 Ranita Pal1
Ranita Pal1 Arpita Poddar
Arpita Poddar Pratim Kumar Chattaraj
Pratim Kumar Chattaraj
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem. , 16 August 2021
Sec. Chemical Physics and Physical Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.730548
This article is part of the Research Topic Atomic Clusters: Theory & Experiments View all 10 articles
Atomic clusters lie somewhere in between isolated atoms and extended solids with distinctly different reactivity patterns. They are known to be useful as catalysts facilitating several reactions of industrial importance. Various machine learning based techniques have been adopted in generating their global minimum energy structures. Bond-stretch isomerism, aromatic stabilization, Rener-Teller effect, improved superhalogen/superalkali properties, and electride characteristics are some of the hallmarks of these clusters. Different all-metal and nonmetal clusters exhibit a variety of aromatic characteristics. Some of these clusters are dynamically stable as exemplified through their fluxional behavior. Several of these cluster cavitands are found to be agents for effective confinement. The confined media cause drastic changes in bonding, reactivity, and other properties, for example, bonding between two noble gas atoms, and remarkable acceleration in the rate of a chemical reaction under confinement. They have potential to be good hydrogen storage materials and also to activate small molecules for various purposes. Many atomic clusters show exceptional opto-electronic, magnetic, and nonlinear optical properties. In this Review article, we intend to highlight all these aspects.
A cluster is defined as a finite aggregation of atoms, starting with as low as two atoms and extending up to an upper bound of some hundred thousand atoms (Herr, 1993; Corrigan and Dehnen, 2017). A theoretical study elucidates that cluster properties strongly depend on the geometry of the isolated cluster and the topology of the cluster sample. F. A. Cotton first used the term cluster for compounds with metal–metal bonds. Nonmetallic atomic clusters came into the limelight later, from both theoretical and experimental studies. The unique size-dependent properties of clusters are distinct from the molecules and bulk solids (Sergeeva et al., 2014). Experimental and theoretical methods on cluster research have seen substantial amount of improvement over the years in discovering a diversity of size-specific phenomena and physicochemical cluster properties (Jena and Castleman, 2010). Various physicochemical properties of metal clusters such as optical, magnetic, thermal, chemical properties differ remarkably from their bulk counterparts (Tsukuda and Hakkinen, 2015). In the last few decades, several studies have been reported on the ligand protected metal clusters, viz. phosphine-protected small Au cluster, thiolate (RS)-protected Au nanoparticles (Brust et al., 1994), etc. For small metal clusters (<∼100 atoms), the electronic structures are not continuous as in the bulk metals, but rather discretized, which is the primary reason for different physicochemical properties and functionalities in small clusters and bulk metal (Sharma et al., 2017). Studies have shown that clusters made of atoms with appropriate size and composition could potentially mimic the chemistry of elemental atoms in periodic table, and hence are known as superatoms (Li et al., 2008). Various experimental techniques such as laser ablation coupled with mass spectrometry, photoelectron spectroscopy have been employed to get insights into the atomic clusters. Along with experimental studies, theoretical investigations are required to get a better understanding of their geometric arrangement and corresponding properties (Srivastava, 2021).
A large number of theoretical studies have been reported dealing with finding the minimum energy structures of pure elemental clusters by using different optimization algorithms. The potential energy surface (PES) of various atomic clusters is explored to minimize their energy functional with the final objective of locating their global minimum (GM). Different optimization methods such as genetic algorithm (Holland, 1992) (GA), basin hopping (BH) algorithm (Wales and Doye, 1997), particle swarm optimization (PSO) algorithm (Bai, 2010), adaptive particle swarm optimization (APSO) (Zhan and Zhang, 2008), simulated annealing (SA) (Woodley et al., 1999), artificial bee colony optimization (ABC) (Karaboga and Basturk, 2007), honey bee mating optimization (HBMO) (Pham et al., 2005), ant colony optimization (ACO) (Colorni et al., 1991), heuristic algorithm combined with the surface and interior operators (HA-SIO), fast annealing evolutionary algorithm (FAEA), firefly algorithm (FA) (Yang, 2010). etc. are increasingly being used to solve the optimization problem in a time- and cost-efficient manner. Various models/empirical potentials (EPs) such as Lennard–Jones (LJ), Born–Mayer, Sutton–Chen, Gupta and Murrell–Mottram potentials can effectively explain the bonding within various clusters. A number of studies performed by Chattaraj et al. reveal that PSO is more efficient than commonly used techniques such as GA, SA, and BH for finding the GM of small clusters (Mitikiri et al., 2018; Jana et al., 2019). Further developments over PSO algorithm have been accomplished. Global optimization of boron clusters (B5 and B6) has been studied using an advanced PSO approach by Mitikiri et al. (2018). Jana et al. (2019) performed a similar study on carbon clusters, Cn (n = 3–6,10) by using a modified PSO algorithm. Mitra et al. (2020) reported the global optimization of Al42- clusters by using firefly algorithm along with DFT.
The concept of trapping atoms and small molecules into the hollow cavity of clusters has shown several applications in biology (Cagle et al., 1996; Thrash et al., 1999; Wilson et al., 1999) and electrical engineering (Cioslowski and Nanayakkara, 1992). Pan et al. (2018) reported the encapsulation of noble gas (Ng) atoms into the B40 host moiety, which is shown to have a fluxional character (Moreno et al., 2014). The dynamical study of the aforementioned system showed that the fluxional character persists even after the encapsulation. Smaller cages such as C20H20 (Cross et al., 1999; Jiménez-Vázquez et al., 2001), C10H16 (Haaland et al., 2004), BN cages (B12N12 and B16N16) (Khatua et al., 2014a), Pb122-, and Sn122- (Sekhar et al., 2017) can act as host molecules to endohedrally trapped noble gas atoms. Cucurbit[n]urils, abbreviated as CB[n]s, n being the number of gycoluril units, can also act as host for different guest molecules including metal cations, organic dyes, drugs, halide ions, etc. (Lagona et al., 2005; Pan et al., 2013a). CB[7] was also reported to bind different guest compounds such as organic dyes (e.g., Stilbenes, naphthalene), viologens, and metal complexes (e.g., Oxaliplatin) applicable in cancer treatment (Wagner et al., 2003). In 2017, Pan et al. (2017) have reported the adsorption of 14 molecules, viz., CH4, C2H2, C2H4, C2H6, F2, Cl2, NO2, NO, CO, CO2, SO2, H2S, N2, H2 endohedrally within the hydrophobic inner cavity of CB[7]. CB[6] is also known to encapsulate noble gas atoms (Pan et al., 2015). Chakraborty et al. (Chakraborty and Chattaraj, 2015) reported the accommodation of noble gas atoms within the BN-doped (3, 3) single-walled carbon nanotubes. The Ng binding ability of BeX (X = SO4, CO3, O) has been reported by Saha et al. (2015). Pan et al. (2014) have explored the stability of Ng-bound SiH3+ cluster ions. An emerging host molecule is the basket-shaped octa acid (OA) cavitand that can encapsulate different gas molecules (Chakraborty et al., 2016). Various steroids (Liu and Gibb, 2008), hydrophobic moieties such as ethane, ethylene, acetylene (Stang and Diederich, 2008; Hu et al., 2009; Florea and Nau, 2011; Zhang et al., 2011; Yang et al., 2015; Chong et al., 2016) have been confined inside OA. The encapsulation of gas molecules, especially the hazardous ones, by the cluster cavity has great applications in environmental chemistry. Encapsulation of greenhouse gases (CO2) (Zhang and Chen, 2009; Jin et al., 2010; Kim et al., 2010; Jin et al., 2011; Mastalerz et al., 2011; Lü et al., 2014), air pollutants (NO2) (Pan et al., 2017), and poisonous gases (CO, NO) using molecular cages has applications in reducing their negative impact on the atmosphere. N2 encapsulation (Msayib et al., 2009; Akhtar et al., 2012; Schneider et al., 2012) is yet another important research topic in environmental chemistry. Chakraborty et al. (2016) reported a set of small gaseous molecules (C2H2, C2H4, C2H6, CO, CO2, NO2, NO, N2, H2−), and rare gas atoms as guest molecules for the OA host system. In fact, OA is a very efficient reaction vessel for accommodating various different guest molecules. Li+, Na+, K+, Be2+, Mg2+, Ca2+, Li3O+, Na3O+, K3O+ and various nucleobases can occupy the basket-shaped octa acid cavitand as reported by Chakraborty and Chattaraj (2018).
In recent days, the conservation of the atmosphere and the desire to save up fuel for the upcoming generations has been a major concern in the scientific community that led to the search for alternatives of fossil fuel. Hydrogen, being renewable, recyclable, environment friendly, and abundantly available in nature, is now a globally acceptable fuel source with the potential to replace fossil fuels in the near future. The challenge, however, is designing compatible storage and transport materials. To that end, hydrogen-storing capacity of metal-organic-frameworks (MOF) (Rosi et al., 2003; Rowsell and Yaghi, 2005), covalent-organic-frameworks (COF) (Kuc et al., 2007; Cabria et al., 2008), clathrate hydrates (Lee et al., 2005; Chattaraj et al., 2011), polymers (McKeown et al., 2006), carbon nanotubes, BN cages, fullerene, grapheme-like materials (Froudakis, 2001; Deng et al., 2004; Heine et al., 2004; Sun et al., 2005; Wu et al., 2008), metal hydrides (Lee et al., 2005) have been explored. Pan et al. (2012a) performed a theoretical study on the H2-storing capability of some Li-doped clusters and super-alkalis. Zhu et al. (2010) have shown cucurbiturils acting as a promising candidate for hydrogen storage. Pan et al. (2013a) have discussed the hydrogen storage capability of the CB[7] system. A different class of compound, alkali-doped carbon materials (graphene sheet and single-walled carbon nanotubes), have been designed for reversible hydrogen storage for transportation purposes by Wei-Qiao Deng et al. (2004). On the other hand, very explosive acetylene can be stored within the porous MOF-505 analogue as reported by Yunxia Hu et al. (2009).
The confinement effect on atoms and molecules has intrigued both theoreticians and experimentalists alike. It brings out interesting changes in the energy levels of the confined systems, their bonding, reactivity, and properties (Grochala et al., 2007; Schettino and Bini, 2007; Sabin and Brandas, 2009; Gubbins et al., 2011; Chakraborty and Chattaraj, 2019; Pal and Chattaraj, 2021). Khatua et al. (2014b) have performed a theoretical investigation on the entrapment of (HF)2 in Cn (n = 60, 70, 80, 90) cages. Although CO and N2 are isoelectric species, the latter is known to be pretty inert owing to its high ionization potential, low electron affinity, and high frontier orbital energy gap (ΔEHOMO-LUMO). Thus, N2 capture in various transition metal complexes has proven to induce bond activation that has various industrial applications (Chatt and Leigh, 1972; Yandulov and Schrock, 2003; Latysheva et al., 2012; Bergman et al., 2013; Hoffman et al., 2013). In this regard, Saha et al. (2017) reported the CO and N2 bound metal supported boron clusters (MB12-, M = Co, Rh, Ir) which form a spinning umbrella-like structure and activate the bound molecules. Boron clusters have found profound applications in material science owing to their ability to act as nanomaterial building blocks. Their property to act as such is due to a bowl-like structure with an outer rim (B9) and an inner well (B3).
Along with the coordination and inorganic cages, various organic cavitands can also be considered host system for encapsulation. One such class of compounds is the cucurbiturils. Chemical reactions catalyzed by host–guest interactions are comparable to those catalyzed by enzymes (Dong et al., 2012). Hennig et al. (2007) successfully induced protease inhibition using the host–guest interaction with CB[7]. In addition to CB[7], ExBox+4 (Barnes et al., 2013) can also act as an organic host molecule that can encapsulate a wide array of guest moieties. Chakraborty et al. (2017) performed a theoretical study on [4+2] cycloaddition reaction confined within CB[7] and ExBox+4 host systems.
In recent times, low dimensional materials are being given more and more attention to be used as host moiety. Graphene has provided us with a plethora of highly efficient devices such as gas sensors, spintronic devices, nanoelectronics, and optoelectronic devices (Chakraborty and Chattaraj, 2017). An inorganic counterpart of graphene is the boron nitride doped system that can be functionalized with OLi4, CLi6, NLi5, BLi7, Al12Be to achieve some interesting properties (Chakraborty and Chattaraj, 2017). The M3O+ (M = Li, Na, K) functionalized graphene nanoflakes (Chakraborty and Chattaraj, 2016a) are known to sequestrate various polar molecules such as CO, NO, and CH3OH. Sequestration of gas molecules such as H2, O2, O3, CO, NO, and H2O through bare boron nitride flakes (BNF) and metal oxide, MO (M = Cu, Ag, Au) functionalized BNF are also reported (Chakraborty and Chattaraj, 2016b). In this review we report some optimization techniques for the generation of minimum energy structures of some selected clusters and also the bonding, reactivity, and different properties of some selected confined systems. Aromatic behavior and electride properties of some clusters are also investigated.
Before optimizing the geometry of any system, we carefully ponder over the requirement of the study and select the level of theory maintaining a parity between the level of accuracy required and the computational cost to be incurred. Most often, the easiest way is to take into consideration the experimental data (if available) and select accordingly. The systems discussed in this article are optimized using the computational chemistry software package, Gaussian 09 (Frisch et al., 2009). We have used B3LYP (Lee et al., 1988; Becke, 1992), BP86 (Perdew, 1986a; Perdew, 1986b; Becke, 1988), ωb97X-D (Chai and Head-Gordon, 2008), PBE (Perdew et al., 1996; Perdew et al., 1997), TPSSTPSS (Staroverov and Scuseria, 2003; Tao et al., 2003), M06, M06-2X (Zhao and Truhlar, 2008), and M05-2X (Zhao et al., 2006) functionals for carrying out DFT calculations of various systems. The exact level of theory (method and basis set) chosen for the individual case studies is mentioned in the Results and Discussion section. Relativistic effects for heavier atoms are taken care of by effective core potentials (ECPs). The stationary states are better understood from the harmonic vibrational frequencies. The minimum energy structure and the transition state (TS) are identified with the presence of zero and one imaginary frequency, respectively.
The atomic charges, nature of interactions present within the systems, and the possible bond formation are analyzed with the help of natural population analysis (NPA) (Reed et al., 1985), Wiberg bond indices (WBI) (Wiberg, 1968) in the NBO scheme (Reed et al., 1988). The electron density topology is mapped using Bader’s quantum theory of atoms-in-molecules (QTAIM) (Bader, 1985) in Multiwfn (Lu and Chen, 2012). Parameters such as electron density [ρ(rc)], total electron energy density [H(rc)], local kinetic energy density [G(rc)], local potential energy density [V(rc)], and Laplacian of electron density [∇2ρ(r)] are computed at the bond critical points (BCPs) and they help analyze the extent of covalent or ionic character present along that bond path. The NCI index reveals the localized binding interaction in a system, and the plot can be visualized as red, blue, or green regions in the NCIPLOT program (Contreras-García et al., 2011) depending on whether the interaction is repulsive, H-bond, or van der Waals, respectively. The nonlinear optical (NLO) properties are evaluated in terms of average linear polarizability (
ADF 2013.01 software (Baerends et al., 2013) is utilized to perform the energy decomposition analysis (EDA) (Morokuma, 1971) with the natural orbitals for chemical valence (NOCV) (Mitoraj et al., 2009). The interaction between two selected fragments of the studied system is represented in terms of three attractive and one repulsive energy terms. The attractive term includes electrostatic energy (ΔEelstat), orbital interaction energy (ΔEorb), dispersion interaction energy (ΔEdisp), while the repulsive term is known as Pauli repulsion energy (ΔEPauli).
In NOCV, the orbital term is represented as the sum of ΔEkorb (pairwise orbital energies) which is related to Δρk(r) (pairwise charge contributions).
Atom-centered density matrix propagation (ADMP) (Iyengar et al., 2001; Schlegel et al., 2001; Schlegel et al., 2002) in Gaussian 09, and Born-Oppenheimer molecular dynamics (BOMD) in deMon2K software (Koster et al., 2011) are used to perform the dynamic study of the systems under discussion in this article.
For the global optimization study using PSO, ADMP-CNN-PSO, and FA, the algorithms are written in Python 3.7 programming language (Van Rossum and Drake, 2009). The single point energies (SPEs) are calculated using Gaussian 09 at the post-processing step. The calculations for all the clusters are performed using the B3LYP (Lee et al., 1988; Becke, 1992) functional of DFT. The basis set 6-311+G(d,p) (McLean and Chandler, 1980; Raghavachari et al., 1980) is used for the boron clusters, 6-311+G(d) for Al42−, C5, and N42-, 6-311G(d) for N64-, and LANL2DZ (Dunning and Hay, 1977; Wadt and Hay, 1985; Hay and Wadt, 1985a; Hay and Wadt, 1985b) with ECPs for Aun (n = 2–8) and AunAgm (2 ≤ (n+m) ≤ 8). The algorithms are executed in a server with two Intel 2.70 GHz Xeon E5-2697 v2 processors (each with 12 cores and 30 threads) and a 256 GB RAM. The software Keras (Chollet, 2015) is used for interfacing with Python 3.7 with convolution neural networks (CNN).
Minimization of a system’s energy functional is the most fundamental step in the determination of its ground state. Reaching the global minimum (GM) geometry, however, poses a number of challenges, the most important being the high probability of getting stuck in local minima in the PES. Swarm intelligence (SI)-based algorithms have turned out to be very effective in searching for optimal solutions in a given search space. Here we discuss three different techniques, PSO combined with DFT, PSO with CNN, and DFT-integrated FA. They do not need to implement any symmetry constraint, or consider bond characterization. DFT-PSO adjusts each particle’s trajectory at every time stamp while following the convergence criteria. We have successfully implemented these techniques to find the GM configurations for small-sized nonmetallic clusters such as Boron (B5 and B6) (Yuan et al., 2014), Carbon (C5) (Jana et al., 2019), and polynitrogen clusters (N42- and N64-) (Mitra et al., 2021), and metallic clusters such as Al42- (Mitra et al., 2020), Aun (n = 2–8), and AunAgm (2 ≤ n+m ≤ 8) (Mitra et al., 2021).
We start off with 14 and 15 random structures for B5 and B6 (Figure 1), respectively, with velocity set at zero and maximum number of iterations set at 1,000. No significant change in bond length is detected after reaching the global best configuration at the end of the PSO run. Following this, the optimization is performed at the B3LYP/6-311+G(d,p) level as the post-processing step that helps align the symmetry of the PSO-obtained final structure and obtain the corresponding exact energy. The post-processing in this case takes only 20 s to complete, and the final geometry obtained is energetically very close (0.0015 eV difference) to that obtained at the end of the PSO run. The zero-point energy (ZPE) corrected energy, free energy, and enthalpy for B5 (C2v) are −123.9873, −124.0135, and −123.9821 a.u., respectively, while those for B6 (C2h) are −148.8100, −148.8381, and −148.8038 a.u., respectively. A comparison drawn between our method and other popular algorithms such as DFT-SA and DFT-BH reveals that while these two require a CPU time of 369.64 and 455.43 min to locate the minimum energy structure of B5, respectively, our method takes only 80.50 min. It is also observed that while the BH and the SA require 600 (unconverged) and 324 number of iterations, respectively, our modified PSO converges after only 138 number of iterations.
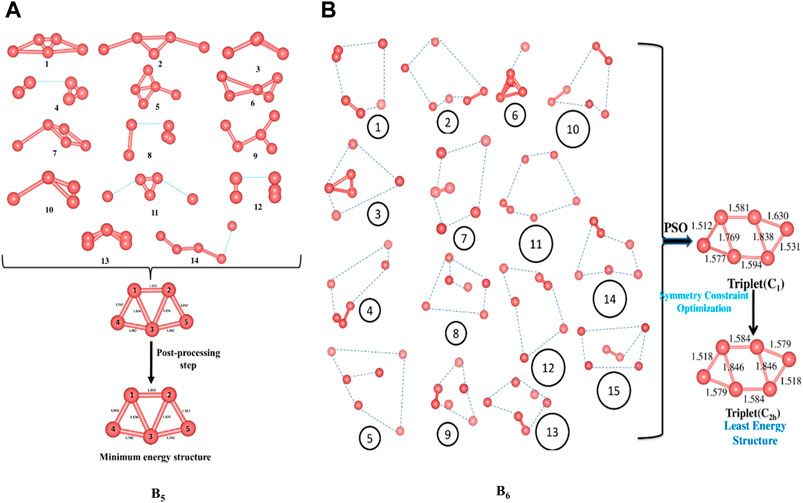
FIGURE 1. Randomly generated configurations of (A) B5 and (B) B6 and their convergence to their respective global minimum energy structures (Bond lengths are provided in Å). (Adapted with permission from Mitikiri et al., 2018. Copyright© 2021, John Wiley & Sons, Inc.).
For the carbon clusters, 10 random configurations are chosen with initial velocity zero and 1,000 number of iterations. For the Cn (n = 3–6) clusters, linear geometries are obtained with D∞h point group as the GM. For n = 4–6, a cyclic isomer for each of them is also obtained with point groups D2h, C2v, and D3h for C4, C5, and C6, respectively, whereas for the relatively larger C10 cluster, the GM geometry is a D10h ring structure. It is to be noted that the geometries and corresponding energies reported here match with those obtained from the experimental reports (Raghavachari and Binkley, 1987; Watts et al., 1992; Hutter and Lüthi, 1994; Pless et al., 1994; Martin and Taylor, 1996; Van Orden and Saykally, 1998). Again, comparing with DFT-SA and DFT-BH, we get encouraging results for our modified PSO approach. The total execution time for our technique is 143.30 min versus 215.98 and 5085.67 min for DFT-SA and DFT-BH, respectively.
For N42- and N64- clusters, convergence takes place after 483 and 627 numbers of iterations with 228 and 323 min of execution time, respectively. The geometries obtained before and after the post-processing step are energetically very close (Figure 2). The N64- cluster, however, shows a higher order saddle point at the post-processing symmetry constrained optimization with point group D6h. In case of the binary gold–silver clusters (AunAgm), AuAg2 has a ring (Cs) doublet GM, those with n+m = 5 have trapezoidal GM, n+m = 6, 7 have triangular 3D geometry (C1), and n+m = 8 failed to converge within the initial coordinates range of [−4, 4].
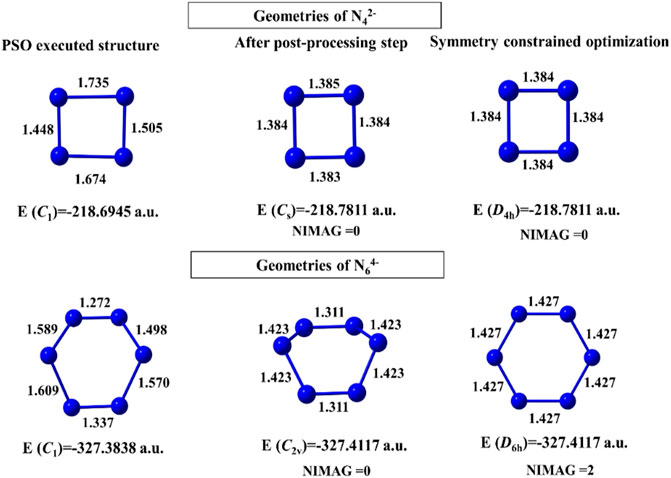
FIGURE 2. Structures of N42- and N64- clusters obtained at the end of the PSO run, post-processing step, and geometry-constrained optimization computed at the B3LYP/6-311 + G(d) level. (Reprinted from Mitra et al., 2021 with permission from Theoretical Chemistry Accounts, Springer Nature. Copyright© 2021, Springer-Verlag GmbH, DE.).
Supervised learning (CNN) is performed on an initial guess set generated with the help of (ADMP) simulation. Their corresponding single point energies (SPEs) are calculated and stored to be read by the PSO to search for the GM geometry. Statistically relevant analysis is derived by making the method 8-fold (each with 30 files containing number of iterations and the SPEs). Remarkably high success rate (∼77–90%) is observed for this combined ADMP, CNN, PSO technique, indicating its efficiency in finding GMs. This technique is tested with the C5 cluster and we have obtained the previously reported (Van Orden and Saykally, 1998) linear geometry as the GM with a higher convergence rate. However, two local minima are also detected due to a premature convergence (Figure 3).
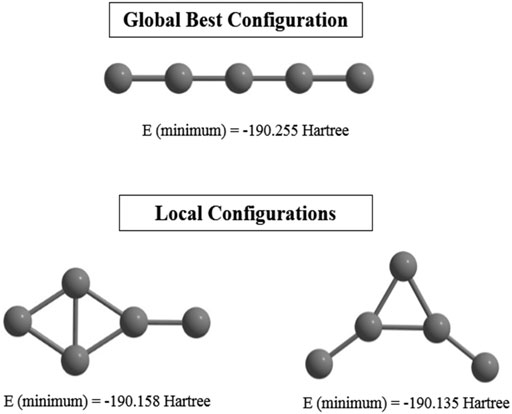
FIGURE 3. Global and local minimum energy structures of C5 cluster obtained at the end of the post-processing step computed at the B3LYP/6-311+ G(d,p) level. (Reprinted from Mitra et al., 2021 with permission from Theoretical Chemistry Accounts, Springer Nature. Copyright© 2021, Springer-Verlag GmbH, DE.).
A comparative study of the DFT-integrated FA algorithm with PSO is performed on Al42- cluster considering planar and nonplanar structures, and it turns out that the former performs better than the PSO. The mean convergence times for the PSO and FA are 68.22 and 61.25 min for the planar, and 85.13 and 74.40 min for the nonplanar approach, respectively. The corresponding success rates are also higher for the FA. Since we know that the GM of Al42- is planar, we have also investigated the search space of only the planar geometry to get a faster convergence since the number of variables decreases in this problem. Again, the modified FA performs better. A relation is also drawn between the stabilization energy of the system and the change in its aromaticity and number of iteration steps it takes to converge to the GM. The energy functional optimization and the NICS (0) value of Al42− are scanned and depicted in Figure 4. It is observed that the aromaticity increases with the decrease in the energy, i.e., with the increase in the stability of the system.
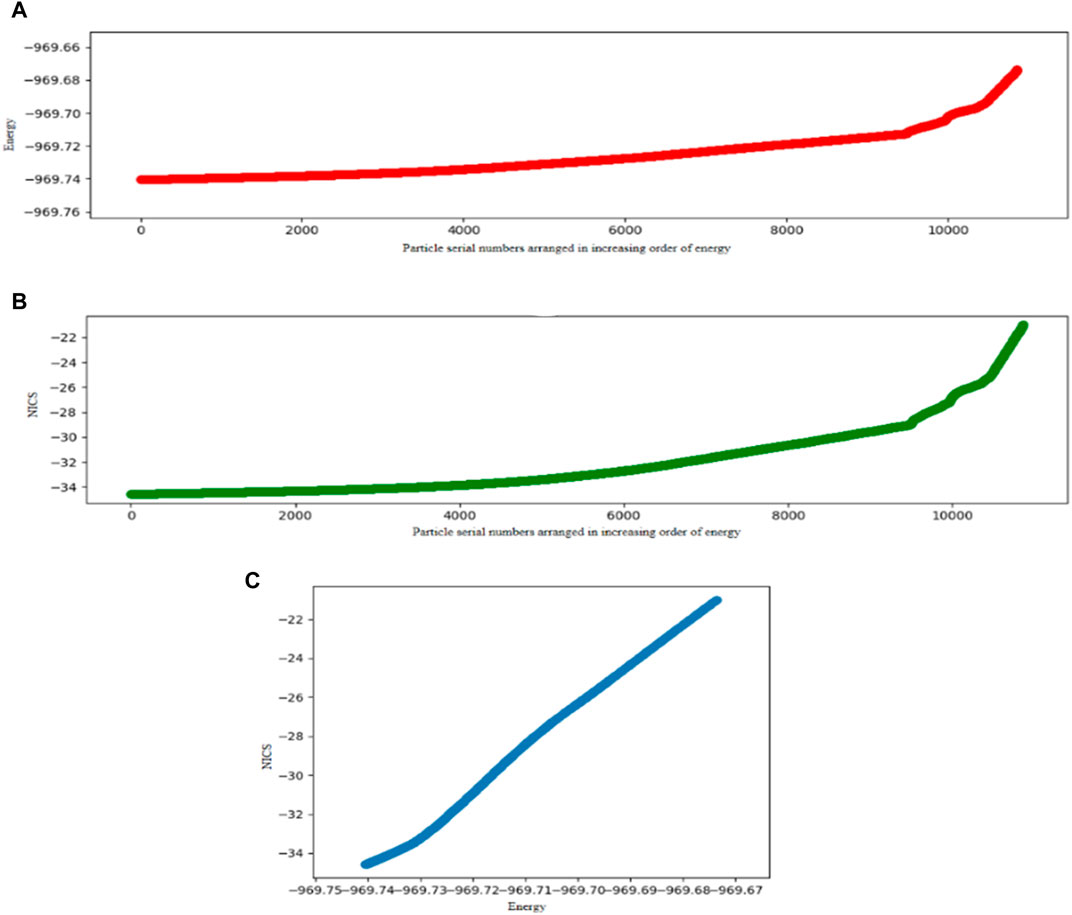
FIGURE 4. Energy profiles (in a.u.) and Nucleus-Independent Chemical Shift (NICS) values on Al42- where 4(A), 4(B), and 4(C) represent plots of Energy vs. Particle serial numbers arranged in an increasing order of energy, NICS vs. Particle serial numbers arranged in an increasing order of energy and NICS vs. Energy, respectively. (Reprinted from Mitra et al., 2020 with permission from Theoretical Chemistry Accounts, Springer Nature. Copyright© 2020, Springer-Verlag GmbH Germany.).
It is well known from Hückel’s (4n+2) π electron theory (Hückel, 1931) and Pauling’s quantum mechanical description (Pauling and Sherman, 1933) that aromaticity of conjugated systems with cyclic and planar geometry is associated with their increased stability. Various structural, energetic, electronic, and magnetic behavior-based parameters are considered to analyze the aromaticity of the systems, among which the Nucleus-Independent Chemical Shift (NICS) (Schleyer et al., 1996) is, perhaps, the most widely used criterion for aromaticity. CDFT also plays an important role in aromaticity determination (Chattaraj et al., 2005; Chattaraj et al., 2006; Chattaraj et al., 2007), and it does so with the help of reactivity descriptors and associated electronic structure principles (Chattaraj et al., 1993; Chattaraj and Maiti, 2001; Pan et al., 2013b; Chakraborty and Chattaraj, 2021). The relative aromaticity index ΔX, where X could be energy (E), polarizability (α), electrophilicity (ω), or hardness (η), is defined as the difference between the respective indices in the cyclic and open (or localized) systems, i.e., ΔX = XCYCLIC – XOPEN(/LOCALIZED). They are observed to show a similar performance as those of the NICS and MCI values. They also provide valuable insights into the stability, reactivity, and electronic properties of the associated cluster.
All-metal aromatic Al42− (Li et al., 2001) and antiaromatic Al44− (Kuznetsov et al., 2003) clusters are investigated at the B3LYP/6-311G(d, p) level of theory to analyze their aromaticity from a CDFT perspective (Chattaraj et al., 2005; Chattaraj et al., 2006; Chattaraj et al., 2007). While Δη > 0 represents aromaticity and Δη < 0 represents antiaromaticity, the reverse is true in the cases of E, α, and ω indices. Their values for the aluminum clusters are compared with those of benzene (C6H6) and cyclobutadiene (C4H4). While Al42− and C6H6 show positive Δη and negative ΔE, Δα, and Δω values indicating an aromatic behavior, C4H4 shows an exact opposite trend to account for its antiaromaticity. For the Al44− cluster, however, we have obtained somewhat contradictory results. ΔE and Δα values indicate the cluster’s antiaromatic nature, whereas Δη, Δω, and NICS values reflect its aromatic nature. It is in conformity with the current knowledge that this cluster exhibits conflicting aromaticity. Experimental synthesis and theoretical studies reported along with ELF analysis (Li et al., 2001; Santos et al., 2005) suggest the cluster to be antiaromatic. Its σ-aromaticity directs the overall aromaticity by dominating over its π-counterpart as studied through NICS (Chen et al., 2003) and magnetic field induced current density (Havenith et al., 2004) analyses. Such conflicting aromaticity, along with other varieties of multiple aromaticity and antiaromaticity, δ- and Φ-aromaticity, bond stretch isomerism, etc. are exhibited by several other all-metal clusters (Zubarev et al., 2009; Zubarev and Boldyrev, 2011). Be32−, Ca32−, and Mg32− clusters are classified as aromatic in terms of the ΔX indices (Roy and Chattaraj, 2008; Giri et al., 2010). Other applications of aromatic clusters are studied by our group through molecular electronic transport (Khatua et al., 2008), hydrogen storage (Havenith et al., 2005; Giri et al., 2011a; Giri et al., 2011b; Das and Chattaraj, 2012; Srinivasu et al., 2012; Pan et al., 2012b), and Zn–Zn and Be–Be bond stabilization (Chattaraj et al., 2008; Roy and Chattaraj, 2008) in the domain of CDFT. The ΔX and NICS parameters show their versatility in quantifying the aromaticity of not just planar and cyclic systems, but also any other nonplanar closed structure. They are both easily computable and ΔX also has a conceptual lucidity since it originates from the electronic structure principles of CDFT.
Certain chemical entities contain loosely bound electrons not directly connected to any atom(s) within the cluster, but trapped within a hollow space (cavity of cage compounds or packing void in crystals) that act as anions. Such entities are known as electrides and they are known for their nonlinear optical (NLO) properties. Besides showing NLO properties, which is considered to be an identifiable character of an electride, it is also widely applicable in electron emission, catalysis, reversible hydrogen storage, super conductivity, etc. (Toda et al., 2007; Xu et al., 2007; Kitano et al., 2012). Organic electrides of crown ethers and cryptands (Ellaboudy et al., 1983; Ward et al., 1988; Dawes et al., 1991; Xie et al., 2000), and inorganic electrides such as [Ca24Al28O64]4+(4e−) (Matsuishi et al., 2003), Y5Si3 (Lu et al., 2016), [Ba2N2](e−), [Li2Ca3N6](2e−) (Qu et al., 2019), [Ca2N]+(e−) (Lee et al., 2013), [Y2C]1.8+·1.8e− (Zhang et al., 2014), Sr5P3 (Wang et al., 2017), and Yb5Sb3 (Lu et al., 2018) are well reported in the literature. A different class of electrides, known as molecular electrides, are basically guest@host complexes containing a significant amount of localized electron cloud within the void of the host. Cavity-containing molecular structures such as decaborane (Muhammad et al., 2009; Muhammad et al., 2011), pyrrole (Chen et al., 2005), tetracyanoquinodimethane (TCNQ) (Li et al., 2009), fullerene cages (Das et al., 2020), C20F20 (Wang et al., 2012), C60F60, extended (3.1.3.1) porphyrin (EP) (Saha and Chattaraj, 2018), and many more are utilized for this purpose. The guest atoms are usually alkali and alkaline earth metals. For a guest@host complex to be characterized as a molecular or cluster electride, certain criteria need to be fulfilled such as the presence of a non-nuclear attractor/maximum (NNA/NNM) (a non-nuclear critical point with a local maximum of electron density), a negative Laplacian of electron density [∇2ρ(rc)], presence of an ELF basin near the NNM, high NLO properties, and a green region in the NCI plot showing accumulation of electron density.
The Mg2EP complex studied at the M06-2X-D3/6-311G(d,p) level of theory (Saha and Chattaraj, 2018) shows the presence of NNA in between the two Mg atoms where the value of ∇2ρ(rc) < 0, with an ELF basin nearby. The electron population at said NNA is 1.02 e with 46% localization. The NLO properties in terms of
The encapsulation of noble gas (Ng) atoms in B40 cavitand along with their structure and interactions of Ng with the Ng and B atoms in the host–guest complex is discussed with the help of DFT-based computations (Pan et al., 2018). Dissociation energy (∆Ediss) and Gibbs free energy change (∆Gdiss) are calculated to study the stability of the encapsulated complexes. NBO, EDA, and NOCV calculations have been done for studying the nature of bonding. The optimized structure of the B40 cage along with the Ng@B40 and Ng2@B40 systems (at ωB97X-D/def2-TZVP level) are depicted in Figure 5. The size of the B40 cage is suitable to accommodate He and Ne atoms at its center, whereas for heavier atom (Ar-Rn) encapsulation, the cavity diameter expands with the help of a certain amount of energy (preparation energy, ΔEprep). Although these complexes are thermochemically unstable, they remain in their encapsulated form on account of their high kinetic barrier. Despite that, the lighter Ng encapsulated complexes have very low ∆Ediss (−1.8 kcal mol−1 for He and −7.1 kcal mol−1 for Ne) as compared with the experimentally identified He@C20H20 complex (−33.8 kcal mol−1). For the heavier Ng atom-encapsulated Ng@B40 systems (Ng = Kr-Rn), the ∆Ediss increases with the size of Ng. The possibility of releasing Ng atoms in the dissociation process is either through B7 or B6 holes for the lighter He-Ar atoms, whereas the heavier ones can only escape through the B7 holes due to their larger size. Decapsulation through the B7 hole has ΔG≠ values ranging within 84.7–206.3 kcal mol−1. The rate constant (k) calculated at 298 K for the dissociation through either the B7 or the B6 hole comes out to be pretty low suggesting that all the Ng@B40 systems are kinetically stable.
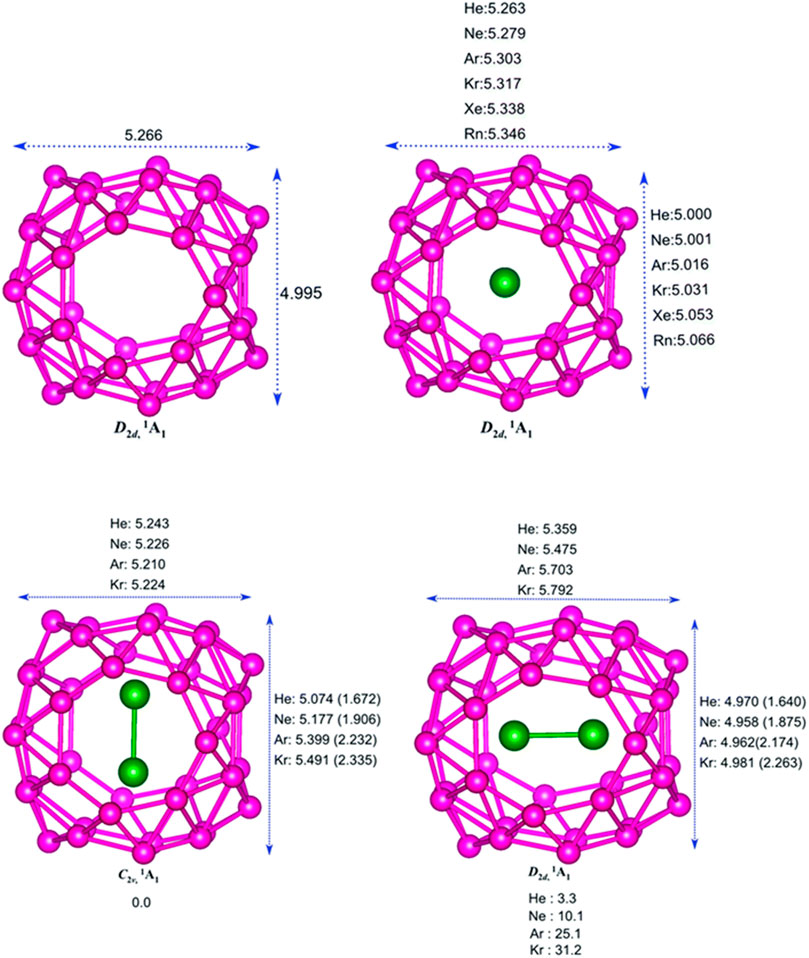
FIGURE 5. Optimized structures of B40, Ng@B40, C2v, and D2d isomers of Ng2@B40 optimized at the ωB97X-D/def2-TZVP level. (Adapted from Pan et al., 2018 with permission from the PCCP Owner Societies.).
In the case of two Ng atoms encapsulation, the inter atomic distance decreases from that of their free state. For Ar2@B40 and Kr2@B40, larger repulsion results in the exergonic dissociation of Ng2@B40 into Ng and Ng@B40. Along with this, large ∆Eprep indicates the nonviability of Ar2B40 and Kr2B40. For even heavier Ng atoms (Xe and Rn), no minimum energy structures are obtained for the corresponding dimer encapsulation. The corresponding transition states of the Ng release process of Ng2@B40 (Ng = He-Ar) suggest that one Ng atom approaches and leaves through the B7 rings, while the other remains near the center of the cavity. The associated ΔG≠ values for the He and Ne dimer encapsulated complexes are high enough for them to be kinetically viable.
The B40 cage exhibits a fluxional behavior due to the continuous interconversion between the B6 and B7 rings caused by the transfer of one B center from B7 to B6. The encapsulated system, Ng@B40, also shows similar dynamic behavior as that of the bare B40. The Ng atom inside the cage does not have any substantial influence on the fluxionality of the cage which is reflected in the free energy barrier values (16.4 kcal mol−1 for the bare cage, and a range of 16–18.9 kcal mol−1 for Ng@B40). Topological analysis of Ng@B40 explains the nature of the interactions therein. ∇2ρ(rc) and H(rc) values being positive at the BCPs of Ng-B bond suggest the presence of noncovalent character in most of the complexes except for Rn@B40, Ar2B40, and Kr2B40, where H(rc) < 0. For the encapsulation of Ng2 (Ng = Ar2 & Kr2) the Ng-Ng bond becomes partially covalent in contrast to their noncovalent character in the free state, except in the cases of He2@B40 and Ne2@B40 where no covalency is imparted. From NBO analysis it has been shown that Ng → B40 charge transfer increases with increasing the size of the Ng atoms. The electron transfer further increases in the case of Ng2 encapsulation. Along He–Rn, an increase in WBI values indicates that the increasing size of Ng atoms increases the degree of covalency between the Ng and B centers. EDA analysis reveals a high positive ∆Epauli which leads to positive ∆Eint suggesting the interaction to be repulsive in case of the heavier noble gas encapsulated B40 systems. Also, both the attractive terms, ∆Eelstat and ∆Eorb, increase with the increasing size of Ng atoms.
Small gas molecules such as C2H2, C2H4, C2H6, CO, CO2, NO2, NO, N2, H2, and Ng atoms (Hen-Xen, n = 1,2) are selected as guest molecules encapsulated in OA cavitand (Figure 6) and analyzed via DFT approach (Chakraborty et al., 2016). The systems under study are optimized at the ωB97X-D/6-311G(d,p) level of theory (LanL2DZ basis set with ECP is used for Xe). There are two possible cavities for the accommodation of the guest atoms, the inner cavity of OA is more suitable as it increases the host–guest interaction. In the case of Ng@OA, due to encapsulation of Ng atoms, no notable distortion is observed in the OA. For Ng2@OA systems, one guest atom can occupy the center of the host cavity, whereas the other one remains in the outer cavity. The Ng–Ng bond distances inside OA are 3.6, 3.7, 3.8, 3.9, and 4.1 Ǻ, respectively, for He2, Ne2, Ar2, Kr2, and Xe2. In the cases of CO@OA and NO@OA, the guest molecules prefer to stay in the inner cavity of OA. Their orientation with respect to the two nearest benzene-like fragments is almost perpendicular, whereas it is parallel to the rest of the benzene fragments of OA. The presence of these π electron clouds close to the encapsulated guests is expected to have a significant impact on the stability of the complexes, the nature of interaction, and dynamical behavior as well. For N2@OA and H2@OA, N2 and H2 remain well inside the inner cavity. Thermochemical study reveals that except for He@OA, in all cases, D0 value is positive indicating the stability of the host–guest complexes concerning their dissociation into the corresponding individual components. Going from lighter to heavier Ng atoms (also for Ng2@OA), D0 value increases, i.e., the host–guest interaction increases. This could most likely be due to the increasing polarizability of the Ng atoms down the group. The dissociation channels for all the encapsulated complexes have positive D0 values. Most of them, however, dissociate spontaneously at room temperature except in the cases of Kr, Kr2, Xe, C2H2, C2H4, C2H6, and N2 guest molecules. Thus, the encapsulation of the mentioned guest molecules is favorable at 298 K. In the cases of NO/NO2@OA and CO/CO2@OA, an increase in D0 value is observed from NO to NO2 and from CO to CO2. It can thus be deduced that the encapsulation of NO2 and CO2 inside OA forms more stable complexes compared with NO and CO, respectively. Having said that, it is to be noted that all the four complexes have favorable dissociation channels at room temperature. The hydrocarbons have better interaction with the OA and hence are not prone to dissociation at ambient temperatures.
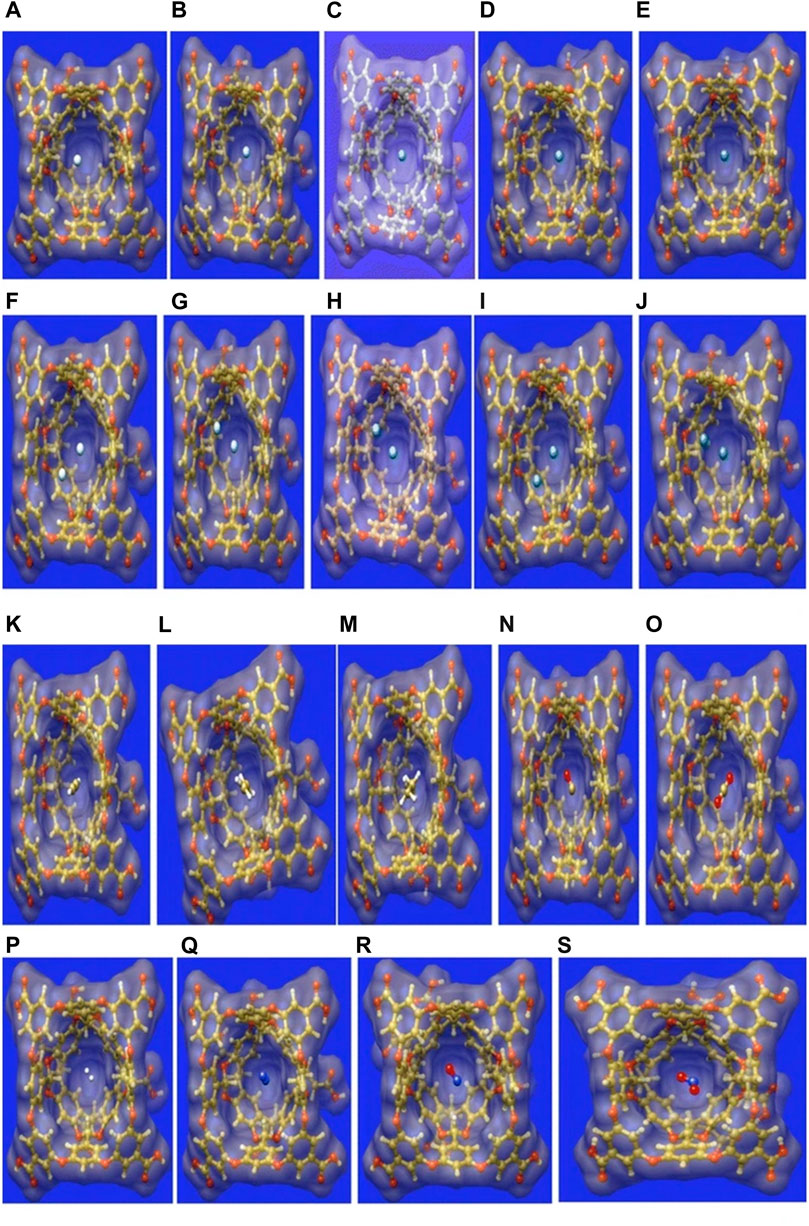
FIGURE 6. The surface representation of the optimized geometries of the guest encapsulated OA, where the guests are: (A) He, (B) Ne, (C) Ar, (D) Kr, (E) Xe, (F) He2, (G) Ne2, (I) Ar2, (J) Kr2, (K) Xe2, (L) C2H2, (M) C2H4, (N) C2H6, (O) CO, (P) CO2, (Q) H2, (R) N2, (S) NO, and (T) NO2. (Adapted from Chakraborty et al., 2016 with permission from Theoretical Chemistry Accounts, Springer Nature. Copyright© 2016, Springer-Verlag Berlin Heidelberg.).
The interaction between the host and guest moieties is analyzed with the help of NBO, NCI, and EDA. All the Ng atoms acquire some positive charges, i.e., transfer of electron density occurs from the Ng atoms to the host OA surface. The donation primarily occurs from the lone pair (LP) of Ng to the C-H antibonding orbital of OA for all the Ng atoms [except Ne where it takes place from LP to antibonding Rydberg state (Ry*)] and for CO, NO, N2, and CO2 molecules. WBI values for Ng–Ng and Ng–OA interactions indicate a purely noncovalent character therein. From this discussion it is clear that the confinement brings about an increase in the reactivity of all the guest atoms/molecules within the OA. NCI isosurfaces show the presence of green surface around the guest molecules indicating van der Waals interaction that stabilizes the host–guest complexes. EDA results show that there exists closed shell type of interaction between guest Ng/H2 and OA. The ΔEdisp and ΔEorb are the largest and the smallest contributors toward the total attractive interaction, respectively. The former increases in magnitude with increasing size of the encapsulated Ng atoms. For the guest hydrocarbons, the contribution from ∆Edisp increases and ΔEelstat decreases with the increasing number of H atoms. This is because the molecules such as C2H2 and C2H4 containing labile electron cloud can accumulate enough positive charge to favorably interact with the electron-rich fragments of the OA. This makes the contribution from ∆Eelstat very important in stabilizing these complexes. The contribution from charge transfer and polarization are very less toward ∆Etot as indicated by the very low values of ΔEorb. A similar type of situation is observed for CO/CO2@OA complexes. For nitrogen-containing guests, the ∆Eorb outweighs the ∆Eelstat contribution toward ΔEtot. Since the main stabilizing factor for all the complexes is ∆Edisp, the nitrogen-containing guest molecules are prone to be affected by the polarization or charge transfer by OA, in comparison with the other guest molecules. ADMP simulation performed at 298 K shows that all the Ng atoms (except Ne) are prone to leaving the cavity, whereas at 50 K they remain within the OA. Polar molecules such as CO, CO2, NO, and NO2 have a higher tendency to stay within the OA than the nonpolar H2. Again, C2H2, C2H4, and N2 containing π electron cloud also prefer to stay inside OA. From the above thermochemical, kinetic, and dynamical analyses, it can be said that OA makes a reasonably good choice for accommodating gas molecules.
Cucurbiturils are methylene-linked macrocyclic molecules having glycoluril unit [=C4H2N4O2=] as a building block. This repeating glycoluril unit can bind with hydrogen with sufficient amount of binding energy. Thus CB[n] can be designed as an effective hydrogen storage compound. The nitrogen and the oxygen centers are found to be the most active centers to bind with hydrogen with positive binding energy. It was found that (CH3)2C4H2N4O2(CH3)2 unit can interact with total 13H2 atoms (Pan et al., 2013a). Since hydrogen has an electric quadrupole moment, a charge–quadrupole interaction plays a pivotal role in binding the hydrogen with the host. NPA charge analysis reveals that the charge transfer occurs from the N and O centers to the
Among the CB[n] family, CB[7] can accommodate five H2 molecules endohedrally. The O centers can adsorb a total of 28
The endohedral adsorption of gas molecules such as C2H2, C2H4, C2H6, CH4, CO, CO2, NO2, NO, N2, H2, F2, Cl2, H2S, and SO2 into the CB[7] cavitand is depicted in Figure 7. Geometry optimizations are performed at the ωB97X-D/6-31G(d,p) and ωB97XD/6-311+G(d,p) levels (Pan et al., 2017). Both the CB[7] cage and the encapsulated gas molecules remain unaffected by the encapsulation. For SO2 encapsulation, the binding enthalpy shows the highest value (14.3 kcal mol−1) followed by Cl2 and C2H2 (11.6 and 10.4 kcal mol−1, respectively). CB[7] also encapsulates C2H4 and C2H6 more favorably than CO2, NO2, and H2S. The binding enthalpies for NO/F2/N2/CO/CH4@CB[7] systems vary from 4.7 to 5.8 kcal mol−1, the highest value corresponding to CH4. From the enthalpy values it is clear that CB[7] can selectively adsorb SO2 among various gas molecules, and hence can be applicable in the SO2 separation process from gas mixtures. ∆G value suggests that C2H6 is less prone to be encapsulated than CO2 inside CB[7]. SO2, Cl2, and C2H2 adsorb with negative ΔG values, whereas those of C2H4 and CO2 are slightly endergonic (0.6–0.7 kcal mol−1). The corresponding ΔG values for the adsorption of C2H6, N2, F2, NO2, NO, and H2S vary within 1.3–2.8 kcal mol−1 at 298 K temperature. EDA results show that for all the discussed complexes, ∆Edisp contributes more toward the stabilization of the host–guest systems. ∆Eelstat term also plays an important role here. In the hydrocarbons, as the number of H atoms increases, ∆Eelstat decreases gradually due to the reduction in the acidic character of the H atoms. Higher ∆Eelstat and lower ∆Epauli values in the SO2 encapsulated complex make the interaction between the host and the guest stronger than for C2H4 and C2H6 analogues. This again validates the higher SO2 selectivity of CB[7].
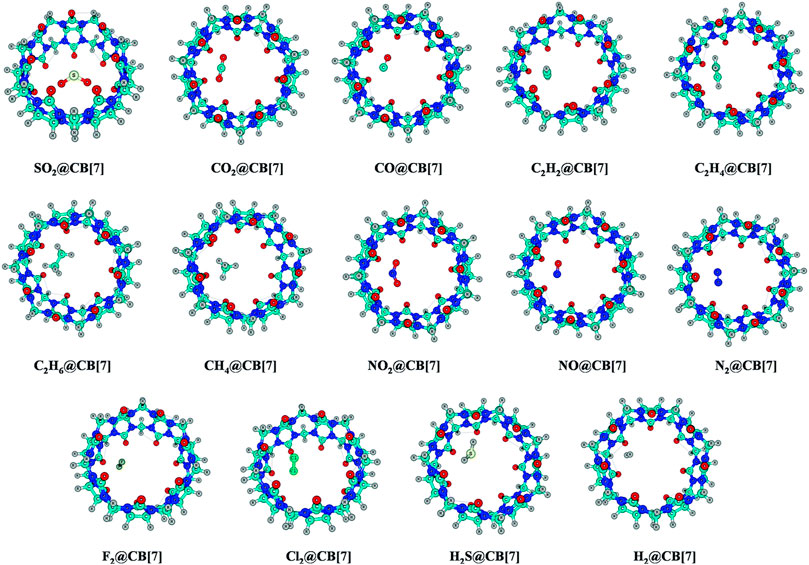
FIGURE 7. Optimized geometries of the guest encapsulated CB[7] systems at the ωB97X-D/6-311+G(d,p) level of theory. (Reproduced from Pan et al., 2017 with permission from the PCCP Owner Societies.).
Cucurbit[6]uril, among the CB[n] family, is found to be a compatible host for the encapsulation of Ngs within its cavity (Pan et al., 2015). The optimized geometries at the ωB97X-D/6-311G(2d,p) level of theory of the Ngn@CB[6] complexes are provided in Figure 8. CB[6] effectively accommodates three Ne atoms, but can only trap two of the large Ar and Kr atoms. No significant distortion in the cage is observed for trapping all the three Ne atoms or for the first atom of Ar and Kr, whereas inserting a second atom deforms the shape of the host. The Ng dissociation process becomes more endothermic as we move from Ne to Kr. At 298 K, the dissociations of all Ngn@CB[6] are exergonic except Kr@CB[6]. At 77 K, apart from the second Ng (Ar and Kr) atom dissociation from Ng2 encapsulated CB[6], all dissociations become endergonic. Kr encapsulation at 298 K and 1 atm pressure is thermochemically favorable, whereas for Ne and Ar encapsulation, high pressure and moderately low temperature are preferred.
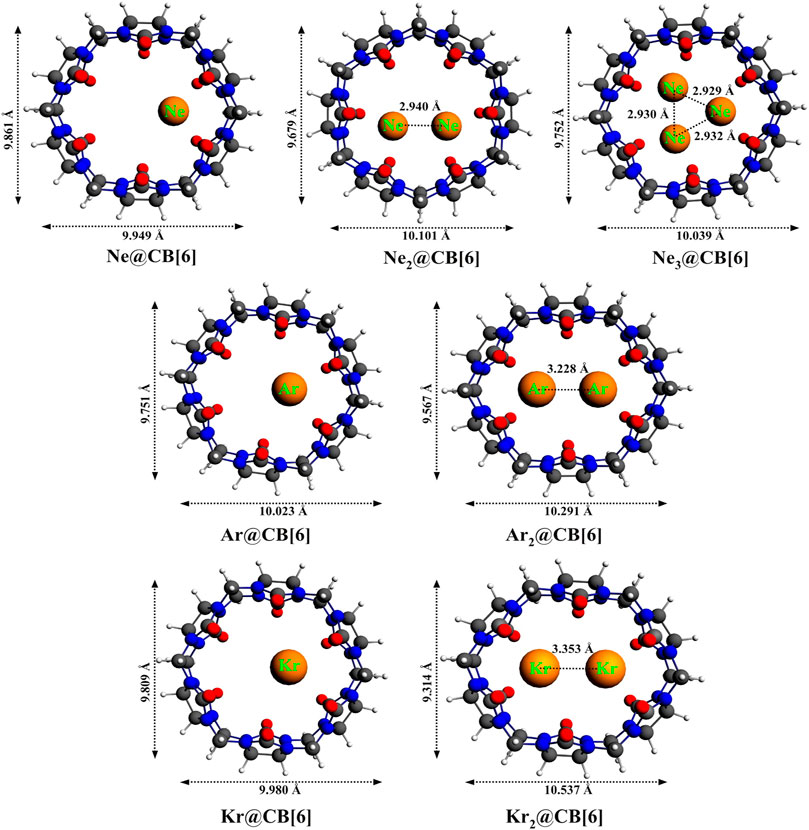
FIGURE 8. Optimized geometries of noble gas encapsulated CB[6] complexes at the ωB97X-D/6-311G(2d,p) level. (Reprinted with permission from Pan et al., 2015. Copyright© 2015, American Chemical Society.).
NPA charge analysis reveals N and O to be negatively charged in Ngn@CB[6], while C and H have positive charges. Slight charge transfer (∼0.01 e-) occurs from Ng→CB[6] moiety. Small ρ(rc) value and positive ∇2ρ(rc) and H(rc) values from topological analysis suggest the interaction to be of closed shell type. ELF analysis shows an absence of electron localization between the Ng-Ng and Ng-cage atoms, corroborating the result obtained from AIM. EDA analysis reveals the contribution from ∆Edisp to be the largest, followed by ∆Eelstat, and the smallest contribution is from ∆Eorb, all of which gradually increases going from Ne to Kr. The green surfaces observed between the Ng and CB[6] units in the NCI isosurface (Figure 9) are an indication of a small van der Waals interaction, which increases with the size of the Ng atoms. The dynamical study (ab initio MD) for 1 ps and at 298 K reveals that Ne and Ar remain inside the cavity, whereas Kr and all the Ng2 in Ng2@CB[6] move toward the open end but do not leave the cage. At 77 K, all guests stay inside the host.
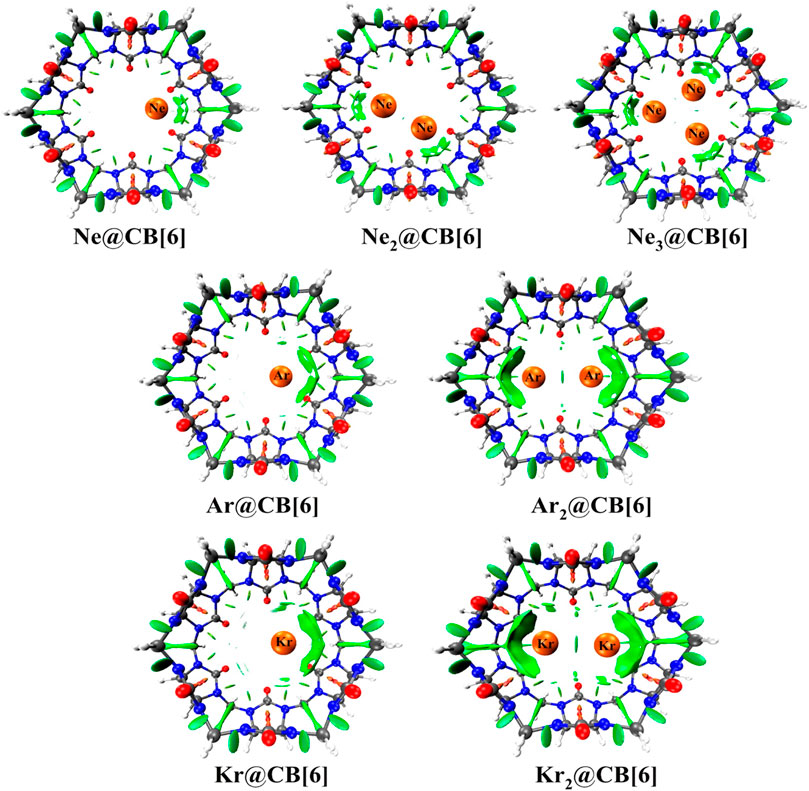
FIGURE 9. NCI plots of Ngn@CB[6] complexes. (Reprinted with permission from Pan et al., 2015. Copyright© 2015, American Chemical Society.).
The activation of small molecules by a metal-supported boron cluster is studied through DFT calculations (Saha et al., 2017). They are known to be applicable in nanomaterial building blocks, automobiles (Norbye, 1971; Jiménez‐Halla et al., 2010) etc. The global minimum energy structures calculated at PBE/def2-TZVPPD (Perdew et al., 1996; Weigend and Ahlrichs, 2005) level of MB12-, CO@MB12-, N2@MB12- clusters and their corresponding TSs for the internal rotation of the B3 ring are provided in Figure 10. The coordination of the small molecules with the MB12- cluster forms an umbrella-shaped structure in which the M-L bonds act like the stick of the umbrella. The coordination of CO with the metal center can take place through both the C and O ends, the former producing a more stable isomer. For both OCMB12_ and NNMB12_, the Ir-L bond has the highest strength, followed by Co and Rh, while for a particular M center, CO forms a stronger bond than N2. ∆G values of these complexes are highly positive which suggest that the corresponding complexes are thermodynamically stable concerning the dissociation process. The O-side bound isomers, however, have low positive ∆G for Co and Ir, and become slightly negative for Rh. They can be made viable by lowering the temperature. The N−N and C–O bonds get lengthened due to complexation in the order COMB12- < N2MB12- < OCMB12- causing a red shift in their bond stretching frequencies which is the highest for the Ir analogues.
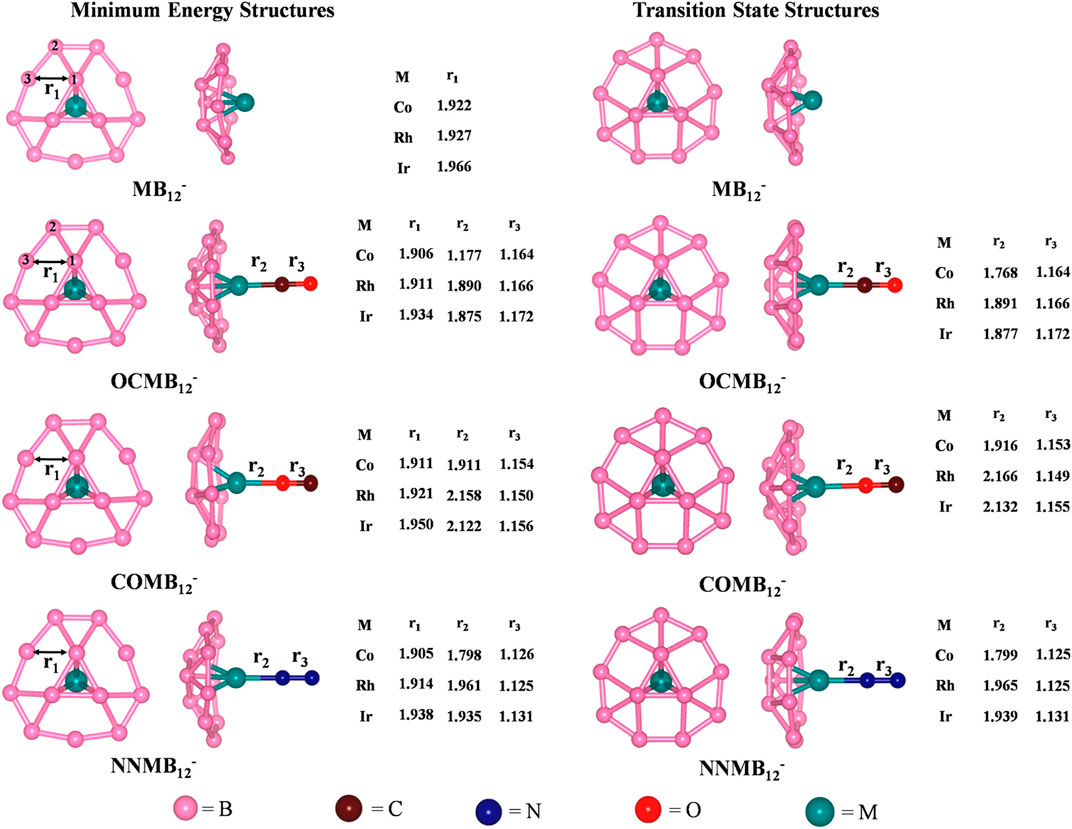
FIGURE 10. Optimized geometries of MB12- and ligand bound MB12- at the PBE/def2-TZVPPD level, and their transition state structures for the internal rotation of the B3 ring. Bond distances are provided in Angstrom unit. (Reprinted with permission from Saha et al., 2017. Copyright© 2017, American Chemical Society.).
NBO analysis reveals that upon complexation, the metal centers get more negatively charged, apart from COIrB12_ and NNIrB12_, where Ir still contains positive charge (less than that in IrB12−). L→M and M→L back transfers take place, and in certain complexes the latter completely compensates (or overcompensates) the former which is indicated by the zero (or negative) charge on the ligand. The Wiberg bond indices suggest that the covalent character follows the order M-C in OCMB12- > M-N in N2MB12- > M-C in COMB12-. From EDA-NOCV, it is seen that the bonding between the metal and the ligand is predominantly orbital and electrostatic interactions (in more or less equal contributions), indicating the L-M bonds to have both covalent and ionic characters. For OCMB12-, however, the contribution from ∆Eelstat is higher than ∆Eorb. Figure 11 shows the deformation densities [Δρ(r)] for the pairwise orbital interactions for the LMB12_ complexes, where a shift in the electron density occurs from the red to the blue region. The Δρ(
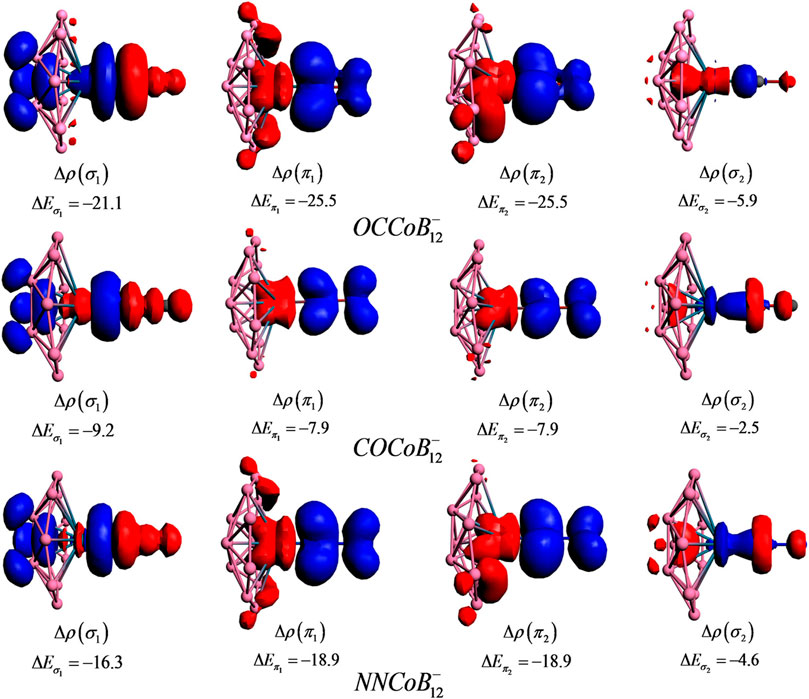
FIGURE 11. Deformation density plots of the pairwise orbital interactions in LCoB12- (OC, CO, and NN) systems at the revPBED3/TZ2P//PBE/def2-TZVPPD level. Energies are provided in kcal/mol. (Reprinted with permission from Saha et al., 2017. Copyright© 2017, American Chemical Society.).
An internal rotation of the inner B3 ring with respect to the outer B9 ring occurs within the MB12- cluster. The energy barrier associated with this rotation is reported (Popov et al., 2014; Liu et al., 2016) to follow the order Co > Rh > Ir. BOMD simulation at 800 K shows the L-M bonds to be intact during the rotation. This makes the complex seem like a spinning umbrella with the L-M bond as the stick.
Clathrate hydrates, a class of inclusion compounds, are known to encapsulate guest compounds within its hydrogen bonded polyhedral cage (Mao et al., 2002; Mao and Mao, 2004; Lee et al., 2010). They constitute a very effective host for hydrogen storage. Four types of clathrate hydrates and their maximum possible hydrogen-encapsulated complexes studied at B3LYP/6-31G(d) level (Chattaraj et al., 2011) are depicted in Figure 12. 512 represents the cavity having 12 pentagonal faces, whereas 5126k (k = 2,4,8) represents 12 pentagonal faces along with k hexagonal faces. Here we discuss the structure, bonding, and stability of the bare and hydrogen-encapsulated complexes from a density functional theory perspective. For nH2@512 complexes it is seen that for the first H2 encapsulation, the process is energetically favorable although the overall nH2 encapsulation is method dependent. Owing to the small size of 512 cavity, it can accommodate a maximum of five H2 molecules, after which a deformation in the cavity is observed. The GM for H2 confinement in the 512 cavity occurs endohedrally. The H2 molecules favor the inside of 512 more than the outside. In the case of 51262 cage, it can also take up a maximum of five H2 molecules. Slight distortion is observed in the system that becomes more noticeable during the third H2 encapsulation which slowly decreases for the fourth and the fifth hydrogen molecule encapsulation. This is reflected in the slightly conflicting trend in the corresponding interaction energies. Now in the case of 51264 clathrate, obtaining the minimum energy structure was difficult. It is fascinating to note that the encapsulation of one H2 into the cage stabilizes the structure although it could not provide with the minimum energy structure. Further incorporation of guest molecules deforms the structure of the system. For 51268, the interaction energy for all the six H2 encapsulation is negative making the process favorable. The large size of the host cavity makes it feasible to accommodate all the six guest molecules efficiently. Positive ∆G value suggests that the complexes are kinetically stable. Finally, it can be concluded that the 512 and 51262 clathrates can encapsulate up to two hydrogen molecules without undergoing any structural distortions, whereas the 51268 clathrate may entrap up to six H2 molecules depending upon the level of theory used. Calculation of CDFT-based reactivity descriptors of the complexes with and without H2 encapsulation suggests that for most of the systems, stability increases with the increase in number of trapped hydrogen molecules. This is concluded from the increasing hardness and decreasing electrophilicity values.
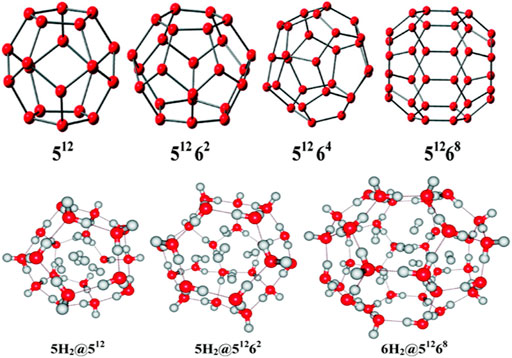
FIGURE 12. Optimized geometries of the clathrate hydrates along with their maximum possible H2 molecule encapsulated complexes. (Adapted with permission from Chattaraj et al., 2011. Copyright© 2011, American Chemical Society.).
Li ion is popularly known to bind well with hydrogen molecule owing to its positive charge (Pan et al., 2012a). Inspired by this, a number of efforts have been made to effectively polarize the Li center of various clusters to increase its hydrogen adsorbing ability. Here we study the H2 storage potential of the Li-doped clusters, M5Li7+ (M = C, Si, Ge), M4Li4 (M = Si, Ge) at the M06/6-311+G(d,p) level, and some super-alkali ions at the M052X/6-311+G(d) level (Figures 13, 14). The Li centers attain a net positive charge due to the high polarizability of the clusters, facilitating electrostatic interactions to bind with the H2 molecules. The negative values of interaction energies and enthalpies indicate the efficacy of these clusters to be good H2 storage materials. The gravimetric wt% of adsorbed H2 are 28.0, 18.3, 9.3, 14.7, and 7.1 for C2Li7+, Si5Li7+, Ge5Li7+, Si4Li4, and Ge4Li4, respectively. For the super-alkali ions, the values range from 13.2 to 40.9%, with the highest being that for BLi6+. On applying electric field, a gradual improvement is observed in the interaction energy value. Thus, in terms of gravimetric wt%, BLi6+ is preferable whereas the interaction energy per H2 molecule suggests B2Li11+ to be the preferred choice for hydrogen storage.
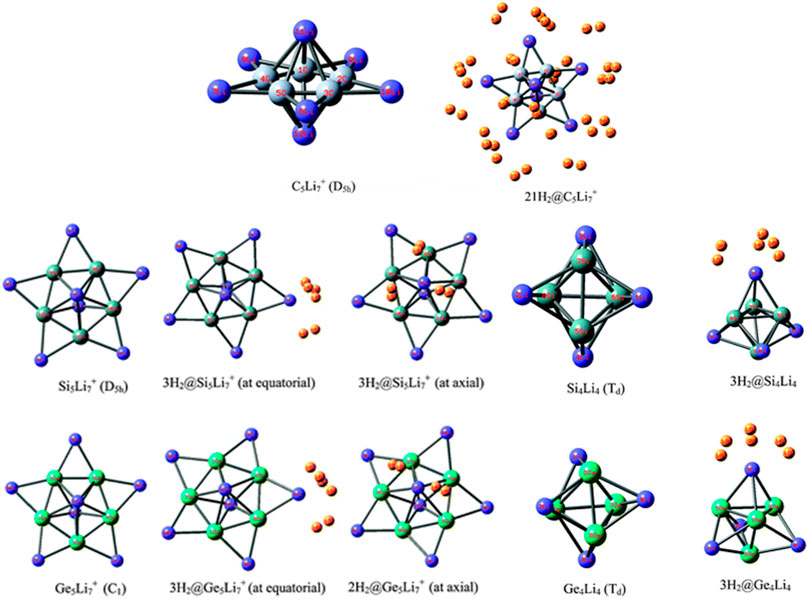
FIGURE 13. Optimized structures of C5Li7+, M5Li7+, M4Li4 (M = Si, Ge) and their H2-trapped analogues at the M06/6-311+G(d,p) level. (Adapted from Pan et al., 2012b with permission from the PCCP Owner Societies.).
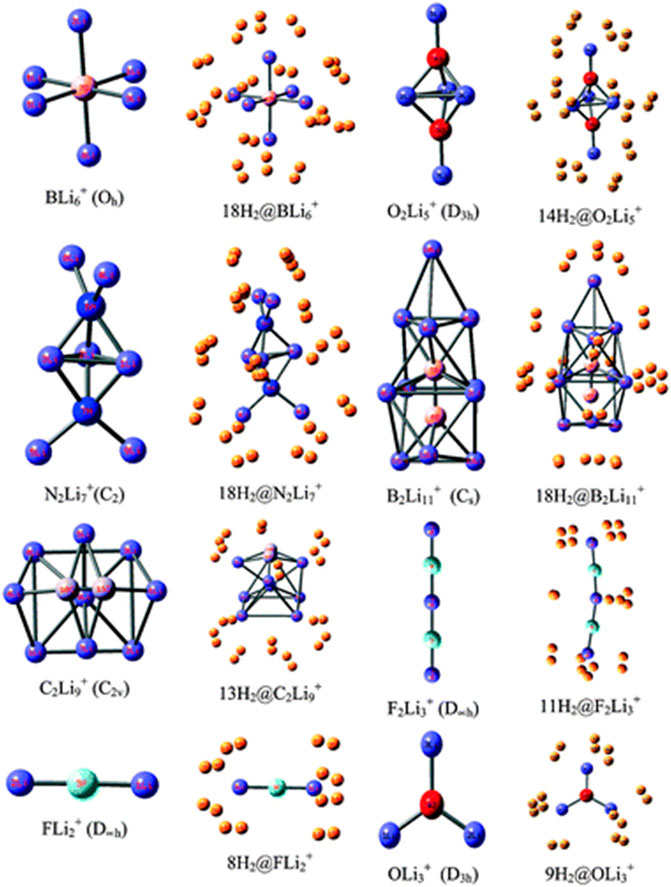
FIGURE 14. Optimized structures of studied super-alkali ions and their hydrogen-trapped analogues at the M052X/6-311+G(d) level. (Adapted from Pan et al., 2012b with permission from the PCCP Owner Societies.).
The influence of encapsulation on the hydrogen bond strength in (HF)2 within the fullerene cages is studied using DFT and ab initio MD (Khatua et al., 2014b). The optimized geometries of (HF)2@Cn (n = 60, 70, 80, 90) complexes wB97X-D/6-31G are depicted in Figure 15. The dissociation energy, enthalpy, and change in free energy are negative for the (HF)2@C60 system which indicates that the encapsulation process is thermodynamically unfavorable, whereas positive values for the rest of the HF encapsulated Cn cages imply them to be favorable (highest being for the C80 cage). Owing to the smaller size of the C60, the HF units orient themselves antiparallelly to reduce repulsion at the cost of hydrogen bond strength. Thus, the energy associated with the HF-HF interaction is observed to be highest in the C60 cage (positive, and hence repulsive in nature). For all the studied cases, upon encapsulation, the hydrogen bond distance reduces from that in the free state, the least being inside the C70 cage. The EDA study reveals that the contribution from ∆Epauli increases and the ∆Eint value decreases with decreasing the Cn cage cavity except for C80 cage. For the C60 cage, a very large value of ∆Epauli makes the overall ∆Eint value positive. On account of the smaller H-bond distance within the C70 and C90 cages compared to the same within the C80 cavity, both the ∆Eelstat and ∆Eorb contribute more to the attractive interaction than those in C80. AIM analysis reveals that for all these confined systems, ∇2ρ(rc) > 0 and H(rc) < 0 implying the partial covalent nature of the hydrogen bonds. The hydrogen bond is mostly covalent in case of (HF)2@C70 ELF analysis that also supports this observation.
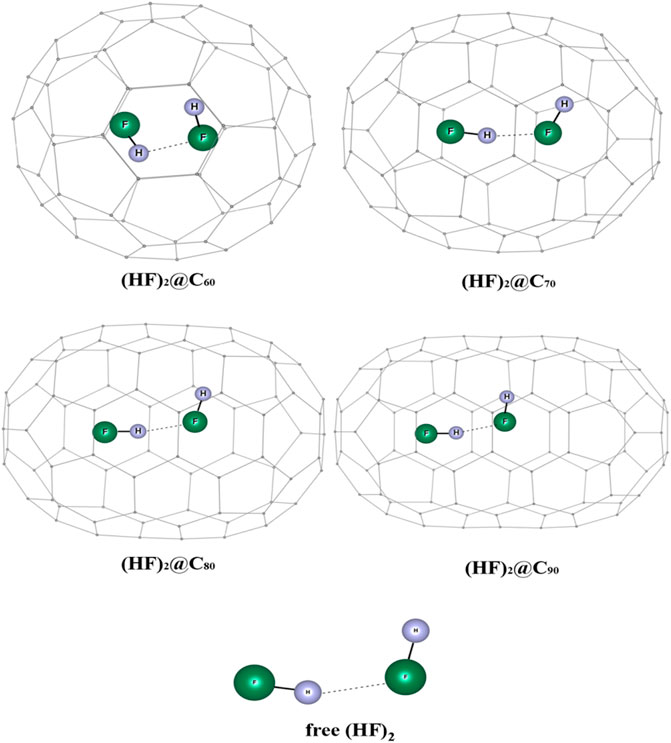
FIGURE 15. Optimized structures of (HF)2@Cn (n = 60, 70, 80, 90) and free (HF)2 at ωB97X-D/6-31G level. (Reprinted from Khatua et al., 2014b with permission from Elsevier. Copyright© 2014, Elsevier B.V.).
There exists an appreciable amount of interest in the field of cluster chemistry, especially in the gas-phase and surface-adsorbed studies, and for good reasons. Common curiosities in this area include the difference between the properties exhibited by the bulk and individual clusters, how the cluster size affects the overall behavior of the bulk, etc. Other important branches of this cluster chemistry include solving their global optimization problem in a fast and cost-effective way, and investigating the effect of confinement on the cluster-encapsulated systems.
The global optimizers discussed in this review are shown to locate the global minimum configurations for small metallic and nonmetallic clusters with less execution time and higher success rate than commonly used optimization algorithms, without having the need to impose any symmetry constraint or any other external restrictions. The only requirement is to adjust the local and global best parameters at each iteration. Comparisons made between our modified PSO with other DFT-integrated BH and SA reveal the superiority of the former with respect to the total execution time and number of iterations the program takes to converge. Again, the DFT-integrated FA turns out to be more efficient than the modified PSO. Furthermore, the ADMP-CNN-PSO technique is well suited for locating the global solution from a huge dataset of initial configurations.
The effect of adsorption and confinement of hydrogen, noble gas atoms, and various other small molecules on their stability, reactivity, nature of interactions, and dynamics are studied from a DFT perspective. The concept of aromaticity is analyzed in terms of CDFT-based descriptors such as E, α, ω, and η, where a lower value of the first three parameters and a higher value of hardness in comparison with that of a reference system characterize an aromatic molecule. The reverse is true for antiaromatic compounds. Certain guest@host complexes containing loosely bound electrons acting as anions and showing high NLO properties, known as molecular electrides, are capable of bond activation in small molecules. Other host–guest complexes exhibit fluxionality. One such example is the B40 cage whose fluxional property remains unaltered even after Ng atoms encapsulation. The complexation ability of the B40 cage is also studied in some sandwich complexes and it is seen than the presence of Xe within the cage enhances its complexation ability. The gas molecules accommodated within the Octa acid cavitand become slightly more reactive compared to their free state. Most of the OA-guest complexes are stable with respect to dissociation. OA can thus be designated as a reasonably good storage material for a variety of small gas molecules. Cucurbiturils form another class of compounds which is well known for its hosting capabilities. CB[6] can act as an efficient noble gas carrier and CB[7] can bind up to 52 hydrogen molecules (8.3 wt%). CB[7] is also found to be highly selective toward the adsorption of SO2 and hence can be used in separating SO2 from a gas mixture. It is also known to accelerate the otherwise slow [4+2] cycloaddition reaction. The binding ability of the transition metal boron cluster (MB12-) with isoelectronic species, CO and N2, is studied along with its fluxionality. Bond activation in both CO and N2 is observed, and the rotation of the ligand-bound complex makes it look like a spinning umbrella. Hydrogen storage capabilities of clathrate hydrates, Li-doped clusters, and super alkali are investigated and it is found that the former can accommodate 2–6 hydrogen molecules, whereas the Li systems show a gravimetric wt% range of 7.1–28.0% for the star-like clusters and 13.2–40.9% for the super-alkali systems. The (HF)2 encapsulation by the fullerene cages describes the confinement effect on the H-bond therein. Apart from C60, all the cages form the complexes in a thermodynamically favorable process. Also, a partial covalent character is observed in the H-bonds upon confinement.
PKC came up with the concept and design of the review, wrote the abstract, reviewed the final manuscript. RP and AP contributed towards the literature survey, writing the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer (SG) declared a past co-authorship with one of the authors (PKC) to the handling Editor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
PKC would like to thank Ambrish Kumar Srivastava for kindly inviting him to contribute an article to the research topic “Atomic Clusters: Theory & Experiments” in the journal, Frontiers in Chemistry. He also thanks DST, New Delhi, for the J. C. Bose National Fellowship, grant number SR/S2/JCB-09/2009, and his students whose work is presented in this article. RP and AP thank CSIR and IIT Kharagpur, respectively, for their fellowships.
Akhtar, F., Liu, Q., Hedin, N., and Bergström, L. (2012). Strong and Binder Free Structured Zeolite Sorbents with Very High CO2-over-N2 Selectivities and High Capacities to Adsorb CO2 Rapidly. Energy Environ. Sci. 5, 7664–7673. doi:10.1039/C2EE21153J
Baerends, E. J., Ziegler, T., Autschbach, J., Bashford, D., Bérces, A., Bickelhaupt, F. M., and Ellis, D. E. (2013). “ADF2013. 01. SCM,” in Theo. Chem (Amsterdam, Netherlands: Vrije Universiteit).
Bai, Q. (2010). Analysis of Particle Swarm Optimization Algorithm. Cis 3, 180. doi:10.5539/cis.v3n1p180
Barnes, J. C., Juríček, M., Strutt, N. L., Frasconi, M., Sampath, S., Giesener, M. A., et al. (2013). ExBox: a Polycyclic Aromatic Hydrocarbon Scavenger. J. Am. Chem. Soc. 135, 183–192. doi:10.1021/ja307360n
Becke, A. D. (1988). Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Becke, A. D. (1992). Density‐functional Thermochemistry. I. The Effect of the Exchange‐only Gradient Correction. J. Chem. Phys. 96, 2155–2160. doi:10.1063/1.462066
Bergman, B., Sandh, G., Lin, S., Larsson, J., and Carpenter, E. J. (2013). Trichodesmium- a Widespread marine Cyanobacterium with Unusual Nitrogen Fixation Properties. FEMS Microbiol. Rev. 37, 286–302. doi:10.1111/j.1574-6976.2012.00352.x
Bhatia, S. K., and Myers, A. L. (2006). Optimum Conditions for Adsorptive Storage. Langmuir 22, 1688–1700. doi:10.1021/la0523816
Brust, M., Walker, M., Bethell, D., Schiffrin, D. J., and Whyman, R. (1994). Synthesis of Thiol-Derivatised Gold Nanoparticles in a Two-phase Liquid-Liquid System. J. Chem. Soc. Chem. Commun. 7, 801–802. doi:10.1039/C39940000801
Cabria, I., López, M. J., and Alonso, J. A. (2008). Hydrogen Storage Capacities of Nanoporous Carbon Calculated by Density Functional and Møller-Plesset Methods. Phys. Rev. B 78, 075415. doi:10.1103/PhysRevB.78.075415
Cagle, D. W., Thrash, T. P., Alford, M., Chibante, L. P. F., Ehrhardt, G. J., and Wilson, L. J. (1996). Synthesis, Characterization, and Neutron Activation of Holmium Metallofullerenes. J. Am. Chem. Soc. 118, 8043–8047. doi:10.1021/ja960841z
Chai, J.-D., and Head-Gordon, M. (2008). Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. doi:10.1039/B810189B
Chakraborty, D., and Chattaraj, P. K. (2019). Bonding, Reactivity, and Dynamics in Confined Systems. J. Phys. Chem. A. 123, 4513–4531. doi:10.1021/acs.jpca.9b00830
Chakraborty, D., and Chattaraj, P. K. (2021). Conceptual Density Functional Theory Based Electronic Structure Principles. Principles. Chem. Sci. 12, 6264–6279. doi:10.1039/D0SC07017C
Chakraborty, D., and Chattaraj, P. K. (2015). Confinement Induced Binding in noble Gas Atoms within a BN-Doped Carbon Nanotube. Chem. Phys. Lett. 621, 29–34. doi:10.1016/j.cplett.2014.12.053
Chakraborty, D., and Chattaraj, P. K. (2017). Effect of Functionalization of boron Nitride Flakes by Main Group Metal Clusters on Their Optoelectronic Properties. J. Phys. Condens. Matter 29, 425201. doi:10.1088/1361-648X/aa8651
Chakraborty, D., and Chattaraj, P. K. (2018). Host-guest Interactions between Octa Acid and Cations/nucleobases. J. Comput. Chem. 39, 161–175. doi:10.1002/jcc.25097
Chakraborty, D., and Chattaraj, P. K. (2016a). Optical Response and Gas Sequestration Properties of Metal Cluster Supported Graphene Nanoflakes. Phys. Chem. Chem. Phys. 18, 18811–18827. doi:10.1039/C6CP02134D
Chakraborty, D., and Chattaraj, P. K. (2016b). Sequestration and Activation of Small Gas Molecules on BN-Flakes and the Effect of Various Metal Oxide Molecules Therein. J. Phys. Chem. C 120, 27782–27799. doi:10.1021/acs.jpcc.6b08404
Chakraborty, D., Das, R., and Chattaraj, P. K. (2017). Does Confinement Always Lead to Thermodynamically And/or Kinetically Favorable Reactions? A Case Study Using Diels-Alder Reactions within ExBox+4 and CB[7]. ChemPhysChem 18, 2162–2170. doi:10.1002/cphc.201700308
Chakraborty, D., Pan, S., and Chattaraj, P. K. (2016). Encapsulation of Small Gas Molecules and Rare Gas Atoms inside the Octa Acid Cavitand. Theor. Chem. Acc. 135, 119. doi:10.1007/s00214-016-1876-y
Chatt, J., and Leigh, G. J. (1972). Nitrogen Fixation. Chem. Soc. Rev. 1, 121–144. doi:10.1039/cs9720100121
Chattaraj, P. K., Bandaru, S., and Mondal, S. (2011). Hydrogen Storage in Clathrate Hydrates. J. Phys. Chem. A. 115, 187–193. doi:10.1021/jp109515a
Chattaraj, P. K., and Maiti, B. (2001). Electronic Structure Principles and the Atomic Shell Structure. J. Chem. Educ. 78, 811–813. doi:10.1021/ed078p811
Chattaraj, P. K., Nath, S., and Sannigrahi, A. (1993). Ab Initio SCF Study of Maximum Hardness and Maximum Molecular Valency Principles. Chem. Phys. Lett. 212, 223–230. doi:10.1016/0009-2614(93)89318-C
Chattaraj, P. K., Roy, D. R., and Duley, S. (2008). Bonding and Aromaticity in an All-Metal sandwich-like Compound, Be82−. Chem. Phys. Lett. 460, 382–385. doi:10.1016/j.cplett.2008.06.005
Chattaraj, P. K., Roy, D. R., Elango, M., and Subramanian, V. (2006). Chemical Reactivity Descriptor Based Aromaticity Indices Applied to and Systems. J. Mol. Struct. THEOCHEM 759, 109–110. doi:10.1016/j.theochem.2005.10.041
Chattaraj, P. K., Roy, D. R., Elango, M., and Subramanian, V. (2005). Stability and Reactivity of All-Metal Aromatic and Antiaromatic Systems in Light of the Principles of Maximum Hardness and Minimum Polarizability. J. Phys. Chem. A. 109, 9590–9597. doi:10.1021/jp0540196
Chattaraj, P. K., Sarkar, U., and Roy, D. R. (2007). Electronic Structure Principles and Aromaticity. J. Chem. Educ. 84, 354. doi:10.1021/ed084p354
Chen, W., Li, Z.-R., Wu, D., Li, Y., Sun, C.-C., and Gu, F. L. (2005). The Structure and the Large Nonlinear Optical Properties of Li@Calix[4]pyrrole. J. Am. Chem. Soc. 127, 10977–10981. doi:10.1021/ja050601w
Chen, Z., Corminboeuf, C., Heine, T., Bohmann, J., and Schleyer, P. V. R. (2003). Do all-metal Antiaromatic Clusters Exist? J. Am. Chem. Soc. 125, 13930–13931. doi:10.1021/ja0361392
Chollet, F. (2015). Keras. GitHub. Available at: https://github.com/fchollet/keras. doi:10.2210/pdb4trn/pdb
Chong, Z. R., Yang, S. H. B., Babu, P., Linga, P., and Li, X.-S. (2016). Review of Natural Gas Hydrates as an Energy Resource: Prospects and Challenges. Appl. Energ. 162, 1633–1652. doi:10.1016/j.apenergy.2014.12.061
Cioslowski, J., and Nanayakkara, A. (1992). Endohedral Fullerites: a New Class of Ferroelectric Materials. Phys. Rev. Lett. 69, 2871–2873. doi:10.1103/PhysRevLett.69.2871
Colorni, A., Dorigo, M., and Maniezzo, V. (1991). “Distributed Optimization by Ant Colonies,” in Proceedings of the first European conference on artificial life. 142. Paris, France: Elsevier Publishing, 134–142.
Contreras-García, J., Johnson, E. R., Keinan, S., Chaudret, R., Piquemal, J.-P., Beratan, D. N., et al. (2011). NCIPLOT: a Program for Plotting Noncovalent Interaction Regions. J. Chem. Theor. Comput. 7, 625–632. doi:10.1021/ct100641a
Cross, R. J., Saunders, M., and Prinzbach, H. (1999). Putting Helium inside Dodecahedrane. Org. Lett. 1, 1479–1481. doi:10.1021/ol991037v
Das, P., and Chattaraj, P. K. (2021a). Comparison between Electride Characteristics of Li3@B40 and Li3@C60. Front. Chem. 9, 638581. doi:10.3389/fchem.2021.638581
Das, P., and Chattaraj, P. K. (2020). Electride Characteristics of Some Binuclear Sandwich Complexes of Alkaline Earth Metals, M2(η5-L)2 (M = Be, Mg; L = C5H5-, N5-, P5-, As5-). J. Phys. Chem. A. 124, 9801–9810. doi:10.1021/acs.jpca.0c08306
Das, P., Saha, R., and Chattaraj, P. K. (2020). Encapsulation of Mg2 inside a C60 Cage Forms an Electride. J. Comput. Chem. 41, 1645–1653. doi:10.1002/jcc.26207
Das, P., and Chattaraj, P. K. (2021b). Substituent Effects on Electride Characteristics of Mg2(η5-C5H5)2: A Theoretical Study. J. Phys. Chem. A. 125,6207–6220. doi:10.1021/acs.jpca.1c04605
Das, R., and Chattaraj, P. K. (2012). A (T-P) Phase Diagram of Hydrogen Storage on (N4C3H)6Li6. J. Phys. Chem. A. 116, 3259–3266. doi:10.1021/jp212472u
Dawes, S. B., Eglin, J. L., Moeggenborg, K. J., Kim, J., and Dye, J. L. (1991). Cesium(+1)(15-crown-5)2.cntdot.e-. A Crystalline Antiferromagnetic Electride. J. Am. Chem. Soc. 113, 1605–1609. doi:10.1021/ja00005a025
Deng, W.-Q., Xu, X., and Goddard, W. A. (2004). New Alkali Doped Pillared Carbon Materials Designed to Achieve Practical Reversible Hydrogen Storage for Transportation. Phys. Rev. Lett. 92, 166103. doi:10.1103/PhysRevLett.92.166103
Dong, Z., Luo, Q., and Liu, J. (2012). Artificial Enzymes Based on Supramolecular Scaffolds. Chem. Soc. Rev. 41 (23), 7890–7908. doi:10.1039/C2CS35207A
Dunning, T. H., and Hay, P. J. (1977). “Gaussian Basis Sets for Molecular Calculations,” in Modern Theoretical Chemistry. Editor H. F. Schaefer (New York: Plenum), Vol. 3, 1–27. doi:10.1007/978-1-4757-0887-5_1
Ellaboudy, A., Dye, J. L., and Smith, P. B. (1983). Cesium 18-crown-6 Compounds. A Crystalline Ceside and a Crystalline Electride. J. Am. Chem. Soc. 105, 6490–6491. doi:10.1021/ja00359a022
Florea, M., and Nau, W. M. (2011). Strong Binding of Hydrocarbons to Cucurbituril Probed by Fluorescent Dye Displacement: A Supramolecular Gas-Sensing Ensemble. Angew. Chem. 123, 9510–9514. doi:10.1002/ange.201104119
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. Wallingford CT: Gaussian, Inc.
Froudakis, G. E. (2001). Hydrogen Interaction with Single-Walled Carbon Nanotubes: A Combined Quantum-Mechanics/molecular-Mechanics Study. Nano Lett. 1, 179–182. doi:10.1021/nl015504p
Giri, S., Bandaru, S., Chakraborty, A., and Chattaraj, P. K. (2011a). Role of Aromaticity and Charge of a System in its Hydrogen Trapping Potential and Vice Versa. Phys. Chem. Chem. Phys. 13, 20602–20614. doi:10.1039/C1CP21752F
Giri, S., Chakraborty, A., and Chattaraj, P. K. (2011b). Potential Use of Some Metal Clusters as Hydrogen Storage Materials-A Conceptual DFT Approach. J. Mol. Model. 17, 777–784. doi:10.1007/s00894-010-0761-1
Giri, S., Roy, D. R., Duley, S., Chakraborty, A., Parthasarathi, R., Elango, M., et al. (2009). Bonding, Aromaticity, and Structure of Trigonal Dianion Metal Clusters. J. Comput. Chem. 31, 1815–1821. doi:10.1002/jcc.21452
Grochala, W., Hoffmann, R., Feng, J., and Ashcroft, N. W. (2007). The Chemical Imagination at Work inVery Tight Places. Angew. Chem. Int. Ed. 46 (20), 3620–3642. doi:10.1002/anie.200602485
Gubbins, K. E., Liu, Y.-C., Moore, J. D., and Palmer, J. C. (2011). The Role of Molecular Modeling in Confined Systems: Impact and Prospects. Phys. Chem. Chem. Phys. 13, 58–85. doi:10.1039/C0CP01475C
Haaland, A., Shorokhov, D. J., and Tverdova, N. V. (2004). Topological Analysis of Electron Densities: Is the Presence of an Atomic Interaction Line in an Equilibrium Geometry a Sufficient Condition for the Existence of a Chemical Bond? Chem. Eur. J. 10, 4416–4421. doi:10.1002/chem.200400663
Havenith, R. W. A., Fowler, P. W., Steiner, E., Shetty, S., Kanhere, D., and Pal, S. (2004). Aromaticity and Antiaromaticity of LixAl4clusters: Ring Current Patterns versus Electron Counting. Phys. Chem. Chem. Phys. 6, 285–288. doi:10.1039/B311559N
Havenith, R. W., Proft, F. De., Fowler, P. W., and Geerlings, P. (2005). σ-Aromaticity in H3+ and Li3+: Insights from Ring-Current Maps. Chem. Phys. Lett. 407, 391–396. doi:10.1016/j.cplett.2005.03.099
Hay, P. J., and Wadt, W. R. (1985b). Ab Initio effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 82, 299–310. doi:10.1063/1.448975
Hay, P. J., and Wadt, W. R. (1985a). Ab Initio effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 82, 270–283. doi:10.1063/1.448799
Heine, T., Zhechkov, L., and Seifert, G. (2004). Hydrogen Storage by Physisorption on Nanostructured Graphite plateletsElectronic Supplementary Information (ESI) Available: Fig. 1S: Potential Energy Surface of H2 Parallel to Benzene at the MP2 Level. See. Phys. Chem. Chem. Phys. 6, 980–984. doi:10.1039/B316209E
Hennig, A., Ghale, G., and Nau, W. M. (2007). Effects of Cucurbit[7]uril on Enzymatic Activity. Chem. Commun. 16, 1614–1616. doi:10.1039/B618703J
Herr, W. A. D. (1993). The Physics of Simple Metal Clusters: Experimental Aspects and Simple Models. Rev. Mod. Phys. 65, 611. doi:10.1103/RevModPhys.65.611
Hoffman, B. M., Lukoyanov, D., Dean, D. R., and Seefeldt, L. C. (2013). Nitrogenase: a Draft Mechanism. Acc. Chem. Res. 46, 587–595. doi:10.1021/ar300267m
Holland, J. H. (1992). Genetic Algorithms. Sci. Am. 267, 66–72. doi:10.1038/scientificamerican0792-66
Hu, Y., Xiang, S., Zhang, W., Zhang, Z., Wang, L., Bai, J., et al. (2009). A New MOF-505 Analog Exhibiting High Acetylene Storage. Chem. Commun. 48, 7551–7553. doi:10.1039/B917046D
Hückel, E. (1931). Quantentheoretische Beiträge Zum Benzolproblem. Z. Phys. 70, 204–286. doi:10.1007/BF01339530
Hutter, J., and Lüthi, H. P. (1994). The Molecular Structure of C6: A Theoretical Investigation. J. Chem. Phys. 101, 2213–2216. doi:10.1063/1.467661
Iyengar, S. S., Schlegel, H. B., Millam, J. M., A. Voth, G. G., Scuseria, G. E., and Frisch, M. J. (2001). Ab Initio molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals. II. Generalizations Based on Mass-Weighting, Idempotency, Energy Conservation and Choice of Initial Conditions. J. Chem. Phys. 115, 10291–10302. doi:10.1063/1.1416876
Jana, G., Mitra, A., Pan, S., Sural, S., and Chattaraj, P. K. (2019). Modified Particle Swarm Optimization Algorithms for the Generation of Stable Structures of Carbon Clusters, Cn (N = 3-6, 10). Front. Chem. 7, 485. doi:10.3389/fchem.2019.00485
Jena, P., and Castleman, A. W. (2010). Introduction to Atomic Clusters. Sci. Technol. At. Mol. Condensed Matter Biol. Syst. 1, 1–36. doi:10.1016/B978-0-444-53440-8.00001-X
J. F. Corrigan, and S. Dehnen (Editors) (2017). Clusters-contemporary Insight in Structure and Bonding (Cham: Springer), 174.
Jiménez-Halla, J. O., Islas, R., Heine, T., and Merino, G. (2010). B19-: an Aromatic Wankel Motor. Angew. Chem. Int. Ed. Engl. 49, 5668–5671. doi:10.1002/anie.201001275
Jiménez-Vázquez, H. A., Tamariz, J., and Cross, R. J. (2001). Binding Energy in and Equilibrium Constant of Formation for the Dodecahedrane Compounds He@C20H20 and Ne@C20H20. J. Phys. Chem. A. 105, 1315–1319. doi:10.1021/jp0027243
Jin, Y., Voss, B. A., Jin, A., Long, H., Noble, R. D., and Zhang, W. (2011). Highly CO2-Selective Organic Molecular Cages: What Determines the CO2Selectivity. J. Am. Chem. Soc. 133, 6650–6658. doi:10.1021/ja110846c
Jin, Y., Voss, B. A., Noble, R. D., and Zhang, W. (2010). A Shape-Persistent Organic Molecular Cage with High Selectivity for the Adsorption of CO2 over N2. Angew. Chem. 122, 6492–6495. doi:10.1002/ange.201001517
Karaboga, D., and Basturk, B. (2007). A Powerful and Efficient Algorithm for Numerical Function Optimization: Artificial Bee Colony (ABC) Algorithm. J. Glob. Optim. 39, 459–471. doi:10.1007/s10898-007-9149-x
Khatua, M., Pan, S., and Chattaraj, P. K. (2014a). Confinement Induced Binding of noble Gas Atoms. J. Chem. Phys. 140, 164306. doi:10.1063/1.4871800
Khatua, M., Pan, S., and Chattaraj, P. K. (2014b). Confinement of (HF)2 in Cn (n= 60, 70, 80, 90).... Cages. Chem. Phys. Lett. 616-617, 49–54. doi:10.1016/j.cplett.2014.10.025
Khatua, S., Roy, D. R., Bultinck, P., Bhattacharjee, M., and Chattaraj, P. K. (2008). Aromaticity in Cyclic Alkali Clusters. Phys. Chem. Chem. Phys. 10, 2461–2474. doi:10.1039/B718176K
Kim, H., Kim, Y., Yoon, M., Lim, S., Park, S. M., Seo, G., et al. (2010). Highly Selective Carbon Dioxide Sorption in an Organic Molecular Porous Material. J. Am. Chem. Soc. 132, 12200–12202. doi:10.1021/ja105211w
Kitano, M., Inoue, Y., Yamazaki, Y., Hayashi, F., Kanbara, S., Matsuishi, S., et al. (2012). Ammonia Synthesis Using a Stable Electride as an Electron Donor and Reversible Hydrogen Store. Nat. Chem 4, 934–940. doi:10.1038/nchem.1476
Klontzas, E., Tylianakis, E., and Froudakis, G. E. (2008). Hydrogen Storage in 3D Covalent Organic Frameworks. A Multiscale Theoretical Investigation. J. Phys. Chem. C 112, 9095–9098. doi:10.1021/jp711326g
Koster, A. M., Geudtner, G., Calaminici, P., Casida, M. E., Dominguez, V. D., Flores-Moreno, R., and Salahub, D. R. (2011). DeMon2K, Version 3. Cinvestav, México: The deMon Developers.
Kuc, A., Zhechkov, L., Patchkovskii, S., Seifert, G., and Heine, T. (2007). Hydrogen Sieving and Storage in Fullerene Intercalated Graphite. Nano Lett. 7, 1–5. doi:10.1021/nl0619148
Kuznetsov, A. E., Birch, K. A., Boldyrev, A. I., Li, X., Zhai, H. J., and Wang, L. S. (2003). All-Metal Antiaromatic Molecule: Rectangular Al44- in the Li3Al4- Anion. Science 300, 622–625. doi:10.1126/science.1082477
Lagona, J., Mukhopadhyay, P., Chakrabarti, S., and Isaacs, L. (2005). The Cucurbit[n]uril Family. Angew. Chem. Int. Ed. 44 (31), 4844–4870. doi:10.1002/anie.200460675
Latysheva, N., Junker, V. L., Palmer, W. J., Codd, G. A., and Barker, D. (2012). The Evolution of Nitrogen Fixation in Cyanobacteria. Bioinformatics 28 (5), 603–606. doi:10.1093/bioinformatics/bts008
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 37, 785–789. doi:10.1103/PhysRevB.37.785
Lee, H., Lee, J.-w., Kim, D. Y., Park, J., Seo, Y.-T., Zeng, H., et al. (2005). Tuning Clathrate Hydrates for Hydrogen Storage. Nature 434, 743–746. doi:10.1038/nature03457
Lee, H., Lee, J.-w., Kim, D. Y., Park, J., Seo, Y.-T., Zeng, H., Moudrakovski, I. L., Ratcliffe, C. I., and Ripmeester, J. A. (2010). “Tuning Clathrate Hydrates for Hydrogen Storage,” in Materials For Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, 285–288. doi:10.1142/9789814317665_0042
Lee, K., Kim, S. W., Toda, Y., Matsuishi, S., and Hosono, H. (2013). Dicalcium Nitride as a Two-Dimensional Electride with an Anionic Electron Layer. Nature 494, 336–340. doi:10.1038/nature11812
Li, F., Zhao, J., Johansson, B., and Sun, L. (2010). Improving Hydrogen Storage Properties of Covalent Organic Frameworks by Substitutional Doping. Int. J. Hydrogen Energ. 35, 266–271. doi:10.1016/j.ijhydene.2009.10.061
Li, S., and Jena, P. (2008). Li-and B-Decorated Cis-Polyacetylene: A Computational Study. Phys. Rev. B. 77, 193101. doi:10.1103/physrevb.77.193101
Li, X., Kuznetsov, A. E., Zhang, H. F., Boldyrev, A. I., and Wang, L. S. (2001). Observation of All-Metal Aromatic Molecules. Science 291, 859–861. doi:10.1126/science.291.5505.859
Li, Y., Wu, D., and Li, Z.-R. (2008). Compounds of Superatom Clusters: Preferred Structures and Significant Nonlinear Optical Properties of the BLi6-X (X = F, LiF2, BeF3, BF4) Motifs. Inorg. Chem. 47, 9773–9778. doi:10.1021/ic800184z
Li, Z.-J., Wang, F.-F., Li, Z.-R., Xu, H.-L., Huang, X.-R., Wu, D., et al. (2009). Large Static First and Second Hyperpolarizabilities Dominated by Excess Electron Transition for Radical Ion Pair Salts M2˙+TCNQ˙−(M = Li, Na, K). Phys. Chem. Chem. Phys. 11, 402–408. doi:10.1039/B809161G
Liu, L., Moreno, D., Osorio, E., Castro, A. C., Pan, S., Chattaraj, P. K., et al. (2016). Structure and Bonding of IrB12−: Converting a Rigid boron B12 Platelet to a Wankel Motor. RSC Adv. 6, 27177–27182. doi:10.1039/C6RA02992B
Liu, S., and Gibb, B. C. (2008). High-definition Self-Assemblies Driven by the Hydrophobic Effect: Synthesis and Properties of a Supramolecular Nanocapsule. Chem. Commun. 32, 3709–3716. doi:10.1039/B805446K
Lü, J., Perez-Krap, C., Suyetin, M., Alsmail, N. H., Yan, Y., Yang, S., et al. (2014). A Robust Binary Supramolecular Organic Framework (SOF) with High CO2 Adsorption and Selectivity. J. Am. Chem. Soc. 136, 12828–12831. doi:10.1021/ja506577g
Lu, T., and Chen, F. (2012). Multiwfn: a Multifunctional Wavefunction Analyzer. J. Comput. Chem. 33, 580–592. doi:10.1002/jcc.22885
Lu, Y., Li, J., Tada, T., Toda, Y., Ueda, S., Yokoyama, T., et al. (2016). Water Durable Electride Y5Si3: Electronic Structure and Catalytic Activity for Ammonia Synthesis. J. Am. Chem. Soc. 138, 3970–3973. doi:10.1021/jacs.6b00124
Lu, Y., Wang, J., Li, J., Wu, J., Kanno, S., Tada, T., et al. (2018). Realization of Mott-insulating Electrides in Dimorphic Yb5Sb3. Phys. Rev. B 98, 125128. doi:10.1103/PhysRevB.98.125128
Mao, W. L., and Mao, H.-k. (2004). Hydrogen Storage in Molecular Compounds. Proc. Natl. Acad. Sci. 101, 708–710. doi:10.1073/pnas.0307449100
Mao, W. L., Mao, H. K., Goncharov, A. F., Struzhkin, V. V., Guo, Q., Hu, J., and Zhao, Y. (2002). Hydrogen Clusters in Clathrate Hydrate. Science 297, 2247–2249. doi:10.1126/science.1075394
Martin, J. M. L., and Taylor, P. R. (1996). Structure and Vibrations of Small Carbon Clusters from Coupled-Cluster Calculations. J. Phys. Chem. 100, 6047–6056. doi:10.1021/jp952471r
Mastalerz, M., Schneider, M. W., Oppel, I. M., and Presly, O. (2011). A Salicylbisimine Cage Compound with High Surface Area and Selective CO2/CH4 Adsorption. Angew. Chem. Int. Ed. 50, 1046–1051. doi:10.1002/anie.201005301
Matsuishi, S., Toda, Y., Miyakawa, M., Hayashi, K., Kamiya, T., Hirano, M., et al. (2003). High-Density Electron Anions in a Nanoporous Single Crystal: [Ca24Al28O64]4+(4e-). Science. 301, 626–629. doi:10.1126/science.1083842
McKeown, N. B., Gahnem, B., Msayib, K. J., Budd, P. M., Tattershall, C. E., Mahmood, K., et al. (2006). Towards Polymer-Based Hydrogen Storage Materials: Engineering Ultramicroporous Cavities within Polymers of Intrinsic Microporosity. Angew. Chem. Int. Ed. 45, 1804–1807. doi:10.1002/anie.200504241
McLean, A. D., and Chandler, G. S. (1980). Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z=11-18. J. Chem. Phys. 72, 5639–5648. doi:10.1063/1.438980
Mitikiri, P., Jana, G., Sural, S., and Chattaraj, P. K. (2018). A Machine Learning Technique toward Generating Minimum Energy Structures of Small boron Clusters. Int. J. Quan. Chem. 118, e25672. doi:10.1002/qua.25672
Mitoraj, M. P., Michalak, A., and Ziegler, T. (2009). A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theor. Comput. 5, 962–975. doi:10.1021/ct800503d
Mitra, A., Jana, G., Agrawal, P., Sural, S., and Chattaraj, P. K. (2020). Integrating Firefly Algorithm with Density Functional Theory for Global Optimization of Al42− Clusters. Theor. Chem. Acc. 139, 1–12. doi:10.1007/s00214-020-2550-y
Mitra, A., Jana, G., Pal, R., Gaikwad, P., Sural, S., and Chattaraj, P. K. (2021). Determination of Stable Structure of a Cluster Using Convolutional Neural Network and Particle Swarm Optimization. Theor. Chem. Acc. 140, 1–12. doi:10.1007/s00214-021-02726-z
Moreno, D., Pan, S., Zeonjuk, L. L., Islas, R., Osorio, E., Martínez-Guajardo, G., et al. (2014). B182−: a Quasi-Planar Bowl Member of the Wankel Motor Family. Chem. Commun. 50, 8140–8143. doi:10.1039/C4CC02225D
Morokuma, K. (1971). Molecular Orbital Studies of Hydrogen Bonds. III. C=O···H-O Hydrogen Bond in H2CO···H2O and H2CO···2H2O. J. Chem. Phys. 55, 1236–1244. doi:10.1063/1.1676210
Msayib, K. J., Book, D., Budd, P. M., Chaukura, N., Harris, K. D. M., Helliwell, M., et al. (2009). Nitrogen and Hydrogen Adsorption by an Organic Microporous Crystal. Angew. Chem. 121, 3323–3327. doi:10.1002/ange.200900234
Muhammad, S., Xu, H., Liao, Y., Kan, Y., and Su, Z. (2009). Quantum Mechanical Design and Structure of the Li@B10H14 Basket with a Remarkably Enhanced Electro-Optical Response. J. Am. Chem. Soc. 131, 11833–11840. doi:10.1021/ja9032023
Muhammad, S., Xu, H., and Su, Z. (2011). Capturing a Synergistic Effect of a Conical Push and an Inward Pull in Fluoro Derivatives of Li@B10H14Basket: Toward a Higher Vertical Ionization Potential and Nonlinear Optical Response. J. Phys. Chem. A. 115, 923–931. doi:10.1021/jp110401f
Norbye, J. P. (1971). The Wankel Engine: Design, Development, Applications. 1st Ed. Philadelphia, PA: Chilton Book Company.
Pal, R., and Chattaraj, P. K. (2021). Possible Effects of Fluxionality of a Cavitand on its Catalytic Activity through Confinement. Phys. Chem. Chem. Phys. 23, 15817–15834. doi:10.1039/D1CP01826D
Pan, S., Ghara, M., Kar, S., Zarate, X., Merino, G., and Chattaraj, P. K. (2018). Noble Gas Encapsulated B40 Cage. Phys. Chem. Chem. Phys. 20, 1953–1963. doi:10.1039/C7CP07890K
Pan, S., Giri, S., and Chattaraj, P. K. (2012a). A Computational Study on the Hydrogen Adsorption Capacity of Various Lithium-Doped boron Hydrides. J. Comput. Chem. 33, 425–434. doi:10.1002/jcc.21985
Pan, S., Jana, G., Gupta, A., Merino, G., and Chattaraj, P. K. (2017). Endohedral Gas Adsorption by Cucurbit[7]uril: a Theoretical Study. Phys. Chem. Chem. Phys. 19, 24448–24452. doi:10.1039/C7CP03984K
Pan, S., Mandal, S., and Chattaraj, P. K. (2015). Cucurbit[6]uril: a Possible Host for noble Gas Atoms. J. Phys. Chem. B 119, 10962–10974. doi:10.1021/acs.jpcb.5b01396
Pan, S., Merino, G., and Chattaraj, P. K. (2012b). The Hydrogen Trapping Potential of Some Li-Doped star-like Clusters and Super-alkali Systems. Phys. Chem. Chem. Phys. 14, 10345–10350. doi:10.1039/C2CP40794A
Pan, S., Mondal, S., and Chattaraj, P. K. (2013a). Cucurbiturils as Promising Hydrogen Storage Materials: a Case Study of Cucurbit[7]uril. New J. Chem. 37, 2492–2499. doi:10.1039/C3NJ00399J
Pan, S., Moreno, D., Merino, G., and Chattaraj, P. K. (2014). Stability of Noble-Gas-Bound SiH3+Clusters. ChemPhysChem 15, 3554–3564. doi:10.1002/cphc.201402370
Pan, S., Solà, M., and Chattaraj, P. K. (2013b). On the Validity of the Maximum Hardness Principle and the Minimum Electrophilicity Principle During Chemical Reactions. J. Phys. Chem. A 117, 1843–1852. doi:10.1021/jp312750n
Pauling, L., and Sherman, J. (1933). The Nature of the Chemical Bond. VI. The Calculation from Thermochemical Data of the Energy of Resonance of Molecules Among Several Electronic Structures. J. Chem. Phys. 1, 606–617. doi:10.1063/1.1749335
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Perdew, J. P., Burke, K., and Ernzerhof, M. (1997). Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396. doi:10.1103/PhysRevLett.78.1396
Perdew, J. P. (1986a). Density-functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 33, 8822–8824. doi:10.1103/PhysRevB.33.8822
Perdew, J. P. (1986b). Erratum: Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 34, 7406. doi:10.1103/PhysRevB.34.7406
Pham, D. T., Ghanbarzadeh, A., Koc, E., Otri, S., Rahim, S., and Zaidi, M. (2005). “The Bees Algorithm,” in Technical Note (UK: Manufacturing Engineering Centre, Cardiff University).
P. J. Stang, and F. Diederich (Editors) (2008). Modern Acetylene Chemistry (Hoboken, New Jersey, USA: John Wiley & Sons).
Pless, V., Suter, H. U., and Engels, B. (1994). Ab Initio study of the Energy Difference between the Cyclic and Linear Forms of the C6 Molecule. J. Chem. Phys. 101, 4042–4048. doi:10.1063/1.467521
Popov, I. A., Li, W.-L., Piazza, Z. A., Boldyrev, A. I., and Wang, L.-S. (2014). Complexes between Planar Boron Clusters and Transition Metals: A Photoelectron Spectroscopy and Ab Initio Study of CoB12- and RhB12-. J. Phys. Chem. A. 118, 8098–8105. doi:10.1021/jp411867q
Qu, J., Zhu, S., Zhang, W., and Zhu, Q. (2019). Electrides with Dinitrogen Ligands. ACS Appl. Mater. Inter. 11, 5256–5263. doi:10.1021/acsami.8b18676
Raghavachari, K., Binkley, J. S., Seeger, R., and Pople, J. A. (1980). Self-Consistent Molecular Orbital Methods. 20. Basis Set for Correlated Wave-Functions. J. Chem. Phys. 72, 650–654. doi:10.1063/1.438955
Raghavachari, K., and Binkley, J. S. (1987). Structure, Stability, and Fragmentation of Small Carbon Clusters. J. Chem. Phys. 87, 2191–2197. doi:10.1063/1.453145
Reed, A. E., Curtiss, L. A., and Weinhold, F. (1988). Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 88, 899–926. doi:10.1021/cr00088a005
Reed, A. E., Weinstock, R. B., and Weinhold, F. (1985). Natural Population Analysis. J. Chem. Phys. 83, 735–746. doi:10.1063/1.449486
Rosi, N. L., Eckert, J., Eddaoudi, M., Vodak, D. T., Kim, J., O'Keeffe, M., et al. (2003). Hydrogen Storage in Microporous Metal-Organic Frameworks. Science 300, 1127–1129. doi:10.1126/science.1083440
Rowsell, J. L. C., and Yaghi, O. M. (2005). Strategies for Hydrogen Storage in Metal-Organic Frameworks. Angew. Chem. Int. Ed. 44, 4670–4679. doi:10.1002/anie.200462786
Roy, D. R., and Chattaraj, P. K. (2008). Reactivity, Selectivity, and Aromaticity of Be32- and its Complexes. J. Phys. Chem. A. 112, 1612–1621. doi:10.1021/jp710820c
Sabin, J. R., and Brandas, E. J. (2009). Advances in Quantum Chemistry: Theory of Confined Quantum Systems-Part One. Cambridge: Academic Press.
Saha, R., and Chattaraj, P. K. (2018). Activation of Small Molecules (H2, CO2, N2O, CH4, and C6H6) by a Porphyrinoid-Based Dimagnesium(I) Complex, an Electride. ACS Omega 3, 17199–17211. doi:10.1021/acsomega.8b03006
Saha, R., Das, P., and Chattaraj, P. K. (2019). A Complex Containing Four Magnesium Atoms and Two Mg-Mg Bonds Behaving as an Electride. Eur. J. Inorg. Chem. 2019, 4105–4111. doi:10.1002/ejic.201900813
Saha, R., Kar, S., Pan, S., Martínez-Guajardo, G., Merino, G., and Chattaraj, P. K. (2017). A Spinning Umbrella: Carbon Monoxide and Dinitrogen Bound MB12- Clusters (M = Co, Rh, Ir). J. Phys. Chem. A. 121, 2971–2979. doi:10.1021/acs.jpca.6b12232
Saha, R., Pan, S., Merino, G., and Chattaraj, P. K. (2015). Comparative Study on the Noble-Gas Binding Ability of BeX Clusters (X = SO4, CO3, O). J. Phys. Chem. A. 119, 6746–6752. doi:10.1021/acs.jpca.5b03888
Santos, J. C., Andres, J., Aizman, A., and Fuentealba, P. (2005). An Aromaticity Scale Based on the Topological Analysis of the Electron Localization Function Including σ and π Contributions. J. Chem. Theor. Comput. 1, 83–86. doi:10.1021/ct0499276
Schettino, V., and Bini, R. (2007). Constraining Molecules at the Closest Approach: Chemistry at High Pressure. Chem. Soc. Rev. 36, 869–880. doi:10.1039/B515964B
Schlegel, H. B., Iyengar, S. S., Li, X., Millam, J. M., Voth, G. A., Scuseria, G. E., et al. (2002). Ab Initio molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals. III. Comparison with Born-Oppenheimer Dynamics. J. Chem. Phys. 117, 8694–8704. doi:10.1063/1.1514582
Schlegel, H. B., Millam, J. M., Iyengar, S. S., Voth, G. A., Daniels, A. D., Scuseria, G. E., et al. (2001). Ab Initiomolecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals. J. Chem. Phys. 114, 9758–9763. doi:10.1063/1.1372182
Schleyer, P. v. R., Maerker, C., Dransfeld, A., Jiao, H., and van Eikema Hommes, N. J. R. (1996). Nucleus-independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 118, 6317–6318. doi:10.1021/ja960582d
Schneider, M. W., Oppel, I. M., Ott, H., Lechner, L. G., Hauswald, H.-J. S., Stoll, R., et al. (2012). Periphery-Substituted [4+6] Salicylbisimine Cage Compounds with Exceptionally High Surface Areas: Influence of the Molecular Structure on Nitrogen Sorption Properties. Chem. Eur. J. 18, 836–847. doi:10.1002/chem.201102857
Sekhar, P., Ghosh, A., Joshi, M., and Ghanty, T. K. (2017). Noble Gas Encapsulated Endohedral Zintl Ions Ng@Pb122- and Ng@Sn122- (Ng = He, Ne, Ar, and Kr): A Theoretical Investigation. J. Phys. Chem. C 121, 11932–11949. doi:10.1021/acs.jpcc.7b03294
Sergeeva, A. P., Popov, I. A., Piazza, Z. A., Li, W.-L., Romanescu, C., Wang, L.-S., et al. (2014). Understanding Boron through Size-Selected Clusters: Structure, Chemical Bonding, and Fluxionality. Acc. Chem. Res. 47, 1349–1358. doi:10.1021/ar400310g
Sharma, S., Kurashige, W., Niihori, Y., and Negishi, Y. (2017). Nanocluster Science. Supra-materials Nanoarchitectonics, 3–32. doi:10.1016/B978-0-323-37829-1.00001-8
Srinivasu, K., Ghosh, S. K., Das, R., Giri, S., and Chattaraj, P. K. (2012). Theoretical Investigation of Hydrogen Adsorption in All-Metal Aromatic Clusters. RSC Adv. 2, 2914–2922. doi:10.1039/C2RA00643J
Srivastava, R. (2021). Application of Optimization Algorithms in Clusters. Front. Chem. 9, 637286. doi:10.3389/fchem.2021.637286
Staroverov, V. N., Scuseria, G. E., Tao, J., and Perdew, J. P. (2003). Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 119, 12129–12137. doi:10.1063/1.1626543
Sun, Q., Wang, Q., and Jena, P. (2005). Storage of Molecular Hydrogen in B−N Cage: Energetics and Thermal Stability. Nano Lett. 5, 1273–1277. doi:10.1021/nl050385p
Tao, J., Perdew, J. P., Staroverov, V. N., and Scuseria, G. E. (2003). Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 91, 146401. doi:10.1103/PhysRevLett.91.146401
Tsukuda, T., and Hakkinen, H. (2015). “Protected Metal Clusters: From Fundamentals to Applications,” in Frontiers of Nanoscience. 1st Ed. Editors R. E. Palmer (Amsterdam: Elsevier), Vol. 9.
Thrash, T. P., Cagle, D. W., Alford, J. M., Wright, K., Ehrhardt, G. J., Mirzadeh, S., et al. (1999). Toward Fullerene-Based Radiopharmaceuticals: High-Yield Neutron Activation of Endohedral 165Ho Metallofullerenes. Chem. Phys. Lett. 308, 329–336. doi:10.1016/S0009-2614(99)00581-3
Toda, Y., Yanagi, H., Ikenaga, E., Kim, J. J., Kobata, M., Ueda, S., et al. (2007). Work Function of a Room-Temperature, Stable Electride [Ca24Al28O64]4+(e-)4. Adv. Mater. 19, 3564–3569. doi:10.1002/adma.200700663
Van Orden, A., and Saykally, R. J. (1998). Small Carbon Clusters: Spectroscopy, Structure, and Energetics. Chem. Rev. 98, 2313–2358. doi:10.1021/cr970086n
Van Rossum, G., and Drake, F. L. (2009). Python 3 Reference Manual. Scotts Valley, CA: CreateSpace. doi:10.1201/9781420049114.ch23
Vitillo, J. G., Regli, L., Chavan, S., Ricchiardi, G., Spoto, G., Dietzel, P. D. C., et al. (2008). Role of Exposed Metal Sites in Hydrogen Storage in MOFs. J. Am. Chem. Soc. 130, 8386–8396. doi:10.1021/ja8007159
Wadt, W. R., and Hay, P. J. (1985). Ab Initio effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 82, 284–298. doi:10.1063/1.448800
Wagner, B. D., Stojanovic, N., Day, A. I., and Blanch, R. J. (2003). Host Properties of Cucurbit[7]uril: Fluorescence Enhancement of Anilinonaphthalene Sulfonates. J. Phys. Chem. B 107, 10741–10746. doi:10.1021/jp034891j
Wales, D. J., and Doye, J. P. K. (1997). Global Optimization by basin-hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. J. Phys. Chem. A. 101, 5111–5116. doi:10.1021/jp970984n
Wang, J.-J., Zhou, Z.-J., Bai, Y., Liu, Z.-B., Li, Y., Wu, D., et al. (2012). The Interaction between Superalkalis (M3O, M = Na, K) and a C20F20 Cage Forming Superalkali Electride Salt Molecules with Excess Electrons inside the C20F20 Cage: Dramatic Superalkali Effect on the Nonlinear Optical Property. J. Mater. Chem. 22, 9652–9657. doi:10.1039/C2JM15405F
Wang, J., Hanzawa, K., Hiramatsu, H., Kim, J., Umezawa, N., Iwanaka, K., et al. (2017). Exploration of Stable Strontium Phosphide-Based Electrides: Theoretical Structure Prediction and Experimental Validation. J. Am. Chem. Soc. 139, 15668–15680. doi:10.1021/jacs.7b06279
Ward, D. L., Huang, R. H., and Dye, J. L. (1988). Structures of Alkalides and Electrides. I. Structure of Potassium cryptand[2.2.2] Electride. Acta Crystallogr. C 44, 1374–1376. doi:10.1107/s0108270188002847
Watts, J. D., Gauss, J., Stanton, J. F., and Bartlett, R. J. (1992). Linear and Cyclic Isomers of C4. A Theoretical Study with Coupled‐cluster Methods and Large Basis Sets. J. Chem. Phys. 97, 8372–8381. doi:10.1063/1.463407
Weigend, F., and Ahlrichs, R. (2005). Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. doi:10.1039/B508541A
Wiberg, K. B. (1968). Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 24, 1083–1096. doi:10.1016/0040-4020(68)88057-3
Wilson, L. J., Cagle, D. W., Thrash, T. P., Kennel, S. J., Mirzadeh, S., Alford, J. M., et al. (1999). Metallofullerene Drug Design. Coord. Chem. Rev. 190–192, 199–207. doi:10.1016/S0010-8545(99)00080-6
Woodley, S. M., Battle, P. D., Gale, J. D., and Richard A. Catlow, C. (1999). The Prediction of Inorganic crystal Structures Using a Genetic Algorithm and Energy Minimisation. Phys. Chem. Chem. Phys. 1, 2535–2542. doi:10.1039/A901227C
Wu, X., Gao, Y., and Zeng, X. C. (2008). Hydrogen Storage in Pillared Li-Dispersed boron Carbide Nanotubes. J. Phys. Chem. C 112, 8458–8463. doi:10.1021/jp710022y
Xie, Q., Huang, R. H., Ichimura, A. S., Phillips, R. C., Pratt, W. P., and Dye, J. L. (2000). Structure and Properties of a New Electride, Rb+(cryptand[2.2.2])e-. J. Am. Chem. Soc. 122, 6971–6978. doi:10.1021/ja9943445
Xu, H.-L., Li, Z.-R., Wu, D., Wang, B.-Q., Li, Y., Gu, F. L., et al. (2007). Structures and Large NLO Responses of New Electrides: Li-Doped Fluorocarbon Chain. J. Am. Chem. Soc. 129, 2967–2970. doi:10.1021/ja068038k
Yandulov, D. V., and Schrock, R. R. (2003). Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum center. Science 301, 76–78. doi:10.1126/science.1085326
Yang, C.-J., Leveen, L., and King, K. (2015). Ethane as a Cleaner Transportation Fuel. Environ. Sci. Technol. 49, 3263–3264. doi:10.1021/acs.est.5b00575
Yang, X. S. (2010). Firefly Algorithm, Stochastic Test Functions and Design Optimisation. Int. J. Bio-Inspired Comput. 2, 78–84. doi:10.1504/IJBIC.2010.032124
Yuan, G.-N., Zhang, L.-N., Liu, L.-Q., and Wang, K. (2014). Passengers' Evacuation in Ships Based on Neighborhood Particle Swarm Optimization. Math. Probl. Eng. 2014, 1–10. doi:10.1155/2014/939723
Zhan, Z.-h., and Zhang, J. (2008). “Adaptive Particle Swarm Optimization,” in International Conference on Ant Colony Optimization and Swarm Intelligence (Berlin, Germany: Springer), 227–234. doi:10.1007/978-3-540-87527-7_21
Zhang, J.-P., and Chen, X.-M. (2009). Optimized Acetylene/carbon Dioxide Sorption in a Dynamic Porous crystal. J. Am. Chem. Soc. 131, 5516–5521. doi:10.1021/ja8089872
Zhang, X., Xiao, Z., Lei, H., Toda, Y., Matsuishi, S., Kamiya, T., et al. (2014). Two-Dimensional Transition-Metal Electride Y2C. Chem. Mater. 26, 6638–6643. doi:10.1021/cm503512h
Zhang, Z., Xiang, S., and Chen, B. (2011). Microporous Metal-Organic Frameworks for Acetylene Storage and Separation. CrystEngComm 13 (20), 5983–5992. doi:10.1039/C1CE05437F
Zhao, Y., Schultz, N. E., and Truhlar, D. G. (2006). Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theor. Comput. 2, 364–382. doi:10.1021/ct0502763
Zhao, Y., and Truhlar, D. G. (2008). The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Account. 120, 215–241. doi:10.1007/s00214-007-0310-x
Zhu, H., Liu, Y., Chen, Y., and Wen, Z. (2010). Cyclodextrins: Promising Candidate media for High-Capacity Hydrogen Adsorption. Appl. Phys. Lett. 96, 054101. doi:10.1063/1.3294631
Zubarev, D. Y., and Boldyrev, A. I. (2011). “Multiple Aromaticity, Multiple Antiaromaticity, and Conflicting Aromaticity in Inorganic Systems,” in Encyclopedia of Inorganic and Bioinorganic Chemistry (Hoboken, NY, USA: John Wiley & Sons). doi:10.1002/9781119951438.eibc0396
Keywords: aromaticity, Electrides, Particle swarm optimization, Firefly algorithm, Confinement, Hydrogen storage, Fluxionality
Citation: Pal R, Poddar A and Chattaraj PK (2021) Atomic Clusters: Structure, Reactivity, Bonding, and Dynamics. Front. Chem. 9:730548. doi: 10.3389/fchem.2021.730548
Received: 25 June 2021; Accepted: 13 July 2021;
Published: 16 August 2021.
Edited by:
Ambrish Kumar Srivastava, Deen Dayal Upadhyay Gorakhpur University, IndiaReviewed by:
Santanab Giri, Haldia Institute of Technology, IndiaCopyright © 2021 Pal, Poddar and Chattaraj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pratim Kumar Chattaraj, cGtjQGNoZW0uaWl0a2dwLmFjLmlu
†ORCID:Pratim Kumar Chattarajorcid.org/0000-0002-5650-7666
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.