 Zakir Ullah
Zakir Ullah Kang Kim1†
Kang Kim1† Moon Il Kim
Moon Il Kim Mi-hyun Kim
Mi-hyun Kim- 1Department of Pharmacy, College of Pharmacy, Gachon Institute of Pharmaceutical Science, Gachon University, Incheon, South Korea
- 2Department of Chemistry, Korea Advanced Institute of Science and Technology, Daejeon, South Korea
- 3Department of BioNano Technology, Gachon University, Seongnam, South Korea
As a non-covalent interaction of a chiral scaffold in catalysis, pnicogen bonding of epi-cinchonidine (epi-CD), a cinchona alkaloid, was simulated to consider whether the interaction can have the potential controlling enantiotopic face like hydrogen bonding. Among five reactive functional groups in epi-CD, two stable complexes of the hydroxyl group (X-epi-CD1) at C17 and of the quinoline ring (X-epi-CD2) at N16 with pnictide family analytes [X = substituted phosphine (PX), i.e., F, Br, Cl, CF3, CN, HO, NO2, and CH3, and pnictide family analytes, i.e., PBr3, BiI3, SbI3, and AsI3] were predicted with intermolecular interaction energies, charge transfer (QMulliken and QNBO), and band gap energies of HOMO–LUMO (Eg) at the B3LYP/6-31G(d,p) level of density functional theory. It was found that the dominant site of pnicogen bonding in epi-CD is the quinoline ring (N16 atom) rather than the hydroxyl group (O36 atom). In addition, the UV-Vis spectra of the complex were calculated by time-dependent density functional theory (TD-DFT) at the B3LYP/6-31+G(d,p) level and compared with experimental measurements. Through these calculations, two intermolecular interactions (H-bond vs. pnicogen bond) of epi-CD were compared.
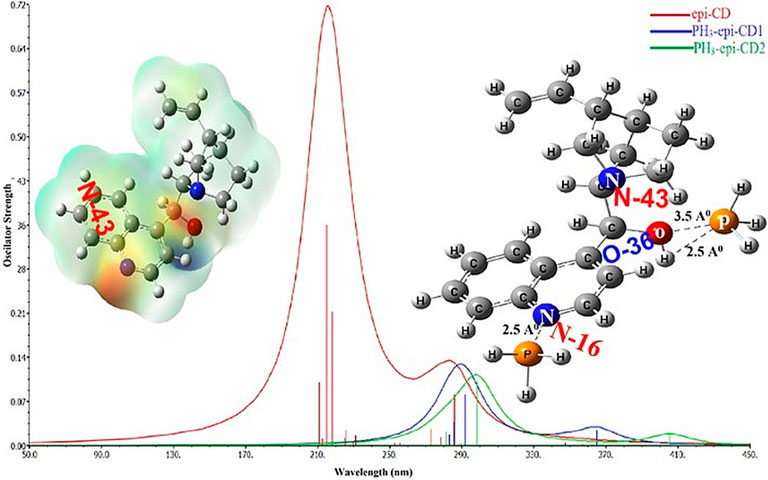
GRAPHICAL ABSTRACT
Introduction
Cinchona alkaloids have played an important role as privileged chiral sources in the history of chemistry due to their diverse chiral skeletons and tunable reactions (like chiral ligands in Sharpless asymmetric dihydroxylation; Maeda et al., 2011; Tian et al., 2004; Szollosi et al., 2011; Sim et al., 2015; Marcelli and Hiemstra, 2010). In addition, they were clinically used as antimalarial or anti-arrhythmic agents (Gurung and De, 2017; Yardley et al., 1971; de Villiers et al., 2012; Vandekerckhove and D’hooghe, 2015; Karle and Bhattacharjee, 1999; Wesche et al., 1990). Recently, cinchona alkaloids have been screened as an anti-diabetic agent (Ezekwesili et al., 2012) and used as the most powerful chiral template for designing new organic catalysts (e.g., bifunctional catalyst, phase-transfer catalyst) (Dalko and Moisan, 2001; Jones, 2001; Lygo and Wainwright, 1997). Reactive sites in cinchona alkaloids and their derivatives were widely studied (Ullah and Itsuno, 2017; Li et al., 2004; Wang et al., 2007; Yeboah et al., 2011; Luo et al., 2009). The highly basic and bulky nitrogen atom of quinuclidine is able to bind with an electrophile or metal to produce a stereotopic face (Figure 1) (Song, 2009a). The vicinal aminoalcohol in cinchona alkaloids has the capability to associate the proximal hydroxyl group at C17 (Lewis acid) with the nitrogen atom of quinuclidine (Lewis base). The methoxy group in the quinolone ring of quinines and quinidines can be converted into the free phenolic −OH group as an H-donor. Similarly, quinoline exhibits π–π stacking, and its vinyl group can act as a nucleophile. However, the delicate differences in the reactivity of their five functional groups have not been sufficiently investigated when compared with intensive use of cinchona alkaloids. In particular, despite their potential controlling asymmetric reactions through the non-covalent interaction (NCI) (Manna and Mugesh, 2012; Pal et al., 2016; Wheeler et al., 2016; Benz et al., 2017; Breugst et al., 2017; Li and Hong, 2018), the scope and limitation of the NCIs in these five functional groups has not been properly studied. Rather than the study, conformational investigations of cinchona are reported (Yanuka et al., 1981; Dijkstra et al., 1989a; Dijkstra et al., 1989b; Caner et al., 2003; Prakash et al., 2011; Wang et al., 2014). Exceptionally, an oxyanion hole between the hydroxyl group at C17 and the nitrogen atom of quinuclidine (N43) was proposed (Bürgi and Baiker, 1998), and a density functional theory (DFT) study of quinine-catalyzed aza-Henry reaction explained the mechanism through the hydrogen bonding interaction (Xue et al., 2016).
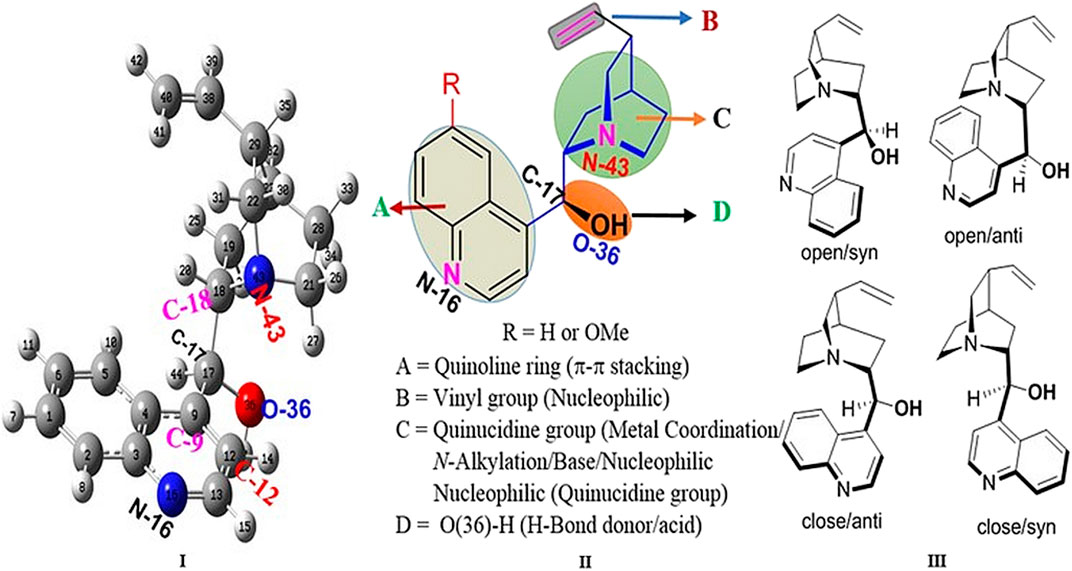
FIGURE 1. Reactive sites of cinchona alkaloids and the representative conformations of epi-cinchonidine (right: syn/anti, open/close; left: optimized geometry with the atomic number).
NCIs are very crucial phenomena happening at the atomic level so that hydrogen bonding and halogen bonding among them have been strongly used for molecular design in various fields (e.g., drug design, catalyst design, porous material architecture design) (Hiemstra and Wynberg, 1981; Shi and Xu, 2002; Li et al., 2005a; Li et al., 2005b; Akiyama et al., 2006; Taylor and Jacobsen, 2006; Doyle and Jacobsen, 2007; Yang and Wong, 2013; Grayson and Houk, 2016a; Cavallo et al., 2016; Qian et al., 2017; Heinen et al., 2018). Recently, new types of non-covalent bonds such as carbon bonding (Escudero-Adán et al., 2015; Hatakeyama et al., 2015), pnicogen bonding (Scheiner, 2011a; Scheiner, 2011b; Scheiner and Adhikari, 2011; Sánchez-Sanz et al., 2012; Alkorta and Elguero, 2013; Alkorta et al., 2013; Scheiner, 2013a; Scheiner, 2013b; Alkorta and Elguero, 2014; Del Bene et al., 2014; Sánchez-Sanz et al., 2015; Scheiner, 2015; Del Bene et al., 2017), chalcogen bonding (Fanfrlík et al., 2014; George et al., 2014), and aerogen bonding (Bauz and Frontera, 2015; Esrafili and Vessally, 2016; Gao et al., 2016; Esrafili and Qasemsolb, 2017) have been investigated, and properties of the bonds have also been applied to catalysis (Manna and Mugesh, 2012; Pal et al., 2016; Wheeler et al., 2016; Benz et al., 2017; Breugst et al., 2017; Li and Hong, 2018). Among the applicable properties, σ-hole, a positive electrostatic region, is the most outstanding. The σ-hole is the interaction between a covalently bonded atom of groups 14–18 as a Lewis acid and a lone pair present in a Lewis base or an anion (Politzer et al., 2013). The formation of a σ-hole bond results from the existence of an electron-deficient region (σ-hole) in the outer lobe of the half-filled p orbital that is involved in the covalent bond. To our knowledge, such new types of non-covalent bonds have not been applied yet to directly control the enantiotopic face of a chiral organocatalyst. If such bonds can be designed onto a chiral scaffold (or catalyst) and can play key interactions in controlling the stereoelectronic environment, the interactions inducing chiral bias of the product can be state-of-the-art based on more diverse available elements and bond strength of a broader range rather than the current interactions (hydrogen bonding, ionic interaction, etc.) in the catalytic enantioselective transformation area. Until today, promising interactions have been conceptually established, and some successful studies were reported through computational study and experimental proofs (Jungbauer and Huber, 2015; Benz et al., 2016; Bulfield and Huber, 2016; Benz et al., 2017; Gliese et al., 2017; Schmauck and Breugst, 2017). However, reported studies are on racemic reactions (Athawale and Manjrekar, 2001; Kacprzak and Gawronski, 2001; Jha and Joshi, 2002; Gaunt et al., 2007; Vakulya et al., 2008; Wang et al., 2011; De Fusco et al., 2012; Pham et al., 2013; Jusseau et al., 2014; Chen et al., 2015; Nath et al., 2015; Scorzelli et al., 2015; Singh and Yeboah, 2016; Viveros-Ceballos et al., 2016; Auria-Luna et al., 2017; Gliese et al., 2017; Guo and Wong, 2017; Szollosi, 2017; Fulton et al., 2018; Li and Hong, 2018; Zhang et al., 2018), or new types of bonds cannot contribute to enantioselectivity (Athawale and Manjrekar, 2001; Kacprzak and Gawronski, 2001; Jha and Joshi, 2002; Gaunt et al., 2007; Vakulya et al., 2008; Wang et al., 2011; De Fusco et al., 2012; Pham et al., 2013; Jusseau et al., 2014; Chen et al., 2015; Nath et al., 2015; Scorzelli et al., 2015; Benz et al., 2016; Bulfield and Huber, 2016; Singh and Yeboah, 2016; Viveros-Ceballos et al., 2016; Auria-Luna et al., 2017; Guo and Wong, 2017; Szollosi, 2017; Fulton et al., 2018; Zhang et al., 2018). Therefore, for future research, the current research background motivated us in applying the new types of NCIs (groups 14–18) into a cinchona alkaloid as a privileged chiral scaffold of the organocatalyst or an organocatalyst itself.
In this paper, we studied the possibility of pnicogen bonding between a Lewis base of epi-cinchonidine (epi-CD), a type of cinchona alkaloid, and covalently bonded P, As, Sb, and Bi of the pnictide family as a Lewis acid. To our knowledge, there are no reports on pnicogen bonding in any chiral scaffold (among chiral ligands, chiral auxiliaries, and chiral catalysts) like cinchona alkaloids so that a theoretical study on pnicogen bonding can be a guidance for reaction designs in the field of catalysis and asymmetric synthesis. To compare hydrogen bonding with pnicogen bonding, epi-CD complexes interacting with methacrylic acid (MA) were simulated to locate the hydrogen bonding in epi-CD. The simulation of pnicogen bonding in epi-CD was designed with PH3 and substituted phosphane derivatives having F, Br, Cl, CF3, CN, HO, NO2, and CH3 as substituents to describe substituent effects on pnicogen bonding.
Computational Methodology
DFT calculations were performed using a hybrid functional [Becke 3-parameter (exchange), Lee, Yang, and Parr with both the local and non-local correlations, B3LYP] (Becke, 1988; Becke, 1993) with well-accepted basis sets: 6-31G(d,p) and 6-31G+(d,p) (Bürgi et al., 2002). The DGDZVP basis set for As and Sb atoms and SDD basis set for Bi atoms were used. All calculations were performed at the default temperature and pressure (298.15 K and 1.00 atm). All calculations were performed using Gaussian 09 (Frisch et al., 2009), and the results were visualized with the GaussView, Gabedit (Allouche, 2011), and GaussSum (O’boyle et al., 2008) computer programs. The term “epi-CD” was used as an abbreviated name for epi-cinchonidine, and epi-CD-X was used for its complexes. epi-CD and its epi-CD-X complexes (where X is an analyte) were neutral with singlet spins (charge = 0 and singlet state). Vibrational frequency calculations were used to confirm that the optimized structures were true minimum on the potential energy surface, as characterized by the absence of imaginary vibrational frequencies. The intermolecular interaction energies and excited-state properties (e.g., UV-Vis spectra) (Salzner, 2008), QMulliken and QNBO, charge analysis, and band gap energies (Eg) were calculated at the above-mentioned level of theory. We simulated QMulliken and QNBO charge analyses with different types of basis sets and concluded that the properties were basis set dependent (Ullah et al., 2013). The band gap energies were estimated from the energy differences for the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO), where the negative of the HOMO was the ionization potential (IP) and the energy of the LUMO was estimated from the electron affinity (EA). Eq. 1 was used to simulate the binding energies of the various optimized structures. The counterpoise method corrected this energy, which was based on Eq. 2 (Boys and Bernardi, 1970):
where ΔEint is the total binding energy of the optimized epi-CD interacting with different analytes. E(Reactant1) is the total energy of isolated epi-CD and E(Reactant2) is that of analytes, while E(Product) is the total energy of a particular epi-CD-X complex. EBSSE is the basis set superposition error energy of epi-CD-X, and ΔEint,CP is the geometrical counterpoise–corrected interaction energy of these complexes. The binding energies of Eqs 1, 2 are related to the relaxed structures with the minimum amount of energy. The B3LYPgCP-D3/6-31G (Brandenburg et al., 2013) energy corrections are also made from Grimme’s web server (http://wwwtc.thch.uni-bonn.de/) (Kruse et al., 2012). ΔEgCP-D3 is the geometrical counterpoise–corrected energy with D3 corrections, whereas E(Reactant1)gCP-D3 is used for epi-CD and the analytes and E(Product)gCP-D3 is the binding energies of the epi-CD complex. In addition, geometry optimization of one example considering the dispersion term also was conducted using B3LYP-D3 with 6-31G(d,p) (Kruse et al., 2012; Brandenburg et al., 2013), and then two geometries resulting from B3LYP and B3LYP-D3 were compared to confirm the acceptability of our chosen method, B3LYP/6-31G. The NCI energy can be estimated as negative, i.e., more negative energy relates to high stability, and vice versa.
Synthetic Description and Spectra Measurement
For explanation of pnicogen bonding of epi-cinchonidine, we synthesized epi-cinchonidine (2) from cheaply available cinchonidine (1) as mentioned in Figure 2. Cinchonidine (1) was subjected to one-pot Mitsunobu inversion followed by saponification with 4-nitrobenzoic acid (PNBA)/diethyl azodicarboxylate (DEAD)/triphenylphosphine (PPh3) and 1 M LiOH (lithium hydroxide) to afford diastereomerically pure epi-cinchonidine (2) in 78% yield (Sidorowicz and Skarżewski, 2011). All spectra data are given in the supplementary material.

FIGURE 2. Synthetic route of epi-cinchonidine.
For the IR and UV-Vis absorption spectra, 0.5 mg/ml epi-CD solution in three different solvents [CHCl3, EtOH, and DW (distilled water)] was prepared according to solubility of analytes (Qin et al., 2009). Analyte solutions were also prepared to the same concentration (1.7 mM). Without additional treatment, the UV-Vis absorption spectra of sole epi-CD solution were measured after the blank test (solvent). After mixing two solutions (analyte: epi-CD = 1 to 1), the equilibrium between the complex and the free form waited, and then UV-Vis absorption spectra were recorded on a NanoDrop 1000 UV-Vis spectrophotometer. The experimental result could be integrated for the comparison with the predicted result.
Results and Discussion
Geometry Selection of epi-CD. In this study, epi-cinchonidine (epi-CD) was chosen among available four cinchona alkaloids due to conformational rigidity (Yanuka et al., 1981; Dijkstra et al., 1989a; Dijkstra et al., 1989b; Caner et al., 2003; Prakash et al., 2011; Wang et al., 2014). For economic geometry selection among an enormous number of conformers, two torsional angles τ1 (C12−C9−C17−C18) and τ2 (C9−C17−C18−N43) were used as criteria of conformer generation based on previous reports on cinchona alkaloids (Bürgi and Baiker, 1998). The τ1 rotation decides the interconversion between closed and open conformations on whether the quinuclidine nitrogen atom of cinchona is close to ring A of quinolone or not. The τ2 rotation determines syn and anti conformations according to the relative position of the hydroxy group (OH) aligning with ring B of quinoline. Even though more stable conformation can change according to solvents and substituents, every conformer used in this study was in the anti opened state for maximizing the space which analytes can be close to. The prominent optimized structural parameters (bond length, bond angle, and dihedral angle) in the geometry of epi-CD were calculated at DFT-B3LYP/6-31G(d,p) and 6-311G levels of theory, and the results are compared with the Cambridge Crystallographic Data Centre (CCDC) data of the epi-cinchonidine derivative (Martin and Zipse, 2005; Klare et al., 2014) in Table 1.
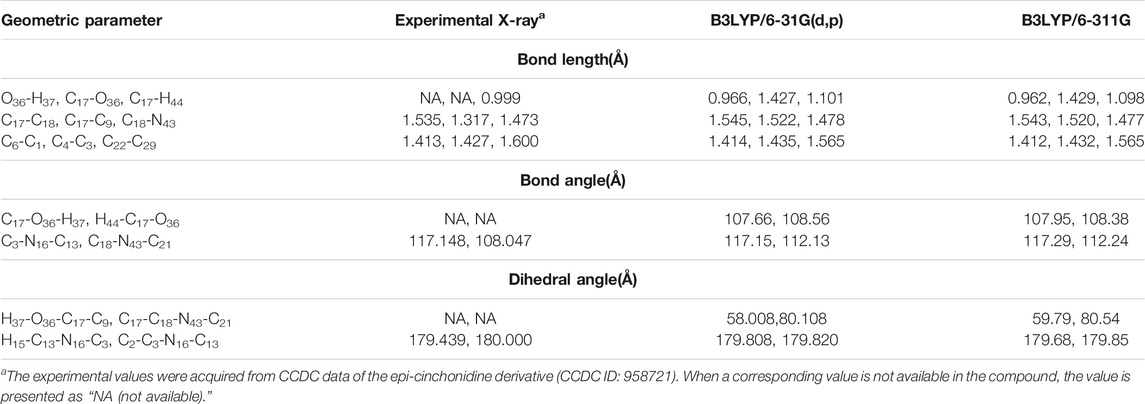
TABLE 1. Optimized geometric parameters of epi-CD.
Charge Analysis and Molecular Electrostatic Potential of epi-CD. Before the reactive site simulation of epi-CD, both Mulliken charge and natural bonding orbital (NBO) charge on the optimized geometry of epi-CD were calculated. The highest negatively charged atom (the most nucleophilic) commonly present was O36 in both charges (NBO: −0.76 e−, Mulliken: −0.55 e−). N16, C40, N43, and C28 were also negatively charged, but the reactivity order was not identical (Mulliken: N16 > N43 >> C40 > C28, NBO: N43 > C28 > C40 > N16) in Supplementary Tables S1, S2. The molecular electrostatic potential (MEP) of epi-CD predicted the nature of the electrophilic and nucleophilic reactions of the molecule to present the charge density, delocalization, and site of the molecular chemical reactivity. Based on known reports depicting five reactive functional groups of cinchona alkaloids (Song, 2009b), the quinoline ring and hydroxyl group (O36 atom), tertiary nitrogen (N43, N16), and olefin region (C40) were expected to show high electron density. In particular, the MEP map presented the quinoline ring as the highest charge density and the region around O36 as the highest electron density.
Orbital energies of the HOMO and the lowest LUMO were also calculated for electric and optical properties like the UV-Vis spectra at the B3LYP/6-31G(d,p) level. The energy gap between the HOMO and the LUMO, indicating the molecular chemical stability and electrical transport property, was 4.20 eV, and the value as the standard was compared with the HOMO–LUMO energy gap of each epi-CD-X complex. In contrast to the MEP map, the frontier molecular orbital analysis could not propose the reactivity of the olefin (C40), and the electron density of the HOMO was distributed on the right side of epi-CD, especially on the C17 hydroxyl group (O36), quinuclidine (N43), and some C–C bonds (Figure 3). In the LUMO, the quinoline ring (N16) and the hydroxyl group (O36) showed prominent electron density. Our experimental reaction with excessive electrophile, methyl iodide, under aqueous basic condition also showed the dialkylated product at only N43 and N16 positions (Supplementary Figure S3).
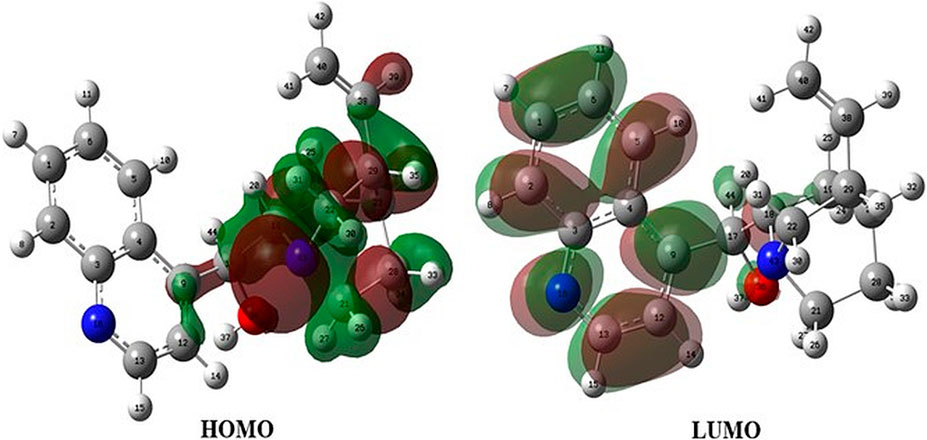
FIGURE 3. HOMO and LUMO of epi-cinchonidine.
Infrared Spectral Characteristics and UV-Vis Spectral Analysis of epi-CD. According to the non-linear molecular vibrational mode formula, epi-CD is expected to have 126 normal modes of vibrations under C1 symmetry. The simulated infrared (IR) spectrum of epi-CD is plotted and compared with the experimental spectrum in Supplementary Figure S4. Comparison of simulated spectra with experimental spectra enabled assignment of the vibrational modes to experimental vibrational bands. Among 126 modes, selected major band peaks and their approximate assignments are listed in Table 2. In particular, the functional group region of the simulated IR spectrum contained seven peaks comprising hydroxyl and CH group stretching. When compared with the C–C/C–O/C–N stretching band, the predicted value of the O–H stretching band (3,737 cm−1) presented a slightly larger gap with the experimental band in the CHCl3 phase (3,433 cm−1). In spite of quantitative limitation, the qualitative agreement between the simulation and the experiment was achieved under our computational method.
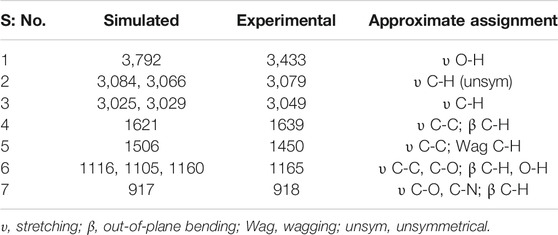
TABLE 2. Simulated IR frequencies (in cm−1) of epi-CD (gas phase) at the B3LYP/6-311+G(d,p) level of theory.
The UV-Vis spectrum of epi-CD describing the electronic transitions was acquired through time-dependent (TD)-DFT calculations at B3LYP/6-31+G(d,p) and 6-311+G(d,p) levels of theory. The simulation was performed both in a vacuum and in CHCl3 as solvent media to predict absorption wavelengths including λmax, electronic excitation values [such as excitation energies (E), oscillator strengths (f), and major contributions of the transitions], and their assignments (electronic transitions) in Table 3. The simulated UV-Vis spectra of epi-CD in double zeta showed two absorption bands at 288.83 and 222.48 nm. The strong absorption band peak was 284 approximate to the simulated spectrum (double zeta: 288.84 nm, triple zeta: 289.14 nm in CHCl3 solution). The verified spectrum through the experimental UV-Vis spectrum was used as the standard for the comparison with spectra of each epi-CD-X complex.
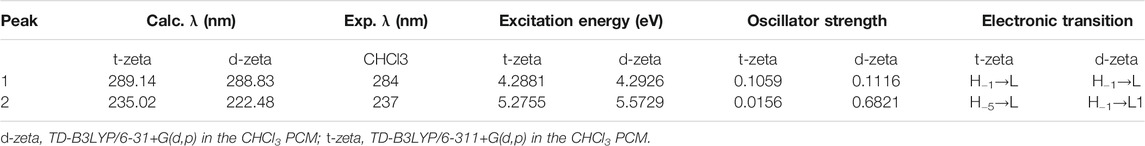
TABLE 3. Experimental and calculated electronic excitations of epi-CD.
Substituent Effect on Pnicogen Bonding of epi-CD-X. During the study, we had three questions, (1) “ Is a pnicogen bond applicable for controlling the enantiotopic face in the chiral scaffold?”, (2) “Can a pnicogen bonding also make an effective interaction as hydrogen bonding or halogen bonding with the chiral scaffold?”, and (3) “If the pnicogen bonding can have an effective force, which functional group is the most promising in a cinchona alkaloid?” To get answers for these questions, the dataset for our simulation was considered, and the efficient design of the simulation was possible through sampling representative analytes. For sampling enough number of cases, the next three factors were considered: 1) the aspect of the pnicogen bonding according to substituent diversity, PH3, and substituted phosphanes with eight kinds of substituents (X = F, Br, Cl, CF3, CN, OH, NO2, and CH3), 2) the spectral aspect of the bonding according to an element type of group 15, and 3) the comparison of the pnicogen bonding with the hydrogen bonding in the epi-CD-X complex. For the purpose, QMulliken and QNBO, HOMO–LUMO interaction, and UV-Vis spectra were simulated for the mixture of epi-CD and analyte (X) at TD-B3LYP/6-31+G(d,p) and B3LYP/6-31+G(d,p) levels of theory.
Firstly, the binding characteristics of the analytes, phosphane derivatives complexed with epi-CD, were investigated. They interacted with two functional groups of epi-CD, the hydroxyl group (X-epi-CD1) and the quinoline ring (X-epi-CD2). The distances of the pnicogen bonding are ca. 2.6–3.6 Å between the hydroxyl group and phosphorus in X-epi-CD1 and ca. 2.1−4.0 Å between quinoline nitrogen (N17) and phosphorus in X-epi-CD2 in Figure 4 and Supplementary Figures S7–S22. Even though the stability of non-covalent bonds partially depends on charge transfer from the electron-donor atom to the σ* antibonding orbital of the acceptor, electrostatic attraction also needs to be considered. In the case of the pnicogen bond, a σ-hole of the pnictide element is not essential so that the electrostatic potential of the whole complex is considered rather than the charge of binding atoms (Scheiner, 2013a). From this point, the amount of charge transfer is simulated from Mulliken and NBO charge analyses (Bibi et al., 2015; Guo and Wong, 2017). In Table 4, the charge transfers (QNBO and QMulliken) between epi-CD and an analyte present less deviation in X-epi-CD1 (up to 0.117 and 0.311 e−) than the values in X-epi-CD2 (up to 0.467 and 0.179 e−). Even though electron-withdrawing substituents tend to present the enhanced charge transfer, the highly steric hindered nitro group showed bigger QNBO and QMulliken in X-epi-CD2 (N16) than in X-epi-CD1 (O36). In mono-substituted phosphine, XPH2, σ*(XP) was the LUMO that withdrew the electrons from the HOMO of N16 of quinoline or hydroxyl (−O36H37) at C17. In NO2PH2 and CNPH2, the LUMO was dπ* and resulted from the binding between the π orbital of the cyano or nitro substituent and the d orbital on phosphorous, and σ*(XP) became LUMO+1. The ΔEint and ΔEint,CP could show a moderately strong pnicogen bond between PH3 and O36/N16 of epi-CD. The strength comparison of the two pnicogen bonds (P···N and P···O) in both ΔEint,CP (O36 vs. N16) and distance (O36 vs. N16) can propose the pnicogen bonding of X-epi-CD2 is generally stronger than the bonding of X-epi-CD1, but the electron-withdrawing substituent (σI) that is less bulky like Cl can make the pattern inversed. In the case of HOPH2, because the electron donating substituent, −OH, has a strong sigma inductive effect and also has the hydrogen bonding interaction, two pnicogen bonds could not be compared and showed out of the pattern. Based on the distances of HOPH2, X-epi-CD1 favored pnicogen bonding without a hydrogen bonding and X-epi-CD2 favored hydrogen bonding with the existence of the hydrogen bonding (Figure 4). When compared with the energy of X-epi-CD1 resulting from hydrogen bonding (Supplementary Table S3), the binding energy of X-epi-CD1 resulting from pnicogen bonding was not inferior.
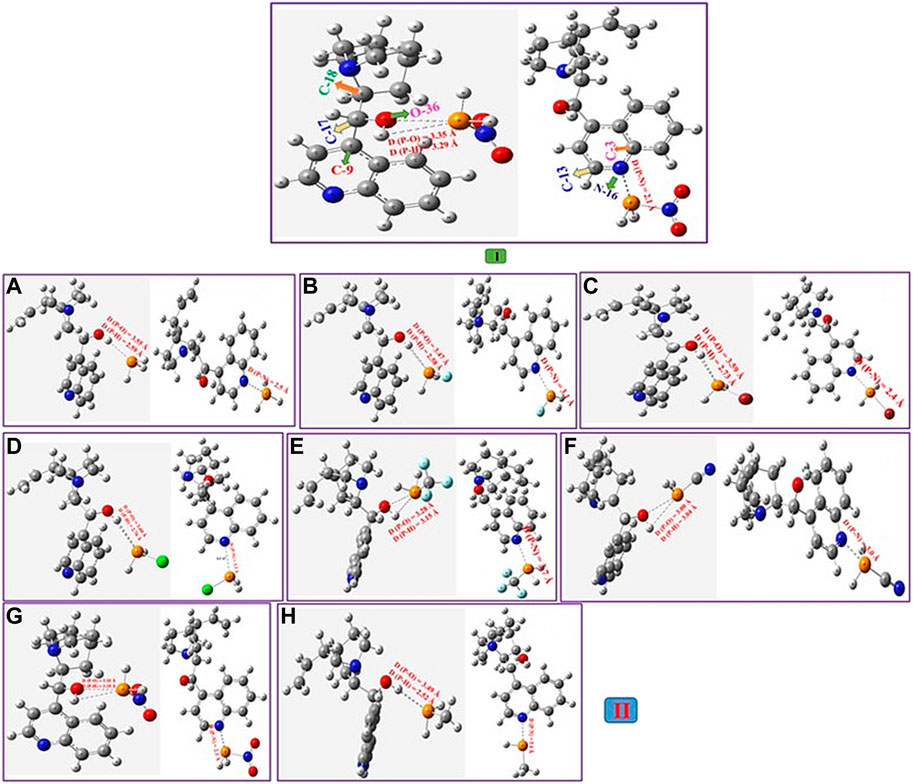
FIGURE 4. Geometry analysis of the epi-cinchonidine complex (epi-CD-X1/epi-CD-X2) with the pnicogen bond.
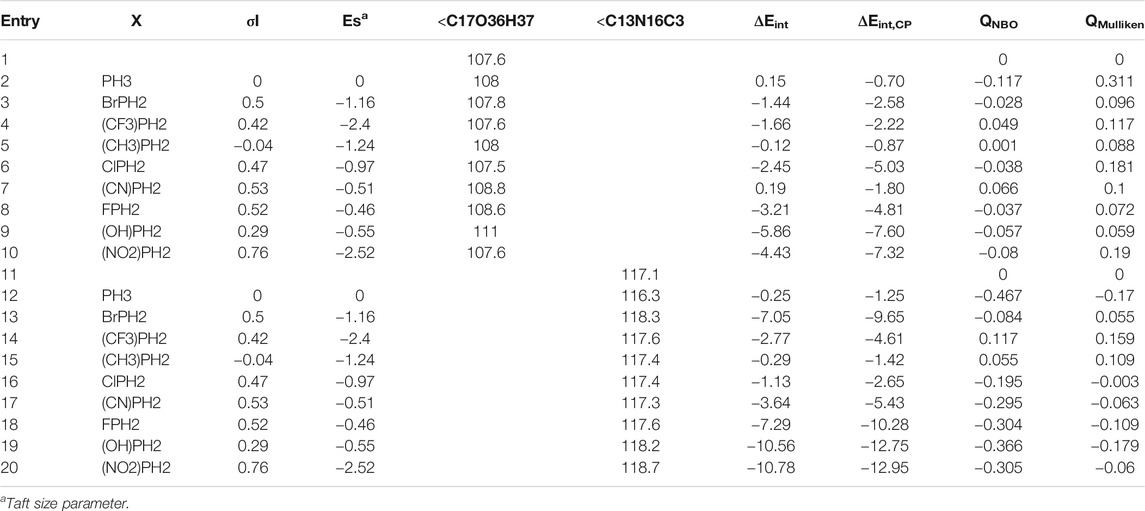
TABLE 4. Optimized geometric parameters, ΔEint, ΔEint, CP, QNBO, and QMulliken of X-epi-CD1/X-epi-CD2.
In sequence, the dipole moment and HOMO–LUMO energy of the X-epi-CD1 complex are further calculated in Table 5, and the substituent effect on interaction energy was compared with the effect on the band gap. Notably, the dipole moment of the complex was sensitive to the substituent type and the number of substituents, but the deviation of band gap was less than the deviation of dipole moment or interaction energy. Mono-substituted analytes were expected to present a higher dipole moment and HOMO–LUMO energy gap than di-substituted and tri-substituted analytes. An electron-withdrawing substituent also made us expect a larger HOMO–LUMO energy gap than PH3 based on the literature (Sarkar et al., 2015). The simulation presented the expected result in CF3, CN, and NO2 (entries 8–19) but did not explain every case. In particular, the high electronegative and small halide substituent (entries 2–7) showed the mismatch between our expectation and simulation. When relative arrangement between the analyte and epi-CD in optimal geometry was described through the angle (<O-H-P) and distances [D(P-O) and D(P-H)], the mismatch can be explained by the confounding effect between expected pnicogen bonding and hydrogen bonding. The reports on P-substituent effects on a pnicogen bond described the interaction energy grew in the order: F > Cl > OH > CF3 > H > CH3 (Scheiner, 2013a), and di- or tri-halogenation does not produce any additional stabilization, in marked contrast to H-bonds. In our X-epi-CD complex, mono-halogenation also showed the biggest band gap as well as ΔEint,CP in NO2, CN, and CF3.
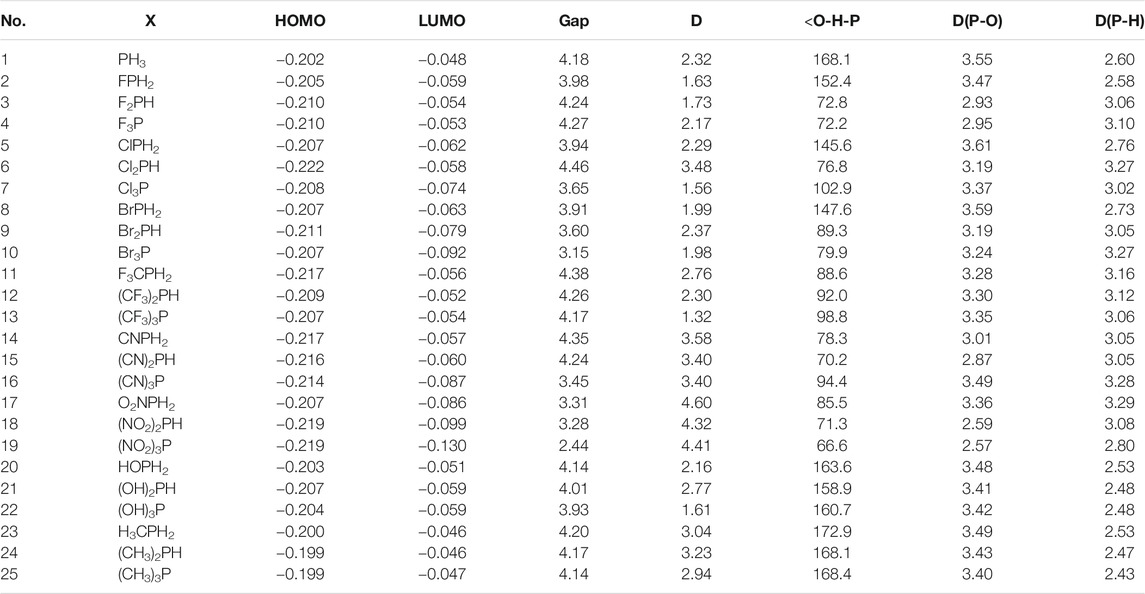
TABLE 5. Orbital energies of the HOMO and LUMO (hartree), band gap (eV), and dipole moment (D) for the X-epi-CD1 complexes.
UV-Vis Spectroscopic Study of epi-CD-X for Pnicogen Bonding. The aspect of the bonding according to an element type of group 15 was studied through UV-Vis spectra prediction and experimental measurement of the spectra (Figure 5). The simulated excitation energies (eV), oscillator strengths, and molecular orbitals of the first allowed singlet transition involved in the excitation for the epi-CD, X-epi-CD1, and X-epi-CD2 complexes are given in Table 6 and Supplementary Figures S22, S23. Under both gas and solution phases, the binding of the pnictide analytes (P, As, Sb, and Bi) with epi-CD at the two positions caused the red-shifted absorption energy peak (λmax). In general, a more prominent increase in λmax was predicted in X-epi-CD1 rather than in X-epi-CD2, and experimental values were closer to the values of X-epi-CD2 than X-epi-CD1 except for phosphoric acid (PA) (entries 11 and 12). It seems that the variation of λmax depends on the stereoelectronic property of an analyte so that the bulkier and lower electron density of the analyte tends to show a larger variation of the red-shift. Even though the aspect of λmax according to an element type of group 15 under the same halogen substituent (BiI3, SbI3, and AsI3) did not exactly match with the atomic diameter, with the difference between metallic and non-metallic elements, BiI3 and AsI3 could present more dramatic variation of red-shift than PBr3 and phosphoric acid. The excited state energies for the first allowed transition states were 2.06, 3.07, 2.47, 3.33, 2.10, and 3.26 eV for the AsI3-epi-CD1, AsI3-epi-CD2, SbI3-epi-CD1, SbI3-epi-CD2, BiI3-epi-CD1, and BiI3-epi-CD2 complexes, respectively. When compared with hydrogen bonding, pnicogen bonding presents a larger variation (entry 2) in Table 6. The interaction with AsI3 and BiI3 was stronger than that with other tested analytes (entries 5, 6, 9, and 10) in our simulation, and the predicted tendency matched with experimental spectra. Due to the excellent interaction, the electronic structure of epi-CD was greatly altered and more outstanding alteration was simulated in X-epi-CD1 between the hydroxyl group (X-epi-CD1) and the quinoline ring (X-epi-CD2). Notably, the experimental data were closer to X-epi-CD2 than X-epi-CD1 so that the data could support the possibility of dominant binding of the corresponding analytes with the quinoline ring.
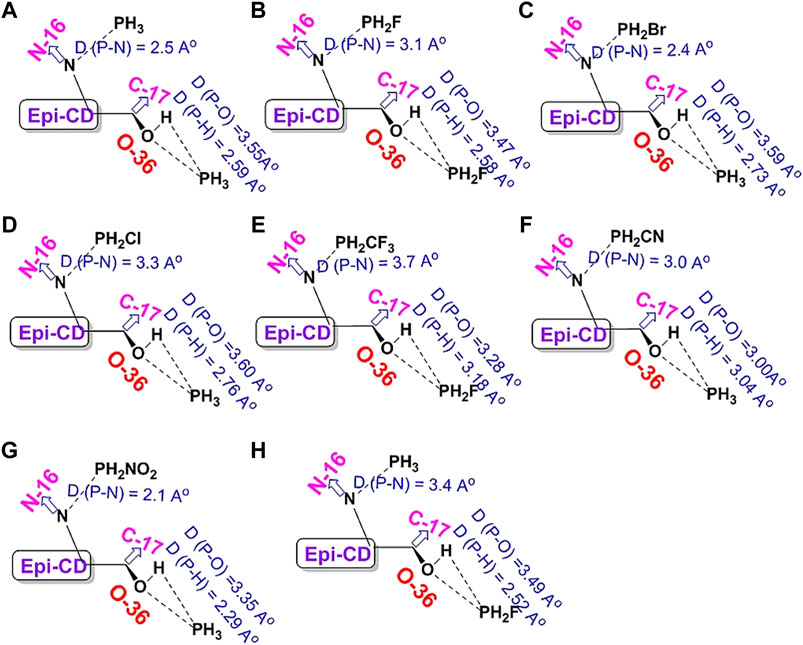
FIGURE 5. Predicted full UV-Vis spectrum of the epi-cinchonidine complex: X-axis = wavelength and Y-axis = oscillator strengths and epsilon. Experimental full UV-Vis spectrum of the epi-cinchonidine complex: X-axis = wavelength and Y-axis = observance.
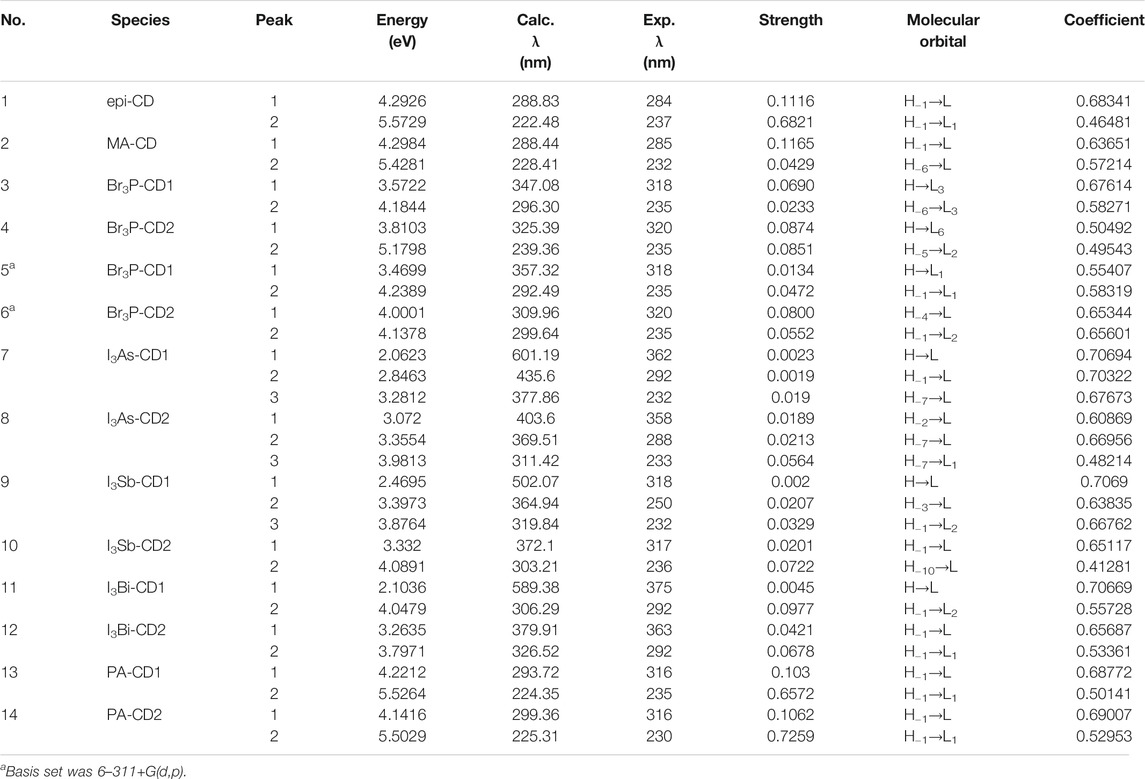
TABLE 6. Calculated excitation energies, oscillator strengths, and molecular orbitals (MOs) of CD and CD-X at the TD-B3LYP/6-31+G(d,p) level of theory.
AIM Analysis of epi-CD-X. The quantum theory of atoms in molecules (AIM) has been widely used to analyze the real space functions and characterize the different types of interactions. Herein, the NCIs of the epi-CD complex with PH2CN, as a representative X-epi-CD, were analyzed according to Bader’s AIM theory (Bader, 1985), using the Multiwfn code (Lu and Chen, 2012). It is known that the isosurface of the reduced density gradient (RDG) is a valuable tool for delicately revealing NCI regions (particularly weak interactions) based on the next dimensionless equation given as follows:
The sign of λ2, the second largest eigenvalue of the Hessian matrix of electron density, discriminates (3, −1) type critical point (CP), which appears in the chemical bond path or between atom pairs that have a weak attractive interaction, from (3, +1) type CP, which appears in the ring center or displays a steric effect in Bader’s AIM theory. As shown in 2D plots of the RDG with the sign of λ2 (Figure 6), RDG isosurfaces having the values of
In general, blue, red, and green (or earth green) colors indicate the strong attractive, strong repulsive, and van der Waals interactions, respectively. In other words, Figures 6A,B indicate the pnicogen bonding interactions between epi-CD and PH2CN show the strength between strong, attractive, and van der Waals interactions. Furthermore, the density of electrons (ρ) and Laplacian of electron density (Δ2ρ) at bond critical points (BCPs) in Figures 7C,D were calculated. For example, in the case of hydrogen bonding, ρ and Δ2ρ typically varied in the range from 0.002 to 0.04 a.u. and from 0.020 to 0.139 a.u., respectively, in AIM analysis (Roohi et al., 2011). In the epi-CD complex, ρ and Δ2ρ were calculated to be 0.00074762 a.u. and Laplacian of electron density was 0.013382 a.u. at BCP1 (PH2CN–OH as X-epi-CD1). In contrast to BCP1, the density of electrons was 0.00011455 a.u. and the Laplacian of electron density at BCP2 was 0.0012619 a.u., respectively, which shows the corresponding interactions (PH2CN–N as X-epi-CD2). The two-dimensional NCI plots of RDG with
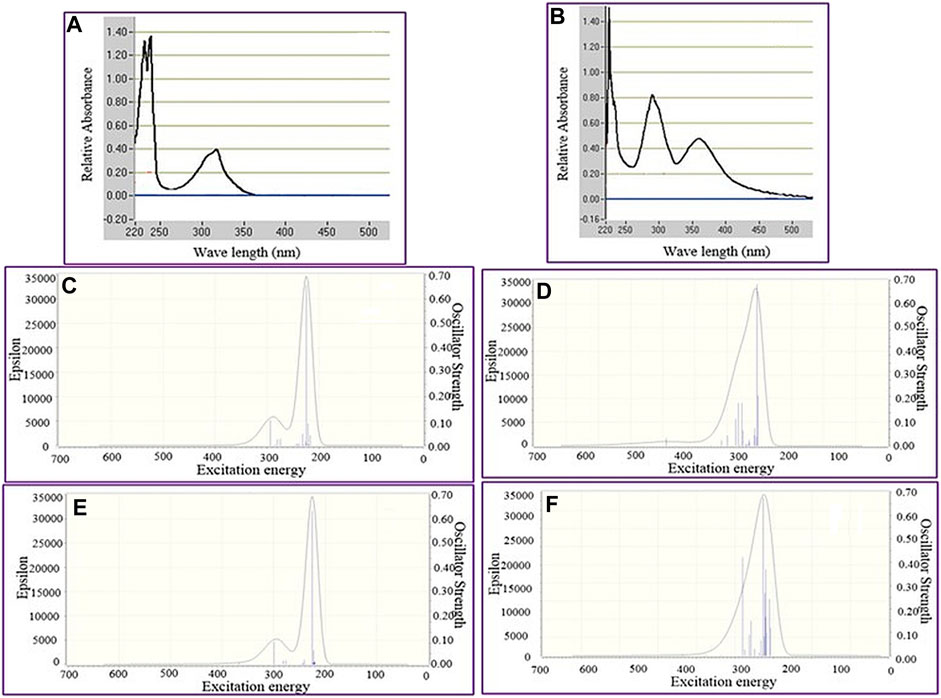
FIGURE 6. AIM analysis of the epi-CD-X complex. RDG isosurface maps: (A) PH2CN–OH as a representative X-epi-CD1 (X: PH2CN, OH: O36H37 at C17) and (B) PH2CN–N as a representative X-epi-CD2 (X: PH2CN, N: N16 of quinoline). Display of critical points: (C) PH2CN–OH (as X-epi-CD1) and (D) PH2CN (as X-epi-CD2). NCI plot of the epi-CD-X complex: (E) PH2CN–OH (as X-epi-CD1) and (F) PH2CN (as X-epi-CD2).
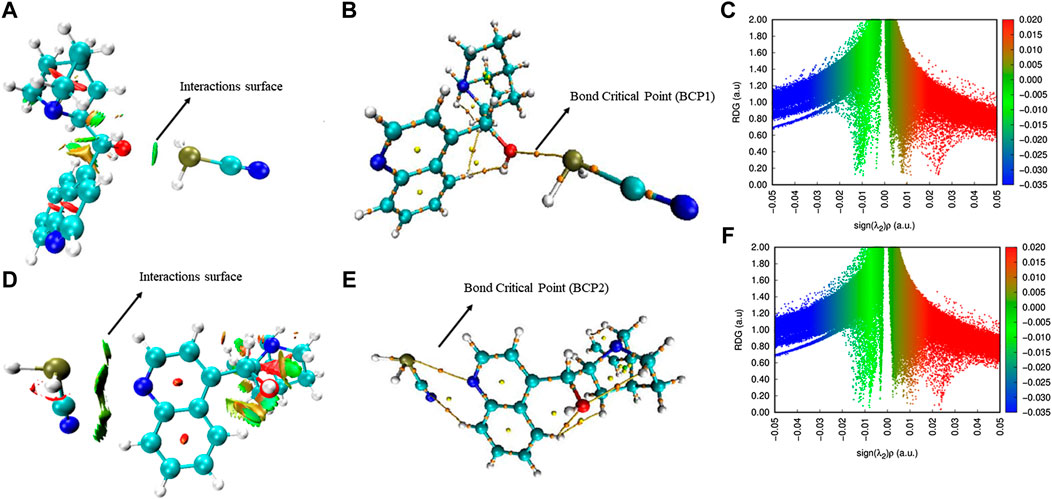
FIGURE 7. Proposed models of the cinchona alkaloid–catalyzed asymmetric reaction.
Conceptual Sketch of X-epi-CD. To improve the insufficiency of the X-epi-CD system, we considered the sketch based on how to revise this system. In the literature (Tanriver et al., 2016), it has been proposed that cinchona alkaloids tend to have complexes with substrates through hydrogen bonds in the hydroxyl group (H-O36) or quinuclidine (H+-N43) and long range interactions at conjugated pi electrons to produce the activation of substrate reactivity as well as enantioselectivity. For example, two models of conjugate addition reaction of thiolate (Wynberg’s and Grayson/Houk’s) elucidated the mediation of two protons between an electrophile/nucleophile and a cinchona alkaloid in the asymmetric catalysis as shown in Figure 7 (left). If such mediation of the hydrogen atom can be replaced with other elements, enantiotopic selectivity of the complexes also can be archived by new types of non-covalent bonds. Through the experimental data as well as simulation study, the dominant site of pnicogen bonding in epi-CD is the quinoline ring (N16 atom) rather than the hydroxyl group (O36 atom) showing P···O distance longer than P···N distance in almost all complexes. Because the nitrogen atom of the quinoline ring is very far from the complex and dense chiral environment of the cinchona alkaloid, it is rarely possible that X-epi-CD2 controls any enantiotopic face regardless of strength of the interaction. However, if the design of an analyte can modify the ratio of X-epi-CD1 (major) to X-epi-CD2 (minor) to reduce X-epi-CD2, the problem of uncontrolled stereoselectivity can be overcome. In particular, the HOMO–LUMO band gap energy as well as binding energy sometimes presented reversed pattern between X-epi-CD1 and X-epi-CD2. For example, the binding energy of ClPH2 (pnicogen bond) is similar to the binding energy of MA (hydrogen bond) at the same position, the hydroxyl group (O36 atom) in Supplementary Table S3. Moreover, AIM analysis of the PH2CN epi-CD1 complex also showed less difference from hydrogen bonding rather than epi-CD2 in ρ and Δ2ρ. It makes us propose the preliminary sketch of enantioselective reaction in which a pnicogen bonding of the cinchona alkaloid assigns enantiotopic selectivity into a substrate. Based on the literature (Yaghoobi and Sohrabi−Mahboub, 2018; LiXueCheng, 2017; Grayson and Houk, 2016b), the plausible idea is that the pnicogen bonding catalyzed the enantioselective aza-Diels–Alder reaction in Figure 7. For an ideal example, the complex X-epi-CD1 can have desirable interactions with a dienophile through pnicogen bonding as well as hydrogen bonding. In that situation, the enantiotopic face can be formed through the triangle composition of the 1) quinuclidine core, 2) chiral OH group of epi-CD, and 3) dienophile (or pnictide atom) because a face can be defined by three points. At that time, a diene can have the bias among front side and back side of the enantiotopic face. After pre-activating the preferred complex X-epi-CD1 (having a P···O bond), a diene and a dienophile can be added into the complex. If the dienophile has a substituent (e.g., NH2) bearing an unshared pair of electrons at the α-position, the strength of the pnicogen bond can be more enhanced through another NCI bond (P-H···N bond) with a dienophile. At that time, the mediation of phosphane (through the pnicogen bond) can make the complex able to discriminate the enantiotopic face as described in Figure 7. In other words, the behind face of dienophile is less favored by the attack of diene. While the σ-hole of halogen bonding is essential at the halogen atom, the pnicogen bond does not require such a strong σ-hole at the pnictogen atom (Scheiner, 2013a). Rather than electrostatic attraction, it is expected that the geometry for charge transfer is critical between atoms of the NCIs. To make the current theoretical reaction more feasible, the four components (epi-CD, analyte, dienophile, and diene) need to be modified to be an entropically favored form.
Conclusion
In this study, at B3LYP/6-31G(d) and B3LYPgCP-D3/6-31G levels of theory, intermolecular interactions of epi-CD with analytes (X) were described by the geometrical parameters and electronic, thermodynamic, and charge analyses. O36 of the hydroxyl group (in X-epi-CD1) and N16 of the quinoline ring (in X-epi-CD2) among Lewis basic atoms of epi-CD could be the interacting atoms of pnicogen bonds. While the dominant force of hydrogen bonding generally is electrostatic attraction, HOMO–LUMO energy, QNBO, QMulliken, AIM analysis, and UV-Vis analysis of the pnicogen bonds elucidated the interaction including the polarization and electron transfer. Based on the results, researchers can further progress pnicogen-based asymmetric catalysis based on our study in the recent future.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, and further inquiries can be directed to the corresponding author.
Author Contributions
M-hK conceived and designed the study with the funding source. ZU and KK conducted every simulation and analyzed the simulation data. AV synthesized the test compound. AV and HL carried out all the spectra measurements and data work. M-hK and ZU wrote the manuscript, and M-hK and AV revised it. M-hK and M-IK provided the computational lab and research work facility. All the authors read and approved the final manuscript.
Funding
This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), which was funded by the Ministry of Education, Science, and Technology (Nos. 2017R1D1A1B03027936 and 2017R1E1A1A01076642). The National Institute of Supercomputing and Network/Korea Institute of Science and Technology Information provided supercomputing resources including technical support (No. KSC-2017-C2-0017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.669515/full#supplementary-material
Abbreviations
CD, cinchonidine; X, phosphine (PH3)-substituted analytes (F, Br, Cl, CF3, CN, HO, NO2, and CH3); HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital; TD, time-dependent; DFT, density functional theory; X-epi-CD1, analytes bonding with the hydroxyl group (O-36); X-epi-CD2, analytes bonding with the quinoline ring (N16); DOS, density of states; IP, ionization potential; EA, electron affinity; PNBA, 4-nitrobenzoic acid; DEAD, diethyl azodicarboxylate; PPh3, triphenylphosphine; DCM, dichloromethane; LiOH, lithium hydroxide, CHCl3, chloroform; EtOH, ethanol; DW, distilled water; MEP, molecular electrostatic potential; NBO, natural bonding orbital; IR, infrared; MA, methyl acrylate, PBr3, phosphorus tribromide; BiI3, bismuth(III) iodide; SbI3, antimony triiodide; AsI3, arsenic triiodide.
References
Akiyama, T., Itoh, J., and Fuchibe, K. (2006). Recent Progress in Chiral Brønsted Acid Catalysis. Adv. Synth. Catal. 348, 999–1010. doi:10.1002/adsc.200606074
Alkorta, I., Elguero, J., and Del Bene, J. E. (2013). Pnicogen-Bonded Cyclic Trimers (PH2X)3 with X = F, Cl, OH, NC, CN, CH3, H, and BH2. J. Phys. Chem. A. 117, 4981–4987. doi:10.1021/jp403651h
Alkorta, I., Sánchez-Sanz, G., Elguero, J., and Del Bene, J. E. (2014). Pnicogen Bonds between X═PH3 (X = O, S, NH, CH2) and Phosphorus and Nitrogen Bases. J. Phys. Chem. A. 118, 1527–1537. doi:10.1021/jp411623h
Bauzá, A., Alkorta, I., Frontera, A., and Elguero, J., (2013). On the Reliability of Pure and Hybrid DFT Methods for the Evaluation of Halogen, Chalcogen, and Pnicogen Bonds Involving Anionic and Neutral Electron Donors. J. Chem. Theor. Comput. 9, 5201–5210. doi:10.1021/ct400818v
Allouche, A.-R. (2011). Gabedit-A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 32, 174–182. doi:10.1002/jcc.21600
Athawale, V., and Manjrekar, N. (2001). Mild Chemo-Enzymatic Synthesis of Polymer-Supported Cinchona Alkaloids and Their Application in Asymmetric Michael Addition. Tetrahedron Lett. 42, 4541–4543. doi:10.1016/s0040-4039(01)00731-6
Auria-Luna, F, Marqués-López, E., Gimeno, M. C, Heiran, R., Mohammadi, S., and Herrera, R. P. (2017). Asymmetric Organocatalytic Synthesis of Substituted Chiral 1,4-Dihydropyridine Derivatives. J. Org. Chem. 82 5516–5523. doi:10.1021/acs.joc.7b00176
Becke, A. D. (1988). Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 38, 3098–3100. doi:10.1103/physreva.38.3098
Becke, A. D. (1993). A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 98, 1372–1377. doi:10.1063/1.464304
Benz, S., Macchione, M., Verolet, Q., Mareda, , Sakai, N., and Matile, S. (2016). Anion Transport with Chalcogen Bonds. J. Am. Chem. Soc. 138, 9093–9096. doi:10.1021/jacs.6b05779
Benz, S., López-Andarias, J., Mareda, J., Sakai, N., and Matile, S. (2017). Catalysis with Chalcogen Bonds. Angew. Chem. Int. Ed. 56, 812–815. doi:10.1002/anie.201611019
Bibi, S., Ullah, H., Ahmad, S. M., Ali Shah, A.U -H., Bilal, S., Tahir, A. A., et al. (2015). Molecular and Electronic Structure Elucidation of Polypyrrole Gas Sensors. J. Phys. Chem. C 119, 15994–16003. doi:10.1021/acs.jpcc.5b03242
Boys, S. F., and Bernardi, F. (1970). The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 19, 553–566. doi:10.1080/00268977000101561
Brandenburg, J. G., Alessio, M., Civalleri, B., Peintinger, M. F., Bredow, T., and Grimme, S. (2013). Geometrical Correction for the Inter- and Intramolecular Basis Set Superposition Error in Periodic Density Functional Theory Calculations. J. Phys. Chem. A. 117, 9282–9292. doi:10.1021/jp406658y
Breugst, M., von der Heiden, D., and Schmauck, J. (2017). Novel Noncovalent Interactions in Catalysis: A Focus on Halogen, Chalcogen, and Anion-π Bonding. Synthesis 49, 3224–3236. doi:10.1055/s-0036-1588838
Bulfield, D., and Huber, S. M. (2016). Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 22, 14434–14450. doi:10.1002/chem.201601844
Bürgi, T., and Baiker, A. (1998). Conformational Behavior of Cinchonidine in Different Solvents: A Combined NMR and Ab Initio Investigation. J. Am. Chem. Soc. 120, 12920–12926. doi:10.1021/ja982466b
Bürgi, T., Vargas, A., and Baiker, A. (2002). VCD Spectroscopy of Chiral Cinchona Modifiers Used in Heterogeneous Enantioselective Hydrogenation: Conformation and Binding of Non-chiral Acids. J. Chem. Soc. Perkin 1 (29), 1596–1601. doi:10.1039/b203251a
Caner, H., Biedermann, P. U., and Agranat, I. (2003). Conformational Spaces of Cinchona Alkaloids. Chirality 15, 637–645. doi:10.1002/chir.10265
Cavallo, G., Metrangolo, P., Milani, R., Pilati, T., Priimagi, A., Resnati, G., et al. (2016). The Halogen Bond Chem. Rev. 116, 2478–2601doi:10.1021/acs.chemrev.5b00484
Chen, Y.-R., Das, U., Liu, M.-H., and Lin, W., (2015). Organocatalytic Enantioselective Direct Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactam to β-Acyl Acrylates and 1,2-Diacylethylenes. J. Org. Chem. 80, 1985–1992. doi:10.1021/jo5027047
Dalko, P. I., and Moisan, L. (2001). Enantioselective Organocatalysis. Angew. Chem. Int. Ed. 40, 3726–3748. doi:10.1002/1521-3773(20011015)40:20<3726::aid-anie3726>3.0.co;2-d
de Villiers, K. A., Gildenhuys, J., and Roex, T. (2012). Iron(III) Protoporphyrin IX Complexes of the AntimalarialCinchonaAlkaloids Quinine and Quinidine. ACS Chem. Biol. 7, 666–671. doi:10.1021/cb200528z
Del Bene, J. E., Alkorta, I., and Elguero, J. (2014). Pnicogen-Bonded Anionic Complexes. J. Phys. Chem. A. 118 (18), 3386–3392. doi:10.1021/jp502667k
Del Bene, J. E., Alkorta, I., Elguero, J., and Sanchez-Sanz, G. (2017). Lone-Pair Hole on P: P··· N Pnicogen Bonds Assisted by Halogen Bonds. J. Phys. Chem. A. 121 (6), 1362–1370. doi:10.1021/acs.jpca.6b12553
Dijkstra, G. D. H., Kellogg, R. M., and Wynberg, H. (1989). Conformational Analysis of Some Chiral Catalysts of the Cinchona1 and Ephedra2 Family. The Alkaloid Catalyzed Addition of Aromatic Thiols to Cyclic α,β-unsaturated Ketones. Recl. Trav. Chim. Pays-bas 108, 195–204. doi:10.1002/recl.19891080507
Dijkstra, G. D. H., Kellogg, R. M., Wynberg, H., Svendsen, J. S., Marko, I., and Sharpless, K. B. (1989). Conformational Study of Cinchona Alkaloids. A Combined NMR, Molecular Mechanics and X-ray Approach. J. Am. Chem. Soc. 111, 8069–8076. doi:10.1021/ja00203a001
Doyle, A. G., and Jacobsen, E. N., (2007). Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 107, 5713–5743. doi:10.1021/cr068373r
Escudero-Adán, E. C., Bauzá, A., Frontera, A., and Ballester, P. (2015). Nature of Noncovalent Carbon-Bonding Interactions Derived from Experimental Charge-Density Analysis. ChemPhysChem 16, 2530–2533. doi:10.1002/cphc.201500437
Esrafili, M. D., and Qasemsolb, S. (2017). Tuning Aerogen Bonds via Anion-π or Lone Pair-π Interaction: a Comparative Ab Initio Study. Struct. Chem. 28, 1255–1264. doi:10.1007/s11224-017-0949-4
Esrafili, M. D., and Vessally, E. (2016). A Theoretical Evidence for Cooperative Enhancement in Aerogen-Bonding Interactions: Open-Chain Clusters of KrOF2 and XeOF2. Chem. Phys. Lett. 662, 80–85. doi:10.1016/j.cplett.2016.09.037
Ezekwesili, C., Ogbunugafor, H., and Ezekwesili–Ofili, J. (2012). Anti-Diabetic Activity of Aqueous Extracts of Vitex Doniana Leaves and Cinchona Calisaya Bark in Alloxan–Induced Diabetic Rats. Int. J. Trop. Dis. 2 (4), 290–300.
Fanfrlík, J., Přáda, A., Padělková, Z., Pecina, A., Macháček, J., Lepšík, M., et al. (2014). The Dominant Role of Chalcogen Bonding in the Crystal Packing of 2D/3D Aromatics. Angew. Chem. Int. Ed. 53, 10139–10142. doi:10.14210.1002/anie.201405901
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Revision D.01. Wallingford CT: Gaussian, Inc.
Fulton, J. L., Horwitz, M. A., Bruske, E. L., and Johnson, J. S. (2018). Asymmetric Organocatalytic Sulfa-Michael Addition to Enone Diesters. J. Org. Chem. 83, 3385–3391. doi:10.1021/acs.joc.8b00007
De Fusco, C., Fuoco, T., Croce, G., and Lattanzi, A. (2012). Noncovalent Organocatalytic Synthesis of Enantioenriched Terminal Aziridines with a Quaternary Stereogenic Center. Org. Lett. 14 (16), 4078–4081. doi:10.1021/ol3017066
Gao, M., Cheng, J., Li, W., Xiao, B., and Li, Q. (2016). The Aerogen-π Bonds Involving π Systems. Chem. Phys. Lett. 651, 50–55. doi:10.1016/j.cplett.2016.03.021
Gaunt, M. J., Johansson, C. C. C., McNally, A., and Vo, N. T. (2007). Enantioselective Organocatalysis. Drug Discov. Today 12, 8–27. doi:10.1016/j.drudis.2006.11.004
George, J., Deringer, V. L., and Dronskowski, R. (2014). Cooperativity of Halogen, Chalcogen, and Pnictogen Bonds in Infinite Molecular Chains by Electronic Structure Theory, J. Phys. Chem. A. 118, 3193–3200. doi:10.1021/jp5015302
Gliese, J.-P., Jungbauer, S. H., and Huber, S. M. (2017). A Halogen-Bonding-Catalyzed Michael Addition Reaction. Chem. Commun. 53, 12052–12055. doi:10.1039/c7cc07175b
Grayson, M. N., and Houk, K. N. (2016). Cinchona Urea-Catalyzed Asymmetric Sulfa-Michael Reactions: The Brønsted Acid−Hydrogen Bonding Model J. Am. Chem. Soc. 138, 9041–9044. doi:10.1021/jacs.6b05074
Grayson, M. N., and Houk, K. N. (2016). Cinchona Alkaloid-Catalyzed Asymmetric Conjugate Additions: The Bifunctional Brønsted Acid-Hydrogen Bonding Model J. Am. Chem. Soc. 138, 1170–1173. doi:10.1021/jacs.5b13275
Guo, J., and Wong, M. W. (2017). Cinchona Alkaloid-Squaramide Catalyzed Sulfa-Michael Addition Reaction: Mode of Bifunctional Activation and Origin of Stereoinduction. J. Org. Chem. 82, 4362–4368. doi:10.1021/acs.joc.7b00388
Hatakeyama, M., Ogata, K., Ishida, T., Kitamura, K., and Nakamura, S. (2015). Theoretical Study on Carbon-Carbon Short Contact of 2.3 Å: Intermediate State between Nonbonding and σ-Covalent Bonding. J. Phys. Chem. A. 119, 781–785. doi:10.1021/jp511096p
Heinen, F., Engelage, E., Dreger, A., Weiss, R., and Huber, S. M. (2018). Iodine (III) Derivatives as Halogen Bonding Organocatalysts. Angew. Chem. Int. Ed. 57 (14)3830–3833. doi:10.1002/anie.201713012
Hiemstra, H., and Wynberg, H. (1981). Addition of Aromatic Thiols to Conjugated Cycloalkenones, Catalyzed by Chiral .beta.-hydroxy Amines. A Mechanistic Study of Homogeneous Catalytic Asymmetric Synthesis. J. Am. Chem. Soc. 103, 417–430. doi:10.1021/ja00392a029
Jha, S. C., and Joshi, N. N. (2002). Catalytic, Enatioselective Michael Addition Reactions. Arkivoc 2002, 167–196. doi:10.3998/ark.5550190.0003.718
Jones, R. (2001). Quaternary Ammonium Salts and Their Use as Phase Transfer Catalysts. New York: Academic Press.
Jungbauer, S. H., and Huber, S. M. (2015). Cationic Multidentate Halogen-Bond Donors in Halide Abstraction Organocatalysis: Catalyst Optimization by Preorganization. J. Am. Chem. Soc. 137, 12110–12120. doi:10.1021/jacs.5b07863
Jusseau, X., Chabaud, L., and Guillou, C. (2014). Synthesis of γ-butenolides and α,β-unsaturated γ-butyrolactams by Addition of Vinylogous Nucleophiles to Michael Acceptors. Tetrahedron 70, 2595–2615. doi:10.1016/j.tet.2014.01.057
Kacprzak, A. K., and Gawronski, J. (2001). Cinchona Alkaloids and Their Derivatives: Versatile Catalysts and Ligands in Asymmetric Synthesis. Synthesis 7, 0961–0998.
Karle, J. M., and Bhattacharjee, A. K. (1999). Stereoelectronic Features of the Cinchona Alkaloids Determine Their Differential Antimalarial Activity. Bioorg. Med. Chem. 7, 1769–1774. doi:10.1016/s0968-0896(99)00120-0
Klare, H., Neudörfl, J. M., and Goldfuss, B. (2014). New Hydrogen-Bonding Organocatalysts: Chiral Cyclophosphazanes and Phosphorus Amides as Catalysts for Asymmetric Michael Additions. Beilstein J. Org. Chem. 10, 224–236. doi:10.3762/bjoc.10.18
Kruse, H., Goerigk, L., and Grimme, S. (2012). Why the Standard B3LYP/6-31G* Model Chemistry Should Not Be Used in DFT Calculations of Molecular Thermochemistry: Understanding and Correcting the Problem. J. Org. Chem. 77, 10824–10834. doi:10.83410.1021/jo302156p
Li, H., and Hong, X. (2018). Computational Studies of Cinchona Alkaloid-Catalyzed Asymmetric Michael Additions. Chin. Chem. Lett. 29, 1585–1590. doi:10.1016/j.cclet.2018.01.030
Li, H., Wang, Y., Tang, L., and Deng, L. (2004). Highly Enantioselective Conjugate Addition of Malonate and β-Ketoester to Nitroalkenes: Asymmetric C−C Bond Formation with New Bifunctional Organic Catalysts Based on Cinchona Alkaloids. J. Am. Chem. Soc. 126, 9906–9907. doi:10.1021/ja047281l
Li, H., Song, J., Liu, X., and Deng, L. (2005). Catalytic Enantioselective C−C Bond Forming Conjugate Additions with Vinyl Sulfones. J. Am. Chem. Soc. 127, 8948–8949. doi:10.1021/ja0511063
Li, H., Wang, Y., Tang, L., Wu, F., Liu, X., Guo, C., et al. (2005). Stereocontrolled Creation of Adjacent Quaternary and Tertiary Stereocenters by a Catalytic Conjugate Addition. Angew. Chem. Int. Ed. 44, 105–108. doi:10.1002/anie.200461923
Li, M., Xue, X.-S., and Cheng, J.-P. (2017). Mechanism and Origins of Stereoinduction in Natural Cinchona Alkaloid Catalyzed Asymmetric Electrophilic Trifluoromethylthiolation of β-Keto Esters with N-Trifluoromethylthiophthalimide as Electrophilic SCF3 Source, ACS Catal. 7, 7977–7986. doi:10.1021/acscatal.7b03007
Lu, T., and Chen, F. (2012). Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 33, 580–592. doi:10.1002/jcc.22885
Luo, J., Xu, L.-W., Hay, R. A. S., and Lu, Y. (2009). Asymmetric Michael Addition Mediated by Novel Cinchona Alkaloid-Derived Bifunctional Catalysts Containing Sulfonamides. Org. Lett. 11, 437–440. doi:10.1021/ol802486m
Lygo, B., and Wainwright, P. G. (1997). A New Class of Asymmetric Phase-Transfer Catalysts Derived from Cinchona Alkaloids - Application in the Enantioselective Synthesis of α-amino Acids. Tetrahedron Lett. 38, 8595–8598. doi:10.1016/s0040-4039(97)10293-3
Maeda, N., Hungerbuhler, K., and Baiker, A. (2011). Asymmetric Hydrogenation on Chirally Modified Pt: Origin of Hydrogen in the N–H–O Interaction Between Cinchonidine and Ketone. J. Am. Chem. Soc. 133 (49), 19567–19569.
Manna, D., and Mugesh, G. (2012). Regioselective Deiodination of Thyroxine by Iodothyronine Deiodinase Mimics: An Unusual Mechanistic Pathway Involving Cooperative Chalcogen and Halogen Bonding. J. Am. Chem. Soc. 134, 4269–4279. doi:10.1021/ja210478k
Marcelli, T., and Hiemstra, H. (2010). Cinchona Alkaloids in Asymmetric Organocatalysis. Synthesis 2010, 1229–1279. doi:10.1055/s-0029-1218699
Martin, F., and Zipse, H. (2005). Charge Distribution in the Water Molecule?A Comparison of Methods. J. Comput. Chem. 26, 97–105. doi:10.1002/jcc.20157
Nath, U., Banerjee, A., Ghosh, B., and Pan, S. C. (2015). Organocatalytic Asymmetric Michael Addition of 1-acetylcyclohexene and 1-acetylcyclopentene to Nitroolefins. Org. Biomol. Chem. 13, 7076–7083. doi:10.1039/c5ob00878f
O'boyle, N. M., Tenderholt, A. L., and Langner, K. M. (2008). Cclib: A Library for Package-independent Computational Chemistry Algorithms. J. Comput. Chem. 29, 839–845. doi:10.1002/jcc.20823
Pal, P., Konar, S., Lama, P., Das, K., Bauzá, A., Frontera, A., et al. (2016). On the Importance of Noncovalent Carbon-Bonding Interactions in the Stabilization of a 1D Co(II) Polymeric Chain as a Precursor of a Novel 2D Coordination Polymer. J. Phys. Chem. B 120, 6803–6811. doi:10.1021/acs.jpcb.6b04046
Pham, T. S., Gönczi, K., Kardos, G., Süle, K., Hegedűs, L., Kállay, M., et al. (2013). Cinchona Based Squaramide Catalysed Enantioselective Michael Addition of α-nitrophosphonates to Aryl Acrylates: Enantioselective Synthesis of Quaternary α-aminophosphonates. Tetrahedron: Asymmetry 24, 1605–1614. doi:10.1016/j.tetasy.2013.10.008
Politzer, P., Murray, J. S., and Clark, T. (2013). Halogen Bonding and Other σ-hole Interactions: a Perspective. Phys. Chem. Chem. Phys. 15, 11178–11189. doi:10.1039/c3cp00054k
Prakash, G. K. S., Wang, F., Ni, C., Shen, J., Haiges, R., Yudin, A. K., et al. (2011). Conformational Study of 9-Dehydro-9-Trifluoromethyl Cinchona Alkaloids via19F NMR Spectroscopy: Emergence of Trifluoromethyl Moiety as a Conformational Stabilizer and a Probe. J. Am. Chem. Soc. 133, 9992–9995. doi:10.1021/ja202373d
Qian, Z., Zhijian, Xu., and Weiliang, Z. (2017). The Underestimated Halogen Bonds Forming With Protein Side Chains in Drug Discovery and Design. J. Chem. Inf. Model. 57 (1), 22–26. doi:10.1021/acs.jcim.6b00628
Qin, W., Vozza, A., and Brouwer, A. M. (2009). Photophysical Properties of Cinchona Organocatalysts in Organic Solvents. J. Phys. Chem. C 113, 11790–11795. doi:10.1021/jp901867h
Roohi, H., Nowroozi, A.-R., and Anjomshoa, E. (2011). H-bonded Complexes of Uracil with Parent Nitrosamine: A Quantum Chemical Study. Comput. Theor. Chem. 965, 211–220. doi:10.1016/j.comptc.2011.01.048
Salzner, U. (2008). Investigation of Charge Carriers in Doped Thiophene Oligomers through Theoretical Modeling of Their UV/Vis Spectra. J. Phys. Chem. A. 112, 5458–5466. doi:10.1021/jp800606m
Sánchez-Sanz, G., Trujillo, C., Alkorta, I., and Elguero, J. (2012). Electron Density Shift Description of Non-bonding Intramolecular Interactions. Comput. Theor. Chem. 991, 124–133. doi:10.1016/j.comptc.2012.04.007
Sánchez-Sanz, G., Trujillo, C., Alkorta, I., and Elguero, J. (2015). Theoretical Study of Cyanophosphines: Pnicogen vs. Dipole-Dipole Interactions. Comput. Theor. Chem. 1053, 305–314. doi:10.1016/j.comptc.2014.07.009
Sarkar, S., Pavan, M. S., and Guru Row, T. N. (2015). Experimental Validation of “pnicogen Bonding” in Nitrogen by Charge Density Analysis. Phys. Chem. Chem. Phys. 17, 2330–2334. doi:10.1039/c4cp04690k
Scheiner, S., and Adhikari, U. (2011). Abilities of Different Electron Donors (D) to Engage in a P···D Noncovalent Interaction. J. Phys. Chem. A. 115, 11101–11110. doi:10.1021/jp2082787
Scheiner, S. (2011). On the Properties of XN Noncovalent Interactions for First-, Second-, and Third-Row X Atoms. J. Chem. Phys. 134, 164313. doi:10.1063/1.3585611
Scheiner, S. (2011). A New Noncovalent Force: Comparison of P··· N Interaction With hydrogen and Halogen Bonds. J. Chem. Phys. 134 (9), 094315. doi:10.1063/1.3562209
Scheiner, S. (2013). The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 46, 280–288. doi:10.1021/ar3001316
Scheiner, S. (2013). Detailed Comparison of the Pnicogen Bond with Chalcogen, Halogen, and Hydrogen Bonds. Int. J. Quan. Chem. 113, 1609–1620. doi:10.1002/qua.24357
Schmauck, J., and Breugst, M. (2017). The Potential of Pnicogen Bonding for Catalysis - a Computational Study. Org. Biomol. Chem. 15, 8037–8045. doi:10.1039/c7ob01599b
Scorzelli, F., Di Mola, A., Palombi, L., and Massa, A. (2015). Isoindolinones as Michael Donors under Phase Transfer Catalysis: Enantioselective Synthesis of Phthalimidines Containing a Tetrasubstituted Carbon Stereocenter. Molecules 20, 8484–8498. doi:10.3390/molecules20058484
Shi, M., and Xu, Y.-M. (2002). Catalytic, Asymmetric Baylis-Hillman Reaction of Imines with Methyl Vinyl Ketone and Methyl Acrylate. Angew. Chem. Int. Ed. 41, 4507–4510. doi:10.1002/1521-3773(20021202)41:23<4507::aid-anie4507>3.0.co;2-i
Sidorowicz, L., and Skarżewski, J. (2011). Easy Access to 9-Epimers of Cinchona Alkaloids: One-Pot Inversion by Mitsunobu Esterification-Saponification. Synthesis 5, 708–710.
Sim, S.-B. D., Wang, M., and Zhao, Y. (2015). Phase-Transfer-Catalyzed Enantioselective α-Hydroxylation of Acyclic and Cyclic Ketones with Oxygen. ACS Catal. 5, 3609–3612. doi:10.1021/acscatal.5b00583
Singh, G., and Yeboah, E. (2016). Recent Applications of Cinchona Alkaloid-Based Catalysts in Asymmetric Addition Reactions. Roc Vol. 6, 47–75. doi:10.2147/roc.s73908
Song, C. (2009). An Overview of Cinchona Alkaloids in Chemistry in Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA.
Song, C. E. (2009). An Overview of Cinchona Alkaloids in Chemistry. Cinchona Alkaloids in Synthesis and Catalysis: Ligands. Immobilization and Organocatalysis 1, 1–9. doi:10.1002/9783527628179
Szőllősi, G., Busygin, I., Hermán, B., Leino, R., Bucsi, I., Murzin, D. Yu., et al. (2011). Inversion of the Enantioselectivity in the Hydrogenation of (E)-2, 3-Diphenylpropenoic Acids Over Pd Modified By Cinchonidine Silyl Ethers. ACS Catal. 1 (10), 1316–1326.
Szollosi, G. (2017). Lenke Kovacs, Viktoria Kozma, Vanessza Judit Kolcsar. Reac. Kinet. Mech. Cat. 121, 293–306. doi:10.1021/cs200199
Tanriver, G., Dedeogl, B., Catak, S., and Aviyente, V. (2016). Computational Studies on Cinchona Alkaloid-Catalyzed Asymmetric Organic Reactions Acc. Chem. Res. 4, 1250–1262. doi:10.1021/acs.accounts.6b00078
Taylor, M. S., and Jacobsen, E. N. (2006). Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 45, 1520–1543. doi:10.1002/anie.200503132
Tian, S.-K., Chen, Y., Hang, J., Tang, L., Mcdaid, P., and Deng, L. (2004). Asymmetric Organic Catalysis with Modified Cinchona Alkaloids. Acc. Chem. Res. 37, 621–631. doi:10.1021/ar030048s
Ullah, M. S., and Itsuno, S. (2017). Synthesis of Cinchona Alkaloid Squaramide Polymers as Bifunctional Chiral Organocatalysts for the Enantioselective Michael Addition of β-ketoesters to Nitroolefins. Mol. Catal. 438, 239–244. doi:10.1016/j.mcat.2017.06.010
Ullah, H., Shah, A.-U-H. A., Bilal, S., and Ayub, K. (2013). DFT Study of Polyaniline NH3, CO2, and CO Gas Sensors: Comparison with Recent Experimental Data. J. Phys. Chem. C 117, 23701–23711. doi:10.1021/jp407132c
Vakulya, B., Varga, S., and Soós, T. (2008). Epi-Cinchona Based Thiourea Organocatalyst Family as an Efficient Asymmetric Michael Addition Promoter: Enantioselective Conjugate Addition of Nitroalkanes to Chalcones and α,β-Unsaturated N-Acylpyrroles. J. Org. Chem. 73, 3475–3480. doi:10.1021/jo702692a
Vandekerckhove, S., and D’hooghe, M. (2015). Quinoline-based Antimalarial Hybrid Compounds. Bioorg. Med. Chem. 23, 5098–5119. doi:10.1016/j.bmc.2014.12.018
Viveros-Ceballos, J., Ordóñez, M., Sayago, F., and Cativiela, C. (2016). Stereoselective Synthesis of α-Amino-C-phosphinic Acids and Derivatives. Molecules 21, 1141. doi:10.3390/molecules21091141
Wang, B., Wu, F., Wang, Y., Liu, X., and Deng, L. (2007). Control of Diastereoselectivity in Tandem Asymmetric Reactions Generating Nonadjacent Stereocenters with Bifunctional Catalysis by Cinchona Alkaloids. J. Am. Chem. Soc. 129, 768–769. doi:10.1021/ja0670409
Wang, X.-F., An, J., Zhang, X.-X., Tan, F., Chen, J.-R., and Xiao, W.-J. (2011). Catalytic Asymmetric Aza-Michael−Michael Addition Cascade: Enantioselective Synthesis of Polysubstituted 4-Aminobenzopyrans. Org. Lett. 13, 808–811. doi:10.1021/ol1031188
Prakash, G. K. S., Wang, F., Rahm, M., Zhang, Z., Ni, C., Shen, J., et al. (2014). The Trifluoromethyl Group as a Conformational Stabilizer and Probe: Conformational Analysis of Cinchona Alkaloid Scaffolds. J. Am. Chem. Soc. 136, 10418–10431. doi:10.1021/ja504376u
Wesche, D. L., and Black, J. (1990). A Comparison of the Antimalarial Activity of the Cinchona Alkaloids Against Plasmodium falciparum In Vitro. J. Trop. Med. Hyg. 93 (3), 153–159.
Wheele, S. E., Seguin, T. J., Guan, Y., and Doney, A. C., (2016). Noncovalent Interactions in Organocatalysis and the Prospect of Computational Catalyst Design. Acc. Chem. Res. 49, 1061–1069. doi:10.1021/acs.accounts.6b00096
Xue, Y., Wang, Y., Cao, Z., Zhou, J., and Chen, Z.-X. (2016). Computational Insight into the Cooperative Role of Non-covalent Interactions in the Aza-Henry Reaction Catalyzed by Quinine Derivatives: Mechanism and Enantioselectivity. Org. Biomol. Chem. 14, 9588–9597. doi:10.1039/c6ob01611a
Yaghoobi, F., and Sohrabi−Mahboub, M. (2018). Theoretical Study on the Aza-Diels-Alder Reaction Catalyzed by PHCl2 Lewis Acid via Pnicogen Bonding. J. Phys. Chem. A. 122, 2781–2791. doi:10.1021/acs.jpca.7b12400
Yang, H., and Wong, M. W. (2013). Oxyanion Hole Stabilization by C-H···O Interaction in a Transition State-A Three-Point Interaction Model for Cinchona Alkaloid-Catalyzed Asymmetric Methanolysis of Meso-Cyclic Anhydrides. J. Am. Chem. Soc. 135, 5808–5818. doi:10.1021/ja4005893
Yanuka, Y., Yosselson-Superstine, S., Geryes, A., and Superstine, E. (1981). Impact of Stable Conformation of Cinchona Alkaloids on Protonation Site. J. Pharm. Sci. 70, 679–682. doi:10.1002/jps.2600700627
Yardley, J. P., Bright, R. E., Rane, L., Rees, R. W., Russell, P. B., and Smith, H. (1971). Antimalarial and Other Biological Activities of Some 2'-alkyl and 2'-aryl Derivatives of Cinchona Alkaloids. J. Med. Chem. 14, 62–65. doi:10.1021/jm00283a020
Yeboah, E. M. O., Yeboah, S. O., and Singh, G. S. (2011). Recent Applications of Cinchona Alkaloids and Their Derivatives as Catalysts in Metal-free Asymmetric Synthesis. Tetrahedron 67, 1725–1762. doi:10.1016/j.tet.2010.12.050
Keywords: pnicogen bonding, DFT calculation, enantiotopic face, HOMO–LUMO, UV-Vis spectroscopy
Citation: Ullah Z, Kim K, Venkanna A, Kim Hs, Kim MI and Kim M-h (2021) Plausible Pnicogen Bonding of epi-Cinchonidine as a Chiral Scaffold in Catalysis. Front. Chem. 9:669515. doi: 10.3389/fchem.2021.669515
Received: 19 February 2021; Accepted: 10 June 2021;
Published: 06 July 2021.
Edited by:
Ranjith Kumar Kankala, Huaqiao University, ChinaReviewed by:
Ataualpa A. C. Braga, University of São Paulo, BrazilSuhana Arshad, Universiti Sains Malaysia, Malaysia
Copyright © 2021 Ullah, Kim, Venkanna, Kim, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mi-hyun Kim, a21oMDUxNUBnYWNob24uYWMua3I=
†These authors share first authorship