 Meigeng Hu
Meigeng Hu Dan Zhao1,2,3†
Dan Zhao1,2,3† Guoxu Ma
Guoxu Ma Haifeng Wu
Haifeng Wu- 1Key Laboratory of Bioactive Substances and Resource Utilization of Chinese Herbal Medicine, Ministry of Education, Beijing Key Laboratory of Innovative Drug Discovery of Traditional Chinese Medicine (Natural Medicine) and Translational Medicine, Institute of Medicinal Plant Development, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
- 2Beijing Key Laboratory of Innovative Drug Discovery of Traditional Chinese Medicine (Natural Medicine) and Translational Medicine, Institute of Medicinal Plant Development, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
- 3Key Laboratory of Efficacy Evaluation of Chinese Medicine Against Glycolipid Metabolic Disorders, State Administration of Traditional Chinese Medicine, Institute of Medicinal Plant Development, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
Phytochemical studies on the rhizomes of Actaea asiatica led to the isolation of seven new cycloartane triterpenes, actaticas A−G (1−7). Their structures were determined by NMR, HRESIMS, and chemical analysis. All the isolates were evaluated for their antiproliferative activity against HT-29 and McF-7 cell lines. The results showed that all the compounds displayed cytotoxicity. All compounds showed significant inhibitory effects with IC50 values of 9.2–26.4 μM.
Introduction
Actaea asiatica H. Hara, a perennial herb belonging to the family Ranunculaceae, is mainly distributed in the southwest and northwest of China. Its roots have been traditionally used among the Tujia folk in Hubei Province for treating headache, sore throat, rheumatic pain, rubella, measles, pertussis, uterine prolapse, and dog bites (Gao et al., 2006a; Gao et al., 2006b; Fan et al., 2007; Gao et al., 2007). Phytochemical studies indicated that the genus Actaea contained cycloartane triterpene glycosides with cytotoxic activities (Kusano et al., 1998; Kusano et al., 1999; Gao et al., 2006b). However, little systematic chemical work on A. asiatica has been carried out so far. In order to find the bioactive constituents from A. asiatica, chemical research were carried out, resulting in the isolation of seven new cycloartane triterpene glycosides, namely, actaticas A–G (1–7) (Figure 1). Their structures were determined by spectroscopic analysis and chemical methods. Herein, structural elucidation of compounds 1–7 was reported as well as their cytotoxic activities.
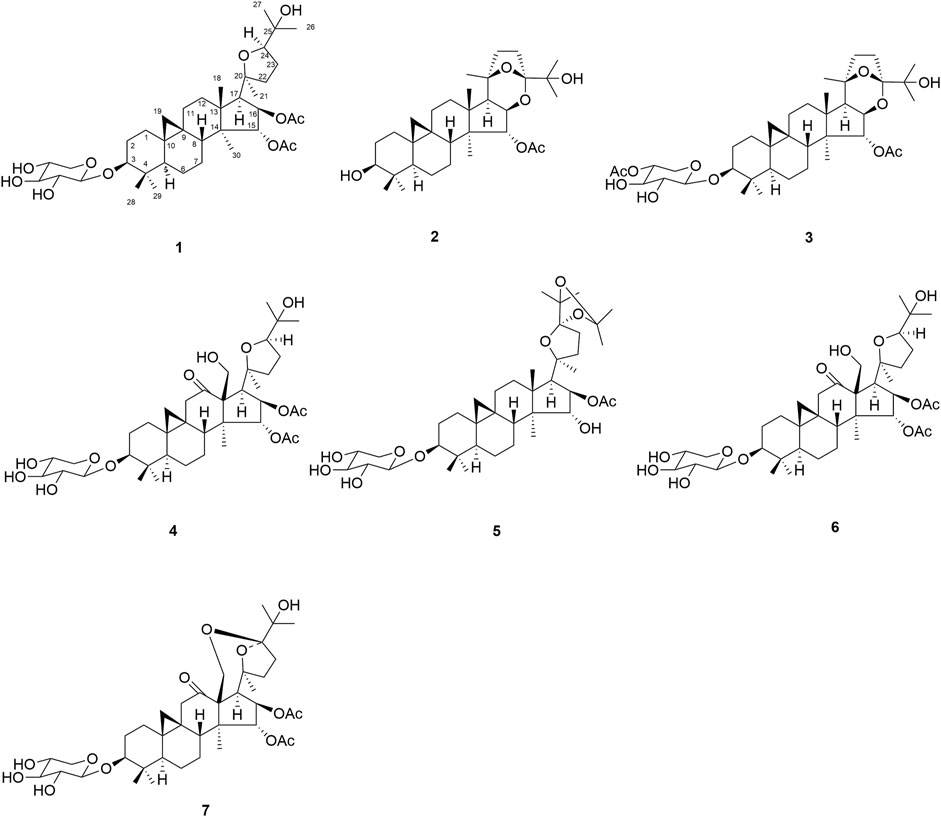
FIGURE 1. Structures of compounds 1–7.
Materials and Methods
General Experimental Procedures
Optical rotations were obtained on a PerkinElmer 341 digital polarimeter. IR spectra were recorded on Shimadzu FTIR-8400S spectrometers. NMR spectra were obtained with a Bruker AV III 600 NMR spectrometer (chemical shift values are presented as δ values with TMS as the internal standard). HR-ESIMS spectra were performed on a LTQ-Obitrap XL spectrometer. Preparative HPLC was performed on a Lumtech K-1001 analytic LC equipped with two pumps of K-501, a UV detector of K-2600, and an YMC Pack C18 column (250 × 10 mm, i.d., 5 μm, YMC Co. Ltd., Japan) eluted with CH3OH-H2O at a flow rate of 2 ml/min. C18 reversed–phase silica gel (40–63 μm, Merk, Darmstadt, Germany), MCI gel (CHP 20P, 75–150 μm, Mitsubishi Chemical Corporation, Tokyo, Japan), and silica gel (100–200 mesh, Qingdao Marine Chemical plant, Qingdao, the People’s Republic of China) were used for column chromatography. Pre-coated silica gel GF254 plates (Zhi Fu Huang Wu Pilot Plant of Silica Gel Development, Yantai, the People’s Republic of China) were used for TLC. All solvents used were of analytical grade (Beijing Chemical Works).
Plant Material
The plants of A. asiatica were collected at Jinfuo Mountain in Chongqing province, the People’s Republic of China, in November 2016, and were authenticated by Professor Sirong Yi. The voucher specimen (CS161108) has been deposited at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences.
Extraction and Isolation
The air-dried powdered rhizomes A. asiatica (6.8 kg) was extracted with 95% EtOH (20 L) three times (each time for 2 h). Removal of the EtOH under reduced pressure yielded the extract (879 g). The residue was suspended in H2O (1.5 L) and partitioned with petroleum ether (3 × 1 L), EtOAc (3 × 1 L), acetone (3 × 1 L), and n-BuOH (3 × 1 L) successively. The EtOAc fraction (510 g) was subjected to CC over silica gel (100–200 mesh, 8 × 100 cm) eluting with a stepwise gradient of CH2Cl2-MeOH (from 1:0 to 0:1) to afford six fractions A–F. Fraction B (29.4 g) was subjected to MCI column chromatography (4 × 80 cm) elution with MeOH-H2O (40:60; 60:40; 70:30; 80:20; 100:0, v/v) giving five subfractions (Fr. B1–B5). Subfraction B3 (911 mg) was chromatographed by semi-preparative HPLC using acetonitrile–H2O (75: 25, v/v) to yield compound 1 (9.4 mg, tR = 26.3 min) and 7 (7.2 mg, tR = 29.5 min). Subfraction B4 (503 mg) was purified through preparative HPLC elution using an acetonitrile–H2O (65: 35, v/v) system to give compound 2 (12.1 mg, tR = 23.0 min). Fraction D (5.8 g) was loaded on an ODS C18 column (2 × 80 cm) eluted with MeOH-H2O (40:60; 60:40; 70:30; 80:20; 100:0, v/v) to give five subfractions (Fr. D1–D5). Subfraction D3 (503 mg) was chromatographed by semi-preparative HPLC using acetonitrile–H2O (70: 30, v/v) to yield compounds 3 (6.1 mg, tR = 18.5 min), 4 (8.7 mg, tR = 21.4 min), and 5 (7.0 mg, tR = 28.3 min). Fraction F (6.7 g) was fractioned on an MCI-gel column chromatography eluted with MeOH–H2O (40:60; 60:40; 70:30; 80:20; 100:0, v/v) to give five subfractions (Fr. F1–F5). Subfraction F3 (223 mg) was chromatographed by preparative HPLC using acetonitrile–H2O (75: 25, v/v) to yield compounds 6 (5.8 mg, tR = 22.7 min).
Actatica A (1): C39H62O11, white amorphous powder; [α]20 D + 21.6 (c = 0.18, MeOH); IR (KBr) νmax: 3,440, 3,397, 2,924, 1,733, 1,457, 1,044 cm−1; UV (MeOH) λmax (log ε): 200 nm; for 1H NMR (600 MHz, pyridine-d5) and 13C-APT (150 MHz, pyridine-d5) spectroscopic data, see Tables 1, 2; HR-ESIMS m/z: 729.4233 (calcd for C39H62O11Na [M + Na]+, 729.4184).

TABLE 1. 1H NMR Spectroscopic Data (600 MHz, in pyridine-d5) for compounds 1–7.
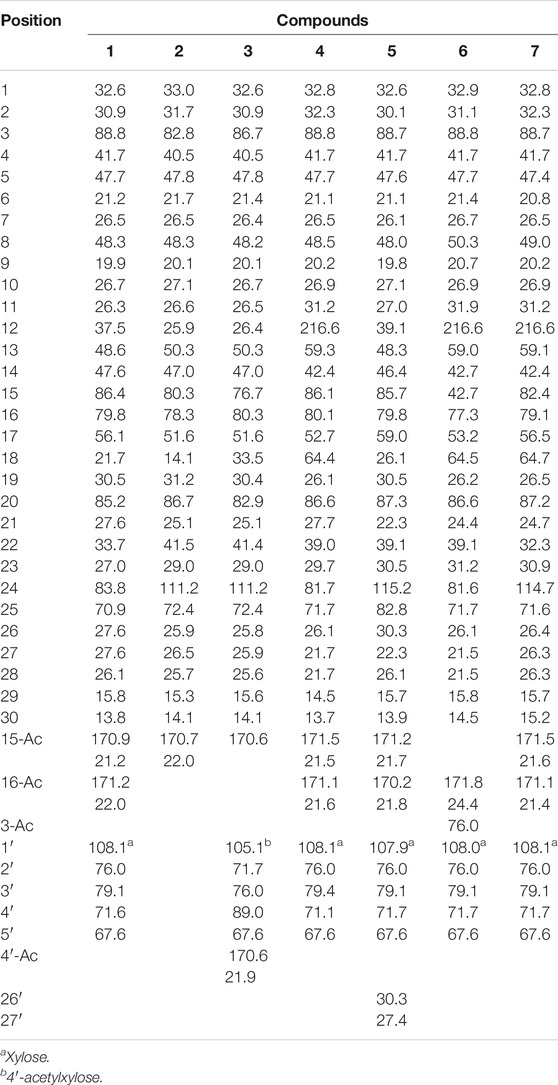
TABLE 2. 13C NMR Data (150 MHz, in pyridine-d5) for compounds 1–7 (δH in ppm, J in Hz).
Actatica B (2): C32H50O6, white amorphous powder; [α]20 D + 19.0 (c = 0.15, MeOH); IR (KBr) νmax: 3,376, 2,957, 1,738, 1,373, 1,032 cm−1; UV (MeOH) λmax (log ε): 201 nm; for 1H NMR (600 MHz, pyridine-d5) and 13C-APT (150 MHz, pyridine-d5) spectroscopic, data see Tables 1, 2; HR-ESIMS m/z: 553.3533 (calcd for C32H50O6Na, 553.3500).
Actatica C (3): C39H60O11, white amorphous powder; [α]20 D+ 35.1 (c = 0.31, MeOH); IR (KBr) νmax: 3,493, 2,928, 1,730, 1,375, 1,044 cm−1; UV (MeOH) λmax (log ε): 201 nm; for 1H NMR (600 MHz, pyridine-d5) and 13C-APT (150 MHz, pyridine-d5) spectroscopic data, see Tables 1, 2; HR-ESIMS m/z: 727.4100 (calcd for C39 H60O11Na, [M + Na]+, 727.4088).
Actatica D (4): C40H62O13, white amorphous powder; [α]20 D + 22.4 (c = 0.22, MeOH); IR (KBr) νmax: 3,439, 2,934, 1,734, 1,264, 1,033, 1,033, 962 cm−1; λmax (log ε): 201 nm; for 1H NMR (600 MHz, pyridine-d5) and 13C-APT (150 MHz, pyridine-d5) spectroscopic data, see Tables 1, 2; HR-ESIMS m/z: 759.3974 (calcd for C40H62O13Na [M + Na]+, 759.3926).
Actatica E (5): C42H66O12, white amorphous powder; [α]20 D + 42.7 (c = 0.35, MeOH); IR (KBr) νmax: 3,449, 2,935, 1,741, 1,375, 1,045 cm−1; λmax (log ε): 200 nm; for 1H NMR (600 MHz, pyridine) and 13C-APT (150 MHz, pyridine-d5) spectroscopic data, see Tables 1, 2; HR-ESIMS m/z: 785.4341 (calcd for C42H66O12Na [M + Na]+, 785.4341).
Actatica F (6): C37H58O11, white amorphous powder; [α]20 D + 23.3 (c = 0.12, MeOH); IR (KBr) νmax: 3,350, 2,820, 1,710, 1,534, 952 cm−1; λmax (log ε): 202 nm; for 1H NMR (600 MHz, pyridine-d5) and 13C-APT (150 MHz, pyridine-d5) spectroscopic data, see Tables 1, 2; HR-ESIMS m/z: 701.3904 ([M + Na]+, calcd for C37H58O11Na, 701.3926).
Actatica G (7): C39H58O13, white amorphous powder; [α]20 D + 22.6 (c = 0.13, MeOH); IR (KBr) νmax: 3,298, 2,781, 1,716, 1,422, 950 cm−1; for 1H NMR (600 MHz, pyridine-d5) and 13C-APT (150 MHz, pyridine-d5) spectroscopic data, see Tables 1, 2; HR-ESIMS m/z: 757.3765 ([M + Na]+, calcd for C39H58O13Na, 757.3770).
Hydrolysis of Compounds
Acid hydrolysis of 1 and 4–7: A solution of compounds 1, 4, 5, 6, and 7 (5 mg) in 2 M HCl (1 ml) was heated at reflux for 24 h. The reaction mixture was neutralized with 2 M NaOH and extracted by partition with EtOAc (5 × 1 ml). 10 ml of water was added to the residue and extracted with CH2Cl2 three times. Sugars were analyzed by TLC and GC analysis and compared with authentic sample of D-sugars. The spots were visualized by spraying with EtOH–H2SO4–anisaldehyde (9:0.5:0.5, v/v), then heated at 150°C. Furthermore, the absolute configurations of the sugars were determined by gas chromatography according to a method previously described (Ma et al., 2016; Li et al., 2017). Compound 3 was dissolved in MeOH (15 ml), then 4% K2CO3 (15 ml) was added and each solution was stirred at room temperature overnight. Each solution was neutralized by 10% AcOH, and extracted with EtOAc (2 × 20 ml). EtOAc extract after removal of solvent, was dissolved in MeOH (10 ml) and refluxed with 0.5 N HCl (3 ml) for 4 h (Yin et al., 2010).
Cytotoxic Assay
The cytotoxicity of compounds 1−7 was evaluated using the MTT procedure with HT-29 and McF-7 cancer cell lines. The cells were incubated in DMEM supplemented with 10% fetal bovine serum and cultured at a density of 1.2 × 104 cells/ml in a 96-well microtiter plate. Five various concentrations of each agent dissolved in dimethyl sulfoxide (DMSO) were then put in the wells. Each concentration was evaluated three times. After incubation under 5% CO2 at 37°C for 48 h, 10 ml of MTT (4 mg/ml) was placed into each well, and the cells were incubated for an additional 4 h. Then, the liquid was taken out, and DMSO (200 ml) was put into the wells. The absorbance was documented with a microplate reader at wave length of 570 nm.
Results and Discussion
Compound 1 was obtained as white amorphous powder. Its IR spectrum showed absorptions of hydroxyl group at 3,440 and 3,397 cm−1 and carbonyl at 1,733 cm−1. The HRESIMS spectrum showed a pseudo-molecular ion at m/z 729.4233 [M + Na]+ in the positive ion mode from which in conjunction with NMR data the molecular formula was established as C39H62O11, consistent with nine degrees of unsaturation. In the 1H NMR spectrum (Table 1) two cyclopropane–methylene protons as an AX system at δH 0.18 and 0.47 (each 1H, d, J = 4.0 Hz, H2-19) together with seven tertiary methyl groups at δH 1.49, 1.47, 1.32, 1.36, 1.35, 1.02, and 1.09, indicated a cycloartane triterpenoid structure (Ju et al., 2002a; Mohamed, 2014; Gan et al., 2015; Wu et al., 2017). The 1H NMR spectrum also showed two oxygenated proton signals at δH 5.49 (d, J = 4.8 Hz) and δH 5.82 (d, J = 4.8 Hz), indicating two acetyl groups at C-15 and C-16. Except for sugar carbons, the 13C-NMR spectrum (Table 2) of 1 displayed 39 carbon resonances including methylene carbon of cyclopropane ring at δC 30.5 (C-19), an oxymethine carbon at δC 88.8 (C-3), an oxygenated quaternary carbon at δC 85.2 (C-20), and an anomeric carbon at δC 108.1, together with acetyl signals at δC 170.9, 171.2, 21.2, and 22.0. The 1H and 13C NMR spectroscopic data of 1 confirmed that the compound was a cycloartane triterpene glycoside (Jung et al., 2002; Wu et al., 2017; Wua et al., 2017).
All proton signals were assigned to the corresponding carbons through direct 1H and 13C correlations in the HSQC spectrum. Inspection of the 1H-1H COSY spectrum showed fragments of C-1/C-2/C-3, C-5/C-6/C-7/C-8, C-11/C-12, C-15/C-16/C-17, and C-22/C-23/C-24. In the HMBC spectrum (Figure 2), the correlations were observed from H-28/29 to C-3 and C-5, H-19 to C-1, C-5, C-6, C-9, and C-11, and H-18 to C-12 and C-17, H-30 to C-8, C-14, C-16 and C-18, H-21 to C-22, H-22 to C-24, and H-24 to C-26 and C-2 fully confirmed the basic skeleton cycloartane triterpene of compound 1, which was consistent with the above deduction. The acetyl groups were connected with C-15 and C-16 supported by the correlations from H-15 to δC 170.9 (the carbonyl carbon of OAc) and H-16 to δC 171.2 (the carbonyl carbon of OAc). The sugar was connected with C-3 based on the key HMBC correlation between H-1′ (δH 4.86, d, J = 7.2 Hz) and C-3 (δC 88.8), which was identified as D-xylose by TLC in comparison with authentic monosaccharides (visualization with ethanol-5% H2SO4 spraying) followed by gas chromatography.
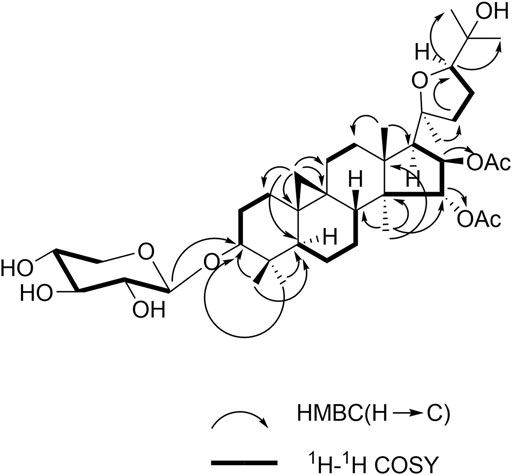
FIGURE 2. Key HMBC and 1H-1H COSY correlations for 1.
The NOESY experiment and coupling constants established the relative configuration of compound 1 (Figure 3), in which correlation of H-3/H-5 showed α-orientation of H-3. The larger coupling constants (3J1,2 > 7.0 Hz) of the anomeric protons indicated the β configuration of the sugar unit. The significant cross peaks from H-15 to H3-18, H-17α to Me-21, H-16 to H3-30, and H-24 to Me-21 were observed, which enabled the establishment of OAc-15α and OAc-16β. Until now, all the isolated cycloartane triterpene share the identical absolute configuration with trans A/B, B/C, C/D rings. Considering the same cycloartane triterpene skeleton and identical carbon signals at C-20/C-24, compound 1 was established as 20S and 24R configurations (Ju et al., 2002a). Therefore, the structure of the compound was identified as shown and given the trivial name actatica A.
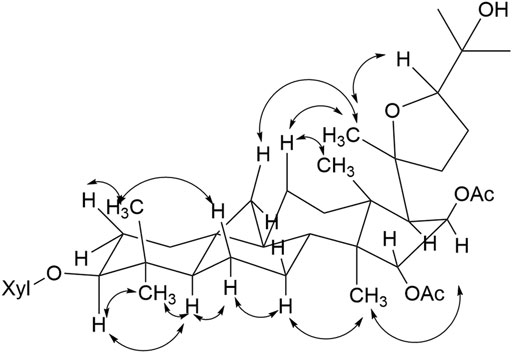
FIGURE 3. Key NOESY correlations of compound 1.
Compound 2 was determined to have the molecular formula C32H50O6, by the observation of the ion peak at m/z 553.3533 (calcd for C32H50O6Na, 553.3500). The 1H-NMR spectrum (Tables 1, 2) displayed signals for seven tertiary methyls (δH 1.67, 1.62, 1.54, 1.54, 1.58, 1.08, and 1.25), two typical signals at δH 0.32 (1H, d, J = 4.2 Hz) and 0.53 (1H, d, J = 4.2 Hz) ascribable to a cyclopropane moiety, indicating that 2 might be a cycloartane-type triterpenoid. The 13C NMR spectrum of 2 displayed 32 carbon signals, three signals attributable to oxygen-bearing quaternary carbons at δ 82.8, 111.2, and 72.4. The NMR data were similar to the reported one (20S, 24S)-16β, 24; 20, 24-diepoxy-9, 19-cycloeanostane-3β, 15α, 18, 25-tetraol-3-O-β-D-xylopyranoside (Mu et al., 2014). The differences were the absence of the sugar at C-3, and the appearance of acetyl group at C-15 in compound 2. In the HMBC spectrum, the correlation observed from H-15 to OAc together with the molecular formula confirmed the deduction above. The α configurations of H-16 and H-17 were confirmed by the NOESY correlations between δH1.86(H-17) and δH1.23 (H3-30), δH4.47 (H-16) and δH2.00 (H-17). Taken together with the 2D-NMR spectra data, compound 2 was characterized and named actatica B.
Compound 3, which was isolated as a white amorphous powder, was assigned as C39H60O11, based on its positive HRESIMS ion at m/z 727.4100 (calcd for C39H60O11Na, [M + Na] +, 727.4088). The 1H NMR spectrum showed that 3 possesses a cyclopropane ring, seven methyl groups, and an AB-type hydroxymethyl group (H2-18). The NMR (Tables 1, 2) spectroscopic data for this compound were analogous to 2, except for the appearance of the anomeric proton at δH 4.81 (d, J = 7.8 Hz) and δC 105.1, 71.7, 76.0, 89.0, 67.6, 170.6, and 22.0. The sugar was identified as a 4′-O-β-D- xylose after acid hydrolysis. Inspection of the 1H-1H COSY spectrum showed fragments of C-1/C-2/C-3, C-5/C-6/C-7/C-8, C-11/C-12, C-15/C-16/C-17/C-18, and C-22/C-23. In the HMBC spectrum, the correlation from H-3 (3.36, dd, J = 11.4, 3.0 Hz) to the anomeric carbon signal at δC 86.7 supported that the sugar unit was attached to C-3. Thus, the structure of 3 was determined as actatica C.
Compound 4 has a molecular formula of C39H60O13 according to the HRESIMS (m/z 759.3974 [M + Na] +, calcd for C39H60O13Na, 759.3926). Its IR spectrum showed strong hydroxyl (3,439, 1,044 cm−1) and carboxyl (1730 cm−1) absorptions. The 1H and 13CNMR spectra indicated that 4 had two acetoxyl groups. Detailed NMR spectral analysis revealed that 4 possessed a cyclopropane ring, six methyl groups, a hydroxymethyl group at C-18, and a D-xylosyl unit at C-3. The 1H and 13C NMR spectra of 4 were similar to those of beesioside J (Ju et al., 2002b), except for a carbonyl group (C=O) connected to C-12 of 4, which causes the downfield chemical shift of C-12 (δC 216.6). The correlation from δH 4.54 (H-11) to δC 216.6 (C=O) according to the HMBC supported the above result. Therefore, compound 4 was tentatively determined and named actatica D.
Compound 5 has the molecular formula C42H66O12 determined by HR-ESIMS (m/z 785.4301, calcd for C42H66O12Na [M + Na] +, 785.4341). In the 1H NMR spectrum (Table 1) two cyclopropane–methylene protons as an AX system at δH 0.21 and 0.59 (each 1H, d, J = 4.0 Hz, H2-19) together with nine tertiary methyl groups indicated a cycloartane triterpenoid structure. The 1H NMR and 13C APT data for this compound were analogous to 1, except for the additional NMR signals at δC 30.3 and 27.4, and δH 1.31 (3H, s), and 1.45 (3H, s). The differences showed that 5 had one more hydroxyisopropyl group connected at C-24. In the HMBC spectrum, the correlations from H-24 to C-26, C-26′, C-27, and C-27′ confirmed the above deduction. Taken together with the NOESY spectra data, compound 5 was established as 24R configurations. Thus, compound 5 was established and named actatica E.
Compound 6 was determined to have the molecular formula of C37H58O11 based on the 13C APT data and by the HRESIMS ion peak at m/z 701.3904 ([M + Na] +, calcd for C37H58O11Na, 701.3926). The 1H-NMR spectrum (Table 1) displayed signals for seven tertiary methyls (δ 1.02, 1.22, 1.23, 1.30, 1.53, and 1.55), two typical signals at δ 0.12 (1H, d, J = 4.2 Hz) and 0.44 (1H, d, J = 4.2 Hz) ascribable to a cyclopropane methylene group, indicating that 6 might be a cycloartane-type triterpenoid. Examination of the 1H and 13C APT data (Tables 1, 2) showed the structure of 6 to be similar to 4. The NMR spectrum showed that compound 6 has only one set of acetyl group data. On the basis of 1H-1H COSY and HSQC and comparison with related 4, all signals were assigned as shown in Tables 1, 2. The correlation from H-16 (δH 5.47) to acetyl carbon (δC 171.8) was observed in HMBC spectrum, which means the acetyl group was connected to C-16. Therefore, compound 6 was clearly determined and named actatica F.
Compound 7 has the molecular formula C39H58O13 according to its HRESIMS result (m/z 757.3765 [M + Na] +, calcd for C39H58O13Na, 757.3770). In the 1H NMR spectrum (Table 1) two cyclopropane-methylene protons as an AX system at δH 0.28 and 0.41 (each 1H, d, J = 4.2 Hz, H2-19) together with nine tertiary methyl groups indicated a cycloartane triterpenoid structure. The 1H NMR and 13C APT data were closely related to those of beesioside I (Tables 1, 2) (Sakurai et al., 1990). The differences showed that 7 had a carbonyl group attached to C-12, which caused C-12 to move to a lower field, and the chemical shift is greatly increased to δC 216.6). The HMBC spectrum shows that δH 4.54 (H-11) is related to δC 216.6 (C = O), confirming the above inference. Moreover, in the NOESY spectrum, correlations were also detected between Me-21/H-22α/H-23α/H-24α, H-22α/H-22β, H-23α/H-23β, H-22β/H-23β, and H-24α/Me-26/Me-27. Considering the same cycloartane triterpene skeletonand identical carbon signals at C-20/C-24, compound 7 enabled a determination of a 20S*, 24R* configuration (Ju et al., 2016). As a result, the structure of 7 was established and named actatica G.
Bioactive Activity
The cytotoxic of all compounds 1–7 were tested for their inhibitory activity against human HT-29 and McF-7 cancer cell lines using MTT assay. All compounds showed significant inhibitory effects with IC50 values of 9.2–26.4 μM (Table 3). Compound 7, with an oxygen bridge between C-18 and C-24, showed the best potency among the isolated constituents. With a tetrahydrofuran fragment connected by C-20 and C-24, compounds 1 and 4–7 showed better activity than 2 and 3.
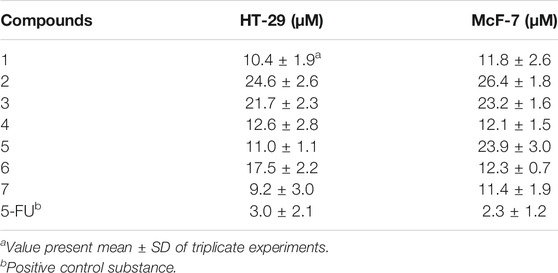
TABLE 3. Cytotoxicity of compounds 1–7 against HT-29 and McF-7 cancer cell lines.
Seven new 9,19-cycloartane glycosides were isolated from the rhizomes of A. asiatica H. Hara. Until now, nearly 200 naturally occurring triterpenes with a 9,19-cycloartane have been reported (Su et al., 2016; Hassan et al., 2020). However, compound 5 with one more hydroxy isopropyl group was first isolated from the genus Actaea. All compounds displayed inhibitory activity against human HT-29 and McF-7 cancer cell lines. Further analysis of the data showed that compounds 1 and 4–7 exhibited better protective effect than other compounds, which indicated that the tetrahydrofuran fragment connected by C-20 and C-24 may affect the inhibitory activity regarding HT-29 and McF-7.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
HW and XC were responsible for study design. MH and DZ were responsible for compound isolation and cytotoxic activity testing. GM, XX, MH, and DZ were responsible for structure elucidation and validation of compound identities. All authors contributed equally to manuscript writing, review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Technological Large Platform for Comprehensive Research and Development of New Drugs in the Twelfth Five-Year “Significant New Drugs Created” Science and Technology Major Projects (No. 2012ZX09301-002-001-026) and CAMS Innovation Fund for Medical Sciences, Grant/Award Numbers: CIFS 2019-I2M-1-005).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Huiling Liu was gratefully acknowledged for her kindness to measuring the NMR spectrum.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.695456/full#supplementary-material
References
Fan, Y. S., Yao, Z., Teng, J., Pan, Q., and Duan, H. Q. (2007). Triterpenoids from Actaea Asiatica with Antitumor Activity. Chin. Traditional Herbal Drugs 38 (2), 167–170. doi:10.3321/j.issn:0253-2670.2007.02.003
Gan, L.-S., Zheng, D.-J., Liu, Q., Zhou, J., Zhang, M.-Z., Yao, W., et al. (2015). Eight New Cycloartane Triterpenoids from Beesia Calthifolia with Hepatoprotective Effects against D-Galactosamine Induced L02 Cell Damage. Bioorg. Med. Chem. Lett. 25 (18), 3845–3849. doi:10.1016/j.bmcl.2015.07.070
Gao, J.-C., Zhang, J.-C., Lu, Z.-J., Zhu, G.-Y., Yang, M.-S., and Xiao, P.-G. (2006b). Chemical Constituents of Actaea Asiatica Hara and Their Anti-osteoporosis Activities. Biochem. Syst. Ecol. 34 (9), 710–713. doi:10.1016/j.bse.2006.02.004
Gao, J.-C., Zhang, J.-C., Zhu, G.-Y., Yang, M.-S., and Xiao, P.-G. (2007). Chromones and Indolinone Alkaloids from Actaea Asiatica Hara. Biochem. Syst. Ecol. 35 (7), 467–469. doi:10.1016/j.bse.2007.01.012
Gao, J., Huang, F., Zhang, J., Zhu, G., Yang, M., and Xiao, P. (2006a). Cytotoxic Cycloartane Triterpene Saponins fromActaeaasiatica. J. Nat. Prod. 69 (10), 1500–1502. doi:10.1021/np060113h
Hassan, A. R., Ashour, A., Amen, Y., Nagata, M., El-Toumy, S. A., and Shimizu, K. (2020). A New Cycloartane Triterpene and Other Phytoconstituents from the Aerial Parts of Euphorbia Dendroides. Nat. Product. Res. 1–9. doi:10.1080/14786419.2020.1800693
Ju, J.-H., Liu, D., Lin, G., Xu, X. D., Han, B., Yang, J.-S., et al. (2002a). Beesiosides A−F, Six New Cycloartane Triterpene Glycosides from Beesia Calthaefolia. J. Nat. Prod. 65 (1), 42–47. doi:10.1021/np010293p
Ju, J.-H., Liu, D., Lin, G., Zhang, Y.-M., Yang, J.-S., Lu, Y., et al. (2002b). Beesiosides G, H, and J−N, Seven New Cycloartane Triterpene Glycosides fromBeesiacalthifolia. J. Nat. Prod. 65 (2), 147–152. doi:10.1021/np010294h
Jung, D.-W., Lee, J. M., and Sung, C. K. (2002). Enzyme-linked Immunosorbent Assay for the Determination of 20(S)-protopanaxatriol. Analytica Chim. Acta 462 (2), 157–163. doi:10.1016/S0003-2670(02)00340-9
Kusano, A., Takahira, M., Shibano, M., Miyase, T., Kusano, G., and Bulletin, P. (1999). Studies on the Constituents of Cimicifuga Species. XXVI. Twelve New Cyclolanostanol Glycosides from the Underground Parts of Cimicifuga Simplex WORMSK. Chem. Pharm. Bull. 47 (4), 511–516. doi:10.1248/cpb.47.511
Kusano, A., Takahira, M., Shibano, M., Miyase, T., Okuyama, T., and Kusano, G. (1998). ChemInform Abstract: Studies on the Constituents of Cimicifuga Species. Part 22. Structures of Two New Cyclolanostanol Xylosides, Cimiacerosides A and B. ChemInform 5 (48), 1003–1013. doi:10.1002/chin.199839202
Li, P., Zhu, N., Hu, M., Wu, H., Yu, T., Wu, T., et al. (2017). New Cucurbitane Triterpenoids with Cytotoxic Activities from Hemsleya Penxianensis. Fitoterapia 120, 158–163. doi:10.1016/j.fitote.2017.06.009
Ma, G.-X., Feng, W., Sun, Z.-H., Li, P.-F., Zhu, N.-L., Yang, J.-S., et al. (2016). New Stigmastane Type of Steroidal Glycosides from the Roots of Vernonia Cumingiana. J. Carbohydr. Chem. 35 (3), 172–179. doi:10.1080/07328303.2016.1170137
Mohamed, G. A. (2014). New Cytotoxic Cycloartane Triterpene from Cassia Italica Aerial Parts. Nat. Product. Res. 28 (13), 976–983. doi:10.1080/14786419.2014.902820
Mu, L.-H., Li, H.-J., Guo, D.-H., Zhao, J.-Y., and Liu, P. (2014). Cycloartane Triterpenes from Beesia Calthaefolia (Maxim.). Fitoterapia 92, 41–45. doi:10.1016/j.fitote.2013.10.005
Sakurai, N., Goto, T., Nagai, M., Inoue, T., and Xiao, P. (1990). Studies on the Constituents of Beesia Calthaefolia, and Souliea Vaginata. III, Breesiodide IV, a Cyclolanostanol Xyloside from the Rhizomes of B. Calthaefolia and S. Vaginata. Heterocycles 30 (2), 897–904. doi:10.3987/COM-89-S78
Su, Y., Chi, W.-C., Wu, L., Wang, Q.-H., and Kuang, H.-X. (2016). Photochemistry and Pharmacology of 9, 19-cyclolanostane Glycosides Isolated from Genus Cimicifuga. Chin. J. Nat. Medicines 14 (10), 721–731. doi:10.1016/S1875-5364(16)30087-5
Wu, H.-F., Liu, X., Zhu, Y.-D., Zhou, J., Gong, Y.-Y., Ma, G.-X., et al. (2017). A New Cycloartane Triterpenoid Glycoside from Souliea Vaginata. Nat. Product. Res. 31 (21), 2484–2490. doi:10.25135/rnp.10.17.06.10310.1080/14786419.2017.1314283
Wua, H., Yang, Z., Wang, Q., Zhub, N., Xub, X., Zou, Q., et al. (2017). A New Cytotoxic Cyclolanostane Triterpenoid Xyloside from Souliea Vaginata. Nat. Prod. Commun. 12 (2), 229–232. doi:10.1177/1934578X1701200222
Keywords: Actaea asiatica, cycloartane triterpenes, antiproliferative activity, HT-29 cell lines, MCF-7 cell lines
Citation: Hu M, Zhao D, Xu X, Ma G, Wu H and Chen X (2021) Actaticas A−G, Cycloartane Triterpenes From Actaea asiatica With Their Antiproliferative Activity. Front. Chem. 9:695456. doi: 10.3389/fchem.2021.695456
Received: 23 April 2021; Accepted: 31 May 2021;
Published: 29 July 2021.
Edited by:
Guigen Li, Texas Tech University, United StatesReviewed by:
Shao-Jiang Song, Shenyang Pharmaceutical University, ChinaGeorge A. O’Doherty, Northeastern University, United States
Copyright © 2021 Hu, Zhao, Xu, Ma, Wu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haifeng Wu, aGZ3dUBpbXBsYWQuYWMuY24=; Xi Chen, Y2hlbnhpQGltcGxhZC5hYy5jbg==
†These authors have contributed equally to this work