 Wensha Xia
Wensha Xia Jianwei Cao
Jianwei Cao Qing Lu
Qing Lu Wensheng Bian
Wensheng Bian- 1Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China
- 2School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing, China
Laser cooling molecules to the ultracold regime is the prerequisite for many novel science and technologies. It is desirable to take advantage of theoretical approaches to explore polyatomic molecular candidates, which are capable of being cooled to the ultracold regime. In this work, we explore two polyatomic candidates, CaNC and SrNC, which are suitable for laser cooling. These molecules possess impressively large permanent dipole moments (∼6 Debye), which is preferred for applications using an external electric field. High-level ab initio calculations are carried out to reveal electronic structures of these molecules, and the calculated spectroscopic constants agree very well with the available experimental data. For each molecule, the Franck-Condon factor matrix is calculated and shows a diagonal distribution. The radiative lifetimes for CaNC and SrNC are estimated to be 15.5 and 15.8 ns, respectively. Based upon the features of various electronic states and by choosing suitable spin-orbit states, we construct two feasible laser cooling schemes for the two molecules, each of which allows scattering nearly 10000 photons for direct laser cooling. These indicate that CaNC and SrNC are excellent ultracold polyatomic candidates with strong polarity.
1 Introduction
Cooling molecules to the ultracold regime is of significant importance for novel science and techniques. Representative applications include precision measurements, quantum information storage, and quantum computing (Tarbutt, 2018; Isaev, 2020). For a long time, however, laser cooling to ultracold regimes has been limited to atoms and diatomic molecules. This obstructs further development of those next-generation techniques. So, it is desirable to extend laser cooling studies to polyatomic molecules. On the adventure of exploring polyatomic molecular candidates for direct laser cooling, the Doyle group and the Berger group suggest several polyatomic molecules suitable for direct Doppler cooling (Isaev and Berger, 2016; Kozyryev et al., 2016). Then, Doyle and co-workers contribute a series of milestone work such as experimentally realizing laser cooling SrOH and CaOCH3 to the ultracold regime (Kozyryev et al., 2017; Baum et al., 2020; Mitra et al., 2020). Further studies have also been carried out such as cooling CaOH to the sub-Doppler temperature, accurate determination of vibrational branching ratios (VBRs) and radiative lifetimes for YbOH (Baum et al., 2021; Vilas N et al., 2022). Other theoretical and experimental groups have also made important contributions to this emerging field (O'Rourke and Hutzler, 2019; Isaev et al., 2017; Liu et al., 2021; Xia et al., 2021; Paul et al., 2019; Ivanov et al., 2020a; Ivanov et al., 2020b; Ivanov et al., 2020c).
It is clear that much still awaits to be explored in searching for polyatomic candidates which can be cooled to the ultracold regime. Molecular candidates for laser cooling should meet criteria proposed by Di Rosa, although the criteria were originally brought up for diatomic molecules (Di Rosa, 2004). Based on the three criteria proposed by Di Rosa, candidate molecules should have a highly diagonal Franck-Condon factor (FCF) matrix or vibrational branching ratios. In addition, the candidate molecule should have a short lifetime for the upper state. Thirdly, it is required that there exists no interference from the intermediate electronic state. Recently, Bian proposed the fourth criterion that there should be no electronic-state crossing nearby the states of interests (Li et al., 2020).
Here, accurate ab initio calculations are carried out for the triatomic molecules, CaNC and SrNC. The results show that CaNC and SrNC meet the four criteria mentioned above, which reveals that they are excellent ultracold polyatomic candidates. We have constructed feasible laser cooling schemes for them, which are able to bring them to the ultracold regime. As will be shown later, these molecules possess impressively large permanent dipole moments (PDM); for instance, the dipole moment of CaNC is more than 6 Debye. To the best of our knowledge, these dipole moments are larger than those of other known ultracold candidates for laser cooling. Such a strong molecular polarity is very useful, particularly in applications applying external electric fields (Isaev et al., 2017). It is worth adding that the CaSH molecule also has large dipole moments (∼5.5 Debye) which has been proposed for direct laser cooling (Augenbraun et al., 2020), although it has a bent configuration.
The previous studies on CaNC and SrNC mainly focus on their spectroscopic properties. For the CaNC molecule, the adiabatic excitation energy has been determined by the laser-induced fluorescence spectroscopy (Douay and Bernath, 1990; Whitham et al., 1990; Steimle et al., 1992; Scurlock et al., 1994) as well as theoretical calculations (Bauschlicher et al., 1985; Nanbu et al., 1997; Ishii et al., 2003; Isaev and Berger, 2016). The vibrational spectroscopic constants of CaNC have been experimentally measured by Andrews and co-workers (Lanzisera and Andrews, 1997). Its vibrational frequencies were also calculated at different levels of theories and basis sets (Lanzisera and Andrews, 1997; Nanbu et al., 1997; Ishii et al., 2003; Isaev and Berger, 2016). In addition, high-lying excited states have been studied by the ultraviolet laser spectroscopy (Greetham and Ellis, 2000a). For the SrNC molecule, less has been explored. Its A2Π→X2Σ+ transition has been studied by the laser spectroscopy (Douay and Bernath, 1990), and the vibrational frequencies for the ground state were experimentally determined by the infrared spectroscopy (Lanzisera and Andrews, 1997). Its high-lying excited states have been studied by the jet-cooled laser-induced fluorescence spectroscopy (Greetham and Ellis, 2000a; Greetham and Ellis, 2000b), although the geometry and spectroscopic information for its first excited state is still unavailable.
So far, the laser cooling studies on CaNC and SrNC have been very limited. Isaev et al. (Isaev and Berger, 2016) carried out an interesting study involving the calculation of FCFs for CaNC, however, the laser cooling scheme was not constructed in that work and a further detailed study is required. In particular, it is important to examine if there are nearby electronic states intervening the first excited state, and to take the spin-orbit coupling effect into account. On the other hand, no laser cooling studies have been carried out for SrNC.
In this work, we carry out detailed theoretical investigations on laser cooling of CaNC and SrNC. The article is organized as follows: The computational details are described in the “Methods and computational details” section. In the “Results and Discussion” section, the potential energy curves (PECs) of CaNC and SrNC are first described. Next, the spectroscopic as well as other molecular properties are summarized and compared to experimental values. Lastly, the cooling schemes are constructed for the two molecules. Conclusions are given at the end of the article.
2 Methods and computational detail
In this work, the potential energy curves are constructed based on ∼35 grid points for both CaNC and SrNC. To calculate the energy of each grid point, the constrained optimization is carried out by scanning the M-N distance while freezing the N-C bond length as 1.183 Å and bond angle as 180°. A relaxed scanning of the M-N (M = Ca or Sr) distance is also performed while optimizing the N-C bond length and the bond angle. The electronic energies are calculated by the internally contracted multi-reference configuration interaction method with the Davidson correction (icMRCI + Q) running after the state-averaged complete active space self-consistent field method (SA-CASSCF) (Werner and Knowles, 1988; Knowles and Werner, 1992). The C2v symmetry is used for the constrained PEC scanning, while the Cs symmetry is used for the un-constrained PEC scanning. The selection of active space is very important for the icMRCI + Q calculations (Yu and Bian, 2012; Feller et al., 2014; El-Kork et al., 2017; Zeid et al., 2020; Li and Bian, 2021; Moussa et al., 2021). Here, the chosen active space includes 7 electrons in 13 orbitals for CaNC or SrNC. The active space includes four a1, four b1, four b2, and one a2 molecular orbitals in the C2v symmetry, which involves the 4s electron on Ca (or 5s electron on Sr) and the π and “lone-pair” electrons on NC for the linear M-NC system (M = Ca or Sr). For the PEC calculations as shown in Figure 1, in total 9 states with equal weights are used for the state-averaged calculations. For the frequency calculations, the lowest 4 states are included for the state-averaged calculation with equal weights. The size of basis sets is also important in improving the accuracy of calculations (Feller et al., 2008; Liu and Bian, 2008; Liu et al., 2009; Yu and Bian, 2011). In this work, the def2-QZVP basis set is used for Ca and the dhf-QZVP basis set is used for Sr (Weigend et al., 2003; Weigend and Baldes, 2010), while the aug-cc-pv5z basis sets are used for N and C (Dunning, 1989; Kendall et al., 1992). The spin-orbit coupling effect is calculated by using the Breit-Pauli operator with the state-interacting method (Berning et al., 2000). The PDM are calculated at the same level of theory and basis sets as PEC calculations.
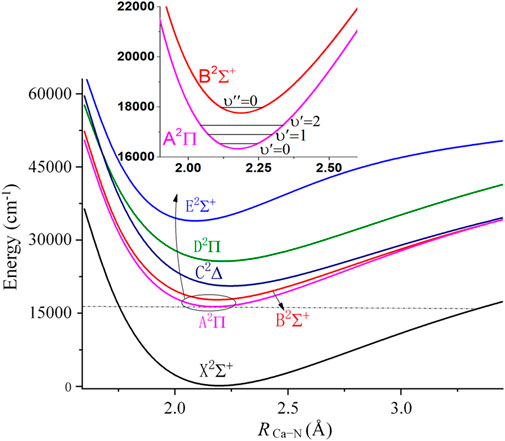
FIGURE 1. Potential energy curves of linear CaNC along RCa-N, with RN-C fixed at 1.183Å. The gray dashed line shows that the minimum of the A state is well below the dissociation limit of the X state.
The local potential energy surfaces around the equilibrium geometries are constructed by fitting ∼50 grid points. Each local potential energy surface is fitted by a 5th order polynomial in the internal displacement coordinate (Lu, 2020; Lu and Peterson, 2020). The energy of each grid point is calculated by the icMRCI + Q method. The active space and basis sets are kept the same as the PEC calculations. The optimized geometries and calculated frequencies are similar using different methods.
The FCFs are calculated as the square of the overlap of electronic and vibrational wavefunctions. The Born-Oppenheimer (BO) approximation is accordingly used. Although explicit considerations of the vibronic coupling effect could increase the accuracy for branching ratio calculations, such as those from the study of CaOH (Zhang et al., 2021), the numerical changes due to the breakdown of BO approximation are expected to be very small, since only heavy atoms and the A (000) state are involved in this work (the hydrogen atom is involved in the CaOH study (Zhang et al., 2021)). Due to the selection rules, the rotational branching can be eliminated by driving the J = 1 → J' = 0 transition for each vibrational level, where J is the rotation quantum number for each vibrational level and the primed label refers to the excited electronic state (Fitch et al., 2021). Based on the calculated FCFs, the radiative lifetime (τ) for the transition is calculated by Isaev et al. (2017)):
where FC is the FCF for each vibronic transition υυ′, and the transition dipole moment (TDM) is calculated at the equilibrium geometry of the ground state using the same level of theory and basis sets as the PEC calculations mentioned above. The ∆E υυ' is the energy difference between the vibronic levels υυ′. g′ and g″ are the degeneracies of upper and lower states, respectively. In addition, the VBRs are calculated following a previous study (Lefebvre-Brion and Field, 2004).
The Doppler temperature (TDoppler) is evaluated by Zeng et al. (2001):
where h and kB are the Planck constant and the Boltzmann constant, respectively. And τ is the radiative lifetime as calculated above.
The recoil temperature (Trecoil) is approximated as (Cohen-Tannoudji, 1998):
where m is the relative molecular mass, and λ is the wavelength for the main transition channel.
The fitting of PECs is carried out by the LEVEL program (Le Roy, 2017). The FCFs are calculated by the ezFCF package (Gozem and Krylov, 2021). All remaining calculations are performed by the Molpro package (Werner et al., 2012; Werner et al., 2015; Werner et al., 2020).
3 Results and discussion
3.1 Potential energy curves
In order to be laser cooled to the ultracold regime, the molecules are required to have no interference from intermediate electronic states. It is well-known that the isocyanide ligand has a large energy gap between the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO), while the HOMO-LUMO energy gap for alkaline earth metals is relatively small. Therefore, the interfering states should mainly result from the metal center. The present calculations show that the three low-lying electronic states possess linear configurations, in accordance with the evidences obtained in the previous studies (Douay and Bernath, 1990; Scurlock et al., 1994; Greetham and Ellis, 2000b). Accordingly, we scan the M-NC (M = Ca or Sr) PECs to examine the interfering states. Figure 1 shows the PECs of the lowest 6 electronic states for the CaNC molecule. The ground state and the first excited state of CaNC are the 2Σ+ and 2Π states, respectively. Higher electronic states include the B2Σ+, C2∆, D2Π, and E2Σ+ states. All states have smooth curves towards the dissociation limit. Based on the molecular orbital coefficients, the unpaired electron is mainly located on the s orbital of the Ca atom for the X2Σ+ state. And it transits to the p orbital of the Ca atom for the A2Π state (The relevant molecular orbitals are shown in Supplementary Figure S1). The second excited state is the B2Σ+ state lying ∼1800 cm−1 above the A2Π state, as shown in the inset panel of Figure 1. This energy separation indicates that the nearby B2Σ+ state should not come into play as an interference state. As a comparison, the energy gap of SrOH between its first and second excited states is ∼1700 cm−1 (Nakagawa et al., 1983), while the molecule has been experimentally cooled to the ultracold regime.
Between the A2Π and X2Σ+ states, the energy gap of CaNC is ∼16000 cm−1. After including the zero-point energy (ZPE) correction, the adiabatic excitation energy is calculated as 16218.3 cm−1 (see Table 1). We have also carried out calculations by scanning the Ca-N distance with all other degrees of freedom optimized. The corresponding adiabatic excitation energy is calculated as 16217.9 cm−1. The little difference between the two adiabatic excitation energies indicates that the N and C atoms move very little during the elongation of the Ca-N bond. Moreover, the calculated adiabatic excitation energy (16218 cm−1) is in excellent agreement with the experimental value of 16229.3 cm−1 (Scurlock et al., 1994). This energy gap is equivalent to ∼617 nm wavelength for the major laser pump. This reveals two additional features of the molecule. First, the excitation energy falls into the region of red color. Therefore, the incidence energy from laser pumps is not high and would not destabilize the molecule. In fact, as shown in Figure 1, the minimum of the A2Π state is well below the dissociation limit of the X2Σ+ state. Secondly, the ∼617 nm wavelength is easier to be accessible by laser gratings compared to its ultraviolet counterpart.
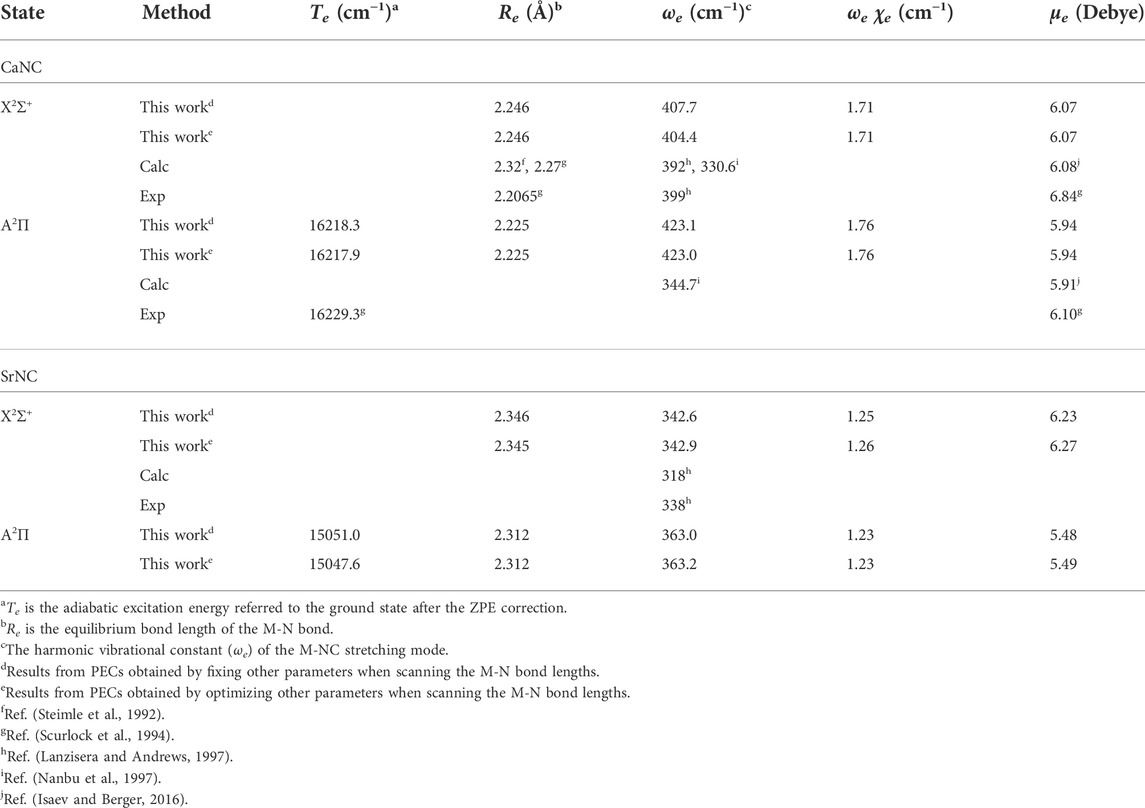
TABLE 1. Spectroscopic constants of the ground state and the first excited state of CaNC and SrNC.
It is worthy to add that the minimum configurations of all calculated states are between 2.0 Å and 2.3 Å. Especially, the minimum configurations of A2Π and X2Σ+ states are very close. The calculated equilibrium Ca-N bond length is 2.246 Å for the X2Σ+ state, and 2.225 Å for the A2Π state. Similar to the adiabatic excitation energy calculations, the relaxation or non-relaxation of other degrees of freedom during the PEC scans makes very little difference to the equilibrium Ca-N bond length. The close values of Ca-N bond lengths between A2Π and X2Σ+ states indicate that the molecular geometry does not have a significant change during the vibronic transition. Accordingly, a diagonal FCF matrix would be expected. This will be discussed in more details in the later sections.
Figure 2 shows the PECs for the lowest 6 electronic states of the SrNC molecule. The electronic structure is similar to that of CaNC, where the 2Σ+ state is the ground state, and higher electronic states are the A2Π, B2Σ+, C2∆, D2Π and E2Σ+ states, respectively. The unpaired electron locates on the s orbital of the Sr atom for the X2Σ+ state, and on the p orbital of the Sr atom for the A2Π state. The second excited state is also a 2Σ+ state and it is ∼1700 cm−1 above the A2Π state (shown in the inset panel of Figure 2). This energy gap is comparable to that of SrOH as mentioned above. The energy separation between A2Π and X2Σ+ state is ∼15000 cm−1. After including the ZPE correction, the adiabatic excitation energy is calculated as 15051.0 cm−1 (based on PECs obtained by fixing other degrees of freedom), which is equivalent to ∼664 nm wavelength for the major laser pump. The adiabatic excitation energy which is based on PECs obtained by optimizing other degrees of freedom is calculated as 15047.6 cm−1. On the other hand, the calculated adiabatic excitation energy of SrNC is lower than that of CaNC. This may be understood as resulting from the smaller HOMO-LUMO energy gap for the Sr atom than that for the Ca atom.
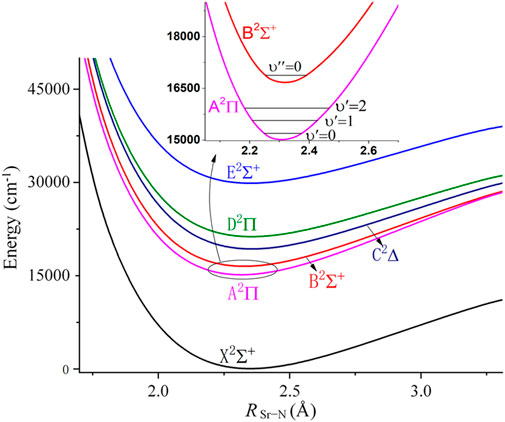
FIGURE 2. Potential energy curves of linear SrNC along RSr-N, with RN-C fixed at 1.183Å.
It should not be ignored that the spin-orbit coupling (SOC) effect may play an important role, especially for molecules involving heavy elements (Fu et al., 2016a; Fu et al., 2016b; Fu et al., 2017; Xia et al., 2017). Figure 3A and Figure 3B show the PECs including SOC for CaNC and SrNC, respectively. It can be seen that the degenerate A2Π state splits after including the SOC effect. The energy gaps between the split states are ∼68 cm−1 and ∼282 cm−1 for CaNC and SrNC, respectively. The adiabatic excitation energy between the ground and the lowest excited state for CaNC is calculated as 16188.1 cm−1, which excellently agrees with the experimental value of 16190.3 cm−1 (Steimle et al., 1992). The excitation energy between the X2Σ1/2+ and A2Π3/2 states also excellently agrees with the experimental value.
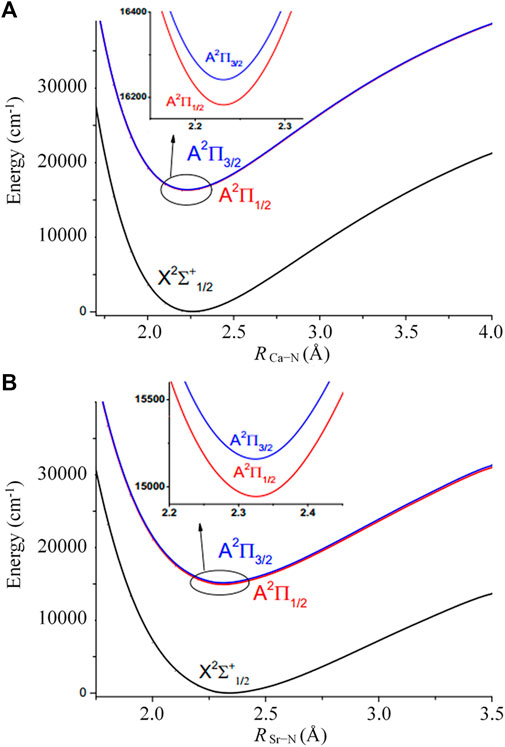
FIGURE 3. Potential energy curves for the X2Σ1/2+, A2Π1/2 and A2Π3/2 states of (A) CaNC and (B) SrNC obtained by fixing other parameters when scanning the M-N bond lengths.
For the SrNC molecule, on the other hand, the adiabatic excitation energy is calculated as 14914.8 cm−1, in excellent agreement with the experimental value of 14903.7 cm−1 (Douay and Bernath, 1990). The adiabatic excitation energy between the X2Σ1/2+ and A2Π3/2 states also very well agrees with the experimental value. The SOC effect is more evident than CaNC in terms of both the energy split of the A2Π state, and the energy shift of the adiabatic excitation energy.
3.2 Spectroscopic constants
Table 1 shows the adiabatic excitation energy (including the ZPE correction), equilibrium M-N bond length (M = Ca, Sr), M-NC harmonic stretching frequency, anharmonic term, and the PDM for CaNC and SrNC. The previous experimental and theoretical values are also listed for comparison.
For the CaNC molecule, the calculated adiabatic excitation energy excellently agrees with the experimental value. This indicates that the selected computation levels of theory and basis sets appropriately determine the geometry equilibria and the electronic states. More importantly, these give an accurate estimation of the laser wavelengths used in the cooling cycles (see the later section for more discussion). In addition, the PDM is calculated as ∼6 Debye, which is in very good agreement with the experimental measurement (∼6.8 Debye). This ensures the quality of the wavefunction. Noticeably, the large electric dipole moment (∼6 Debye) implies the Ca-N bond has a strong ionic bond character, rendering this molecule attractive for laser cooling applications (Isaev and Berger, 2016). As an interesting comparison, the CaCCH molecule (Xia et al., 2021), which is isoelectronic to CaNC, has an electric dipole moment only ∼3 Debye. In addition, the Ca-N bond length in CaNC is calculated as 2.246 Å, shorter than the Ca-C bond length in CaCCH (2.361 Å). These imply that the Ca-NC interaction is more ionic than the Ca-CCH interaction.
In terms of geometry and spectroscopic constants, the equilibrium Ca-N bond length of the X2Σ+ state agrees well with the experimental measurement. The corresponding bond length for the A2Π state is calculated as 2.225 Å. The harmonic stretching frequency of the Ca-NC mode is calculated as 404.4 cm−1, in very good agreement with experimental value (399 cm−1) (Lanzisera and Andrews, 1997). The anharmonic term is found to be very small. These indicate that the matrix effect to the vibrations is also small. Importantly, the FCF calculation relies on the geometry constants as well as harmonic frequencies. Therefore, the very good agreement of these constants with experimental measurements provides a reliable basis for FCF calculations.
For the SrNC molecule, less experimental data is available. The calculated stretching frequency of the Sr-NC mode is in very good agreement with experimental values. Unfortunately, no experimental data is available in terms of the geometry constants. Yet given the very good agreement achieved for spectroscopic constants of CaNC, and the very good agreement of Sr-NC stretching frequency, the corresponding calculated results should be an accurate estimation for the geometry constants. Similar to the CaNC molecule, the anharmonic term is found very small for the Sr-NC stretching.
The PDM of SrNC at its ground electronic state is calculated as 6.23 Debye. This is slightly larger than the PDM of CaNC at its ground state. For the first excited state, the PDMs for both molecules decrease, together with a shrink of the metal-N bond lengths. These might suggest that metal-NC interaction becomes less ionic when the electron is promoted to the higher orbital. The similar observation can also be found for the molecule pair of CaCCH and SrCCH (Xia et al., 2021).
Table 2 shows the spectroscopic constants of the CaNC and SrNC molecules with the SOC correction included. For the CaNC molecule, the SOC constant is calculated as 68.3 cm−1, in very good agreement with the experimental value. For the SrNC molecule, as expected, the SOC constant increases to 283.6 cm−1. The adiabatic excitation energy including SOC for CaNC is red-shifted by 31.1 cm−1, and the adiabatic excitation energy including SOC for SrNC is red-shifted by 135.0 cm−1.
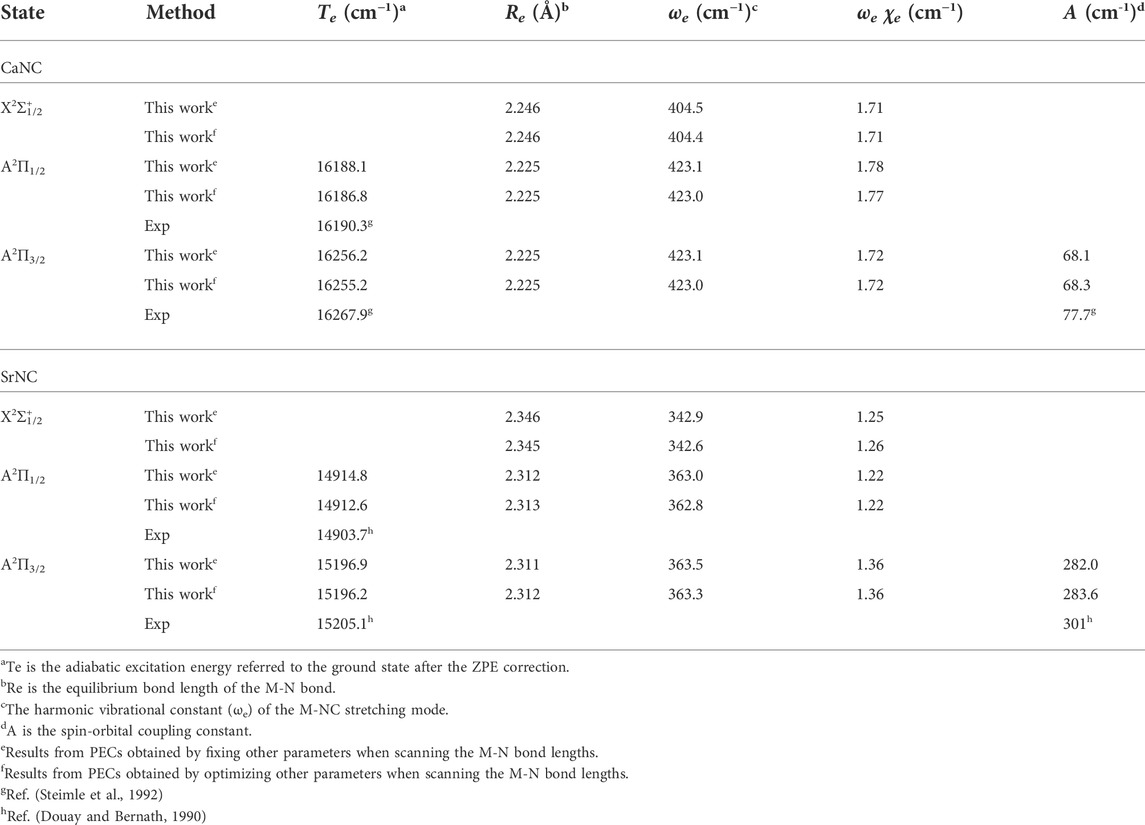
TABLE 2. The calculated spectroscopic constants of several Ω states of CaNC and SrNC.
It should be stressed at the end of this sub-section that the CaNC and SrNC stand out from other ultracold polyatomic molecular candidates in terms of their significantly strong molecular polarity. To the best of our knowledge, they have larger PDM (∼6 Debye) than other ultracold polyatomic molecular candidates currently known. For comparisons, the PDM for CaF (Childs et al., 1984) and CaCCH (Xia et al., 2021) are ∼3 Debye. More comparisons can be found in the supplementary material (Supplementary Table S1). Such a large dipole moment would facilitate the trapping of cold molecules for further applications (Grimm et al., 2000), because the molecules would respond more sensitively to the external electric field. In addition, the dipole-dipole interaction between the MNC (M = Ca or Sr) molecules should be more evident than their peers. Comparing to the atom-atom interaction, on the other hand, the dipole-dipole interaction is a long-range van der Waals interaction. Previous studies have shown that van der Waals interactions play important roles in molecular reaction dynamics (Skouteris et al., 1999; Shen et al., 2017; Cao et al., 2019; Wu et al., 2019). Hence, the long-range dipole-dipole interaction might lead to discoveries of new quantum effects, based on which novel technologies could be developed (Chen and Yan, 2019).
3.3 Laser cooling schemes
It requires several conditions for a molecule to be favored for direct laser cooling: a diagonal FCF matrix, a short lifetime for the excited state, no interference from intermediate electronic states, and no state-crossing nearby the state of interests. From the above discussions, we have shown that there are no intermediate electronic states interfering. In addition, the geometry and spectroscopic constants suggest a diagonal FCF matrix. Table 3 shows the FCFs and VBRs for the A2Π (000) → X2Σ+ (υ1υ2υ3) transitions, where υ1, υ2, and υ3 stand for the M-NC stretching mode, bending mode, and the MN-C stretching mode, respectively (M = Ca or Sr). The (000) represents the ground vibrational state. The FCF for the main transition (A2Π (000) → X2Σ+ (000)) is calculated as 0.9101 for the CaNC molecule. The corresponding FCF for SrNC is calculated as 0.9528. For both molecules, the FCFs (A2Π (000) → X2Σ+ (υ1υ2υ3)) show a rapid decay pattern, which is desirable for designing laser cooling cycles. The VBRs show very similar patterns to those of FCFs. The accumulated FCFs for CaNC and SrNC are 0.99991 and 0.99994, respectively by utilizing 6 and 4 laser pumps. These indicate that nearly 10000 photons can be scattered for each molecule, and thus the molecules can be cooled to the ultracold regime. It is worth mentioning that the vibronic effects will alter the FCFs to some extent and would have to be calculated to accurately give FCFs (for example, to the level of 10−5 or so) (Paul et al., 2019; Zhang et al., 2021).
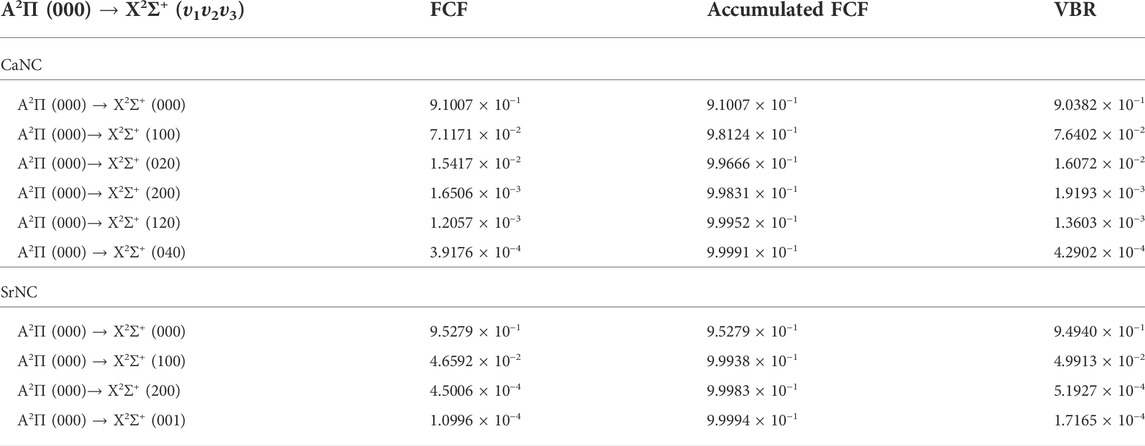
TABLE 3. The calculated Franck-Condon factors (FCFs) and vibrational branching ratios (VBRs) of the A2Π (000) → X2Σ+ (υ1υ2υ3) transitions for CaNC and SrNC.
The radiative lifetime τ should be short in favor of vertical transitions. The corresponding lifetimes for CaNC and SrNC are calculated as 15.5 and 15.8 ns, respectively. As a reference, the YbF molecule has been experimentally cooled to 100 μK and its radiative lifetime was measured as 28 ns (Lim et al., 2018). Obviously, the short radiative lifetimes meet the requirement for selecting laser-cooling candidates.
In addition to questing the feasibility of direct laser cooling, it is also interesting to know the temperature limit towards which molecules can be cooled. The Doppler temperature is evaluated accordingly. For the CaNC and SrNC molecules, the Doppler temperatures are evaluated as 246.6 and 242.0 µK, respectively. As a comparison, the Doppler temperatures for the isoelectronic molecules CaCCH and SrCCH are 153.5 and 141.1 µK, respectively. Another quantity usually used to characterize the temperature limit by laser cooling is the recoil temperature. The recoil temperatures for CaNC and SrNC are estimated as 762.0 and 381.2 nK, respectively, while for CaCCH and SrCCH the recoil temperatures are 706.5 and 337.7 nK, respectively.
Overall, the laser cooling schemes are constructed and shown in Figure 4 and Figure 5. The transitions are between X2Σ+ (υ1υ2υ3) and A2Π (000) states. Given the excellent agreement between experimental and calculated values in terms of adiabatic excitation energies and vibrational frequencies, the wavelengths for the laser pumps can be accurately determined. The wavelengths for laser pumps range from 617.1 to 650.4 nm for CaNC, and from 663.9 to 771.1 nm for SrNC. The main transition is between the X2Σ+ (000) and A2Π (000) states, so that the excited molecules statistically prefer returning to the X2Σ+ (000) state after scattering photons. The repump lasers re-excite molecules back to the A2Π (000) state to reduce the leakage to other vibrational states. The transition arrows shown in Figure 4 and Figure 5 are qualitatively represented by red colors with different levels of transparency, as the wavelengths of laser pumps fall in the region from red to near-infrared. The energy levels are qualitatively placed. The FCFs are shown with three decimals for better readability.
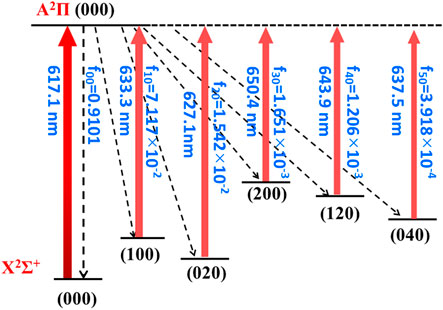
FIGURE 4. The constructed cooling scheme for CaNC using the A2Π (000) → X2Σ+ (υ1υ2υ3) transitions. The arrow colors qualitatively represent the laser wavelengths by different levels of transparency. The rotational branching can be eliminated by driving the J = 1 → J' = 0 transition.
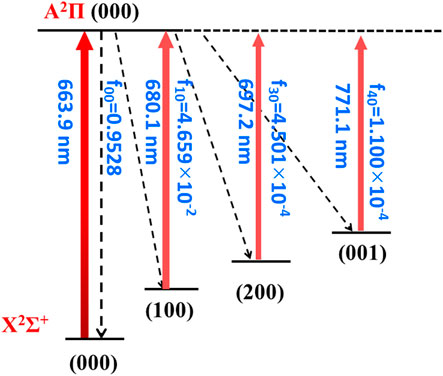
FIGURE 5. The constructed cooling scheme for SrNC using the A2Π (000) → X2Σ+ (υ1υ2υ3) transitions. The arrow colors qualitatively represent the laser wavelengths by different levels of transparency. The rotational branching can be eliminated by driving the J = 1 → J' = 0 transition.
4 Conclusion
In this work, we establish two polyatomic candidates, CaNC and SrNC, which are suitable for laser cooling to the ultracold regime. These molecules possess impressively large electric dipole moments, which are preferred for applications using an external electric field. High-level ab initio calculations are carried out to reveal electronic structures of these molecules, and the calculated spectroscopic constants are in very good agreement with the available experimental data. For each molecule, the obtained Franck-Condon factor matrix is highly diagonal. The estimated radiative lifetimes for CaNC and SrNC are 15.5 and 15.8 ns, respectively, which are short enough for rapid and efficient laser cooling. By choosing suitable spin-orbit states, we construct feasible laser cooling schemes for CaNC and SrNC, each of which allows scattering nearly 10000 photons for direct laser cooling. These calculated results indicate that CaNC and SrNC are excellent ultracold polyatomic candidates with strong polarity. We hope the present work could stimulate experimental research interests in these two promising molecules.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
WX and QL carried out the calculations. JC, WX, QL and WB analyzed the data, interpreted the results, developed the theoretical scheme and wrote the paper. WB supervised the research.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 21773251, 21973098, and 22003068), Beijing Municipal Natural Science Foundation (No. 2214065) and the Beijing National Laboratory for Molecular Sciences.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.1009986/full#supplementary-material
References
Augenbraun, B. L., Doyle, J. M., Zelevinsky, T., and Kozyryev, I. (2020). Molecular asymmetry and optical cycling: Laser cooling asymmetric top molecules. Phys. Rev. X 10 (3), 031022. doi:10.1103/physrevx.10.031022
Baum, L., Vilas, N. B., Hallas, C., Augenbraun, B. L., Raval, S., Mitra, D., et al. (2020). 1D Magneto-optical trap of polyatomic molecules. Phys. Rev. Lett. 124 (13), 133201. doi:10.1103/physrevlett.124.133201
Baum, L., Vilas, N. B., Hallas, C., Augenbraun, B. L., Raval, S., Mitra, D., et al. (2021). Establishing a nearly closed cycling transition in a polyatomic molecule. Phys. Rev. A . Coll. Park. 103 (4), 043111. doi:10.1103/physreva.103.043111
Bauschlicher, C. W., Langhoff, S. R., and Partridge, H. (1985). Ab initio study of BeCN, MgCN, CaCN and BaCN. Chem. Phys. Lett. 115 (2), 124–129. doi:10.1016/0009-2614(85)80663-1
Berning, A., Schweizer, M., Werner, H-J., Knowles, P. J., and Palmieri, P. (2000). Spin-orbit matrix elements for internally contracted multireference configuration interaction wavefunctions. Mol. Phys. 98 (21), 1823–1833. doi:10.1080/00268970050177710
Cao, J., Li, F., Xia, W., and Bian, W. (2019). van der Waals interactions in bimolecular reactions. Chin. J. Chem. Phys. 32 (2), 157–166. doi:10.1063/1674-0068/cjcp1901007
Chen, T., and Yan, B. (2019). Laser cooling and trapping of polar molecules. Acta Phys. Sin. 68 (4), 043701. doi:10.7498/aps.68.20181655
Childs, W. J., Goodman, L. S., Nielsen, U., and Pfeufer, V. (1984). Electric‐dipole moment of CaF (X 2Σ+) by molecular beam, laser‐rf, double‐resonance study of Stark splittings. J. Chem. Phys. 80 (6), 2283–2287. doi:10.1063/1.447005
Cohen-Tannoudji, C. N. (1998). Nobel lecture: Manipulating atoms with photons. Rev. Mod. Phys. 70 (3), 707–719. doi:10.1103/revmodphys.70.707
Di Rosa, M. D. (2004). Laser-cooling molecules. Eur. Phys. J. D - Atomic, Mol. Opt. Plasma Phys. 31 (2), 395–402.
Douay, M., and Bernath, P. F. (1990). Laser spectroscopy of CaNC and SrNC. Chem. Phys. Lett. 174 (3), 230–234. doi:10.1016/0009-2614(90)85337-c
Dunning, T. H. (1989). Gaussian basis sets for use in correlated molecular calculations. I the atoms boron through neon and hydrogen. J. Chem. Phys. 90 (2), 1007–1023. doi:10.1063/1.456153
El-Kork, N., Abu el kher, N., Korjieh, F., Chtay, J. A., and Korek, M. (2017). Electronic structure of the polar molecules XF (X: Be, Mg, Ca) with rovibrational and dipole moment calculations. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 177, 170–196. doi:10.1016/j.saa.2017.01.035
Feller, D., Peterson, K. A., and Davidson, E. R. (2014). A systematic approach to vertically excited states of ethylene using configuration interaction and coupled cluster techniques. J. Chem. Phys. 141 (10), 104302. doi:10.1063/1.4894482
Feller, D., Peterson, K. A., and Dixon, D. A. (2008). A survey of factors contributing to accurate theoretical predictions of atomization energies and molecular structures. J. Chem. Phys. 129 (20), 204105. doi:10.1063/1.3008061
Fitch, N. J., and Tarbutt, M. R. (2021). “Chapter three - laser-cooled molecules,” in Advances in atomic, molecular, and optical Physics. Editors L. F. Dimauro, H. Perrin, and S. F. Yelin (Academic Press), 70, 157–262.
Fu, M., Cao, J., Ma, H., and Bian, W. (2016). Laser cooling of copper monofluoride: A theoretical study including spin–orbit coupling. RSC Adv. 6 (102), 100568–100576. doi:10.1039/c6ra07835d
Fu, M., Ma, H., Cao, J., and Bian, W. (2016). Extensive theoretical study on electronically excited states of calcium monochloride: Molecular laser cooling and production of ultracold chlorine atoms. J. Chem. Phys. 144 (18), 184302. doi:10.1063/1.4948631
Fu, M., Ma, H., Cao, J., and Bian, W. (2017). Laser cooling of CaBr molecules and production of ultracold Br atoms: A theoretical study including spin–orbit coupling. J. Chem. Phys. 146 (13), 134309. doi:10.1063/1.4979566
Gozem, S., and Krylov, A. I. (2021). The ezSpectra suite: An easy-to-use toolkit for spectroscopy modeling. WIREs Comput. Mol. Sci., e1546.
Greetham, G. M., and Ellis, A. M. (2000). The C̃–X̃ electronic spectrum of the SrNC free radical: A jet-cooled investigation. Chem. Phys. Lett. 332 (3), 303–307. doi:10.1016/s0009-2614(00)01254-9
Greetham, G. M., and Ellis, A. M. (2000). Ultraviolet laser spectroscopy of jet-cooled CaNC and SrNC free radicals: Observation of bent excited electronic states. J. Chem. Phys. 113 (20), 8945–8952. doi:10.1063/1.1319344
Grimm, R., Weidemüller, M., and Ovchinnikov, Y. B. (2000). “Optical dipole traps for neutral atoms,” in Advances in atomic, molecular, and optical Physics. Editors B. Bederson, and H. Walther (Academic Press), 42, 95–170.
Isaev, T. A., and Berger, R. (2016). Polyatomic candidates for cooling of molecules with lasers from simple theoretical concepts. Phys. Rev. Lett. 116 (6), 063006. doi:10.1103/physrevlett.116.063006
Isaev, T. A. (2020). Direct laser cooling of molecules. Phys. -Usp. 63 (3), 289–302. doi:10.3367/ufne.2018.12.038509
Isaev, T. A., Zaitsevskii, A. V., and Eliav, E. (2017). Laser-coolable polyatomic molecules with heavy nuclei. J. Phys. B At. Mol. Opt. Phys. 50 (22), 225101. doi:10.1088/1361-6455/aa8f34
Ishii, K., Taketsugu, T., and Hirano, T. (2003). Theoretical study on the potential energy surfaces of CaNC and CaCN. Chem. Phys. Lett. 374 (5), 506–512. doi:10.1016/s0009-2614(03)00738-3
Ivanov, M. V., Bangerter, F. H., Wójcik, P., and Krylov, A. I. (2020). Toward ultracold organic chemistry: Prospects of laser cooling large organic molecules. J. Phys. Chem. Lett. 11 (16), 6670–6676. doi:10.1021/acs.jpclett.0c01960
Ivanov, M. V., Gulania, S., and Krylov, A. I. (2020). Two cycling centers in one molecule: Communication by through-bond interactions and entanglement of the unpaired electrons. J. Phys. Chem. Lett. 11 (4), 1297–1304. doi:10.1021/acs.jpclett.0c00021
Ivanov, M. V., Jagau, T-C., Zhu, G-Z., Hudson, E. R., and Krylov, A. I. (2020). In search of molecular ions for optical cycling: A difficult road. Phys. Chem. Chem. Phys. 22 (30), 17075–17090. doi:10.1039/d0cp02921a
Kendall, R. A., Dunning, T. H., and Harrison, R. J. (1992). Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96 (9), 6796–6806. doi:10.1063/1.462569
Knowles, P. J., and Werner, H-J. (1992). Internally contracted multiconfiguration-reference configuration interaction calculations for excited states. Theor. Chim. Acta 84 (1), 95–103. doi:10.1007/bf01117405
Kozyryev, I., Baum, L., Matsuda, K., Augenbraun, B. L., Anderegg, L., Sedlack, A. P., et al. (2017). Sisyphus laser cooling of a polyatomic molecule. Phys. Rev. Lett. 118 (17), 173201. doi:10.1103/physrevlett.118.173201
Kozyryev, I., Baum, L., Matsuda, K., and Doyle, J. M. (2016). Proposal for laser cooling of complex polyatomic molecules. Chemphyschem 17 (22), 3641–3648. doi:10.1002/cphc.201601051
Lanzisera, D. V., and Andrews, L. (1997). Reactions of laser-ablated Mg, Ca, Sr, and Ba atoms with hydrogen cyanide in excess Argon. Matrix infrared spectra and density functional calculations on novel isocyanide products. J. Phys. Chem. A 101 (50), 9666–9672. doi:10.1021/jp972098i
Le Roy, R. J. (2017). Level: A computer program for solving the radial schrödinger equation for bound and quasibound levels. J. Quantitative Spectrosc. Radiat. Transf. 186, 167–178. doi:10.1016/j.jqsrt.2016.05.028
Lefebvre-Brion, F., and Field, R. (2004). The spectra and dynamics of diatomic molecules, revised and enlarged. Elsevier.
Li, D., and Bian, W. (2021). Excellent ultracold molecular candidates from group VA hydrides: Whether do nearby electronic states interfere? Front. Chem. 9, 778292. doi:10.3389/fchem.2021.778292
Li, D., Fu, M., Ma, H., Bian, W., Du, Z., and Chen, C. (2020). A theoretical study on laser cooling feasibility of group IVA hydrides XH (X = Si, Ge, Sn, and Pb): The role of electronic state crossing. Front. Chem. 8, 20. doi:10.3389/fchem.2020.00020
Lim, J., Almond, J. R., Trigatzis, M. A., Devlin, J. A., Fitch, N. J., Sauer, B. E., et al. (2018). Laser cooled YbF molecules for measuring the electron's electric dipole moment. Phys. Rev. Lett. 120 (12), 123201. doi:10.1103/physrevlett.120.123201
Liu, K., and Bian, W. (2008). Extensive theoretical study on the low-lying electronic states of silicon monofluoride cation including spin-orbit coupling. J. Comput. Chem. 29 (2), 256–265. doi:10.1002/jcc.20785
Liu, K., Yu, L., and Bian, W. (2009). Extensive theoretical study on various low-lying electronic states of silicon monochloride cation including spin-orbit coupling. J. Phys. Chem. A 113 (9), 1678–1685. doi:10.1021/jp809618y
Liu, L., Yang, C-L., Sun, Z-P., Wang, M-S., and Ma, X-G. (2021). Direct laser cooling schemes for the triatomic SOH and SeOH molecules based on ab initio electronic properties. Phys. Chem. Chem. Phys. 23 (3), 2392–2397. doi:10.1039/d0cp04963h
Lu, Q., and Peterson, K. A. (2020). Coupled cluster spectroscopic properties of the coinage metal nitrosyls, M–NO (M = Cu, Ag, Au). Theor. Chem. Acc. 139 (4), 81. doi:10.1007/s00214-020-02597-w
Lu, Q. (2020). Accurate ab initio vibronic spectroscopy of the CCP and CCAs radicals. Chem. Phys. Lett. 739, 137017. doi:10.1016/j.cplett.2019.137017
Mitra, D., Vilas Nathaniel, B., Hallas, C., Anderegg, L., Augenbraun Benjamin, L., Baum, L., et al. (2020). Direct laser cooling of a symmetric top molecule. Science 369 (6509), 1366–1369. doi:10.1126/science.abc5357
Moussa, A., El-Kork, N., and Korek, M. (2021). Laser cooling and electronic structure studies of CaK and its ions CaK±. New J. Phys. 23 (1), 013017. doi:10.1088/1367-2630/abd50d
Nakagawa, J., Wormsbecher, R. F., and Harris, D. O. (1983). High-resolution laser excitation spectra of linear triatomic molecules: Analysis of the B2Σ+-X2Σ+ system of SrOH and SrOD. J. Mol. Spectrosc. 97 (1), 37–64. doi:10.1016/0022-2852(83)90336-3
Nanbu, S., Minamino, S., and Aoyagi, M. (1997). Theoretical study of the potential energy surfaces and dynamics of CaNC/CaCN. J. Chem. Phys. 106 (19), 8073–8083. doi:10.1063/1.473815
O'Rourke, M. J., and Hutzler, N. R. (2019). Hypermetallic polar molecules for precision measurements. Phys. Rev. A . Coll. Park. 100 (2), 022502. doi:10.1103/physreva.100.022502
Paul, A. C., Sharma, K., Reza, M. A., Telfah, H., Miller, T. A., and Liu, J. (2019). Laser-induced fluorescence and dispersed-fluorescence spectroscopy of the Ã2E−X̃2A1 transition of jet-cooled calcium methoxide (CaOCH3) radicals. J. Chem. Phys. 151 (13), 134303. doi:10.1063/1.5104278
Scurlock, C. T., Fletcher, D. A., and Steimle, T. C. (1994). An analysis of the à 2Π–X̃ 2Σ+ band system of isotopically substituted calcium isocyanide CaNC. J. Chem. Phys. 101 (9), 7255–7261. doi:10.1063/1.468283
Shen, Z., Ma, H., Zhang, C., Fu, M., Wu, Y., Bian, W., et al. (2017). Dynamical importance of van der Waals saddle and excited potential surface in C(1D)+D2 complex-forming reaction. Nat. Commun. 8, 14094. doi:10.1038/ncomms14094
Skouteris, D., Manolopoulos David, E., Bian, W., Werner, H-J., Lai, L-H., and Liu, K. (1999). van der Waals interactions in the Cl + HD reaction. Science 286 (5445), 1713–1716. doi:10.1126/science.286.5445.1713
Steimle, T. C., Fletcher, D. A., Jung, K. Y., and Scurlock, C. T. (1992). Molecular beam optical Stark spectroscopy of calcium monocyanide. J. Chem. Phys. 97 (5), 2909–2919. doi:10.1063/1.463032
Tarbutt, M. R. (2018). Laser cooling of molecules. Contemp. Phys. 59 (4), 356–376. doi:10.1080/00107514.2018.1576338
Vilas N, B., Hallas, C., Anderegg, L., Robichaud, P., Winnicki, A., Mitra, D., et al. (2022). Magneto-Optical trapping and sub-Doppler cooling of a polyatomic molecule. Nature 606, 70–74. arXiv. doi:10.1038/s41586-022-04620-5
Weigend, F., and Baldes, A. (2010). Segmented contracted basis sets for one- and two-component Dirac–Fock effective core potentials. J. Chem. Phys. 133 (17), 174102. doi:10.1063/1.3495681
Weigend, F., Furche, F., and Ahlrichs, R. (2003). Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys. 119 (24), 12753–12762. doi:10.1063/1.1627293
Werner, H-J., Knowles, P. J., Knizia, G., Manby, F. R., Schütz, M., Celani, P., et al. (2015). MOLPRO, 2015 , a package of ab initio programs.
Werner, H-J., Knowles, P. J., Knizia, G., Manby, F. R., and Schütz, M. (2012). Molpro: A general-purpose quantum chemistry program package. WIREs. Comput. Mol. Sci. 2 (2), 242–253. doi:10.1002/wcms.82
Werner, H-J., Knowles, P. J., Manby, F. R., Black, J. A., Doll, K., Heßelmann, A., et al. (2020). The Molpro quantum chemistry package. J. Chem. Phys. 152 (14), 144107. doi:10.1063/5.0005081
Werner, H. J., and Knowles, P. J. (1988). An efficient internally contracted multiconfiguration–reference configuration interaction method. J. Chem. Phys. 89 (9), 5803–5814. doi:10.1063/1.455556
Whitham, C. J., Soep, B., Visticot, J. P., and Keller, A. (1990). Observation and spectroscopy of metallic free radicals produced by reactive collisions during a supersonic expansion. J. Chem. Phys. 93 (2), 991–1000. doi:10.1063/1.459126
Wu, Y., Cao, J., Ma, H., Zhang, C., Bian, W., Nunez-Reyes, D., et al. (2019). Conical intersection–regulated intermediates in bimolecular reactions: Insights from C(1D) + HD dynamics. Sci. Adv. 5 (4), eaaw0446. doi:10.1126/sciadv.aaw0446
Xia, W., Fu, M., Ma, H., and Bian, W. (2017). A theoretical study on laser cooling of silicon monofluoride. Chem. Phys. 485-486, 29–34. doi:10.1016/j.chemphys.2017.01.007
Xia, W., Ma, H., and Bian, W. (2021). Production of ultracold CaCCH and SrCCH molecules by direct laser cooling: A theoretical study based on accurate ab initio calculations. J. Chem. Phys. 155 (20), 204304. doi:10.1063/5.0072013
Yu, L., and Bian, W. (2012). Electronically excited-state properties and predissociation mechanisms of phosphorus monofluoride: A theoretical study including spin–orbit coupling. J. Chem. Phys. 137 (1), 014313. doi:10.1063/1.4731635
Yu, L., and Bian, W. (2011). Extensive theoretical study on electronically excited states and predissociation mechanisms of sulfur monoxide including spin–orbit coupling. J. Comput. Chem. 32 (8), 1577–1588. doi:10.1002/jcc.21737
Zeid, I., Al Abdallah, R., El-Kork, N., and Korek, M. (2020). Ab-initio calculations of the electronic structure of the alkaline Earth hydride anions XH− (X = Mg, Ca, Sr and Ba) toward laser cooling experiment. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 224, 117461. doi:10.1016/j.saa.2019.117461
Zeng, J., Long, G., and Deng, F. (2001). Recent progress in quantum mechanics. Beijing: Tsinghua University Press.
Keywords: molecular laser cooling, ultracold polyatomic molecules, large polarity, ab initio, Franck-Condon factor
Citation: Xia W, Cao J, Lu Q and Bian W (2022) Production of ultracold polyatomic molecules with strong polarity by laser cooling: A detailed theoretical study on CaNC and SrNC. Front. Chem. 10:1009986. doi: 10.3389/fchem.2022.1009986
Received: 02 August 2022; Accepted: 22 August 2022;
Published: 23 September 2022.
Edited by:
Luca Evangelisti, University of Bologna, ItalyReviewed by:
Jacek Antoni Klos, University of Maryland, United StatesInga Ulusoy, Heidelberg University, Germany
Copyright © 2022 Xia, Cao, Lu and Bian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Lu, cWluZ2x1QGljY2FzLmFjLmNu; Wensheng Bian, YmlhbkBpY2Nhcy5hYy5jbg==
†These authors contributed equally to this work.