 Tomoya Kusama
Tomoya Kusama Shuzo Hirata*
Shuzo Hirata*- Department of Engineering Science, University of Electro-Communications, Tokyo, Japan
The suppression of thermally driven triplet deactivation is crucial for efficient persistent room-temperature phosphorescence (pRTP). However, the mechanism by which triplet deactivation occurs in metal-free molecular solids at room temperature (RT) remains unclear. Herein, we report a large pRTP intensity change in a molecular guest that depended on the reversible amorphous–crystal phase change in the molecular host, and we confirm the large contribution made by the rigidity of the host in suppressing intermolecular triplet quenching in the guest. (S)-(−)-2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl ((S)-BINAP) was doped as a guest into a highly purified (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl ((S)-H8-BINAP) host. It was possible to reversibly form the amorphous and crystalline states of the solid by cooling to RT from various temperatures. The RTP yield (Φp) originating from the (S)-BINAP was 6.7% in the crystalline state of the (S)-H8-BINAP host, whereas it decreased to 0.31% in the amorphous state. Arrhenius plots showing the rate of nonradiative deactivation from the lowest triplet excited state (T1) of the amorphous and crystalline solids indicated that the large difference in Φp between the crystalline and amorphous states was mostly due to the discrepancy in the magnitude of quenching of intermolecular triplet energy transfer from the (S)-BINAP guest to the (S)-H8-BINAP host. Controlled analyses of the T1 energy of the guest and host, and of the reorganization energy of the intermolecular triplet energy transfer from the guest to the host, confirmed that the large difference in intermolecular triplet quenching was due to the discrepancy in the magnitude of the diffusion constant of the (S)-H8-BINAP host between its amorphous and crystalline states. Quantification of both the T1 energy and the diffusion constant of molecules used in solid materials is crucial for a meaningful discussion of the intermolecular triplet deactivation of various metal-free solid materials.
Introduction
Room-temperature phosphorescence (RTP) with an emission lifetime of more than 100 ms—i.e., persistent RTP (pRTP)—from metal-free molecular solids occurs after ceasing exposure to fluorescence-independent excitation light (Clapp. 1939; Zhang et al., 2007; Hirata et al., 2013). Because autofluorescence-independent pRTP can be detected using small-scale and low-cost photo detectors, chemicals and materials with pRTP characteristics are crucial for state-of-the-art security, sensing, and bioimaging applications (Deng et al., 2013; Zhang et al., 2014; Fateminia et al., 2017; Zhen et al., 2017; Louis et al., 2019; Zhou and Yan, 2019). Efficient pRTP produces much brighter persistent emission compared with the general long persistent luminescence from materials that has been reported previously (Bhattacharjee and Hirata, 2020). Therefore, photophysical insight into chemicals and/or materials to access the efficiency and brightness of pRTP is crucial. Because pRTP from metal-free chromophores is a slow process (Hirata, 2017), the suppression of thermo-driven triplet deactivation is necessary for efficient pRTP. However, the mechanism by which the triplet deactivation of metal-free molecular solids occurs at RT remains unclear.
Triplet deactivation includes both intramolecular radiation-less transition from the lowest triplet excited state (T1) to the ground state (S0) and the T1 quenching caused by intermolecular interactions. The rate of the intermolecular radiation-less transition at RT (knr(RT)) is related to spin–orbit coupling including vibrations and the energy gap between T1 and the S0 of the target chromophores (Schlag et al., 1971; Metz et al., 1972; Metz, 1973). However, the rate of the T1 quenching caused by intermolecular interactions (kq(RT)) is based on charge transfer theory (Köhler and Bässler, 2011). In the 1980s, knr(RT) and kq(RT) were considered separately using benzophenone as a guest in polymer matrices, and the knr(RT) of benzophenone was found to be almost independent of temperature from 77 K to RT (Horie and Mita, 1982; Horie et al., 1984). Recently, cooperative analysis of optical measurements and quantum chemical calculations have confirmed that the knr(RT) values of a variety of heavy atom-free chromophores are less than 100 s−1 (Bhattacharjee et al., 2021; Hirata and Bhattacharjee et al., 2021). This indicates that most triplet deactivation of materials is caused by the kq(RT) (Hirata et al., 2020; Hirata and ; Bhattacharjee et al., 2021). With regard to the kq(RT) of host–guest molecular solids, it is generally necessary for the host molecules to have a larger T1 energy than the metal-based phosphorescence guests to suppress the kq(RT) (Adachi et al., 2001). However, for heavy atom-free and/or metal-free guests, a long-lived T1 at RT generally requires the host molecules to have a considerably greater T1 than the guests to significantly suppress the kq(RT) (Hirata et al., 2013; Totani et al., 2013). Contrary to general discussions about the contribution made by the T1 energy difference between the host molecules and the guest chromophores to the kq(RT), triplet deactivation due to the rigidity of the materials has often been reported, although it is still regarded as phenomenological (Zhao et al., 2020). Although recent analyses suggest that rigidity—including intermolecular interactions—might not be related to knr(RT) but to kq(RT), investigations into the physical factors governing rigidity and the T1 energy difference between the host and the guest have not been reported.
Herein, we report the dependency of the thermo-reversible intensity change of green pRTP of a guest on the amorphous–crystalline phase change of a host molecule, and determine the contribution made by the rigidity of the host in the suppression of endothermic intermolecular triplet quenching. (S)-(−)-2,2′-Bis(diphenylphosphino)-1,1′-binaphthyl ((S)-BINAP) was doped into a highly purified (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl ((S)-H8-BINAP) host. The host–guest material in the crystalline state produced green pRTP under ambient conditions, and the RTP yield (Φp(RT)) was 6.7%. However, the Φp(RT) of the host–guest material in the amorphous state decreased to 0.31%, even when the materials were stored in a high vacuum. Detailed optical measurements indicated that the large Φp(RT) in the crystalline state was caused by both the increase in the triplet generation yield of the (S)-BINAP guest and the large decrease of kq(RT) in the crystalline state compared with in the amorphous state. The comparable T1 energy values of the (S)-H8-BINAP host in both the amorphous and crystalline states suggests that the decrease in kq(RT) was caused by the suppressed molecular diffusion of the (S)-H8-BINAP host molecules in the crystalline state. The suppressed molecular diffusion in the crystalline (S)-H8-BINAP host was observed using molecular dynamic simulation. Thus, suppressed molecular diffusion is a crucial factor for minimizing the endothermic intermolecular triplet energy transfer that induces triplet quenching.
Results and Discussion
Thermo-Reversible Intensity Change of Persistent Room-temperature Phosphorescence
Commercially available (S)-H8-BINAP crystals often generate green pRTP after ultraviolet (UV) excitation has ceased (Supplementary Figure S1A) because of the presence of (S)-BINAP as an impurity (Supplementary Figure S2). After careful repeated purification of the (S)-H8-BINAP using silica column chromatography, however, we noted that the purified crystals of (S)-H8-BINAP did not produce pRTP (Supplementary Figure S1B) (Hirata et al., 2020). First (S)-BINAP powder was dissolved in molten pure (S)-H8-BINAP at 220°C. Next, the molten materials were placed on a quartz substrate on a hotplate at 250°C which is higher temperature than a melting point of 208°C of (S)-H8-BINAP host (Supplementary Figure S3), and the substrate was quenched to room temperature (RT) to prepare an amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP film. The resulting amorphous film of 5 wt% (S)-BINAP-doped (S)-H8-BINAP produced markedly weak pRTP after ceasing UV excitation (Figure 1B; top). The weak pRTP of the amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP film did not increase under vacuum. The amorphous form of 5 wt% (S)-BINAP-doped (S)-H8-BINAP has a glass transition temperature of 104°C and crystallizes at 165°C (Figure 1A; top). After crystallization at 165°C for 30 min, the produced 5 wt% (S)-BINAP-doped (S)-H8-BINAP crystalline films (Figure 1A; lower) produced much brighter green pRTP compared with the amorphous form after exposure to excitation light of 330 nm had ceased (Figure 1B; lower).
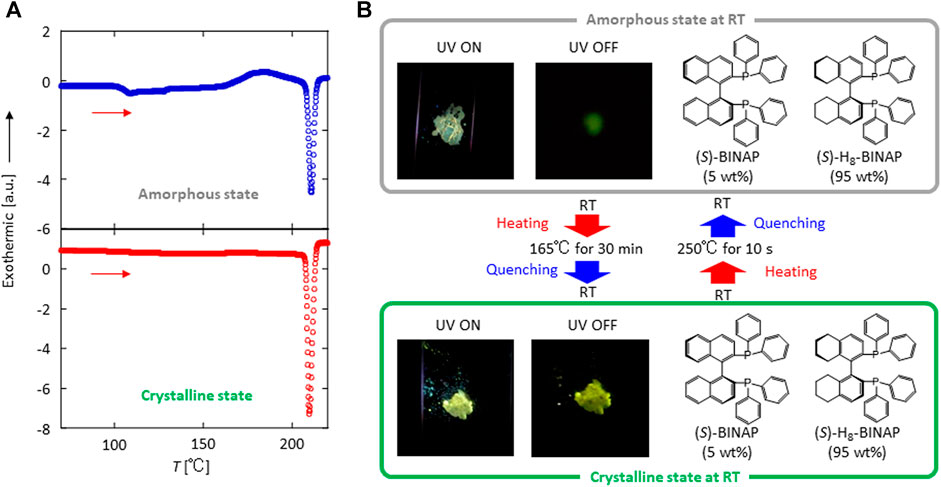
FIGURE 1. Reversible crystal–amorphous phase change at RT and pRTP characteristics of the (S)-BINAP doped (S)-H8-BINAP solid. (A) Differential scanning calorimetry characteristics of amorphous state (upper) and crystalline state (lower) when heated at 5°C/min. (B) Emission behavior during and immediately after excitation by exposure to radiation (330 nm) of amorphous (upper) and crystalline (lower) solids. ((S)-BINAP = (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (S)-H8-BINAP = (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl; pRTP = persistent room-temperature phosphorescence).
More detailed optical measurements were performed to investigate the differences in the pRTP characteristics between the crystalline and amorphous states of 5 wt% (S)-BINAP-doped (S)-H8-BINAP solid. Both the crystalline and amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP films absorbed radiation at 330 nm ((i) and (ii) in Figure 2A). The amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP film produced a fluorescence spectrum beginning at approximately 400 nm ((iii) in Figure 2A), and the steady-state RT emission yield was 2.5%. Immediately after excitation by exposure to UV radiation ceased, a weak green pRTP remained ((v) in Figure 2A). The quantum yield of phosphorescence at RT (Φp(RT)) was estimated to be 0.31% by comparing the intensity of the steady-state RT emission spectrum ((iii) in Figure 2A) with the RTP spectral intensity immediately after ceasing excitation ((v) in Figure 2A). The RTP decay had multi-exponential characteristics (gray in Figure 2B). For the crystalline 5 wt% (S)-BINAP-doped (S)-H8-BINAP films, the larger increase of absorbance at wavelengths less than 350 nm resulted from the absorption of (S)-BINAP and (S)-H8-BINAP ((ii) in Figure 2A). An increase of the baseline at wavelengths longer than 400 nm was caused by light scattering arising from the polycrystalline films. In contrast to the amorphous state, the crystalline 5 wt% (S)-BINAP-doped (S)-H8-BINAP film had two large peaks at 510 and 545 nm under steady-state UV excitation at 330 nm in addition to fluorescence spectra commencing at approximately 400 nm ((iv) in Figure 2A). The steady-state RT emission yield was 9.8%. After ceasing excitation, a large RTP spectra with peaks at 510 and 545 nm remained ((vi) in Figure 2A). Φp(RT) was determined to be 6.7% by comparing the spectral intensity of steady-state RT emission ((iv) in Figure 2A) with the RTP spectral intensity immediately after ceasing excitation ((vi) in Figure 2A). The RTP had single exponential decay characteristics (green in Figure 2B). Thus, the intensity of the pRTP produced by the 5 wt% (S)-BINAP-doped (S)-H8-BINAP solid differed by approximately 20 times when comparing the crystal and amorphous states of the materials. The large Φp(RT) of (S)-BINAP-doped (S)-H8-BINAP crystalline films does not change at concentrations of (S)-BINAP from 5 wt% to 15 wt%, but substantially decreased at higher concentrations of (S)-BINAP (Supplementary Figure S4 and Supplementary Table S1).
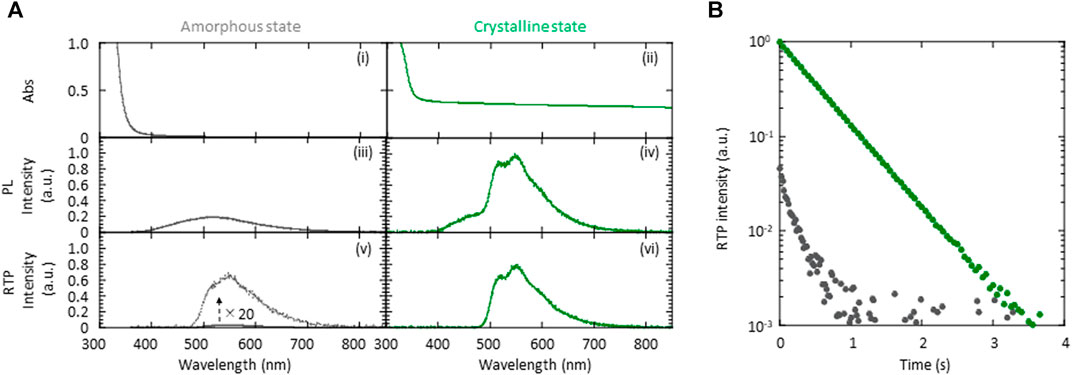
FIGURE 2. Absorption and emission characteristics of 5 wt% (S)-BINAP-doped (S)-H8-BINAP films. (A) Absorption (i, ii), steady-state RT emission (iii, iv), and RTP (v, vi) spectra of amorphous (gray) and crystalline (green) films. (B) RTP decay characteristics of amorphous (gray) and crystalline (green) films. The RTP intensity immediately after ceasing excitation in the crystalline state was normalized to one and the initial intensity of RTP immediately after ceasing excitation in the amorphous state was set by considering the difference in Φp(RT) between the crystalline and amorphous states. In (A) and (B), the excitation wavelength used for emission measurements was 330 nm. (RT = room-temperature; RTP = room-temperature phosphorescence; (S)-BINAP = (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (S)-H8-BINAP = (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl).
Fundamental Photophysical Properties
The absorption and emission characteristics of the (S)-BINAP guest and the (S)-H8-BINAP host were measured to determine the mechanism by which green pRTP was generated by the 5 wt% (S)-BINAP-doped (S)-H8-BINAP films. When dissolved in 2-methyltetrahydrofuran (2Me-THF), the (S)-BINAP guest produced an absorption spectrum at wavelengths of less than 350 nm, and the molar absorption coefficient (ε) at 330 nm was 6.1 × 103 M−1 cm−1 (Figure 3A; (i)). The neat amorphous film of (S)-H8-BINAP had a long small tail of absorption in the 350–400 nm range, and an ε at 330 nm of 6.4 × 102 M−1 cm−1 when the density of the amorphous (S)-H8-BINAP solid was considered to be approximately 1 (Figure 3A; (ii)). The comparison of ε between the (S)-BINAP guest and the (S)-H8-BINAP host indicated that the (S)-BINAP guest absorbed 33% of the excitation light at 330 nm when the amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP films were irradiated with excitation light. In the neat crystalline film of (S)-H8-BINAP, light scattering increased from the baseline at wavelengths longer than 350 nm, whereas (S)-H8-BINAP exhibited marked absorption at wavelengths less than 350 nm (Figure 3A; (iii)).
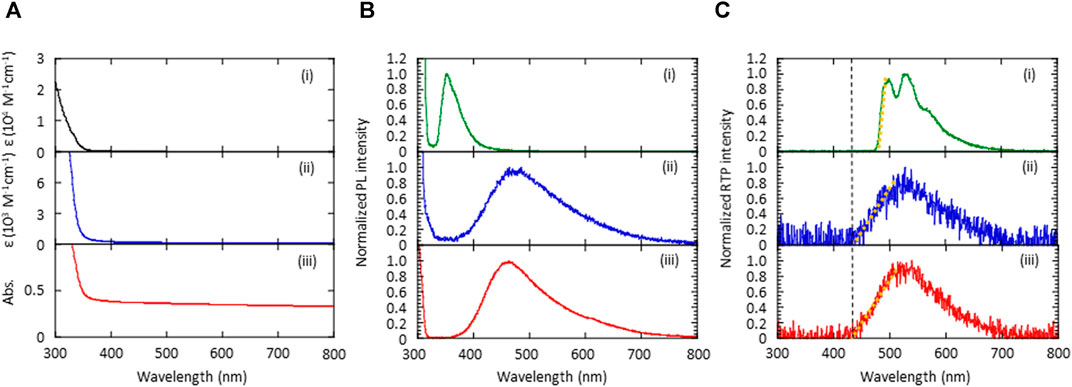
FIGURE 3. Absorption and emission spectra of (S)-BINAP molecularly dispersed in 2Me-THF (i), an amorphous (S)-H8-BINAP film (ii), and a crystalline (S)-H8-BINAP film (iii). (A) Absorption spectra. (B) Fluorescence spectra obtained by excitation at 300 nm at RT. (C) Phosphorescence spectra obtained after ceasing excitation at 300 nm at 77 K. The orange dotted lines in the graph were used to determine the onset energy of T1. (2Me-THF = 2-methyltetrahydrofuran; RT = room-temperature; (S)-BINAP = (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (S)-H8-BINAP = (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl).
The (S)-BINAP guest dissolved in 2Me-THF produced a fluorescence spectrum with a peak at 353 nm (Figure 3B; (i)). The amorphous and crystalline (S)-H8-BINAP hosts produced broad fluorescence spectra with onset energies at approximately 400 nm (Figure 3B; 2) and (iii)). Thus, the energy of the lowest singlet excited state (S1) of the amorphous and crystalline (S)-H8-BINAP hosts was slightly smaller than that of the molecularly dispersed (S)-BINAP. However, the T1 energy of the molecularly dispersed (S)-BINAP was much smaller than that of the amorphous and crystalline (S)-H8-BINAP hosts. The (S)-BINAP molecularly dispersed in 2Me-THF produced green persistent phosphorescence at 77 K immediately after ceasing excitation at 330 nm (Figure 3C; (i)). The spectral shape produced by the phosphorescence and the energy of the molecularly dispersed (S)-BINAP was comparable to those of the amorphous and crystalline 5 wt% (S)-BINAP-doped (S)-H8-BINAP films. Because neat solid (S)-BINAP does not produce RTP at all, even when it is in a vacuum, the green pRTP of the amorphous and crystalline 5 wt% (S)-BINAP-doped (S)-H8-BINAP films was caused by the molecularly dispersed (S)-BINAP. Although high-concentration doping is not generally appropriate when different chromophores are doped into different crystalline molecules, the similar sizes and structures of (S)-BINAP and (S)-H8-BINAP may allow the efficient replacement of (S)-H8-BINAP by (S)-BINAP in a (S)-H8-BINAP crystalline lattice. At 77K, both the amorphous and crystalline neat films of (S)-H8-BINAP produced broad phosphorescence spectra with onset energies of approximately 425 nm (Figure 3C; 2) and (iii)). The comparable S1 and T1 energies between the amorphous and crystalline (S)-H8-BINAP hosts do not satisfactorily explain the large difference in the Φp(RT) between the (S)-BINAP in the amorphous (S)-H8-BINAP host and that in the crystalline (S)-H8-BINAP host. Appropriate discussions about Φp(RT) generally require investigation of both the generation yield of T1 and the T1-S0 processes (Hirata, 2017). Therefore, the following two sections comprise a discussion of these two points.
Triplet Generation Scheme
The T1 generation mechanism and the yield of the (S)-BINAP doped into the (S)-H8-BINAP host were investigated. Again, we note that the (S)-BINAP dissolved in 2Me-THF produced fluorescence with a peak wavelength at 353 nm (Figure 3B, (i)). However, the peak disappeared, and a broad-fluorescence spectra with an onset energy at approximately 400 nm appeared when the (S)-BINAP was doped into the amorphous and crystalline (S)-H8-BINAP hosts ((iii) and (iv) of Figure 2A, respectively). Those fluorescence spectra are comparable to the fluorescence spectra of the amorphous and crystalline (S)-H8-BINAP hosts ((ii) and (iii) of Figure 3B, respectively). The S1 energy of (S)-BINAP was transferred to the amorphous and crystalline (S)-H8-BINAP hosts via fluorescence resonance energy transfer (FRET) ((i) in Figure 4A). The generated S1 energy of the (S)-H8-BINAP hosts allowed intersystem crossing (ISC) from S1 to generate T1 in the (S)-H8-BINAP hosts ((ii) in Figure 4A). Finally, the generated T1 in the (S)-H8-BINAP hosts was effectively trapped by the T1 of the (S)-BINAP, because the T1 of the (S)-BINAP was much less than that of the (S)-H8-BINAP hosts ((iii) in Figure 4A).
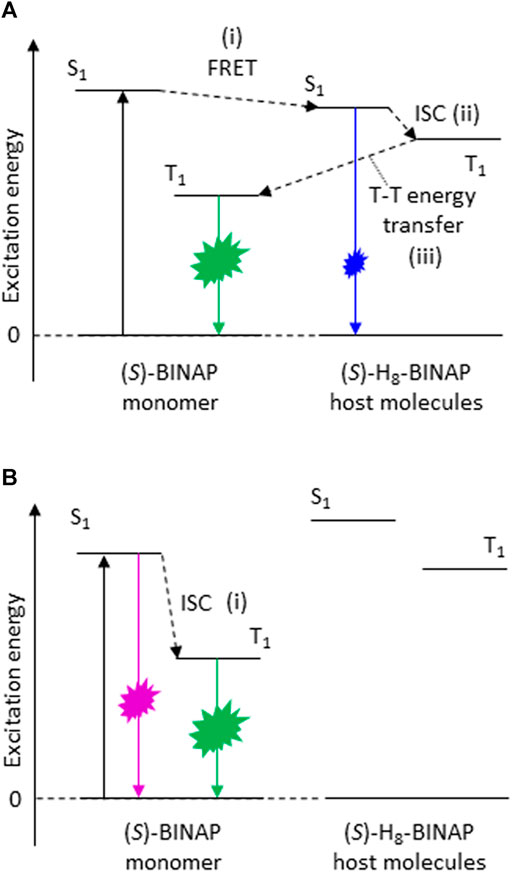
FIGURE 4. Scheme showing T1 generation in (S)-BINAP. (A) In (S)-H8-BINAP hosts. (B) In amorphous β-estradiol host. ((S)-BINAP = (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (S)-H8-BINAP = (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl).
The T1 generation yield of the (S)-BINAP in the (S)-H8-BINAP host (Φt) via the processes from (i) to (iii) in Figure 4A has the following relationship with Φp(77 K):
where kp is the rate constant of phosphorescence of the (S)-BINAP guest. (S)-BINAP doped into an amorphous β-estradiol host produced a fluorescence spectrum at 355 nm (blue in Supplementary Figure S2A), and generated green pRTP (green in Supplementary Figure S5A,B). In addition, the fluorescence energy of the (S)-BINAP doped into the amorphous β-estradiol was comparable to that of the (S)-BINAP dissolved in 2Me-THF. Therefore, the T1 of the (S)-BINAP doped into the amorphous β-estradiol was formed via the ISC from S1 to the triplet states ((i) in Figure 4B). The Φp(RT) of the (S)-BINAP doped into the amorphous β-estradiol can be expressed as (Hirata, 2017):
where Φisc is the ISC yield from S1 to the triplet states. The Φisc of the molecularly dispersed (S)-BINAP was quantified as 0.29 using a transient absorption technique (Supplementary Figure S6; Supplementary Table S2) (Bhattacharjee and Hirata, 2020). Because 0.3 wt% (S)-BINAP doped into β-estradiol had an Φp(RT) of 0.052 and a τp(RT) of 0.72 s (Supplementary Figure S5C), the kp of (S)-BINAP was quantified as 0.25 s−1 according to the values of Φisc, Φp(RT), and τp(RT) in Eq. (2). The optically determined kp was comparable to 0.22 s−1, which was the kp value calculated using the optimized T1 geometry of (S)-BINAP. In addition, the locally excited transition characteristics between T1 and S0, which can be used to explain the optically observed vibrational shape of the pRTP spectra of (S)-BINAP, were calculated for the optimized T1 geometry of (S)-BINAP (Supplementary Figure S7A). The calculated T1-S0 transition energy based on the optimized T1 geometry of (S)-BINAP provides a simple statistical explanation for the green color of the pRTP of the (S)-BINAP (Supplementary Figure S7B). The Φp(77K) and τp(77K) values of the amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP film were 0.023 and 0.63 s, respectively. The optically determined kp, the Φp(77K), and the τp(77K) were substituted into Eq. 1 to determine the Φt = 0.15 of the amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP film (Table 1). Similarly, the Φp(77K) and τp(77K) values of the crystalline 5 wt% (S)-BINAP-doped (S)-H8-BINAP film were 0.125 and 0.75 s, respectively. The kp, the Φp(RT), and the τp(RT) were substituted into Eq. 1 to determine the Φt = 0.68 of the crystalline 5 wt% (S)-BINAP-doped (S)-H8-BINAP film (Table 1). Hence the approximate 4.5 times difference in Φt between the crystalline and amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP films. However, the difference in Φt was still small compared with the approximate 20 times difference in Φp(RT) between the crystalline and amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP films. Therefore, further investigation about the mechanism of transition from T1 to S0 in the films is required.
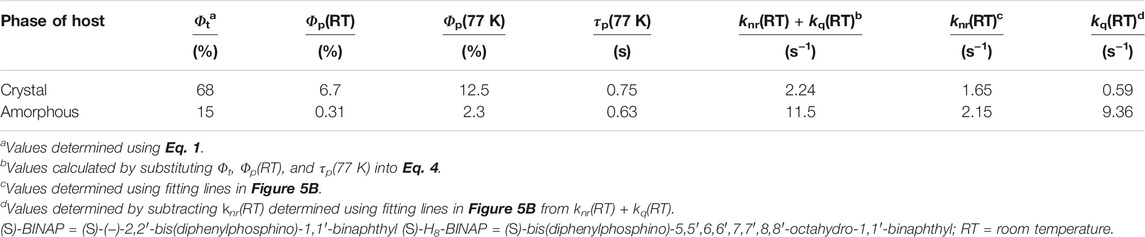
TABLE 1. Photophysical values relating to phosphorescence of 5 wt% (S)-BINAP-doped (S)-H8-BINAP solids in crystalline and amorphous states.
Large Different Triplet Deactivation Depending on the Phase Change of the Host
To investigate the large difference in Φp(RT) between the amorphous and crystalline states of the 5 wt% (S)-BINAP-doped (S)-H8-BINAP, the temperature dependence of the emission characteristics of the two materials was determined. The fluorescence characteristics were almost independent of temperature in both the amorphous and crystalline states (Supplementary Figure S8), whereas there was a large difference between the temperature dependence of the phosphorescence characteristics of the amorphous and crystalline states (Figure 5A and Supplementary Figure S9) of the 5 wt% (S)-BINAP-doped (S)-H8-BINAP solid. The Φp of the crystalline 5 wt% (S)-BINAP-doped (S)-H8-BINAP decreased from 12.5 to 6.7% as the temperature increased from 77 K to RT (red in Figure 5A). In contrast, the Φp of the amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP decreased markedly from 2.3 to 0.31% as the temperature increased from 77 K to RT (blue in Figure 5A). The phosphorescence lifetime at T (τp(T)) is generally expressed as (Hirata, 2017):
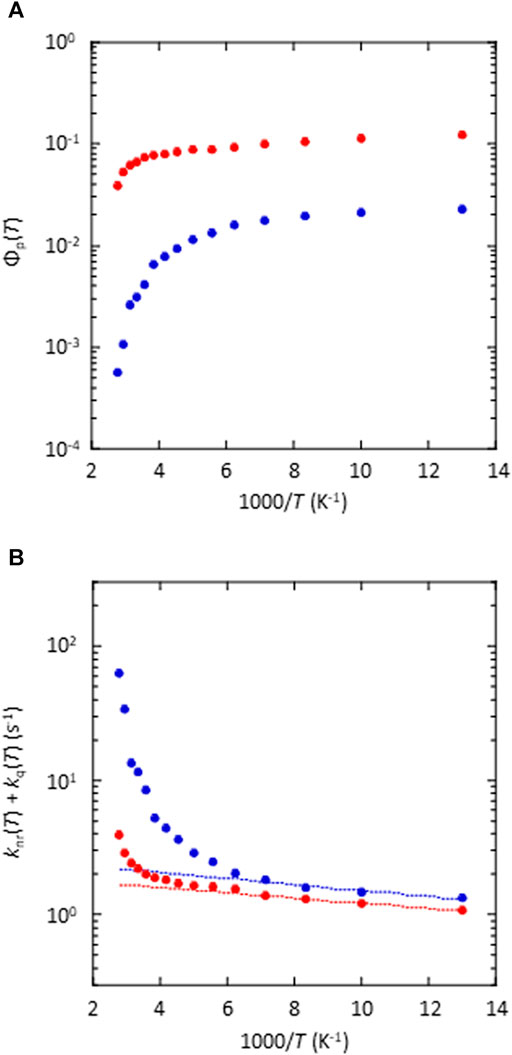
FIGURE 5. Temperature dependence of phosphorescence and nonradiative deactivation of amorphous (blue) and crystalline (red) 5 wt% (S)-BINAP-doped (S)-H8-BINAP. (A) Temperature dependence of Φp(T). The excitation wavelength was 330 nm. (B) Temperature dependence of knr(T) + kq(T). The dashed lines are fitting lines for knr(T), which was determined according to the exponential function for data from 77K to 140 K. ((S)-BINAP = (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (S)-H8-BINAP = (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl).
The following equation can be produced from Eq. 1 and Eq. 3:
For the amorphous 5 wt% (S)-BINAP-doped (S)-H8-BINAP film, Φt = 0.15, kp = 0.25 s−1, and Φp(T) (blue plots in Figure 5A) were substituted into Eq. 4 to determine the knr(T) + kq(T) of the molecularly dispersed (S)-BINAP in the amorphous (S)-H8-BINAP host (blue plots in Figure 5B). For the 5 wt% (S)-BINAP doped into the crystalline (S)-H8-BINAP film, Φt = 0.68, kp = 0.25 s−1, and Φp(T) (red plots in Figure 5A) were substituted into Eq. 4 to determine the knr(T) + kq(T) of the molecularly dispersed (S)-BINAP in the crystalline (S)-H8-BINAP host (red plots in Figure 5B).
The knr(T) + kq(T) of (S)-BINAP did not increase significantly when (S)-BINAP was doped into the crystalline (S)-H8-BINAP host (red in Figure 5B). However, the knr(T) + kq(T) of (S)-BINAP increased markedly in the amorphous (S)-H8-BINAP host (blue in Figure 5B). We note that knr(T) + kq(T) was comparable in the low-temperature range, whereas it was markedly different in the high-temperature range. In previous research, the very small increase in knr(T) + kq(T) in the low-temperature range was often caused by knr(T) (Hirata et al., 2013; Kwon et al., 2015). The lack of a marked increase in knr(T) as the temperature increases has been confirmed from experimental and theoretical viewpoints using a variety of heavy atom-free chemical backbones (Bhattacharjee et al., 2021; Hirata and Bhattacharjee, 2021). Therefore, the lack of a marked increase in knr(T) in (S)-BINAP is plausible. Indeed, the theoretical calculations that depend on spin–orbit coupling with consideration for vibrations support the idea that the knr(T) in (S)-BINAP hardly increased from 77 K to RT (Supplementary Figure S10). Therefore, knr(T) is almost independent of intermolecular interactions between (S)-BINAP and (S)-H8-BINAP. Because the comparable knr(T) values of (S)-BINAP in different phase conditions were estimated (Figure 5B), the large difference in knr(T) + kq(T) at high temperatures was caused by intermolecular processes, i.e., kq(T).
Discussion of the Driving Force Behind Intermolecular Triplet Quenching
Triplet quenching caused by endothermic triplet–triplet energy transfer has been reported for metal-free and/or heavy atom-free chromophores with small kp and knr(RT) values, even when the T1 energy of the host is much larger than that of the guest, and the solid materials are under high vacuum (Hirata et al., 2013; Totani et al., 2013). In research on organic light emitting diodes, kq(RT) values of varying magnitudes are often discussed in terms of the difference in the T1 energy between the guest and host (ΔE) (Adachi et al., 2001). However, the absence of a significant difference between the onset energy values of the phosphorescence spectra of the amorphous and crystalline states of (S)-H8-BINAP (Figure 3C; (ii) and (iii)) indicates that the T1 energy of the neat (S)-H8-BINAP solid hardly changed between those states. Because the onset energy of phosphorescence of the (S)-BINAP guest also hardly changed between the amorphous and crystalline states of (S)-H8-BINAP host, the ΔE of the 5 wt% (S)-BINAP-doped (S)-H8-BINAP was comparable between those states. Therefore, other physical viewpoints are crucial for explaining the difference in kq(RT) values between the amorphous and crystalline states of 5 wt% (S)-BINAP-doped (S)-H8-BINAP solid.
It may be necessary to consider other diffusion-limited processes to determine potential factors that contribute to kq(RT). Although endothermic processes are not generally rapid, the diffusion process in a solid state is potentially slower than a slow endothermic process. Because this condition satisfies the definition of a diffusion-limited process, the observable rate constant kq(RT) can be expressed using the formula for a general diffusion-limited process (Fukuzumi et al., 2003; Chakraborty et al., 2006):
where kdiff(T) is the diffusion rate constant at T and ket(T) is the rate constant of triplet deactivation via triplet–triplet energy transfer from guest to host. kdiff(T) is generally expressed as: (Horie et al., 1984)
where R is the reaction radius between guest and host, D(T) is the diffusion coefficient at T K, N is the Avogadro number, and [C] is the concentration of host. Because Dexter energy transfer is often discussed using the double electron transfer model according to Marcus-based theory (Dexter, 1953; Marcus, 1956; Closs et al., 1989; Köhler and Bässler, 2011), ket(T) may be expressed based on the following Marcus formula:
where ν is the frequency of the motion in the reactant potential well between guest and host, λ is the reorganization energy for triplet–triplet energy transfer, and k is the Boltzman constant. Eq. 5 and Eq. 6 indicate that D(RT) is almost proportional to kq(RT) when kdiff(RT) is much less than ket(RT). Although the relationship between kq(RT) and ΔE is mostly considered with regard to solid molecular materials in the field of organic light emitting diodes (Adachi et al., 2001; Hirata et al., 2013; Notsuka et al., 2017), Eqs. 5–7 indicate that an increase in λ and a decrease in D(RT) also contribute to the suppression of kq(RT). Therefore, consideration of ΔE, λ, and D(RT) is necessary for a discussion of the difference in kq(RT) values between amorphous and crystalline states.
If λ in the crystalline state is much larger than it is in the amorphous state due to more potential intermolecular interactions, the difference in the number of intermolecular interactions causes a large difference in kq(RT), even when ΔE is comparable. Under the condition that (S)-BINAP had intermolecular interactions with the (S)-H8-BINAP molecules in the crystalline (S)-H8-BINAP lattice (λc), the reorganization energy with regard to the energy transfer from T1 of (S)-BINAP to T1 of (S)-H8-BINAP was calculated to enable further discussion of the contribution made by λ to kq(RT) (Figure 6A; (ii)). Initially, a molecule of (S)-H8-BINAP in the crystalline lattice of (S)-H8-BINAP was replaced by (S)-BINAP (Supplementary Figure S11A; (I)). The S0 and T1 geometries of the replaced (S)-BINAP were optimized without changing any of the coordinates of the (S)-H8-BINAP molecules around the (S)-BINAP (Supplementary Figure S11A, (II)). Subsequently, the S0 and T1 geometries of the (S)-H8-BINAP molecule in the crystalline lattice of (S)-H8-BINAP were optimized without changing any of the coordinates of the (S)-H8-BINAP molecules around the target (S)-H8-BINAP (Supplementary Figure S11B; (III)). The four geometries were used to determine λc, and λc = 1.59 eV was calculated using the Amsterdam Modeling Suite (AMS) 2020 software package with the B3LYP functional and the TZP basis set (Supplementary Material, Section 4). The reorganization energy relating to the energy transfer from T1 of (S)-BINAP to T1 of (S)-H8-BINAP under the condition that (S)-BINAP and (S)-H8-BINAP had no intermolecular interactions (λm) was also calculated (Figure 6A; (i) and Supplementary Figure S12). The S0 and T1 geometries were calculated for isolated molecules of (S)-BINAP and (S)-H8-BINAP. The four geometries were used to determine λm, and λm = 1.84 eV was calculated using the AMS 2020 software package with the B3LYP functional and the TZP basis set (Supplementary Material, Section 4). Although a larger λ in the crystalline state had been logically considered because intermolecular interactions may cause more energy to coordinate change for reorganization, λc was slightly smaller than λm. For example, the reorganization energy for charge transfer between molecules in a crystalline semiconductor is 0.1 eV (Liu et al., 2017). However, this is often less than that calculated for isolated molecules without any intermolecular interactions (Narushima et al., 2019). Therefore, the local rigidity in molecular solids may be hardly related to the reorganization energy for charge transfer. Therefore, ΔE and λ do not properly explain the large increase in kq(RT) in the amorphous, compared with the crystalline, 5 wt% (S)-BINAP-doped (S)-H8-BINAP.
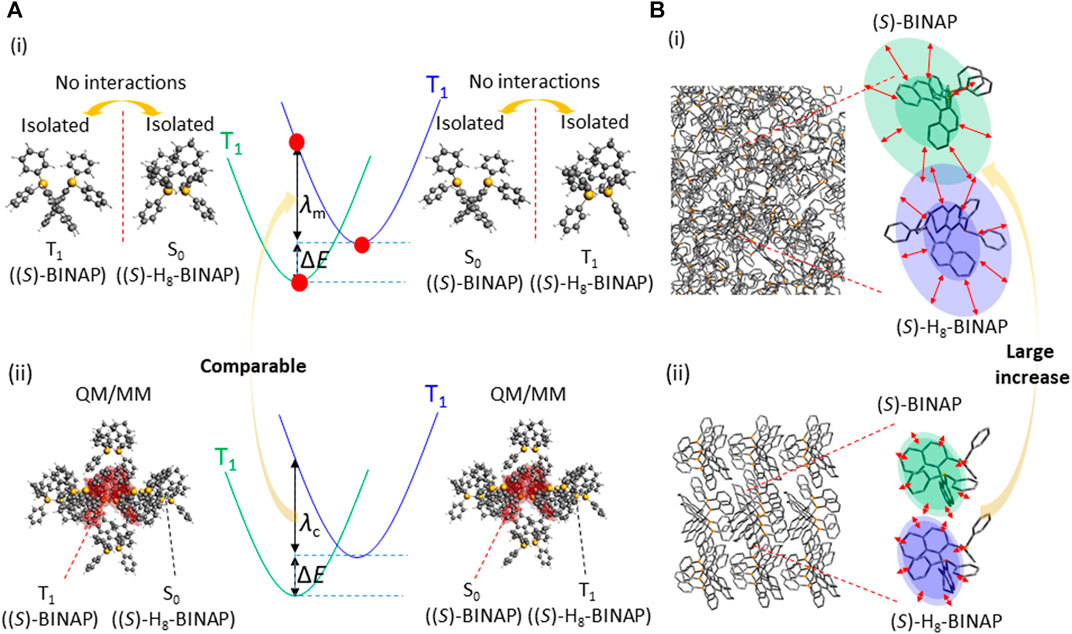
FIGURE 6. Physical behavior relating to kq(RT) except for ΔE from the perspective of intermolecular interactions between an (S)-BINAP guest and the (S)-H8-BINAP host molecules. (A) Reorganization energy for endothermic triplet–triplet energy transfer. (i) represents a case in which (S)-BINAP and (S)-H8-BINAP have no intermolecular interactions. (ii) represents a case in which a molecule in a crystalline lattice of (S)-H8-BINAP is replaced by (S)-BINAP. (B) The diffusion constant; (i) and (ii) represent the amorphous and crystalline states, respectively. The red arrows indicate different magnitudes of the diffusion constant at RT between the amorphous and crystalline states. (RT = room temperature; (S)-BINAP = (S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (S)-H8-BINAP = (S)-bis(diphenylphosphino)-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-binaphthyl).
Another potential reason to change kq(RT) independently of ΔE and λ may be D(RT). Diffusion depends on Brownian motion. It is a different physical phenomenon from the vibrations and coordination changes that take place in molecules during reorganization (Brown, 1828; Einstein, 1905). The theory underlying Brownian motion was established in the early 1900s (von Smoluchowski, 1906). Because molecules in an amorphous state generally have a large free volume compared with those in a crystalline state, the local diffusion of molecules is allowed, even when the solids are below the glass transition temperature (Horie and Mita, 1982; Horie et al., 1984). The D(RT) of a side group in a representative amorphous polymer—i.e., polymethylmethacrylate—was reported to have a magnitude of 10−14 cm2/s (Horie et al., 1984), which is less than the D(RT) of a general liquid molecule, i.e., 1/108 (Monguzzi et al., 2013). The D(RT) in the amorphous state may be large compared with that in the crystalline state for solid materials at RT. To enable triplet–triplet energy transfer, two molecular orbitals must overlap. In the amorphous state, a large D(RT) increases the possibility of the overlap of two molecular orbitals enabling triplet–triplet energy transfer (Figure 6B; (i)). However, stronger intermolecular interactions decrease D(RT), reducing the possibility of orbital overlap (Figure 6B; (ii)). Molecular dynamics (MD) analysis indicates that the simulated D(RT) of (S)-H8-BINAP in the amorphous state is approximately 10 times larger than it is in the crystalline state (Supplementary Figure S13). The magnitude of the difference in the calculated D(RT) between the amorphous and crystalline states is comparable to that of the kq(RT) between those states. Thus, the similar increase of magnitude of the enhanced D(RT) of crystalline (S)-H8-BINAP solid compared with amorphous (S)-H8-BINAP solid was observed between optically estimated D(RT) and simulated D(RT). To date there has been no discussion of ΔE, λ, and D(RT) with regard to kq(RT). By investigating large differences in kq(RT) between two solid materials with comparable ΔE and λ values, we attempted to confirm that the control of D(RT) is also a dominant factor in increasing the triplet quenching caused by endothermic triplet energy transfer. Therefore, more detailed discussions about molecular diffusion in solid materials are essential. The contribution of D(RT) to kq(RT) has not been addressed in research into organic electronics. The contribution made by ΔE to kq(RT) has not been investigated in research on aggregated chromophores that generate pRTP, and the relationship between rigidity and vibrations and intermolecular interactions remains unclear. The controlled results reported in the present paper indicate that discussions about both ΔE and D(RT) are crucial for the intrinsic control of kq(RT) and the long-lived RT triplet state of heavy atom-free and/or metal-free solid materials.
Conclusion
In the present study, a thermo-reversible phase change of an (S)-H8-BINAP host doped with (S)-BINAP was induced. The (S)-BINAP produced a pRTP spectrum that was comparable to the spectra of the (S)-H8-BINAP host in the two phases. The molecularly dispersed (S)-BINAP had a Φp(RT) of 6.7% in the crystalline (S)-H8-BINAP host and a Φp(RT) of 0.31% in the amorphous host. Detailed photophysical analyses indicated that the triplet generation of (S)-BINAP and the magnitude of the difference in kq(RT) between the (S)-BINAP guest and the (S)-H8-BINAP host both differed between the amorphous and crystalline states. This caused a large difference in the Φp(RT) of (S)-BINAP between those states. Differing ΔE, λ, and D(RT) values were considered as a potential explanation for the difference in kq(RT) values between the amorphous and crystalline states. Comparable ΔE and λ values in both the amorphous and crystalline states indicated that the D(RT) is the reason for the large discrepancy in kq(RT). The large difference in the magnitude of D(RT) between the amorphous and crystalline states was supported by MD simulations. The suppression of vibrations by intermolecular interactions is often used to explain the decrease in nonradiative deactivation from T1. However, analysis of ΔE, λ, and D(RT) from all perspectives clarifies that the small D(RT) in the solid state makes a large contribution to the control of kq(RT), and intermolecular interactions could be used to suppress molecular diffusion in the solid state, thereby reducing the kq(RT). Because the significantly low rate of diffusion in molecular solids is also logically related to solid-state chemical reactions, analysis of slow local diffusion may be crucial to the control of chemical conversion in the solid state (Kubota et al., 2019). Because an accurate estimation of the small diffusion constant in molecular solids is difficult, the analysis introduced in the present paper is important for the construction of a variety of materials that allow long-lived RT triplet excitons, and for the control of electron transfer and chemical reactions in molecular solids.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
TK and SH wrote the critical parts of the manuscript, performed the photophysical experiments, and analyzed the photophysical data. SH performed the quantum chemical and molecular dynamics calculations.
Funding
The present work was supported by the JST FOREST Program (grant number JPMJFR201T, Japan) and JSPS KAKENHI (grant numbers JP18H04507, JP20H04664, JP21H02011, and JP21K18928).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.788577/full#supplementary-material
References
Adachi, C., Kwong, R. C., Djurovich, P., Adamovich, V., Baldo, M. A., Thompson, M. E., et al. (2001). Endothermic Energy Transfer: A Mechanism for Generating Very Efficient High-Energy Phosphorescent Emission in Organic Materials. Appl. Phys. Lett. 79, 2082–2084. doi:10.1063/1.1400076
Bhattacharjee, I., Hayashi, K., and Hirata, S. (2021). Key of Suppressed Triplet Nonradiative Transition-dependent Chemical Backbone for Spatial Self-Tunable Afterglow. JACS Au 1, 945–954. doi:10.1021/jacsau.1c00132
Bhattacharjee, I., and Hirata, S. (2020). Highly Efficient Persistent Room‐Temperature Phosphorescence from Heavy Atom‐Free Molecules Triggered by Hidden Long Phosphorescent Antenna. Adv. Mater. 32, 2001348. doi:10.1002/adma.202001348
Brown, R. (1828). XXVII. A Brief Account of Microscopical Observations Made in the Months of June, July and August 1827, on the Particles Contained in the Pollen of Plants; and on the General Existence of Active Molecules in Organic and Inorganic Bodies. Philosophical Mag. 4, 161–173. doi:10.1080/14786442808674769
Chakraborty, A., Chakrabarty, D., Seth, D., Hazra, P., and Sarkar, N. (2006). Photo-Induced Intermolecular Electron Transfer from Electron Donating Solvents to Coumarin Dyes in Bile Salt Aggregates: Role of Diffusion in Electron Transfer Reaction. Spectrochimica Acta A: Mol. Biomol. Spectrosc. 63, 594–602. doi:10.1016/j.saa.2005.06.006
Clapp, D. B. (1939). The Phosphorescence of Tetraphenylmethane and Certain Related Substances. J. Am. Chem. Soc. 61, 523–524. doi:10.1021/ja01871a504
Closs, G. L., Johnson, M. D., Miller, J. R., and Piotrowiak, P. (1989). A Connection between Intramolecular Long-Range Electron, Hole, and Triplet Energy Transfers. J. Am. Chem. Soc. 111, 3751–3753. doi:10.1021/ja00192a044
Deng, Y., Zhao, D., Chen, X., Wang, F., Song, H., and Shen, D. (2013). Long Lifetime Pure Organic Phosphorescence Based on Water Soluble Carbon Dots. Chem. Commun. 49, 5751–5753. doi:10.1039/C3CC42600A
Dexter, D. L. (1953). A Theory of Sensitized Luminescence in Solids. J. Chem. Phys. 21, 836–850. doi:10.1063/1.1699044
Einstein, A. (1905). Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann. Phys. 322, 549–560. doi:10.1002/andp.19053220806
Fateminia, S. M. A., Mao, Z., Xu, S., Yang, Z., Chi, Z., and Liu, B. (2017). Organic Nanocrystals with Bright Red Persistent Room-Temperature Phosphorescence for Biological Applications. Angew. Chem. Int. Ed. 56, 12160–12164. doi:10.1002/ange.20170594510.1002/anie.201705945
Fukuzumi, S., Ohkubo, K., Imahori, H., and Guldi, D. M. (2003). Driving Force Dependence of Intermolecular Electron-Transfer Reactions of Fullerenes. Chem. Eur. J. 9, 1585–1593. doi:10.1002/chem.200390182
Hirata, S., and Bhattacharjee, I. (2021). Vibrational Radiationless Transition from Triplet States of Chromophores at Room Temperature. J. Phys. Chem. A. 125, 885–894. doi:10.1021/acs.jpca.0c09410
Hirata, S., Hara, H., and Bhattacharjee, I. (2020). Phosphorescence Quenching of Heavy-atom-free Dopant Chromophores Triggered by Thermally Activated Triplet Exciton Diffusion of a Conjugated Crystalline Host. J. Phys. Chem. C 124, 25121–25132. doi:10.1021/acs.jpcc.0c07864
Hirata, S. (2017). Recent Advances in Materials with Room-Temperature Phosphorescence: Photophysics for Triplet Exciton Stabilization. Adv. Opt. Mater. 5, 1700116. doi:10.1002/adom.201700116
Hirata, S., Totani, K., Zhang, J., Yamashita, T., Kaji, H., Marder, S. R., et al. (2013). Efficient Persistent Room Temperature Phosphorescence in Organic Amorphous Materials under Ambient Conditions. Adv. Funct. Mater. 23, 3386–3397. doi:10.1002/adfm.201203706
Horie, K., and Mita, I. (1982). Photochemistry in Polymer Solids. Decay of Benzophenone Phosphorescence in Poly(methyl Methacrylate). Chem. Phys. Lett. 93, 61–65. doi:10.1016/0009-2614(82)85056-2
Horie, K., Morishita, K., and Mita, I. (1984). Photochemistry in Polymer Solids. 3. Kinetics for Nonexponential Decay of Benzophenone Phosphorescence in Acrylic and Methacrylic Polymers. Macromolecules 17, 1746–1750. doi:10.1021/ma00139a020
Köhler, A., and Bässler, H. (2011). What Controls Triplet Exciton Transfer in Organic Semiconductors? J. Mater. Chem. 21, 4003–4011. doi:10.1039/C0JM02886J
Kubota, K., Pang, Y., Miura, A., and Ito, H. (2019). Redox Reactions of Small Organic Molecules Using Ball Milling and Piezoelectric Materials. Science 366, 1500–1504. doi:10.1126/science.aay8224
Kwon, M. S., Yu, Y., Coburn, C., Phillips, A. W., Chung, K., Shanker, A., et al. (2015). Suppressing Molecular Motions for Enhanced Room-Temperature Phosphorescence of Metal-free Organic Materials. Nat. Commun. 6, 8947. doi:10.1038/ncomms9947
Liu, C., Huang, K., Park, W.-T., Li, M., Yang, T., Liu, X., et al. (2017). A Unified Understanding of Charge Transport in Organic Semiconductors: the Importance of Attenuated Delocalization for the Carriers. Mater. Horiz. 4, 608–618. doi:10.1039/C7MH00091J
Louis, M., Thomas, H., Gmelch, M., Haft, A., Fries, F., and Reineke, S. (2019). Blue‐Light‐Absorbing Thin Films Showing Ultralong Room‐Temperature Phosphorescence. Adv. Mater. 31, 1807887. doi:10.1002/adma.201807887
Marcus, R. A. (1956). On the Theory of Oxidation‐Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 24, 966–978. doi:10.1063/1.1742723
Metz, F., Friedrich, S., and Hohlneicher, G. (1972). What Is the Leading Mechanism for the Nonradiative Decay of the Lowest Triplet State of Aromatic Hydrocarbons? Chem. Phys. Lett. 16, 353–358. doi:10.1016/0009-2614(72)80291-4
Metz, F. (1973). Position-Dependent Deuterium Effect on Relative Rate Constants for ISC Processes in Aromatic Hydrocarbons. Chem. Phys. Lett. 22, 186–190. doi:10.1016/0009-2614(73)80567-6
Monguzzi, A., Bianchi, F., Bianchi, A., Mauri, M., Simonutti, R., Ruffo, R., et al. (2013). High Efficiency Up-Converting Single Phase Elastomers for Photon Managing Applications. Adv. Energ. Mater. 3, 680–686. doi:10.1002/aenm.201200897
Narushima, K., Kiyota, Y., Mori, T., Hirata, S., and Vacha, M. (2019). Suppressed Triplet Exciton Diffusion Due to Small Orbital Overlap as a Key Design Factor for Ultralong-Lived Room-Temperature Phosphorescence in Molecular Crystals. Adv. Mater. 31, 1807268. doi:10.1002/adma.201807268
Notsuka, N., Kabe, R., Goushi, K., and Adachi, C. (2017). Confinement of Long‐Lived Triplet Excitons in Organic Semiconducting Host-Guest Systems. Adv. Funct. Mater. 27, 1703902. doi:10.1002/adfm.201703902
Schlag, E. W., Schneider, S., and Fischer, S. F. (1971). Lifetimes in Excited States. Annu. Rev. Phys. Chem. 22, 465–526. doi:10.1146/annurev.pc.22.100171.002341
Totani, K., Okada, Y., Hirata, S., Vacha, M., and Watanabe, T. (2013). Thermoresponsive Persistent Phosphorescent Color Change Using Efficient Thermally Activated Reverse Energy Transfer with a Large Energy Difference. Adv. Opt. Mater. 1, 283–288. doi:10.1002/adom.201300013
von Smoluchowski, M. (1906). Zur kinetischen Theorie der Brownschen Molekularbewegung und der Suspensionen. Ann. Phys. 326, 756–780. doi:10.1002/andp.19063261405
Zhang, G., Chen, J., Payne, S. J., Kooi, S. E., Demas, J. N., and Fraser, C. L. (2007). Multi-Emissive Difluoroboron Dibenzoylmethane Polylactide Exhibiting Intense Fluorescence and Oxygen-Sensitive Room-Temperature Phosphorescence. J. Am. Chem. Soc. 129, 8942–8943. doi:10.1021/ja0720255
Zhang, X., Xie, T., Cui, M., Yang, L., Sun, X., Jiang, J., et al. (2014). General Design Strategy for Aromatic Ketone-Based Single-Component Dual-Emissive Materials. ACS Appl. Mater. Inter. 6, 2279–2284. doi:10.1021/am405209w
Zhao, W., He, Z., and Tang, B. Z. (2020). Room-Temperature Phosphorescence from Organic Aggregates. Nat. Rev. Mater. 5, 869–885. doi:10.1038/s41578-020-0223-z
Zhen, X., Tao, Y., An, Z., Chen, P., Xu, C., Chen, R., et al. (2017). Ultralong Phosphorescence of Water-Soluble Organic Nanoparticles for In Vivo Afterglow Imaging. Adv. Mater. 29, 1606665. doi:10.1002/adma.201606665
Keywords: persistent room-temperature phosphorescence, triplet quenching, nonradiative deactivation, phase change, diffusion constant, reorganization energy
Citation: Kusama T and Hirata S (2021) Thermo-Reversible Persistent Phosphorescence Modulation Reveals the Large Contribution Made by Rigidity to the Suppression of Endothermic Intermolecular Triplet Quenching. Front. Chem. 9:788577. doi: 10.3389/fchem.2021.788577
Received: 02 October 2021; Accepted: 26 October 2021;
Published: 16 November 2021.
Edited by:
Rene A. Nome, State University of Campinas, BrazilCopyright © 2021 Kusama and Hirata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuzo Hirata, c2h1em9oaXJhdGFAdWVjLmFjLmpw