 Marcin Sarewicz
Marcin Sarewicz Sebastian Pintscher
Sebastian Pintscher Artur Osyczka
Artur Osyczka- Department of Molecular Biophysics, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków, Poland
Cytochrome bc1 (mitochondrial complex III) catalyzes electron transfer from quinols to cytochrome c and couples this reaction with proton translocation across lipid membrane; thus, it contributes to the generation of protonmotive force used for the synthesis of ATP. The energetic efficiency of the enzyme relies on a bifurcation reaction taking place at the Qo site which upon oxidation of ubiquinol directs one electron to the Rieske 2Fe2S cluster and the other to heme bL. The molecular mechanism of this reaction remains unclear. A semiquinone spin-coupled to the reduced 2Fe2S cluster (SQo-2Fe2S) was identified as a state associated with the operation of the Qo site. To get insights into the mechanism of the formation of this state, we first constructed a mutant in which one of the histidine ligands of the iron ion of heme bLRhodobacter capsulatus cytochrome bc1 was replaced by asparagine (H198N). This converted the low-spin, low-potential heme into the high-spin, high-potential species which is unable to support enzymatic turnover. We performed a comparative analysis of redox titrations of antimycin-supplemented bacterial photosynthetic membranes containing native enzyme and the mutant. The titrations revealed that H198N failed to generate detectable amounts of SQo-2Fe2S under neither equilibrium (in dark) nor nonequilibrium (in light), whereas the native enzyme generated clearly detectable SQo-2Fe2S in light. This provided further support for the mechanism in which the back electron transfer from heme bL to a ubiquinone bound at the Qo site is mainly responsible for the formation of semiquinone trapped in the SQo-2Fe2S state in R. capusulatus cytochrome bc1.
Introduction
The cytochrome bc1 complex (Cytbc1) is one of the enzymes involved in energy conversion that takes place in mitochondria and many prokaryotic respiratory chains and anoxygenic photosynthesis (Berry et al., 2000; Crofts, 2004; Sarewicz and Osyczka, 2015). Structurally and functionally very similar to Cytbc1—cytochrome b6f is a central enzyme of oxygenic photosynthesis in cyanobacteria and plants (Darrouzet et al., 2004; Baniulis et al., 2008; Hasan et al., 2013; Cramer and Hasan, 2016). Cytbc1 catalyzes electron transfer from membrane-soluble electron carriers–quinone (Q) derivatives (usually ubiquinones) (Al-Attar and de Vries, 2013) to water-soluble electron carrier: cytochrome c (Bertini et al., 2006). The energy released during enzymatic reduction of cytochrome c by ubiquinol (UQH2) is used to transfer protons across the membrane, contributing to building up the protonmotive force (Hunte et al., 2003). Cytbc1 is not operating as a typical proton pump that uses special proton channels but it utilizes quinone molecules to transport protons across the lipid bilayers (Al-Attar and de Vries, 2013). This transport is carried out by coupling two opposite redox reactions of quinones at the two catalytic sites that are located within the enzyme structure at the opposite sides of the membrane (Mitchell, 1975; Crofts et al., 1983). The UQH2 oxidation site (Qo site) is located close to the p side of the membrane (the positively charged side), while the UQ-reduction site (Qi site) is located close to the n side of the membrane (the negatively charged side) (Xia et al., 1997; Crofts and Berry, 1998). The Qo and Qi sites are electronically connected by two low-potential, low-spin hemes b: heme bL [Em,7 ∼ −120 mV in R. capsulatus (Zhang et al., 2008)], which is adjacent to the Qo site and heme bH (Em,7 ∼ + 60 mV), which is adjacent to the Qi site. The oxidation of UQH2 at the Qo site is accompanied by the release of two protons to the bulk water at the p side. Oppositely, the reduction taking place at the Qi site is associated with uptake of two protons from the n side of the membrane. This way the protons are transported through the membrane along with pairs of diffusing UQH2/UQ molecules.
The reduction of UQ at the Qi site is a sequential process, involving two consecutive electron transfers from the same cofactor (heme bH). A stable semiquinone is an intermediate of this reaction (Robertson et al., 1984; Jünemann et al., 1998; Dikanov et al., 2004) and, as implicated from recent studies, a mechanism of its stabilization might involve polarization of charges within the ring (Pintscher et al., 2020). On the other hand, the oxidation of UQH2 at the Qo site directs two electrons derived from the UQH2 into two separate cofactor chains. In this so-called electron bifurcation reaction one electron is transferred on the Rieske [2Fe-2S] cluster (2Fe2S) and further, through cytochrome c1 onto a water-soluble cytochrome c (or c2 in some bacteria, such as the Rhodobacter strains). The second electron is transferred to heme bL and subsequently through heme bH it reaches the Qi site where UQ is reduced first to semiquinone form (USQi) and then to UQH2.
Despite a long history of studying the mechanism of electron bifurcation, the involvement of a semiquinone intermediate (USQo) in this reaction is still unclear and thus a matter of intense discussion (Osyczka et al., 2004; Osyczka et al., 2005; Cape et al., 2007; Zhang et al., 2007; Zhu et al., 2007; Crofts et al., 2013; Sarewicz et al., 2013; Crofts et al., 2017; Sarewicz et al., 2021). A general agreement has been reached that a superoxide generation by Cytbc1 results from a reaction of USQo with molecular oxygen (Boveris and Cadenas, 1975; Ksenzenko et al., 1983; Muller et al., 2002; Borek et al., 2008; Dröse and Brandt, 2008; Sarewicz et al., 2010; Pagacz et al., 2021). However, trapping the intermediate USQo radical during the UQH2 oxidation and its detection by electron paramagnetic resonance (EPR) has been proven difficult and obtained results have often been disputable or contradictory (de Vries et al., 1981; Jünemann et al., 1998; Cape et al., 2006; Cape et al., 2007; Zhang et al., 2007; Zhu et al., 2007; Sarewicz et al., 2013; Vennam et al., 2013; Victoria et al., 2013; Pietras et al., 2016; Crofts et al., 2017; Sarewicz et al., 2017; Sarewicz et al., 2018; Bujnowicz et al., 2019). Additionally, the inability to generate USQo in the equilibrium redox titrations (Takamiya and Dutton, 1979; Sarewicz et al., 2018) implicated a concept of a high instability of the semiquinone and its reactivity was proposed to be a reason for superoxide production by Cytbc1.
Detection of a small amount of USQo under nonequilibrium conditions in isolated, antimycin-inhibited Cytbc1 has been reported in a few studies (Cape et al., 2007; Zhang et al., 2007; Vennam et al., 2013; Victoria et al., 2013). The presence of USQo in Cytbc1 samples was assigned to a stigmatellin-sensitive, X-band EPR signal which is typical of an organic radical with a nearly isotropic g value ∼2.0 (Cape et al., 2007; Vennam et al., 2013). Although experimental evidences were not presented, generation of this signal was assumed to be associated with the oxidation of UQH2 by the 2Fe2S cluster under conditions in which the resulting USQo was unable to donate electron to heme bL as this heme, in antimycin-inhibited enzyme, was expected to be in the reduced state after two consecutive electron transfers from the Qo site (Cape et al., 2007; Crofts et al., 2017).
Our previous work reported an unusual EPR signal with an average g value less than 2, which originated from the Qo site of the antimycin-inhibited Cytbc1, isolated from Rhodobacter capsulatus (Sarewicz et al., 2013; Sarewicz et al., 2017; Bujnowicz et al., 2019). This signal was detected only when the Qo site was able to catalyze the UQH2 oxidation and the cytochrome c reduction before the system reached equilibrium. The signal was not present in the samples containing specific inhibitors of the Qo site (stigmatellin, myxothiazol, famoxadone, azoxystrobin, kresoxim-methyl, etc.), nor in the mutants with disabled Qo site (like, for example, cytb:G158W). Simulations of the EPR spectra suggested that this signal results from the spin-spin exchange interactions between USQo and the reduced 2Fe2S (it is thus referred to as SQo-2Fe2S) with approximated J constant ∼3.6 GHz (Sarewicz et al., 2013). This suggestion was further supported by the observations that the g values of the SQo-2Fe2S state were frequency-dependent—they changed upon increasing the microwave frequency from X-band ∼9.4 to Q-band ∼33.5 GHz. At X-band, the most prominent “derivative-shaped” transition of SQo-2Fe2S in Cytbc1 was detected at g = ∼1.94 and was found to shift to ∼1.96 at Q-band. Remarkably, the SQo-2Fe2S signal was also detected in isolated but non-inhibited spinach cytochrome b6f during the oxidation of synthetic decylplastoquinol and the reduction of plastocyanin (Sarewicz et al., 2017).
Our subsequent work revealed that the SQo-2Fe2S signal can be generated in antimycin-supplemented native chromatophore membranes isolated from R. capsulatus, in which the reactions at the Qo site are triggered by the photosynthetic reaction center (RC) (Sarewicz et al., 2018). In these experiments, the characteristic transitions of the SQo-2Fe2S state with a readily detectable maximum in continuous wave X-band spectra at g = 1.95 were detected during the redox-titration of chromatophores in relatively narrow ranges of the external redox potential (Eh). However, the signal appears only when the chromatophores are illuminated by continuous light during the titrations, which keeps the RC activated to supply substrates for the Qo site. Similar titrations performed in dark, i.e. without activation of RC (equilibrium conditions), do not lead to the generation of SQo-2Fe2S. Clearly, in membranes, similarly to isolated Cytbc1, the SQo-2Fe2S state corresponds to one of the states of the enzyme that can be populated to detectable levels only under nonequilibrium conditions.
One of the most important observations regarding the enzymatic state of Rhodobacter capsulatus Cytbc1 is that the SQo-2Fe2S state is detected in samples in which heme bL remains oxidized (Sarewicz et al., 2013; Sarewicz et al., 2018). This suggests that in those experiments, the reaction sequence leading to the formation of the SQo-2Fe2S state begins with the electron transfer from the reduced heme bL to the UQ molecule bound at the Qo site. The resulting SQo then interacts with the reduced 2Fe2S that is present at the Qo site. However, a direct evidence supporting this mechanism has not yet been provided. Furthermore, the SQo can, in principle, also be formed in another reaction: electron transfer from UQH2 to oxidized 2Fe2S, which would also result in the formation of the SQo-2Fe2S state if the subsequent electron transfer from SQo to heme bL is (somehow) blocked. In this work, we addressed this issue with a series of experiments performed with the histidine ligand mutant of R. capsulatus in which the heme bL was inactivated. The mutation of choice was patterned after the previous study with closely related Rhodobacter sphaeroides where replacing the histidine ligand with asparagine (cytb:H198N) was found to result in a loss of spectral properties of heme bL typical of WT Cytbc1 and in a complete loss of the enzymatic activity (Yang et al., 2008). We found that in R. capsulatus the H198N mutation converted the low-spin heme bL into the high-spin heme unable to transfer electron from SQo to heme bH, and such conditions were tested for the efficiency of SQo-2Fe2S generation in chromatophores in dark or illuminated by light. The tests failed to detect the spin-spin–coupled SQo-2Fe2S state in H198N even in chromatophores activated by light, which we considered an indication that electron transfer from heme bL to the UQ molecule at the Qo site is mainly responsible for creating conditions that ultimately lead to the formation of R. capsulatus.
Materials and Methods
Construction of the H198N Mutant
The H198N mutant of Rhodobacter capsulatus containing the H198N mutation in cytochrome b subunit was generated using the genetic system described previously (Atta-Asafo-Adjei and Daldal, 1991). Mutation H198N was constructed by the QuikChange site-directed mutagenesis kit from Stratagene using pPET1-ST (Czapla et al., 2012b) as the template, and the mutagenic forward H198N-F: 5′-TTC TTC TCG CTG AAC TAT CTG CTG CCC TTC G -3′ and reverse H198N-R 5′-GGG CAG CAG ATA GTT CAG CGA GAA GAA GCG G -3′ oligonucleotides. After sequencing, the XmaI/SfuI fragment of pPET1-ST-H198N plasmid bearing the desired mutation and no other mutations was exchanged with its wild-type counterparts in pMTS1. This created the expression vector pMTS1-ST-H198N that was introduced into R. capsulatus MT-RBC1 strain (petABC-operon deletion background) via triparental crosses (Atta-Asafo-Adjei and Daldal, 1991). The presence of H198N mutation was confirmed by sequencing the plasmid DNA isolated from the mutated R. capsulatus strain (H198N mutant).
Isolation of Chromatophores
Chromatophores from R. capsulatus strains were prepared as described in detail by Sarewicz et al. (2018). Briefly, bacterial cultures were grown heterotrophically under semiaerobic conditions on the MPYE medium. After 48 h of growing the bacteria cells were centrifuged for 30 min at 6,641 g and the pellet resuspended in buffer containing 50 mM MOPS (pH 7), 100 mM KCl, and 1 mM EDTA, followed by the addition of protease inhibitors (benzamidine, PMSF, and 6-aminocaproic acid). The cells’ suspension was then passed two times through French Press® with pressure maintained between 11 and 15 MPa. Bacterial lysate was then centrifuged for 30 min at 24,104g and the obtained supernatant was then ultra-centrifuged at 244,062g for 1.5 h. The pellet containing isolated chromatophores was resuspended and homogenized in 50 mM bicine buffer (pH 8) containing 100 mM KCl and 1 mM EDTA.
Redox Titrations
The prepared chromatophores membranes containing WT and mutant H198N Cytbc1 were supplemented with antimycin at a final concentration of 0.15 mM and redox titrated under anaerobic conditions in dark. The solutions contained the following redox mediators: 50 µM 2,3,5,6-tetramethyl-1,4-phenylenediamine (DAD), 50 µM 1,2-naphthoquinone (NQ), 50 µM 1,2-naphthoquinone-4-sulfonate (NQS), 12.5 µM phenazine ethosulfate (PES), 25 µM phenazine methosulfate (PMS), 50 µM duroquinone (DQ), 12.5 µM indigotrisulfonate (ITS), 50 µM anthraquinone-2-sulfonic acid (AQS), 50 µM 2-hydroxy-1,4-naphthoquinone (HNQ), and 50 µM benzyl viologen (BV). The external redox potential (Eh) of the buffer was adjusted by injection of small volumes of sodium dithionite or potassium ferricyanide solutions. In dark equilibrium redox titration for each Eh about ∼200 µL of the chromatophore solution was collected and injected to an argon-flushed EPR quartz tube and then subsequently frozen in cold isopentane. The illuminated samples were prepared by inserting the EPR tube containing chromatophores to a home-built cylindrical chamber blocked at the bottom with a piston connected to an electromagnet. The chamber contained 43 diodes (OSRICA3131A by Optosupply), having a single emission band at 850 nm, that were built in the walls of the chamber. The tubes were illuminated for 5 s and then were released to cold isopentane bath by pulling the piston of the electromagnet. A freezing time was estimated to be ∼ 1–2 s. With this method a maximum level of SQo-2Fe2S is reached after 2 s of illumination, as determined for chromatophores containing antimycin-inhibited WT Cytbc1. A decay time constant of the SQo-2Fe2S state after switching off the light is about 1.8 s−1, which means that not more than ∼60% of the SQo-2Fe2S state can be trapped after 1–2 s of the deadtime associated with the freezing of the samples.
We note that isolation of chromatophores required special care to prevent loss of cytochrome c2 during the cell centrifugations with an excessive RCF. This is because the formation of the SQo-2Fe2S center in light-activated chromatophores was found to be sensitive to the coupling of RC with Cytbc1 by cytochrome c2. This effect is illustrated in Supplementary Figure S1, which demonstrates a decrease or even complete disappearance of the SQo-2Fe2S state in chromatophores having a shortage of cytochrome c2. A partial restoration of the signal is possible upon the addition of external horse cytochrome c to the chromatophores followed by the sonication of the samples.
Preparation of Isolated WT and H198N Enzymes
Cytbc1 complexes were isolated from detergent-solubilized chromatophores by Strep-tag® affinity chromatography, using the procedure described by Czapla et al. (2012a). Purity of the isolated proteins was checked with SDS–PAGE (Supplementary Figure S2). Cytbc1 samples were concentrated on centrifugal filter units and dialyzed against buffer composed of 50 mM Tris (pH 8), 100 mM NaCl, 1 mM EDTA, 20% glycerol (v/v), and 0.01% (m/m) DDM. The concentration of the isolated cytochrome bc1 was determined as described by Valkova-Valchanova et al. (1998).
EPR Measurements
All the measurements were performed using a Bruker ElexSys E580 spectrometer equipped with a super-HighQ (SQHE0511) resonator and ESR900 cryostat unit (Oxford Instruments). The chromatophore membranes were measured using the parameters described by Sarewicz et al. (2018). Briefly, the temperature of the measured samples was 20 K, microwave power of 2 mW and frequency 9.4 GHz, modulation amplitude of 14.36 G, sweep width of 926.8 G, sweep time of 40.96 s, and time constant of 81.92 ms. The number of scans was usually 1–3 depending on the signal-to-noise (S/N) ratio. The registered spectra were processed and analyzed using the Eleana EPR program.
The isolated WT and H198N Cytbc1 complexes were measured as follows. The X-band EPR spectra of low- and high-field transitions of hemes were measured at 10 K, with 2 mW microwave power, frequency of 9.4 GHz, and modulation amplitude of 6 G. The X-band EPR spectra of the 2Fe2S cluster was measured at 30 K with 0.65 mW microwave power and modulation amplitude of 16 G.
Results and Discussion
H198N Mutant Incorporates a High-Potential, High-Spin Heme bL
Cytbc1 contains the low-potential [Em,7 = ∼ −120 mV (Zhang et al., 2008)], low-spin (L-S) heme bL which is axially coordinated by two histidine side chains: H97 and H198 (R. capsulatus numbering of cytb subunit) (Berry et al., 2004). This heme is diamagnetic in the reduced state and, together with a higher potential (Em = +60 mV) heme bH, contributes to the absorbance band with a maximum at ∼560 nm of the fully reduced WT Cytbc1 (Figure 1A, left, black). A replacement of histidine 198 to non-liganding asparagine is expected to significantly alter properties of heme bL or even prevent its incorporation into the protein, as previously reported for the ligand mutants of hemes b in R. capsulatus and R. sphaeroides (Yang et al., 2008). Indeed, the optical spectrum of the H198N mutant in R. capsulatus constructed in this work (RcH198N) exhibits a significant decrease in the absorbance at ∼560 nm (Figure 1A, right, black), consistent with the previous observation on the analogical H198N mutant in Rhodobacter sphaeroides (RsH198N) (Yang et al., 2008). Such a decrease suggests that one of the L-S hemes b might be missing, as was concluded for the RsH198N mutant. However, along with the disappearance of the absorption band at ∼560 nm we note an additional absorption maximum at ∼590 nm present in both the ascorbate- and dithionite-reduced samples containing RcH198N. This band was not seen in WT and suggested that it is associated with a change in the heme ligand in the RcH198N mutant.
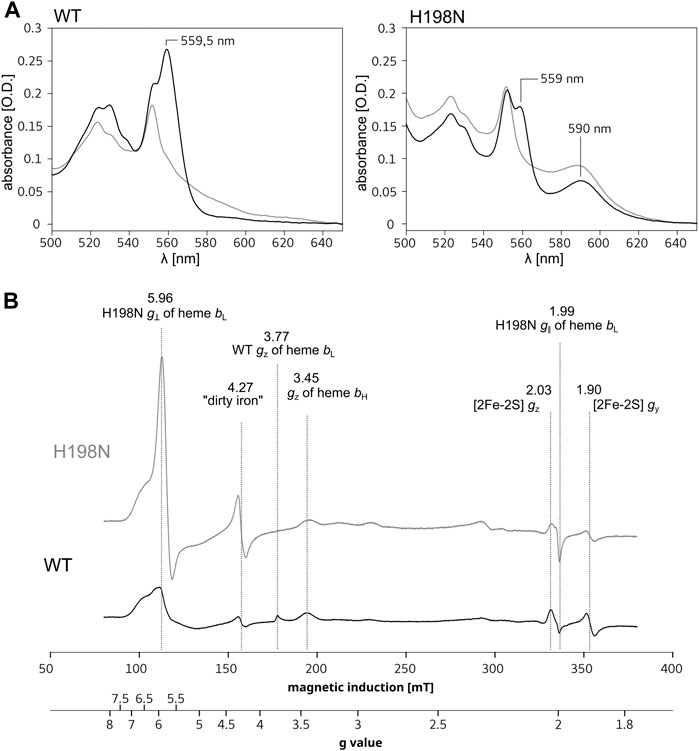
FIGURE 1. Comparison of spectra for isolated WT and H198N mutants of Cytbc1. (A) Optical spectra of WT (left) and H198N mutant (right) measured for ascorbate (gray) and dithionite-reduced (black) Cytbc1. Maximum at ∼560 nm originates from absorption of hemes b. Additional absorption band at ∼590 nm is likely to originate from heme bL with changed ligand in H198N mutant. (B) X-band EPR spectra of air-oxidized WT (black) and H198N mutant (gray) measured at 10 K. In the range between ∼70 and ∼160 mT the contribution from high-spin iron centers is detected. In the range between ∼160 and 200 mT, the gz transitions of HALS hemes b transitions are detected, while the reduced 2Fe2S contributes to transitions at the magnetic field above ∼320 mT.
Low-temperature X-band EPR spectra for isolated WT and the H198N mutant for air-oxidized samples are compared in Figure 1B. WT Cytbc1 shows the gz transitions typical of highly anisotropic low-spin (HALS) hemes at g = 3.77 and 3.45 (Figure 1B, black), originating from the oxidized, low-spin hemes bL and bH, respectively (Salerno, 1984; Finnegan et al., 1996). From those two transitions, only the g = 3.45 originating from heme bH was detected in H198N mutant (Figure 1B, gray), indicating that this heme was properly assembled into the protein core and retained its native bis–his axial ligation. The missing g = 3.77 transition was consistent with the expected lack of HALS heme bL with his–his axial ligation. Instead, a relatively intense transition at g = ∼5.96 and a much weaker at g = ∼2 were detected revealing that in RcH198N, the HALS heme bL was converted into an axial, high-spin (H-S) form. Given the fact that other centers contribute to the EPR signal in the region of g = 5.96, the heterogeneity, if any, of the spin state of heme bL in the mutant cannot be evaluated.
To get insights into the redox properties of hemes b in the H198N mutant, we analyzed the relaxation properties of a semiquinone formed at the Qi site, which, as reported recently, strongly depend on whether the electron preferentially stays on heme bH or heme bL, thus can be used to estimate the relative difference in equilibrium redox potentials between these hemes (Pintscher et al., 2018). The results, discussed in detail in SI (Supplementary Figures S3 and S4), revealed that heme bL in the mutant has a redox potential much more positive than the native HALS heme bH (Pintscher et al., 2018). Furthermore, the g = 5.96 transition disappeared when the RcH198N Cytbc1 was reduced by a synthetic substrate—decylubiquinol (Supplementary Figure S5), which suggests that heme bL in RcH198N mutant has a much higher redox midpoint potential than heme bL of the wild type.
H198N Mutant Binds Properly UQ to the Qo Site
The shape of the CW EPR spectra of the reduced 2Fe2S cluster in chromatophores is sensitive to the type of molecule that occupies the Qo site. In particular, the relatively narrow gx transition at 1.80 indicates that UQ is bound at this site and interacts with the cluster, whereas a more broad gx = 1.77 indicates the presence of UQH2 in the Qo site (Robertson et al., 1993; Ding et al., 1995; Cooley et al., 2004; Sharp et al., 1999). This change in gx transition is so characteristic that it was often used in evaluating the effects of mutations on the binding of ubiquinone molecules to the catalytic site, or in the redox titrations of ubiquinones in the membranes (Cooley et al., 2004; Osyczka et al., 2006). As shown in Figure 2A, the CW EPR spectra of the ascorbate-reduced 2Fe2S cluster in chromatophores containing WT and H198N Cytbc1 were similar and both exhibited clear gx = 1.80 transition when the Q pool was fully oxidized (at high Eh). Furthermore, reducing the Q pool, upon decreasing the external redox potential, changed the gx from 1.80 to ∼1.77 in both WT and the mutant protein (Figure 2B). These observations indicate that UQ and UQH2 can bind to the Qo site of the mutant in the same way as in WT [see references Cooley et al. (2004), Osyczka et al. (2006) for details]. This feature, along with the specifically altered redox properties of heme bL described earlier, made H198N an attractive mutant for the mechanistic analysis of the formation of the SQo-2Fe2S state.
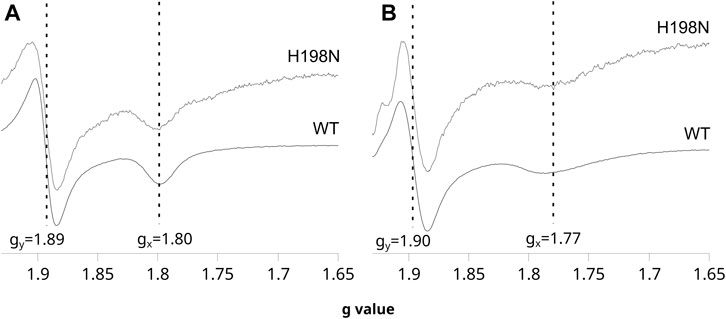
FIGURE 2. Sensitivity of the EPR spectra of the 2Fe2S cluster to changes in redox state of the Q pool for WT and the H198N mutant at pH 8. (A) The spectra measured for chromatophores poised at + 200 mV (Q pool oxidized) in dark. (B) The spectra measured for chromatophores poised at + 20 mV (Q pool half reduced). The vertical lines indicate the positions of gy and gx transitions of the 2Fe2S cluster, which shifts to higher and lower g values, respectively, upon the reduction of the Q pool. The spectra of the mutant were magnified ∼5 times to normalize the gy signals to the same level as in WT.
Controlling the Efficiency of SQo-2Fe2S Generation in Illuminated Chromatophores
In our previous studies we established a general methodology for monitoring the formation of the SQo-2Fe2S state in chromatophore membranes (Sarewicz et al., 2018). We found that this state can be detected by CW EPR during the redox titrations of chromatophores containing antimycin-inhibited Cytbc1 only under nonequilibrium conditions when the photosynthetic reaction center and allied Cytbc1 were kept activated by continuous light during the titration. The same experiments performed in dark (equilibrium conditions) do not lead to the formation of SQo-2Fe2S at detectable levels (Sarewicz et al., 2018).
Given that in these experiments, the formation of SQo-2Fe2S strongly depends on the activation level of RC, any comparative redox titrations of chromatophores illuminated by continuous light (nonequilibrium conditions) require careful controls to assure that conditions for the generation of the SQo-2Fe2S signal and its detection were equally met in each of the studied cases. We found that the most important factor influencing the efficiency of SQo-2Fe2S generation in chromatophores was the optical transparency for the light that activates RC. This effect is shown in Figure 3, which compares the two EPR spectra obtained for the concentrated (gray) and diluted (black) samples of the same batch of chromatophores (containing 40 and 20 μM cytochrome c1, respectively).
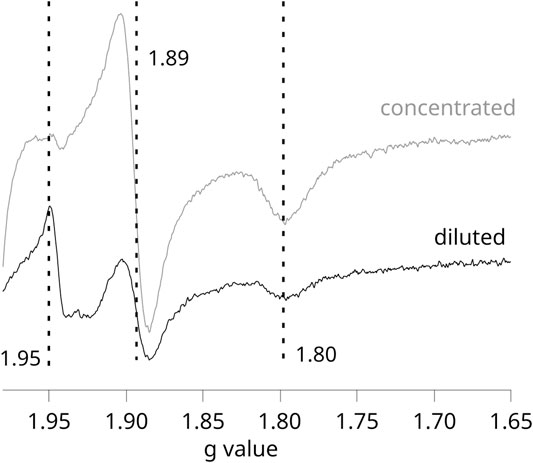
FIGURE 3. X-band EPR spectra measured for illuminated chromatophores containing WT Cytbc1 at pH 8 and Eh = +77 mV. Gray and black spectra were obtained for samples containing 40 and 20 μM, respectively. Dashed vertical lines show gy = 1.89 and gx = 1.80 transitions of 2Fe2S cluster. The maximum at 1.95 originates from SQo-2Fe2S.
This example emphasizes that the SQo-2Fe2S signal (g = 1.95) can be lost in the more concentrated sample despite improvement in the signal-to-noise ratio, and this is related to the fact that the activation of RC is much weaker due to increased opacity of the solution. The weaker light activation of RC in the more concentrated samples was also reflected in the measured UQ/UQH2 average redox midpoint potential, which, despite illumination of the sample, approached those detected in dark. Thus, the titrations of the SQo-2Fe2S signal on illuminated chromatophores required sufficiently diluted samples. However, after diluting the samples, a measurement of the redox state of hemes b by EPR is compromised due to the fact that the amplitudes of these hemes are relatively very weak. We found that at the concentrations of Cytbc1 at which hemes b became clearly detectable by EPR, the SQo-2Fe2S signal started to disappear due to increase of the opacity of the samples. Therefore, the experiments were limited to a relatively narrow range of Cytbc1 concentrations that allowed efficient activation of RC and, at the same time, a detection of relatively good-quality EPR spectra.
Light-Activated RC Interacts With H198N in Chromatophores and Imposes a Shift in Apparent Midpoint Potential of the 2Fe2S Cluster
In view of the observation that the efficiency of SQo-2Fe2S generation in the light-activated WT chromatophores strongly depends on the optical transparency of the samples (see the paragraph above), it became apparent that prior to comparing WT and the H198N mutant, it was necessary to verify that RC in both WT and the H198N mutant containing chromatophores were sufficiently well activated by light and, consequently, both the systems were pushed out of equilibrium. Furthermore, it was necessary to apply a verification method that was independent of the analysis of the SQo-2Fe2S signal. One such possibility relates to the fact that light activation affects the redox state of the membranous Q pool. Changes in the redox state of the Q pool can be monitored by measuring a progressive decrease in the amplitude of the normalized gx = 1.80 transition of the 2Fe2S cluster as the Q pool becomes progressively reduced (Sarewicz et al., 2018). Illumination leads to the electron transfer from the cytochrome c2 pool to the Q pool and the extent of the transfer is reflected in a shift in the Nernst curve of UQ/UQH2 couple toward higher values. Thus, the shift confirms the activation level of RC.
Figure 4A shows that in chromatophores containing WT Cytbc1 at a concentration equal to 20 μM cytochrome c1 (corresponding to black spectrum of Figure 3), the obtained equilibrium redox potential of UQ/UQH2 couple in dark shifts by approximately +60 mV upon the activation of RC by light. Clearly, the optical transparency of the chromatophores in this case was enough to allow sufficient RC activation. In a more concentrated sample (corresponding to the gray spectrum of Figure 3) the measured UQ/UQH2 average redox midpoint potential was similar in light and in dark, and the lack of the shift (lack of activation of RC) was concomitant with the lack of the SQo-2Fe2S signal (data not shown).
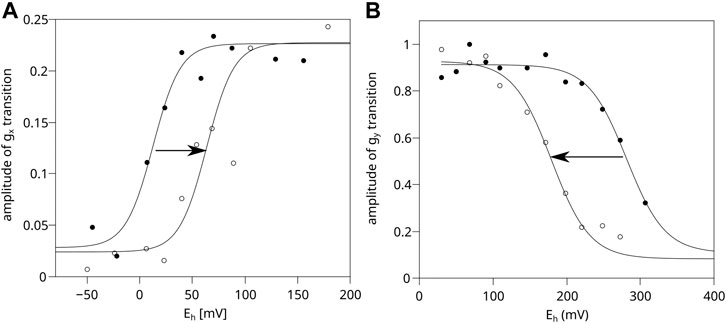
FIGURE 4. Comparison of efficiency of light activation of RC in chromatophores containing WT and H198N mutant of Cytbc1. (A) Amplitude of the normalized gx transition of the 2Fe2S cluster of WT Cytbc1 which is proportional to the amount of UQ in the Q pool in dark (closed circles) and in illuminated samples (open circles). The experiential data points were simulated using an appropriate Nernst equation with n = 2. (B) Amplitude of the gy transition of the 2Fe2S cluster measured for the illuminated chromatophores containing H198N mutant of Cytbc1 (open circles) and chromatophores in dark (closed circles).
In the case of the H198N mutant, the expression level of Cytbc1 was found to be lower by approximately 2–3 folds than WT (data not shown). Thus, after concentrating the H198N chromatophores to reach 20 μM of cytochrome c1, the samples became too opaque to allow efficient activation of RC. As a result, the illumination of the samples did not lead to a shift of the UQ/UQH2 Nernst curve toward a higher value (data not shown). Therefore, the chromatophores containing H198N must have been diluted to 10 μM of cytochrome c1 to reach the optical transparency similar to that of WT chromatophores. This, in turn, exerted a negative effect of the quality of the measured EPR spectra of 2Fe2S cluster, and the reliable measurement of gx transition was not possible, precluding the possibility of monitoring activation level by determining the shift in the redox state of Q pool.
Instead, we monitored the extent of 2Fe2S oxidation upon activation of RC by comparing the amplitude of the relatively strong gy transition of the 2Fe2S cluster, as a function of Eh for illuminated chromatophores with those measured in dark. Illumination of H198N chromatophores imposed a shift of the Nernst curve with the midpoint potential of the 2Fe2S cluster from approximately +300 mV (in dark) to approximately +180 mV (in light) (Figure 4B, dashed and solid lines, respectively). This shift was an expected effect if RC had been efficiently activated and able to oxidize the whole chain of high-potential cofactors connecting RC and Cytbc1 (i.e., cytochrome c2/c1 and 2Fe2S). We note that similar shift in the 2Fe2S cluster can be observed in WT chromatophores; however, Cytbc1 must be inhibited by myxothiazol (i.e., it is not observed in non-inhibited Cytbc1). This inhibitor binds at the Qo site and prevents oxidation of UQH2 at this site; however, it does not impair the movement of the 2Fe2S cluster nor its interaction with other components of the high-potential chains (Kim et al., 1998; Darrouzet et al., 2000; Esser et al., 2004; Osyczka et al., 2004; Sarewicz et al., 2009; Cieluch et al., 2010). Thus, the observed shift in H198N chromatophores not only confirms the activation of RC, but also indicates that the components of the high-potential chain in this mutant are functionally linked with RC. At the same time, these components remain decoupled from the low-potential chain of Cytbc1 because of the enzymatic incompetence of mutated heme bL (with the net effect similar to the presence of myxothiazol in the Qo site of the native enzyme). We note that the observed shift in the 2Fe2S cluster induced by light activation of RC is interesting itself, and it will be discussed elsewhere.
H198N Fails to Generate Detectable Amounts of SQo-2Fe2S Spin-Coupled State
Having established the conditions for meaningful comparison of WT and H198N chromatophores (see the previous paragraphs), we performed a series of redox titrations that screened for the SQo-2Fe2S state in the H198N mutant.
The results for WT chromatophores were consistent with the previous observations (Sarewicz et al., 2018) (Figure 5A). When titrations were performed in light at pH 8, the SQo-2Fe2S was detected within the range of Eh below +150 mV and above 0 mV (with the maximum amplitude at Eh ∼ +66 mV) (closed circles). The SQo-2Fe2S was not detected in this range of Eh when similar titrations were performed in dark (open circles).
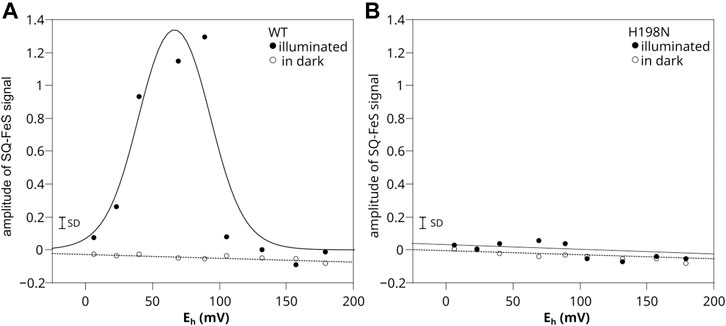
FIGURE 5. Comparison of SQo-2Fe2S generation in chromatophores containing WT (A) and H198N mutant (B) of Cytbc1. Amplitude of SQo-2Fe2S was measured for illuminated samples (closed circles) and titrated in dark (open circles). Optical transparencies were the same for A and B, and the concentrations of cytochrome c1 were 20 and 10 μM, respectively. The solid line in A represents the fit of fSQo-2Fe2S = fA/{1 + exp [0.039n (Eh − E1)] + exp [0.039n (E2 − Eh)]} function described in detail by Sarewicz et al. (2018). The fit yielded E1 = +92 ± 8 mV and E2 = +40 ± 7 mV for n = 2 and fA = 1.7 ± 0.4. The small bar denoted marked as SD shows the typical uncertainty of the amplitude reads and includes the baseline and the noise of the EPR spectrum. It does not include uncertainty related to changes in efficiency of SQo-2Fe2S generation dominated by variations in light activation and/or changes in the freezing time.
Remarkably, in titrations performed with chromatophores containing H198N instead of WT Cytbc1, the SQo-2Fe2S was not detected at all, regardless of whether the experiment was conducted in light or in dark (Figure 5B). A corollary of this result on the mechanisms of the formation of the SQo-2Fe2S state in antimycin-inhibited WT Cytbc1 is discussed below.
The H198N Mutation Implicates That Semireverse Mechanisms of SQo-2Fe2S Formation Are Dominant in WT Cytbc1
Let us consider two possible mechanisms of the formation of SQo-2Fe2S in antimycin-inhibited Cytbc1 shown schematically in Figure 6. In each of these two scenarios it is assumed that heme bH is already reduced after the oxidation of the first UQH2 molecule at the Qo site. This heme cannot be reoxidized by electron transfer to the Qi site, because this site is occupied by antimycin.
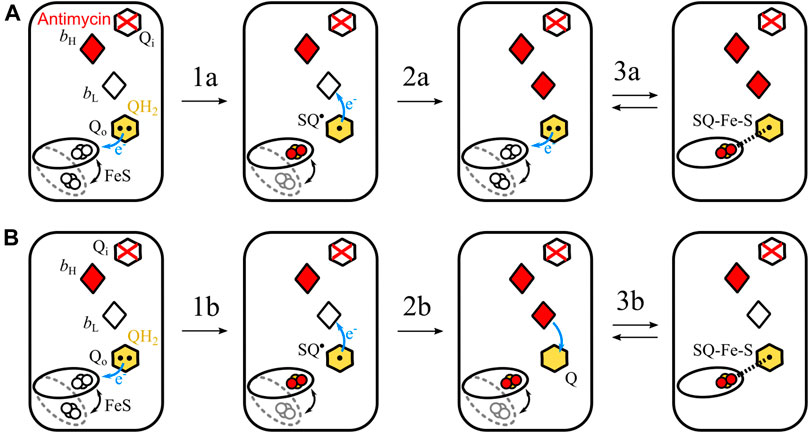
FIGURE 6. Simplified scheme of two possible reactions of SQo-2Fe2S formation. (A) The semiforward mechanism of UQH2 oxidation by the 2Fe2S cluster at the time when heme bL is unable to accept electron (because of being already reduced) from resulting SQo, leading to the creation of the SQo-2Fe2S state. (B) The semireverse mechanism assumes that the reduced heme bL donates electrons to UQ at the time when the reduced 2Fe2S is close to the Qo site. This leads to the creation of the SQo-2Fe2S state. According to the mechanism shown in A, the redox-active heme bL is not necessary, while the mechanism shown in B requires heme bL for the generation of the SQo-2Fe2S state.
The first mechanism assumes the sequence of reactions schematically shown in Figure 6A. The UQH2 binds at the Qo site where it undergoes one-electron oxidation by 2Fe2S forming an unstable semiquinone, SQo (transition 1a). This semiquinone immediately donates electron to heme bL and the resulting UQ molecule is then replaced with next UQH2 (transition 2a in Figure 6A). As the Qi site is blocked by antimycin, both hemes bH and bL cannot be reoxidized by electron transfer to the Qi site. Thus, SQo formed upon the oxidation of the second UQH2 molecule by the 2Fe2S cluster is unable to donate electron to heme bL and SQo and the reduced cluster forms spin-coupled state (transition 3a). Because this reaction corresponds to uncompleted oxidation of UQH2 in the forward direction of the operation of the Qo site, it is referred to as a semiforward reaction.
The second scenario assumes the sequence of reactions schematically shown in Figure 6B. The UQH2 molecule at the Qo site undergoes oxidation and two electrons are transferred to the 2Fe2S cluster and heme bL, leading to UQ bound at the Qo site. Since the electron transfer from heme bL to heme bH is not possible, the electron from heme bL is transferred back to the UQ molecule forming SQo. At the same time the reduced 2Fe2S cluster is still at the position close to the Qo site and is able to interact with SQo. As a result the SQo-2Fe2S state is generated. Because this reaction corresponds to uncompleted reduction of UQH2 in the reverse direction of the operation of the Qo site, it is referred to as a semireverse reaction.
The role of heme bL in the formation of SQo-2Fe2S is different for both the mechanisms. The semiforward reaction occurs when heme bL is unable to accept the electron from SQo; thus, the redox-active heme bL is not required for the formation of the SQo-2Fe2S state. Conversely, the semireverse reaction obligatorily requires presence of the redox-active heme bL to enable back electron transfer to the Qo site to form the SQo-2Fe2S.
In view of these considerations, H198N provided an attractive opportunity to test the contribution of the semireverse reaction to the formation of the SQo-2Fe2S state. The high-spin, high-potential heme bL in this mutant, once accepting the electron, will preferably stay reduced, and thus is unable to pass electron further to heme bH at a significant rate. This means that its redox activity required for the catalytic turnover is lost. At the same time, this heme is unable to transfer electron back to the Qo site. It follows that the H198N mutant provides conditions that should favor the formation of the SQo-2Fe2S if the semiforward reaction is a dominant mechanism, but should discourage the formation of SQo-2Fe2S if the semireverse reaction is dominant. Thus, if the SQo-2Fe2S state was detected in the H198N mutant of Cytbc1, then both semiforward and semireverse would be potentially responsible for the formation of this state. This was clearly not the case. The observation that the H198N mutation leads to complete disappearance of the SQo-2Fe2S signal under the tested conditions implies that the semireverse reaction can be considered the dominant mechanism for the formation of this state in WT Cytbc1. This mechanism involves electron transfer from heme bL back to the ubiquinone bound to the Qo site followed by its interaction with the reduced 2Fe2S cluster.
We note that this scenario prevails even if heme bL was able to pass electron to heme bH at a significant rate in H198N, which, given that antimycin was present at the Qi site, would eventually lead to a reduction of both hemes bL and bH. In this case, as long as heme bL was unable to transfer electron back to the Qo site, the SQo-2Fe2S signal would still be absent if occurrence of the semireverse reaction was required to generate it.
We also note that our conclusion requires an assumption that proton paths and proton transfers are not different in the H198N mutant and WT Cytbc1. In addition, one should bear in mind that there is still a possibility that the SQo-2Fe2S state can be generated in the H198N mutant but it was not detected in our experiments. This would mean that either the efficiency of its formation, i.e., contribution of the semiforward mechanism is much lower than the contribution of the semireverse mechanism, or the lifetime of the SQo-2Fe2S state under conditions of altered ligand of heme bL is much shorter than the lifetime of this state in WT. If so, then the decay of the SQo-2Fe2S state must have taken place within the deadtime of the method (i.e., before a sample became frozen, see the Materials and Methods). If this alternative explanation was true, it would immediately suggest a change in a mechanism of SQo-2Fe2S formation when properties of heme bL were altered in the mutant. In other words, the presence of oxidized heme bL in antimycin-inhibited Cytbc1 from R. capsulatus would greatly stabilize the SQo-2Fe2S state, whereas the reduced or disabled heme bL would eliminate or greatly destabilize this state.
Summary and Conclusion
In this work we constructed a mutant H198N that targeted one of the His ligands of heme bL. We found that this mutation leads to the conversion of HALS heme bL into the high-spin and high-potential form, which was indicated from the analysis of optical and EPR spectra of isolated enzymes. While H198N binds properly the substrates at the Qo site, the specific redox properties of heme bL in this mutant made the heme unable to transfer electrons to heme bH on a catalytically relevant timescale. Consequently, the enzymatic activity of Cytbc1 in H198N was lost. We then used this mutant to test the formation of the SQo-2Fe2S state under nonequilibrium conditions (light-activated chromatophores) when heme bL cannot perform normal electron-relay function between the Qo site and heme bH.
Careful analysis of various chromatophore samples established that the SQo-2Fe2S signal can be efficiently generated in illuminated chromatophores containing antimycin-inhibited WT Cytbc1, provided that the samples are optically transparent and cytochrome c2 molecules were not lost during the preparation of the chromatophores. Monitoring of the apparent shift in the midpoint potential of the UQ/UQH2 couple induced by light activation of RC provides convenient means for verifying the effectiveness of setting the nonequilibrium conditions in the analyzed samples, independently of the analysis of the SQo-2Fe2S signal. In H198N chromatophores, for which this method turned out to be unsuitable, imposing the nonequilibrium conditions was found to result in a large, negative shift of the apparent Em of the 2Fe2S cluster (a similar shift in the apparent redox midpoint potential of the 2Fe2S cluster was observed in WT Cytbc1 inhibited by myxothiazol). This provided an alternative method for verifying the effectiveness of light activation of RC and additionally confirmed their efficient coupling with Cytbc1via cytochrome c2 resulting in the oxidation of 2Fe2S.
Comparative redox titrations of light-induced WT and H198N chromatophores revealed that the H198N mutant failed to form detectable amounts of the SQo-2Fe2S state under the same conditions at which WT Cytbc1 generated a large amount of SQo-2Fe2S. This failure implicates that the semireverse mechanism that leads to the SQo-2Fe2S state is the dominant over the semiforward reaction in WT Cytbc1 (Sarewicz et al., 2010). According to the semireverse mechanism, the electron transfer from the low-spin heme bL to the UQ bound at the Qo site (semireverse reaction) results in the formation of the semiquinone that is spin-coupled to the reduced 2Fe2S, forming the SQo-2Fe2S state detectable by EPR.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
MS designed experiments, performed EPR measurements, contributed to construction of illumination chamber, analyzed data, and wrote the manuscript. SP isolated and analyzed spectral properties of the mutant protein, and participated in writing the manuscript. LB designed electronic parts of illumination chamber and performed EPR measurements. MW isolated chromatophores and performed redox titrations. AO discussed data and wrote the manuscript.
Funding
This work was funded by National Science Centre, Poland, Grant No. 2015/18/A/NZ1/00046 to A.O.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Ewelina Cieluch for preparing the H198N mutant of cytochrome bc1. We would like to thank Dr. Robert Ekiert for SDS page analysis of isolated proteins.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.658877/full#supplementary-material
Abbreviations
Cytbc1, cytochrome bc1; Q, quinone (general); QH2, quinol (general); UQ, ubiquinone; UQH2, ubihydroquinone; RC, reaction center; 2Fe2S, [2Fe-2S] Rieske iron–sulfur cluster; SQo, semiquinone at the Qo site; SQo-2Fe2S, semiquinone spin-coupled to the reduced 2Fe2S.
References
Al-Attar, S., and de Vries, S. (2013). Energy Transduction by Respiratory Metallo-Enzymes: From Molecular Mechanism to Cell Physiology. Coord. Chem. Rev. 257, 64–80. doi:10.1016/j.ccr.2012.05.022
Atta-Asafo-Adjei, E., and Daldal, F. (1991). Size of the Amino Acid Side Chain at Position 158 of Cytochrome B Is Critical for an Active Cytochrome Bc1 Complex and for Photosynthetic Growth of Rhodobacter Capsulatus. Proc. Natl. Acad. Sci. 88, 492–496. doi:10.1073/pnas.88.2.492
Baniulis, D., Yamashita, E., Zhang, H., Hasan, S. S., and Cramer, W. A. (2008). Structure-Function of the Cytochrome b6f Complex. Photochem. Photobiol. 84, 1349–1358. doi:10.1111/j.1751-1097.2008.00444.x
Berry, E. A., Guergova-Kuras, M., Huang, L.-S., and Crofts, A. R. (2000). Structure and Function of CytochromebcComplexes. Annu. Rev. Biochem. 69, 1005–1075. doi:10.1146/annurev.biochem.69.1.1005
Berry, E. A., Huang, L.-S., Saechao, L. K., Pon, N. G., Valkova-Valchanova, M., and Daldal, F. (2004). X-Ray Structure of Rhodobacter Capsulatus Cytochrome Bc1: Comparison with its Mitochondrial and Chloroplast Counterparts. Photosynth. Res. 81, 251–275. doi:10.1023/b:pres.0000036888.18223.0e
Bertini, I., Cavallaro, G., and Rosato, A. (2006). Cytochrome C: Occurrence and Functions. Chem. Rev. 106, 90–115. doi:10.1021/cr050241v
Borek, A., Sarewicz, M., and Osyczka, A. (2008). Movement of the Iron−Sulfur Head Domain of Cytochromebc1Transiently Opens the Catalytic QoSite for Reaction with Oxygen. Biochemistry 47, 12365–12370. doi:10.1021/bi801207f
Boveris, A., and Cadenas, E. (1975). Mitochondrial Production of Superoxide Anions and its Relationship to the Antimycin Insensitive Respiration. FEBS Lett. 54, 311–314. doi:10.1016/0014-5793(75)80928-8
Bujnowicz, Ł., Borek, A., Kuleta, P., Sarewicz, M., Osyczka, A., and Osyczka, A. (2019). Suppression of Superoxide Production by a Spin‐spin Coupling between Semiquinone and the Rieske Cluster in Cytochrome bc1. FEBS Lett. 593, 3–12. doi:10.1002/1873-3468.13296
Cape, J. L., Bowman, M. K., and Kramer, D. M. (2007). A Semiquinone Intermediate Generated at the Qo Site of the Cytochrome bc1 Complex: Importance for the Q-Cycle and Superoxide Production. Proc. Natl. Acad. Sci. 104, 7887–7892. doi:10.1073/pnas.0702621104
Cape, J. L., Bowman, M. K., and Kramer, D. M. (2006). Understanding the Cytochrome bc Complexes by what They Don't Do. The Q-Cycle at 30. Trends Plant Sci. 11, 46–55. doi:10.1016/j.tplants.2005.11.007
Cieluch, E., Pietryga, K., Sarewicz, M., and Osyczka, A. (2010). Visualizing Changes in Electron Distribution in Coupled Chains of Cytochrome bc(1) by Modifying Barrier for Electron Transfer between the FeS Cluster and Heme c(1). Biochim. Biophys. Acta (Bba) - Bioenerg. 1797, 296–303. doi:10.1016/j.bbabio.2009.11.003
Cooley, J. W., Roberts, A. G., Bowman, M. K., Kramer, D. M., and Daldal, F. (2004). The Raised Midpoint Potential of the [2Fe2S] Cluster of Cytochrome bc1 is Mediated by Both the QoSite Occupants and the Head Domain Position of the Fe−S Protein Subunit. Biochemistry 43, 2217–2227. doi:10.1021/bi035938u
Cramer, W. A., and Hasan, S. S. (2016). “Structure-Function of the Cytochrome B 6 F Lipoprotein Complex,” in Cytochrome Complexes Evol. Struct. Energy Transduction, Signal. Editors W. A. Cramer, and T. Kallas (Dordrecht: Springer), 177–207. doi:10.1007/978-94-017-7481-9_9
Crofts, A. R., and Berry, E. A. (1998). Structure and Function of the Cytochrome Bc1 Complex of Mitochondria and Photosynthetic Bacteria. Curr. Opin. Struct. Biol. 8, 501–509. doi:10.1016/s0959-440x(98)80129-2
Crofts, A. R., Hong, S., Wilson, C., Burton, R., Victoria, D., Harrison, C., et al. (2013). The Mechanism of Ubihydroquinone Oxidation at the Qo-Site of the Cytochrome bc1 Complex. Biochim. Biophys. Acta (Bba) - Bioenerg. 1827, 1362–1377. doi:10.1016/j.bbabio.2013.01.009
Crofts, A. R., Meinhardt, S. W., Jones, K. R., and Snozzi, M. (1983). The Role of the Quinone Pool in the Cyclic Electron-Transfer Chain of Rhodopseudomonas Sphaeroides A Modified Q-Cycle Mechanism. Biochim. Biophys. Acta (Bba) - Bioenerg. 723, 202–218. doi:10.1016/0005-2728(83)90120-2
Crofts, A. R., Rose, S. W., Burton, R. L., Desai, A. V., Kenis, P. J. A., and Dikanov, S. A. (2017). The Q-Cycle Mechanism of the bc1 Complex: A Biologist's Perspective on Atomistic Studies. J. Phys. Chem. B 121, 3701–3717. doi:10.1021/acs.jpcb.6b10524
Crofts, A. R. (2004). The Cytochromebc1Complex: Function in the Context of Structure. Annu. Rev. Physiol. 66, 689–733. doi:10.1146/annurev.physiol.66.032102.150251
Czapla, M., Borek, A., Sarewicz, M., and Osyczka, A. (2012a). Enzymatic Activities of Isolated Cytochrome Bc1-like Complexes Containing Fused Cytochrome B Subunits with Asymmetrically Inactivated Segments of Electron Transfer Chains. Biochemistry 51, 829–835. doi:10.1021/bi2016316
Czapla, M., Borek, A., Sarewicz, M., and Osyczka, A. (2012b). Fusing Two Cytochromes B of Rhodobacter Capsulatus Cytochrome Bc 1 Using Various Linkers Defines a Set of Protein Templates for Asymmetric Mutagenesis. Protein Eng. Des. Sel. 25. doi:10.1093/protein/gzr055
Darrouzet, E., Cooley, J. W., and Daldal, F. (2004). The Cytochrome bc(1) Complex and its Homologue the b(6) f Complex: Similarities and Differences. Photosynth. Res. 79, 25–44. doi:10.1023/b:pres.0000011926.47778.4e
Darrouzet, E., Valkova-Valchanova, M., Moser, C. C., Dutton, P. L., and Daldal, F. (2000). Uncovering the [2Fe2S] Domain Movement in Cytochrome Bc1 and its Implications for Energy Conversion. Proc. Natl. Acad. Sci. 97, 4567–4572. doi:10.1073/pnas.97.9.4567
de Vries, S., Albracht, S. P., Berden, J. A., and Slater, E. C. (1981). A New Species of Bound Ubisemiquinone Anion in QH2: Cytochrome C Oxidoreductase. J. Biol. Chem. 256, 11996–11998. doi:10.1016/s0021-9258(18)43222-x
Dikanov, S. A., Samoilova, R. I., Kolling, D. R. J., Holland, J. T., and Crofts, A. R. (2004). Hydrogen Bonds Involved in Binding the Qi-Site Semiquinone in the Bc1 Complex, Identified through Deuterium Exchange Using Pulsed EPR. J. Biol. Chem. 279, 15814–15823. doi:10.1074/jbc.m313417200
Ding, H., Moser, C. C., Robertson, D. E., Tokito, M. K., Daldal, F., and Dutton, P. L. (1995). Ubiquinone Pair in the Qo Site Central to the Primary Energy Conversion Reactions of Cytochrome Bc1 Complex. Biochemistry 34, 15979–15996. doi:10.1021/bi00049a012
Dröse, S., and Brandt, U. (2008). The Mechanism of Mitochondrial Superoxide Production by the Cytochrome Bc1 Complex. J. Biol. Chem. 283, 21649–21654. doi:10.1074/jbc.m803236200
Esser, L., Quinn, B., Li, Y.-F., Zhang, M., Elberry, M., Yu, L., et al. (2004). Crystallographic Studies of Quinol Oxidation Site Inhibitors: A Modified Classification of Inhibitors for the Cytochrome Bc 1 Complex. J. Mol. Biol. 341, 281–302. doi:10.1016/j.jmb.2004.05.065
Finnegan, M. G., Knaff, D. B., Qin, H., Gray, K. A., Daldal, F., Yu, L., et al. (1996). Axial Heme Ligation in the Cytochrome Bc1 Complexes of Mitochondrial and Photosynthetic Membranes. A Near-Infrared Magnetic Circular Dichroism and Electron Paramagnetic Resonance Study. Biochim. Biophys. Acta (Bba) - Bioenerg. 1274, 9–20. doi:10.1016/0005-2728(95)00155-7
Hasan, S. S., Yamashita, E., and Cramer, W. A. (2013). Transmembrane Signaling and Assembly of the Cytochrome B6f-Lipidic Charge Transfer Complex. Biochim. Biophys. Acta (Bba) - Bioenerg. 1827, 1295–1308. doi:10.1016/j.bbabio.2013.03.002
Hunte, C., Palsdottir, H., and Trumpower, B. L. (2003). Protonmotive Pathways and Mechanisms in the Cytochrome Bc 1 Complex. FEBS Lett. 545, 39–46. doi:10.1016/s0014-5793(03)00391-0
Jünemann, S., Heathcote, P., and Rich, P. R. (1998). On the Mechanism of Quinol Oxidation in Thebc1 Complex. J. Biol. Chem. 273, 21603–21607. doi:10.1074/jbc.273.34.21603
Kim, H., Xia, D., Yu, C.-A., Xia, J.-Z., Kachurin, A. M., Zhang, L., et al. (1998). Inhibitor Binding Changes Domain Mobility in the Iron-Sulfur Protein of the Mitochondrial Bc1 Complex from Bovine Heart. Proc. Natl. Acad. Sci. 95, 8026–8033. doi:10.1073/pnas.95.14.8026
Ksenzenko, M., Konstantinov, A. A., Khomutov, G. B., Tikhonov, A. N., and Ruuge, E. K. (1983). Effect of Electron Transfer Inhibitors on Superoxide Generation in the Cytochrome Bc 1 Site of the Mitochondrial Respiratory Chain. FEBS Lett. 155, 19–24. doi:10.1016/0014-5793(83)80200-2
Mitchell, P. (1975). The Protonmotive Q Cycle: A General Formulation. FEBS Lett. 59, 137–139. doi:10.1016/0014-5793(75)80359-0
Muller, F., Crofts, A. R., and Kramer, D. M. (2002). Multiple Q-Cycle Bypass Reactions at the QoSite of the Cytochromebc1Complex†. Biochemistry 41, 7866–7874. doi:10.1021/bi025581e
Osyczka, A., Moser, C. C., Daldal, F., and Dutton, P. L. (2004). Reversible Redox Energy Coupling in Electron Transfer Chains. Nature 427, 607–612. doi:10.1038/nature02242
Osyczka, A., Moser, C. C., and Dutton, P. L. (2005). Fixing the Q Cycle. Trends Biochem. Sci. 30, 176–182. doi:10.1016/j.tibs.2005.02.001
Osyczka, A., Zhang, H., Mathé, C., Rich, P. R., Moser, C. C., and Dutton, P. L. (2006). Role of the PEWY Glutamate in Hydroquinone−Quinone Oxidation−Reduction Catalysis in the Qo Site of Cytochrome Bc1. Biochemistry 45, 10492–10503. doi:10.1021/bi060013a
Pagacz, J., Broniec, A., Wolska, M., Osyczka, A., and Borek, A. (2021). ROS Signaling Capacity of Cytochrome bc1: Opposing Effects of Adaptive and Pathogenic Mitochondrial Mutations. Free Radic. Biol. Med. 163, 243–254. doi:10.1016/j.freeradbiomed.2020.12.019
Pietras, R., Sarewicz, M., and Osyczka, A. (2016). Distinct Properties of Semiquinone Species Detected at the Ubiquinol Oxidation Qo Site of Cytochrome Bc1 and Their Mechanistic Implications. J. R. Soc. Interf. 13, 1–11. doi:10.1098/rsif.2016.0133
Pintscher, S., Pietras, R., Sarewicz, M., and Osyczka, A. (2018). Electron Sweep across Four B-Hemes of Cytochrome Bc1 Revealed by Unusual Paramagnetic Properties of the Qi Semiquinone Intermediate. Biochim. Biophys. Acta (Bba) - Bioenerg. 1859, 459–469. doi:10.1016/j.bbabio.2018.03.010
Pintscher, S., Wójcik-Augustyn, A., Sarewicz, M., and Osyczka, A. (2020). Charge Polarization Imposed by the Binding Site Facilitates Enzymatic Redox Reactions of Quinone. Biochim. Biophys. Acta (Bba) - Bioenerg. 1861, 148216. doi:10.1016/j.bbabio.2020.148216
Robertson, D. E., Ding, H., Chelminski, P. R., Slaughter, C., Hsu, J., Moomaw, C., et al. (1993). Hydroubiquinone-Cytochrome C2 Oxidoreductase from Rhodobacter Capsulatus: Definition of a Minimal, Functional Isolated Preparation. Biochemistry 32, 1310–1317. doi:10.1021/bi00056a016
Robertson, D. E., Prince, R. C., Bowyer, J. R., Matsuura, K., Dutton, P. L., and Ohnishi, T. (1984). Thermodynamic Properties of the Semiquinone and its Binding Site in the Ubiquinol-Cytochrome C (C2) Oxidoreductase of Respiratory and Photosynthetic Systems. J. Biol. Chem. 259, 1758–1763. doi:10.1016/s0021-9258(17)43472-7
Salerno, J. C. (1984). Cytochrome Electron Spin Resonance Line Shapes, Ligand Fields, and Components Stoichiometry in Ubiquinol-Cytochrome C Oxidoreductase. J. Biol. Chem. 259, 2331–2336. doi:10.1016/s0021-9258(17)43356-4
Sarewicz, M., Borek, A., Cieluch, E., Świerczek, M., and Osyczka, A. (2010). Discrimination between Two Possible Reaction Sequences that Create Potential Risk of Generation of Deleterious Radicals by Cytochrome Bc1. Biochim. Biophys. Acta (Bba) - Bioenerg. 1797, 1820–1827. doi:10.1016/j.bbabio.2010.07.005
Sarewicz, M., Bujnowicz, Ł., Bhaduri, S., Singh, S. K., Cramer, W. A., and Osyczka, A. (2017). Metastable Radical State, Nonreactive with Oxygen, Is Inherent to Catalysis by Respiratory and Photosynthetic Cytochromes Bc1/b6f. Proc. Natl. Acad. Sci. USA 114, 1323–1328. doi:10.1073/pnas.1618840114
Sarewicz, M., Bujnowicz, Ł., and Osyczka, A. (2018). Generation of semiquinone-[2Fe-2S]+ Spin-Coupled Center at the Qo Site of Cytochrome Bc1 in Redox-Poised, Illuminated Photosynthetic Membranes from Rhodobacter Capsulatus. Biochim. Biophys. Acta (Bba) - Bioenerg. 1859, 145–153. doi:10.1016/j.bbabio.2017.11.006
Sarewicz, M., Dutka, M., Froncisz, W., and Osyczka, A. (2009). Magnetic Interactions Sense Changes in Distance between HemebLand the Iron−Sulfur Cluster in Cytochromebc1. Biochemistry 48, 5708–5720. doi:10.1021/bi900511b
Sarewicz, M., Dutka, M., Pintscher, S., and Osyczka, A. (2013). Triplet State of the Semiquinone-Rieske Cluster as an Intermediate of Electronic Bifurcation Catalyzed by Cytochrome Bc1. Biochemistry 52, 6388–6395. doi:10.1021/bi400624m
Sarewicz, M., and Osyczka, A. (2015). Electronic Connection between the Quinone and Cytochrome C Redox Pools and its Role in Regulation of Mitochondrial Electron Transport and Redox Signaling. Physiol. Rev. 95, 219–243. doi:10.1152/physrev.00006.2014
Sarewicz, M., Pintscher, S., Pietras, R., Borek, A., Bujnowicz, Ł., Hanke, G., et al. (2021). Catalytic Reactions and Energy Conservation in the Cytochrome Bc 1 and B 6 F Complexes of Energy-Transducing Membranes. Chem. Rev. 121, 2020–2108. doi:10.1021/acs.chemrev.0c00712
Sharp, R. E., Palmitessa, A., Gibney, B. R., White, J. L., Moser, C. C., Daldal, F., et al. (1999). Ubiquinone Binding Capacity of theRhodobacter capsulatusCytochromebc1Complex: Effect of Diphenylamine, a Weak Binding QOSite Inhibitor. Biochemistry 38, 3440–3446. doi:10.1021/bi982639+
Takamiya, K.-I., and Dutton, P. L. (1979). Ubiquinone in Rhodopseudomonas Sphaeroides. Some Thermodynamic Properties. Biochim. Biophys. Acta (Bba) - Bioenerg. 546, 1–16. doi:10.1016/0005-2728(79)90166-x
Valkova-Valchanova, M. B., Saribas, A. S., Gibney, B. R., Dutton, P. L., and Daldal, F. (1998). Isolation and Characterization of a Two-Subunit Cytochromeb−c1Subcomplex fromRhodobacter Capsulatusand Reconstitution of its Ubihydroquinone Oxidation (Qo) Site with Purified Fe-S Protein Subunit. Biochemistry 37, 16242–16251. doi:10.1021/bi981651z
Vennam, P. R., Fisher, N., Krzyaniak, M. D., Kramer, D. M., and Bowman, M. K. (2013). A Caged, Destabilized, Free Radical Intermediate in the Q-Cycle. ChemBioChem. 14, 1745–1753. doi:10.1002/cbic.201300265
Victoria, D., Burton, R., and Crofts, A. R. (2013). Role of the -PEWY-Glutamate in Catalysis at the Qo-Site of the Cyt Bc1 Complex. Biochim. Biophys. Acta (Bba) - Bioenerg. 1827, 365–386. doi:10.1016/j.bbabio.2012.10.012
Xia, D., Yu, C.-A., Kim, H., Xia, J.-Z., Kachurin, A. M., Zhang, L., et al. (1997). Crystal Structure of the Cytochrome bc1Complex from Bovine Heart Mitochondria. Science 277, 60–66. doi:10.1126/science.277.5322.60
Yang, S., Ma, H.-W., Yu, L., and Yu, C.-A. (2008). On the Mechanism of Quinol Oxidation at the QP Site in the Cytochrome Bc1 Complex. J. Biol. Chem. 283, 28767–28776. doi:10.1074/jbc.m803013200
Zhang, H., Chobot, S. E., Osyczka, A., Wraight, C. A., Dutton, P. L., and Moser, C. C. (2008). Quinone and Non-quinone Redox Couples in Complex III. J. Bioenerg. Biomembr. 40, 493–499. doi:10.1007/s10863-008-9174-6
Zhang, H., Osyczka, A., Dutton, P. L., and Moser, C. C. (2007). Exposing the Complex III Qo Semiquinone Radical. Biochim. Biophys. Acta (Bba) - Bioenerg. 1767, 883–887. doi:10.1016/j.bbabio.2007.04.004
Keywords: cytochrome bc1 (complex III), electron transfer, quinol oxidation, semiquinone, electron paramagnetic resonance
Citation: Sarewicz M, Pintscher S, Bujnowicz Ł, Wolska M and Artur Osyczka (2021) The High-Spin Heme bL Mutant Exposes Dominant Reaction Leading to the Formation of the Semiquinone Spin-Coupled to the [2Fe-2S]+ Cluster at the Qo Site of Rhodobacter capsulatus Cytochrome bc1. Front. Chem. 9:658877. doi: 10.3389/fchem.2021.658877
Received: 26 January 2021; Accepted: 19 April 2021;
Published: 07 May 2021.
Edited by:
Petra Hellwig, Université de Strasbourg, FranceReviewed by:
Frauke Baymann, CNRS UMR7281 Bioénergétique et Ingénierie des Protéines, FranceDi Xia, National Institutes of Health (NIH), United States
Copyright © 2021 Sarewicz, Pintscher, Bujnowicz, Wolska and Artur Osyczka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Artur Osyczka, YXJ0dXIub3N5Y3prYUB1ai5lZHUucGw=
†These authors have contributed equally to this work