 Yimin Zhang
Yimin Zhang Daqiang Chen
Daqiang Chen Weite Meng
Weite Meng Shunfang Li
Shunfang Li Sheng Meng
Sheng Meng- 1Key Laboratory of Material Physics, Ministry of Education, School of Physics and Microelectronics, Zhengzhou University, Zhengzhou, China
- 2Beijing National Laboratory for Condensed Matter Physics and Institute of Physics, Chinese Academy of Sciences, Beijing, China
- 3School of Chemical Engineering, Anhui University of Science and Technology, Huainan, China
- 4School of Physical Sciences, University of Chinese Academy of Sciences, Beijing, China
A promising route to realize solar-to-chemical energy conversion resorts to water splitting using plasmon photocatalysis. However, the ultrafast carrier dynamics and underlying mechanism in such processes has seldom been investigated, especially when the single-atom catalyst is introduced. Here, from the perspective of quantum dynamics at the atomic length scale and femtosecond time scale, we probe the carrier and structural dynamics of plasmon-assisted water splitting on an Ag-alloyed Pt single-atom catalyst, represented by the Ag19Pt nanocluster. The substitution of an Ag atom by the Pt atom at the tip of the tetrahedron Ag20 enhances the interaction between water and the nanoparticle. The excitation of localized surface plasmons in the Ag19Pt cluster strengthens the charge separation and electron transfer upon illumination. These facts cooperatively turn on more than one charge transfer channels and give rise to enhanced charge transfer from the metal nanoparticle to the water molecule, resulting in rapid plasmon-induced water splitting. These results provide atomistic insights and guidelines for the design of efficient single-atom photocatalysts for plasmon-assisted water splitting.
Introduction
Photoinduced water splitting is a feasible way to mitigate the energy crisis and the associated environmental issues (Lewis, 2007). Given the high atom utilization efficiency, unique electronic structure, precisely identified active site, and excellent catalytic activity and selectivity, single-atom catalysts (SACs) have emerged as a new frontier in heterogeneous catalysis including photocatalytic water splitting in recent years (Qiao et al., 2011; Lang et al., 2020; Zhuo et al., 2020). As reported by Yang et al., compared to atomically dispersed Pd and Rh, single Pt sites anchored on TiO2 exhibit excellent efficiency, high stability, and photo-corrosion resistance for solar-driven water splitting (Xing et al., 2014). Afterward, a series of single-atom catalysts on different substrates had sparked tremendous attention in photoinduced water splitting (Sui et al., 2017; Wu et al., 2018; Hejazi et al., 2020). For instance, Schmuki et al. found that the rate of water splitting on the Pt site of the thin TiO2 layer was enhanced 150 times higher than that on Pt nanoparticles (Hejazi et al., 2020). However, these processes face great challenges in suppressing the recombination of photogenerated carriers and further enhancing the efficiency of charge transfer and chemical reactions. One way to address this weakness is to combine the advantages of SACs and plasmonic excitations in metal clusters to strengthen the light-matter interactions, thanks to the superior optical absorption and extended lifetime of excited carriers of the latter (Nie and Emory, 1997; Xu et al., 1999; Prodan et al., 2003).
Plasmonic metal clusters such as Au, Ag, Cu, and Al can concentrate and channel the energy of solar light into the absorbates after plasmon excitation, which have been prevalently utilized in chemical and solar energy conversion, especially in plasmon-driven photocatalysis including O2 dissociation (Christopher et al., 2011; Christopher et al., 2012; Seemala et al., 2019) and H2O splitting (Liu et al., 2011; Primo et al., 2011; Thimsen et al., 2011; Warren and Thimsen, 2012; Qian et al., 2014; Sigle et al., 2015; Yan et al., 2016; Yan et al., 2018). However, plasmon-induced photocatalysis using SACs has seldom been investigated, especially its underlying carrier dynamics and reaction mechanism. Mark et al. reported a theoretical study about the plasmon-mediated N2 dissociation on a single-atom Fe-functionalized Au cluster (Martirez and Carter, 2017). It was revealed that the strong localized surface plasmon of Au and the active Fe site worked together to lower the dissociation barrier after the consecutive resonance energy transfer. However, the carrier dynamics and underlying charge transfer mechanism was absent. Zhou et al. quantitatively explored the hot carriers and thermal contributions in a plasmon-assisted ammonia photolysis using atomically dispersed Ru on Cu nanoparticles under light irradiation (Zhou et al., 2018). However, the detailed role of the Ru site in the reaction process remains elusive and needs to be further investigated.
Here, we investigate plasmon-induced water splitting on the Ag cluster doped by a single Pt atom at the single-molecule level using real-time time-dependent density functional theory (rt-TDDFT). Through the carrier and structural dynamics analysis, we find that the introduction of single Pt atom improves light absorption and electronic level alignment as given by the strong light-matter interactions. Specially, it opens up different charge transfer channels to magnify the charge transfer rates to water molecules, enabling high-efficiency water splitting. The aforementioned findings offer new prospects for solar water splitting and for the design of optimal photocatalysts with high efficiency.
Results and Discussion
Atomic Configuration and Absorption Spectrum. Here, the tetrahedral Ag20 cluster is used as a model system, while larger clusters such as Ag55 are also tested. The reasons for choosing Ag20 are as follows: first, the tetrahedral Ag20 nanoparticle is structurally stable among many other clusters (Wang et al., 2003). Second, it has been widely used as a good model system for investigating the interactions between small molecules and metal clusters under illumination (Zhao et al., 2006a; Zhao et al., 2006b). Third, the tetrahedral Au20 clusters have already been obtained experimentally on ultrathin NaCl films (Li et al., 2020), which gives guidance for the synthesis of tetrahedral Ag20 on the supported substrates. After the relaxation of the adsorption geometry of the representative high-symmetry configurations, we selected the most stable atomic structures for Ag19Pt–H2O and Ag20–H2O, as shown in Figures 1A,B. Here, compared to the initial distance of 3 Å, the distance of Pt–O is 2.11 Å, less than that for Ag–O (2.24 Å), implying that there exists a stronger interaction between the Ag19Pt and H2O molecule than Ag20. In addition, the ∠H–O–H bond angle in water is 107.6° and 108.3° for Ag19Pt-H2O, Ag20-H2O, respectively, compared to the value of 104.5° in intact water molecules, suggesting that the water molecule is activated after adsorption.
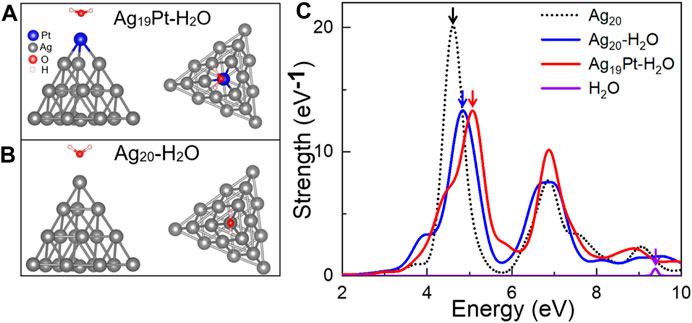
FIGURE 1. Atomic configuration and absorption spectra. The top and side views of (A) Ag19Pt–H2O and (B) Ag20–H2O after geometry optimization. (C) Absorption spectra of Ag20, Ag20–H2O, Ag19Pt–H2O, and H2O, respectively. The colored arrows denote the position of the corresponding absorption peak.
The absorption spectra of Ag19Pt–H2O, Ag20–H2O, Ag20, and a freestanding H2O molecule have been calculated and are shown in Figure 1C. The absorption peak of freestanding H2O is located at >8 eV, corresponding to the transition from the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) of water. Compared to the highest absorption peak located at 4.62 eV for Ag20, Ag19Pt–H2O and Ag20–H2O complexes both display a blue shift where the major absorption peak moves to 5.07 and 4.83 eV, respectively. This means that the Ag19Pt cluster has a stronger electronic coupling between the metal cluster and H2O molecule than the Ag20 cluster, which can also be confirmed by the photo absorption spectra calculated for Ag55Pt–H2O and Ag55–H2O, as shown in Supplementary Figure S1.
Ultrafast Molecular Dynamics After Photoexcitation. To investigate the photoinduced response, we calculate the time-dependent changes in the bond length of O–H for the two complexes mentioned earlier upon laser illumination under different maximum field strength Emax, as shown in Figures 2A,B. The couplings between atomic and electronic motions are governed by the Ehrenfest approximation (Alonso et al., 2008). Given that the equilibrium O–H bond length of gaseous H2O is 0.98 Å, we assume that the interaction between the OH and H atom can be considered negligible when the H–OH distance is >2.0 Å. So, for simplicity, we take the O–H bond length of 2.0 Å as the criterion for determining the breaking of the O–H bond. Obviously, one finds that without laser illumination, the O–H bond length exhibits a stable oscillation around 0.98 Å and the bond does not break, indicating that water splitting does not occur directly after adsorption on the metal clusters. Upon illumination, the O–H bond length increases with Emax for the two systems. In particular, the O–H bond breaks at 40 fs with Emax = 0.5 V/Å for Ag19Pt–H2O, while it does not break but only shows an elongation of 0.2 Å for Ag20–H2O under the same field strength. These results suggest that photocatalytic water splitting can be attributed to the cooperation of the photoexcitation and the introduction of the Pt atom.
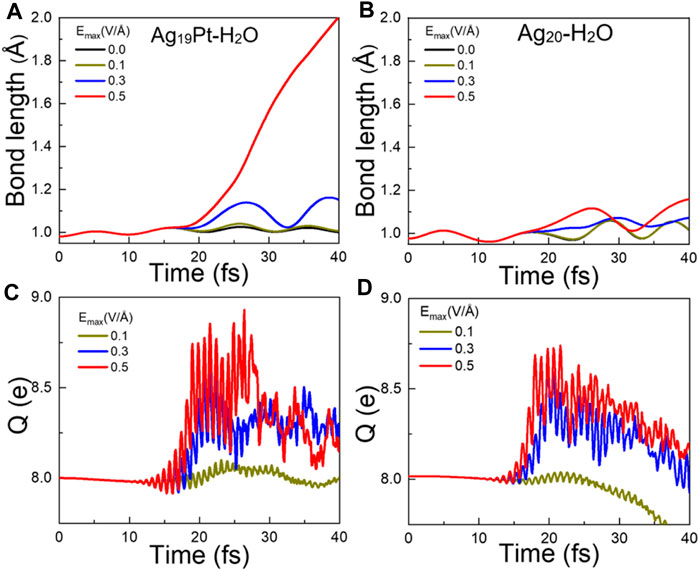
FIGURE 2. Ultrafast dynamic responses after photoexcitation. Time evolved bond length of O–H for (A) Ag19Pt–H2O and (B) Ag20–H2O under different field strengths Emax. Time-evolved total charge (Q) located on H2O for (C) Ag19Pt–H2O and (D) Ag20–H2O under different field strengths.
In order to uncover the underlying mechanism of water splitting, we first compare the time-evolved total charge Q located on the H2O molecule under different field strengths for the two complexes, as shown in Figures 2C,D. For the two systems under irradiation, Q rises quickly when the field strength increases and then gradually decays with an oscillation to a value around 8 e. Here, 8 e corresponds to the initial total charge of a freestanding H2O molecule, that is, there exists a charge transfer ΔQ (ΔQ = Q − 8) from the metal cluster to the H2O molecule if Q > 8. In particular, compared to the charge transfer ΔQ = 0.75 e to the H2O molecule for Ag20–H2O at Emax = 0.5 V/Å, a charge transfer ΔQ = 0.92 e takes place for the Ag19Pt–H2O cluster under the same field strength, implying that the introduction of the Pt atom enhances the amount of charges transferred to water.
Time-Evolved Kohn–Sham States. To further explore the difference in charge transfer for the two systems, the time-evolved occupation of Kohn–Sham (KS) states and the corresponding projected local density of states (LDOS) under a field strength of 0.1 V/Å are calculated, as shown in Figure 3. Here, the occupation of KS states is calculated by projecting the time-dependent KS state onto KS orbitals at time t = 0. At first, there exists an obvious oscillation in the electronic occupation for the KS states near the Fermi level for the two systems, as shown in Figures 3A,C, indicating charge density oscillations around the metal cluster surface (Townsend and Bryant, 2011). Then, the electrons at the deep energy region can be photoexcited to high-energy levels, implying the rapid plasmon decay into hot electron–hole pairs following photoexcitation (Townsend and Bryant, 2014; Ma et al., 2015). It can be seen that the energy region of excitation almost extends all over the range of 0–5 eV for the Ag19Pt–H2O cluster (Figure 3A), while it mainly locates at 3.5–5 eV for Ag20–H2O (Figure 3C), further confirming that more charge transfer to the H2O molecule is favored in the Ag19Pt–H2O system. In order to explain the difference in charge density oscillations for the two systems, we calculated the charge density at t = 40 fs under different field strengths, as shown in Supplementary Figure S2, indicating that the introduction of a single Pt atom alters the distribution of charge oscillations within and around the metal surface. For the sake of analysis, a weak laser pulse with a field strength of 0.1 V/Å is used. Similar results are observed under stronger laser pulses with the relative contribution of different excitation channels varying, such as the case of Emax = 0.5 V/Å, as shown in Supplementary Figure S3.
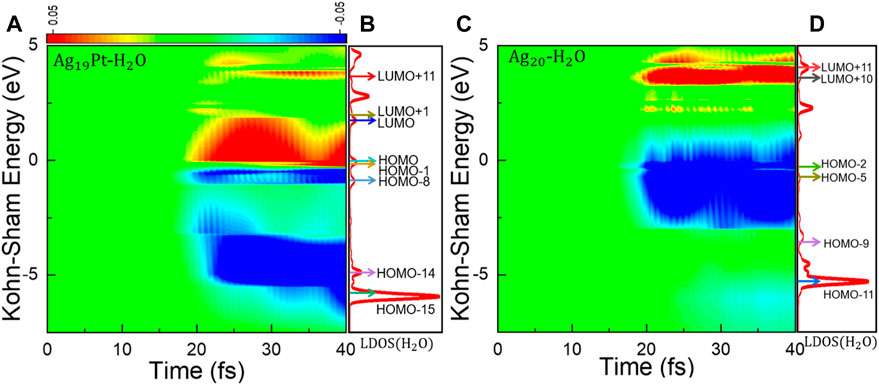
FIGURE 3. Distribution of Kohn–Sham energy levels and changes in occupation. (A) Time-dependent changes in the occupation of the KS states and (B) projected local density of states (LDOS) on the H2O species at t = 0 fs for Ag19Pt–H2O with a field strength of 0.1 V/Å. The eight arrows from the bottom to top denote the energy levels of the eight KS states, that is, HOMO-15, HOMO-14, HOMO-8, HOMO-1, HOMO, LUMO, LUMO+1, and LUMO+11, respectively. (C) Time-dependent changes in the occupation of the KS states and (D) local projected density of states (LDOS) on the H2O species at t = 0 fs for Ag20–H2O under the same condition. The six arrows from the bottom to top denote the energy levels of the six KS states, that is, HOMO-11, HOMO-9, HOMO-5, HOMO-2, LUMO+10, and LUMO+11, respectively.
From the LDOS shown in Figures 3B,D, we can find that the introduction of the Pt atom displays a metallic character for the Ag19Pt–H2O cluster, while a semiconducting behavior is identified for Ag20–H2O, suggesting a qualitative change in the electronic structure. Furthermore, the LDOS located on H2O species is very diffusive, suggesting strong electronic couplings between water and the nanoparticle in both cases.
Time-Dependent Occupation of Kohn–Sham States. To offer a direct description of ultrafast carrier dynamics, the time evolution of the occupation of Kohn–Sham states are shown in Figure 4. In Figure 4A, the change of occupation for the HOMO-14 and HOMO-15 (corresponding to the HOMO level of the H2O species as shown in Figure 4B) is 15% and 7.5%, respectively, which means that the intramolecular charge transfer exists for Ag19Pt–H2O. In contrast, the change of occupation for HOMO-9 and HOMO-11 (corresponding to the HOMO level of water as shown in Figure 4D) is 0, ruling out the intramolecular charge transfer channels for the Ag20–H2O case. In other words, the introduction of the Pt atom opens up additional charge transfer channels to facilitate the plasmon-induced chemical reaction, that is, an intramolecular charge transfer in the case of Ag19Pt–H2O. Moreover, there is a contrary variation trend between LUMO, LUMO+1, LUMO+11 and HOMO, HOMO-1, and HOMO-8 for Ag19Pt–H2O (Figure 4A), implying a charge transfer among these orbitals, so does the orbitals between LUMO+11, LUMO+16 and HOMO-2, HOMO-5 for the Ag20–H2O case. These channels stand for the hot electron generation and intermolecular charge transfer pathways between the metal nanoparticle and water.
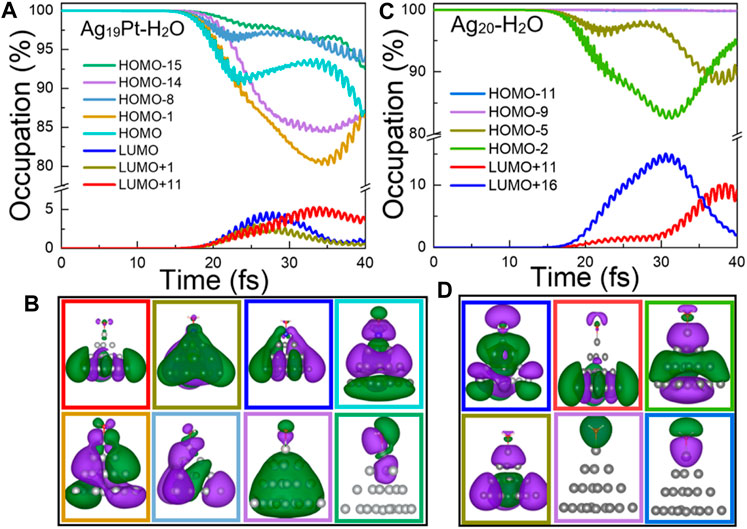
FIGURE 4. Time-evolved occupation of Kohn–Sham states and wave function plots. (A) Time-evolved occupation and (B) the corresponding wave functions of the eight states shown in different colors for Ag19Pt–H2O under a field strength of 0.1 V/Å. (C) Time-evolved occupation and (D) the corresponding wave functions of the six states shown in different colors for Ag20–H2O under the same condition. Here, the isosurface value for the wave functions is 0.02 Å−3.
Charge Transfer Mechanisms. To confirm the underlying charge transfer mechanisms for the two systems, we calculated the time-evolved transition coefficients from all occupied states to the orbitals mentioned earlier, as shown in Figure 5. The charge transfer mechanism for the two systems is analyzed as follows.
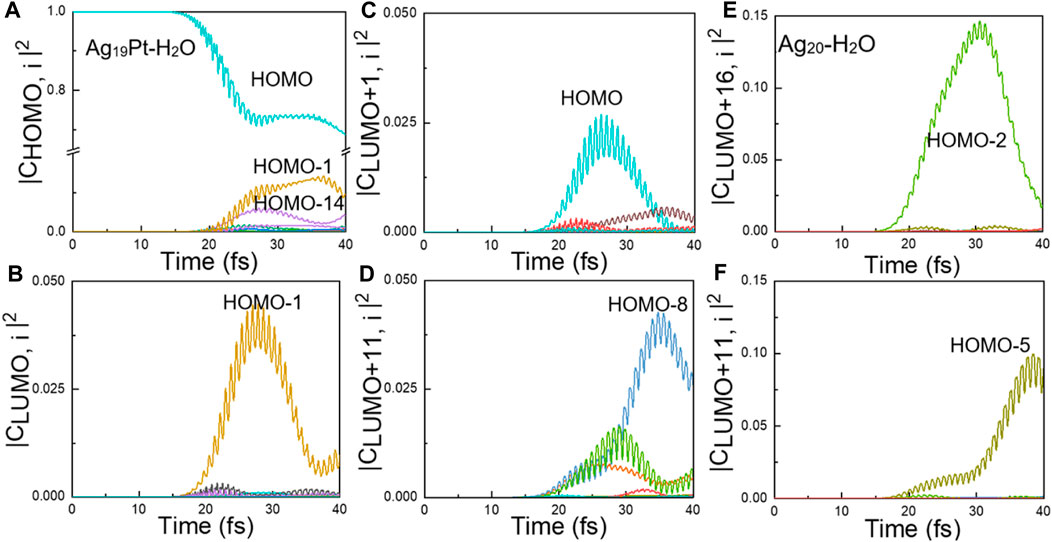
FIGURE 5. Ultrafast electron dynamics. Time-evolved transition coefficients from all the occupied states i to the Kohn–Sham state of (A) HOMO, (B) LUMO, (C) LUMO+1, and (D) LUMO+11 for Ag19Pt–H2O, respectively. The index i (i = 0–18) denotes the numerical order of the occupied states, while LUMO + j (j = 0–19) corresponds to the unoccupied states. Time-evolved transition coefficients from all the occupied states i to the state (E) LUMO+11 and (F) LUMO+16 for Ag20–H2O, respectively. The index i (i = 0–13) denotes the numerical order of the occupied states, while LUMO + j (j = 0–19) corresponds to the unoccupied states. Only the important orbitals that significantly contribute to the density change are labeled.
The Ag19Pt–H2O Case. Given the fact that the total number of electrons in Ag19Pt–H2O is an odd number, the HOMO of the system is half-occupied in spin-unpolarized calculations. Figure 5A reveals that the main contribution to the HOMO level stems from the HOMO-1 level. In addition, the main wave function component of the two orbitals is dominantly originated from the metal nanoparticle, which can be deduced from Figure 4B. Similar results can be obtained by the same analysis, as shown in Figures 4B, 5B,C, that is, the HOMO-1 and HOMO levels make a dominant contribution to the excitation to the LUMO and LUMO+1 state, where the corresponding wave functions are mainly distributed on the metal nanoparticle. Therefore, the channels of indirect charge transfer can be open via inelastic electron tunneling (Christopher et al., 2011; Brongersma et al., 2015; Linic et al., 2015; Thrall et al., 2013; Mukherjee et al., 2013; Mukherjee et al., 2014; Zhang et al., 2018; Yan et al., 2015). Second, there is a significant charge transfer from the HOMO-8 to LUMO+11 states, as shown in Figure 5D. Through the analysis given in Figure 4B, the orbital component of HOMO-8 is mainly contributed by the metal cluster, while the LUMO+11 orbital is mainly distributed on the water molecule. Meanwhile, the energy gap of (LUMO+11)-(HOMO-8) is equal to the photoenergy corresponding to the highest absorption peak for Ag19Pt–H2O, implying that this is a direct charge transfer via localized surface plasmon resonances (Yan et al., 2011; Kale et al., 2013; Kale et al., 2014; Yan et al., 2016; Kumar et al., 2019). In summary, the introduction of the Pt atom opens up more channels of charge transfer including intramolecular, indirect, and direct charge transfer pathways, resulting in efficient charge transfer and subsequent water splitting.
The Ag20–H2O Case. Figures 5E,F show that there exists obvious charge transfer from the HOMO-5 and HOMO-2 states to the LUMO+11 and LUMO+16 states, respectively. In addition, the analysis of the wave function component, as shown in Figure 4D, shows that the HOMO-5 and HOMO-2 states are mainly located on the metal nanoparticle, while the LUMO+11 and LUMO+16 states are mainly located on the H2O molecule. Meanwhile, the energy gap between the HOMO-5, HOMO-2 and LUMO+11, LUMO+16 states equals to the photoenergy of the major peak in the photoabsorption spectra for the Ag20–H2O system. These facts suggest that the direct charge transfer is the main charge transfer mechanism for the Ag20–H2O case.
Conclusion
In summary, we probe the difference in the underlying charge transfer mechanism for plasmon-driven water splitting for two representative systems: Ag19Pt–H2O and Ag20–H2O. Through the analysis of non-adiabatic molecular dynamic trajectories, we find that water may split within 40 fs after photoexcitation in the Ag19Pt–H2O case, where the O–H bond is only slightly elongated and does not break in the simulations for the Ag20–H2O system. Our ultrafast carrier dynamic analysis finds that there is more charge transferred to the H2O molecule in the Ag19Pt–H2O system than in the Ag20–H2O system. More importantly, it can be inferred that the introduction of single Pt atom in Ag19Pt–H2O strengthens the optical absorption as well as the interactions between the water and metal nanoparticle, opening up more charge transfer channels than that in Ag20–H2O, resulting in successful water splitting events. These results provide a new microscopic picture for solar water splitting and may facilitate the design of high-efficiency single-atom photocatalysts.
Methods
Numerical Calculations
Most of the calculations are carried out using the real-space TDDFT code OCTOPUS (Castro et al., 2006; Andrade et al., 2012; Andrade et al., 2015), with local density approximation (LDA) for the exchange correlation functional. The simulation zone is defined by assigning a sphere around each atom with a radius of 5.0 Å and a spacing of 0.25 Å between the grid points. Hartwigsen–Goedecker–Hutter pseudopotentials are used to represent the interactions between valence electrons and the atomic cores (Troullier and Martins, 1991). A time step of 0.002 fs is used in the calculations. An electromagnetic pulse (δ function) is used for the optical absorption spectrum. In the simulations, the H2O molecule is initially placed 3.0 Å away from the tip of Ag19Pt and Ag20, as shown in Figures 1A,B. The laser pulse is a Gaussian wave packet, E (ω, t) = Emax exp
Ground-state DFT simulations were performed with the Vienna Ab initio Simulation Package (VASP) (Kresse and Furthmüller, 1996) to obtain ground-state properties, using a projector-augmented wave (PAW) pseudopotential in conjunction with the Perdew–Burke–Ernzerhof (PBE) functional (Perdew et al., 1996), and the plane-wave basis set with an energy cutoff at 400 eV. The atomic structure of two systems was positioned in a cubic supercell of 30 × 30 × 30 Å3 along three directions and fully relaxed until the force on each atom was less than 0.02 eV/Å. A Monkhorst–Pack k-point mesh of 1 × 1×1 was adopted for the calculations. There are other methods which can also deal with the excited state dynamics including plasmon excitations of metal nanoparticles and their couplings with molecules (Shao et al., 2006; Guan et al., 2018; Lian et al., 2018; You et al., 2021).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
SM designed and directed the research. The calculations were performed by YZ with the help of DC, WM, SL and SM. YZ, SL, and SM analyzed the data. DC contributed the analysis codes. The manuscript was written by YZ, SL, and SM with input from all authors.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge financial support from NSFC (11934003, 12025407, 11774396, 91850120, and 11674289), MOST (grant 2016YFA0300902), and CAS (XDB330301).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.742794/full#supplementary-material
References
Alonso, J. L., Andrade, X., Echenique, P., Falceto, F., Prada-Gracia, D., and Rubio, A. (2008). Efficient Formalism for Large-Scale Ab Initio Molecular Dynamics Based on Time-dependent Density Functional Theory. Phys. Rev. Lett. 101, 096403. doi:10.1103/PhysRevLett.101.096403
Andrade, X., Alberdi-Rodriguez, J., Strubbe, D. A., Oliveira, M. J. T., Nogueira, F., Castro, A., et al. (2012). Time-dependent Density-Functional Theory in Massively Parallel Computer Architectures: the OCTOPUS Project. J. Phys. Condens. Matter 24, 233202. doi:10.1088/0953-8984/24/23/233202
Andrade, X., Strubbe, D., De Giovannini, U., Larsen, A. H., Oliveira, M. J. T., Alberdi-Rodriguez, J., et al. (2015). Real-space Grids and the Octopus Code as Tools for the Development of New Simulation Approaches for Electronic Systems. Phys. Chem. Chem. Phys. 17, 31371–31396. doi:10.1039/c5cp00351b
Brongersma, M. L., Halas, N. J., and Nordlander, P. (2015). Plasmon-induced Hot Carrier Science and Technology. Nat. Nanotech 10, 25–34. doi:10.1038/nnano.2014.311
Castro, A., Appel, H., Oliveira, M., Rozzi, C. A., Andrade, X., Lorenzen, F., et al. (2006). octopus: a Tool for the Application of Time-dependent Density Functional Theory. Phys. Stat. Sol. (B) 243, 2465–2488. doi:10.1002/pssb.200642067
Christopher, P., Xin, H., and Linic, S. (2011). Visible-light-enhanced Catalytic Oxidation Reactions on Plasmonic Silver Nanostructures. Nat. Chem. 3, 467–472. doi:10.1038/nchem.1032
Christopher, P., Xin, H., Marimuthu, A., and Linic, S. (2012). Singular Characteristics and Unique Chemical Bond Activation Mechanisms of Photocatalytic Reactions on Plasmonic Nanostructures. Nat. Mater. 11, 1044–1050. doi:10.1038/nmat3454
Guan, M.-X., Lian, C., and Meng, S. (2018). Real-time Time Dependent Density Functional Theory with Numerical Atomic Orbital Basis Set: Methodology and Applications. Acta Physica Sinica 67, 120201. doi:10.7498/aps.67.20180487
Hejazi, S., Mohajernia, S., Osuagwu, B., Zoppellaro, G., Andryskova, P., Tomanec, O., et al. (2020). On the Controlled Loading of Single Platinum Atoms as a Co-catalyst on TiO2 Anatase for Optimized Photocatalytic H2 Generation. Adv. Mater. 32, e1908505. doi:10.1002/adma.201908505
Kale, M. J., Avanesian, T., and Christopher, P. (2013). Direct Photocatalysis by Plasmonic Nanostructures. ACS Catal. 4, 116–128. doi:10.1021/cs400993w
Kale, M. J., Avanesian, T., Xin, H., Yan, J., and Christopher, P. (2014). Controlling Catalytic Selectivity on Metal Nanoparticles by Direct Photoexcitation of Adsorbate-Metal Bonds. Nano Lett. 14, 5405–5412. doi:10.1021/nl502571b
Kresse, G., and Furthmüller, J. (1996). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186. doi:10.1103/physrevb.54.11169
Kumar, P. V., Rossi, T. P., Marti-Dafcik, D., Reichmuth, D., Kuisma, M., Erhart, P., et al. (2019). Plasmon-Induced Direct Hot-Carrier Transfer at Metal-Acceptor Interfaces. ACS Nano 13, 3188–3195. doi:10.1021/acsnano.8b08703
Lang, R., Du, X., Huang, Y., Jiang, X., Zhang, Q., Guo, Y., et al. (2020). Single-Atom Catalysts Based on the Metal-Oxide Interaction. Chem. Rev. 120, 11986–12043. doi:10.1021/acs.chemrev.0c00797
Lewis, N. S. (2007). Toward Cost-Effective Solar Energy Use. Science 315, 798–801. doi:10.1126/science.1137014
Li, Z., Chen, H.-Y. T., Schouteden, K., Picot, T., Liao, T.-W., Seliverstov, A., et al. (2020). Unraveling the Atomic Structure, Ripening Behavior, and Electronic Structure of Supported Au20 Clusters. Sci. Adv. 6, eaay4289. doi:10.1126/sciadv.aay4289
Lian, C., Guan, M., Hu, S., Zhang, J., and Meng, S. (2018). Photoexcitation in Solids: First-Principles Quantum Simulations by Real-Time TDDFT. Adv. Theor. Simulations 1, 1800055. doi:10.1002/adts.201800055
Linic, S., Aslam, U., Boerigter, C., and Morabito, M. (2015). Photochemical Transformations on Plasmonic Metal Nanoparticles. Nat. Mater 14, 567–576. doi:10.1038/nmat4281
Liu, Z., Hou, W., Pavaskar, P., Aykol, M., and Cronin, S. B. (2011). Plasmon Resonant Enhancement of Photocatalytic Water Splitting under Visible Illumination. Nano Lett. 11, 1111–1116. doi:10.1021/nl104005n
Ma, J., Wang, Z., and Wang, L. W. (2015). Interplay between Plasmon and Single-Particle Excitations in a Metal Nanocluster. Nat. Commun. 6, 10107. doi:10.1038/ncomms10107
Martirez, J. M. P., and Carter, E. A. (2017). Excited-State N2 Dissociation Pathway on Fe-Functionalized Au. J. Am. Chem. Soc. 139, 4390–4398. doi:10.1021/jacs.6b12301
Mukherjee, S., Libisch, F., Large, N., Neumann, O., Brown, L. V., Cheng, J., et al. (2013). Hot Electrons Do the Impossible: Plasmon-Induced Dissociation of H2 on Au. Nano Lett. 13, 240–247. doi:10.1021/nl303940z
Mukherjee, S., Zhou, L., Goodman, A. M., Large, N., Ayala-Orozco, C., Zhang, Y., et al. (2014). Hot-electron-induced Dissociation of H2 on Gold Nanoparticles Supported on SiO2. J. Am. Chem. Soc. 136, 64–67. doi:10.1021/ja411017b
Nie, S., and Emory, S. R. (1997). Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science 275, 1102–1106. doi:10.1126/science.275.5303.1102
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Primo, A., Corma, A., and García, H. (2011). Titania Supported Gold Nanoparticles as Photocatalyst. Phys. Chem. Chem. Phys. 13, 886–910. doi:10.1039/c0cp00917b
Prodan, E., Radloff, C., Halas, N. J., and Nordlander, P. (2003). A Hybridization Model for the Plasmon Response of Complex Nanostructures. Science 302, 419–422. doi:10.1126/science.1089171
Qian, K., Sweeny, B. C., Johnston-Peck, A. C., Niu, W., Graham, J. O., DuChene, J. S., et al. (2014). Surface Plasmon-Driven Water Reduction: Gold Nanoparticle Size Matters. J. Am. Chem. Soc. 136, 9842–9845. doi:10.1021/ja504097v
Qiao, B., Wang, A., Yang, X., Allard, L. F., Jiang, Z., Cui, Y., et al. (2011). Single-atom Catalysis of CO Oxidation Using Pt1/FeOx. Nat. Chem. 3, 634–641. doi:10.1038/nchem.1095
Seemala, B., Therrien, A. J., Lou, M., Li, K., Finzel, J. P., Qi, J., et al. (2019). Plasmon-Mediated Catalytic O2 Dissociation on Ag Nanostructures: Hot Electrons or Near Fields? ACS Energ. Lett. 4, 1803–1809. doi:10.1021/acsenergylett.9b00990
Shao, Y., Molnar, L. F., Jung, Y., Kussmann, J., Ochsenfeld, C., Brown, S. T., et al. (2006). Advances in Methods and Algorithms in a Modern Quantum Chemistry Program Package. Phys. Chem. Chem. Phys. 8, 3172–3191. doi:10.1039/b517914a
Sigle, D. O., Zhang, L., Ithurria, S., Dubertret, B., and Baumberg, J. J. (2015). Ultrathin CdSe in Plasmonic Nanogaps for Enhanced Photocatalytic Water Splitting. J. Phys. Chem. Lett. 6, 1099–1103. doi:10.1021/acs.jpclett.5b00279
Sui, Y., Liu, S., Li, T., Liu, Q., Jiang, T., Guo, Y., et al. (2017). Atomically Dispersed Pt on Specific TiO2 Facets for Photocatalytic H2 Evolution. J. Catal. 353, 250–255. doi:10.1016/j.jcat.2017.07.024
Thimsen, E., Le Formal, F., Grätzel, M., and Warren, S. C. (2011). Influence of Plasmonic Au Nanoparticles on the Photoactivity of Fe2O3 Electrodes for Water Splitting. Nano Lett. 11, 35–43. doi:10.1021/nl1022354
Thrall, E. S., Preska Steinberg, A., Wu, X., and Brus, L. E. (2013). The Role of Photon Energy and Semiconductor Substrate in the Plasmon-Mediated Photooxidation of Citrate by Silver Nanoparticles. J. Phys. Chem. C 117, 26238–26247. doi:10.1021/jp409586z
Townsend, E., and Bryant, G. W. (2011). Plasmonic Properties of Metallic Nanoparticles: The Effects of Size Quantization. Nano Lett. 12, 429–434. doi:10.1021/nl2037613
Townsend, E., and Bryant, G. W. (2014). Which Resonances in Small Metallic Nanoparticles Are Plasmonic? J. Opt. 16, 114022. doi:10.1088/2040-8978/16/11/114022
Troullier, N., and Martins, J. L. (1991). Efficient Pseudopotentials for Plane-Wave Calculations. Phys. Rev. B 43, 1993–2006. doi:10.1103/physrevb.43.1993
Wang, J., Wang, G., and Zhao, J. (2003). Structures and Electronic Properties of Cu20, Ag20, and Au20 Clusters with Density Functional Method. Chem. Phys. Lett. 380, 716–720. doi:10.1016/j.cplett.2003.09.062
Warren, S. C., and Thimsen, E. (2012). Plasmonic Solar Water Splitting. Energy Environ. Sci. 5, 5133–5146. doi:10.1039/c1ee02875h
Wu, X., Zhang, H., Dong, J., Qiu, M., Kong, J., Zhang, Y., et al. (2018). Surface Step Decoration of Isolated Atom as Electron Pumping: Atomic-Level Insights into Visible-Light Hydrogen Evolution. Nano Energy 45, 109–117. doi:10.1016/j.nanoen.2017.12.039
Xing, J., Chen, J. F., Li, Y. H., Yuan, W. T., Zhou, Y., Zheng, L. R., et al. (2014). Stable Isolated Metal Atoms as Active Sites for Photocatalytic Hydrogen Evolution. Chem. Eur. J. 20, 2138–2144. doi:10.1002/chem.201303366
Xu, H., Bjerneld, E. J., Käll, M., and Börjesson, L. (1999). Spectroscopy of Single Hemoglobin Molecules by Surface Enhanced Raman Scattering. Phys. Rev. Lett. 83, 4357–4360. doi:10.1103/physrevlett.83.4357
Yan, J., Jacobsen, K. W., and Thygesen, K. S. (2011). First-Principles Study of Surface Plasmons on Ag(111) and H/Ag(111). Phys. Rev. B 84, 235430. doi:10.1103/physrevb.84.235430
Yan, L., Ding, Z., Song, P., Wang, F., and Meng, S. (2015). Plasmon-Induced Dynamics of H2 Splitting on a Silver Atomic Chain. Appl. Phys. Lett. 107, 083102. doi:10.1063/1.4929611
Yan, L., Wang, F., and Meng, S. (2016). Quantum Mode Selectivity of Plasmon-Induced Water Splitting on Gold Nanoparticles. ACS Nano 10, 5452–5458. doi:10.1021/acsnano.6b01840
Yan, L., Xu, J., Wang, F., and Meng, S. (2018). Plasmon-Induced Ultrafast Hydrogen Production in Liquid Water. J. Phys. Chem. Lett. 9, 63–69. doi:10.1021/acs.jpclett.7b02957
You, P., Chen, D., Lian, C., Zhang, C., and Meng, S. (2021). First-principles Dynamics of Photoexcited Molecules and Materials towards a Quantum Description. WIREs Comput. Mol. Sci. 11, e1492. doi:10.1002/wcms.1492
Zhang, Y., Nelson, T., Tretiak, S., Guo, H., and Schatz, G. C. (2018). Plasmonic Hot-Carrier-Mediated Tunable Photochemical Reactions. ACS Nano 12, 8415–8422. doi:10.1021/acsnano.8b03830
Zhao, L., Jensen, L., and Schatz, G. C. (2006a). Pyridine−Ag20 Cluster: A Model System for Studying Surface-Enhanced Raman Scattering. J. Am. Chem. Soc. 128, 2911–2919. doi:10.1021/ja0556326
Zhao, L. L., Jensen, L., and Schatz, G. C. (2006b). Surface-Enhanced Raman Scattering of Pyrazine at the Junction between Two Ag20Nanoclusters. Nano Lett. 6, 1229–1234. doi:10.1021/nl0607378
Zhou, L., Swearer, D. F., Zhang, C., Robatjazi, H., Zhao, H., Henderson, L., et al. (2018). Quantifying Hot Carrier and thermal Contributions in Plasmonic Photocatalysis. Science 362, 69–72. doi:10.1126/science.aat6967
Keywords: photocatalytic water splitting, localized surface plasmon, single-atom catalyst, charge transfer, time-dependent density functional theory
Citation: Zhang Y, Chen D, Meng W, Li S and Meng S (2021) Plasmon-Induced Water Splitting on Ag-Alloyed Pt Single-Atom Catalysts. Front. Chem. 9:742794. doi: 10.3389/fchem.2021.742794
Received: 16 July 2021; Accepted: 02 September 2021;
Published: 25 October 2021.
Edited by:
Kaiying Wang, University of South-Eastern Norway, NorwayReviewed by:
Can Li, Dalian Institute of Chemical Physics (CAS), ChinaChaoqun Cheng, University of South-Eastern Norway, Norway
Copyright © 2021 Zhang, Chen, Meng, Li and Meng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shunfang Li, c2ZsaXp6dUB6enUuZWR1LmNu; Sheng Meng, c21lbmdAaXBoeS5hYy5jbg==