 Patrick K. Tamukong
Patrick K. Tamukong Mark R. Hoffmann
Mark R. Hoffmann- Chemistry Department, University of North Dakota, Grand Forks, ND, United States
The generalized Van Vleck second order multireference perturbation theory (GVVPT2) method was used to investigate the low-lying electronic states of Ni2. Because the nickel atom has an excitation energy of only 0.025 eV to its first excited state (the least in the first row of transition elements), Ni2 has a particularly large number of low-lying states. Full potential energy curves (PECs) of more than a dozen low-lying electronic states of Ni2, resulting from the atomic combinations 3F4 + 3F4 and 3D3 + 3D3, were computed. In agreement with previous theoretical studies, we found the lowest lying states of Ni2 to correlate with the 3D3 + 3D3 dissociation limit, and the holes in the d-subshells were in the subspace of delta orbitals (i.e., the so-dubbed δδ-states). In particular, the ground state was determined as X 1Γg and had spectroscopic constants: bond length (Re) = 2.26 Å, harmonic frequency (ωe) = 276.0 cm−1, and binding energy (De) = 1.75 eV; whereas the 1 1Σg+ excited state (with spectroscopic constants: Re = 2.26 Å, ωe = 276.8 cm−1, and De = 1.75) of the 3D3 + 3D3 dissociation channel lay at only 16.4 cm−1 (0.002 eV) above the ground state at the equilibrium geometry. Inclusion of scalar relativistic effects through the spin-free exact two component (sf-X2C) method reduced the bond lengths of both of these two states to 2.20 Å, and increased their binding energies to 1.95 eV and harmonic frequencies to 296.0 cm−1 for X 1Γg and 297.0 cm−1 for 1 1Σg+. These values are in good agreement with experimental values of Re = 2.1545 ± 0.0004 Å, ωe = 280 ± 20 cm−1, and D0 = 2.042 ± 0.002 eV for the ground state. All states considered within the 3F4 + 3F4 dissociation channel proved to be energetically high-lying and van der Waals-like in nature. In contrast to most previous theoretical studies of Ni2, full PECs of all considered electronic states of the molecule were produced.
Introduction
Since Ni2 has few holes in otherwise complete subshells, one might expect theoretical studies of Ni2 to be less complicated than for other first row transition metal dimers, like Cr2 where one has many more possibilities of distributing 12 electrons in 12 valence orbitals. Reported information on Ni2, however, proves the contrary. For example, the exact symmetry of the ground electronic state of Ni2 is still debated: Different studies have reported different space and spin symmetries for the molecule’s ground term.
Experimental data on Ni2 is sparse and the true ground state of the molecule is not unequivocally accepted. From the analysis of electronic absorption bands of Ni2 in the visible spectral region in argon matrices, De Vore et al. (De Vore et al., 1975) determined ωe = 192 cm−1, whereas a frequency of 380.9 cm−1 was found in solid argon matrix (Ahmed and Nixon, 1979). The latter result was later criticized by Rasanen et al. (Rasanen et al., 1987) In photoelectron spectroscopic studies of Ni2-, ωe = 280 ± 20 cm−1 was determined for the lowest electronic state of Ni2 (Ho et al., 1993). Second and third law analyses of information derived from a combination of Knudsen effusion and mass‐spectrometric techniques led to a binding energy of D0 = 2.03 ± 0.30 eV (second law result) and D0 = 2.36 ± 0.22 eV (third law result) for ground state Ni2 (Kant, 1964). By using time-delayed resonant two-photon ionization, Morse et al. (Morse et al., 1984) determined D0 = 2.068 ± 0.010 eV and Re = 2.200 ± 0.007 Å for the lowest state of Ni2, but assigned as either 3Γu or 1Γg. Also from two-photon ionization studies on supersonic jet-cooled Ni2 in argon carrier gas, Pinegar et al. (Pinegar et al., 1995) determined D0 = 2.042 ± 0.002 eV and Re = 2.1545 ± 0.0004 Å for the lowest state of Ni2 but were unable to ascertain the symmetry of this state.
Theoretical studies of the electronic states of Ni2 are complicated by the fact that the first excited state of the Ni atom, 3D3 (3d94s1), is only 0.025 eV above the 3F4 (3d84s2) ground atomic state, which is the least excitation energy of any of the first row of transition elements (Harrison, 2000; Kramida et al., 2013). This low promotion energy supports the existence of several low-lying molecular states of the Ni2 molecule resulting from the 3F4 + 3F4 and 3D3 + 3D3 atomic combinations. For example, limited configuration interaction (CI) calculations (Shim et al., 1979) found 84 states of Ni2, corresponding to the 3F4 + 3F4 dissociation limit, to lie within an energy range of only 300 K (0.026 eV), and 45 states, also within a narrow energy range, to correlate with the 3D3 + 3D3 dissociation asymptote. Melius et al. (Melius et al., 1976) also noted that the manifold of electronic states within 0.50 eV of the ground state of Ni2 was dense and complex.
Since the fully filled 4s-subshell of the 3F4 (3d84s2) ground state of Ni discourages significant bonding interaction, bonding in low-lying states of Ni2 results largely from the coupling of excited state Ni atoms. In particular, the lowest states of the Ni2 molecule can be expected to correlate with the 3D3 (3d94s1) + 3D3 (3d94s1) dissociation channel. At the generalized valence bond (GVB) and polarization CI (POL-CI) level of theory, (Upton and Goddard III, 1978) 30 of 45 low-lying states of Ni2, within the 3D3 + 3D3 dissociation channel, were found (Shim et al., 1979) to be singlets and triplets ordered energetically as
δδ (6 states) < πδ (8 states) < δσ (4 states) < ππ (6 states) < πσ (4 states) < σσ (2 states), with the six lowest states (of symmetries 1Γg, 1Σg+, 3Σg−, 1Σu−, 3Γu, and 3Σu+) being virtually degenerate and having an average equilibrium bond length, Re, of 2.04 Å, and binding energy, De, of 2.29 eV. The designations δδ, πδ, et cetera, specify the positions of the holes in the dominant configurations of the 3d-orbitals at each atomic center which has a 3d94s1 configuration (Shim et al., 1979). In brief, the correct energy spacing of the low-lying states of the Ni dimer is problematic for both theory and experiment.
The determination of energy ordering and spacing is no less complicated for the Ni atom, and provides insight into necessary levels of experimental and theoretical approaches. The 3F4 (3d84s2) → 3D3 (3d94s1) excitation energy has been determined by at least one experimental study (Moore, 1952) to be negative (-0.029 eV). This negative value was supported by ab initio wave function and density functional theory (DFT) calculations (Bauschlicher et al., 1982; Raghavachari and Trucks, 1989; Russo et al., 1994). A more recent study, (Schultz et al., 2005) that employed several functionals at all five rungs of Jacob’s ladder of DFT functionals, predicted the ground state configuration of the Ni atom as 3d94s1 (3D3) with most of the functionals when using a triple-ζ quality basis set. On the other hand, multireference studies (Andersson and Roos, 1992; Murphy and Messmer, 1992) predicted a 3d84s2 (3F4) ground state configuration for the Ni atom. Illustrating additional complexity, Upton and Goddard (Upton and Goddard III, 1978) found that averaging over J components (where J is the sum of spin and orbital angular momenta of the atom) of each state places the 3D3 (3d94s1) state lower energetically than the 3F4 (3d84s2) state.
One of the earliest theoretical studies (Cooper et al., 1972) on Ni2 employed the extended Hückel molecular orbital method and found a 3Σg- ground state with Re = 2.21 Å, ωe = 370 cm−1, and De = 2.45 eV, with dominant configuration 3dδg4 3dδu4 3dπu4 3dπg2 3dσg2 3dσu2 4sσg2. On the other hand, the self-consistent field (SCF) scattered-wave (Xα-sw) method (Rösch and Rhodin, 1974) found a 1Σg+ ground state with configuration 3dδg4 3dδu4 3dπu4 3dπg4 3dσg2 4sσg2; whereas the generalized valence bond (GVB) method (Melius et al., 1976) also predicted a 1Σg+ ground state for Ni2, but with configuration 3dδg4 3dδu2 3dπu4 3dπg4 3dσg2 3dσu2 4sσg2. A Hartree-Fock followed by limited CI study, (Shim et al., 1979) which explored a variety of states of Ni2 resulting from the 3F4 + 3F4 and 3D3 + 3D3 atomic combinations, found the ground state to be 1Σg+ with the same configuration as was reported by the GVB study(Melius et al., 1976). The states 1Γg and 1Σu- were reported to be in close proximity to the 1Σg+ state in the CI study. Other theoretical studies found the six δδ–hole states (i.e., 1Γg, 1Σg+, 3Σg-, 1Σu-, 3Γu, 3Σu+), resulting from the 3D3 + 3D3 atomic coupling, to be quasidegenerate(Basch et al., 1980; Noell et al., 1980; Wood et al., 1980).
Using an effective core potential basis set specifically optimized for the Ni atom in the 3D3 state within the generalized valence bond CI (GVBCI) method, Noell et al. (Noell et al., 1980) found the splitting of the six δδ–hole states of Ni2 to be quite small (≤0.1 eV), with the lowest states being 1Γg and 1Σg+. Inclusion of polarization configurations involving single and double excitations to the virtual space (POLSDCI) placed the triplet states (3Σg-, 3Γu, 3Σu+) approximately 0.07 eV below the singlets. At the singles and doubles CI (SDCI) level of theory, these authors found the energy splitting of the six lowest δδ–hole states of Ni2 to be less than 0.009 eV, with an average bond length of 2.26 Å and binding energy of 1.88 eV.
With a basis set similar to that used by Noell et al., (Noell et al., 1980) a contemporaneous restricted Hartree-Fock (RHF) and CI with single and double excitations (CISD) study predicted a 3Σu+ ground state for Ni2, with spectroscopic data: Re = 2.33 Å, ωe = 211 cm−1, and De = 1.43 eV(Basch et al., 1980). Calculations by these authors at the same levels of theory using an all electron basis set corroborated the prediction of the ground state symmetry as 3Σu+. On the other hand, a local spin density method (Harris and Jones, 1979) predicted a 3Σg- ground state with Re = 2.18 Å, ωe = 320 cm−1, and De = 2.70 eV. A slightly earlier CASSCF/CASPT2 study (Pou-Amérigo et al., 1994) that used an atomic natural orbital (ANO) type contraction of the (21s15p10d6f4g) primitive basis to [6s5p4d3f2g] for calculations without correlation of the semi-core 3s3p electrons, and a [10s9p8d3f2g] contracted basis for calculations involving the correlation of 3s3p electrons, likewise found the six lowest δδ–hole states of Ni2 to lie within a particularly narrow energy gap (0.04 eV) with the triplet states higher in energy than the singlets. However, after inclusion of scalar relativistic effects, the ground term was predicted as 1Γg, with the 1Σg+ term lying only 0.01 eV higher at the equilibrium geometry. Correlating the 3s3p electrons in these calculations predicted the 1Γg and 1Σg+ states to be degenerate to the reported accuracy, with slightly improved spectroscopic constants relative to reference experimental values [i.e., Re = 2.23 Å, De = 2.06 eV; and ωe = 293 cm−1 for 1Γg versus ωe = 294 cm−1 for 1Σg+ compared with experimental values of Re = 2.1545 ± 0.0004 Å, (Pinegar et al., 1995) D0 = 2.042 ± 0.002 eV, (Pinegar et al., 1995) and ωe = 280 ± 20 cm−1 (Ho et al., 1993)].
DFT studies of Ni2 have also been inconclusive. Yanagisawa et al. (Yanagisawa et al., 2000) used various DFT functionals to study the 3Σg- and 3Σu+ states of Ni2 and found B3LYP (Becke, 1993) to predict the 3Σu+ state to lie lower than 3Σg- whereas the rest of the functionals predicted the latter state to lie lower at the equilibrium geometry. However, the 3Σg- state that they found had a configuration that corresponded to the ππ–hole manifold rather than the δδ. Gutsev et al. (Gutsev and Bauschlicher, 2003) also found a 3Σg- ground state, with the same configuration as did Yanagisawa et al., (Yanagisawa et al., 2000) when using a variety of hybrid functionals. Contrarily, Diaconu et al. (Diaconu et al., 2004) found a singlet δδ–hole ground state (with a mixture of 1Γg and 1Σg+) for Ni2 when using B3LYP with the (14s11p6d3f)/[8s6p4d1f] basis set, whereas use of the Stuttgart RSC ECP basis set (Dolg et al., 1987) with the same functional gave a triplet δδ–hole (with a mixture of 3Σg- and 3Γu symmetries) that lay 0.001 eV lower than the singlet δδ–hole state at the equilibrium geometry. Using functionals at all levels of Jacob’s ladder of DFT functionals, Schultz et al. (Schultz et al., 2005) also found different functionals to predict different ground state symmetries for Ni2, with all local spin density approximation (LSDA) functionals predicting a 3Σg- ground state and all generalized gradient approximation (GGA) and meta GGA functionals predicting a 3Πu ground state; whereas hybrid GGA and hybrid meta GGA functionals found either 3Σu+ or 3Σg- to lie lowest energetically. Du et al. (Du et al., 2008) used various functionals to study the low-lying states of Ni2. Their results that agreed best with experiment were obtained when using BLYP, which predicted a triplet σδ–hole (3dz2 σu*1 3dx2-y2 δu*1) ground state. The space symmetry of this state was not reported. With the B3P86 functional, (Perdew, 1986; Becke, 1993) a quintet ground state was predicted for Ni2, (Shi-Ying and Zheng-He, 2008) although the space symmetry was not reported. The Perdew-Burke-Ernzerhof (PBE) exchange correlation functional (Perdew et al., 1996) predicted a 3Σg- ground state for Ni2, (Kamal et al., 2012) with spectroscopic constants Re = 2.93 Å, De = 3.09 eV, and ωe = 334.08 cm−1, which showed significant deviations from experimental values.
Some of the most recent wave function based calculations on Ni2 include those due to Dong et al. (Dong et al., 2013) using the symmetry-adapted-cluster configuration interaction (SAC-CI) method (Nakatsuji, 1979) and Cheskidov et al. (Cheskidov et al., 2012) using the average coupled pair functional (ACPF), (Gdanitz and Ahlrichs, 1988) average quadratic coupled cluster (AQCC), (Szalay and Bartlett, 1993) internally contracted single and double multireference configuration interaction (MRCI or MRCI with Davidson corrections, i.e., MRCI + Q), (Werner and Knowles, 1990) and N-electron valence state second-order perturbation theory (NEVPT2) (Angeli et al., 2001) methods. The study by Dong et al. (Dong et al., 2013) predicted a 3B1u ground state (with Re = 2.56 Å) for Ni2 in
The above synopsis of previous work on Ni2 shows the difficulties involved in studying not only the spectroscopic constants, but even the ordering of the low-lying electronic states of Ni2. Although wave function methods generally support δδ–hole states (1Γg, 1Σg+, 3Σg-, 1Σu-, 3Γu, 3Σu+) as lying lowest energetically, the methods predict different ground state symmetries with some finding all six states to be degenerate. Similarly, experimental spectroscopic data have been obtained but most of the studies could not ascertain the ground state symmetry of the molecule. Our current study exploits the ability of the GVVPT2 method (Khait et al., 2002; Jiang et al., 2009) to describe well full PECs of ground- and excited-electronic states of complicated transition element dimers, such as has already been demonstrated on other problematic transition metal molecules [e.g., Cr2 and Y2 (Tamukong et al., 2012; Tamukong et al., 2014)]. It should be noted that of all previous theoretical work described above on electronic states of Ni2, only six of the articles reported full PECs of the states they investigated. Consequently, where other data is available, this study also provides further assessment of the capabilities of the GVVPT2 method for difficult transition metal dimers, including the bond breaking regions. We have constructed full PECs of 21 states of Ni2. All (nonrelativistic) calculations used the Dunning-type cc-pVTZ basis set (Balabanov and Peterson, 2005), and calculations were performed using
Methods
GVVPT2
The GVVPT2 method for electron correlation in molecules has been thoroughly described elsewhere, (Khait et al., 2002; Jiang et al., 2009) as has its application to some challenging systems (Mbote et al., 2010; Khait et al., 2012; Tamukong et al., 2012; Mokambe et al., 2013; Tamukong et al., 2014). Here, salient features of the GVVPT2 method relevant to the present study are reviewed. In GVVPT, the total Hilbert space of many-electron functions [e.g., configuration state functions (CSFs)] with appropriate molecular space and spin symmetry (L) is partitioned into a model space (LM), and an external space (LQ) whose electronic configurations are derived from configurations generated from the model space by single and double electron excitations into virtual orbitals, L = LM ⊕ LQ. The model space is further partitioned into a primary subspace, spanned by a set of reference functions that are linear combinations of CSFs (typically the lowest MCSCF or CASSCF states), and a secondary subspace. States within the primary subspace are then perturbatively corrected through primary-external (P–Q) interactions whereas the secondary subspace serves as a buffer energetically separating the primary and external subspaces. This energy buffer circumvents most intruder state problems that plague many multireference perturbation theory techniques. An effective Hamiltonian matrix
where the Cmi denote components of eigenvectors of the unperturbed model Hamiltonian;
Spin-Free Exact Two Component Method
Matrix formulations of two-component relativistic methods have had significant successes in molecular electronic structure calculations, following Dyall’s seminal Normalized Elimination of Small Component (NESC) paper (Dyall, 1997) in 1997. For a summary of development of matrix two-component methods, we refer the reader to Ref. (Liu, 2014). In this work, we adopt the spin-free version of exact two component (sf-X2C) (Liu, 2010; Cheng and Gauss, 2011; Li et al., 2012; Li et al., 2013) that was previously used with GVVPT2 (Tamukong et al., 2014). The following Hamiltonian, written in second quantization form, incorporates scalar relativistic effects through the sf-X2C approach
where the first term is the one-electron spin-free part of the exact two-component (X2C) Hamiltonian, while the second is the unmodified Coulombic two-electron term. The sf-X2C Hamiltonian for positive energy states derives from a modified Dirac Hamiltonian, hD which is decomposed into spin-free (sf) and spin-dependent (sd) parts.
where V is the matrix representation of the external nuclear attraction potential operator; T is the matrix representation of the kinetic energy operator; α is the fine-structure constant; while W is the matrix representation of the operator
The spin-free (sf) part of W, that is Wsf, describes scalar relativistic effects whereas the spin-dependent (sd) part, Wsd, incorporates spin-orbit coupling effects.
Computational Details
Unperturbed wavefunctions for GVVPT2 calculations were obtained from multiconfigurational self-consistent field (MCSCF) reference functions, using the local code “undmol.” The macroconfiguration approach (Khait et al., 2004) was used in all MCSCF and GVVPT2 calculations. In the macroconfiguration scheme, active orbitals are partitioned into orbital groups based on physical and mathematical intuition, such as orbital type or energy, and electrons are assigned to active orbital groups. Each unique assignment of electrons to active orbital groups defines a macroconfiguration, κ(n). Orbital rotations within each active orbital group are redundant (in MCSCF calculations), as all possible distributions of electrons within each group leading to the desired total spin and space symmetry are allowed. Each κ(n) generates a unique set of configurations, and hence configuration state functions (CSFs) that are orthogonal to those of all other κ(n). Additionally, the macroconfiguration approach permits a large number of noninteracting electronic configuration pairs to be efficiently screened by recognizing that all matrix elements resulting from configurations that differ by more than two electrons must be identically zero. The macroconfiguration formalism also provides an efficient way of generating excited configurations.
In all calculations, the active space consisted of 3d and 4s-derived molecular orbitals (MOs) of Ni2 (Figure 1 shows representative 3d and 4s-derived MOs that were used to constitute the active spaces in this study). Depending on the specific state being investigated, some of the 3d-derived MOs and/or 4s-derived MOs were restricted to be doubly occupied in the MCSCF calculations, but were included with the 3s- and 3p-derived MOs in the active core and correlated at the GVVPT2 level of theory. For example, in all MCSCF calculations of δδ–hole states the 3dσ and 3dπ MOs were placed in the active core and only correlated at the GVVPT2 level. Similarly, the 3dσ and 3dπ electrons were kept doubly occupied in MCSCF calculations of ππ–hole states while only the 3dσ electrons were kept doubly occupied in MCSCF calculations of δπ–hole states, whereas the 4sσ, or 4sσ + 3dπ, or 4sσ + 3dσ orbitals were kept doubly occupied in MCSCF calculations of states within the 3F4 (3d84s2) + 3F4 (3d84s2) dissociation channel. The remaining orbitals in the active space were partitioned into sets (or so-called orbital groups), leading to configurations that describe δδ–, δπ–, δσ–, ππ–, πσ–, or σσ–hole states from the 3D3 (3d94s1) + 3D3 (3d94s1) atomic combination or configurations that describe 3D3(3d94s1) +3F4(3d84s2) and 3F4(3d84s2) + 3F4(3d84s2) atomic couplings. In the present study, we indicate the positions of holes within the subspace of active 3d–derived MOs by specifying the active orbital types that qualitatively describe where holes exist (e.g., δδ–, δπ–, ππ–) as did Shim et al. (Shim et al., 1979) and Noel et al. (Noell et al., 1980)
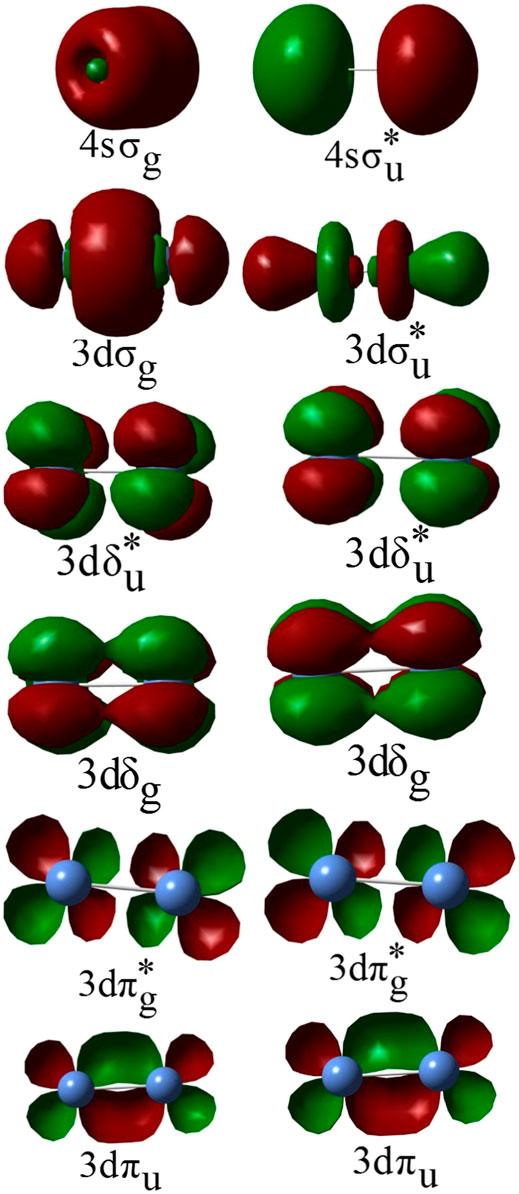
FIGURE 1. Example of MCSCF 3d- and 4s-derived molecular orbitals of the Ni2 molecule.
All δδ–states were computed using a single reference macroconfiguration,
where the superscripts denote the number of electrons in each orbital group. The semi-core 3s3p electrons were correlated together with those derived from 3dz2, 3dxz and 3dyz at the GVVPT2 level. For four of the computed δδ–states, additional calculations were also performed in which the 3s3p were kept doubly occupied throughout (i.e., at both the MCSCF and GVVPT2 levels). When using the cc-pVTZ basis set, (Balabanov and Peterson, 2005) reference κ1(n) generated 8 model and 27,891,120 total CSFs (in D2h symmetry) for the 1 3Σu+ and 1 3Γu states; 8 model and 15,290,666 total space CSFs for the 11Σu−, 11Σg−, and 11Γu states; 10 model and 27,982,592 all space CSFs for the 13Σg- and 13Σu- states; and 12 model versus 15,270,687 all space CSFs for the X 1Γg and 1 1Σg+ states. Without correlating the 3s3p electrons at the GVVPT2 level, the dimensions were: 12 model space CSFs versus 3,593,707 total CSFs for the X 1Γg and 1 1Σg+ states; and 8 model versus 6,434,550 all space CSFs for the 13Σu+ and 13Γu states. Relativistic calculations on the X 1Γg and 1 1Σg+ states utilized the same reference macroconfiguration, κ1(n). A
which gave rise to 4 model space and 7,518,688 all space CSFs, using the cc-pVTZ basis set. The ππ–states were computed using reference
which is similar to κ1(n) but with delta replaced with pi orbitals. This macroconfiguration gave rise to 12 model space and 15,267,629 all space CSFs for the 11Δg and 21Σg+ states when using the cc-pVTZ basis set and D2h molecular space symmetry. The δπ–states were computed from
κ4(n) generated 16 model space and 27,178,852 total space CSFs for the 11Φg and 11Πg states versus 20 model and 50,736,846 all space CSFs for the 13Φg and 13Πg states, when using the cc-pVTZ basis set.
Within the 3F4 + 3F4 manifold, the 31Σg+, 13Σg+, 2 3Σg+, 13Δu, and 2 3Σu+ δπ–states were computed using
In these calculations, the 3s, 3p, 3dz2, and 4s electrons were kept doubly occupied at the MCSCF level but correlated at the GVVPT2 level of theory. Reference κ5(n) resulted in 40 model versus 55,053,638 total CSFs for the 31Σg+ state; 36 model and 103,306,512 all space CSFs for the 13Σg+ and 2 3Σg+ states; and 40 model versus 103,312,902 all space CSFs for the 13Δu and 2 3Σu+ states when using the cc-pVTZ basis set.
Two δπ–states, 2 1Γg and 2 1Δg, were computed within the 3F4 + 3F4 manifold using
Reference κ6(n) generated 12 model space and 15,270,687 all space CSFs for the computed states using the cc-pVTZ basis set. Two quintet δπσ–states (i.e., 15Φu and 15Πu) were also computed within the 3F4 + 3F4 manifold, using
This reference, κ7(n), led to 12 model versus 69,738,914 total CSFs for the computed quintet states. Lastly, a limited study was done on two quintet states within the 3D3(3d94s1) + 3F4(3d84s2) manifold at short bond lengths. While reference
can describe the 15Δg and 2 5Δg δπσ–states resulting from the 3D3(3d94s1) + 3F4(3d84s2) manifold of molecular states adequately at short internuclear distances, it cannot be expected to do so at long internuclear distances and our studies were restricted to the shorter bond lengths for these two states. Reference κ8(n) generated 12 model CSFs versus 69,740,135 total space CSFs.
The reference macroconfigurations described above were used to define the active space, while all lower energy MOs were doubly occupied in MCSCF calculations. Initial MOs for MCSCF calculations were obtained from approximate natural orbitals of second-order restricted Møller−Plesset perturbation (RMP2) calculations from a closed-shell Hartree−Fock (HF) reference. At the GVVPT2 level, 3s, 3p, and all 3d and/or 4s electrons were correlated whether they were or were not at the MCSCF level. For comparison purposes, a few of the GVVPT2 calculations were performed without correlating the 3s and 3p electrons. Calculations that accounted for scalar relativistic effects employed the sf-X2C method described above. Finally, to aid in interpretation, the effective bond order (EBO) was computed, and used the following expression
where η is the EBO, χi is the EBO for the ith CSF, while ci2 is its corresponding weight. For each CSF used to estimate EBO, χi was determined as
where nb and nab are the numbers of bonding and antibonding electrons, respectively. Vibrational frequencies were obtained by 3-point finite differencing near the minima of the curves.
Results and Discussion
Where indicated, the letter “R” in parentheses following a molecular term denotes that scalar relativistic effects were included in the calculations, while the expression “no 3s3p” in parentheses after a molecular term symbol denotes that 3s and 3p electrons were not correlated in GVVPT2 calculations.
The δδ–Hole States
PECs of the δδ–hole states are shown in Figure 2 and the data describing them are in Table 1. In agreement with results from other high level ab initio methods, the lowest states of Ni2 were found to be δδ–hole states of the 3D3 (3d94s1) + 3D3 (3d94s1) manifold. In particular, the ground state was found to be X 1Γg, with the 11Σg+ state lying only 16.40 cm−1 (0.002 eV) higher at the equilibrium geometry. After including scalar relativistic effects, the energy gap between these states slightly increased to 23.39 cm−1 at equilibrium, with the X 1Γg term having spectroscopic constants: Re = 2.20 Å, De = 1.95 eV, and ωe = 296 cm−1. These results are in good agreement with experimental data (Re = 2.1545 ± 0.0004 Å, (Cooper et al., 1972)
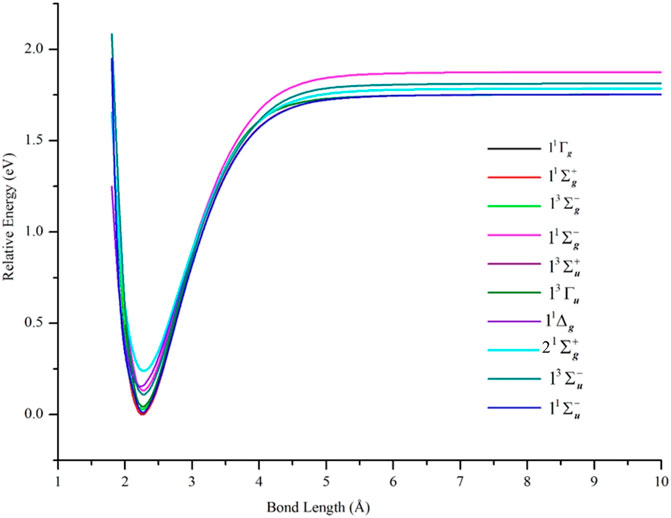
FIGURE 2. PECs of low-lying electronic states of Ni2 computed at the GVVPT2 level of theory using the cc-pVTZ basis set. All energies are plotted relative to the lowest energy value of the ground X 1Γg term. For all states, the holes are in the 3d delta orbitals (δδ–holes) except for 11Δg and 2 1Σg+ states which are ππ–hole states.
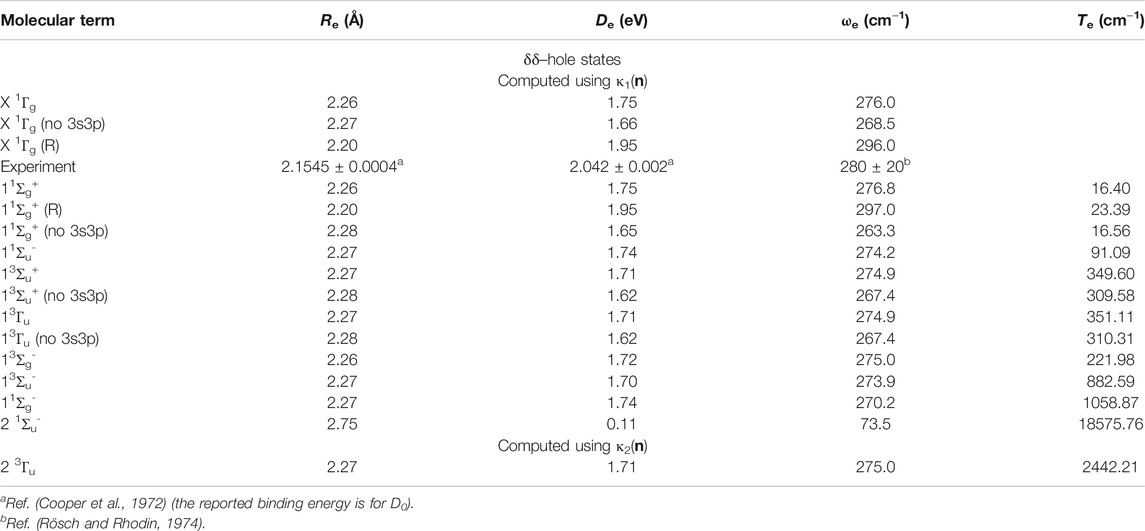
TABLE 1. Equilibrium bond lengths, Re (Å), binding energies, De (eV), harmonic frequencies, ωe (cm−1), and adiabatic transition energies, Te (cm−1), of electronic states of Ni2 calculated at the GVVPT2 level of theory using the cc-pVTZ basis set (or cc-pVTZ-DK for scalar relativistic calculations).
The semi-core 3s3p electrons were found to be important in the description of low-lying states of Ni2. The inclusion of 3s3p electron correlation at the GVVPT2 level increased the binding energies by 0.09 eV for X 1Γg, 0.10 eV for 11Σg+, and 0.09 eV for 13Σu+ and 13Γu states in non-relativistic calculations. As can be seen in Figure 3 and Table 1, the effects of the 3s3p electrons on the equilibrium bond lengths and harmonic frequencies for these states are minimal whereas inclusion of such core-valence correlation raises, for example, the binding energy of X 1Γg from 1.66 to 1.75 eV compared to a reference D0 value of 2.042 ± 0.002 eV (Cooper et al., 1972). Scalar relativistic effects shortened the bond length of X 1Γg by 0.06 Å and further increased the bond energy by 0.20 to 1.95 eV which agreed even better with the reference experimental values (see Figure 4 and Table 1). The 3s3p electrons did not have any effect on the EBOs of X 1Γg and 11Σg+; the EBOs were determined as 0.962 and 0.959 at 2.27 Å for X 1Γg and 11Σg+, respectively, when the 3s3p electrons were not correlated compared to 0.963 vs. 0.960 when the semi-core electrons (3s3p) were correlated. Note the quasidegeneracy in the X 1Γg and 11Σg+ states. For example, in Figure 4, the blue and green curves for the X 1Γg and 11Σg+ states, respectively, lie on top of each other (only the green is visible). Also, the black and red curves for the X 1Γg (R) and 11Σg+ (R) states lie on each other (only the red curve is visible).
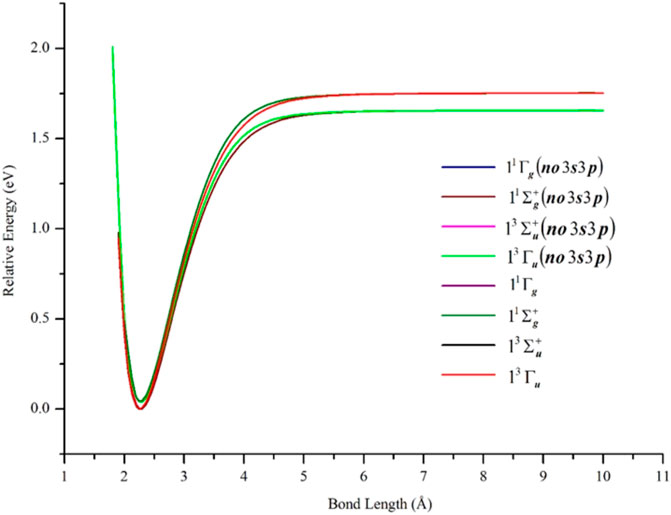
FIGURE 3. PECs of low-lying δδ–hole electronic states of Ni2 computed at the GVVPT2 level of theory, with and without the correlation of 3s3p semi-core electrons, using the cc-pVTZ basis set. All energies are plotted relative to the lowest energy value of the ground X 1Γg term.
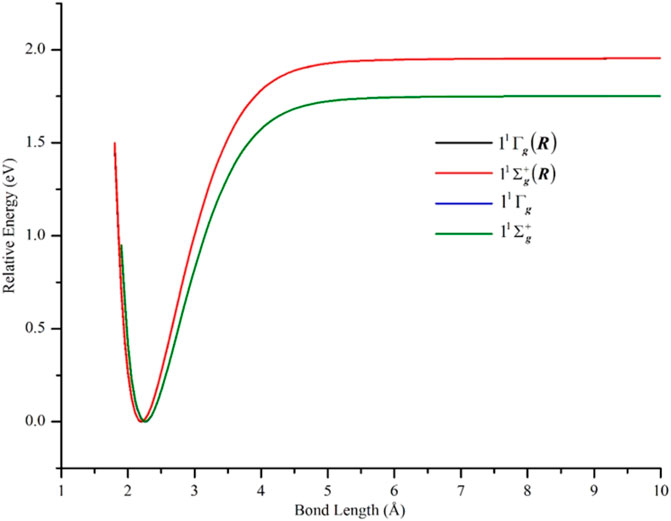
FIGURE 4. PECs of the lowest-lying δδ–hole X 1Γg and 11Σg+ states of Ni2 computed at the GVVPT2 level of theory, with and without scalar relativity included, using the cc-pVTZ (or cc-pVTZ-DK) basis set. Non-relativistic energies are plotted relative to the lowest energy value of the ground X 1Γg term while relativistic energies are plotted relative to the lowest energy of the X 1Γg (R) term.
The 11Σu- state, which was predicted as the ground state of Ni2 at the ACPF level of theory (Cheskidov et al., 2012) and found to lie quite close to a 1Σg+ ground state in a limited CI study (Ahmed and Nixon, 1979), was found at the GVVPT2 level to lie 91.09 cm−1 above the X 1Γg state at equilibrium. The 21Σu- state, however, lay much higher energetically (18,575.76 cm−1 above the ground state at equilibrium).
As can be seen in Table 1, GVVPT2 predicted the triplet δδ–hole states, 13Σg-, 13Σu+, and 13Γu, to lie energetically in the order 13Σg-< 13Σu+ < 13Γu. Cheskidov et al. (Cheskidov et al., 2012) found this same ordering at the ACPF, AQCC, MRCI and MRCI + Q levels of theory, whereas their NEVPT2 calculations predicted the ordering 13Σu+ < 13Γu < 13Σg-, with the 13Σg- state lying at least 139 cm−1 higher than the other two states. It should be noted that the vertical excitation energies in Ref. (Cheskidov et al., 2012) were not determined at the equilibrium geometries of the computed states. The 13Σu+ state, which was predicted as the ground state of Ni2 in some previous wavefunction (Wood et al., 1980; Schultz et al., 2005) and DFT (Diaconu et al., 2004; Kramida et al., 2013) studies, was found in our current study to lie 349.60 cm−1 above the X 1Γg state at the equilibrium geometry. Likewise, the 13Σg- state reported in other studies (Murphy and Messmer, 1992; Diaconu et al., 2004; Kramida et al., 2013) as the ground state of Ni2 lay 221.98 cm−1 higher at equilibrium. The 13Σu- state was found to have a bond length and bond energy comparable to those of 13Σg-, 13Σu+, and 13Γu but lying at least 531.48 cm−1 higher in energy. The EBOs for these triplet states were 0.971 for 13Σg-, 0.933 for 13Σu+ and 13Γu, and 0.923 for 13Σu- in the vicinity of their equilibrium geometries (again computed with 10 leading CSFs). The major configurations for the δδ–hole triplet states involved a doubly occupied 4sσg bonding orbital.
The 13Γu state, in which both δ–holes were in the 3dx2-y2 δg and δu∗ orbitals, was computed using reference κ2(n). As can be seen in Table 1, this state was found to have spectroscopic constants comparable to other δδ–hole triplet states but lay much higher energetically (2442.21 cm−1 above the ground state at equilibrium). The present results suggest that the 3dδ orbitals are indeed split in the bonding interaction. Since they are nondegenerate, the Aufbau principle suggests that energetically low-lying orbitals (the bonding 3dδ orbitals) be occupied before higher ones. Moreover, Hund’s rule suggests that orbitals with similar energies (in this case, 3dδu∗ orbitals) be singly occupied before electron pairing occurs. This seems to bring qualitative understanding into why the 13Σg-, 13Σu+, and 13Γu states in which the 3dx2-y2δu∗ and 3dxyδu∗ are singly occupied lie energetically lower than the 2 3Γu state for which the 3dx2-y2δg and 3dx2-y2δu∗ are singly occupied.
The δπ–Hole and ππ–Hole States
The PECs of the computed ππ–hole states (11Δg and 21Σg+) are shown in Figure 2, while those for the δπ–hole states (11Φg, 11Πg, 13Πg, and 13Φg) are shown in Figure 5 and compared with the ground state PEC. The data describing these curves are in Table 2. GVVPT2 predicted the ππ–hole states to lie higher in energy than the δπ–hole states, in agreement with previous studies (Ahmed and Nixon, 1979; Rasanen et al., 1987; Russo et al., 1994). For all four δπ–hole states studied, the major CSFs involved a doubly occupied 4sσg bonding orbital. Thus, the main configurations of the 11Φg and 11Πg states involved an unpaired spin-increasing electron in the 3dδ subspace and an unpaired spin-decreasing electron in the 3dπ subspace, e.g., 3dδg2 3dδu∗2 3dδg2 3dδu∗(↑) 3dπu2 3dπg∗2 3dπu2 3dπg∗(↓) 4sσg2, whereas the major configurations of the 13Φg and 13Πg states were similar to those of the corresponding singlet states but with two unpaired spin-increasing electrons: one in each of the 3dδ and 3dπ subspaces, e.g., 3dδg2 3dδu∗2 3dδg2 3dδu∗(↑) 3dπu2 3dπg∗2 3dπu2 3dπg∗(↑) 4sσg2.
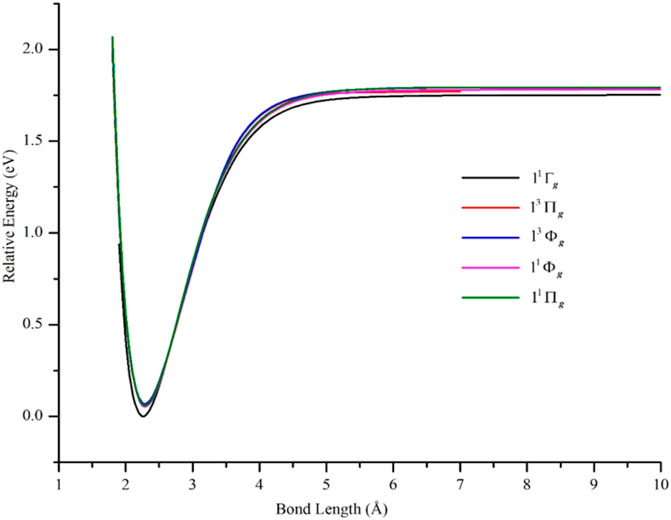
FIGURE 5. PECs of low-lying δπ–hole electronic states of Ni2 computed at the GVVPT2 level of theory using the cc-pVTZ basis set compared with the PEC of the X 1Γg ground term. All energies are plotted relative to the lowest energy value of the X 1Γg ground term.
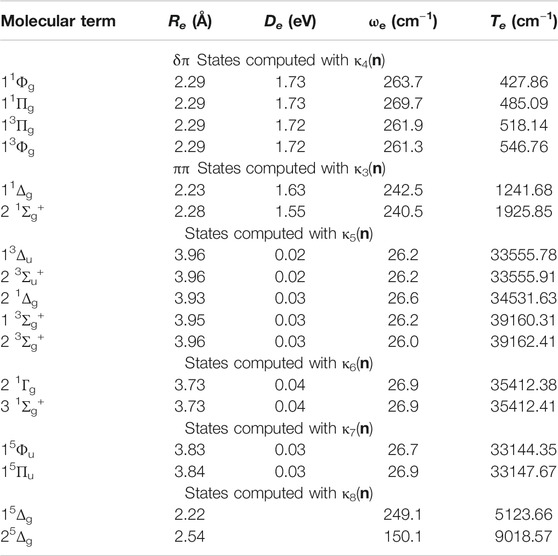
TABLE 2. Equilibrium bond lengths, Re (Å), binding energies, De (eV), harmonic frequencies, ωe (cm−1), and adiabatic transition energies, Te (cm−1), of electronic states of Ni2 calculated at the GVVPT2 level of theory using the cc-pVTZ basis set and reference macroconfigurations κ3(n) to κ8(n).
At the equilibrium geometries, the EBOs were 0.930 for the singlet 11Φg and 11Πg states and 0.933 for the 13Πg and 13Φg states. GVVPT2 predicted the four δπ–hole states to lie energetically in the order 11Φg < 11Πg < 13Πg < 13Φg, in agreement with the Ref. (Cheskidov et al., 2012) study at the scalar relativistic ACPF, AQCC, MRCI, and MRCI + Q levels of theory. However, our calculations found all three states considered in Ref. (Cheskidov et al., 2012) (i.e., 11Πg, 13Πg, and 13Φg) to lie some 500 cm−1 closer to the ground state; e.g., at the scalar relativistic MRCI + Q level, the 13Φg state was reported (Cheskidov et al., 2012) as lying 1238 cm−1 above the ground state at 2.5 Å while non-relativistic GVVPT2 calculations predicted this state to lie 546.76 cm−1 above the ground state at equilibrium. Based on our observation that including scalar relativistic effects increased the energy gap between the 11Σg+ and X 1Γg states, it is likely that including such effects in GVVPT2 calculations on the δπ–hole states might lead to increases in corresponding adiabatic transition energies. It is not anticipated, however, that such effects would lead to any change in the energy ordering of the states.
Although lying higher in energy than the δπ–hole states, the ππ–hole states were found to have slightly shorter bond lengths and larger bond strengths than the δπ–hole states. The 1 1Δg state was 0.06 Å shorter while the 2 1Σg+ state was 0.01 Å shorter in bond length than the δπ–hole states. At 2.24 Å, the EBOs of 1 1Δg and 2 1Σg+ were 1.108 and 1.084 respectively (computed with a dozen CSFs); these were slightly higher than the EBOs of all computed δδ–hole and δπ–hole Ni2 states. Near equilibrium, the major configurations of these ππ–hole states involved a doubly occupied 4sσg bonding orbital and a configuration of the 3dπ subspace that had the two π–holes in the same π–orbital; e.g., 3dπu2 3dπg∗2 3dπu2 3dπg∗0 4sσg2.
States of the 3F4 + 3F4 and 3F4 + 3D3 Manifolds
Figure 6 contains PECs of states belonging to the 3F4 + 3F4 manifold. The data describing these curves are in Table 2. Irrespective of how the model space was partitioned into macroconfigurations [i.e., κi(n)], all such states were found to be van der Waals-like with interaction energies ≤0.04 eV. For example, near the equilibrium geometry (i.e., 3.77 Å), the 2 1Γg and 3 1Σg+ states had EBO of only 0.005, while the 15Φu and 15Πu states had EBOs of 0.003 and 0.00, respectively, at 3.84 Å. These latter quintet states were computed using reference κ7(n) and found to have the lowest total energies among the computed states of the 3F4 + 3F4 manifold; the 15Πu state is 3.312 cm−1 less stable than the 15Φu state at equilibrium. The rest of the PECs in Figure 6 were plotted relative to the 15Φu PEC.
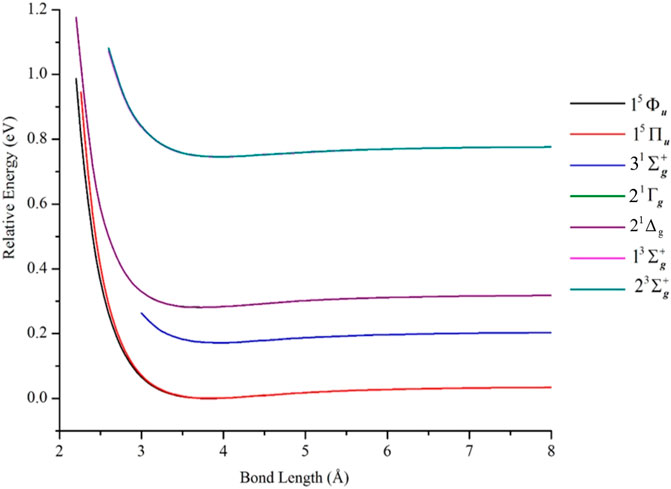
FIGURE 6. PECs of electronic states of Ni2 from the 3F4 (3d84s2) + 3F4 (3d84s2) manifold, computed at the GVVPT2 level of theory using the cc-pVTZ basis set and reference macroconfigurations κ5(n) to κ7(n). All energies are plotted relative to the lowest energy value of the 15Φu term.
Lastly, the 15Δg and 2 5Δg states of the 3F4 + 3D3 manifold were investigated, at short bond lengths only, using reference macroconfiguration κ8(n). The data for these states are included in Table 2. Whereas the 3F4 (3d84s2) + 3F4 (3d84s2) states are van der Waals-like, the short bond length (2.22 Å) and high frequency (249.1 cm−1) of the 15Δg state is suggestive of significant bonding interaction. At 2.22 Å, the major configuration for this state was 3dx2-y2δg2 3dx2-y2δu∗2 3dxyδg2 3dxyδu∗1 3dπu2 3dπg∗1 3dπu2 3dπg∗1 4sσg2 4sσu∗1, contributing 50% by weight to the total wave function. At this geometry, the EBO was found to be 1.186. However, one should approach these results for 3F4 + 3D3 derived states with some caution, as the macroconfiguration used cannot describe their dissociation limits.
Conclusion
The GVVPT2 method was used to study low-lying electronic states of Ni2. The results indicate, in general, that bonding in these states involves predominantly the doubly occupied 4sσg bonding orbital with the 3d–3d electrons pairwise spin coupled (e.g., consistent with antiferromagnetism). This statement is corroborated by EBOs that were found to be approximately 1.0 for most states studied and, moreover, states that did not allow this type of interaction [e.g., belonging to the 3F4 (3d84s2) + 3F4 (3d84s2) manifold] were found to be bound only by weak polarization forces with bond orders close to zero. For computed states of the 3D3 (3d94s1) + 3D3 (3d94s1) dissociation limit, all major configurations involved a doubly occupied 4sσg bonding orbital and a vacant 4sσu∗ antibonding orbital. The energy ordering of the computed states of the 3D3 (3d94s1) + 3D3 (3d94s1) manifold is in agreement with previous studies that found the δδ–hole states to lie lowest in energy followed by the δπ–hole and then the ππ–hole states (Rasanen et al., 1987). For the investigated δδ–hole states, the singlets were more stable than the triplet states at the GVVPT2 level of theory. As expected, based on previous GVVPT2 studies of transition metal dimers, all computed PECs are smooth and without unphysical artifacts (e.g., wiggles).
The ground state of Ni2 was predicted as X 1Γg in agreement with previous results from other high level ab initio methods (Cheskidov et al., 2012; Pou-Amérigo et al., 1994). The determined equilibrium spectroscopic constants of the X 1Γg state, using a cc-pVTZ (or cc-pVTZ-DK) basis, were within the uncertainties of experimental results. The lowest 1Σg+ state was found to be only 0.002 eV (16.40 cm−1) higher than the ground state at the equilibrium geometry. Core-valence correlation was found to be important in the description of low-lying states of Ni2 where the inclusion of 3s3p electron correlation at the GVVPT2 level was shown to improve harmonic frequencies and bond energies (e.g., by an increase of 7.5 cm−1 in frequency and 0.09 eV in bond energy when 3s3p electron correlation was included in GVVPT2 calculations of the X 1Γg state). Scalar relativistic effects were also shown to be important, especially for dissociation energy, where spectroscopic constants from relativistic calculations were predicted to agree better with reference data (e.g., a decrease of 0.06 Å in bond length, increase of 0.20 eV in bond energy, and an increase of 20.0 cm−1 in harmonic frequency when including scalar relativistic effects in calculations of the X 1Γg state). In our previous study (Tamukong et al., 2014) on electronic states of Y2, Mn2, and Tc2, we did not find scalar relativistic effects to be as important for the Mn2 molecule as they have proven to be in the present study. The inclusion of spin-orbit coupling effects was previously found (Cheskidov et al., 2012; Rasanen et al., 1987; Pou-Amérigo et al., 1994) to mix the low-lying states of Ni2, leading to a 0g+[1Σg+(δδ) + 3Σg-(δδ)] ground state. Since the two curves are close and nearly parallel for all internuclear separations in our study, we expect that including such effects after our scalar relativistic GVVPT2 treatment should lead to a similar mixing of states (i.e., and without change in spectroscopic constants).
The states investigated within the 3F4 (3d84s2) + 3D3 (3d94s1) manifold suggested significant bonding interaction, giving large harmonic frequencies and short bond lengths in comparison with states correlating with the 3F4 (3d84s2) + 3F4 (3d84s2) dissociation limit. Further work on Ni2 should explore the 3F4 (3d84s2) + 3D3 (3d94s1) manifold more thoroughly, including possible expansion of the active space. It should be noted, however, that in the present study, we did not observe any significant electron excitations from the valence orbitals to 4p-dominated virtual orbitals. Also, while spin-free relativistic effects are noticeable (especially for bond dissociation energies), the parallelity and small energy separations for states in these manifolds are very similar to those in the 3D3 (3d94s1) + 3D3 (3d94s1) dissociation channel and, again, we do not expect that spin-dependent relativistic effects would change PECs appreciably, although current capabilities of our GVVPT2 code did not let us explore this.
In summary, the present study showed that Ni2 does not form strong bonds with atomic configurations in which the 4s subshell is fully filled. This observation is consistent with other studies, by us and by others, of transition elements of the first row. Bonding in these systems is generally favored by atomic configurations that involve at least one of the participating atoms in an excited state (3dn+14s1). For example, in our previous study (Tamukong et al., 2012) of the low-lying electronic states of Sc2, Cr2, and Mn2, the lowest states of Sc2 were shown to correlate with the 2D (3d14s2) + 4F (3d24s1) dissociation asymptote, while those of Cr2 correlated with the 7S (3d54s1) + 7S (3d54s1) dissociation limit. However, bonding in transition metal dimers is subtle and the most stable states of Mn2 were obtained from weakly coupled 5D (3d54s2) + 5D (3d54s2) ground state Mn atoms, similar to the 3F4 (3d84s2) + 3F4 (3d84s2) coupling of ground state Ni atoms. Our results provide further evidence that GVVPT2, with sf-X2C treatment of relativistic effects, predict electronic excitation and bond energy trends in the first row transition metals consistent with experiment and the highest level ab initio calculations. In the present case, the quasidegeneracy of the 3F and 3D states of the Ni atom demonstrates that GVVPT2 can successfully be used for a system with an extraordinarily dense manifold of states, which generally requires computationally intensive variational treatments.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
MH and PT contributed to conception and design of the study. PT performed numerical calculations. MH and PT interpreted data and organized presentation. PT wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
The authors gratefully acknowledge the National Science Foundation (Grant No. EPS 0814442) for the financial support under which the project was started, and to North Dakota EPSCoR for support in later stages.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahmed, F., and Nixon, E. R. (1979). The A→X System of Ni2 in Argon Matrices. J. Chem. Phys. 71 (8), 3547–3549. doi:10.1063/1.438750
Andersson, K., and Roos, B. O. (1992). Excitation Energies in the Nickel Atom Studied with the Complete Active Space Scf Method and Second-Order Perturbation Theory. Chem. Phys. Lett. 191 (6), 507–514. doi:10.1016/0009-2614(92)85581-t
Angeli, C., Cimiraglia, R., Evangelisti, S., Leininger, T., and Malrieu, J.-P. (2001). Introduction Ofn-Electron Valence States for Multireference Perturbation Theory. J. Chem. Phys. 114 (23), 10252–10264. doi:10.1063/1.1361246
Balabanov, N. B., and Peterson, K. A. (2005). Systematically Convergent Basis Sets for Transition Metals. I. All-Electron Correlation Consistent Basis Sets for the 3d Elements Sc–Zn. J. Chem. Phys. 123 (6), 064107. doi:10.1063/1.1998907
Basch, H., Newton, M. D., and Moskowitz, J. W. (1980). The Electronic Structure of Small Nickel Atom Clusters. J. Chem. Phys. 73 (9), 4492–4510. doi:10.1063/1.440687
Bauschlicher, C. W., Walch, S. P., and Partridge, H. (1982). On Correlation in the First Row Transition Metal Atoms. J. Chem. Phys. 76 (2), 1033–1039. doi:10.1063/1.443095
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98 (7), 5648–5652. doi:10.1063/1.464913
Cheng, L., and Gauss, J. (2011). Analytic Energy Gradients for the Spin-free Exact Two-Component Theory Using an Exact Block Diagonalization for the One-Electron Dirac Hamiltonian. J. Chem. Phys. 135 (8), 084114. doi:10.1063/1.3624397
Cheskidov, A. V., Buchachenko, A. A., and Bezrukov, D. S. (2012). Ab Initio Spin-Orbit Calculations on the Lowest States of the Nickel Dimer. J. Chem. Phys. 136 (21), 214304. doi:10.1063/1.4721624
Cooper, W. F., Clarke, G. A., and Hare, C. R. (1972). Molecular Orbital Theory of the Diatomic Molecules of the First Row Transition Metals. J. Phys. Chem. 76 (16), 2268–2273. doi:10.1021/j100660a016
De Vore, T. C., Ewing, A., Franzen, H. F., and Calder, V. (1975). The Visible Absorption Spectra of Mn2, Fe2, and Ni2 in Argon Matrices. Chem. Phys. Lett. 35 (1), 78–81. doi:10.1016/0009-2614(75)85592-8
Diaconu, C. V., Cho, A. E., Doll, J. D., and Freeman, D. L. (2004). Broken-Symmetry Unrestricted Hybrid Density Functional Calculations on Nickel Dimer and Nickel Hydride. J. Chem. Phys. 121 (20), 10026–10040. doi:10.1063/1.1798992
Dolg, M., Wedig, U., Stoll, H., and Preuss, H. (1987). Energy‐adjustedabinitiopseudopotentials for the First Row Transition Elements. J. Chem. Phys. 86 (2), 866–872. doi:10.1063/1.452288
Dong, C. D., Lefkidis, G., and Hübner, W. (2013). Quantum Isobaric Process in Ni2. J. Supercond. Nov. Magn. 26 (5), 1589–1594. doi:10.1007/s10948-012-1948-8
Du, J., Sun, X., and Wang, H. (2008). The Confirmation of Accurate Combination of Functional and Basis Set for Transition-Metal Dimers: Fe2, Co2, Ni2, Ru2, Rh2, Pd2, Os2, Ir2, and Pt2. Int. J. Quan. Chem. 108 (9), 1505–1517. doi:10.1002/qua.21684
Dyall, K. G. (1997). Interfacing Relativistic and Nonrelativistic Methods. I. Normalized Elimination of the Small Component in the Modified Dirac Equation. J. Chem. Phys. 106 (23), 9618–9626. doi:10.1063/1.473860
Gdanitz, R. J., and Ahlrichs, R. (1988). The Averaged Coupled-Pair Functional (ACPF): A Size-Extensive Modification of MR CI(SD). Chem. Phys. Lett. 143 (5), 413–420. doi:10.1016/0009-2614(88)87388-3
Gutsev, G. L., and Bauschlicher, C. W. (2003). Chemical Bonding, Electron Affinity, and Ionization Energies of the Homonuclear 3d Metal Dimers. J. Phys. Chem. A. 107 (23), 4755–4767. doi:10.1021/jp030146v
Harris, J., and Jones, R. O. (1979). Density Functional Theory and Molecular Bonding. III. Iron-Series Dimers. J. Chem. Phys. 70 (2), 830–841. doi:10.1063/1.437516
Harrison, J. F. (2000). Electronic Structure of Diatomic Molecules Composed of a First-Row Transition Metal and Main-Group Element (H−F). Chem. Rev. 100 (2), 679–716. doi:10.1021/cr980411m
Ho, J., Polak, M. L., Ervin, K. M., and Lineberger, W. C. (1993). Photoelectron Spectroscopy of Nickel Group Dimers: Ni2-, Pd2-, and Pt2-. J. Chem. Phys. 99 (11), 8542–8551. doi:10.1063/1.465577
Jiang, W., Khait, Y. G., and Hoffmann, M. R. (2009). Configuration-Driven Unitary Group Approach for Generalized Van Vleck Variant Multireference Perturbation Theory. J. Phys. Chem. A. 113 (16), 4374–4380. doi:10.1021/jp811082p
Kamal, C., Banerjee, A., Ghanty, T. K., and Chakrabarti, A. (2012). Interesting Periodic Variations in Physical and Chemical Properties of Homonuclear Diatomic Molecules. Int. J. Quan. Chem. 112 (4), 1097–1106. doi:10.1002/qua.23088
Kant, A. (1964). Dissociation Energies of Diatomic Molecules of the Transition Elements. I. Nickel. J. Chem. Phys. 41 (6), 1872–1876. doi:10.1063/1.1726169
Khait, Y. G., Song, J., and Hoffmann, M. R. (2002). Explication and Revision of Generalized Van Vleck Perturbation Theory for Molecular Electronic Structure. J. Chem. Phys. 117 (9), 4133–4145. doi:10.1063/1.1497642
Khait, Y. G., Song, J., and Hoffmann, M. R. (2004). Macroconfigurations in Molecular Electronic Structure Theory. Int. J. Quan. Chem. 99 (4), 210–220. doi:10.1002/qua.10852
Khait, Y. G., Theis, D., and Hoffmann, M. R. (2012). Nonadiabatic Coupling Terms for the GVVPT2 Variant of Multireference Perturbation Theory. Chem. Phys. 401, 88–94. doi:10.1016/j.chemphys.2011.09.014
Kramida, A., Ralchenko, Y., and Reader, J. (2013). NIST Atomic Spectra Database (Ver. 5.1) [Online]. Gaithersburg, MD: National Institute of Standards and TechnologyAvailable at: http://physics.nist.gov/asd (Accessed Feb 7, 2014).
Li, Z., Suo, B., Zhang, Y., Xiao, Y., and Liu, W. (2013). Combining Spin-Adapted Open-Shell TD-DFT with Spin-Orbit Coupling. Mol. Phys. 111 (24), 3741–3755. doi:10.1080/00268976.2013.785611
Li, Z., Xiao, Y., and Liu, W. (2012). On the Spin Separation of Algebraic Two-Component Relativistic Hamiltonians. J. Chem. Phys. 137 (15), 154114. doi:10.1063/1.4758987
Liu, W. (2010). Ideas of Relativistic Quantum Chemistry. Mol. Phys. 108 (13), 1679–1706. doi:10.1080/00268971003781571
Liu, W. (2014). Advances in Relativistic Molecular Quantum Mechanics. Phys. Rep. 537 (2), 58–89. doi:10.1016/j.physrep.2013.11.006
Mbote, Y. E. B., Khait, Y. G., Hardel, C., and Hoffmann, M. R. (2010). Multireference Generalized Van Vleck Perturbation Theory (GVVPT2) Study of the NCO + HCNO Reaction: Insight into Intermediates†. J. Phys. Chem. A. 114 (33), 8831–8836. doi:10.1021/jp102051p
Melius, C. F., Moskowitz, J. W., Mortola, A. P., Baillie, M. B., and Ratner, M. A. (1976). A Molecular Complex Model for the Chemisorption of Hydrogen on a Nickel Surface. Surf. Sci. 59 (1), 279–292. doi:10.1016/0039-6028(76)90305-8
Mokambe, R. M., Hicks, J. M., Kerker, D., Jiang, W., Theis, D., Chen, Z., et al. (2013). GVVPT2 Multireference Perturbation Theory Study of Selenium Oxides. Mol. Phys. 111 (9-11), 1078–1091. doi:10.1080/00268976.2013.809163
Morse, M. D., Hansen, G. P., Langridge‐Smith, P. R. R., Zheng, L. S., Geusic, M. E., Michalopoulos, D. L., et al. (1984). Spectroscopic Studies of the Jet‐cooled Nickel Dimer. J. Chem. Phys. 80 (11), 5400–5405. doi:10.1063/1.446646
Murphy, R. B., and Messmer, R. P. (1992). Correlation in First-Row Transition Metal Atoms Using Generalized Mo/ller-Plesset Perturbation Theory. J. Chem. Phys. 97 (7), 4974–4985. doi:10.1063/1.463850
Nakatsuji, H. (1979). Cluster Expansion of the Wavefunction. Electron Correlations in Ground and Excited States by Sac (Symmetry-Adapted-Cluster) and Sac Ci Theories. Chem. Phys. Lett. 67 (2), 329–333. doi:10.1016/0009-2614(79)85172-6
Noell, J. O., Newton, M. D., Hay, P. J., Martin, R. L., and Bobrowicz, F. W. (1980). An Ab Initio Study of the Bonding in Diatomic Nickel. J. Chem. Phys. 73 (5), 2360–2371. doi:10.1063/1.440386
Perdew, J. P. (1986). Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 33 (12), 8822–8824. doi:10.1103/physrevb.33.8822
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77 (18), 3865–3868. doi:10.1103/physrevlett.77.3865
Pinegar, J. C., Langenberg, J. D., Arrington, C. A., Spain, E. M., and Morse, M. D. (1995). Ni2 Revisited: Reassignment of the Ground Electronic State. J. Chem. Phys. 102 (2), 666–674. doi:10.1063/1.469562
Pou-Amérigo, R., Merchan, M., Nebot‐Gil, I., Malmqvist, P. Å., and Roos, B. O. (1994). The Chemical Bonds in CuH, Cu2, NiH, and Ni2 Studied with Multiconfigurational Second Order Perturbation Theory. J. Chem. Phys. 101 (6), 4893–4902.
Raghavachari, K., and Trucks, G. W. (1989). Highly Correlated Systems. Excitation Energies of First Row Transition Metals Sc-Cu. J. Chem. Phys. 91 (2), 1062–1065. doi:10.1063/1.457230
Rasanen, M., Heimbrook, L. A., and Bondybey, V. E. (1987). Rare Gas Matrix Studies of the Products of Vaporization of Nickel. J. Mol. Struct. 157 (1), 129–140. doi:10.1016/0022-2860(87)87088-6
Rösch, N., and Rhodin, T. N. (1974). Bonding of Ethylene to Diatomic Nickel According to a Self-Consistent-Field,Xα, Scattered-Wave Model. Phys. Rev. Lett. 32 (21), 1189–1192. doi:10.1103/physrevlett.32.1189
Russo, T. V., Martin, R. L., and Hay, P. J. (1994). Density Functional Calculations on First‐row Transition Metals. J. Chem. Phys. 101 (9), 7729–7737. doi:10.1063/1.468265
Schultz, N. E., Zhao, Y., and Truhlar, D. G. (2005). Databases for Transition Element Bonding: Metal−Metal Bond Energies and Bond Lengths and Their Use to Test Hybrid, Hybrid Meta, and Meta Density Functionals and Generalized Gradient Approximations. J. Phys. Chem. A. 109 (19), 4388–4403. doi:10.1021/jp0504468
Shi-Ying, Y., and Zheng-He, Z. (2008). Spin Polarization Effect of Ni 2 Molecule. Chin. Phys. B 17 (12), 4498–4503. doi:10.1088/1674-1056/17/12/028
Shim, I., Dahl, J. P., and Johansen, H. (1979). Ab Initio Hartree-Fock and Configuration-Interaction Treatment of the Interaction between Two Nickel Atoms. Int. J. Quan. Chem. 15 (3), 311–331. doi:10.1002/qua.560150306
Szalay, P. G., and Bartlett, R. J. (1993). Multi-Reference Averaged Quadratic Coupled-Cluster Method: A Size-Extensive Modification of Multi-Reference CI. Chem. Phys. Lett. 214 (5), 481–488. doi:10.1016/0009-2614(93)85670-j
Tamukong, P. K., Hoffmann, M. R., Li, Z., and Liu, W. (2014). Relativistic GVVPT2 Multireference Perturbation Theory Description of the Electronic States of Y2and Tc2. J. Phys. Chem. A. 118 (8), 1489–1501. doi:10.1021/jp409426n
Tamukong, P. K., Theis, D., Khait, Y. G., and Hoffmann, M. R. (2012). GVVPT2 Multireference Perturbation Theory Description of Diatomic Scandium, Chromium, and Manganese. J. Phys. Chem. A. 116 (18), 4590–4601. doi:10.1021/jp300401u
Upton, T. H., and Goddard, W. A. (1978). The Electronic States of Ni2 and Ni2+. J. Am. Chem. Soc. 100 (18), 5659–5668. doi:10.1021/ja00486a014
Werner, H.-J., and Knowles, P. J. (1990). A Comparison of Variational and Non-variational Internally Contracted Multiconfiguration-Reference Configuration Interaction Calculations. Theor. Chim. Acta 78 (3), 175–187.
Wood, C., Doran, M., Hillier, I. H., and Guest, M. F. (1980). Theoretical Study of the Electronic Structure of the Transition Metal Dimers, Sc2, Cr2, Mo2 and Ni2. Faraday Symp. Chem. Soc. 14, 159–169. doi:10.1039/fs9801400159
Keywords: nickel dimer, metal dimers, multireference perturbation theory, generalized Van Vleck perturbation theory, electronic structure calculations
Citation: Tamukong PK and Hoffmann MR (2021) Low-Lying Electronic States of the Nickel Dimer. Front. Chem. 9:678930. doi: 10.3389/fchem.2021.678930
Received: 11 March 2021; Accepted: 27 April 2021;
Published: 13 May 2021.
Edited by:
Wolfgang Schöfberger, Johannes Kepler University of Linz, AustriaReviewed by:
Ivan Cernusak, Comenius University, SlovakiaSudip Chattopadhyay, Indian Institute of Engineering Science and Technology, Shibpur, India
Copyright © 2021 Tamukong and Hoffmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark R. Hoffmann, bWFyay5ob2ZmbWFubkB1bmQuZWR1