 Jun Li1,2Zhiyong Liu3Mingye Hong4Changli Sun1,2
Jun Li1,2Zhiyong Liu3Mingye Hong4Changli Sun1,2 Tianyu Zhang3
Tianyu Zhang3 Hua Zhang4
Hua Zhang4 Jianhua Ju1,2,5
Jianhua Ju1,2,5 Junying Ma1,2,5*
Junying Ma1,2,5*- 1CAS Key Laboratory of Tropical Marine Bio-Resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, RNAM Center for Marine Microbiology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 2Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou, China
- 3Tuberculosis Research Laboratory, State Key Laboratory of Respiratory Disease, Guangdong-Hong Kong-Macao Joint Laboratory of Respiratory Infectious Diseases, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China
- 4Guangdong Provincial Key Laboratory of Medical Molecular Diagnostics, Institute of Laboratory Medicine, Guangdong Medical University, Dongguan, China
- 5College of Oceanology, University of Chinese Academy of Sciences, Qingdao, China
Tuberculosis (TB) is still a global disease threatening people’s lives. With the emergence of multi-drug-resistant Mycobacterium tuberculosis the prevention and control of tuberculosis faces new challenges, and the burden of tuberculosis treatment is increasing among the world. Ilamycins are novel cyclopeptides with potent anti-TB activities, which have a unique target protein against M. tuberculosis and drug-resistant strains. Herein, ilamycin F, a major secondary metabolite isolated from the marine-derived mutant strain Streptomyces atratus SCSIO ZH16 ΔilaR, is used as a scaffold to semi-synthesize eighteen new ilamycin derivatives (ilamycin NJL1–NJL18, 1–18). Our study reveals that four of ilamycin NJLs (1, 6, 8, and 10) have slightly stronger anti-TB activities against Mtb H37Rv (minimum inhibitory concentration, 1.6–1.7 μM) compared with that of ilamycin F on day 14th, but obviously display more potent activities than ilamycin F on day 3rd, indicating anti-TB activities of these derivatives with fast-onset effect. In addition, cytotoxic assays show most ilamycin NJLs with low cytotoxicity except ilamycin NJL1 (1). These findings will promote the further exploration of structure-activity relationships for ilamycins and the development of anti-TB drugs.
Introduction
Tuberculosis (TB) is an infectious disease caused by the pathogen Mycobacterium tuberculosis (Mtb), which is the leading cause of death from a single infectious agent. Globally, it is estimated that 10 million new cases and 1.2 million deaths occurred in 2019 due to TB infections (World Health Organization, 2020). With the appearance of drug-resistant strains, multidrug resistance (MRD) TB and extensive drug resistance (XDR) TB have resulted in a major challenge to the prevention and treatment of TB in the world, especially in developing countries (Saravanan et al., 2018). Standard treatment of TB is a long course, including a 2-months induction phase and a 4-months consolidation phase, thus it is important that drugs with fast-onset action can contribute to shorten treatment in clinical trials (Horsburgh et al., 2015). Hence, there is an urgent demand for development of novel anti-TB drugs with unique targets and fast-onset action.
With the development of drug-resistant tuberculosis, the discovery of new drugs or the drug-repurposed for tuberculosis is increasing recently (Furin et al., 2019). Ilamycins, also named rufomycins, comprise a representative of cycloheptapeptides with strong anti-TB activity, which were isolated from Streptomyces atratus and S. islandicus (Takita et al., 1962; Cary et al., 1971; Ma et al., 2017; Sun et al., 2020; Zhou et al., 2020). Previous studies identified the target of rufomycins, caseinolytic protein C1 (ClpC1), which was different from that of the current therapeutic drugs (Sassetti et al., 2003; Lee et al., 2016; Choules et al., 2018; Choules et al., 2019; Wolf et al., 2019). Therefore, compounds of this family are a promising drug-lead for the treatment of MRD- and XRD-TB. Moreover, for the structure-activity relationship (SAR) studies, Eli Lilly and Company firstly synthesized a series of ilamycin derivatives in 2000 (Lambooy, 2000). Anti-TB assays showed that 6 of the derivatives exhibited strong inhibitory activities against Mtb H37Ra (Figure 1A).
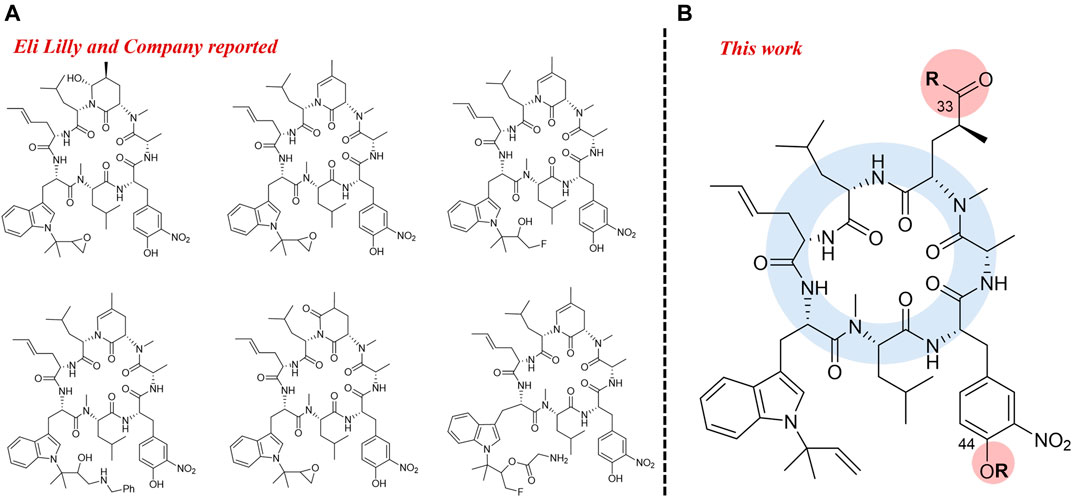
FIGURE 1. Semi-synthesis of rufomycin active derivatives from Eli Lilly and Company (A) Modified position from this work (B).
In recent years, our group has been focusing on the discovery and the biosynthesis of anti-infective antibiotics. Ilamycin F, isolated from a genetic engineered mutant of the deep South China Sea-derived strain Streptomyces atratus SCSIO ZH16, had a strong anti-TB activity against M. tuberculosis H37Rv with minimum inhibitory concentration (MIC) value of 1.2 μM (Ma et al., 2017). As the main metabolite of the mutant strain S. atratus SCSIO ZH16 ΔilaR, the yield of ilamycin F is about 400–500 mg/L in its mutant. In this regard, ilamycin F is ideally utilized as a starting material for preparing new ilamycin derivatives, which will facilitate to further investigate their SAR and discover more efficient anti-TB drug leads. Herein, we report the preparation and characterization of eighteen new ilamycin F derivatives (ilamycin NJL1–NJL18) on C-33 and C-44 of ilamycin F (Figure 1B). Several semi-synthesized derivatives display potent anti-TB activity against M. tuberculosis H37Rv with fast-onset effect and low cytotoxicity.
Results and Discussion
Semi-Synthesis of Ilamycin NJLs
Ilamycin F has two types of functional groups for modification, C-33 carboxyl group and C-44 hydroxyl group. With the aim of synthesizing new derivatives, several modifications in ilamycin F were introduced by acylation and esterification (Scheme 1). Ilamycin NJL1–NJL12 (1–12) were concisely synthesized by 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC)/1-hydroxybenzotriazole (HOBt) assisted amidation at C-33 carboxyl group using l-amino acid methyl esters and benzylamine derivatives (Figure 2). The yields ranged from 74 to 83%. Moreover, according to the twin drug strategy (Contreras et al., 2008; Ibacache et al., 2018), isoniazid and N-deacetyl-linezolid, two anti-TB substrates, were respectively coupled with ilamycin F to generate ilamycin NJL13–NJL14 (13–14) using the aforementioned amidation method in 76–77% yield (Figure 2). The strategy was proposed to produce synergic effect by binding two targets.
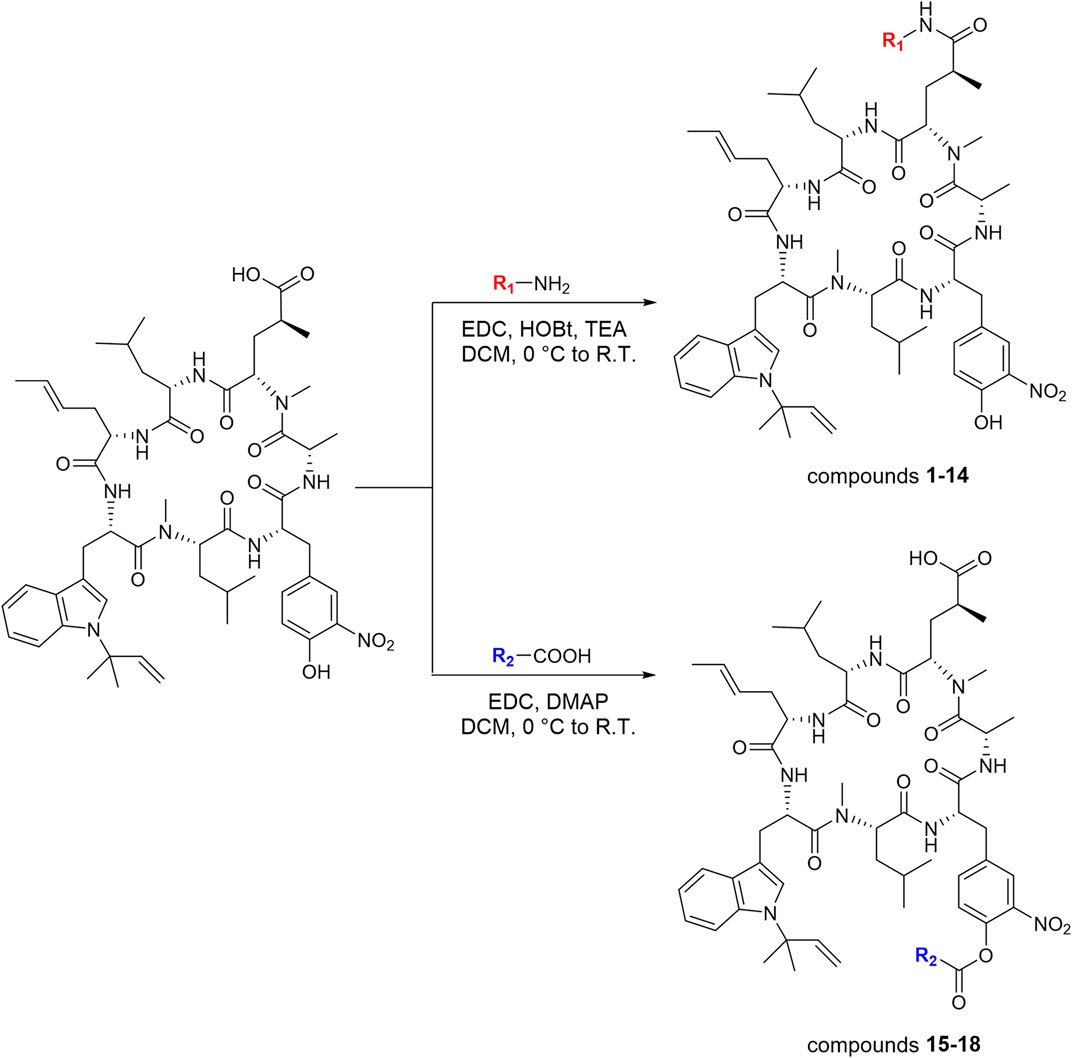
SCHEME 1. Synthetic routes of ilamycin NJLs (compounds 1–18).
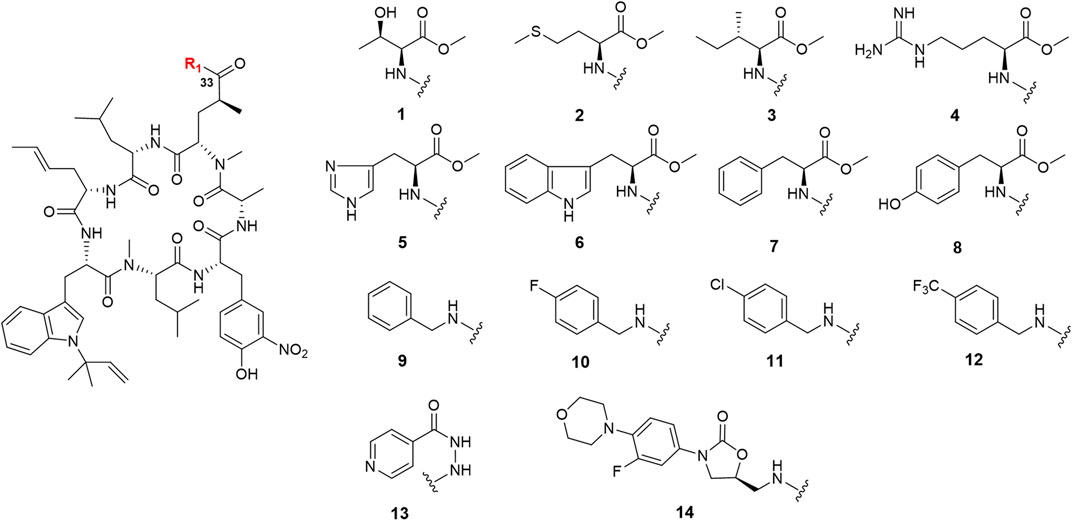
FIGURE 2. Structures of ilamycin NJLs modified at C-33 (compounds 1–14).
The C-44 hydroxyl group of ilamycin F is another position for derivatization. Although various ether and aliphatic sidechains at C-44 were created through etherification or esterification by Eli Lilly and Company, all derivatives displayed low anti-TB activities (Lambooy, 2000). To further study SAR of ilamycin F, heteroaromatic rings, p-fluorophenylacetic acid and 3-(methylthio) propionic acid were introduced at the C-44 hydroxyl group (Scheme 1). Ilamycin NJL15–NJL18 (15–18) could be successfully obtained in the presence of EDC and 4-dimethylaminopyridine (DMAP) (Figure 3), but owing to low nucleophilicity of the C-44 hydroxyl group, the yields of the reaction were only given to 32–40%.
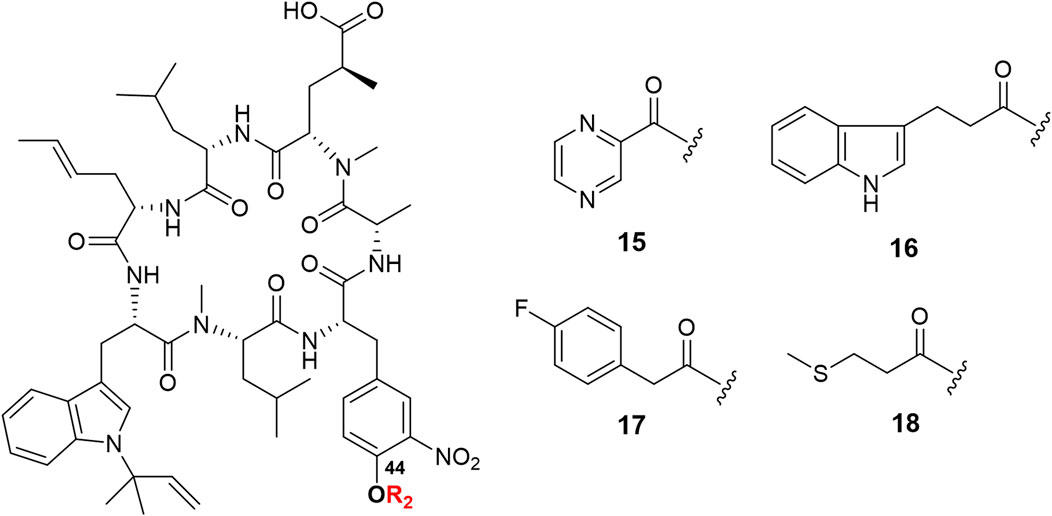
FIGURE 3. Structures of ilamycin NJLs modified at C-44 (compounds 15–18).
Bioactivities of Ilamycin NJLs
The anti-TB activities of ilamycin NJL1–NJL18 (1–18) were evaluated against M. tuberculosis H37Rv, which has pathogenic and still popularly used in virulent laboratory (Camus et al., 2002; Målen et al., 2011). As depicted in Table 1, although basic amino acid derivatives (compounds 4–5) at the C-33 had weak activities, the modification of neutral amino acid derivatives, in comparison with ilamycin F, displayed potent anti-TB activities with fast-onset effect. Compounds 1–3, 6, and 8, showed efficient activities on day 3rd, which was higher than that of ilamycin F with 4–19 folds. Importantly, compounds 1, 6, and 8 had slightly stronger activities than that of ilamycin F on day 14th (MIC, 1.6–1.7 μM), speculating that their modification did not change the interaction with targets, but would obviously facilitate to promoting penetration of membranes in Mtb. Moreover, benzylamine derivatives of ilamycin F, compounds 9–11, were also exhibited the fast-onset effect on day 3rd except compound 12. This finding indicated that benzylamine modified with a larger substituent significantly affected anti-TB activity of ilamycin derivatives. Although showed a similar activity with that of ilamycin F on day 14th, compound 10 was 2-fold more potent MIC value than that of 9 and 11, which might result from the promotion of its lipophilicity by fluorine substituent and improved the penetration to cell membranes (Smart, 2001; Purser et al., 2008). However, the activities of compounds 13–14 were significantly decreased under the twin drug strategy, when a carboxyl group at the C-33 was replaced by isoniazid or N-deacetyl-linezolid. This result indicated that the construction of compounds 13–14 affected the binding to their targets, and exhibited no synergistic effect on anti-TB activity. Additionally, compounds 15–18 modified at the C-44 also showed lower activities. The similar groups coupling at the C-33 with beneficial effects could not produce the same promotion at the C-44 hydroxyl group of ilamycin F. The results suggested that the hydroxyl group at C-44 might serve as a pharmacophore, which was critical in achieving anti-TB activity and also consistence with our previous discoveries (Sun et al., 2020).
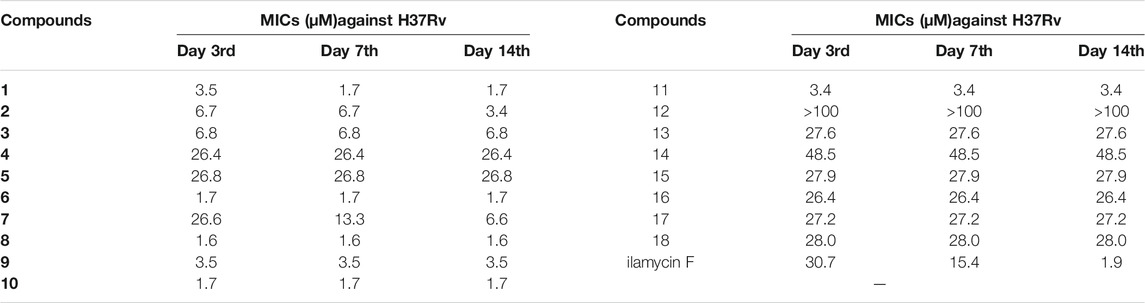
TABLE 1. Anti-tubercular activity of ilamycin NJLs (1–18) against M. tuberculosis H37Rv.
To evaluate the application potential of these compounds, the cytotoxicity of ilamycin NJLs (1–18) was evaluated in vitro using five human cancer cell lines, including breast adenocarcinoma (MCF-7), cervical carcinoma (HeLa), hepatocellular carcinoma (HepG2), lung cancer (A549), colon cancer (HCT116); two normal cell lines including human hepatic cell line (L02) and human umbilical vein endothelial cell line (Huvec-12). Although most compounds (2–18) showed no or weak cytotoxicity, compound 1 exhibited a moderate IC50 value (5.7–9.0 μM) against MCF-7, A549, HCT116, and L02, which had a 3–8-fold promotion compared with that of ilamyicn F (Table 2). The result indicated that threonine methyl ester modified in C-33 carboxyl group of ilamycin F was favorable for the cytotoxic activity.
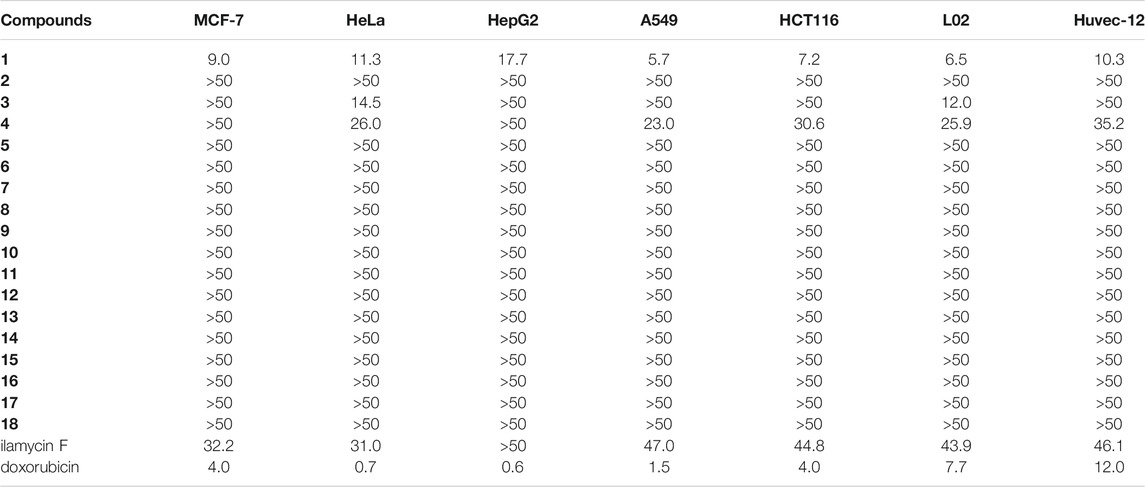
TABLE 2. IC50 value (μM) of ilamycin NJLs (1–18) against five human cancer cell lines and two normal cell lines.
Conclusion
In this study, ilamycin F, a starting material isolated from marine-derived mutant S. atratus ZH16 ΔilaR, was employed to semi-synthesize eighteen ilamycin F derivatives (ilamcyin NJL1–NJL18). Their inhibitory effects on M. tuberculosis H37Rv were tested in vitro. Our study revealed that compounds 1, 6, 8, and 10 exhibited slightly stronger anti-TB activity (1.6–1.7 μM) with that of ilamycin F on day 14th, but displayed a 9–19-fold increased anti-TB activities compared with that of ilamycin F on day 3rd (MICs 1.6–3.5 μM), which indicated their rapid suppression effect on M. tuberculosis. In addition, most ilamycin NJLs had low cytotoxicity except compound 1 displayed a moderate cytotoxic activity (IC50, 5.7–11.3 μM) against five human cancer cell lines and two normal cell lines. Our results will be beneficial to further exploration for SAR of ilamycins and promote the development of anti-TB drugs.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
JL conducted the chemical experiments and drafted the manuscript. ZL and TZ did the antitubercular assay. MH and HZ performed the cytotoxicity assays. CS carried out the NMR analysis. JM and JJ initiated the project and revised the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (82022067, 31870046, and 81973372), Key Science and Technology Project of Hainan Province (ZDKJ202018), Key-Area Research and Development Program of Guangdong Province (2020B1111030005), Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0406), Guangdong Provincial-level Special Funds for Promoting High-quality Economic Development (2020032), Key Science and Technology Project of Sichuan Province (19SYXHZ0025).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer (SF) declared a past co-authorship with one of the authors (TZ) to the handling Editor.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.774555/full#supplementary-material
References
Camus, J.-C., Pryor, M. J., Médigue, C., and Cole, S. T. (2002). Re-Annotation of the Genome Sequence of Mycobacterium Tuberculosis H37Rv. Microbiology. 148, 2967–2973. doi:10.1099/00221287-148-10-2967
Cary, L. W., Takita, T., and Ohnishi, M. (1971). A Study of the Secondary Structure of Ilamycin B1by 300 MHz Proton Magnetic Resonance. FEBS Lett. 17, 145–148. doi:10.1016/0014-5793(71)80584-7
Choules, M. P., Klein, L. L., Lankin, D. C., McAlpine, J. B., Cho, S.-H., Cheng, J., et al. (2018). Residual Complexity Does Impact Organic Chemistry and Drug Discovery: the Case of Rufomyazine and Rufomycin. J. Org. Chem. 83, 6664–6672. doi:10.1021/acs.joc.8b00988
Choules, M. P., Wolf, N. M., Lee, H., Anderson, J. R., Grzelak, E. M., Wang, Y., et al. (2019). Rufomycin Targets ClpC1 Proteolysis in Mycobacterium Tuberculosis and M. Abscessus. Antimicrob. Agents Chemother. Agents Ch. 63, e02204–18. doi:10.1128/AAC.02204-18
Contreras, J.-M., and Sippl, W. (2008). Homo and Heterodimer Ligands: the Twin Drug Approach. The Practice of Medicinal Chemistry. 3rd ed. Netherlands: Elsevier, 380–414. doi:10.1016/B978-0-12-374194-3.00018-4
Furin, J., Cox, H., and Pai, M. (2019). Tuberculosis. The Lancet. 393, 1642–1656. doi:10.1016/S0140-6736(19)30308-3
Horsburgh, C. R., Barry, C. E., and Lange, C. (2015). Treatment of Tuberculosis. N. Engl. J. Med. 373, 2149–2160. doi:10.1056/NEJMra1413919
Ibacache, J., Faundes, J., Montoya, M., Mejías, S., and Valderrama, J. (2018). Preparation of Novel Homodimers Derived From Cytotoxic Isoquinolinequinones. A Twin Drug Approach. Molecules. 23, 439–448. doi:10.3390/molecules23020439
Lambooy, P. K. (2000). Process for the Isolation of Rufomycin Factors. United States: Eli Lilly and Company. Available online at: http://europepmc.org/article/PAT/WO0078798.
Lee, H., and Suh, J.-W. (2016). Anti-tuberculosis lead Molecules From Natural Products Targeting Mycobacterium Tuberculosis ClpC1. J. Ind. Microbiol. Biot. 43, 205–212. doi:10.1007/s10295-015-1709-3
Målen, H., De Souza, G. A., Pathak, S., Søfteland, T., and Wiker, H. G. (2011). Comparison of Membrane Proteins of Mycobacterium TuberculosisH37Rv and H37Ra Strains. BMC Microbiol. 11, 1–10. doi:10.1186/1471-2180-11-18
Ma, J., Huang, H., Xie, Y., Liu, Z., Zhao, J., Zhang, C., et al. (2017). Biosynthesis of Ilamycins Featuring Unusual Building Blocks and Engineered Production of Enhanced Anti-Tuberculosis Agents. Nat. Commun. 8, 1–10. doi:10.1038/s41467-017-00419-5
Purser, S., Moore, P. R., Swallow, S., and Gouverneur, V. (2008). Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 37, 320–330. doi:10.1039/B610213C
Saravanan, M., Niguse, S., Abdulkader, M., Tsegay, E., Hailekiros, H., Gebrekidan, A., et al. (2018). Review on Emergence of Drug-Resistant Tuberculosis (MDR & XDR-TB) and its Molecular Diagnosis in Ethiopia. Microb. Pathogenesis. 117, 237–242. doi:10.1016/j.micpath.2018.02.047
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes Required for Mycobacterial Growth Defined by High Density Mutagenesis. Mol. Microbiol. 48, 77–84. doi:10.1046/j.1365-2958.2003.03425.x
Smart, B. E. (2001). Fluorine Substituent Effects (On Bioactivity). J. Fluorine Chem. 109, 3–11. doi:10.1016/S0022-1139(01)00375-X
Sun, C., Liu, Z., Zhu, X., Fan, Z., Huang, X., Wu, Q., et al. (2020). Antitubercular Ilamycins From Marine-Derived Streptomyces Atratus SCSIO ZH16 ΔilaR. J. Nat. Prod. 83, 1646–1657. doi:10.1021/acs.jnatprod.0c00151
Takita, T., Ohi, K., Okami, Y., Maeda, K., and Umezawa, H. (1962). New Antibiotics, Ilamycins. J. Antibiot. (Tokyo). 15, 46–48. doi:10.11554/antibioticsa.15.1_46
Wolf, N. M., Lee, H., Choules, M. P., Pauli, G. F., Phansalkar, R., Anderson, J. R., et al. (2019). High-resolution Structure of ClpC1-Rufomycin and Ligand Binding Studies Provide a Framework to Design and Optimize Anti-Tuberculosis Leads. ACS Infect. Dis. 5, 829–840. doi:10.1021/acsinfecdis.8b00276
World Health Organization (2020). Global Tuberculosis Report. Available online at: https://www.who.int/teams/global-tuberculosis-programme/data.
Keywords: streptomycetes, cyclopeptide, antitubercular activity, ilamycin, semi-synthesis, derivatization
Citation: Li J, Liu Z, Hong M, Sun C, Zhang T, Zhang H, Ju J and Ma J (2021) Semi-Synthesis of Marine-Derived Ilamycin F Derivatives and Their Antitubercular Activities. Front. Chem. 9:774555. doi: 10.3389/fchem.2021.774555
Received: 12 September 2021; Accepted: 27 September 2021;
Published: 29 October 2021.
Edited by:
Yuanyuan Lu, China Pharmaceutical University, ChinaReviewed by:
Scott Gary Franzblau, University of Illinois at Chicago, United StatesRama P. Tripathi, Csir-Central Drug Research Institute, Uttar Pradesh, India
Copyright © 2021 Li, Liu, Hong, Sun, Zhang, Zhang, Ju and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junying Ma, bWFqdW55aW5nQHNjc2lvLmFjLmNu