 Fei Yu
Fei Yu Ming Cai
Ming Cai Liang Shao
Liang Shao Jihong Zhang
Jihong Zhang- Medical School of Kunming University of Science and Technology, Kunming, China
Kinase dysregulation is greatly associated with cell proliferation, migration and survival, indicating the importance of kinases as therapeutic targets for anticancer drug development. However, traditional kinase inhibitors binding to catalytic or allosteric sites are associated with significant challenges. The emergence of resistance and targeting difficult-to-degrade and multi-domain proteins are significant limiting factors affecting the efficacy of targeted anticancer drugs. The next-generation treatment approaches seem to have overcome these concerns, and the use of proteolysis targeting chimera (PROTAC) technology is one such method. PROTACs bind to proteins of interest and recruit E3 ligase for degrading the whole target protein via the ubiquitin-proteasome pathway. This review provides a detailed summary of the most recent signs of progress in PROTACs targeting different kinases, primarily focusing on new chemical entities in medicinal chemistry.
Introduction
Cancer is a condition in which cells demonstrate abnormal growth and invasion to adjacent tissues. According to a World Health Organization (WHO) report, more than 15% of overall global deaths are due to cancer (Ward et al., 2020). Chemotherapy was first used to treat tumorous lesions in the 20th century. Several selective small-molecular anticancer drugs have been developed in the past few decades (Singh et al., 2011; Ward et al., 2020).
Small molecules have greatly improved cancer treatment, but the tumor cells are inherently resistant to certain anticancer mechanisms or acquire resistance to therapeutic agents. Alternative treatment options must be devised for patients resistant to anticancer treatment.
Recently, the use of proteolysis-targeting chimera (PROTAC) to discover and develop new drugs has been gaining significant traction. The cell cycle can be modulated by regulating the protein function. Inhibitors can modulate protein function partially, whereas PROTACs induce complete protein degradation, affecting all functions of that protein (Pettersson and Crews, 2019). A PROTAC comprises an E3 ubiquitin ligase ligand, a protein of interest (POI) ligand, and a linker. It employs heterobifunctional small molecules to recruit an E3 ubiquitin ligase and the POI (Coleman and Crews, 2018) to cause a ternary complex. The E3 ligase-mediated ubiquitination of the POI leads to proteasomal degradation (Figure 1).
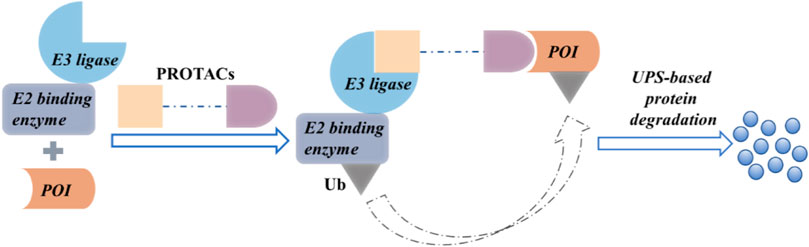
FIGURE 1. The proposed mechanism of action of PROTACs.
PROTAC technology has been well received following the development of the first PROTAC molecule by Crews et al. (Sakamoto et al., 2001; Yin and Hu, 2020; Zhou X. et al., 2020; Martín-Acosta and Xiao, 2021; Zeng et al., 2021). ARV-110 is the first oral small-molecule PROTAC degrader that targets androgen receptors (ARs). It is being tested in clinical trials to treat metastatic castration-resistant prostate cancer. Academic and industrial research have been greatly encouraged by these advances.
In this review, we mainly summarize advances in the use of PROTACs targeting protein kinases. These findings will help drive new approaches to overcome drug resistance in cancer therapy.
Recent Progress of PROTACs Targeting Protein Kinase
Advances in modern biological approaches have enabled the discovery of more than 500 protein kinases. These molecules affect the survival, migration, and proliferation of cells, indicating their potential as therapeutic targets for anticancer drugs. To date, more than 40 small-molecule inhibitors were approved by the US Food and Drug Administration for cancer therapy. Most of these small molecules target protein kinases, which modulate cell signaling by catalyzing the phosphorylation relevant proteins (Bedard et al., 2020).
Proteolysis Targeting Chimeras Targeting BCR-ABL
The leukemogenic ability of BCR-ABL in chronic myeloid leukemia (CML) is well recognized, making it a potential anticancer drug target. Imatinib was the first tyrosine kinase inhibitor (TKI) targeting BCR-ABL (Schindler, 2000; Deininger et al., 2005; Reddy and Aggarwal, 2012). Subsequently, multiple ABL kinase inhibitors, including ponatinib (Gozgit et al., 2012), dasatinib (Shah et al., 2004), nilotinib (Weisberg et al., 2006), and bosutinib (Boschelli et al., 2001) have been approved for clinical use. Although TKI use has improved CML treatment outcomes, long-term therapy and drug resistance are significant challenges.
Degradation of BCR-ABL may be beneficial in CML treatment. Nagar et al. developed PROTACs comprising linkers of variable lengths in term of the co-crystal structure of the c-ABL tyrosine kinase domain-imatinib complex (Nagar et al., 2003). Lai et al. conjugated dasatinib or bosutinib to a cereblon (CRBN) E3 ubiquitin ligase ligand for successful protein degradation of BCR-ABL (Lai et al., 2016). Shimokawa et al. reported a new anticancer agent, SNIPER(ABL)-062 (1, Figure 2), that degrades oncogenic proteins. The compound demonstrated strong binding affinities with cIAP1/2, ABL1 and XIAP, leading to complete BCR-ABL degradation and inhibition of the BCR-ABL-mediated signaling pathways (Shimokawa et al., 2017). Further, Demizo et al. reported a conjugate of imatinib derivative and bestatin that recruited the E3 ubiquitin ligase cIAP1 to induce BCR-ABL degradation (Demizu et al., 2016).
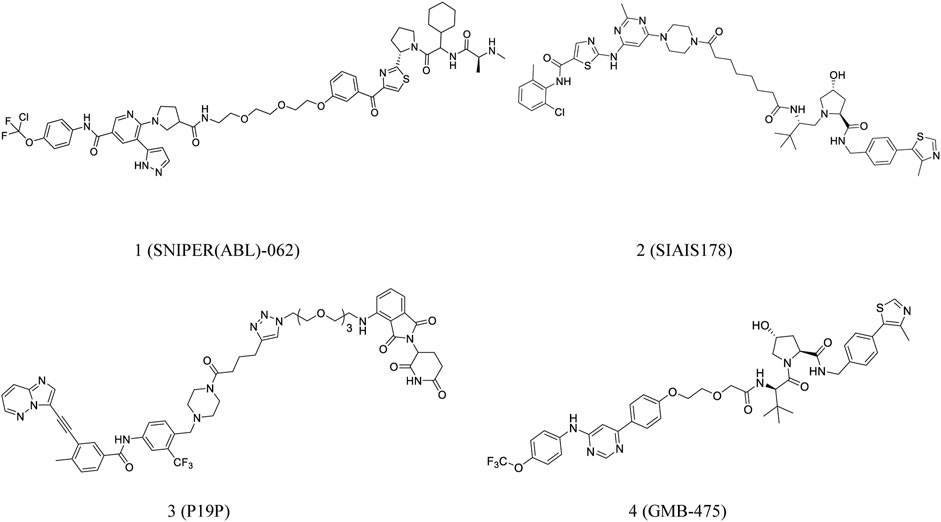
FIGURE 2. Chemical structures of PROTACs targeting BCR-ABL.
As mentioned earlier, a PROTAC consists of a target-protein ligand and an E3 ubiquitin ligase ligand coupled using a flexible linker (Bondeson et al., 2018; Cyrus et al., 2011). Zhao et al. speculated that a significant interaction between BCR-ABL and E3 ligase could be induced using an optimized linker. Based on this assumption, they developed SIAIS178 (2, Figure 2), a strong BCR-ABL degrader. SIAIS178 demonstrated appreciable selectivity and considerably inhibited the growth of BCR-ABL+ leukemic cells in vitro. It also induced substantial regression of K562 xenograft tumors in vivo (Zhao et al., 2019).
Importantly, overexpression of BCR-ABL and mutation of T315I can significantly contribute to drug resistance (Yang and Fu, 2015). However, there are no approved drugs targeting T315I-mutant cells. Yang et al. reported a series of PROTACs targeting all three binding sites of dasatinib, ponatinib, and asciminib in the BCR-ABL protein. P19P (3, Figure 2) degrades dasatinib-resistant T315I and asciminib-resistant V468F mutations in BCR-ABL and reduces the adverse effects of ponatinib (Yang et al., 2020). Moreover, GMB-475 (4, Figure 2), discovered by Crews et al., induced rapid proteasomal degradation and demonstrated increased sensitivity. It also inhibited certain BCR-ABL1 kinase domain point mutations (Burslem et al., 2019).
Proteolysis Targeting Chimeras Targeting Phosphatidylinositol 3-Kinase/Akt
Phosphatidylinositol 3-kinases (PI3Ks) are intracellular lipid kinases that play essential roles in cell survival, proliferation, growth, differentiation, and migration (Vanhaesebroeck et al., 2012; Thorpe et al., 2015). There are three classes of PI3Ks, of which Class I is widely investigated (Liu et al., 2009). Most cancer cells demonstrate hyperactivating mutations of type I PI3Ks, which drive tumor cell proliferation and survival (Sheppard et al., 2012). PI3Ks are potential anticancer targets, and agents inhibiting them are preferred in chemotherapy (Yadav et al., 2016).
PROTACs are also used for PI3K degradation (Hines et al., 2013; Guo et al., 2017). Li et al. developed new PI3K-PROTACs by associating pomalidomide and piperazine derivative using different linkers. Many of these compounds could significantly inhibit PI3Ka with IC50 values at the nanomolar level. The most potent compound B (5, Figure 3) showed an IC50 of 18 nM against PI3Ka. These compounds were also able to inhibit HepG2 cell proliferation (Li et al., 2018).

FIGURE 3. Chemical structures of PROTACs targeting PI3K/Akt.
PI3K or Akt inhibitors activate and increase SGK3 expression in patients with ER+ breast cancer (BC) receiving long-term treatment. SGK3, instead of Akt, activates mTORC1 by inducing TSC2 phosphorylation (Bago et al., 2016). These findings implied SGK3 as an effective target to overcome resistance to PI3K/Akt inhibition in cancer treatment.
Tovell et al. reported SGK3-PROTAC1 (6, Figure 3), which selectively degrades SGK3. In HEK293 cells, the PROTAC did not degrade the SGK1 and SGK2 isoforms or other proteins. SGK3-PROTAC1, at submicromolar concentrations, induced proteasomal-mediated degradation in several cancer cell lines. It was able to cause significant degradation within 2 h (Tovell et al., 2019).
Proteolysis Targeting Chimeras Targeting Bruton’s Tyrosine Kinase
Bruton’s tyrosine kinase (BTK) promotes B-cell growth, maturation, migration, and apoptosis (Campbell et al., 2018). Cancer, autoimmunity, or inflammation may be a result of dysregulated BTK pathways (Pal Singh et al., 2018). Studies have verified BTK as an essential target for agents against chronic lymphocytic leukemia (CLL). Research on BTKs is advancing considerably, with the BTK inhibitors ibrutinib (Advani et al., 2013), acalabrutinib (Byrd et al., 2016) and zanubrutinib (Guo et al., 2019) approved for use by the FDA. However, drug resistance and off-target effect are limitations of BTK inhibitors (Woyach et al., 2014). As such, there is an immediate need to develop drugs with new molecular entities and mechanisms targeting BTK or its mutants.
Zorba et al. synthesized 11 PROTACs and investigated their BTK-degrading abilities. They found that the length of a linker is crucial for promoting BTK and CRBN interaction (Zorba et al., 2018).
Recently, Buhimschi et al. reported the discovery of MT802 (7, Figure 4), which has a BTK-specific ligand and CRBN ligand connected by a PEG linker. The compound effectively degraded BTK in cells. Compared to ibrutinib, this compound had fewer off-target kinase binding and more effective BTK degradation (Buhimschi et al., 2018).
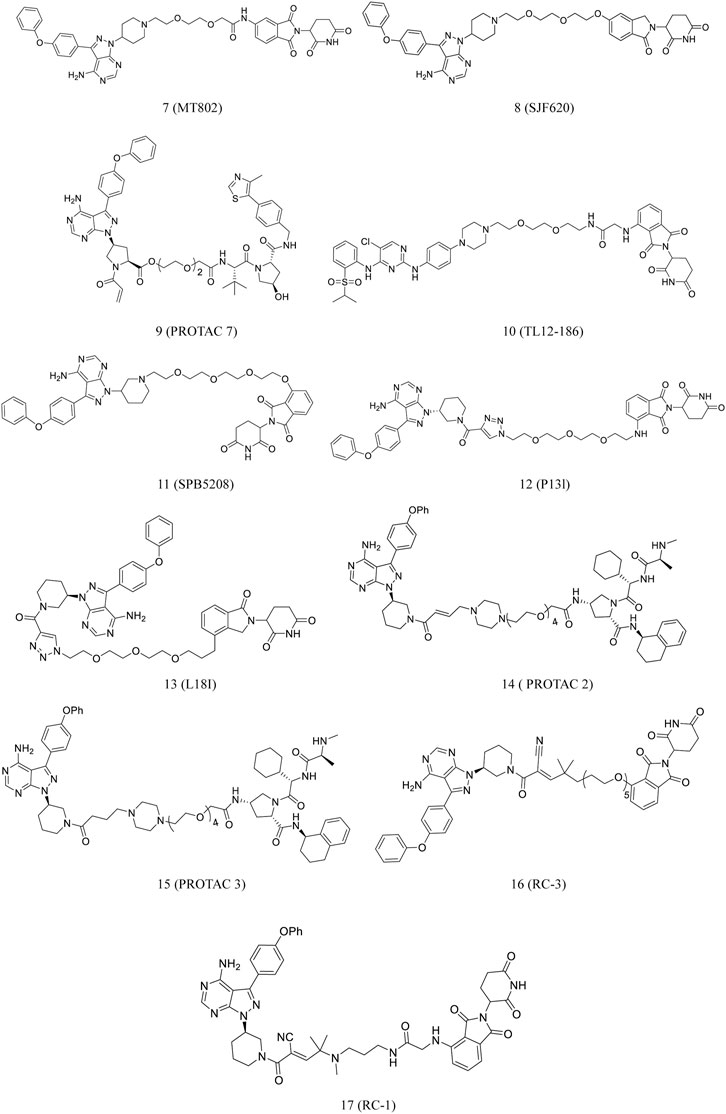
FIGURE 4. Chemical structures of PROTACs targeting BTK.
Pharmacokinetic data on MT802 in mice, however, demonstrated its unsuitability for further in vivo studies. Jaime-Figueroa et al. modified the CRBN ligand structure of MT802 to synthesize SJF620 (8, Figure 4), demonstrating a superior pharmacokinetic profile than MT802 in mice (Jaime-Figueroa et al., 2020).
Pan et al. designed and synthesized PROTAC 7 (9, Figure 4) based on their previous work. The compound significantly reduced BTK protein level and degraded the BLK protein (Chen J. et al., 2018; Xue et al., 2020).
TL12-186 (10, Figure 4), a PROTAC targeting CRBN, was developed by Huang et al. The compound demonstrated >90% inhibition of 193 kinases at a tested level of 1 μM. TL12-186 considerably downregulated the expressions of 14 out of the 7,559 identified proteins, including BTK in MOLM-14 cells, by at least 25% (Huang et al., 2018).
Liu et al. synthesized SPB5208 (11, Figure 4), a BTK-degrading PROTAC, by linking ibrutinib and thalidomide. The compound reduced the activity of BTK enzyme with high selectivity and effectively suppressed the proliferation of cancer cell in vitro. Studies on the mechanism of action revealed that it degraded BTK JeKo-1 cells in a proteasome- and CRBN-dependent fashion. Additionally, the earlier findings suggested that this compound caused significant BTK protein degradation in ICR mice in a manner of dose-dependent (Liu et al., 2020).
Ibrutinib was conjugated to pomalidomide to synthesize P13l (12. Figure 4). The compound developed by Sun et al. induced 73% degradation of BTK in RAMOS cells at 10 nM. BTK degradation began at about 4 h after treatment with PI3I and was completed by 24 h. In addition, the compound could effectively degrade BTK in MCL and MM cell lines (Sun et al., 2018). Mantle cell lymphoma (MCL) accounts for more than 6–8% of all non-Hodgkin lymphomas (NHLs) worldwide (Sun et al., 2018). The tumor cells with drug-resistant were obtained from patients with MCL in the course of ibrutinib treatment. The relapse C481S missense BTK mutation could have contributed to this resistance (Chiron et al., 2014). During the growth suppression of diffuse large B-cell lymphoma (DLBCL), the BTK C481S mutant lead to resistance to ibrutinib (Sun et al., 2018). Based on these findings, Sun et al. developed L18I (13, Figure 4), which could induce the degradation of ibrutinib-resistant C481S BTK in HBL-1 cells at 30 nM. Excitingly, L18I also caused an apparent antitumor effect in mice culture with C481S BTK HBL-1 cells (Sun et al., 2019).
To date, most reported PROTACs reported operate through covalent or noncovalent binding. Irreversible covalent inhibitors have strong target affinities and high target occupancies and have been successful in the clinical setting However, irreversible bindings may reduce the potency by negating the catalytic nature of the PROTAC activity (Gabizon et al., 2020).
Dittus et al. selected the covalent inhibitor ibrutinib and a reversibly binding analog to develop PROTAC 2 (14, Figure 4) and PROTAC 3 (15, Figure 4), which were investigated for their BTK degradation capabilities (Dittus et al., 2017). PROTAC 2 did not degrade the covalently bound targets, whereas PROTAC 3 degraded its target protein. This finding highlights the importance of catalysis for successful PROTAC-mediated degradation (Tinworth et al., 2019).
Is there an effective way to bind PROTACs covalently to the target, and at certain conditions, is this covalent bond reversible? Theoretically, the selectivity, enhanced potency, and long duration of action that accompany covalent bond formation can promote reversible covalent PROTACs (Serafimova et al., 2012; Bradshaw et al., 2015). Moreover, reversible covalent PROTACs could combine the advantages of covalent binding, including the selectivity and increased potency, while keeping the reversibility, which is necessary for catalytic properties of chemical reactions of a PROTAC’s efficacy.
Gabizon et al. tested this hypothesis by designing acrylamide- and cyanoacrylamide-based reversible covalent PROTACs. BTK was selected as the target, and irreversible covalent, noncovalent, and reversible covalent PROTACs were systematically analyzed. Cyanoacrylamide-containing PROTACs were reported to be much more potent than acrylamide analogs and equivalent noncovalent PROTACs. Based on these data, a highly potent, selective, reversible covalent RC-3 (16, Figure 4) was synthesized. The compound demonstrated increased selectivity for the BTK protein than its noncovalent and irreversible covalent counterparts (Gabizon et al., 2020). These findings suggested the potential use of PROTACs as a plethora of challenging targets.
PROTACs are high-molecular-weight compounds, resulting in poor membrane permeabilities and, subsequently, low intracellular concentrations compared with other drugs. As such, they demonstrate low target occupancy and depend heavily on substoichiometric protein degradation for therapeutic efficacy (Guo et al., 2020).
Guo et al. discovered that a cyanoacrylamide-based reversible covalent binder to BTK significantly increased target engagement and the accumulation of drug in cells (Guo et al., 2020). It is known that cyanoacrylamides reversibly react with thiols with millimolar dissociation equilibrium constants (Kd) and rapid kinetics (Krishnan et al., 2014; Bradshaw et al., 2015; Jiang et al., 2017). Based on these findings, they developed RC-1 (17, Figure 4), a reversible covalent BTK PROTAC, which was the most potent BTK degrader reported at that time (Zorba et al., 2018; Tinworth et al., 2019). The compound effectively degraded BTK at 8–40 nM. It also induced BTK degradation in MOLM-14 cells and also degraded BTK regardless of its mutation status, which was surprising. This reversible covalent strategy can be generalized to other PROTACs to develop a new approach to enhance the efficacy of a PROTAC (Guo et al., 2020).
Proteolysis Targeting Chimeras Targeting Receptor Tyrosine Kinases
Following the identification of the cDNA encoding the epidermal growth factor receptor, studies were conducted to understand critical roles of receptor tyrosine kinases (RTK) in signaling pathways governing fundamental cellular processes and processes that regulate intercellular communications.
Dysregulation of protein kinases is well recognized in cancer cells (Gossage and Eisen, 2010). EGFR, as a kind of RTK, is critically associated with the regulation of cell apoptosis, proliferation, metabolism, and survival (Yarden and Sliwkowski, 2001). Overexpression and activating mutations of EGFR are related to several cancer types. Unregulated activation of EGFR leads to abnormal cell growth and downstream signaling. These adverse effect may lead to the endocytosis of EGFR signal and intracellular terminal transport (Rosell et al., 2009; Yewale et al., 2013).
Recently, some therapeutic agents targeting EGFR with small molecules and antibodies were developed (Maennling et al., 2019). This protein has been extensively investigated in oncogenic and normal signaling, and EGFR-based anticancer drugs are commonly used therapeutically (Ayati et al., 2020). To date, over ten EGFR inhibitors have been given to patients with non-small-cell lung cancer by FDA, but the EGFR-mutant variants may compromise drug efficacy (Ayati et al., 2020). Unlike kinase activity inhibition, EGFR degradation results in complete, lasting downstream signal inactivation. PROTACs degraded several RTKs, including c-Met, HER2, EGFR, and multiple mutants of EGFR and c-Met.
Konecny et al. synthesized PROTAC 1 by conjugating lapatinib, an EGFR-binding element, to an E3 ligase binding ligand (18, Figure 5). At low nanomolar concentrations, the compound demonstrated cell-membrane penetration and EGFR degradation (Konecny et al., 2006; Buckley et al., 2012; Burslem et al., 2018).
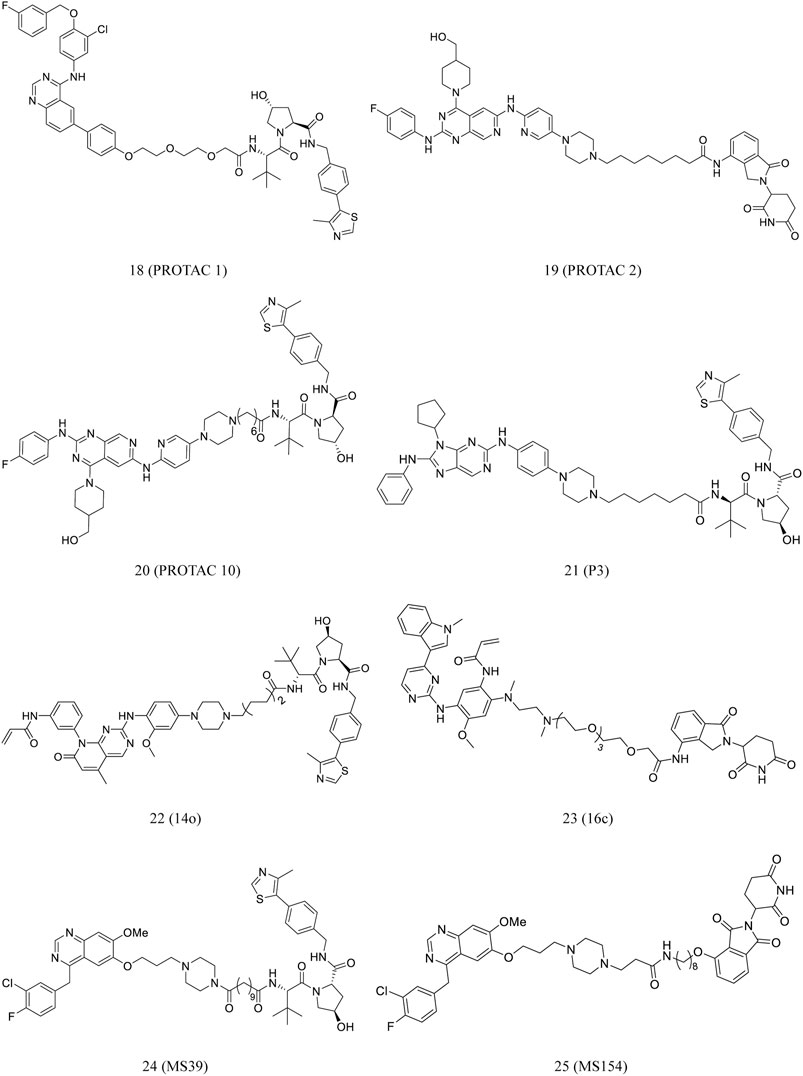
FIGURE 5. Chemical structures of PROTACs targeting RTK.
A new series of EGFR degraders containing the pyrido[3,4-d]pyrimidine moiety were proposed and developed by the Zhang et al. In HCC827 cells, PROTACs 2 (19, Figure 5) and 10 (20, Figure 5) effectively degraded EGFR. Furthermore, they significantly induced HCC827 cell apoptosis (Zhang H. et al., 2020).
Gefitinib, a first-generation EGFR TKI, targets mutated and overactive EGFR and provides considerable clinical benefits (Mok et al., 2009). However, acquired resistance is known to develop following short-term treatment with the compound (Yu et al., 2013). To overcome T790M EGFR mutation-induced drug resistance, second and third generation EGFR inhibitors were developed (Sos et al., 2010). Osimertinib, a third-generation EGFR inhibitor, was approved by FDA and used in the patients with metastatic NSCLC in 2015. Regarding the mechanism of drug action, the acrylamide moiety in osimertinib undergoes a Michael addition reaction with the Cys797 residue to get inhibitory activity against the EGFRT790M mutants (Finlay et al., 2014; Jänne et al., 2015). Unfortunately, newly acquired drug resistance has been identified (Thress et al., 2015; Bersanelli et al., 2016; Zheng et al., 2017; Chen L. et al., 2018).
EGFRL858R/T790M serves as an ideal and relatively safe target for PROTACs because it is selectively expressed in tumor cells with acquired drug resistance, but it can be found in normal cells. Multiple selective EGFRL858R/T790M mutant PROTAC degraders were proposed and developed by Zhang et al. Of these degraders, 14o (22, Figure 5) is the most potential as PROTAC to effectively and selectively degrade EGFRL858R/T790M (Zhang X. et al., 2020).
Zhao et al. reported a series of EGFR-targeting small-molecule PROTACs, which demonstrated potent efficacies. Of these PROTACs, P3 (21, Figure 5) displayed intense antiproliferative activity against the cell lines of H1975 and HCC827. In this process, the IC50 values of P3 (21, Figure 5) against H1975 and HCC827 was 203.01 and 0.83 nM, respectively. Moreover, It was reported that P3 treatment significantly degraded EGFRL858R/T790M and EGFRdel19 (Zhao et al., 2020).
He et al. developed a class of small-molecule EGFR degraders by conjugating lenalidomide with osimertinib through different linkers. Compound 16c (23, Figure 5) effectively degraded EGFR in PC9 cells, with the maximum degradation rate at 68%. Furthermore, it promoted PC9 cell apoptosis and arrest in the G0/G1 phase (He et al., 2020).
Cheng et al. developed a novel gefitinib-based VHL-recruiting EGFR degrader (MS39, 24, Figure 5) and a first-in-class gefitinib-based CRBN-recruiting EGFR degrader (MS154, 25, Figure 5). Both compounds selectively induced degradation of mutant EGFR and downstream signaling inhibition in cells, whereas no apparent effect was noted on the wild-type protein (Cheng et al., 2020).
Proteolysis Targeting Chimeras Targeting Focal Adhesion Kinase
Focal adhesion kinase (FAK), a cytoplasmic tyrosine kinase that was first reported in 1992 (Mitra et al., 2005). FAK promotes tumor growth, invasion, and metastasis through kinase-dependent manner. It was found that FAK acts as a kinase and serves as a scaffold for some signal proteins (Hanks et al., 2003; Parsons, 2003; Sulzmaier et al., 2014; Lee et al., 2015; Aboubakar Nana et al., 2019). It was suggested that the increased FAK expression and activity in the primary and metastatic cancer tissues played a critical role in the poor survival of patients (Sulzmaier et al., 2014).
FAK inhibition in metastatic cancers can potentially prevent tumor progression. FAK inhibitors regulate cancer cell angiogenesis, invasion, and migration, and ECs in tumor angiogenesis. Several studies have been conducted to investigate the clinical effect of small-molecule FAK inhibitors (Chambers et al., 2002; Parsons et al., 2008; Lu and Sun, 2020).
Unfortunately, FAK has both kinase-dependent and kinase-independent functions, implying that FAK inhibitors can antagonize its kinase-dependent functions but not kinase-independent functions. However, PROTACs targeting FAK can inhibit both kinase-dependent and kinase-independent functions of the kinase (Gao et al., 2019).
According to clinical FAK inhibitor defactinib, Luzzio et al. designed certain FAK-targeting PROTACs. Guided by earlier investigations on the SAR, the aminomethyl part of defactinib was used as a linker incorporation (Luzzio et al., 2017). The most promising degrader, PROTAC-3 (26, Figure 6), is superior to defactinib in inhibiting FAK signaling and cell migration and invasion in human triple-negative breast cancer cells. Moreover, PROTAC-3 was reported to improve selectivity over defactinib, as it only binds to FAK with less than 1% of the control compound remaining (Cromm et al., 2018).
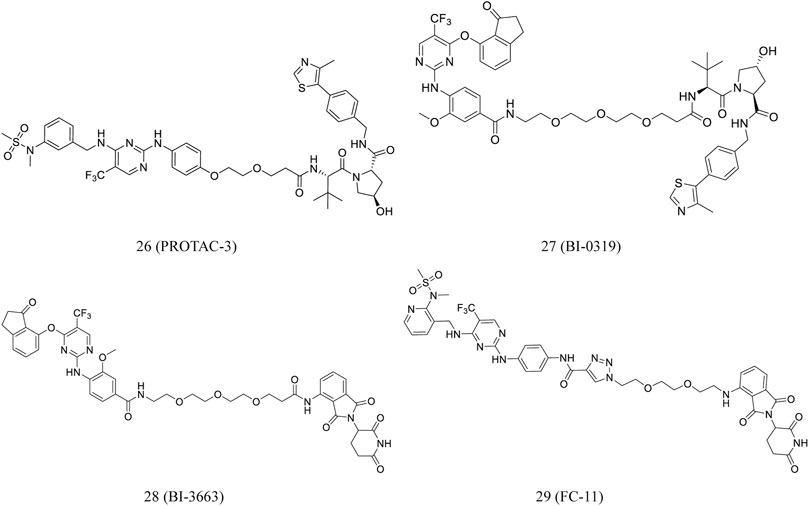
FIGURE 6. Chemical structures of PROTACs targeting FAK.
Human hepatocellular carcinoma (HCC) cells overexpress focal adhesion tyrosine kinase (PTK2). Highly selective PROTACs targeting PTK2 degradation were developed by Popow et al. Structure-guided conjugation of a highly selective PTK2 inhibitor to either a CRBN or VHL ligand by polyethylene glycol linkers led to the development of selective PTK2 degraders BI-0319 (27, Figure 6) and BI-3663 (28, Figure 6), respectively. Both compounds could selectively degrade the PTK2 protein. However, further research suggested that BI-3663- and BI-0319-mediated PTK2 depletion is unlikely sufficient to investigate the inhibition effect of kinase activity under the condition of affecting proliferation in vitro (Popow et al., 2019).
Based on the FAK inhibitor and CRBN E3 ligand, Gao et al. developed a FAK PROTAC library. FC-11 (29, Figure 6), a novel FAK-targeting PROTAC, rapidly degraded FAK after 8 h of treatment in various cell lines. Furthermore, the FAK proteins can be completely recovered after being washed away by PROTAC molecules (Gao et al., 2019).
Proteolysis Targeting Chimeras Targeting Cyclin-dependent Kinase
The previous study reported that in the cyclin dependent kinases (CDKs) family, CDK1-4, CDK11, and CDK6 regulate the cell cycle, whereas CDK7-9 mainly regulated transcription (Bellan et al., 2004). In the earlier studies, clinical trials have been investigating the effects of small-molecule CDK inhibitors on various cancers, such as acute myeloid leukemia (AML), BC, NSCLC, and prostate cancer (PC) (Malumbres et al., 2008; Heathcote et al., 2010; Parry et al., 2010; Siemeister et al., 2012; Cicenas et al., 2014; Heptinstall et al., 2018). In 2015, palbociclib, the first selective CDK4/6 inhibitor, was approved for the treatment of metastatic BC by the FDA (Bedard et al., 2020).
Furthermore, terminally differentiated cells demonstrate increased CDK9 expression (Bagella et al., 1998). Some studies found that selective CDK9 inhibitors had the potential to treat a variety of human diseases, including cancer. A lot of evidence suggested that CDK9 is a potential tumor therapeutic target. The previous studies illustrated that the adverse effects and toxicities limited the wide application of CDK9 inhibitors in clinic (Diab et al., 2007; Tong et al., 2010). Therefore, there is a need to develop feasible treatment strategies for CDK9-induced malignant tumors (Wu et al., 2020).
PROTAC 3 (30, Figure 7) was developed by conjugating an aminopyrazole derivative with thalidomide. CDK9 can be degraded by this compound in a dose-dependent manner. The previous study found that in CDK family members, this compound only selectively degrades CDK9 in HCT116 cells (Robb et al., 2017).
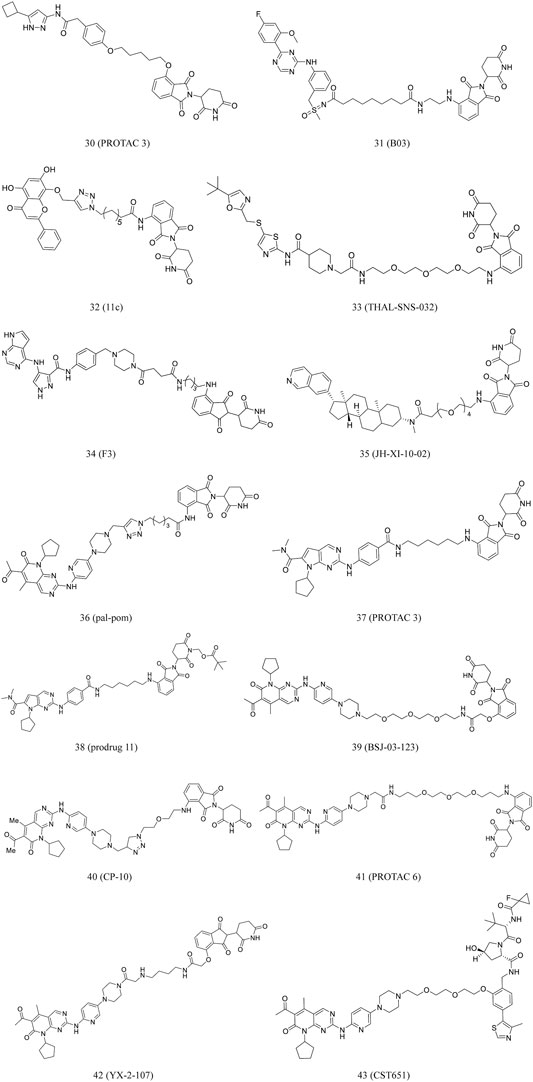
FIGURE 7. Chemical structures of PROTACs targeting CDK.
Qiu et al. converted the CDK9 inhibitor BAY-1143572 into some PROTACs, which induced CDK9 degradation in AML cells at low nanomolar concentrations. The most potent compound, B03 (31, Figure 7), inhibited cell growth much more effectively than BAY-1143572 alone, with no evident inhibition of other kinases. Moreover, it could induce CDK9 degradation in vivo (Qiu et al., 2021).
Wogonin, as a product obtained from Scutellaria baicalensis, is a potent and selective inhibitor of CDK9 (IC50 = 0.19 μM). In vivo studies found that wogonin induced apoptosis and suppressed the growth of human cancer xenografts (Polier et al., 2011; Ding et al., 2012). By using this scaffold, Bian et al. discovered CDK9-targeting PROTAC 11c (32, Figure 7) by recruiting ubiquitin E3 ligase and CRBN (Bian et al., 2018). Western blot results revealed that 11c selectively downregulated the intracellular CDK9 level in a concentration-dependent manner (Bian et al., 2018). The researchers also found that 11c has the ability to promote proteasome- and CRBN-dependent degradation. This study contributed to the development of PROTACs derived from natural products for cancer treatment.
Olson et al. developed THAL-SNS-032 (33, Figure 7) by conjugating selective ATP competitive CDK9 inhibitor and selective CDK9 degradation agent composed of thalidomide. Surprisingly, it was found that the compound induced highly selective and rapid degradation of CDK9 without affecting the levels of other related targets (Olson et al., 2018).
Zhou et al. have been investigating small-molecule degraders targeting CDKs. They identified F3 (34, Figure 7), a dual degrader, that could potentially degrade both CDK9 and CDK2. It inhibited cell proliferation through effective blockade of the S and G2/M phases of PC-3 cells. Furthermore, it degraded CDK2/9 activities in various cancer cell lines, suggesting its anticancer potential (Zhou F. et al., 2020).
CDK8 modulates gene transcription and, therefore, plays an important role in the function of stem cells, inflammation and immune response (Carlsten et al., 2013; Allen and Taatjes, 2015). In melanoma tissues, CDK8 was overexpressed, indicating its essential role in cell proliferation (Kapoor et al., 2010). CDK8 knockdown suppressed cell growth and promoted cell cycle arrest, and hence apoptosis, in tumor models (Firestein et al., 2008). These findings indicate that small-molecule-induced inhibition of CDK may be a promising therapeutic strategy for cancer.
Hatcher et al. conducted a study to investigated highly selective CDK8 inhibitor, JH-VIII-49. The compound is an analog of cortistatin A with slightly reduced efficacy against CDK8 in biochemical and cellular tests. According to this scaffold, they further developed JH-XI-10-02 (35, Figure 7), a bivalent degrader that can induce significant degradation of CDK8 via recruiting the E3 ligase in Jurkat cells (Hatcher et al., 2018).
In cancer treatment, CDK4/6 are attractive targets, and palbociclib, ribociclib, and abemaciclib, are dual inhibitors. The FDA approved CDK4/6 inhibitors for advanced or metastatic BC to enhance. progression-free survival by FDA (Bedard et al., 2020).
A series of imide-based degrader molecules with the potential to inhibit CDK4/6-induced cellular effects were developed by Jiang et al. These compounds successfully reduced pRB levels, promoted G1 arrest, and induced antiproliferative activity in the treated cells (Jiang et al., 2019). Similarly, Zhao and Burgess developed CDK4/6-targeting PROTACs synthesized by conjugating pomalidomide to palbociclib or ribociclib. The most potent compound, pal-pom (36, Figure 7), was remarkably potent than other CDK8-9-targeting compounds (Zhao and Burgess, 2019).
CDK2 overexpression and its complex with cyclins are positively associated with dysregulation of the cell cycle (Smalley et al., 2008). CDK 2/4/6-targeting inhibitors were regarded as a feasible chemotherapeutic regimen.
According to a ribociclib derivative and a CRBN ligand, Wei et al. developed PROTAC 3 (37, Figure 7). This compound could simultaneously and effectively degrade CDK 2/4/6 and their complexes in melanomas. Furthermore, in various types of cancer cells, it can quickly start the cell cycle and cause apoptosis. The structure of PROTAC 3 was modified to obtain its prodrug 11 (38, Figure 7), which demonstrated good oral bioavailability in animal studies (Wei et al., 2021).
The FDA has approved several CDK4 and CDK6 inhibitors for effective treatment of BC. Recent reports have highlighted the central role of CDK6 in transcriptional regulation (Kollmann et al., 2016). However, it is challenging to investigate its kinase-independent functions due to lack of selectivity. Brand et al. reported BSJ-03-123 (39, Figure 7), a degradation agent based on phthalimide (Su et al., 2019). It was found that this degradation agent can achieve proteomic selectivity of CDK6 degradation by using protein interface determinants (Brand et al., 2019).
Su et al. developed a compound library of CDK6 degraders by linking palbociclib (CDK inhibitor) and pomalidomide (E3 ligase CRBN recruiter). These PROTAC degraders strongly inhibited proliferation of hematopoietic cancer cells and degraded copy-amplified or mutated CDK6.
Moreover, to understand the binding mechanism between PROTAC and target, researchers evaluated binding affinity, spatial orientation, and linker length. The most potent PROTAC CP-10 (40, Figure 7) prevented the proliferation of several hematopoietic cancer cells. Additionally, it was reported that this compound can degrade CDK6 from mutation and overexpression (Su et al., 2019).
In contrast to findings of Su et al. (Su et al., 2019), which suggested that VHL or IAP PROTACs could not degrade CDK4 and CDK6, Anderson et al. observed that palbociclib-based PROTACs recruiting different E3 ligases demonstrated preferential CDK6 vs. CDK4 degradation selectivity (Anderson et al., 2020).
Rana et al. reported the development of five PROTACs synthesized by conjugating palbociclib to pomalidomide using flexible linkers of varying lengths and compositions. PROTAC 6 (41, Figure 7) demonstrated potent, selective degradation of CDK6, sparing other CDKs (Rana et al., 2019).
CDK6 silencing seems to be more effective than palbociclib in suppressing Ph+ ALL in mice models. De Domici et al. developed PROTACs that induced CDK6 degradation in vitro. YX-2-107 (42, Figure 7) could promote the rapid and preferential degradation of CDK6 over CDK4 in Ph+ ALL cells, as well as inhibit the activity of phospho-RB, FOXM1, and proliferation (De Dominici et al., 2020).
VHL-based PROTACs, developed by Steinebach et al., are highly active, selective CDK-6 inhibitor with demonstrated efficacy in several human and murine cancer cells. Of these PROTACs, CST651 (43, Figure 7) selectively induced CDK6 degradation, and in this process, when the effective concentration was 0.1 μM, it did not affect the viability of MDA-MB-231 cells. The compound demonstrated effective and durable degradation of human and mouse cells. It also suppressed the proliferation of myeloma, leukemia, and BC cells. Moreover, CST651 could significantly impair cell migration and reduce wound healing by 29% (Steinebach et al., 2020).
Proteolysis Targeting Chimeras Targeting MEK1/2
The previous study suggested that the RAS-RAF-MEK-ERK was involved in several cellular processes, such as proliferation, differentiation, apoptosis, migration, and metabolism (Scholl et al., 2007; Roskoski, 2012). Additionally, MEK1 and MEK2 played important roles in regulating the activity of ERK. ERK signaling hyperactivation induced by MEK1 or MEK2 receptor mutations is related to human cancers. To date, four drugs targeting MEK1/2, selumetinib, binimetinib, cobimetinib, trametinib, were approved by the FDA for treatment of cancer. However, drug resistance to MEK1/2 inhibitors in patients gradually appeared in clinical use, implying the need for new therapeutic strategies.
Wei et al. reported MS432 (44, Figure 8), a first-in-class PROTAC of MEK1/2, developed by conjugated the MEK1/2 inhibitor PD0325901. The compound effectively degraded MEK1 and MEK2 in a concentration- and time-dependent and durable manner and prevented the phosphorylation of ERK in cellular experiments. In addition, the compound was well exposed in the plasma of mice, indicating its suitability for in vivo efficacy studies (Wei et al., 2019).
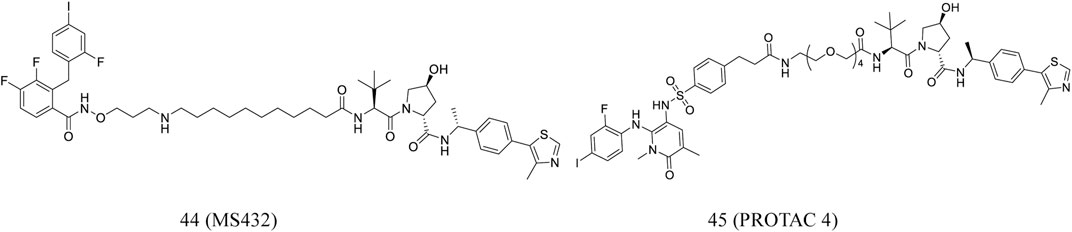
FIGURE 8. Chemical structures of PROTACs targeting MEK.
Vollmer et al. designed PROTAC 4 (45, Figure 8) according to PEG linker and allosteric MEK inhibitors. Although PROTAC 4 is less effective in inhibiting ERK1/2 phosphorylation, it is more potent in inhibiting A375 cell proliferation (Vollmer et al., 2020).
Perspective
PROTACS can disrupt enzymatic and non-enzymatic functions of target proteins, demonstrating a novel approach to address acquired drug resistance. The aforementioned findings indicate the significant progress of PROTAC technology in recent years.
This strategy is particularly suitable for protein kinases. Abnormal functions of protein kinases have been closely related to cancers, highlighting the potential importance of these kinases as therapeutic targets for anti-cancer drug development. Nowadays, both multi-targeted and highly selective kinase inhibitors are used for the treatment of cancers. However, the mutant kinases and emergence of drug resistance are still major factors limiting the efficacy of kinase inhibitors.
In the past few decades, numerous ligands selectively targeting kinases as competitive or allosteric inhibitors have been developed, which provides an appropriate basis for PROTAC molecular designing. In addition, the kinase degradation mediated by PROTACs can also be used to regulate phosphorylation, expanding the applications of PROTACs.
Although the benefits of PROTACs are significant, they still have certain limitations, which need further investigation. Further insights into the action mechanisms involved in these challenges will enable scientists to address the limitations and develop next-generation PROTACs. The emergence of novel PROTACs, including reversible covalent protein-degraders, indicates their potential to be used as antitumor therapeutic agents targeting protein kinases in addition to small-molecule inhibitors and monoclonal antibodies.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81960670) and Science and Technology Plan Project of Science and Technology Department of Yunnan Province (Grant No. 202001AS070012, 2018FB145, 2019FB125).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aboubakar Nana, F., Lecocq, M., Ladjemi, M. Z., Detry, B., Dupasquier, S., Feron, O., et al. (2019). Therapeutic Potential of Focal Adhesion Kinase Inhibition in Small Cell Lung Cancer. Mol. Cancer Ther. 18 (1), 17–27. doi:10.1158/1535-7163.MCT-18-0328
Advani, R. H., Buggy, J. J., Sharman, J. P., Smith, S. M., Boyd, T. E., Grant, B., et al. (2013). Bruton Tyrosine Kinase Inhibitor Ibrutinib (PCI-32765) has Significant Activity in Patients With Relapsed/Refractory B-Cell Malignancies. J. Clin. Oncol. 31 (1), 88–94. doi:10.1200/JCO.2012.42.7906
Allen, B. L., and Taatjes, D. J. (2015). The Mediator Complex: A Central Integrator of Transcription. Nat. Rev. Mol. Cell Biol. 16 (3), 155–166. doi:10.1038/nrm3951
Anderson, N. A., Cryan, J., Ahmed, A., Dai, H., McGonagle, G. A., Rozier, C., et al. (2020). Selective CDK6 Degradation Mediated by Cereblon, VHL, and Novel IAP-Recruiting PROTACs. Bioorg. Med. Chem. Lett. 30 (9), 127106. doi:10.1016/j.bmcl.2020.127106
Ayati, A., Moghimi, S., Salarinejad, S., Safavi, M., Pouramiri, B., and Foroumadi, A. (2020). A Review on Progression of Epidermal Growth Factor Receptor (EGFR) Inhibitors as an Efficient Approach in Cancer Targeted Therapy. Bioorg. Chem. 99, 103811. doi:10.1016/j.bioorg.2020.103811
Bagella, L., MacLachlan, T. K., Buono, R. J., Pisano, M. M., Giordano, A., and De Luca, A. (1998). Cloning of Murine CDK9/PITALRE and its Tissue-specific Expression in Development. J. Cell. Physiol. 177 (2), 206–213. doi:10.1002/(SICI)1097-4652(199811)177:2<206::AID-JCP2>3.0.CO;2-R
Bago, R., Sommer, E., Castel, P., Crafter, C., Bailey, F. P., Shpiro, N., et al. (2016). The hVps34‐ SGK 3 Pathway Alleviates Sustained PI3K/Akt Inhibition by Stimulating mTORC 1 and Tumour Growth. EMBO J. 35 (17), 1902–1922. doi:10.15252/embj.201693929
Bedard, P. L., Hyman, D. M., Davids, M. S., and Siu, L. L. (2020). Small Molecules, Big Impact: 20 Years of Targeted Therapy in Oncology. Lancet 395 (10229), 1078–1088. doi:10.1016/S0140-6736(20)30164-1
Bellan, C., De Falco, G., Lazzi, S., Micheli, P., Vicidomini, S., Schürfeld, K., et al. (2004). CDK9/CYCLIN T1 Expression during normal Lymphoid Differentiation and Malignant Transformation. J. Pathol. 203 (4), 946–952. doi:10.1002/path.1588
Bersanelli, M., Minari, R., Bordi, P., Gnetti, L., Bozzetti, C., Squadrilli, A., et al. (2016). L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR -Mutated NSCLC. J. Thorac. Oncol. 11 (10), e121–e123. doi:10.1016/j.jtho.2016.05.019
Bian, J., Ren, J., Li, Y., Wang, J., Xu, X., Feng, Y., et al. (2018). Discovery of Wogonin-Based PROTACs Against CDK9 and Capable of Achieving Antitumor Activity. Bioorg. Chem. 81, 373–381. doi:10.1016/j.bioorg.2018.08.028
Bondeson, D. P., Smith, B. E., Burslem, G. M., Buhimschi, A. D., Hines, J., Jaime-Figueroa, S., et al. (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 25 (1), 78–87. doi:10.1016/j.chembiol.2017.09.010
Boschelli, D. H., Ye, F., Wang, Y. D., Dutia, M., Johnson, S. L., Wu, B., et al. (2001). Optimization of 4-Phenylamino-3-Quinolinecarbonitriles as Potent Inhibitors of Src Kinase Activity. J. Med. Chem. 44 (23), 3965–3977. doi:10.1021/jm0102250
Bradshaw, J. M., McFarland, J. M., Paavilainen, V. O., Bisconte, A., Tam, D., Phan, V. T., et al. (2015). Prolonged and Tunable Residence Time Using Reversible Covalent Kinase Inhibitors. Nat. Chem. Biol. 11 (7), 525–531. doi:10.1038/nchembio.1817
Brand, M., Jiang, B., Bauer, S., Donovan, K. A., Liang, Y., Wang, E. S., et al. (2019). Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 26 (2), 300–306.e9. doi:10.1016/j.chembiol.2018.11.006
Buckley, D. L., Gustafson, J. L., Van Molle, I., Roth, A. G., Tae, H. S., Gareiss, P. C., et al. (2012). Small-Molecule Inhibitors of the Interaction Between the E3 Ligase VHL and HIF1α. Angew. Chem. Int. Ed. 51 (46), 11463–11467. doi:10.1002/anie.201206231
Buhimschi, A. D., Armstrong, H. A., Toure, M., Jaime-Figueroa, S., Chen, T. L., Lehman, A. M., et al. (2018). Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 57 (26), 3564–3575. doi:10.1021/acs.biochem.8b00391
Burslem, G. M., Schultz, A. R., Bondeson, D. P., Eide, C. A., Savage Stevens, S. L., Druker, B. J., et al. (2019). Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 79 (18), 4744–4753. doi:10.1158/0008-5472.CAN-19-1236
Burslem, G. M., Smith, B. E., Lai, A. C., Jaime-Figueroa, S., McQuaid, D. C., Bondeson, D. P., et al. (2018). The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 25 (1), 67–77.e3. doi:10.1016/j.chembiol.2017.09.009
Byrd, J. C., Harrington, B., O’Brien, S., Jones, J. A., Schuh, A., Devereux, S., et al. (2016). Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 374 (4), 323–332. doi:10.1056/NEJMoa1509981
Campbell, R., Chong, G., and Hawkes, E. (2018). Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, beyond Hematological Malignancies. J. Clin. Med. 7 (4), 62. doi:10.3390/jcm7040062
Carlsten, J. O. P., Zhu, X., and Gustafsson, C. M. (2013). The Multitalented Mediator Complex. Trends Biochem. Sci. 38 (11), 531–537. doi:10.1016/j.tibs.2013.08.007
Chambers, A. F., Groom, A. C., and MacDonald, I. C. (2002). Dissemination and Growth of Cancer Cells in Metastatic Sites. Nat. Rev. Cancer 2 (8), 563–572. doi:10.1038/nrc865
Chen, J., Wang, X., He, F., and Pan, Z. (2018). Development of a Selective Labeling Probe for Bruton’s Tyrosine Kinase Quantification in Live Cells. Bioconjug. Chem. 29 (5), 1640–1645. doi:10.1021/acs.bioconjchem.8b00137
Chen, L., Fu, W., Zheng, L., Liu, Z., and Liang, G. (2018). Recent Progress of Small-Molecule Epidermal Growth Factor Receptor (EGFR) Inhibitors against C797S Resistance in Non-Small-Cell Lung Cancer. J. Med. Chem. 61 (10), 4290–4300. doi:10.1021/acs.jmedchem.7b01310
Cheng, M., Yu, X., Lu, K., Xie, L., Wang, L., Meng, F., et al. (2020). Discovery of Potent and Selective Epidermal Growth Factor Receptor (EGFR) Bifunctional Small-Molecule Degraders. J. Med. Chem. 63 (3), 1216–1232. doi:10.1021/acs.jmedchem.9b01566
Chiron, D., Di Liberto, M., Martin, P., Huang, X., Sharman, J., Blecua, P., et al. (2014). Cell-Cycle Reprogramming for PI3K Inhibition Overrides a Relapse-specific C481S BTK Mutation Revealed by Longitudinal Functional Genomics in Mantle Cell Lymphoma. Cancer Discov. 4 (9), 1022–1035. doi:10.1158/2159-8290.CD-14-0098
Cicenas, J., Kalyan, K., Sorokinas, A., Jatulyte, A., Valiunas, D., Kaupinis, A., et al. (2014). Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers 6 (4), 2224–2242. doi:10.3390/cancers6042224
Coleman, K. G., and Crews, C. M. (2018). Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annu. Rev. Cancer Biol. 2, 41–58. doi:10.1146/annurev-cancerbio-030617-050430
Cromm, P. M., Samarasinghe, K. T. G., Hines, J., and Crews, C. M. (2018). Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 140 (49), 17019–17026. doi:10.1021/jacs.8b08008
Cyrus, K., Wehenkel, M., Choi, E.-Y., Han, H.-J., Lee, H., Swanson, H., et al. (2011). Impact of Linker Length on the Activity of PROTACs. Mol. Biosyst. 7 (2), 359–364. doi:10.1039/c0mb00074d
De Dominici, M., Porazzi, P., Xiao, Y., Chao, A., Tang, H.-Y., Kumar, G., et al. (2020). Selective Inhibition of Ph-Positive ALL Cell Growth Through Kinase-Dependent and -Independent Effects by CDK6-Specific PROTACs. Blood 135 (18), 1560–1573. doi:10.1182/blood.2019003604
Deininger, M., Buchdunger, E., and Druker, B. J. (2005). The Development of Imatinib as a Therapeutic Agent for Chronic Myeloid Leukemia. Blood 105 (7), 2640–2653. doi:10.1182/blood-2004-08-3097
Demizu, Y., Shibata, N., Hattori, T., Ohoka, N., Motoi, H., Misawa, T., et al. (2016). Development of BCR-ABL Degradation Inducers via the Conjugation of an Imatinib Derivative and a cIAP1 Ligand. Bioorg. Med. Chem. Lett. 26 (20), 4865–4869. doi:10.1016/j.bmcl.2016.09.041
Diab, S., Eckhardt, S., Tan, A., Frenette, G., Gore, L., Depinto, W., et al. (2007). A Phase I Study of R547, a Novel, Selective Inhibitor of Cell Cycle and Transcriptional Cyclin Dependent Kinases (CDKs). J. Clin. Oncol. 25, 3528. doi:10.1200/jco.2007.25.18_suppl.3528
Ding, J., Polier, G., Köhler, R., Giaisi, M., Krammer, P. H., and Li-Weber, M. (2012). Wogonin and Related Natural Flavones Overcome Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Protein Resistance of Tumors by Down-Regulation of c-FLIP Protein and Up-Regulation of TRAIL Receptor 2 Expression*. J. Biol. Chem. 287 (1), 641–649. doi:10.1074/jbc.M111.286526
Dittus, L., Werner, T., Muelbaier, M., and Bantscheff, M. (2017). Differential Kinobeads Profiling for Target Identification of Irreversible Kinase Inhibitors. ACS Chem. Biol. 12 (10), 2515–2521. doi:10.1021/acschembio.7b00617
Finlay, M. R. V., Anderton, M., Ashton, S., Ballard, P., Bethel, P. A., Box, M. R., et al. (2014). Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations that Spares the Wild Type Form of the Receptor. J. Med. Chem. 57 (20), 8249–8267. doi:10.1021/jm500973a
Firestein, R., Bass, A. J., Kim, S. Y., Dunn, I. F., Silver, S. J., Guney, I., et al. (2008). CDK8 is a Colorectal Cancer Oncogene That Regulates β-Catenin Activity. Nature 455 (7212), 547–551. doi:10.1038/nature07179
Gabizon, R., Shraga, A., Gehrtz, P., Livnah, E., Shorer, Y., Gurwicz, N., et al. (2020). Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 142 (27), 11734–11742. doi:10.1021/jacs.9b13907
Gao, H., Wu, Y., Sun, Y., Yang, Y., Zhou, G., and Rao, Y. (2019). Design, Synthesis, and Evaluation of Highly Potent FAK-Targeting PROTACs. ACS Med. Chem. Lett. 11 (10), 1855–1862. doi:10.1021/acsmedchemlett.9b00372
Gossage, L., and Eisen, T. (2010). Targeting Multiple Kinase Pathways: A Change in Paradigm. Clin. Cancer Res. 16 (7), 1973–1978. doi:10.1158/1078-0432.CCR-09-3182
Gozgit, J. M., Wong, M. J., Moran, L., Wardwell, S., Mohemmad, Q. K., Narasimhan, N. I., et al. (2012). Ponatinib (AP24534), a Multitargeted Pan-FGFR Inhibitor with Activity in Multiple FGFR-Amplified or Mutated Cancer Models. Mol. Cancer Ther. 11 (3), 690–699. doi:10.1158/1535-7163.MCT-11-0450
Guo, W.-H., Qi, X., Yu, X., Liu, Y., Chung, C.-I., Bai, F., et al. (2020). Enhancing Intracellular Accumulation and Target Engagement of PROTACs With Reversible Covalent Chemistry. Nat. Commun. 11 (1), 4268. doi:10.1038/s41467-020-17997-6
Guo, W., You, X., Wang, X., Wang, L., and Chen, Y. (2017). A Synthetic Peptide Hijacks the Catalytic Subunit of Class I PI3K to Suppress the Growth of Cancer Cells. Cancer Lett. 405, 1–9. doi:10.1016/j.canlet.2017.07.015
Guo, Y., Liu, Y., Hu, N., Yu, D., Zhou, C., Shi, G., et al. (2019). Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 62 (17), 7923–7940. doi:10.1021/acs.jmedchem.9b00687
Hanks, S. K., Ryzhova, L., Shin, N. Y., and Brábek, J. (2003). Focal Adhesion Kinase Signaling Activities and Their Implications in the Control of Cell Survival and Motility. Front. Biosci. 8, d982–996. doi:10.2741/1114
Hatcher, J. M., Wang, E. S., Johannessen, L., Kwiatkowski, N., Sim, T., and Gray, N. S. (2018). Development of Highly Potent and Selective Steroidal Inhibitors and Degraders of CDK8. ACS Med. Chem. Lett. 9 (6), 540–545. doi:10.1021/acsmedchemlett.8b00011
He, K., Zhang, Z., Wang, W., Zheng, X., Wang, X., and Zhang, X. (2020). Discovery and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) as an EGFR Degraders Based on Osimertinib and Lenalidomide. Bioorg. Med. Chem. Lett. 30 (12), 127167. doi:10.1016/j.bmcl.2020.127167
Heathcote, D. A., Patel, H., Kroll, S. H. B., Hazel, P., Periyasamy, M., Alikian, M., et al. (2010). A Novel Pyrazolo[1,5-A]pyrimidine Is a Potent Inhibitor of Cyclin-Dependent Protein Kinases 1, 2, and 9, Which Demonstrates Antitumor Effects in Human Tumor Xenografts Following Oral Administration. J. Med. Chem. 53 (24), 8508–8522. doi:10.1021/jm100732t
Heptinstall, A. B., Adiyasa, I., Cano, C., and Hardcastle, I. R. (2018). Recent Advances in CDK Inhibitors for Cancer Therapy. Future Med. Chem. 10 (11), 1369–1388. doi:10.4155/fmc-2017-0246
Hines, J., Gough, J. D., Corson, T. W., and Crews, C. M. (2013). Posttranslational Protein Knockdown Coupled to Receptor Tyrosine Kinase Activation With phosphoPROTACs. Proc. Natl. Acad. Sci. 110 (22), 8942–8947. doi:10.1073/pnas.1217206110
Huang, H.-T., Dobrovolsky, D., Paulk, J., Yang, G., Weisberg, E. L., Doctor, Z. M., et al. (2018). A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cel Chem. Biol. 25 (1), 88–99. doi:10.1016/j.chembiol.2017.10.005
Jaime-Figueroa, S., Buhimschi, A. D., Toure, M., Hines, J., and Crews, C. M. (2020). Design, Synthesis and Biological Evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK Degraders With Improved Pharmacokinetic Properties. Bioorg. Med. Chem. Lett. 30 (3), 126877. doi:10.1016/j.bmcl.2019.126877
Jänne, P. A., Yang, J. C.-H., Kim, D.-W., Planchard, D., Ohe, Y., Ramalingam, S. S., et al. (2015). AZD9291 in EGFR Inhibitor-Resistant Non-Small-Cell Lung Cancer. N. Engl. J. Med. 372 (18), 1689–1699. doi:10.1056/NEJMoa1411817
Jiang, B., Wang, E. S., Donovan, K. A., Liang, Y., Fischer, E. S., Zhang, T., et al. (2019). Development of Dual and Selective Degraders of Cyclin‐Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. 58 (19), 6321–6326. doi:10.1002/anie.201901336
Jiang, X., Chen, J., Bajić, A., Zhang, C., Song, X., Carroll, S. L., et al. (2017). Quantitative Real-Time Imaging of Glutathione. Nat. Commun. 8, 16087. doi:10.1038/ncomms16087
Kapoor, A., Goldberg, M. S., Cumberland, L. K., Ratnakumar, K., Segura, M. F., Emanuel, P. O., et al. (2010). The Histone Variant macroH2A Suppresses Melanoma Progression Through Regulation of CDK8. Nature 468 (7327), 1105–1109. doi:10.1038/nature09590
Kollmann, K., Heller, G., Schneckenleithner, C., Warsch, W., Scheicher, R., Ott, R. G., et al. (2016). A Kinase-Independent Function of CDK6 Links the Cell Cycle to Tumor Angiogenesis. Cancer Cell 30 (2), 359–360. doi:10.1016/j.ccell.2016.07.003
Konecny, G. E., Pegram, M. D., Venkatesan, N., Finn, R., Yang, G., Rahmeh, M., et al. (2006). Activity of the Dual Kinase Inhibitor Lapatinib (GW572016) Against HER-2-Overexpressing and Trastuzumab-Treated Breast Cancer Cells. Cancer Res. 66 (3), 1630–1639. doi:10.1158/0008-5472.CAN-05-1182
Krishnan, S., Miller, R. M., Tian, B., Mullins, R. D., Jacobson, M. P., and Taunton, J. (2014). Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc. 136 (36), 12624–12630. doi:10.1021/ja505194w
Lai, A. C., Toure, M., Hellerschmied, D., Salami, J., Jaime-Figueroa, S., Ko, E., et al. (2016). Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 55 (2), 807–810. doi:10.1002/anie.201507634
Lee, B. Y., Timpson, P., Horvath, L. G., and Daly, R. J. (2015). FAK Signaling in Human Cancer as a Target for Therapeutics. Pharmacol. Ther. 146, 132–149. doi:10.1016/j.pharmthera.2014.10.001
Li, W., Gao, C., Zhao, L., Yuan, Z., Chen, Y., and Jiang, Y. (2018). Phthalimide Conjugations for the Degradation of Oncogenic PI3K. Eur. J. Med. Chem. 151, 237–247. doi:10.1016/j.ejmech.2018.03.066
Liu, P., Cheng, H., Roberts, T. M., and Zhao, J. J. (2009). Targeting the Phosphoinositide 3-kinase Pathway in Cancer. Nat. Rev. Drug Discov. 8 (8), 627–644. doi:10.1038/nrd2926
Liu, S., Da, Y., Wang, F., Yan, R., Shu, Y., Lin, P., et al. (2020). Targeted Selective Degradation of Bruton’s Tyrosine Kinase by PROTACs. Med. Chem. Res. 29 (4), 802–808. doi:10.1007/s00044-020-02526-3
Lu, Y., and Sun, H. (2020). Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK). J. Med. Chem. 63 (23), 14382–14403. doi:10.1021/acs.jmedchem.0c01248
Luzzio, M. J., Freeman-Cook, K. D., Bhattacharya, S. K., Hayward, M. M., Hulford, C. A., Autry, C. L., et al. (2017). Sulfonyl Amide Derivatives for the Treatment of Abnormal Cell Growth. 20170327485.
Maennling, A. E., Tur, M. K., Niebert, M., Klockenbring, T., Zeppernick, F., Gattenlöhner, S., et al. (2019). Molecular Targeting Therapy Against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers 11 (12), 1826. doi:10.3390/cancers11121826
Malumbres, M., Pevarello, P., Barbacid, M., and Bischoff, J. R. (2008). CDK Inhibitors in Cancer Therapy: What is Next?. Trends Pharmacol. Sci. 29 (1), 16–21. doi:10.1016/j.tips.2007.10.012
Martín-Acosta, P., and Xiao, X. (2021). PROTACs to Address the Challenges Facing Small Molecule Inhibitors. Eur. J. Med. Chem. 210, 112993. doi:10.1016/j.ejmech.2020.112993
Mitra, S. K., Hanson, D. A., and Schlaepfer, D. D. (2005). Focal Adhesion Kinase: In Command and Control of Cell Motility. Nat. Rev. Mol. Cel. Biol. 6 (1), 56–68. doi:10.1038/nrm1549
Mok, T. S., Wu, Y.-L., Thongprasert, S., Yang, C.-H., Chu, D.-T., Saijo, N., et al. (2009). Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 361 (10), 947–957. doi:10.1056/NEJMoa0810699
Nagar, B., Hantschel, O., Young, M. A., Scheffzek, K., Veach, D., Bornmann, W., et al. (2003). Structural Basis for the Autoinhibition of C-Abl Tyrosine Kinase. Cell 112 (6), 859–871. doi:10.1016/s0092-8674(03)00194-6
Olson, C. M., Jiang, B., Erb, M. A., Liang, Y., Doctor, Z. M., Zhang, Z., et al. (2018). Pharmacological Perturbation of CDK9 Using Selective CDK9 Inhibition or Degradation. Nat. Chem. Biol. 14 (2), 163–170. doi:10.1038/nchembio.2538
Pal Singh, S., Dammeijer, F., and Hendriks, R. W. (2018). Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 17 (1), 57. doi:10.1186/s12943-018-0779-z
Parry, D., Guzi, T., Shanahan, F., Davis, N., Prabhavalkar, D., Wiswell, D., et al. (2010). Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 9 (8), 2344–2353. doi:10.1158/1535-7163.MCT-10-0324
Parsons, J. T. (2003). Focal Adhesion Kinase: the First Ten Years. J. Cell Sci. 116 (Pt 8), 1409–1416. doi:10.1242/jcs.00373
Parsons, J. T., Slack-Davis, J., Tilghman, R., and Roberts, W. G. (2008). Focal Adhesion Kinase: Targeting Adhesion Signaling Pathways for Therapeutic Intervention. Clin. Cancer Res. 14 (3), 627–632. doi:10.1158/1078-0432.CCR-07-2220
Pettersson, M., and Crews, C. M. (2019). PROteolysis TArgeting Chimeras (PROTACs) - Past, Present and Future. Drug Discov. Today Tech. 31, 15–27. doi:10.1016/j.ddtec.2019.01.002
Polier, G., Ding, J., Konkimalla, B. V., Eick, D., Ribeiro, N., Köhler, R., et al. (2011). Wogonin and Related Natural Flavones are Inhibitors of CDK9 that Induce Apoptosis in Cancer Cells by Transcriptional Suppression of Mcl-1. Cell Death Dis. 2 (7), e182. doi:10.1038/cddis.2011.66
Popow, J., Arnhof, H., Bader, G., Berger, H., Ciulli, A., Covini, D., et al. (2019). Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J. Med. Chem. 62 (5), 2508–2520. doi:10.1021/acs.jmedchem.8b01826
Qiu, X., Li, Y., Yu, B., Ren, J., Huang, H., Wang, M., et al. (2021). Discovery of Selective CDK9 Degraders with Enhancing Antiproliferative Activity through PROTAC Conversion. Eur. J. Med. Chem. 211, 113091. doi:10.1016/j.ejmech.2020.113091
Rana, S., Bendjennat, M., Kour, S., King, H. M., Kizhake, S., Zahid, M., et al. (2019). Selective Degradation of CDK6 by a Palbociclib Based PROTAC. Bioorg. Med. Chem. Lett. 29 (11), 1375–1379. doi:10.1016/j.bmcl.2019.03.035
Reddy, E. P., and Aggarwal, A. K. (2012). The Ins and Outs of Bcr-Abl Inhibition. Genes Cancer 3 (5–6), 447–454. doi:10.1177/1947601912462126
Robb, C. M., Contreras, J. I., Kour, S., Taylor, M. A., Abid, M., Sonawane, Y. A., et al. (2017). Chemically Induced Degradation of CDK9 by a Proteolysis Targeting Chimera (PROTAC). Chem. Commun. 53 (54), 7577–7580. doi:10.1039/c7cc03879h
Rosell, R., Moran, T., Queralt, C., Porta, R., Cardenal, F., Camps, C., et al. (2009). Screening for Epidermal Growth Factor Receptor Mutations in Lung Cancer. N. Engl. J. Med. 361 (10), 958–967. doi:10.1056/NEJMoa0904554
Roskoski, R. (2012). MEK1/2 Dual-Specificity Protein Kinases: Structure and Regulation. Biochem. Biophys. Res. Commun. 417 (1), 5–10. doi:10.1016/j.bbrc.2011.11.145
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. 98 (15), 8554–8559. doi:10.1073/pnas.141230798
Schindler, T. (2000). Structural Mechanism for STI-571 Inhibition of Abelson Tyrosine Kinase. Science 289 (5486), 1938–1942. doi:10.1126/science.289.5486.1938
Scholl, F. A., Dumesic, P. A., Barragan, D. I., Harada, K., Bissonauth, V., Charron, J., et al. (2007). Mek1/2 MAPK Kinases are Essential for Mammalian Development, Homeostasis, and Raf-Induced Hyperplasia. Dev. Cell 12 (4), 615–629. doi:10.1016/j.devcel.2007.03.009
Serafimova, I. M., Pufall, M. A., Krishnan, S., Duda, K., Cohen, M. S., Maglathlin, R. L., et al. (2012). Reversible Targeting of Noncatalytic Cysteines With Chemically Tuned Electrophiles. Nat. Chem. Biol. 8 (5), 471–476. doi:10.1038/nchembio.925
Shah, N. P., Tran, C., Lee, F. Y., Chen, P., Norris, D., and Sawyers, C. L. (2004). Overriding Imatinib Resistance With a Novel ABL Kinase Inhibitor. Science 305 (5682), 399–401. doi:10.1126/science.1099480
Sheppard, K., Kinross, K. M., Solomon, B., Pearson, R. B., and Phillips, W. A. (2012). Targeting PI3 Kinase/AKT/mTOR Signaling in Cancer. Crit. Rev. Oncog. 17 (1), 69–95. doi:10.1615/critrevoncog.v17.i1.60
Shimokawa, K., Shibata, N., Sameshima, T., Miyamoto, N., Ujikawa, O., Nara, H., et al. (2017). Targeting the Allosteric Site of Oncoprotein BCR-ABL as an Alternative Strategy for Effective Target Protein Degradation. ACS Med. Chem. Lett. 8 (10), 1042–1047. doi:10.1021/acsmedchemlett.7b00247
Siemeister, G., Lücking, U., Wengner, A. M., Lienau, P., Steinke, W., Schatz, C., et al. (2012). BAY 1000394, a Novel Cyclin-Dependent Kinase Inhibitor, With Potent Antitumor Activity in Mono- and in Combination Treatment upon Oral Application. Mol. Cancer Ther. 11 (10), 2265–2273. doi:10.1158/1535-7163.MCT-12-0286
Singh, J., Petter, R. C., Baillie, T. A., and Whitty, A. (2011). The Resurgence of Covalent Drugs. Nat. Rev. Drug Discov. 10 (4), 307–317. doi:10.1038/nrd3410
Smalley, K. S. M., Contractor, R., Nguyen, T. K., Xiao, M., Edwards, R., Muthusamy, V., et al. (2008). Identification of a Novel Subgroup of Melanomas with KIT/Cyclin-Dependent Kinase-4 Overexpression. Cancer Res. 68 (14), 5743–5752. doi:10.1158/0008-5472.CAN-08-0235
Sos, M. L., Rode, H. B., Heynck, S., Peifer, M., Fischer, F., Klüter, S., et al. (2010). Chemogenomic Profiling Provides Insights Into the Limited Activity of Irreversible EGFR Inhibitors in Tumor Cells Expressing the T790M EGFR Resistance Mutation. Cancer Res. 70 (3), 868–874. doi:10.1158/0008-5472.CAN-09-3106
Steinebach, C., Ng, Y. L. D., Sosič, I., Lee, C.-S., Chen, S., Lindner, S., et al. (2020). Systematic Exploration of Different E3 Ubiquitin Ligases: An Approach towards Potent and Selective CDK6 Degraders. Chem. Sci. 11 (13), 3474–3486. doi:10.1039/d0sc00167h
Su, S., Yang, Z., Gao, H., Yang, H., Zhu, S., An, Z., et al. (2019). Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 62 (16), 7575–7582. doi:10.1021/acs.jmedchem.9b00871
Sulzmaier, F. J., Jean, C., and Schlaepfer, D. D. (2014). FAK in Cancer: Mechanistic Findings and Clinical Applications. Nat. Rev. Cancer 14 (9), 598–610. doi:10.1038/nrc3792
Sun, Y., Ding, N., Song, Y., Yang, Z., Liu, W., Zhu, J., et al. (2019). Degradation of Bruton’s Tyrosine Kinase Mutants by PROTACs for Potential Treatment of Ibrutinib-Resistant Non-Hodgkin Lymphomas. Leukemia 33 (8), 2105–2110. doi:10.1038/s41375-019-0440-x
Sun, Y., Zhao, X., Ding, N., Gao, H., Wu, Y., Yang, Y., et al. (2018). PROTAC-Induced BTK Degradation as a Novel Therapy for Mutated BTK C481S Induced Ibrutinib-Resistant B-Cell Malignancies. Cell Res. 28 (7), 779–781. doi:10.1038/s41422-018-0055-1
Thorpe, L. M., Yuzugullu, H., and Zhao, J. J. (2015). PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer 15 (1), 7–24. doi:10.1038/nrc3860
Thress, K. S., Paweletz, C. P., Felip, E., Cho, B. C., Stetson, D., Dougherty, B., et al. (2015). Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non-Small Cell Lung Cancer Harboring EGFR T790M. Nat. Med. 21 (6), 560–562. doi:10.1038/nm.3854
Tinworth, C. P., Lithgow, H., Dittus, L., Bassi, Z. I., Hughes, S. E., Muelbaier, M., et al. (2019). PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase is Inhibited by Covalent Binding. ACS Chem. Biol. 14 (3), 342–347. doi:10.1021/acschembio.8b01094
Tong, W.-G., Chen, R., Plunkett, W., Siegel, D., Sinha, R., Harvey, R. D., et al. (2010). Phase I and Pharmacologic Study of SNS-032, a Potent and Selective Cdk2, 7, and 9 Inhibitor, in Patients With Advanced Chronic Lymphocytic Leukemia and Multiple Myeloma. J. Clin. Oncol. 28 (18), 3015–3022. doi:10.1200/JCO.2009.26.1347
Tovell, H., Testa, A., Zhou, H., Shpiro, N., Crafter, C., Ciulli, A., et al. (2019). Design and Characterization of SGK3-PROTAC1, an Isoform Specific SGK3 Kinase PROTAC Degrader. ACS Chem. Biol. 14 (9), 2024–2034. doi:10.1021/acschembio.9b00505
Vanhaesebroeck, B., Stephens, L., and Hawkins, P. (2012). PI3K Signalling: The Path to Discovery and Understanding. Nat. Rev. Mol. Cell Biol. 13 (3), 195–203. doi:10.1038/nrm3290
Vollmer, S., Cunoosamy, D., Lv, H., Feng, H., Li, X., Nan, Z., et al. (2020). Design, Synthesis, and Biological Evaluation of MEK PROTACs. J. Med. Chem. 63 (1), 157–162. doi:10.1021/acs.jmedchem.9b00810
Ward, R. A., Fawell, S., Floc’h, N., Flemington, V., McKerrecher, D., and Smith, P. D. (2020). Challenges and Opportunities in Cancer Drug Resistance. Chem. Rev. 121, 3297–3351. doi:10.1021/acs.chemrev.0c00383
Wei, J., Hu, J., Wang, L., Xie, L., Jin, M. S., Chen, X., et al. (2019). Discovery of a First-in-Class Mitogen-Activated Protein Kinase Kinase 1/2 Degrader. J. Med. Chem. 62 (23), 10897–10911. doi:10.1021/acs.jmedchem.9b01528
Wei, M., Zhao, R., Cao, Y., Wei, Y., Li, M., Dong, Z., et al. (2021). First Orally Bioavailable Prodrug of Proteolysis Targeting Chimera (PROTAC) Degrades Cyclin-Dependent Kinases 2/4/6 In Vivo. Eur. J. Med. Chem. 209, 112903. doi:10.1016/j.ejmech.2020.112903
Weisberg, E., Manley, P., Mestan, J., Cowan-Jacob, S., Ray, A., and Griffin, J. D. (2006). AMN107 (Nilotinib): A Novel and Selective Inhibitor of BCR-ABL. Br. J. Cancer 94 (12), 1765–1769. doi:10.1038/sj.bjc.6603170
Woyach, J. A., Furman, R. R., Liu, T.-M., Ozer, H. G., Zapatka, M., Ruppert, A. S., et al. (2014). Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 370 (24), 2286–2294. doi:10.1056/NEJMoa1400029
Wu, T., Qin, Z., Tian, Y., Wang, J., Xu, C., Li, Z., et al. (2020). Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An Update. J. Med. Chem. 63 (22), 13228–13257. doi:10.1021/acs.jmedchem.0c00744
Xue, G., Chen, J., Liu, L., Zhou, D., Zuo, Y., Fu, T., et al. (2020). Protein Degradation Through Covalent Inhibitor-Based PROTACs. Chem. Commun. 56 (10), 1521–1524. doi:10.1039/c9cc08238g
Yadav, R. R., Guru, S. K., Joshi, P., Mahajan, G., Mintoo, M. J., Kumar, V., et al. (2016). 6-Aryl Substituted 4-(4-Cyanomethyl) Phenylamino Quinazolines as a New Class of Isoform-Selective PI3K-Alpha Inhibitors. Eur. J. Med. Chem. 122, 731–743. doi:10.1016/j.ejmech.2016.07.006
Yang, K., and Fu, L.-w. (2015). Mechanisms of Resistance to BCR-ABL TKIs and the Therapeutic Strategies: A Review. Crit. Rev. Oncol. Hematol. 93 (3), 277–292. doi:10.1016/j.critrevonc.2014.11.001
Yang, Y., Gao, H., Sun, X., Sun, Y., Qiu, Y., Weng, Q., et al. (2020). Global PROTAC Toolbox for Degrading BCR-ABL Overcomes Drug-Resistant Mutants and Adverse Effects. J. Med. Chem. 63 (15), 8567–8583. doi:10.1021/acs.jmedchem.0c00967
Yarden, Y., and Sliwkowski, M. X. (2001). Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2 (2), 127–137. doi:10.1038/35052073
Yewale, C., Baradia, D., Vhora, I., Patil, S., and Misra, A. (2013). Epidermal Growth Factor Receptor Targeting in Cancer: A Review of Trends and Strategies. Biomaterials 34 (34), 8690–8707. doi:10.1016/j.biomaterials.2013.07.100
Yin, L., and Hu, Q. (2020). Chimera Induced Protein Degradation: PROTACs and Beyond. Eur. J. Med. Chem. 206, 112494. doi:10.1016/j.ejmech.2020.112494
Yu, H. A., Arcila, M. E., Rekhtman, N., Sima, C. S., Zakowski, M. F., Pao, W., et al. (2013). Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients With EGFR-Mutant Lung Cancers. Clin. Cancer Res. 19 (8), 2240–2247. doi:10.1158/1078-0432.CCR-12-2246
Zeng, S., Huang, W., Zheng, X., Cheng, L., Zhang, Z., Wang, J., et al. (2021). Proteolysis Targeting Chimera (PROTAC) in Drug Discovery Paradigm: Recent Progress and Future Challenges. Eur. J. Med. Chem. 210, 112981. doi:10.1016/j.ejmech.2020.112981
Zhang, H., Zhao, H.-Y., Xi, X.-X., Liu, Y.-J., Xin, M., Mao, S., et al. (2020). Discovery of Potent Epidermal Growth Factor Receptor (EGFR) Degraders by Proteolysis Targeting Chimera (PROTAC). Eur. J. Med. Chem. 189, 112061. doi:10.1016/j.ejmech.2020.112061
Zhang, X., Xu, F., Tong, L., Zhang, T., Xie, H., Lu, X., et al. (2020). Design and Synthesis of Selective Degraders of EGFRL858R/T790M Mutant. Eur. J. Med. Chem. 192, 112199. doi:10.1016/j.ejmech.2020.112199
Zhao, B., and Burgess, K. (2019). PROTACs Suppression of CDK4/6, Crucial Kinases for Cell Cycle Regulation in Cancer. Chem. Commun. 55 (18), 2704–2707. doi:10.1039/c9cc00163h
Zhao, H.-Y., Yang, X.-Y., Lei, H., Xi, X.-X., Lu, S.-M., Zhang, J.-J., et al. (2020). Discovery of Potent Small Molecule PROTACs Targeting Mutant EGFR. Eur. J. Med. Chem. 208, 112781. doi:10.1016/j.ejmech.2020.112781
Zhao, Q., Ren, C., Liu, L., Chen, J., Shao, Y., Sun, N., et al. (2019). Discovery of SIAIS178 as an Effective BCR-ABL Degrader by Recruiting Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. J. Med. Chem. 62 (20), 9281–9298. doi:10.1021/acs.jmedchem.9b01264
Zheng, D., Hu, M., Bai, Y., Zhu, X., Lu, X., Wu, C., et al. (2017). EGFR G796D Mutation Mediates Resistance to Osimertinib. Oncotarget 8 (30), 49671–49679. doi:10.18632/oncotarget.17913
Zhou, F., Chen, L., Cao, C., Yu, J., Luo, X., Zhou, P., et al. (2020). Development of Selective Mono or Dual PROTAC Degrader Probe of CDK Isoforms. Eur. J. Med. Chem. 187, 111952. doi:10.1016/j.ejmech.2019.111952
Zhou, X., Dong, R., Zhang, J.-Y., Zheng, X., and Sun, L.-P. (2020). PROTAC: A Promising Technology for Cancer Treatment. Eur. J. Med. Chem. 203, 112539. doi:10.1016/j.ejmech.2020.112539
Keywords: protac, protein kinase, inhibitors, degradation, anti-cancer
Citation: Yu F, Cai M, Shao L and Zhang J (2021) Targeting Protein Kinases Degradation by PROTACs. Front. Chem. 9:679120. doi: 10.3389/fchem.2021.679120
Received: 11 March 2021; Accepted: 15 June 2021;
Published: 30 June 2021.
Edited by:
Wolfgang Sippl, Martin Luther University of Halle-Wittenberg, GermanyReviewed by:
Michael Gütschow, University of Bonn, GermanyDina Robaa, Martin Luther University of Halle-Wittenberg, Germany
Copyright © 2021 Yu, Cai, Shao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jihong Zhang, emhqaWhvbmcyMDAwQGt1c3QuZWR1LmNu