicity and improve water solubility and oral absorption (Alvarez et al., 2016). Quinoline groups are often present in natural alkaloids with a wide range of biological activities and there are many drugs that carry quinoline on the market (Figure 3). Molecules containing quinoline scaffolds can even enhance the cytotoxicity of doxorubicin against multidrug-resistant cancer cell lines at non-toxic concentrations (Chen et al., 2014). Quinoline analogues have also shown anticancer activity by different mechanisms, including alkylating agents, tyrosine kinase inhibitors, and microtubulin inhibitors (Kaur et al., 2010).
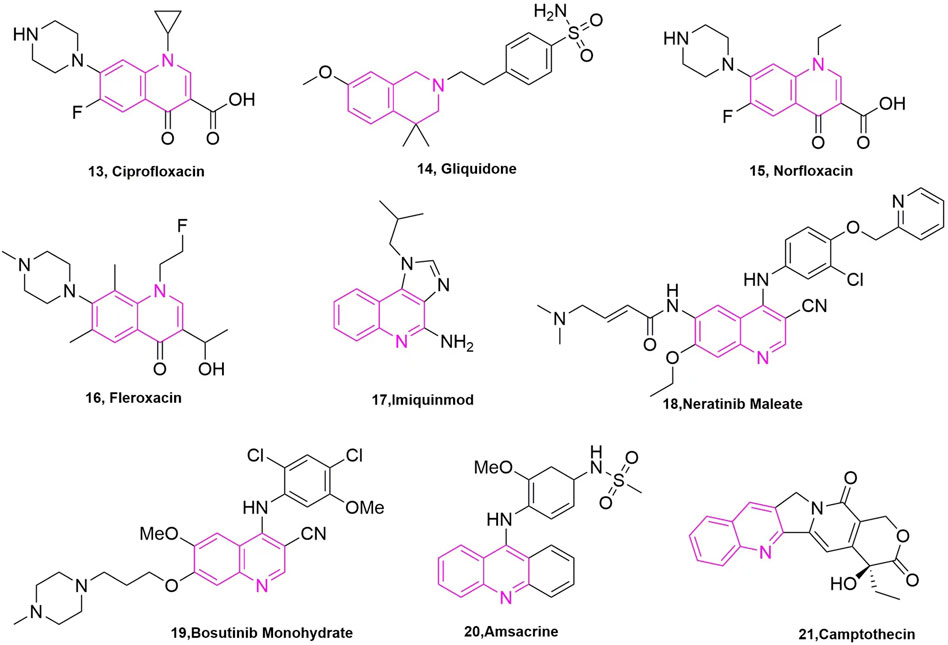
FIGURE 3. The marketed drugs carrying quinoline’s structure.
Meanwhile, extensive structure-activity relationship (SAR) studies were performed on CA-4, which demonstrated the 3,4,5-trimethoxy substitution pattern on the A-ring, 4-methoxy-substituted B-ring and cis-configuration double bond as the basis for the polymerization-inhibiting activity of microtubulin (Stefanski et al., 2018). We have mapped the mechanism of action of these compounds (CA-4 analogs) based on the SAR of CA-4 (Figure 4). Among the modifications of the CA-4 structure, some researchers have modified it using quinoline and iso-quinoline structures. Their study also further demonstrates that the quinoline fraction may be an alternative to the conventional 3,4,5-trimethoxyphenyl fraction when bound to the colchicine site (Li et al., 2019). In addition, the use of other heterocycles to replace the B-ring has been pursued by various researchers (Solomon et al., 2019). They designed and synthesized some derivatives that carry CA-4 quinoline analogues and interact with the colchicine binding site. The results showed that the successfully modified compounds had comparable activity to CA-4, and some had higher activity than CA-4. In addition, we searched PubMed, Web of science and Scoups for 30 related articles published between 1992 and 2022 (Figure 5). This paper mainly reviews the design, synthesis and validation of pharmacological activities of quinoline fragment-containing CA-4 derivatives in these articles, including the experiments involved and the corresponding experimental results. Finally, the similarities and differences among these articles are pointed out, and the outlook is proposed.
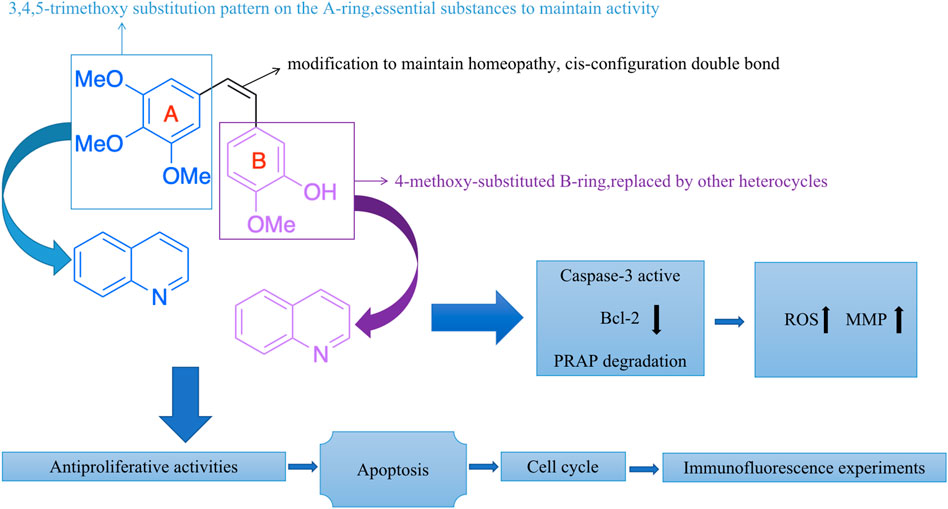
FIGURE 4. The mechanism of action of these compounds (CA-4 analogs).
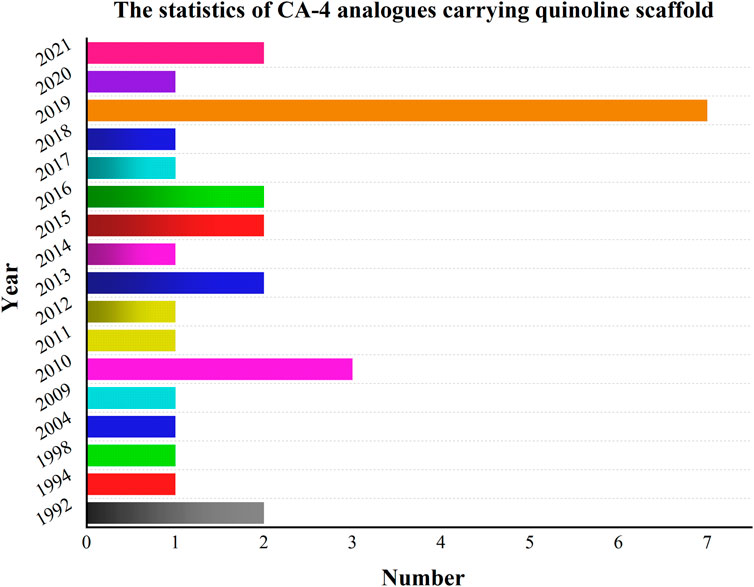
FIGURE 5. Relevant articles have been published from 1992 to 2022.
2 CA-4 analogues carrying quinoline scaffold
2.1 Modification of CA-4
2.1.1 Modification of A-ring
Kuo et al. (1993) started with a tricyclic chemical structural pattern, which was the first and similar to CA-4 analogues carrying quinoline scaffold. They designed and synthesized a series of 1,6,7,8-substituted 2-(4′-substituted phenyl)-4-quinolones, which had been evaluated as cytotoxic compounds and as antimitotic agents that interact with tubulin. These compounds were assayed for their cytotoxicity in vitro against six tumor cell lines, which included human lung carcinoma cell lines (A-549), ileocecal carcinoma cell lines (HCT-8), and melanoma cell lines (RPMI-7951), and epidermoid carcinoma of the nasopharynx cell lines (KB) and two murine leukaemia cell lines (P-388 and L1210). In these compounds, compound 23 (Figure 6), compound 25 (Figure 6), and compound 26 (Figure 6) showed potent cytotoxicity (Monks et al., 1991), with EC50 values < 1.0 μg/ml in all cancer lines. They then tested the effects of these three compounds on tubulin interactions, which used the COMPARE algorithm (Bai et al., 1991) assay and compound 25 showed the best with an IC50 value of 2.7 ± 0.04 μM. In addition, these compounds could inhibit the binding of radiolabelled colchicine to tubulin, compared to colchicine, podophyllotoxin, and CA-4, and compound 25 showed the best of them. Even so, the three compounds were not as potent as natural products.
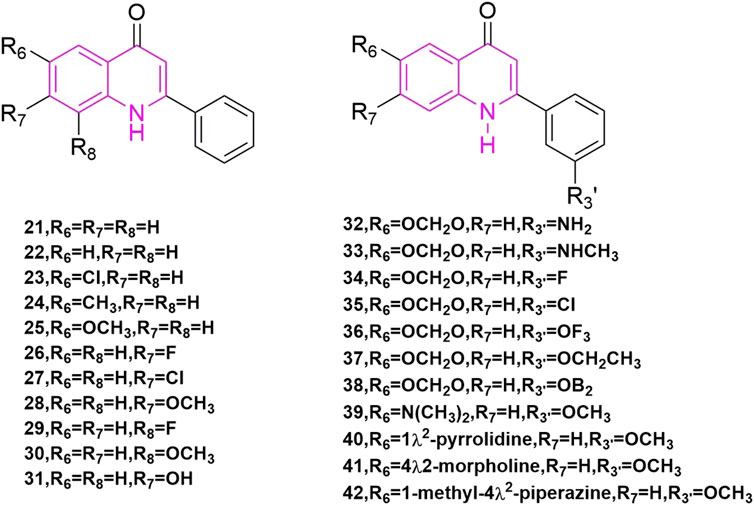
FIGURE 6. The structures of the compounds designed by Kuo et al. (1993) and Li et al. (1994b).
Li et al. (1994b) is a continuation of Kuo et al., they began studying 2-phen-yl-quinolones and designing and synthesizing a new series of compounds, which illustrated strong inhibitory effects against a variety of human tumor cell lines, including those derived from solid tumors. Compound 28 (Figure 6) illustrated great activity and also inhibited tubulin polymerization (Li et al., 1994a), with activity comparable to those of the potent antimitotic natural products colchicine, podophyllotoxin, and CA-4. Compounds 21, 23, 24, 27–29, 33, 40, and 41 (Figure 6) demonstrated strong cytotoxic effects to most of the tumor cell lines with GI50 values in the micromolar to nanomolar range. Also, compounds 42 are just to enrich the structure of quinoline compounds.
Xia et al. (1998) designed and synthesized a novel series of 6,7,2′,3′,4-substituted-1,2,3,4-tetrahydro-2-phenyl-4-quinolones based on the structure of natural anti-mitotic drugs. They evaluated cytotoxicity (Rubinstein et al., 1990) in six cancer cell lines, including HCT-8, breast cancer cell lines (MCF-7), A-549, KB, renal cancer cell lines (CAKI-1), and melanoma cancer cell lines (SKMEL-2). In these compounds, compounds 43, and 44, 45 (Figure 7) illustrated great cytotoxic effects with ED50 values in the nanomolar or sub nanomolar range. Compound 44 demonstrated the best activity, pyrrolidinylrrolinyl ring at the 6-position, similar with Li’s research before.
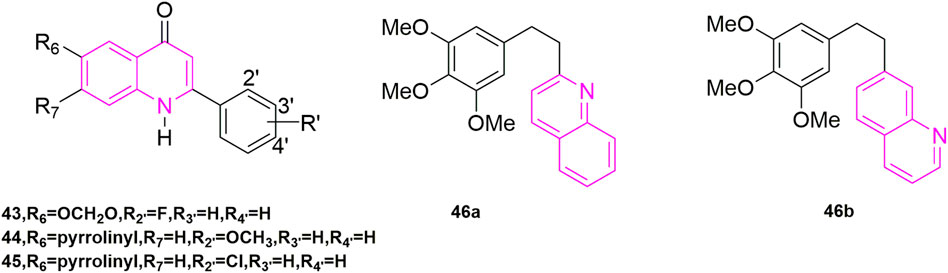
FIGURE 7. The structures of the compounds designed by Xia et al. (1998) and Perez-Melero et al. (2004).
2.1.2 Modification of B-ring and cis-double bond
By studying the conformational relationship of CA-4, Perez-Melero et al. (2004) proposed using 2-naphthyl moiety to replace the 3-hydroxy-4-methoxyphenyl ring of CA-4, which could not observably reduce cytotoxicity and inhibit tubulin polymerization activity. Then they came up with using 6- or 7-quinolyl systems to substitute 3-hydroxy-4-methoxyphenyl ring, keeping the 3,4,5-trimethoxyphenyl moiety as A-ring at the same time (46a, 46b, Figure 7), getting a new family of analogues of CA-4. Compound 46a was highly cytotoxic but did not inhibit tubulin polymerization. While compound 46b was similar to the naphthalene-containing compound previously described and had powerful cytotoxicity [−log (IC50) M = 6.8] and tubulin inhibition ability. Other compounds demonstrated weak inhibition of tubulin compared to CA-4. In addition, the presence of N atoms also made these compounds more water-soluble than CA-4, which pointed out the way to solve the water-soluble problem of CA-4.
Ferlin et al. (2010) designed and synthesized a series of new substituted 7-phenyl-3H-pyrrolo [3,2-f] quinolin-9-ones, which starting from the study of the antiproliferative activity (Gasparotto et al., 2007) of this structure of pyrroloquinoline. By evaluating its antiproliferative activities, the best active compound 47 (Figure 8) was found to be highly selective for human leukaemia cells (GI50 = 0.45 ± 0.10 μM), and it also blocked Jurkat cells in the G2/M phase in a concentration-dependent manner. At a concentration of 0.06 μM for 24 h compared to the control the number of G2/M cells increased from 9.4% to 77% in the control group. G1 cells decreased from 47% to 3% in the control group, while S-phase cells decreased from 43% to 20% (Gasparotto et al., 2007). Such results were reflected in the other compounds as well. To verify that subsequent experiments were relevant, apoptosis experiments were conducted, in which they selected Jurkat cells treated with compound 47 for 24 h and 48 h to observe the extent of apoptosis (Martin et al., 1995; Vermes et al., 1995). The effectiveness led them to further investigate the mechanism of apoptosis, so they carried out mitochondrial depolarization experiments and confirmed that compound 47 could dose-dependently increase the mitochondrial transmembrane potential. To investigate the relationship between mitochondrial depolarization and reactive oxygen species, they used compound 47 to treat cells to see if reactive oxygen species production increased, and the results confirmed that it was as expected and induced reactive oxygen species (ROS) production in the cells (Zamzami et al., 1995). This was the same increase in mitochondrial transmembrane potential as in the mitochondrial depolarization assay. Finally, they found that Jurket cells treated with compound 47 activated caspase-3 in a concentration-dependent manner in vitro, while an increase in Poly ADP-ribose polymerase (PARP) fragments and a decrease in Bcl-2 protein were also observed in Western blot experiments. In their experiments, compound 47 induced apoptosis by activating caspase-3, cleavage PARP, and downregulating Bcl-2 protein (Earnshaw et al., 1999; Denault and Salvesen, 2002).
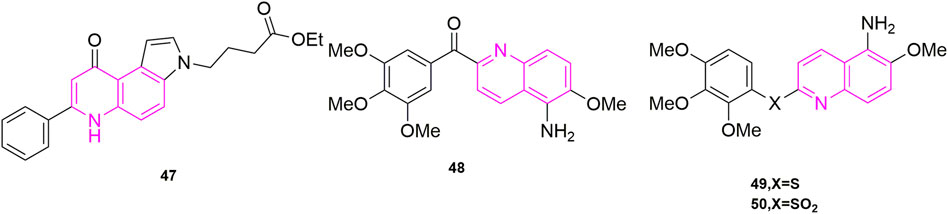
FIGURE 8. The structures of the compounds designed by Ferlin et al. (2010), Nien et al. (2010), and Lee et al. (2011).
Nien et al. (2010) designed and synthesized a series of new aryl quinoline derivatives by coupling the quinoline nucleus with 3,4,5-trimethoxybenzoyl from the pharmacological activity of the parent quinoline nucleus and combining it with CA-4 conformational relationship. In vitro, antiproliferative activityexperiments were also performed, and five cells were selected, including KB, non-small-cell lung carcinoma cell lines (H460), human colorectal carcinoma cell lines (HT29), and human gastric cancer cell lines (MKN45), as well as the MDR-positive cancer cell lines, KB-vin10 cells, that overexpressed P-GP 170/MDR. Compound 48 (Figure 8), which contained 5-amino-2-aroylquinolines, was shown to be the most potent of the five cells (IC50 = 0.2–0.7 nM), surpassing CA-4. To further understand its potentially active structure, molecular docking was performed, and the results showed that the presence of hydrogen bonds increased the potential of compound 48 and contributed to its effectiveness. To investigate the anti-microtubule protein activity and the ability of compound 36 to compete for colchicine binding sites (Kuo et al., 2004), CA-4 was selected as a control, and compound 48 was effective in inhibiting microtubule aggregation (IC50 = 1.6 μM) compared to CA-4 (IC50 = 2.1 μM), which was positively correlated with its anti-proliferative activity. The results of the [3H]-colchicine binding assay showed that compound 48 bound tightly to the colchicine binding domain with a binding affinity comparable to that of CA-4.
Lee et al. (2011) used amine, sulphide, and sulfone group substitution of compound carbonyls from compound 48 (Figure 8) based on Nien’s study. The results showed that compounds 49 (mean IC50 = 42 nM) and 50 (mean IC50 = 12 nM) (Figure 9) containing sulfide and sulfone groups between 3,4,5-trimethoxyphenyl ring and 5-amino-6-methoxyquinoline showed significant antiproliferative activity against KB, HT29 and MKN45 cells. Compound 49 inhibited microtubulin polymerization (Kuo et al., 2004; Liou et al., 2004) (IC50 = 2.0 μM), similar to CA-4. They went on to study the C-5 substituent of the 3′,4′,5′-trimethoxybenzoyl-6-methoxyquinoline derivative and confirmed that compound 49 containing the hydroxyl group had excellent antiproliferative activity (IC50 = 3.4 nM) and microtubule-stabilizing potency of 1.5 μM, greater than that of CA-4 (IC50 = 1.9 μM). It also blocked blood vessels and when HUVECs were treated with compound 49 at 15, 30, 60, and 120 nM, compound 49 was able to destroy the formed capillaries in a concentration-dependent manner. In addition, compounds 49 and 50 showed significant efficacy against MDR/MRP-related drug-resistant cell lines (KB-vin10, KB-S1, and KB-7D) with mean IC50 values of 2.6–6.7 nM, respectively.
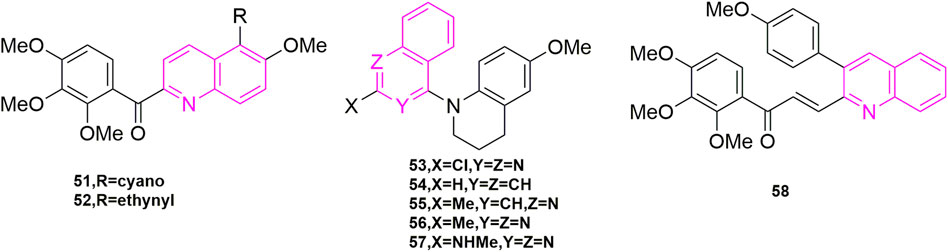
FIGURE 9. The structures of the compounds designed by Lee et al. (2012), Wang et al. (2013b), and Tseng et al. (2015).
Lee et al. (2012) continued his previous studies, also starting with compound 49, and continued to investigate the C-5 substituents of 3,4′,5′-trimethoxybenzoyl-6-methoxyquinoline derivatives, choosing cyano, acetylenic and various aryl groups that are less used by major chemists to design and synthesize a new series of new aryl quinoline derivatives. Three human cancer cell lines, KB, HT29, and MKN45 cells were assayed for antiproliferative activity. Compound 51 and compound 52 (Figure 9) exhibited significant antiproliferative activity with mean IC50 values of 30 and 57 nM, comparing to colchicine. Interestingly, when they attempted to validate the anti-microtubulin activity and colchicine binding activity of compounds 51 and 52 using colchicine and CA-4 as controls, the binding affinity of cyano 51 and alkyne 52 to the colchicine binding site (Nien et al., 2010) was introduced at 47% and 44% respectively with a 2-fold reduction in activity compared to CA-4, suggesting that both compounds may be bound to another binding site of colchicine binding. In addition, compound 51 and compound 52 were selected to observe the anti-microtubulin activity at concentrations of 1.25, 2.5, 5, 10, and 20 μM, respectively, and showed complete inhibition of microtubulin activity at a concentration of 10 μM. Then they each also inhibited MAP-rich microtubulin aggregation in a concentration-dependent manner. Finally, the reason whether the introduction of cyano and alkyne groups is related to the mode of microtubule protein interactions is unclear at present.
Wang et al. (2013b) also continued his previous research by exploring three design options using methyl 6-chloro-2-(N-(4-methoxyphenyl)-N-methyl) amino nicotinate as the lead compound. The first was that they wanted to explore how to affect the growth inhibitory activity of tumor cells through some specific modifications, the second was that they opted for a conformational restriction strategy that explored feasible binding conformations and possible ligand-target interactions, and the third was to enhance the affinity to the binding site on microtubule proteins to produce good antitumor activity. A total of 24 new compounds were designed and synthesized using these three strategies, of which compound 53 (Figure 9) was the most active and showed high cytotoxicity (Perez-Sayans et al., 2010; Hung et al., 2012) (GI50 = 1.5–1.7 nM), especially against P-gp-expressing multidrug-resistant cell lines (vincristine-resistant KB) (KB-vin), in antiproliferative activity against four human tumor cell lines [A549, KB, KB-vin, and prostate cancer (DU145)] using paclitaxel as control (GI50). Analogues 54, 55, 56, and 57 (Figure 9) were also comparable [GI50 = (0.011–0.19 μM)]. In further studies, the active compounds 55–57 significantly inhibited microtubulin polymerization (IC50 = 0.92–1.0 μM) and strongly inhibited colchicine binding to microtubulin (75%–99%), using CA-4 (IC50 = 0.96 μM) as a control. In contrast to the previously designed pharmacological mechanism studies, they involved the study of drug solubility (Log P parameter) and in vivo metabolic stability (Roehm et al., 1991; Sun et al., 2012), in which five compounds, compounds 54, 55, 56 (3.21–7.67 μg/ml) exhibited better water solubility than the other two compounds (1.0 μg/ml), while with propranolol (t1/2 = 3–5 h) and terfenadine (t1/2 = 3 h) were used as positive controls to further assess the metabolic stability of compounds 54–56 by in vitro human liver microsomal culture assays. Compound 54 (t1/2 = 7.89 min) and compound 53 (t1/2 = 10.59 min) were unstable and rapid metabolism may help to reduce drug toxicity in vivo and could be used as novel anticancer candidates.
Tseng et al. (2015) designed and synthesized a series of novel 3-phenylquinolinone derivatives against breast cancer and performed antiproliferative activities in vitro on three breast cancer cell lines (MCF-7, MDA-MB-231, and SKBR-3) and one non-cancerous normal epithelial cell line (H184B5F5/M10). Among them, (E)-3-[3-(4-methoxyphenyl) qui-nolin-2-yl]-1-(3,4,5-trimethoxyphenyl) prop-2-en-1-one (compound 58, Figure 9) showed anti-proliferative activity against MCF-7 (IC50 = 1.05 μM), MDA-MB-231 (IC50 = 0.75 μM) and SKBR-3 (IC50 = 0.78 μM) with growth inhibitory activity and no significant cytotoxicity against normal H184B5F5/M10 cell lines (IC50 > 10 μM). Therefore, a series of pharmacological mechanism studies were subsequently performed. MDA-MB-231 cells were selected for cell cycle experiments and compound 58 at 1, 5, and 10 μM concentrations, with CIL-102 used as a positive control, were treated together for 12, 24, and 36 h. As shown, 28% and 36% of cells were arrested in the G2/M phase when treated with 1 μM and 5 μM compound 58, respectively, indicating that the induced cell cycle was concentration and time-dependent. Western blot experiments showed, downregulation of CDC25c, CDK1, and the cell cycle protein cyclin B1 in cells treated with compound 58. To further investigate the mechanism, they selected CLI-102 as a control (10 μM) and selected the same concentration of compound 58 as in the cell cycle experiments (1, 5, and 10 μM), performed immunofluorescence experiments, and they found that the cells exhibited filamentous structures and a reduced round tube state in the cytoplasm. In addition, they went on to perform Western blot experiments to observe the microtubulin polymerization inhibition assay, and as shown, the microtubulin polymer form showed a concentration-dependent decrease, indication inhibition of microtubulin polymerization. They concluded that the inhibition of cell growth by compound 58 was due to apoptosis and they analysed the effect of compound 58 on the anti-apoptotic protein Bcl-2 and the pro-apoptotic proteins Bad and Bax. Compound 58 had no significant effect on the expression of apoptotic protein Bad, but upregulated apoptotic protein Bax and downregulated anti-apoptotic protein Bcl-2. In parallel they also evaluated caspase-3, caspase-8 and PARP in compound 58 treated MDA-MB-231 cells by Western blot assay. From this experiment they confirmed that compound 58 induces cell cycle arrest in the G2/M phase of PARP by activating caspase-3, caspase-8, which leads to cell death.
Chaudhary et al. (2016) started with the structure of CA-4 to ensure that the A and B ring remain Z-form. Like other chemists, they also considered various linkers to replace the double bond. Among their considerations, they selected the 2-aminoimidazole backbone, a backbone that is also a valuable molecular motif in medicinal chemistry. They designed and synthesized a series of 4,5-diaryl-2-aminoimidazoles and in performing experimental studies for antiproliferative activities, they found that compound 59 (Figure 12) showed good activity against five types of human cancer cells, which are MCF-7 (IC50 = 3 ± 2 nM), HeLa (IC50 = 10 ± 1 nM), hepatocellular carcinoma (HuH-7, IC50 = 335 ± 10 nM), MDA-MB-231(IC50 = 96 ± 13 nM) and drug-resistant mouse breast cancer (EMT6/AR1, IC50 = 350 ± 7 nM). In these cells, compound 59 was more potent than CA-4 (IC50 = 18 ± 2, 25 ± 2, 430 ± 9, 332 ± 32, and 495 ± 11 nM), and they speculated that it might be that the quinoline ring played a role in addition to the 2-aminoimidazole backbone. It might be due to the biological target—the N-/NH and C2-amine functional groups of the 2-aminoimidazole and quinoline N rings in the ligand play the role of hydrogen bonding to stabilize the ligand. Immunofluorescence experiments were performed immediately after and they found that compound 59 mildly depolymerized microtubules at a concentration of 5 nM and strongly depolymerized MCF-7 cells at 10 nM, which found that the compound blocked the single-stage spindle in mitosis. Meanwhile, they performed cell cycle experiments using MCF-7 cells using CA-4 as a control, and 40% and 74% of the cells were in the G2/M phase at 5 and 10 nM of compound 59 concentrations, which compared to CA-4 (20% and 60%). To verify the reliability of the results, they stained the cells with Ser-10, which showed a concentration-dependent increase in the number of staining group proteins compared to the carrier cells. They evaluated the effect of compound 59 on MDA-MB-231 wound healing in migrating tumour cells, using CA-4, a well-known antivascular agent, as an example. Cells in the Control group had complete wound closure after 18 h. The results showed that compound 59 strongly inhibited the migration of MDA-MB-231 (50 and 150 nM, 56% ± 3% and 52% ± 4% inhibition) cells compared to CA-4 (50 nM, 8% ± 1% inhibition). Finally, they verified that the compound 59 is bound to the colchicine binding site (Cortese et al., 1977; Sackett, 1993) of microtubulin by measuring the fluorescence intensity of compound 59, selected from CA-4 mycotoxin, paclitaxel, and vincristine.
Kumari et al. (2019) wanted to further elucidate the specific molecular mechanism and antitumor activity of compound 59 (Figure 10) based on the previous study by Chaudhary. Fibroblasts (L929) and non-tumorigenic mammary epithelial cells (MCF-10A) with IC50 values of 147 ± 45, 136 ± 30, and 3,316 ± 493 nM, respectively. In addition, they performed EB1 gene transfection and EB1 comet number calculations and they found that compound 59 strongly depolymerized microtubules of HeLa cells in interphase and mitotic cells, and also observed that reduction in the number of comets in cells treated with 5, 10, and 20 nM compound 59. To further clarify the mechanism, they investigated the binding of compound 59 to EB1 with microtubules in vitro, and the results were due to a reduction in the number of comet cells due to a reduction in the number of microtubules in the cell body, not due to a reduction in EB1 binding to intracellular microtubules. They concluded not only that compound 59 inhibited intermediate microtubule dynamics in HeLa cells, but also that it could also interfere with mitotic spindle dynamics in the cells. They also evaluated chromosomes by examining the effect of compound 59 on chromosome movement by time-lapse imaging of live HeLa cells expressing histone 2B, using cells treated with a concentration of 10 and 20 nM, which showed that the compound 59 inhibited chromosome movement and that these chromosomes could not be aligned on tumid phase plate, and even an increase in chromosome consistency index (CI) was also observed as well as mitotic defects and chromosome misalignment, which could be responsible for the diminished microtubule dynamics. Subsequently, to investigate whether misaligned chromosomes activate the spindle assembly checkpoint (SAC), SAC activation was next examined after compound 59 treatment, which showed that cells treated with compound 59 were blocked at mid mitosis and were unable to enter the ate phase due to SAC activation, and in addition, they observed that compound 59 inhibited mitotic spindle assembly and induced multistage spindle formation. It is known that there are two ways of SAC activation, one is cell slippage through mitosis and the other is apoptosis. To confirm apoptosis, they did Western blot experiments with HeLa cell-associated proteins and showed that compound 59 can activate caspase-3 and caspase-9 to induce cleavage of PREP-1, which is consistent with the previously observed apoptosis. In addition, they also did mitochondrial membrane potential experiments and found that the compound could cause apoptosis by causing a decrease in mitochondrial membrane potential. These two forms of apoptosis will be of further value for subsequent studies. Finally, they were also the third study to perform animal experiments. They used NOD-SCID mice and according to Chaudhary’s study, which used compound 59 and selected MCF-7 cells to determine the effect of xenografts in mice. The results of the study showed 49% reduction in tumor volume in mice treated with compound 59 compared to the control group, with no significant effect on body weight, suggesting a lesser side effect and a worthwhile candidate to investigate for the treatment of cancer.
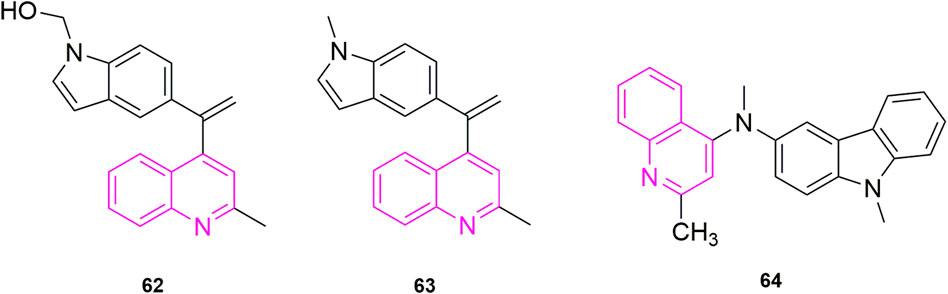
FIGURE 10. The structures of the compounds designed by Li et al. (2019) and Khelifi et al. (2017).
Ibrahim et al. (2020) started from the analysis of the structure of CA-4 and proposed the same idea as major chemists to use heterocycles to replace the unstable double-built structure in CA-4, for example. Some chemists used quinoline to replace the B ring showing compounds with 10 times more powerful activity than CA-4. So, they designed two models of compounds, the first model was synthesized using the 3,4,5-trimethoxyphenyl fraction and the anthraquinone fraction as various electronic substituents for bioelectronic isomeric substitution of the B ring of CA-4. The second model is to add structural rigidity by introducing a C-ring (a hydrazone open linker and its cyclic form) to replace the carbene bond, and this prevents CA-4 from isomerizing in vivo. These compounds were subsequently tested for anti-value-added activity in four human cancer cells: MCF-7, human leukaemia cells (HL-60), HCT-116, and HeLa. Most of the compounds showed significant antiproliferative activity in the sub-micromolar range, with the most potent compound being compound 65 (Figure 11) (7-tert-butyl-substituted quinoline) (IC50 = 0.02–0.04 μM), and they later did antiproliferative activity assays (IC50 > 35 μM) in MCF-10A, showing that the compound against MCF-7 cells with good selectivity. Then they further did in vitro microtubulin polymerization inhibition assay and colchicine binding site assay, and the results were as expected, compound 65 effectively inhibited microtubulin polymerization and inhibited the colchicine binding site. With the results from the previous step, they continued with the cell cycle assay, selecting compound 65 at a concentration of 50 and 250 nM to treat MCF-7 cells, and the percentage of cells in the G2/M phase increased to 23.7% and 35.6% within 48 h compared to the control, a trend that further increased as time progressed. It indicates that compound 65 can induce MCF-7 cells blocked in the G2/M phase in a concentration-dependent manner. At the same time, they also did an apoptosis assay and MCF-7 cells treated with 50 nM and 250 nM significantly increased to 15% and 29% compared to the control (0.8%), indicating that the sub-compound can induce apoptosis in a concentration-dependent manner. The above results make compound 65 worthies of further investigation as an effective chemotherapeutic agent for targeting microtubulin.
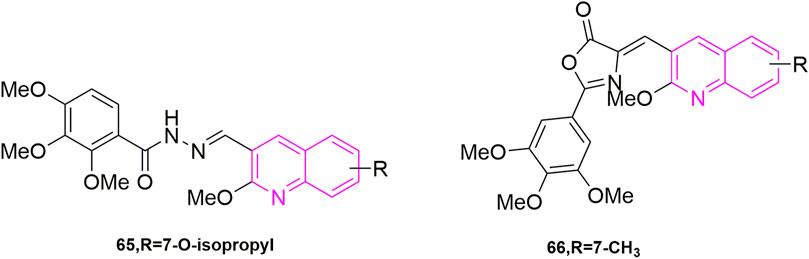
FIGURE 11. The structures of the compounds designed by Ibrahim et al. (2020, 2021).
Another study was carried out by Ibrahim et al. (2021), who further optimized CA-4 from its structure by inserting rigid oxazolones and imidazolines between the A and B ring to maintain cis-activity, and then maintained 3,4,5-trimethoxyphenyl by altering the electron group substitution effect. The designed compounds were subjected to antiproliferative activity assays against four MCF-7, HL-60, HCT-116, HeLa) using CA-4 as a control, where compound 66 (Figure 11) stood out with the optimal effect (IC50 = 0.019–0.042 μM), and more excitingly the compound was selective for cancer cells by verifying that MCF-10A (IC50 > 50 μM), compared to an IC50 value of 6.1 μM for CA-4. They have since performed more detailed mechanistic studies, such as the next microtubulin polymerization inhibition assay, where they used CA-4 as a control (IC50 = 2.17 μM) and found that compound 66 (IC50 = 1.21 μM) strongly inhibited microtubulin polymerization. They then proceeded to use compound 66 concentrations at 1 and 10 μM on the ability of colchicine to compete with microtubulin, using CA-4 as a control (86% and 97%), with 79% and 87% inhibition of compound 66, this result suggests that the odd compound is involved in microtubulin polymerization inhibition via the colchicine binding site. After that they continued with cell cycle experiments, wanting to understand the potential anti-value-added activity of compound 66, and selected 50 and 250 nM to observe the extent of cell block in the G2/M phase at 0, 24, 48, and 72 h. After 48 h, the percentage of the G2/M phase was 28.4% and 38.3% compared to the control (9.2%). After 72 h it was 33% and 40.8%. In addition, they found that the compound also induced phase apoptosis, they also looked at the percentage of apoptosis in cells at 24, 48, and 72 h. They mainly looked at apoptosis at 0 and 250 nM and CA-4 at 50 nM with an increase of 15%, 21%, and 29% respectively, in that order, compared to the control (1%). They then performed Western blot experiments and confirmed that the compound triggered apoptosis in MCF-7 cells by down-regulating the expression level of the anti-apoptotic protein Bcl-2 and up-regulating the expression of the pro-apoptotic protein Bax, while activating caspase-9 and could participate in the mitochondrial apoptotic pathway, causing changes in mitochondrial membrane potential. The final cell scratching assay also confirmed that the compound could effectively inhibit cancer cell migration. In conclusion, this compound 66 is a drug worthy of further investigation for development as a microtubulin inhibitor.
2.2 Modification of iso-CA-4
Khelifi et al. (2017) study found that compounds using a quinoline ring to replace iso-CA-4 and 3,4,5-trimethoxy (A ring) in CA-4, while containing 3-hydroxy-4-methoxy on the B ring were the best actor. They tested five human cancer cell lines (IC50 < 10 nM), namely human astrocytoma cell lines (U87), acute phase chronic myeloid leukaemia cell lines (K562), Adriamycin-resistant K562 (K562R), A-549 and human colon cancer cells (HCT-116). To further investigate the mechanism, they continued to do immunofluorescence experiments using A549 cells, after treating the cells with compound 60 (Figure 12) for 24 h, mitotic spindle formation was disrupted at 1 nM concentration, and at 5 nM concentration, the microtubule system had been severely disrupted and even multinucleated cells appeared. They performed further cell cycle experiments, selecting compound 60 at concentrations of 0.5, 1, 5, and 10 nM, using dimethyl sulfoxide (DMSO) as a control. It can be observed that the compound 60 blocked the A549 cells in the G2/M phase in a concentration-dependent manner, with a 10 nM concentration blocking the entire population of A549 in G2/M phase (Provot et al., 2013). The final molecular docking showed that when compound 60 was bound to microtubulin, the nitrogen atom of the quinoline A ring could form a hydrogen bond with the Cys241 residue of the B subunit of microtubulin, which was noted that the colchicine binding site to microtubulin showed a binding pattern comparable to that previously used with the quinazoline analogues (iso-CoQ) and iso-CA-4.
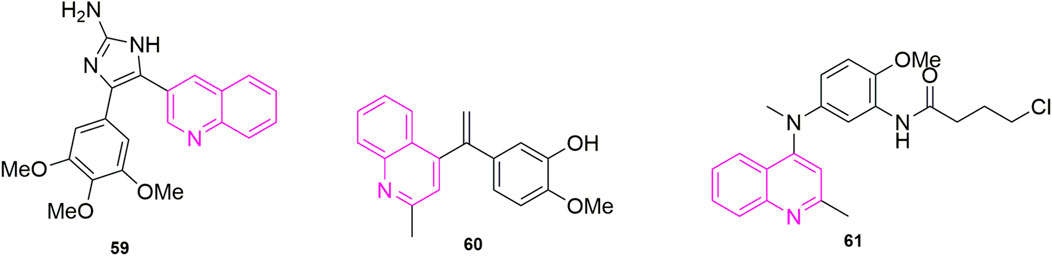
FIGURE 12. The structures of the compounds designed by Chaudhary et al. (2016), Khelifi et al. (2017), and Tseng et al. (2015).
Zhou et al. (2017) resynthesized a series of novel 4-anilinoquinoline derivatives based on their original synthesis of 4-anilino cyclic coumarin derivatives by replacing the coumarin ring with a quinoline ring and performed antiproliferative activity (Sri Ramya et al., 2017) studies in the human colon, lung, ovarian and breast cancer cells, where compound 61 (Figure 12) exhibited potent cytotoxicity (IC50 = 1.5–3.9 nM). In addition, they performed cell cycle assays, immunofluorescence assays, microtubule kinetic analysis and colchicine binding site competition assays sequentially to verify the pharmacological mechanism of the antitumor activity of the compound, which were also the first to study the antitumor activity of quinoline analogues in animal models. In the cell cycle assay, compound 61 was selected at a concentration of 3, 10, and 30 nM, the cells present in the G2/M phase (Wang et al., 2015) increased from 15.34% to 80.67%. In immunofluorescence experiments, they chose colchicine and paclitaxel as controls and treated with human ovarian cancer (A27280) cells with 5 nM and 50 nM of compound 61, and it was seen that the compound 61 significantly disrupted microtubule formation at 50 nM, which was similar to the colchicine effect and even more targeted to microtubule proteins. In the microtubule kinetic analysis assay, compound 61 was taken at a concentration of 0.4, 2, and 10 μM and the control sample was chosen to be colchicine. Compound 61 inhibited microtubule protein polymerizations in a concentration-dependent manner, at 10 μM being significantly stronger than colchicine. In the colchicine binding site (Fortin et al., 2010) competition assay, compound 61 was selected at a concentration of 1, 5, and 25 μM, in Human hepatocellular carcinomas cell lines (HepG2), a dose-dependent inhibition of β-duct formation, leading to the disappearance of the adduct bands, which could be observed by Western blot experiments. Finally, they established an HCT-116 xenograft model in nude mice and selected 5-fluorouracil as a positive control drug (Wang et al., 2013a). They found that compound 61 significantly inhibited tumor growth, with an average inhibition rate of 54.3% already at day 18. The above results laid the clinical foundation for compound 61 to become an anti-tumor drug.
Starting from the structure of iso-CA-4, Li et al. (2019) designed and synthesized a new series of quinoline-indole derivatives by replacing the 3,4,5- trimethoxyphenyl and isovanillin in the structure with quinoline and indole rings, respectively, and selected five human cancer cells for evaluation of antiproliferative activity, which were HepG2, KB, HCT-8, MDA-MB-231, and mouse hepatocellular carcinoma cells (H22). The best activities were compound 62 (Figure 10) and compound 63 (Figure 10) (IC50 = 2 nM–11 nM), which were more active than CA-4 (IC50 = 11 nM–14 nM). To elucidate whether compound 63, the best active compound, targeted the microtubule protein-microscopic system, they went on to evaluate the effect of the compound on microtubule dynamics in K562 cells, using paclitaxel and colchicine as control, which found that compound 50 had 90.5% binding to colchicine at 5 μM. Further, they evaluated whether the compound 63 can disrupt the cell cycle distribution performed cell cycle experiments, using DMSO as control, the cell stays in G2/M phase increased to 23.21% compared to the control (10.51%). While they performed apoptosis experiments, selected compound 63 concentration at 1, 2, and 4 nM concentration, which was the first time in quinoline analogues to use Flow cytometry to analyse the article of apoptosis. The result that the percentage of apoptotic cells in the control group was 5.96% and after 48 h the total number of apoptotic cells reached 25.7%, 54.9%, and 72.8% respectively, indicating that compound 63 induced apoptosis in K562 cells in a concentration-dependent manner. Furthermore, to further understand whether the compound-induced apoptosis was involved in mitochondrial membrane integrity disruption, JC-1 assay was performed. Again, the concentration of compound 63 was selected as 1, 2, and 4 nM concentration and it was observed from the result that the collapse of mitochondrial membrane potential increased from 6.17% to 28.99%, 45.10%, and 73.55% respectively. This confirmed that apoptosis could disrupt mitochondrial membrane potential integrity. Later they performed immunofluorescence analysis to investigate whether the compound could disrupt microtubule dynamics in the cells, and after treatment of K562 cells with compound 62 at a concentration of 1, 2, and 4 nM for 24 h, which was evident that the microtubule network in the cells was disrupted, and these results indicated that compound 62 could disrupt the microtubule network. Based on the unique anti-vascular activity of CA-4, they then evaluated the anti-vascular activity of compound 62 in human umbilical vein endothelial cells (HUVEC) by performing cell scratching experiments, which could observe that compound 62 could inhibit HUVEC cord formation in a concentration-dependent manner. They designed the compound 62 as the second person to perform animal experiments in quinoline compound design, and they evaluated the antitumor activity of compound 62 and compound 63 by inoculating H22 cells into the right side of mice to establish a mouse model of hepatocellular carcinoma xenograft. They chose paclitaxel, CA-4, and CA-4P as positive controls, and both compounds at a dose of 20 mg/kg/day showed 57.3% and 63.7% tumor reduction at 21 days after treatment, even more than CA-4 (51%) and CA-4P (62.7%), and neither compound 62 and 63 had a significant effect on the body weight of the mice compared to the control drug.
Khelifi et al. (2019) based on the previous design of replacing the 3,4,5-trimethoxyphenyl in CA-4 or iso-CA-4 with a quinoline or quinazoline ring to maintain good antitumor activity, designed a new series of compounds based on this by replacing the B ring with a carbazole or indole ring, and they first performed antiproliferative activity assay, in which compound 64 (Figure 10) showed the strongest activity (IC50 = 70 pM). Thereafter, antiproliferative activity assays were performed in six more cells, which were A2780, cisplatin-resistant human ovarian tumor (A2780R), human pancreatic cancer (MiaPaca2), K562R, K562R and breast cancer cells (JIM-T1) continued evaluation, which found that the activity of compound 64 was all located within sub-nanomolar levels. Thereafter they performed cell cycle experiments and selected compound 64 at concentrations of 1/5/10 nM, compared to the control DMSO (23%), which blocked the HCT-116 cell cycle at G2/M by 25%, 80%, and 82%, respectively. Somewhat similar to previous methods, they assessed the ability of compound 64 to induce apoptosis by a specific apoptosis assay that cleaved pro-cysteine aspartate to active cysteine aspartate. HCT-116 cells were incubated with 0.5, 1, 5, and 10 nm of compound 64 for 24 h. The activity of caspase-3 and caspase-7 was assessed using a standard cysteines assay, using DMSO as a control. The proteolytic activity of cystoplasties was significantly increased in HCT-116 cells treated with compound 64, implying that compound 64 induced apoptosis at a low concentration of 5 nM. Finally, they did in vitro antiproliferative activity assays, in which flattened endothelial cell aggregates form a reticular vascular network consisting of capillary-like vessels when grown on a stromal gel. After 2, 3, and 5 h, compound 64 was added to the tubular structure at a concentration of 10 nM, and it was observed that compound 64 rapidly disrupted the integrity of the vascular network. Their results also revealed a highly cytotoxic HUVEC showing antiproliferative activity at extended times (72 h) with a GI50 value of 3.23 ± 0.28 nM. This did not correlate with the disruption of the vasculature observed in its short time, suggesting that this compound is expected to be further screened as an anti-vascular active drug.
3 Similarities and differences in pharmacological experiments
The introduction of quinoline heterocycles, and the insertion of rigid heterocycles between the A and B rings seem to be a common interest of all major researchers in all designed quinoline compounds. By observing the designed compounds, it is found that the double bond is substituted with a heterocyclic ring, and the B ring is substituted with a quinoline ring to obtain a compound with higher activity, such as compound 47 (MCF-7, IC50 = 3 ± 2 nM). Some of them also modify the compounds designed by their predecessors, including the substitution of substituents or modification of linkage bonds, all to more active compounds, such as compound 53 and 57, which had mentioned above.
Among the compounds they designed, the human cancer cells they used the most were KB, which was used by five investigators, followed by MCF-7, HCT-116, and HeLa, which were used by three investigators each (Figure 13). Of course, after the antiproliferative activity assay in the human cancer cells they selected, they continued to select the compound with the best activity for the next mechanistic study. Their mechanistic studies included cell cycle assays, immunofluorescence assays, apoptosis assays, microtubulin polymerization inhibition assays, competitive colchicine binding assays, mitochondrial machine reactive oxygen mechanism assays, Western blot assays, cell scratching assays, and cell colony assays as well as molecular docking assays. In their pharmacological experiments, tubulin polymerization inhibition experiments were involved, indicating that this pharmacological mechanism experiment is an important means to prove the effective activity of the compounds. Some experiments have explored deeper pharmacological mechanisms, such as EB1 gene transfection and EB1 comet count, as well as the alteration of a series of apoptosis induced after SAG activation, such as compound 47.

FIGURE 13. The researchers used all cell lines.
In most cell cycle experiments, the compound with the best quinoline derivative activity blocked the selected cancer cells in the G2/M phase in a concentration-dependent manner. Similarly in apoptosis experiments, the percentage of apoptotic cells increases with increasing compound concentration. In immunofluorescence experiments, the microtubule state disrupted by the compounds is usually seen, even with multinucleation. In microtubule protein polymerization inhibition assays, most quinoline compounds exhibit inhibition of microtubule proteins, some even beyond CA-4. In mitochondrial mechanism assays, compounds cause a collapse of the mitochondrial membrane potential, which leads to apoptosis. In Western blot experiments, compounds triggered apoptosis in cancer cells by downregulating the expression level of the anti-apoptotic protein Bcl-2 and upregulating the expression of the pro-apoptotic protein Bax, while activating caspase-9 also triggered apoptosis. Cell scratching experiments also illustrated that quinoline-like microtubulin inhibitors can inhibit the migration of cancer cells. For the selected derivatives with activity beyond CA-4, the researchers further conducted animal experiments, taking subcutaneous inoculation of cancer cells and observing the size of tumor cells in mice after drug treatment, as well as observing changes in body weight of mice, and they found that most of the drugs with good activity had a manageable effect on body weight of mice. These pharmacological experiments can provide strong evidence for the follow-up research on these drugs.
4 Concluding remarks
This paper reviews all the designed and synthesized CA-4 analogues containing quinoline structures from 1992 to 2022, from the beginning of design to synthesis to pharmacological activity studies. Among these numerous quinoline-based derivatives, several good compounds deserve further push, such as compound 46, which has been studied in detail from design to synthesis and then in vitro, and the results of the study are in line with expectations and are ready to be studied for consideration to enter the clinical study part. In addition, we did statistics on quinoline derivatives and found that the most used cell in the antiproliferative activity assay was the KB. Therefore, we suggest that the future design of quinoline derivatives can be considered to use this cell for the study. One point worth mentioning is that various researchers have only made modifications to the structure and stability, including modifications to the A and B ring, insertion of rigid thickened heterocyclic quinolines or indoles between the A and B ring, and substitution of 3,4,5-trimethoxyphenyl for other heterocycles. Fortunately, the modified derivatives have maintained better activity, and some even surpassed CA-4. However, for the modified toxicity of the compounds, only three investigators have conducted experiments on two non-human cells and confirmed the high selectivity of their designed compounds. Therefore, we suggest that the selectivity for normal cells can be improved on the existing basis. We encourage other researchers to also select potent compounds from mechanistic studies beyond the other mechanistic pathways studied above, not limited to apoptosis. For example, we propose the use of potent compounds in combination with cell signaling pathways for deeper studies. Finally, these quinoline-containing derivatives enrich the diversity of tubulin inhibitors, and also lay a foundation for the druggability of these compounds to go to the clinic.
Author contributions
CW; writing—original draft preparation, CW and JC; writing—review and editing, SY, LS, and YZ; supervision, Figure, JM, JZ, RZ; Table, DX; All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Natural Science Foundation of Shandong (ZR2021QH156) and the Medical and Health Science and Technology Development Plan Project of Shandong (202113051140).
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1040333/full#supplementary-material
References
Alvarez, R., Aramburu, L., Puebla, P., Caballero, E., Gonzalez, M., Vicente, A., et al. (2016). Pyridine based antitumour compounds acting at the colchicine site. Curr. Med. Chem. 23 (11), 1100–1130. doi:10.2174/092986732311160420104823
Bai, R. L., Paull, K. D., Herald, C. L., Malspeis, L., Pettit, G. R., and Hamel, E. (1991). Halichondrin B and homohalichondrin B, marine natural products binding in the vinca domain of tubulin. Discovery of tubulin-based mechanism of action by analysis of differential cytotoxicity data. J. Biol. Chem. 266 (24), 15882–15889. doi:10.1016/s0021-9258(18)98491-7
Chaudhary, V., Venghateri, J. B., Dhaked, H. P., Bhoyar, A. S., Guchhait, S. K., and Panda, D. (2016). Novel combretastatin-2-aminoimidazole analogues as potent tubulin assembly inhibitors: Exploration of unique pharmacophoric impact of bridging skeleton and aryl moiety. J. Med. Chem. 59 (7), 3439–3451. doi:10.1021/acs.jmedchem.6b00101
Chen, M., Chen, H., Ma, J., Liu, X., and Zhang, S. (2014). Synthesis and anticancer activity of novel quinoline-docetaxel analogues. Bioorg. Med. Chem. Lett. 24 (13), 2867–2870. doi:10.1016/j.bmcl.2014.04.091
Cortese, F., Bhattacharyya, B., and Wolff, J. (1977). Podophyllotoxin as a probe for the colchicine binding site of tubulin. J. Biol. Chem. 252 (4), 1134–1140. doi:10.1016/s0021-9258(17)40631-4
Denault, J. B., and Salvesen, G. S. (2002). Caspases: Keys in the ignition of cell death. Chem. Rev. 102 (12), 4489–4500. doi:10.1021/cr010183n
Earnshaw, W. C., Martins, L. M., and Kaufmann, S. H. (1999). Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68, 383–424. doi:10.1146/annurev.biochem.68.1.383
Ferlin, M. G., Bortolozzi, R., Brun, P., Castagliuolo, I., Hamel, E., Basso, G., et al. (2010). Synthesis and in vitro evaluation of 3h-pyrrolo[3, 2-f]-quinolin-9-one derivatives that show potent and selective anti-leukemic activity. ChemMedChem 5 (8), 1373–1385. doi:10.1002/cmdc.201000180
Fortin, S., Lacroix, J., Cote, M. F., Moreau, E., Petitclerc, E., and René, C. G. (2010). Quick and simple detection technique to assess the binding of antimicrotubule agents to the colchicine-binding site. Biol. Proced. Online 12 (1), 113–117. doi:10.1007/s12575-010-9029-5
Gasparotto, V., Castagliuolo, I., and Ferlin, M. G. (2007). 3-substituted 7-phenyl-pyrroloquinolinones show potent cytotoxic activity in human cancer cell lines. J. Med. Chem. 50 (22), 5509–5513. doi:10.1021/jm070534b
Gigant, B., Wang, C., Ravelli, R. B., Roussi, F., Steinmetz, M. O., Curmi, P. A., et al. (2005). Structural basis for the regulation of tubulin by vinblastine. Nature 435 (7041), 519–522. doi:10.1038/nature03566
Honore, S., Pasquier, E., and Braguer, D. (2005). Understanding microtubule dynamics for improved cancer therapy. Cell. Mol. Life Sci. 62 (24), 3039–3056. doi:10.1007/s00018-005-5330-x
Hung, H. Y., Ohkoshi, E., Goto, M., Bastow, K. F., Nakagawa-Goto, K., and Lee, K. H. (2012). Antitumor agents. 293. Nontoxic dimethyl-4, 4'-dimethoxy-5, 6, 5', 6'-dimethylenedioxybiphenyl-2, 2'-dicarboxylate (DDB) analogues chemosensitize multidrug-resistant cancer cells to clinical anticancer drugs. J. Med. Chem. 55 (11), 5413–5424. doi:10.1021/jm300378k
Ibrahim, T. S., Hawwas, M. M., Malebari, A. M., Taher, E. S., Omar, A. M., Neamatallah, T., et al. (2021). Discovery of novel quinoline-based analogues of combretastatin A-4 as tubulin polymerisation inhibitors with apoptosis inducing activity and potent anticancer effect. J. Enzyme Inhib. Med. Chem. 36 (1), 802–818. doi:10.1080/14756366.2021.1899168
Ibrahim, T. S., Hawwas, M. M., Malebari, A. M., Taher, E. S., Omar, A. M., O'Boyle, N. M., et al. (2020). Potent quinoline-containing combretastatin A-4 analogues: Design, synthesis, antiproliferative, and anti-tubulin activity. Pharm. (Basel) 13 (11), 393. doi:10.3390/ph13110393
Kaur, K., Jain, M., Reddy, R. P., and Jain, R. (2010). Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 45 (8), 3245–3264. doi:10.1016/j.ejmech.2010.04.011
Khelifi, I., Naret, T., Hamze, A., Bignon, J., Levaique, H., Garcia Alvarez, M. C., et al. (2019). N, N-bis-heteroaryl methylamines: Potent anti-mitotic and highly cytotoxic agents. Eur. J. Med. Chem. 168, 176–188. doi:10.1016/j.ejmech.2019.02.038
Khelifi, I., Naret, T., Renko, D., Hamze, A., Bernadat, G., Bignon, J., et al. (2017). Design, synthesis and anticancer properties of IsoCombretaQuinolines as potent tubulin assembly inhibitors. Eur. J. Med. Chem. 127, 1025–1034. doi:10.1016/j.ejmech.2016.11.012
Kumari, A., Srivastava, S., Manne, R. K., Sisodiya, S., Santra, M. K., Guchhait, S. K., et al. (2019). C12, a combretastatin-A4 analog, exerts anticancer activity by targeting microtubules. Biochem. Pharmacol. 170, 113663. doi:10.1016/j.bcp.2019.113663
Kuo, C. C., Hsieh, H. P., Pan, W. Y., Chen, C. P., Liou, J. P., Lee, S. J., et al. (2004). BPR0L075, a novel synthetic indole compound with antimitotic activity in human cancer cells, exerts effective antitumoral activity in vivo. Cancer Res. 64 (13), 4621–4628. doi:10.1158/0008-5472.CAN-03-3474
Kuo, S. C., Lee, H. Z., Juang, J. P., Lin, Y. T., Wu, T. S., Chang, J. J., et al. (1993). Synthesis and cytotoxicity of 1, 6, 7, 8-substituted 2-(4'-substituted phenyl)-4-quinolones and related compounds: Identification as antimitotic agents interacting with tubulin. J. Med. Chem. 36 (9), 1146–1156. doi:10.1021/jm00061a005
Lee, H. Y., Chang, J. Y., Nien, C. Y., Kuo, C. C., Shih, K. H., Wu, C. H., et al. (2011). 5-Amino-2-aroylquinolines as highly potent tubulin polymerization inhibitors. Part 2. The impact of bridging groups at position C-2. J. Med. Chem. 54 (24), 8517–8525. doi:10.1021/jm201031f
Lee, H. Y., Lee, L. W., Nien, C. Y., Kuo, C. C., Lin, P. Y., Chang, C. Y., et al. (2012). Application of Suzuki arylation, Sonogashira ethynylation and Rosenmund-von Braun cyanation in the exploration of substitution effects on the anticancer activity of 2-aroylquinolines. Org. Biomol. Chem. 10 (48), 9593–9600. doi:10.1039/c2ob26614h
Li, L., Wang, H. K., Kuo, S. C., Wu, T. S., Lednicer, D., Lin, C. M., et al. (1994a). Antitumor agents. 150. 2', 3', 4', 5', 5, 6, 7-substituted 2-phenyl-4-quinolones and related compounds: Their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 37 (8), 1126–1135. doi:10.1021/jm00034a010
Li, L., Wang, H. K., Kuo, S. C., Wu, T. S., Mauger, A., Lin, C. M., et al. (1994b). Antitumor agents. 155. Synthesis and biological evaluation of 3', 6, 7-substituted 2-phenyl-4-quinolones as antimicrotubule agents. J. Med. Chem. 37 (20), 3400–3407. doi:10.1021/jm00046a025
Li, W., Shuai, W., Sun, H., Xu, F., Bi, Y., Xu, J., et al. (2019). Design, synthesis and biological evaluation of quinoline-indole derivatives as anti-tubulin agents targeting the colchicine binding site. Eur. J. Med. Chem. 163, 428–442. doi:10.1016/j.ejmech.2018.11.070
Liou, J. P., Chang, J. Y., Chang, C. W., Chang, C. Y., Mahindroo, N., Kuo, F. M., et al. (2004). Synthesis and structure-activity relationships of 3-aminobenzophenones as antimitotic agents. J. Med. Chem. 47 (11), 2897–2905. doi:10.1021/jm0305974
Lu, Y., Chen, J., Xiao, M., Li, W., and Miller, D. D. (2012). An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 29 (11), 2943–2971. doi:10.1007/s11095-012-0828-z
Martin, S. J., Reutelingsperger, C. P., McGahon, A. J., Rader, J. A., van Schie, R. C., LaFace, D. M., et al. (1995). Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of bcl-2 and abl. J. Exp. Med. 182 (5), 1545–1556. doi:10.1084/jem.182.5.1545
Monks, A., Scudiero, D., Skehan, P., Shoemaker, R., Paull, K., Vistica, D., et al. (1991). Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. JNCI J. Natl. Cancer Inst. 83 (11), 757–766. doi:10.1093/jnci/83.11.757
Nien, C. Y., Chen, Y. C., Kuo, C. C., Hsieh, H. P., Chang, C. Y., Wu, J. S., et al. (2010). 5-Amino-2-aroylquinolines as highly potent tubulin polymerization inhibitors. J. Med. Chem. 53 (5), 2309–2313. doi:10.1021/jm900685y
Perez-Melero, C., Maya, A. B., del Rey, B., Pelaez, R., Caballero, E., and Medarde, M. (2004). A new family of quinoline and quinoxaline analogues of combretastatins. Bioorg. Med. Chem. Lett. 14 (14), 3771–3774. doi:10.1016/j.bmcl.2004.04.098
Perez-Sayans, M., Somoza-Martin, J. M., Barros-Angueira, F., Diz, P. G., Rey, J. M., and Garcia-Garcia, A. (2010). Multidrug resistance in oral squamous cell carcinoma: The role of vacuolar ATPases. Cancer Lett. 295 (2), 135–143. doi:10.1016/j.canlet.2010.03.019
Provot, O., Hamze, A., Peyrat, J. F., Brion, J. D., and Alami, M. (2013). Discovery and hit to lead optimization of novel combretastatin A-4 analogues: Dependence of C-linker length and hybridization. Anticancer. Agents Med. Chem. 13 (10), 1614–1635. doi:10.2174/187152061310131206162302
Pryor, D. E., O'Brate, A., Bilcer, G., Diaz, J. F., Wang, Y., Wang, Y., et al. (2002). The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 41 (29), 9109–9115. doi:10.1021/bi020211b
Roehm, N. W., Rodgers, G. H., Hatfield, S. M., and Glasebrook, A. L. (1991). An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 142 (2), 257–265. doi:10.1016/0022-1759(91)90114-u
Rubinstein, L. V., Shoemaker, R. H., Paull, K. D., Simon, R. M., Tosini, S., Skehan, P., et al. (1990). Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. JNCI J. Natl. Cancer Inst. 82 (13), 1113–1117. doi:10.1093/jnci/82.13.1113
Sackett, D. L. (1993). Podophyllotoxin, steganacin and combretastatin: Natural products that bind at the colchicine site of tubulin. Pharmacol. Ther. 59 (2), 163–228. doi:10.1016/0163-7258(93)90044-e
Solomon, V. R., Pundir, S., and Lee, H. (2019). Examination of novel 4-aminoquinoline derivatives designed and synthesized by a hybrid pharmacophore approach to enhance their anticancer activities. Sci. Rep. 9 (1), 6315. doi:10.1038/s41598-019-42816-4
Sri Ramya, P. V., Angapelly, S., Guntuku, L., Singh Digwal, C., Nagendra Babu, B., Naidu, V. G. M., et al. (2017). Synthesis and biological evaluation of curcumin inspired indole analogues as tubulin polymerization inhibitors. Eur. J. Med. Chem. 127, 100–114. doi:10.1016/j.ejmech.2016.12.043
Stefanski, T., Mikstacka, R., Kurczab, R., Dutkiewicz, Z., Kucinska, M., Murias, M., et al. (2018). Design, synthesis, and biological evaluation of novel combretastatin A-4 thio derivatives as microtubule targeting agents. Eur. J. Med. Chem. 144, 797–816. doi:10.1016/j.ejmech.2017.11.050
Sun, L. Q., Zhu, L., Qian, K., Qin, B., Huang, L., Chen, C. H., et al. (2012). Design, synthesis, and preclinical evaluations of novel 4-substituted 1, 5-diarylanilines as potent HIV-1 non-nucleoside reverse transcriptase inhibitor (NNRTI) drug candidates. J. Med. Chem. 55 (16), 7219–7229. doi:10.1021/jm3007678
Tseng, C. H., Tzeng, C. C., Hsu, C. Y., Cheng, C. M., Yang, C. N., and Chen, Y. L. (2015). Discovery of 3-phenylquinolinylchalcone derivatives as potent and selective anticancer agents against breast cancers. Eur. J. Med. Chem. 97, 306–319. doi:10.1016/j.ejmech.2015.04.054
Vermes, I., Haanen, C., Steffens-Nakken, H., and Reutelingsperger, C. (1995). A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184 (1), 39–51. doi:10.1016/0022-1759(95)00072-i
Wang, C., Zhang, Y., Wu, Y., and Xing, D. (2021). Developments of CRBN-based PROTACs as potential therapeutic agents. Eur. J. Med. Chem. 225, 113749. doi:10.1016/j.ejmech.2021.113749
Wang, G., Peng, F., Cao, D., Yang, Z., Han, X., Liu, J., et al. (2013a). Design, synthesis and biological evaluation of millepachine derivatives as a new class of tubulin polymerization inhibitors. Bioorg. Med. Chem. 21 (21), 6844–6854. doi:10.1016/j.bmc.2013.02.002
Wang, S., Fang, K., Dong, G., Chen, S., Liu, N., Miao, Z., et al. (2015). Scaffold diversity inspired by the natural product evodiamine: Discovery of highly potent and multitargeting antitumor agents. J. Med. Chem. 58 (16), 6678–6696. doi:10.1021/acs.jmedchem.5b00910
Wang, X. F., Wang, S. B., Ohkoshi, E., Wang, L. T., Hamel, E., Qian, K., et al. (2013b). N-aryl-6-methoxy-1, 2, 3, 4-tetrahydroquinolines: A novel class of antitumor agents targeting the colchicine site on tubulin. Eur. J. Med. Chem. 67, 196–207. doi:10.1016/j.ejmech.2013.06.041
Xia, Y., Yang, Z. Y., Xia, P., Bastow, K. F., Tachibana, Y., Kuo, S. C., et al. (1998). Antitumor agents. 181. Synthesis and biological evaluation of 6, 7, 2', 3', 4'-substituted-1, 2, 3, 4-tetrahydro-2-phenyl-4-quinolones as a new class of antimitotic antitumor agents. J. Med. Chem. 41 (7), 1155–1162. doi:10.1021/jm9707479
Zamzami, N., Marchetti, P., Castedo, M., Decaudin, D., Macho, A., Hirsch, T., et al. (1995). Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 182 (2), 367–377. doi:10.1084/jem.182.2.367
Zhou, Y., Yan, W., Cao, D., Shao, M., Li, D., Wang, F., et al. (2017). Design, synthesis and biological evaluation of 4-anilinoquinoline derivatives as novel potent tubulin depolymerization agents. Eur. J. Med. Chem. 138, 1114–1125. doi:10.1016/j.ejmech.2017.07.040
Glossary
CA-4 Combretastatin A-4
CBSI Colchicine binding site inhibitor
A-549 Human lung carcinoma cell lines
HCT-8 Ileocecal carcinoma cell lines
RPMI-7951 Melanoma cell lines
KB Oral epidermoid carcinoma cells
P-388 and L1210 Two murine leukaemia cell lines
MCF-7 Breast cancer cell lines
CAKI-1 Renal cancer cell lines
SKMEL-2 Melanoma cancer cell lines
ROS Reactive oxygen species
PRAP Poly ADP-ribose polymerase
HT-29 Human colorectal carcinoma cell lines
MKN45 Human gastric cancer cell lines
H460 Non-small-cell lung carcinoma cell lines
MDR Multiple drug resistance
MRP Multi-drug resistant protein
KB-vin P-gp-expressing multidrug-resistant cell lines (vincristine-resistant KB)
DU145 Prostate cancer cell lines
H184B5F5/M10 Non-cancerous normal epithelial cell lines
MDA-MB-231 Breast cancer cell lines
SKBR-3 Breast cancer cell lines
CIL-102 A known inhibitor of tubulin polymerization
HuH-7 Hepatocellular carcinoma cell lines
EMT6/AR1 Drug-resistant mouse breast cancer cell lines
U87 Human astrocytoma cell lines
K562 Acute phase chronic myeloid leukaemia cell lines
HCT-116 Human colon cancer cells
A27280 Human ovarian cancer cell lines
HepG2 Human hepatocellular carcinomas cell lines
H22 Mouse hepatocellular carcinoma cell lines
HUVECs Human umbilical vein endothelial cell lines
A2780R Cisplatin-resistant human ovarian tumor cell lines
MiaPaca2 Human pancreatic cancer cell lines
JIM-T1 Breast cancer cell lines
L929 Fibroblasts cell lines
MCF-10A Non-tumorigenic mammary epithelial cell lines
CI Chromosome consistency index
SAC Spindle assembly checkpoint
Keywords: inhibitor of tubulin polymerization, CA-4, CBSI, quinoline, antitumor
Citation: Wang C, Chang J, Yang S, Shi L, Zhang Y, Liu W, Meng J, Zeng J, Zhang R and Xing D (2022) Advances in antitumor research of CA-4 analogs carrying quinoline scaffold. Front. Chem. 10:1040333. doi: 10.3389/fchem.2022.1040333
Received: 09 September 2022; Accepted: 10 October 2022;
Published: 28 October 2022.
Edited by:
Xi Zheng, Rutgers, The State University of New Jersey, United StatesReviewed by:
Hadia Almahli, University of Cambridge, United KingdomFarhad Eisvand, Mashhad University of Medical Sciences, Iran
Copyright © 2022 Wang, Chang, Yang, Shi, Zhang, Liu, Meng, Zeng, Zhang and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao Wang, d2FuZ2NoYW8yMDA4NjkyNUAxMjYuY29t; Dongming Xing, eGRtX3RzaW5naHVhQDE2My5jb20=
†These authors share first authorship