
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 26 May 2021
Sec. Medicinal and Pharmaceutical Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.674967
This article is part of the Research TopicApplication of PROTACs as a Novel Strategy for Drug DiscoveryView all 7 articles
 Hanqiao Xu1,2†Nobumichi Ohoka3†Hidetomo Yokoo1Kanako Nemoto1Takashi Ohtsuki2Hiroshi Matsufuji2
Hanqiao Xu1,2†Nobumichi Ohoka3†Hidetomo Yokoo1Kanako Nemoto1Takashi Ohtsuki2Hiroshi Matsufuji2 Mikihiko Naito4Takao Inoue3Genichiro Tsuji1*
Mikihiko Naito4Takao Inoue3Genichiro Tsuji1* Yosuke Demizu1,5*
Yosuke Demizu1,5*Liver X receptors (LXRs) belong to the nuclear hormone receptor superfamily and function as ligand-dependent transcription factors that regulate cholesterol homeostasis, lipid homeostasis, and immune responses. LXR antagonists are promising treatments for hypercholesterolemia and diabetes. However, effective LXR antagonists and inhibitors are yet to be developed. Thus, we aimed to develop LXR degraders (proteolysis targeting chimeras PROTACs against LXR) as a complementary strategy to provide a similar effect to LXR inhibition. In this study, we report the development of GW3965-PEG5-VH032 (3), a PROTAC capable of effectively degrading LXRβ protein. Compound 3 induced the ubiquitin-proteasome system-dependent degradation of the LXRβ protein, which requires VHL E3 ligase. We hope that PROTACs targeting LXR proteins will become novel therapeutic agents for LXR-related diseases.
Liver X receptor (LXR) is a ligand-dependent transcription factor belonging to the nuclear hormone receptor superfamily (Edwards et al., 2002). Two isoforms, LXRα and LXRβ, have high amino acid sequence homology (78%) but different expression distributions. LXRα is mainly expressed in the liver, intestines, macrophages, and kidneys, whereas LXRβ is ubiquitously expressed in various tissues (Zhu and Li, 2009). Ligand-unbound LXR forms a repressor complex at the LXR target gene promoter. When the ligand binds, it dissociates from the corepressor complex and recruits coactivators such as thyroid hormone receptor-associated protein (TRAP220/DRIP-2) to the target promoter (Wagner et al., 2003; Phelan et al., 2008). LXRs play a pivotal role in the transcriptional regulation of cholesterol homeostasis, fatty acid metabolism, glycolysis, immune responses, and inflammatory responses (Janowski et al., 1996; Wang and Tontonoz, 2018).
Oxidized cholesterol derivatives (oxysterols) such as (22R)-22-hydroxycholesterol, (20S)-22-hydroxycholesterol, and (24S)-24,25-epoxycholesterol are known to be endogenous ligands for LXR, and they activate both LXRα and LXRβ (Baranowski, 2008). In addition, a variety of synthetic LXR ligands have been reported, including isoform-selective ligands (Kick et al., 2016; Kirchgessner et al., 2016). LXR agonists have potential applications as cholesterol-lowering drugs and treatments for atherosclerosis. However, their clinical use is limited because they promote hepatic steatosis by increasing hepatic lipid synthesis (Grefhorst et al., 2002; Viennois et al., 2012). On the other hand, inverse agonists (Griffett et al., 2013; Flaveny et al., 2015) and antagonists (Noguchi-Yachide et al., 2009; Moriwaki et al., 2014; Renga et al., 2015) for LXRs have been developed because inhibiting excessive activation of LXR in the liver is an appropriate strategy to improve hepatic lipid metabolism. However, they have issues with their weak activity and difficulty in chemical synthesis. Thus, the development of complementary strategies could help realize the full potential of LXR inhibition.
Proteolysis targeting chimera (PROTAC) technology has been receiving much attention as a novel strategy to degrade proteins of interest (POI) (Pavia and Crews, 2019; Sun et al., 2019). PROTACs are bifunctional molecules with a ligand for the POI and a ligand for an E3 ligase. PROTACs cross-link between the POI and E3 ligase, which in turn degrades POI by the ubiquitin-proteasome system (UPS). Hence, PROTACs are expected to be a promising tool for suppressing the function of disease-related proteins in drug discovery. Therefore, we hypothesized that we could produce promising compounds with LXR inhibitory activity by developing PROTACs using reported agonists (Gustafson et al., 2015). Herein, we report the design and synthesis of LXR-agonist-based PROTACs which exhibit LXR degradation activity via the UPS.
For PROTAC design, a potent LXRα/LXRβ agonist GW3965 (Collins et al., 2002) was selected because the binding mode between LXRα and GW3965 has previously been determined by X-ray diffraction (Fradera et al., 2010). In the co-crystal structure, the carboxyl group of GW3965 is oriented toward the outside of the protein (Figure 1A). Based on this structural information, we designed a GW3965-based PROTAC by linking the carboxyl group with an E3 ligase ligand via a polyethylene glycol linker (PEG3–PEG6). Two representative types of E3 ligase ligands, pomalidomide binding to cereblon (CRBN) and VH032 binding to Von Hippel-Lindau (VHL), were selected (Figure 1B).
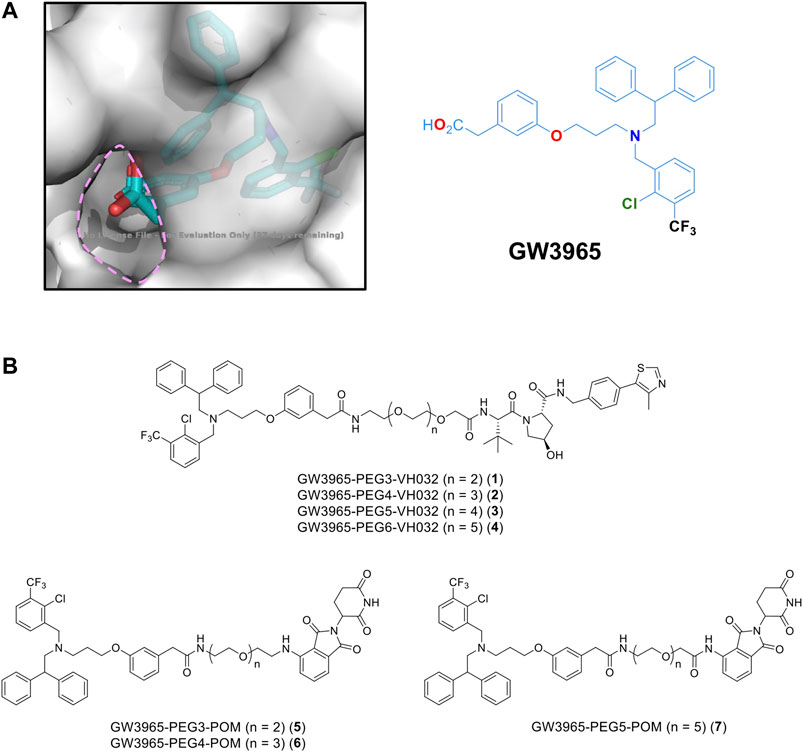
FIGURE 1. (A) X-ray crystal structure of a GW3965-LXRα complex (PDB: 3IPQ). (B) Designated PROTACs in this study.
The representative synthetic route for VH032-based PROTACs, GW3965-(PEG3–PEG6)-VH032, 1–4 is shown in Scheme 1. Ligand GW3965 was conjugated with E3 ligase ligand VH032 with PEG linkers of different lengths via a condensation reaction using HATU/DIPEA or EDCI. Other molecules, including pomalidomide-based PROTAC, were also synthesized in a similar manner, as shown in the Supplementary Material.
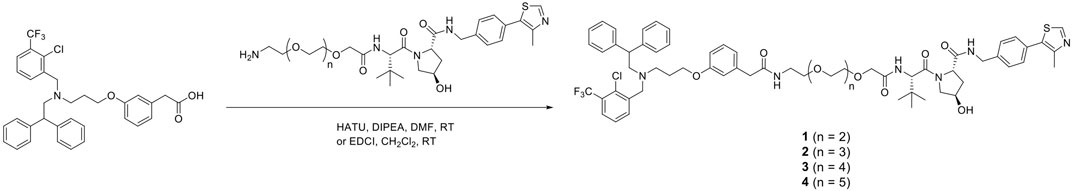
SCHEME 1. Synthesis of PROTACs for LXR (GW3965-PEG-VH032, 1–4).
The degradation activities of the synthesized chimeric compounds against target proteins, LXRα and LXRβ, that bind to GW3965 were evaluated by western blot using HuH-7 human hepatoma cells expressing the target proteins. Since we could not obtain the appropriate antibodies to detect endogenous LXRα, only the results for LXRβ are shown. We first evaluated the LXRβ reduction activities of a series of chimeric compounds containing different E3 ligands (pomalidomide for CRBN and VH032 for VHL) or different linker lengths (PEG3, PEG4, and PEG5). Compound 3 showed the most potent activity among them (Figure 2A and Supplementary Figure S8). To investigate the optimal linker length in the VHL series, compound 4 with PEG6 linker was synthesized. The reduction activity was almost lost with this linker extension, suggesting that the PEG5 length is optimal (Figure 2B). Compound 3 effectively reduced LXRβ protein levels even after 8 h (Figure 2C). The LXRβ binding affinity (EC50) of compound 3 was determined using a time-resolved fluorescence energy transfer (TR-FRET) assay with GW3965 as a positive control. This confirmed that the EC50 values of compound 3 (EC50 = 31 ± 4.4 nM) were comparable to that of GW3965 (EC50 = 20 ± 7.2 nM) (Table 1). As observed in the results of compound 3 and 7 (Figure 2A), the protein degradation efficacy by PROTAC molecules was often suppressed at higher concentrations, which is known as a hook effect (Bondeson et al., 2015). This effect is explained by the inhibition of ternary complex formation (E3-PROTAC-target) by an excess amount of bivalent compounds such as PROTACs.
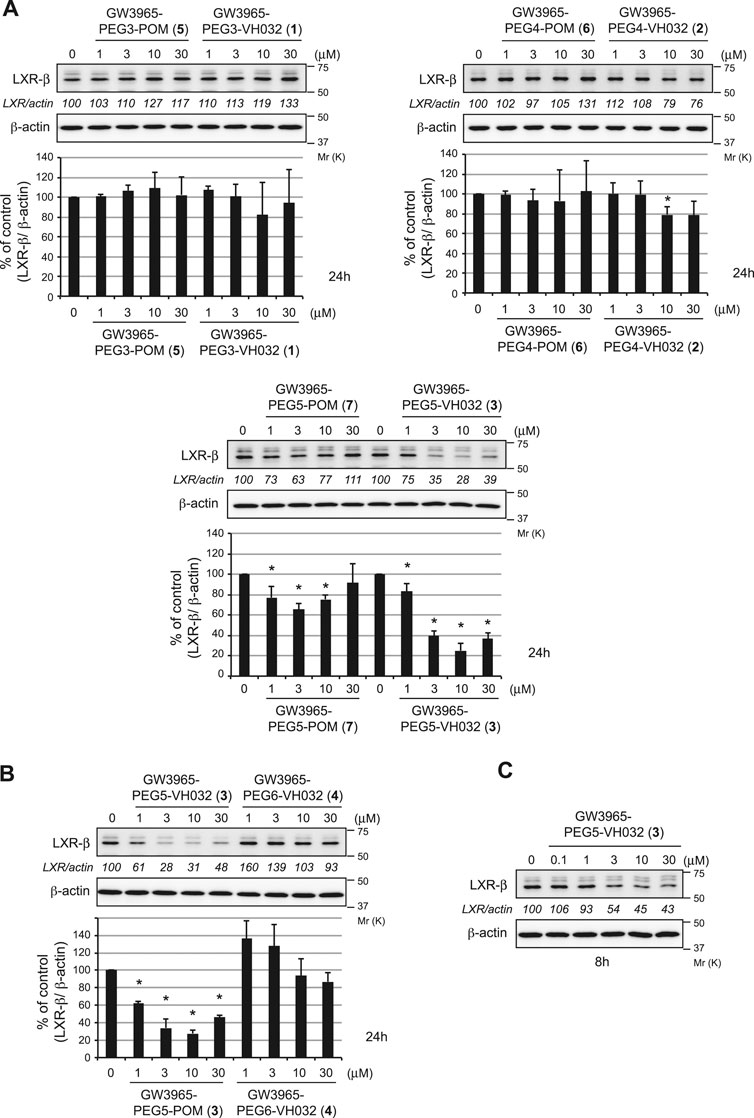
FIGURE 2. Degradation of the LXRβ protein by the synthesized compounds. HuH-7 cells that had been cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) were treated with the indicated compounds for 24 h (A, B) or 8 h (C). Immunoblots of the cell lysates that had been stained with the indicated antibodies are shown (representative data are shown). The numbers below the LXRβ panels represent LXR/actin normalized by designating the expression from the vehicle control condition as 100%. Data in the bar graph are the mean ± S.D. (error bars) of three independent experiments. Asterisks indicate p < 0.05 compared with vehicle control.

TABLE 1. Binding affinities (EC50; half maximal effective concentration) of compounds against LXRβ determined by TR-FRET coactivator assays.
To investigate the mechanism of LXRβ reduction by compound 3, we examined the effect of UPS inhibitors (Figure 3A). Compound 3-induced decrease in the LXRβ protein was abrogated by co-treatment with a proteasome inhibitor, MG132, and a ubiquitin-activating inhibitor, MLN7243, indicating that the compound induces UPS-dependent degradation of the LXRβ protein. To confirm whether VHL is required for the degradation of the LXRβ protein by compound 3, we examined the effect of silencing the E3 ligase by short interfering RNA (siRNA) (Figure 3B). The depletion of VHL by siRNAs completely suppressed the degradation of the LXRβ protein by compound 3, indicating that VHL is required for degradation.
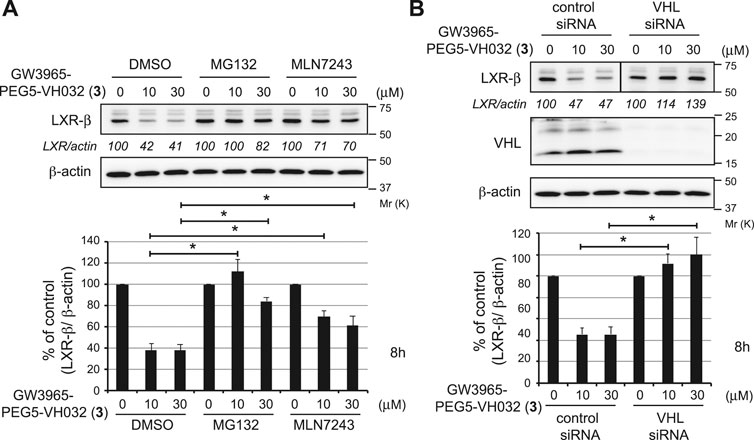
FIGURE 3. Mechanism of LXRβ reduction by GW3965-PEG5-VH032 (3). (A) Effect of UPS inhibitors on GW3965-PEG5-VH032-induced reduction of the LXRβ protein. HuH-7 cells that had been cultured in DMEM containing 10% FBS were treated with the indicated concentrations of GW3965-PEG5-VH032 in the presence or absence of 10 μm of MG132 or MLN7243 for 8 h (B) VHL E3 ligase is required for the degradation of the LXRβ protein by GW3965-PEG5-VH032. HuH-7 cells were transfected with the VHL siRNA for 42 h and treated with the indicated concentrations of GW3965-PEG5-VH032 for 8 h. A mixture of three different siRNAs against VHL was used to suppress expression. Immunoblots of cell lysates that had been stained with the indicated antibodies are shown (representative data are shown). The numbers below the LXRβ panels represent LXR/actin normalized by designating the expression from the vehicle control condition as 100%. Data in the bar graph are the mean ± S.D. (error bars) of three independent experiments. Asterisks indicate p < 0.05.
Herein, we report the synthesis of a PROTAC for LXR degradation as an effective inhibitory molecule. In the molecular design, the linking position of chimeric compounds was determined based on the structural information from X-ray crystallography of LXRα and its agonist GW3965. For the E3 ligase ligand in the PROTAC, VH032 and pomalidomide were introduced into chimeric compounds. The LXRβ degradation activity of the synthesized PROTACs was evaluated by western blot using HuH-7 human hepatoma cells, and it was found that the activity of VH032-based PROTACs (GW3965-PEG-VH032) was more potent than that of pomalidomide-based PROTACs (GW3965-PEG-POM) between the PEG3-PEG5 linkers. To investigate the effect of the linker length on the degradation activity, a series of VH032-type PROTACs with PEG3–PEG6 were examined, which revealed that the PROTAC with PEG5 (GW3965-PEG5-VH032, 3) exhibits the most potent activity for LXRβ degradation among them. Compound 3 was confirmed to bind to LXRβ, inducing its degradation. LXRβ degradation by this molecule occurs via the ubiquitin-proteasome system mediated by VHL E3 ligase. The degraders developed in this study have potential as novel therapeutic agents for LXR-related diseases. Therefore, our results suggest that agonist-based PROTACs could be a new approach to create PROTACs, even in the absence of an appropriate antagonist as a binding ligand for the POI.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
HX and HY carried out the collection of experimental data. NO carried out the experiments and wrote the manuscript, KN conducted the initial experiments. TO, HM, MN, and TI reviewed and edited the article. GT and YD directed the project and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported in part by grants from Japan Agency for Medical Research and Development (20mk0101120j0003 to YD, 20ak0101073j0604 to MN, 20ak0101073j0704 and 20fk0108297j0001 to NO, and 20ak0101073j0904 to YD); Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science and Technology (JSPS/MEXT KAKENHI Grants Number JP17K08385 to YD, JP18K06567 to NO, and JP18H05502 to MN and YD); TERUMO FOUNDATION for life sciences and ARTS (to YD); Takeda Science Foundation (to YD); the Naito Foundation (to YD); the Sumitomo Foundation (to YD); Japan Foundation of Applied Enzymology (to YD); and the Novartis Foundation (Japan) for the Promotion of Science (to YD).
MN is a project professor supported by Eisai and a scientific advisor of Ubience.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.674967/full#supplementary-material
Baranowski, M. (2008). Biological Role of Liver X Receptors. J. Physiol. Pharmacol. 59 (Suppl. 7), 31–55.
Bondeson, D. P., Mares, A., Smith, I. E. D., Ko, E., Campos, S., Miah, A. H., et al. (2015). Catalytic In Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 11, 611–617. doi:10.1038/nchembio.1858
Collins, J. L., Fivush, A. M., Watson, M. A., Galardi, C. M., Lewis, M. C., Moore, L. B., et al. (2002). Identification of a Nonsteroidal Liver X Receptor Agonist through Parallel Array Synthesis of Tertiary Amines. J. Med. Chem. 45, 1963–1966. doi:10.1021/jm0255116
Edwards, P. A., Kast, H. R., and Anisfeld, A. M. (2002). BAREing it All: the Adoption of LXR and FXR and Their Roles in Lipid Homeostasis. J. Lipid Res. 43, 2–12. doi:10.1016/S0022-2275(20)30180-2
Flaveny, C. A., Colin, A., Griffett, K., Ei-Gendy, B. E. I-D., Kazantzis, M., Sengupta, M., et al. (2015). Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 28, 42–56. doi:10.1016/j.ccell.2015.05.007
Fradera, X., Vu, D., Nimz, O., Skene, R., Hosfield, D., Wynands, R., et al. (2010). X-Ray Structures of the LXRα LBD in its Homodimeric Form and Implications for Heterodimer Signaling. J. Mol. Biol. 28, 120–132. doi:10.1016/j.jmb.2010.04.005
Grefhorst, A., Elzinga, B. M., Voshol, P. J., Plo sch, T., Kok, T., Bloks, V. W., et al. (2002). Stimulation of Lipogenesis by Pharmacological Activation of the Liver X Receptor Leads to Production of Large, Triglyceride-Rich Very Low Density Lipoprotein Particles. J. Biol. Chem. 277, 34182–34190. doi:10.1074/jbc.m204887200
Griffett, K., Solt, L. A., El-Gendy, B. E.-D. M., Kamenecka, T. M., and Burris, T. P. (2013). A Liver-Selective LXR Inverse Agonist that Suppresses Hepatic Steatosis. ACS Chem. Biol. 8, 559–567. doi:10.1021/cb300541g
Gustafson, J. L., Neklesa, T. K., Cox, C. S., Roth, A. G., Buckley, D. L., Tae, H. S., et al. (2015). Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed. 54, 9659–9662. doi:10.1002/anie.201503720
Janowski, B. A., Willy, P. J., Devi, T. R., Falck, J. R., and Mangelsdorf, D. J. (1996). An Oxysterol Signalling Pathway Mediated by the Nuclear Receptor LXRα. Nature 383, 728–731. doi:10.1038/383728a0
Kick, E. K., Busch, B. B., Martin, R., Stevens, W. C., Bollu, V., Xie, Y., et al. (2016). Discovery of Highly Potent Liver X Receptor β Agonists. ACS Med. Chem. Lett. 7, 1207–1212. doi:10.1021/acsmedchemlett.6b00234
Kirchgessner, T. G., Sleph, P., Ostrowski, J., Lupisella, J., Ryan, C. S., Liu, X., et al. (2016). Beneficial and Adverse Effects of an LXR Agonist on Human Lipid and Lipoprotein Metabolism and Circulating Neutrophils. Cell Metab. 24, 223–233. doi:10.1016/j.cmet.2016.07.016
Moriwaki, S., Murakami, H., Takahashi, N., Uemura, T., Taketani, K., Hoshino, S., et al. (2014). Yamogenin in Fenugreek Inhibits Lipid Accumulation through the Suppression of Gene Expression in Fatty Acid Synthesis in Hepatocytes. Biosci. Biotechnol. Biochem. 78, 1231–1236. doi:10.1080/09168451.2014.915736
Noguchi-Yachide, T., Miyachi, H., Aoyama, H., Aoyama, A., Makishima, M., and Hashimoto, Y. (2007). Structural Development of Liver X Receptor (LXR) Antagonists Derived from Thalidomide-Related Glucosidase Inhibitors. Chem. Pharm. Bull. 55, 1750–1754. doi:10.1248/cpb.55.1750
Pavia, S.-L., and Crews, C. M. (2019). Targeted Protein Degradation: Elements of PROTAC Design. Curr. Opin. Chem. Biol. 50, 111–119. doi:10.1016/j.cbpa.2019.02.022
Phelan, C. A., Weaver, J. M., Steger, D. J., Joshi, S., Maslany, J. T., Collins, J. L., et al. (2008). Selective Partial Agonism of Liver X Receptor α Is Related to Differential Corepressor Recruitment. Mol. Endocrinol. 22, 2241–2249. doi:10.1210/me.2008-0041
Renga, B., Festa, C., Marino, S. D., Miccoc, S. D., Valeria D’Auria, M. V., Bifulco, G., et al. (2015). Molecular Decodification of Gymnemic Acids from Gymnema Sylvestre. Discovery of a new Class of liver X Receptor Antagonists. Steroids 96, 121–131. doi:10.1016/j.steroids.2015.01.024
Sun, X., Gao, H., Yang, Y., He, M., Wu, Y., Song, Y., et al. (2019). PROTACs: Great Opportunities for Academia and Industry. Sig Transduct Target. Ther. 4, 64. doi:10.1038/s41392-019-0101-6
Viennois, E., Mouzat, K., Dufour, J., Morel, L., Lobaccaro, J.-M., and Baron, S. (2012). Selective Liver X Receptor Modulators (SLiMs): What Use in Human Health?. Mol. Cell Endocrinol. 351, 129–141. doi:10.1016/j.mce.2011.08.036
Wagner, B. L., Valledor, A. F., Shao, G., Daige, C. L., Bischoff, E. D., Petrowski, M., et al. (2003). Promoter-specific Roles for Liver X Receptor/corepressor Complexes in the Regulation of ABCA1 and SREBP1 Gene Expression. Mol. Cell. Biol. 23, 5780–5789. doi:10.1128/mcb.23.16.5780-5789.2003
Wang, B., and Tontonoz, P. (2018). Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat. Rev. Endocrinol. 14, 452–463. doi:10.1038/s41574-018-0037-x
Keywords: liver X receptor, PROTAC, ubiquitin-proteasome system, von Hippel-Lindau, protein degradation
Citation: Xu H, Ohoka N, Yokoo H, Nemoto K, Ohtsuki T, Matsufuji H, Naito M, Inoue T, Tsuji G and Demizu Y (2021) Development of Agonist-Based PROTACs Targeting Liver X Receptor. Front. Chem. 9:674967. doi: 10.3389/fchem.2021.674967
Received: 02 March 2021; Accepted: 05 May 2021;
Published: 26 May 2021.
Edited by:
Hany S. Ibrahim, Egyptian Russian University, EgyptCopyright © 2021 Xu, Ohoka, Yokoo, Nemoto, Ohtsuki, Matsufuji, Naito, Inoue, Tsuji and Demizu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Genichiro Tsuji, Z3RzdWppQG5paHMuZ28uanA=; Yosuke Demizu, ZGVtaXp1QG5paHMuZ28uanA=
†These authors contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.