 Giovanni Meloni
Giovanni Meloni Andrea Giustini2
Andrea Giustini2- 1Department of Chemistry, University of San Francisco, San Francisco, CA, United States
- 2Department of Physical and Chemical Sciences, Università degli Studi de L’Aquila, L’Aquila, Italy
With the aim of finding a suitable synthesizable superalkali species, using the B3LYP/6-31G* density functional level of theory we provide results for the interaction between the buckminsterfullerene C60 and the superalkali Li3F2. We show that this endofullerene is stable and provides a closed environment in which the superalkali can exist and interact with CO2. It is worthwhile to mention that the optimized Li3F2 structure inside C60 is not the most stable C2v isomer found for the “free” superalkali but the D3h geometry. The binding energy at 0 K between C60 and Li3F2 (D3h) is computed to be 119 kJ mol−1. Once CO2 is introduced in the endofullerene, it is activated, and the
Introduction
In 1985, Kroto and co-workers discovered an extremely stable cluster consisting of 60 carbon atoms during a study of long-chain carbon molecules. (Kroto et al., 1985). This cluster, called fullerene, has a football shape with 12 pentagonal and 20 hexagonal rings. (Kroto et al., 1985). Shortly after, a study presenting successful formation of fullerenes with a lanthanum atom trapped in the cavity of C60, called endofullerene, C60La, was published. (Heath et al., 1985). Another consequent experiment proved the stability of C60La+ against H2, O2, NO, and NH3. (Weiss et al., 1988). This suggested that the lanthanum atom can be “protected” by being encapsulated in the fullerene. Since then, a number of studies focusing on novel properties of C60 and its interactions with other species have been carried out. (Ruoff et al., 1993; Diederich and Gómez-López, 1999; Bakry et al., 2007; Mignolet et al., 2013; Wang et al., 2014; Elliott et al., 2018; Kouřil et al., 2018) Some investigations concentrated on medical applications, hydrogen storage, and various endofullerene. (Wang et al., 2013; Srivastava et al., 2016; Srivastava et al., 2017; Elliott et al., 2018; Kouřil et al., 2018). Due to the fullerene's unique cage-like cavity, a procedure called molecular surgery can be performed to entrap an atom or molecule. (Murata et al., 2006; Krachmalnicoff et al., 2016). Utilizing this technique, encapsulation of molecular hydrogen and HF were successfully achieved. (Murata et al., 2006; Krachmalnicoff et al., 2016). In addition, a recent paper by Jana and Chattaraj (2020) describes the effects of a molecular reaction environment, dodecahedrane, on the He dimer bonding. Also, theoretical studies of a new type of endofullerenes with superalkali have been performed. (Srivastava et al., 2016; Srivastava et al., 2017) Superalkalis are clusters with very low adiabatic ionization energies. (Gutsev and Boldyrev, 1982; Gutsev and Boldyrev, 1983; Gutsev and Boldyrev, 1987; Lia et al., 1988; Gutsev and Boldyrev, 1990; Tong et al., 2013a; Tong et al., 2013b). The first and most common superalkalis have the formula Mk+1L, where M is an alkali atom with valence k and L is an electronegative atom. (Gutsev and Boldyrev, 1985; Gutsev and Boldyrev, 1987; Zhao et al., 2017). Nambiar et al. (2021) showed the importance of these compounds through a density functional computational study to improve the efficacy of redox reactions.
The concentration of carbon dioxide (CO2) in the atmosphere has been increasing constantly since the early 20th century with rapid industrialization. (D’Amato and Akdis, 2020; Kuzovkin and Semenov, 2020). This is a globally recognized issue as CO2 contributes significantly to the greenhouse effect and the acidification of the oceans. (Crowley and Berner, 2001; Hönisch et al., 2012). Increment of atmospheric temperature causes climate change that threatens the overall ecosystem. To capture CO2 molecules present in the air, various methods have been employed such as packed column of monoethanolamine and metal-organic frameworks. (Lv et al., 2015; Li et al., 2019; Li et al., 2020). The next step is activating CO2 molecules and converting them to value-added chemicals such as hydrocarbon fuels. (Hu et al., 2013). The activation of CO2 is extremely complex due to its stability and much efforts have been made by researchers to directly convert it into liquid hydrocarbons, useful for the aviation sector as novel jet fuels (Boreriboon et al., 2018; Vogt et al., 2019; Yao et al., 2020) or into oxygenates, such as ethanol. (Song et al., 2016; Bai et al., 2017; Wang et al., 2018). This conversion, whether it involves a direct CO2 hydrogenation route or not, entails the usage of metal-based catalysts to ensure an overall reasonable efficiency. (Yao et al., 2020).
In our previous studies, successful activation of CO2 with a superalkali species, Li3F2, were presented. (Park and Meloni, 2017). The computational study showed charge transfer from Li3F2 to CO2, which indicates migration of the unpaired electron from Li3F2 to CO2. The activated CO2 showed geometric change such as bent
Computational Methods
Geometries and total electronic energies of the investigated species were calculated at the B3LYP/6-31G* level of theory (Becke, 1988; Lee et al., 1988) using the computational software Gaussian09. (Frisch et al., 2016). B3LYP is one of the most commonly used density functional theory (DFT) methods that employs a three-parameter exchange functional developed by Becke (1992) and Becke (1993) with a correlational functional proposed by Lee, Yang, and Parr (LYP) Becke (1988) to approximate the exchange-correlation energy. The B3LYP/6-31G* level has been employed to study endofullerene systems because it yields reliable geometries and energies. (Wang et al., 2013; Srivastava et al., 2016; Srivastava et al., 2017). Partial atomic charges are calculated based on the Mulliken population analysis (Mulliken, 1955) and natural bond orbital (NBO) population analysis. (Reed et al., 1985).
The adiabatic ionization energy (AIE) is calculated by taking the zero-point energy corrected electronic energy difference between the optimized neutral and cation, whereas the adiabatic electron affinity (AEA) is obtained by subtracting the zero-point-energy corrected electronic energy of the optimized anion and neutral. All the optimized structures have real vibrational frequencies and their Cartesian coordinates have been reported in the Supplementary Material.
Results and Discussion
The main intent of this computational investigation is to study the interactions relevant to the reduction of CO2 by the superalkali Li3F2 inside our molecular reaction vessel, i.e., C60, and see how this environment affects the CO2 activation. The system is fairly large and, therefore, computationally challenging to investigate. We have analyzed the possible interactions between the fullerene and the two reactants, CO2 and Li3F2. All the computed energetics are reported in Table 1 together with the available literature (experimental and computed) values.
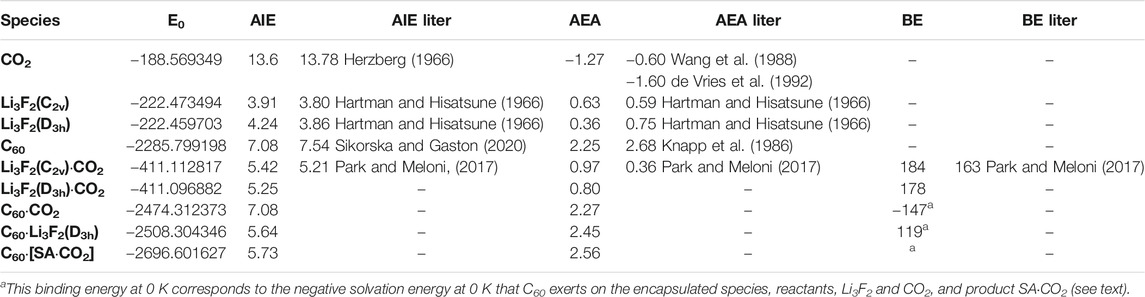
TABLE 1. Energetics of all the species relevant to this study calculated at the B3LYP/6-31G* level of theory. The zero-point-energy corrected total electronic energy (E0) is in Hartree, the adiabatic ionization energy (AIE) and adiabatic electron affinity are in eV, and the binding energy at 0 K is in kJ mol−1. [SA⋅CO2] stays for the endo superalkali⋅CO2 complex.
Figure 1 reports the optimized geometries for CO2 and CO2−, Li3F2(C2v), Li3F2(D3h) and their cations. The structures of CO2 and CO2− reproduce well the literature experimental values for both bond distances and bond angles. In fact, for CO2 we have rC-O = 1.17 Å (1.16 Å) (Herzberg, 1966) and for CO2− we have rC-O = 1.25 Å (1.25 Å) (Hartman and Hisatsune, 1966) and ∠OCO° = 134° (127 ± 7°) . (Hartman and Hisatsune, 1966). Both the geometries of the lowest energy Li3F2(C2v) isomer, trigonal bipyramidal Li3F2(D3h), and their cations are in agreement with our previous work. (Park and Meloni, 2017; Cochran and Meloni, 2014). Figure 2 shows the two superalkali isomers reducing the CO2. The geometry for the previously studied C2v isomer interacting with CO2 is in agreement with our previous results, (Park and Meloni, 2017), whereas the D3h⋅CO2 species is reported for the first time. When the D3h structure reacts with CO2, the trigonal bipyramidal geometry is distorted by increasing two “equatorial” Li-Li distances, maintaining only the Li(1)-Li(2) distance of 2.30 Å, with Li(3) being closer to the CO2, Li(3)-O(7) = Li(3)-O(8) = 2.03 Å and increasing the axial F-F distance from 2.40 to 2.68 Å. The ∠OCO° bond angle is 128° and the C-O bond length is 1.26 Å. The Li3F2 isomers have similar binding energy with CO2, with the C2v isomer presenting a stronger interaction of 184 kJ mol−1. These clusters can be defined as “free” or “naked” because they are isolated in the gas phase. The presented energy values are calculated at the B3LYP/6-31G* level and are within 10% from the literature reported quantities, whether they are experimental or computed at very high level of theory.
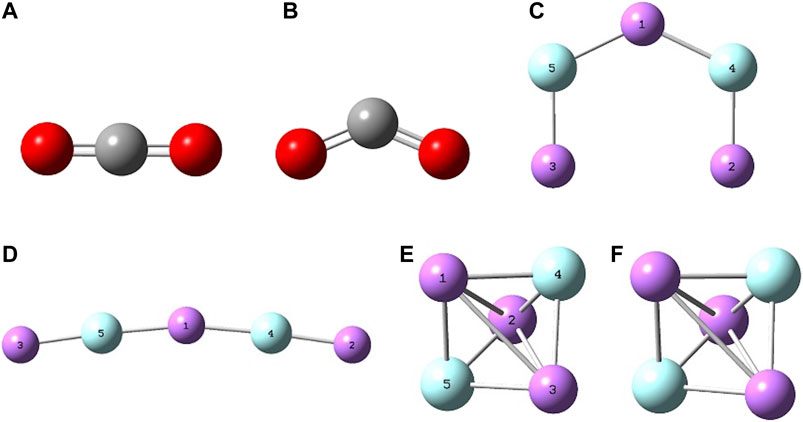
FIGURE 1. B3LYP/6-31G* optimized geometries of (A) CO2, (B) CO2−, (C) Li3F2(C2v), (D) Li3F2+(C2v), (E) Li3F2(D3h), (F) Li3F2+(D3h).
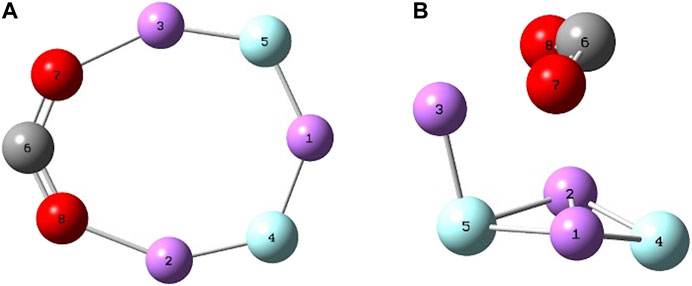
FIGURE 2. B3LYP/6-31G* optimized geometries of (A) Li3F2(C2v), and (B) Li3F2(D3h) reducing CO2.
When a molecule is inserted in the fullerene (yielding an endofullerene), the chemical system is not free, but it will be subjected to the interactions with the carbon cage (“solvation effects”). In Figure 3, the two endofullerenes with CO2 and Li3F2 are shown. In the case of carbon dioxide, it is clear from the energetics presented in Table 1 that CO2 is destabilized by C60 having a negative binding energy at 0 K of −147 kJ mol−1 or a solvation energy at 0 K of 147 kJ mol−1, calculated as E0(CO2) + E0(C60)—E0(C60⋅CO2). The CO2 occupies the center of the C60, aligned with the C3 axis passing through a hexagonal face, minimizing its interactions with the C cage. The solvation energy is more properly defined as the Gibbs free energy change associated with the transfer of a molecule from the gas phase into a solvent, i.e., it provides the relative equilibrium populations of a species between gas phase and the solvent. Therefore, we should also know the entropy change connected with this process. The values that we are reporting in this investigation are at 0 K, so that
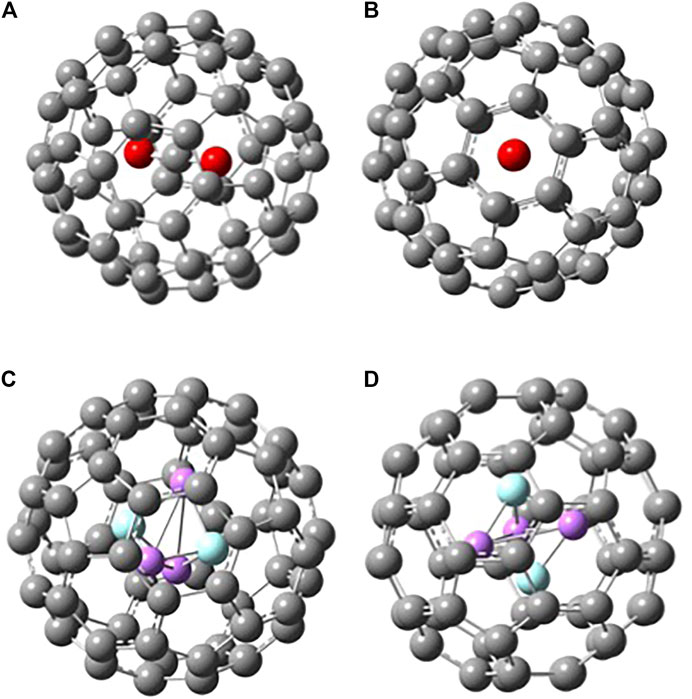
FIGURE 3. Two different views of B3LYP/6-31G* optimized structures of (A)-(B) C60 · CO2 and (C)-(D) C60 · Li3F2(D3h).
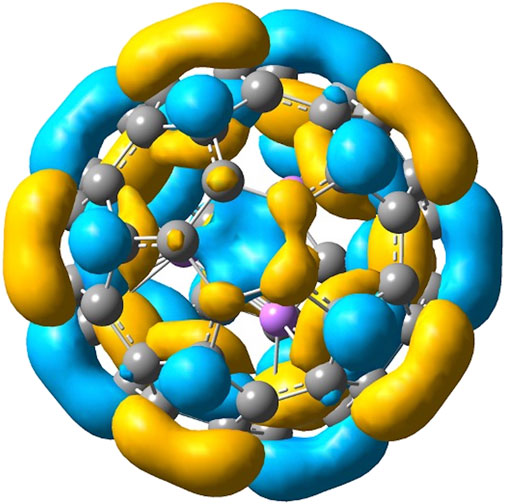
FIGURE 4. HOMO of C60 · Li3F2(D3h).
The insertion of CO2 within C60⋅Li3F2(D3h) produces an unexpected result (Figure 5). In previous computational studies, (Zhao et al., 2017; Park and Meloni, 2017; Sikorska and Gaston, 2020), naked superalkali have been shown to be capable of reducing carbon dioxide by transferring an electron and yielding an activated bent CO2−. In this investigation, we show that our molecular vessel C60 forces the reaction to proceed in a different way (Figure 6). From an analysis of the optimized geometries inside fullerene, it is clear that CO2 is activated by showing a ∠OCO° bond angle of 132° and C-O bond lengths of 1.20 Å, 0.03 Å longer than rC-O in CO2 but 0.06 Å shorter than rC-O in the free Li3F2(D3h)⋅CO2 species. The activation of CO2 is achieved by a F transfer from Li3F2 to CO2 with the formation of a C-F bond of 1.38 Å, almost identical to the rC-F of 1.382 Å in CH3F. (Demaison et al., 1999). This moiety FCO2 does not resemble either the fluorocarboxyl radical FCO2 for which rC-O is 1.234 Å, rC-F is 1.310 Å, and ∠OCO° bond angle is 118.8°, (Zelinger et al., 2003), or the fluoroformate ion FCO2− for which rC-O is 1.234 Å, rC-F is 1.46 Å, and ∠OCO° bond angle is 135.9°. (Arnold et al., 1995; Thomas et al., 2018). In addition, both fluorocarboxyl radical and fluoroformate ion are planar, whereas the endo-reaction species (Figure 6) resembles a non-planar (trigonal pyramidal) FCO2 that interacts with what it looks like a FLi3 species. All the attempts to optimize this structure outside C60 as free endo-Li3F2⋅CO2 returned a Li3F2(D3h)⋅CO2 geometry. Unfortunately, this prevents us from quantifying the interaction of Li3F2(D3h) with CO2 inside C60. In fact, the reaction we need is
from which the interaction of Li3F2(D3h) with CO2 can be derived if we were able to find the [SA⋅CO2] reaction product as a free species and then its binding energy (or negative solvation energy) with C60, i.e.,
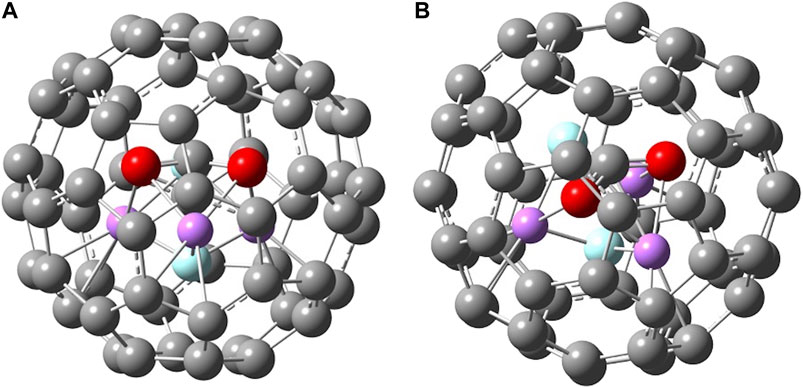
FIGURE 5. Two different views of B3LYP/6-31G* optimized geometry of C60 · Li3F2(D3h) · CO2.
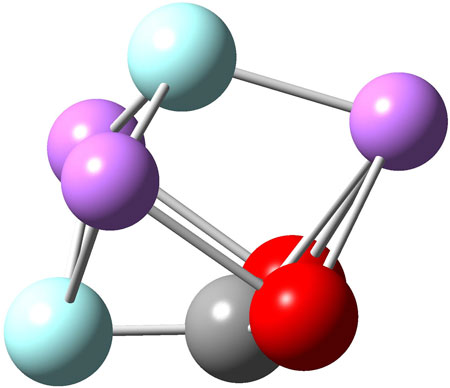
FIGURE 6. B3LYP/6-31G* optimized geometry of the Li3F2.CO2 complex inside C60.
In fact, the interaction of Li3F2(D3h) with CO2 inside fullerene can be calculated as:
In other words, this expression tells us that the interaction between endo-Li3F2(D3h) and endo-CO2, i.e., the BE of superalkali-CO2 in fullerene, is equal to the enthalpy of reaction (1) plus the solvation energies of CO2 and Li3F2(D3h) minus the solvation energy of [SA⋅CO2]. Because we cannot derive this last solvation energy absolute value due to the impossibility of optimizing the free endo-[SA⋅CO2] species, we can estimate this interaction by performing a single-point energy calculation of the free [SA⋅CO2] optimized inside C60. This structure necessarily represents a higher energy structure than a real minimum and, therefore, the estimated BE of superalkali-CO2 in fullerene would denote an upper bound providing us some insights on this interaction. From this computation we get an upper bound for
Conclusion
The activation of CO2 by the Li3F2 superalkali within C60 has been investigated at the B3LYP/6-31G* level of theory. C60 has been utilized as a reaction vessel and its interaction with the reactants, superalkali and carbon dioxide, have been computed. C60 is capable of forcing a superalkali geometry, which does not present the global minimum in the gas phase. Specifically, Li3F2 takes the D3h structure. C60 has a stabilizing effect on the superalkali but a destabilizing effect on the CO2, as it can be deduced by the binding energies of these two systems, BE(C60⋅CO2) = −147 kJ mol−1 and BE(C60⋅Li3F2) = 119 kJ mol−1. Upon interaction of Li3F2(D3h) with CO2 inside fullerene, CO2 is clearly activated showing a ∠OCO° bond angle of 132° and C-O bond lengths of 1.20 Å, 0.03 Å longer than rC-O in CO2 but 0.06 Å shorter than rC-O in the free Li3F2(D3h)⋅CO2 species. The activation of CO2 is achieved by a F transfer from Li3F2 to CO2 with the formation of a C-F bond of 1.38 Å. Due to the impossibility of optimizing a free superalkali-CO2 complex, [SA⋅CO2], resembling the one optimized within the C60, a single-point energy calculation has been performed on the free [SA⋅CO2]. This energy has been utilized to provide an upper bound for the binding energy of Li3F2(D3h) with CO2 within C60 of −965 kJ mol−1, showing that C60 destabilizes the reaction product.
Data Availability Statement
The original contribution presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This research was supported by the American Chemical Society Petroleum Research under the fund grant number #56067-UR6.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.712960/full#supplementary-material
References
Arnold, D. W., Bradforth, S. E., Kim, E. H., and Neumark, D. M. (1995). Study of Halogen–Carbon Dioxide Clusters and the Fluoroformyloxyl Radical by Photodetachment of X−(CO2) (X=I,Cl,Br) and FCO2−. J. Chem. Phys. 102 (9), 3493–3509. doi:10.1063/1.468575
Bai, S., Shao, Q., Wang, P., Dai, Q., Wang, X., and Huang, X. (2017). Highly Active and Selective Hydrogenation of CO2 to Ethanol by Ordered Pd–Cu Nanoparticles. J. Am. Chem. Soc. 139 (20), 6827–6830. doi:10.1021/jacs.7b03101
Bakry, R., Vallant, R. M., Najam-ul-Haq, M., Rainer, M., Szabo, Z., Huck, C. W., et al. (2007). Medicinal Applications of Fullerenes. Int. J. Nanomed. 2 (4), 639–649.
Becke, A. D. (1992). A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 98, 1372–1377.
Becke, A. D. (1988). Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 38, 3098–3100. doi:10.1103/physreva.38.3098
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98, 5648–5652. doi:10.1063/1.464913
Boreriboon, N., Jiang, X., Song, C., and Prasassarakich, P. (2018). Higher Hydrocarbons Synthesis from CO2 Hydrogenation over K- and La-Promoted Fe–Cu/TiO2 Catalysts. Top. Catal. 61 (15), 1551–1562. doi:10.1007/s11244-018-1023-1
Cochran, E., and Meloni, G. (2014). Hypervalence in Monoxides and Dioxides of Superalkali Clusters. J. Chem. Phys. 140, 204319. doi:10.1063/1.4879658
D’Amato, G., and Akdis, C. A. (2020). Global Warming, Climate Change, Air Pollution and Allergies. Allergy 75 (9), 2158–2160.
de Vries, J., Steger, H., Kamke, B., Menzel, C., Weisser, B., Kamke, W., et al. (1992). Single-photon Ionization of C60- and C70-Fullerene with Synchrotron Radiation: Determination of the Ionization Potential of C60. Chem. Phys. Lett. 188 (3), 159–162. doi:10.1016/0009-2614(92)90001-4
Demaison, J., Breidung, J., Thiel, W., and Papousek, D. (1999). The Equilibrium Structure of Methyl Fluoride. Struct. Chem. 10 (2), 129–133. doi:10.1023/a:1022085314343
Diederich, F., and Gómez-López, M. (1999). Supramolecular Fullerene Chemistry. Chem. Soc. Rev. 28, 263–277. doi:10.1039/a804248i
Elliott, S. J., Bengs, C., Kouril, K., Meier, B., Alom, S., Whitby, R. J., et al. (2018). NMR Lineshapes and Scalar Relaxation of the Water-Endofullerene H217O@C60. ChemPhysChem 19, 251–255. doi:10.1002/cphc.201701330
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 09., Revision A.02. Wallingford, CT.: Gaussian. Inc.
Gutsev, G. L., and Boldyrev, A. I. (1983). An Explanation of the High Electron Affinities of the 5d-Metal Hexafluorides. Chem. Phys. Lett. 101, 441–445. doi:10.1016/0009-2614(83)87510-1
Gutsev, G. L., and Boldyrev, A. I. (1982). DVM Xα Calculations on the Electronic Structure of “Superalkali” Cations. Chem. Phy. Lett. 92, 262–266. doi:10.1016/0009-2614(82)80272-8
Gutsev, G. L., and Boldyrev, A. I. (1987). The Electronic Structure of Superhalogens and Superalkalies. Russ. Chem. Rev. 56, 519–531. doi:10.1070/rc1987v056n06abeh003287
Gutsev, G. L., and Boldyrev, A. I. (1985). The Theoretical Investigation of the Electron Affinity of Chemical Compounds. Adv. Chem. Phys. 61, 169–221.
Gutsev, G. L., and Boldyrev, A. I. (1990). Theoretical Estimation of the Maximal Value of the First, Second and Higher Electron Affinity of Chemical Compounds. J. Phys. Chem. 94, 2256–2259. doi:10.1021/j100369a012
Haketa, N., Yokoyama, K., Tanaka, H., and Kudo, H. (2002). Theoretical Study on the Geometric and Electronic Structure of the Lithium-Rich LinF(n-1) (N=2-5) Clusters. J. Mol. Struct. THEOCHEM. 577, 55–67. doi:10.1016/s0166-1280(01)00655-8
Hartman, K. O., and Hisatsune, I. C. (1966). Infrared Spectrum of Carbon Dioxide Anion Radical. J. Chem. Phys. 44 (5), 1913–1918. doi:10.1063/1.1726961
Heath, J. R., O'Brien, S. C., Zhang, Q., Liu, Y., Curl, R. F., Tittel, F. K., et al. (1985). Lanthanum Complexes of Speroidal Carbon Shells. J. Am. Chem. Soc. 107, 7779–7780. doi:10.1021/ja00311a102
Herzberg, G. (1966). Electronic Spectra and Electronic Structure of Polyatomic Molecules. New York: Van Nostrand.
Hönisch, B., Ridgwell, A., Schmidt, D. N., Thomas, E., Gibbs, S. J., Sluijs, A., et al. (2012). The Geological Record of Ocean Acidification. Science 335 (6072), 1058–1063. doi:10.1126/science.1208277
Hu, B., Guild, C., and Suib, S. L. (2013). Thermal, Electrochemical, and Photochemical Conversion of CO2 to Fuels and Value-Added Products. J. Co2Util. 1, 18–27. doi:10.1016/j.jcou.2013.03.004
Jana, G., and Chattaraj, P. K. (2020). Effect of Substitution on the Bonding in He Dimer Confined within Dodecahedrane: A Computational Study. J. Comput. Chem. 41, 2398–2405. doi:10.1002/jcc.26403
Knapp, M., Echt, O., Kreisle, D., Märk, T. D., and Recknagel, E. (1986). Formation of long-lived CO2−, N2O−, and their dimer anions, by electron attachment to van der waals clusters. Chem. Phys. Lett. 126 (3), 225–231. doi:10.1016/s0009-2614(86)80074-4
Kouřil, K., Meier, B., Alom, S., Whitby, R. J., and Levitt, M. H. (2018). Alignment of 17O-Enriched Water–Endofullerene H2O@C60 in a Liquid crystal Matrix. Faraday Discuss. 212, 517–532. doi:10.1039/c8fd00095f
Krachmalnicoff, A., Bounds, R., Mamone, S., Alom, S., Concistrè, M., Meier, B., et al. (2016). The Dipolar Endofullerene HF@C60. Nat. Chem. 8, 953–957. doi:10.1038/nchem.2563
Kroto, H. W., Heath, J. R., O’Brien, S. C., Curl, R. F., and Smalley, R. E. (1985). C60 : Buckminsterfullerene. Nature 318, 162–163. doi:10.1038/318162a0
Kuzovkin, V. V., and Semenov, S. M. (2020). Growth Rate of Carbon Dioxide Concentration in the Atmospheric Surface Layer in the Late 20th Century and Early 21st Century. Russ. Meteorol. Hydrol. 45 (3), 207–210. doi:10.3103/s1068373920030097
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 37, 785–789. doi:10.1103/physrevb.37.785
Li, K., Feron, P. H. M., Jones, T. W., Jiang, K., Bennett, R. D., and Hollenkamp, A. F. (2020). Energy Harvesting from Amine-Based CO2 Capture: Proof-Of-Concept Based on Mono-Ethanolamine. Fuel 263, 116661. doi:10.1016/j.fuel.2019.116661
Li, Z., Wang, L., Li, C., Cui, Y., Li, S., Yang, G., et al. (2019). Absorption of Carbon Dioxide Using Ethanolamine-Based Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 7 (12), 10403–10414. doi:10.1021/acssuschemeng.9b00555
Lia, S. G., Bartmess, J. E., Liebman, J. F., Homes, J. L., Levin, R. D., and Mallard, W. G., Gas-Phase Ion and Neutral Thermochemistry. J. Phys. Chem. Ref. Data, Suppl, 1. 1988, 17, 1–861.
Liu, X., Song, Y., Geng, W., Li, H., Xiao, L., and Wu, W. (2016). Cu-Mo2C/MCM-41: An Efficient Catalyst for the Selective Synthesis of Methanol from CO2. Catalysts 6 (5). doi:10.3390/catal6050075
Luc, W., Collins, C., Wang, S., Xin, H., He, K., Kang, Y., et al. (2017). Ag–Sn Bimetallic Catalyst with a Core–Shell Structure for CO2 Reduction. J. Am. Chem. Soc. 139 (5), 1885–1893. doi:10.1021/jacs.6b10435
Lv, B., Guo, B., Zhou, Z., and Jing, G. (2015). Mechanisms of CO2 Capture into Monoethanolamine Solution with Different CO2 Loading during the Absorption/Desorption Processes. Environ. Sci. Technol. 49 (17), 10728–10735. doi:10.1021/acs.est.5b02356
Mignolet, B., Johansson, J. O., Campbell, E. E. B., and Remacle, F. (2013). Probing Rapidly-Ionizing Super-atom Molecular Orbitals in C60 : A Computational and Femtosecond Photoelectron Spectroscopy Study. ChemPhysChem 14, 3332–3340. doi:10.1002/cphc.201300585
Mulliken, R. S. (1955). Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I. J. Chem. Phys. 23 (10), 1833–1840. doi:10.1063/1.1740588
Murata, M., Murata, Y., and Komatsu, K. (2006). Synthesis and Properties of Endohedral C60 Encapsulating Molecular Hydrogen. J. Am. Chem. Soc. 128, 8024–8033. doi:10.1021/ja061857k
Nambiar, S. R., Jana, G., and Chattaraj, P. K. (2021). Can Superalkalis and Superhalogens Improve the Efficacy of. Redox Reactions? Chem. Phys. Lett. 762, 138131. doi:10.1016/j.cplett.2020.138131
Park, H., and Meloni, G. (2017). Reduction of Carbon Dioxide with a Superalkali. Dalton Trans. 46, 11942–11949. doi:10.1039/c7dt02331f
Reed, A. E., Weinstock, R. B., and Weinhold, F. (1985). Natural Population Analysis. J. Chem. Phys. 83, 735–746. doi:10.1063/1.449486
Ruoff, R. S., Tse, D. S., Malhotra, R., and Lorents, D. C. (1993). Solubility of C60 in a Variety of Solvents. J. Phys. Chem. 97, 3379–3383. doi:10.1021/j100115a049
Sikorska, C., and Gaston, N. (2020). N4Mg6M (M = Li, Na, K) Superalkalis for CO2 Activation. J. Chem. Phys. 153 (14), 144301. doi:10.1063/5.0025545
Song, Y., Peng, R., Hensley, D. K., Bonnesen, P. V., Liang, L., Wu, Z., et al. (2016). High-Selectivity Electrochemical Conversion of CO2 to Ethanol Using a Copper Nanoparticle/N-Doped Graphene Electrode. ChemistrySelect 1 (19), 6055–6061. doi:10.1002/slct.201601169
Srivastava, A. K., Kumar, A., and Misra, N. (2017). Superalkali@C60-superhalogen: Structure and Nonlinear Optical Properties of a New Class of Endofullerene Complexes. Chem. Phys. Lett. 682, 20–25. doi:10.1016/j.cplett.2017.05.070
Srivastava, A. K., Pandey, S. K., and Misra, N. (2016). Prediction of superalkali@C60 Endofullerenes, Their Enhanced Stability and Interesting Properties. Chem. Phys. Lett. 655-656, 71–75. doi:10.1016/j.cplett.2016.05.039
Thomas, D. A., Mucha, E., Gewinner, S., Schöllkopf, W., Meijer, G., and von Helden, G. (2018). Vibrational Spectroscopy of Fluoroformate, FCO2–, Trapped in Helium Nanodroplets. J. Phys. Chem. Lett. 9 (9), 2305–2310. doi:10.1021/acs.jpclett.8b00664
Tong, J., Wu, D., Li, Y., Wang, Y., and Wu, Z. (2013). Superalkali Character of Alkali-Monocyclic (pseudo)Oxocarbon Clusters. Dalton Trans. 42, 9982–9989. doi:10.1039/c3dt50224d
Tong, J., Wu, Z., Li, Y., and Wu, D. (2013). Prediction and Characterization of Novel Polynuclear Superalkali Cations. Dalton Trans. 42, 577–584. doi:10.1039/c2dt31429k
Vogt, C., Monai, M., Kramer, G. J., and Weckhuysen, B. M. (2019). The Renaissance of the Sabatier Reaction and its Applications on Earth and in Space. Nat. Catal. 2 (3), 188–197. doi:10.1038/s41929-019-0244-4
Wang, K., Liu, Z., Wang, X., and Cui, X. (2014). Enhancement of Hydrogen Binding Affinity with Low Ionization Energy Li2F Coating on C60 to Improve Hydrogen Storage Capacity. Int. J. Hydrog. Energ. 39, 15639–15645. doi:10.1016/j.ijhydene.2014.07.132
Wang, L.-S., Reutt, J. E., Lee, Y. T., and Shirley, D. A. (1988). High Resolution UV Photoelectron Spectroscopy of CO2+, COS+ and CS2+ Using Supersonic Molecular Beams. J. Electron. Spectros. Relat. Phenomena 47, 167–186. doi:10.1016/0368-2048(88)85010-2
Wang, L., Wang, L., Zhang, J., Liu, X., Wang, H., Zhang, W., et al. (2018). Selective Hydrogenation of CO2 to Ethanol over Cobalt Catalysts. Angew. Chem. Int. Ed. 57 (21), 6104–6108. doi:10.1002/anie.201800729
Wang, S.-J., Li, Y., Wang, Y.-F., Wu, D., and Li, Z.-R. (2013). Structures and Nonlinear Optical Properties of the Endohedral Metallofullerene-Superhalogen Compounds Li@C60-BX4 (X=F, Cl, Br). Phys. Chem. Chem. Phys. 15, 12903–12910. doi:10.1039/c3cp51443a
Weiss, F. D., Elkind, J. L., O'Brien, S. C., Curl, R. F., and Smalley, R. E. (1988). Photophysics of Metal Complexes of Spheroidal Carbon Shells. J. Am. Chem. Soc. 110, 4464–4465. doi:10.1021/ja00221a085
Yao, B., Xiao, T., Makgae, O. A., Jie, X., Gonzalez-Cortes, S., Guan, S., et al. (2020). Transforming Carbon Dioxide into Jet Fuel Using an Organic Combustion-Synthesized Fe-Mn-K Catalyst. Nat. Commun. 11 (1), 6395. doi:10.1038/s41467-020-20214-z
Yokoyama, K., Haketa, N., Tanaka, H., Furukawa, K., and Kudo, H. (2000). Ionization Energies of Hyperlithiated Li3F Molecule and LinF(n-1) (N=3, 4) Clusters. Chem.Phys. Lett. 330, 339–346. doi:10.1016/s0009-2614(00)01109-x
Zelinger, Z., Dréan, P., Walters, A., Moreno, J. R. A., Bogey, M., Pernice, H., et al. (2003). Gas-phase Detection of the FCO2 Radical by Millimeter Wave and High Resolution Infrared Spectroscopy Assisted by Ab Initio Calculations. J. Chem. Phys. 118 (3), 1214–1220. doi:10.1063/1.1528607
Keywords: CO2 activation, superalkali, endofullerene, ionization energy, solvation energy
Citation: Meloni G, Giustini A and Park H (2021) CO2 Activation Within a Superalkali-Doped Fullerene. Front. Chem. 9:712960. doi: 10.3389/fchem.2021.712960
Received: 21 May 2021; Accepted: 28 June 2021;
Published: 14 July 2021.
Edited by:
Ambrish Kumar Srivastava, Deen Dayal Upadhyay Gorakhpur University, IndiaReviewed by:
Gourhari Jana, Indian Institute of Technology Bombay, IndiaAmlan Kusum Roy, Indian Institute of Science Education and Research Kolkata, India
Copyright © 2021 Meloni, Giustini and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Meloni, Z21lbG9uaUB1c2ZjYS5lZHU=