 Qian Xu1†
Qian Xu1† Zhe-Shan Quan
Zhe-Shan Quan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Chem., 29 April 2021
Sec. Medicinal and Pharmaceutical Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.650569
Natural products and their derivatives are important sources for drug discovery; however, they usually have poor solubility and low activity and require structural modification. Amino acids are highly soluble in water and have a wide range of activities. The introduction of amino acids into natural products is expected to improve the performance of these products and minimize their adverse effects. Therefore, this review summarizes the application of amino acids in the structural modification of natural products and provides a theoretical basis for the structural modification of natural products in the future. The articles were divided into six types based on the backbone structures of the natural products, and the related applications of amino acids in the structural modification of natural products were discussed in detail.
Amino acids are the basic units of proteins and the primary substance supporting biological life activities. They are closely related to the activities of a living body, playing very important roles. They are essential organic molecules in any organism. The amino acid chemical structure contains an amino group (-NH2), a carboxylic acid group (-COOH), and a unique side chain on the carbon that connects the amino group to the carboxyl group. Amino acids have many special physiological functions, playing roles in protein synthesis, metabolism, body development, osmotic pressure stability, and neurotransmission (Wu, 2009).
In addition to these functions, amino acids are also widely used in the food industry. For example, the main raw material of the sweetener aspartame is L-phenylalanine (Liu et al., 2019). Naturally occurring amino acids can be used to enhance the flavor of food and enhance nutrition (Ikeda, 2003). Additionally, amino acids are widely used in the medical and healthcare industries (Carubelli et al., 2015; Nitz et al., 2019). They also have a wide range of pharmacological activities, such as antitumor (Barnham et al., 1996), anti-HIV (Chen A. et al., 2006), and anti-fatigue (You et al., 2011) effects; in addition, they are used to cure chronic liver diseases (Kawaguchi et al., 2011).
The structures of amino acids are simple and diverse, and their pharmacological activities are extensive. These features are commonly employed in drug synthesis and structural modification. It has been reported that after a compound is combined with an amino acid, the pharmacological activity of the compound is enhanced (Ma et al., 2007; Zhang et al., 2008), water solubility is improved (Drag-Zalesinska et al., 2009; Kim et al., 2009), and cytotoxicity is reduced (Anbharasi et al., 2010).
Natural products and their molecular frameworks are valuable sources of medicinal chemistry and drug discovery (Lee, 2004). In the past decades, drugs that directly or indirectly replace derivatives and analogs of natural products have played important roles in the fight against diseases (Rodrigues et al., 2016). Although the biological activities of natural products are extensive, they are not strong and often have the disadvantages of low bioavailability and poor solubility (Shi et al., 2010). Therefore, to enhance their physchem properties and ADME, it is important to make necessary structural modifications to these products. Interestingly, amino acids can improve these shortcomings, and therefore, they have become the preferred natural product structural modifiers. Many natural products have shown improved biological activity and physicochemical properties following the introduction of amino acids (Ma et al., 2000; Brady et al., 2002), and successful modification results have been obtained. Glycolic acid (1) is the product of bile acid linked to glycine that shows anticancer activity by targeting the pump resistance-related and non-pump resistance-related pathways (Lo et al., 2008); it is currently undergoing Phase III clinical studies (Figure 1). Furthermore, HAO472 (2) is a TFA salt compound composed of oridonin and alanine that possesses potential activity to treat acute myelogenous leukemia and is presently undergoing a Phase I clinical trial in China conducted by Hengrui Medicine Co. Ltd. (Figure 1) (Sun et al., 2014). Additionally, valaciclovir hydrochloride (3), an adenine linked to valine via a glycol linker, is an antiviral drug that has been marketed for the treatment of herpes simplex, herpes zoster, and herpes B (Figure 1) (LaVail et al., 2003).
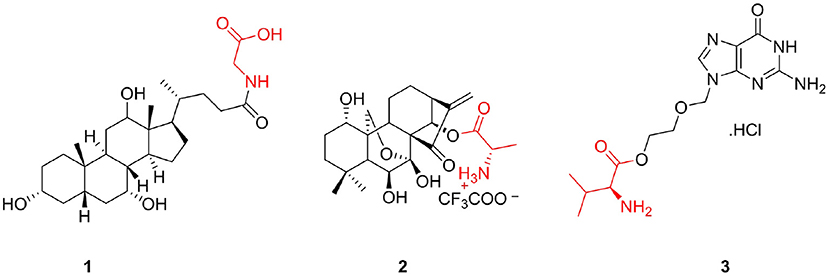
Figure 1. Successful cases of natural product modification with amino acids.
Natural products constitute a potential starting point for new drug design; thus, this review summarizes the structural modification of natural products by amino acid as has been reported between 2010 and 2020. Relevant articles were retrieved from various databases and analyzed. This review is expected to provide a theoretical basis for the structural modification of natural products in the future.
Numerous natural products exist; thus, to facilitate the classification and management of literature, this review classifies all relevant articles into six categories based on the lead framework, including phenylpropanoids, quinones, flavonoids, terpenes, alkaloids, and polyphenols.
Ferulic and isoferulic acids are cinnamic acid compounds containing phenolic hydroxyl groups and exert various biological effects. Gobec et al. (2018) discovered compound 4, a tripeptide derivative of isoferulic acid, which is presently the most potent desmuramylpeptide NOD2 agonist, and the EC50 value for NOD2 is 46 nM. Compound 4 can cooperate with lipopolysaccharide to increase the release of pro-inflammatory cytokines in human peripheral blood mononuclear cells, and it induces ovalbumin-specific IgG titers in a mouse model of adjuvant. Further, Hamed et al. (2020) synthesized a series of N-feruloyl dipeptides and conducted in-depth studies of these compounds as potential anticancer agents against 10 cancer cell lines from different tissue origins. Based on this, compound 5 showed the strongest cytotoxic activity for all cell lines with IC50 values ranging from 2.1 to 7.9 μM, and its activity was stronger than that of ferulic acid (IC50 > 30 μM) (Figure 2A).
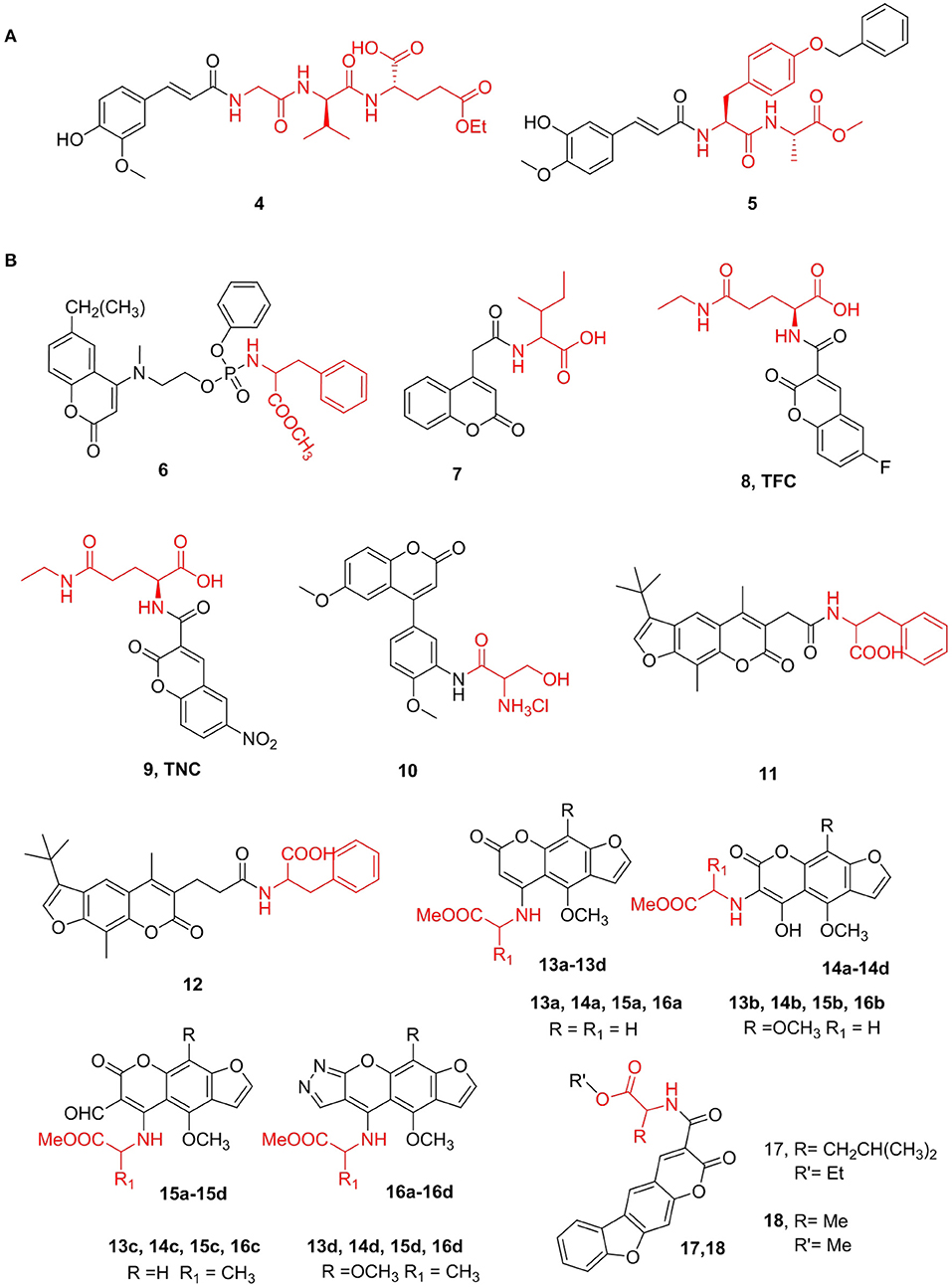
Figure 2. (A) Cinnamic acid derivatives. (B) Coumarins amino acid derivatives.
Coumarin compounds are an important class of natural heterocyclic compounds with antioxidant, antibacterial, anti-inflammatory, and anticancer properties. Since these compounds have a wide range of pharmacological activities and simple chemical structures, they are favored by many medicinal chemistry researchers. Ji et al. (2016) synthesized novel coumarin phosphoramide derivatives and screened for chitin synthase inhibitory and antibacterial activities in vitro. It was found that most of these compounds have a good inhibitory effect on chitin synthase. Among them, compound 6, with an IC50 value of 0.08 mM, was shown to be the strongest chitin synthase inhibitor. Additionally, antifungal experiments have shown that these compounds have moderate or even excellent antibacterial activity against common clinical pathogens. Further, Patagar et al. (2019) designed and synthesized substituted coumarin-4-acetyl amino acids and determined their inhibitory activity against DHODH, a dual-target compound for malaria and cancer. The DHODH inhibitory activity of the newly synthesized compound was then determined. Compared with the standard product, compound 7 showed obviously the highest 70.6% inhibition of DHODH at 0.5 μM, and other compounds with terminal COOH showed moderate inhibition (Figure 2B).
Zhang et al. (2014) synthesized ethyl 6-fluorocoumarin-3-carboxy-L-theanine (8) and ethyl 6-nitrocoumarin-3-carboxy-L-theanine (9). These two compounds enhance the activities of the anticancer drugs cytarabine, vincristine, and methotrexate in inhibiting the growth of lung cancer cells and have no toxic effects on normal human embryonic lung fibroblasts and peripheral blood lymphocytes. TFC (IC50 LLC = 0.125 mM and IC50A549 = 0.99 mM) and TNC (IC50 LLC = 0.09 mM and IC50A549 = 0.064 mM) have shown strong inhibitory effects on the growth of highly metastatic LCC and A549 tumors in tumor-bearing mice and are non-toxic to mice. Additionally, Malysheva et al. (2014) obtained novel 4-arylcoumarin derivatives and found that water-soluble salts (10) show considerable cytotoxicity against the HaCaT cell line (IC50 = 0.07 μM) (Figure 2B).
In addition to the amino acids in furanocoumarin, all synthetic compounds have been tested as EGFR and VEGFR-2 kinase inhibitors. The EGFR enzyme inhibition IC50 values of all compounds range from 20.2 to 57.6 nM, while the EGFR-2 enzyme inhibition (IC50) ranges from 1.2 to 58.9 nM. In general, compound 16d had the strongest inhibitory effect on the two targets. Francisco et al. (2012) synthesized two new tetracyclic benzofuranocoumarin (benzopsoralen) analogs (compounds 17 and 18) and found that they significantly inhibited the proliferation of MDA MB 231, HeLa, and TCC-SUP with GI50 of three tumor cell lines is between 0.043 and 0.205 μM, which is thought to be mainly linked to the inhibition of CYP2A6 (Figure 2B).
Lignans are a kind of natural product formed by the oxidative polymerization of phenylpropanoids, containing two molecules of C6–C3 structure. Podophyllotoxin (PPT), a natural antimitotic product isolated from Podophyllum peltatum L (Imbert, 1998), exhibits various interesting biological activities, such as antitumor and antiviral activities (Garcia and Azambuja, 2004). However, PPT displays many side effects, including damage to normal tissues, leading to the development of lower cytotoxicity and higher efficiency PPT derivatives.
Hu et al. (2011) synthesized a series of novel compounds at the C-4 of PPT and evaluated their cytotoxicity in the K562 cell line in vitro. Compound 19 exhibited better cytotoxicity (IC50 = 2.1 μM) than those of PPT and etoposide, indicating that the antitumor effect might be because of the induction of apoptosis. Further, Wu et al. (2018) synthesized a series of PPT derivatives and tested their cytotoxicity in several cancer and L02 cell lines. Interestingly, some N-Boc amino acid target demonstrated high selectivity, especially compound 20, which showed a high degree of selectivity for both cancer and normal cells. The IC50 values of compound 20 in A549, MCF-7, HepG2, and L02 cells were 9.5, 132.6, 96.4, and 160.2 nM, respectively, showing low cytotoxicity and high selectivity. Moreover, compound 20 induced apoptosis in A549 cells and prevented the transition of these cells from the S phase to the G2 phase; however, it had no significant effect on L02 cells. Han et al. (2016) screened an effective podophyllotoxin derivative, namely 21 (IC50 in MCF-7 cell line = 0.9 μM), with low cytotoxicity to non-cancer cells, including human embryonic kidney cells (293T) (IC50 = 54.4 μM). Additionally, this derivative can cause a significant cell cycle arrest at the G2/M phase and induce MCF-7 cell apoptosis more than PPT. Moreover, the expression of cell cycle protein CDK1 was upregulated, while a protein required for mitotic initiation, Cyclin B1, was downregulated. This result showed that 21 was an effective tubulin polymerization inhibitor, and its effect is equivalent to that of positive control colchicine (Figure 3A).
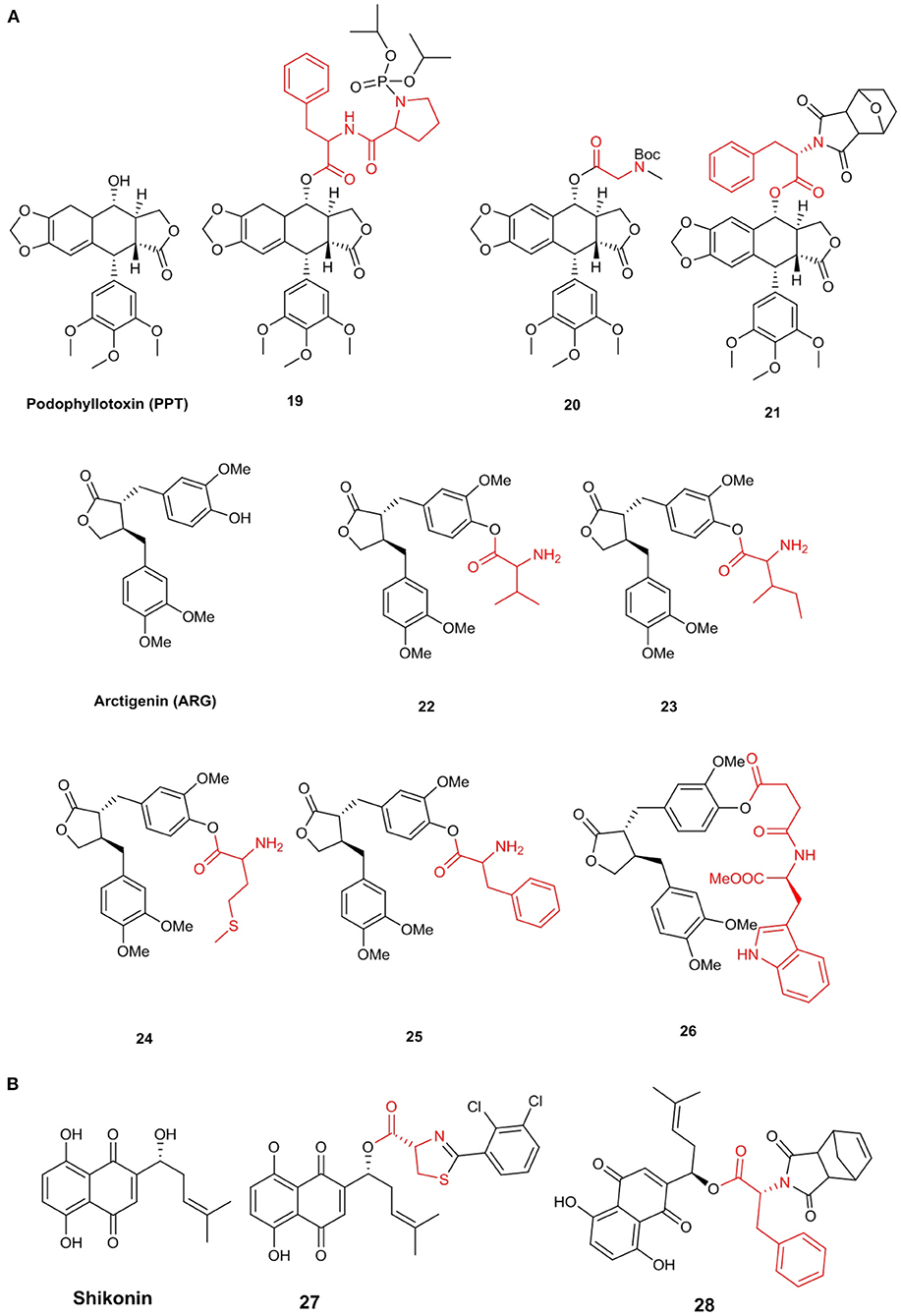
Figure 3. (A) Lignans amino acid derivatives. (B) Shikonin amino acid derivatives.
Arctigenin (ARG) is a lignan-class compound isolated from Arctium lappa L. According to reports, it has a variety of biological activities, such as antitumor, anti-inflammatory, and antiviral activities. Although ARG has a variety of pharmacological activities in vitro, it is insoluble in the human body and thus cannot be absorbed, limiting its clinical application. Therefore, it is very important to use chemical or biological methods to modify ARG into derivatives with better solubility and higher bioavailability (Figure 3A).
Cai et al. (2016) synthesized a series of ARG amino acid derivatives, all of which have better solubility and nitrite elimination ability than those of ARG. This compound and its two amino acid derivatives without a Boc protection group (22, 23) were selected for in vivo antitumor activity evaluation. In H22 tumor-bearing mice, the tumor inhibition rates of compounds 22 and 23 were 69.3 and 43.6% at a dosage of 40 mg/kg, respectively, which was significantly higher than that of ARG. Besides, compared with the positive group, these compounds caused less damage to the liver, kidney, and immune organs of mice. Additionally, they synthesized five other amino acid derivatives (Cai et al., 2018) and measured the antitumor activity of these compounds. The inhibition rates of compounds 24 and 25 were 55.9 and 51.4% at a dosage of 40 mg/kg, respectively, which were twice than that of ARG. Moreover, the compounds were found to reduce damage to internal organs. Further, Zhang H. B. et al. (2018) prepared a series of ARG amino acid derivatives, measured their anti-Toxoplasma gondii activities in vitro, and calculated their selectivity indices. These derivatives showed greater selectivity indices than that of ARG and good anti-Toxoplasma gondii activities. The selectivity index of compound 26 was 3.2, which showed low toxicity to host cells and high anti-Toxoplasma activity (Figure 3A).
They are widely distributed in nature and are divided into four types: benzoquinone, naphthoquinone, phenanthrenequinone, and anthraquinone.
Shikonin is an active naphthoquinone compound isolated from the root of the traditional Chinese Medicine Lithospermum erythrorhizon Sieb. et Zucc. (Chen et al., 2002). Shikonin has received extensive attention from medicinal chemistry researchers due to its particularly good anticancer activity (Lin et al., 2013). However, the side effects and cytotoxicity of shikonin limited its application as a new clinical anticancer drug (Cui et al., 2008). Lin et al. (2015) synthesized a series of shikonin derivatives containing a L-cysteine residue and determined their anticancer activities. Among them, compound 27 showed better antiproliferative activity with IC50 = 3.1_μM in HeLa cells than that of shikonin (IC50 = 5.8 μM). In addition, this compound may cause cell cycle arrest in the G2/M phase, lead to apoptosis, reduce the adhesion ability of HeLa cells, and bind well to tubulin at the paclitaxel binding site, resulting in tubulin polymerization and destruction mitotic. Further, Baloch et al. (2015) synthesized a series of other shikonin derivatives and evaluated their antiproliferative activities against five cancer cell lines, including A549, HeLa, MCF-7, HepG2, and BGC, with IC50 values ranging from 1.3 to 18.5 μM. Compared to shikonin itself and other compounds, compound 28 showed a better antiproliferative effect against all cancer cell lines and could induce apoptosis in a dose- and time-dependent manner, as well as arrest the cell cycle in the G2/M phase (Figure 3B).
Anthraquinone is currently the most important quinone compound with various biological activities, such as purgative, antibacterial, and anticancer activities. Since anthraquinone compounds have a polyaromatic ring-condensed structure, their solubility is extremely poor (<0.1 g/100 mL). Lee et al. (2012) synthesized a series of symmetrically dispersed 1,8-diamidoanthraquinones, evaluated their antiproliferative and telomerase inhibitory effects via a TRAP assay, and measured hTERT expression using SEAP. In particular, among all derivatives, compounds 29 and 30, which are linked to valine and proline methyl esters, respectively, showed the most potent hTERT repressing activities, with IC50 both <10 μM. Aloe-emodin is a type of hydroxyanthraquinone found in aloe leaves (Pecere et al., 2000). Thimmegowda et al. (2015) focused on improving the water solubility of aloe-emodin and synthesized a series of amino acid derivatives, hoping to improve the efficacy of aloe-emodin. They found that the antitumor activity of derivative 31 against HepG2 (IC50 = 4.79 μM) and NCI-H460 (IC50 = 19.0 μM) tumor cell lines was better than that of aloe-emodin, which may be due to the increase in the number of hydroxyl groups and oxygen atoms of fatty acid esters. These groups and atoms increase solubility in water through hydrogen bonds and increase cell communication. Furthermore, Huang et al. (2017) synthesized the phospholipid amino acid alizarin and conducted an in vitro study on its inhibitory effect against tumor cell proliferation. All newly synthesized derivatives showed high cytotoxicity compared with alizarin and low cytotoxicity against the human normal liver HL-7702 cell line. Among them, compound 32 had the strongest killing effect on SK-OV-3 cells, with IC50 = 7.1 μM, slightly lower than that of doxorubicin. The anticancer activity of this compound depended on the apoptotic death of cancer cells by regulating members of the Bcl-2 family and arrest of the SK-OV-3 cell cycle in the G2 phase. Furthermore, Zhang T. J. et al. (2018) designed and synthesized a series of N-(9,10-anthraquinone-2-carbonyl) amino acid derivatives as novel xanthine oxidase inhibitors by mimicking the compound febuxostat. Among them, L-phenylalanine derivative 33 (IC50 = 3.0 μM) and D-phenylalanine derivative 34 (IC50 = 2.9 μM) demonstrated the strongest effect, and both were better than allopurinol (IC50 = 8.1 μM). Compared with the L-enantiomer, D-amino acid derivatives have the same or higher effect (Figure 4).
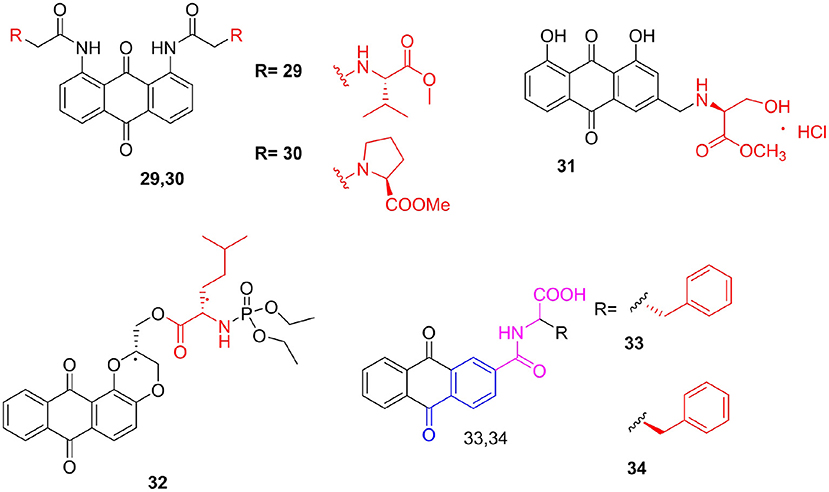
Figure 4. Anthraquinones amino acid derivatives.
The basic nucleus of flavonoids is 2-phenylchromone. Flavonoids are widespread in nature and high in quantity in some plants and berries. They can be divided into flavones, isoflavones, chalcones, and other flavonoids according to their structure. These compounds, similar to the above-mentioned anthraquinones, have low solubility due to their long conjugated structures.
Song X. et al. (2014) prepared a series of chrysin-like amino acid derivatives and measured their antiproliferative activities in gastric cancer MGC-803 cells. They found that compound 35 (IC50 = 3.8 μM) had the strongest inhibitory effect on the growth of MGC-803 cells. This compound induced apoptosis of MGC-803 cells in a dose-dependent manner, increasing Bax and reducing Bcl-xl protein expressions. Baicalein, a natural flavonoid isolated from Scutellaria baicalensis Georgi (Lamiaceae), has been shown to have multiple biological activities. However, this flavonoid has poor solubility and low bioavailability, which limits its clinical application. Li et al. (2013) prepared baicalein amino acid derivatives and tested their in vitro cytotoxic activities. Compounds 36 (IC50 = 9.6 μM) and 37 (IC50 = 13.5 μM) showed significant increases in cytotoxicity compared with baicalein (IC50 = 40.2 μM) in HepG2 cells (Figure 5).
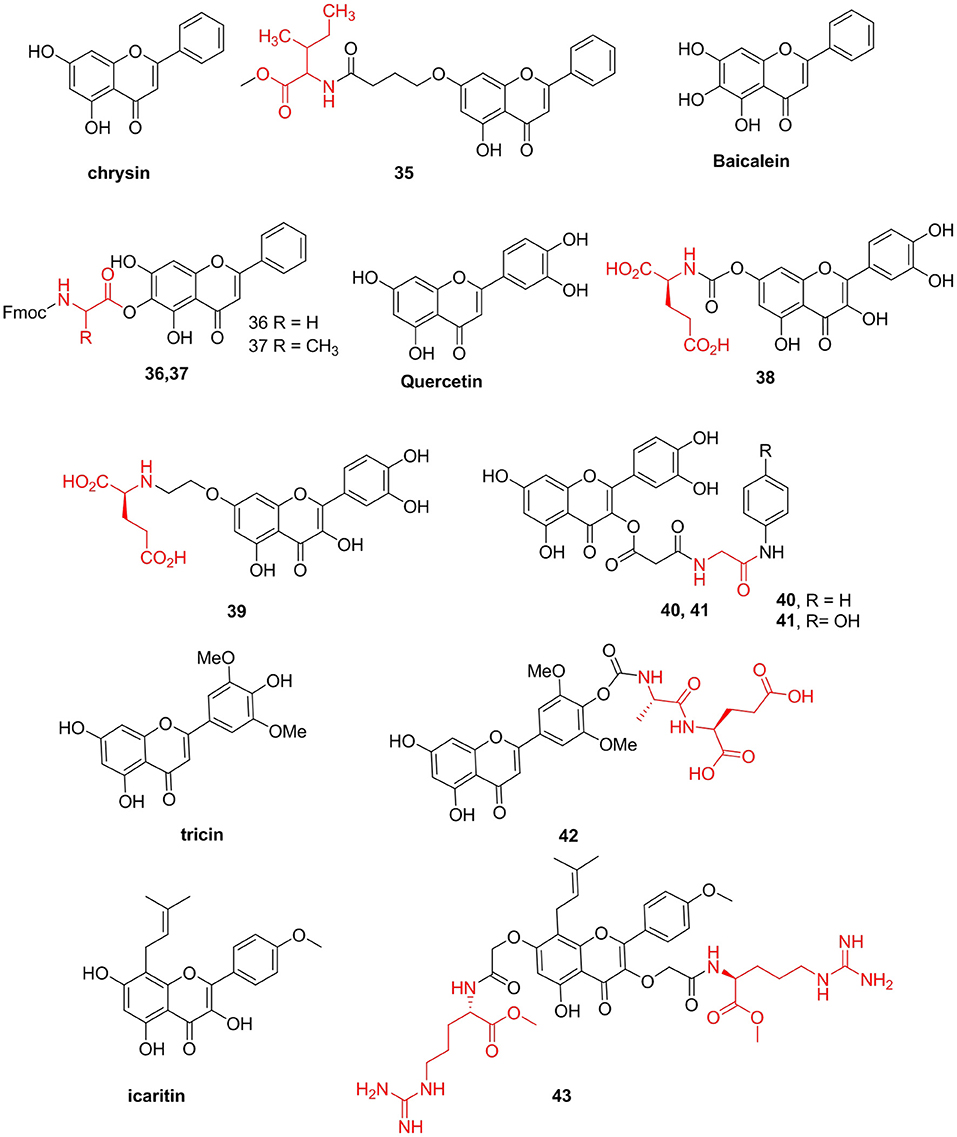
Figure 5. Flavonoids amino acid derivatives.
Furthermore, Kim et al. (2014) prepared quercetin–amino acid derivatives and evaluated their MDR-modulatory effects. A quercetin–glutamic acid derivative 38, with IC50 values of 0.8–0.9 μM, was as effective as verapamil in reversing MDR and sensitizing MDR MES-SA/Dx5 cells to various anticancer drugs. The MDR reversal activity of this derivative is due to its hydrolyzable property. Additionally, there is evidence that the intact quercetin–glutamic acid derivative has stronger MDR-reversal activity than that of quercetin. Kim et al. (2017) synthesized quercetin derivatives with a glutamic acid attached at the 7-O position via a non-hydrolyzable linker. Derivative 39 showed significantly higher activities than those of quercetin. Compared with that of quercetin (1.9-folds), the structurally modified quercetin derivative (22.3-folds) with a non-cleavable linker showed significantly improved MDR-reversal activities. However, the quercetin derivatives were not as effective as verapamil in Pgp inhibition, thereby reversing MDR (Figure 5).
Furthermore, the natural product simocyclinone D8 (SD8) inhibits DNA gyrase via a unique mechanism. Verghese et al. (2013) synthesized flavone-based analogs inspired by the complex compound SD8. However, these analogs do not act as catalytic inhibitors, as SD8 does. Analogs 40 (IC50 = 48.6 μM) and 41 (IC50 = 84.7 μM) were determined to be DNA intercalators and topoisomerase poisons.
Tricin has demonstrated diverse biological activities, including anti-inflammatory (Moscatelli et al., 2006) and anticancer (Jeong et al., 2007) activities. To improve its bioavailability and oral availability, Ninomiya and his team prepared tricin-amino acid derivatives as prodrugs and investigated their cell permeability, stability in vitro, and oral availability in vivo. Among the prodrugs, compound 42 exhibited enhanced permeability with an excellent relative permeation rate of 6.3, stability in MDCK cells (t1/2 = 165 min), and increased bioavailability after oral administration with the maximum concentration of tricin being 2,497 ng/mL in the 100 mg/kg group. Additionally, icaritin exhibits various pharmacological activities, including anticancer and osteoporosis treatment (Chen et al., 2012; Zhao et al., 2015). Because this prenylflavone is very safe, it would be a good lead compound for further modification to obtain potent compounds with remarkably improved antimicrobial activities and low toxicity. Thus, Lin et al. (2017) designed a series of new hemiflavonoid-based small molecules that mimic antimicrobial peptides from icaritin to fight drug-resistant gram-positive bacterial infections. They found that compound 43 containing two arginine residues showed remarkable antibacterial activity (MICs = 1.56–3.13 μg/mL) against gram-positive bacteria MRSA. This compound has low cytotoxicity and selectivity >511, comparable to that of several antibiotics in clinical trials. Their data showed for the first time that icaritin derivatives effectively kill bacteria. Compound 43 showed rapid bactericidal activity by destroying bacterial membranes and circumventing the development of bacterial resistance (Figure 5).
Additionally, scutellarin is a flavonoid extracted from the Compositae plant Erigeron breviscapus. It is mainly used for the treatment of cardiovascular and cerebrovascular diseases (Lin et al., 2007). Results of recent pharmacodynamics and pharmacokinetic studies show that the actual form of absorption and pharmacodynamics of scutellarein in the body after oral administration is the compound itself. However, scutellarin aglycone still has defects such as low absolute bioavailability (7% in rats) and difficulty in intestinal absorption (Chen X. et al., 2006). Thus, Fu et al. (2011) synthesized L-amino acid ester derivatives of scutellarin aglycone and found that the water solubility levels of derivatives 44 and 45 were 1,796 and 4,100 μg/mL, respectively, which were much greater than that of scutellarin (14.4 μg/mL). Furthermore, Jiang et al. (2013) used scutellarin methyl ester and L-amino acid tert-butyl ester hydrochloride as raw materials to prepare a series of scutellarin methyl ester prodrugs and evaluated water solubility and the in vitro stability of the target compound. Their results indicated that compounds 46 (aqueous solubility = 123.1 μg/mL) and 47 (426.5 μg/mL) had better solubility and in vitro stability than those of scutellarin, respectively. Cha et al. (2014) also synthesized a series of 4'-L-amino acid derivatives of scutellarin methyl ester and reported that the designed target compounds had higher enzymatic and chemical stability and aqueous solubility. Among these compounds, compound 48 had the highest PappAPtoBL value (1.9 × 10−6 cm/s) and lowest ER (PappBLtoAP/PappAPtoBL = 0.56) and protected PC12 cells from oxidative damage induced by H2O2 by reducing the loss of MMP and reducing the generation of ROS induced by H2O2 (Figure 6A).
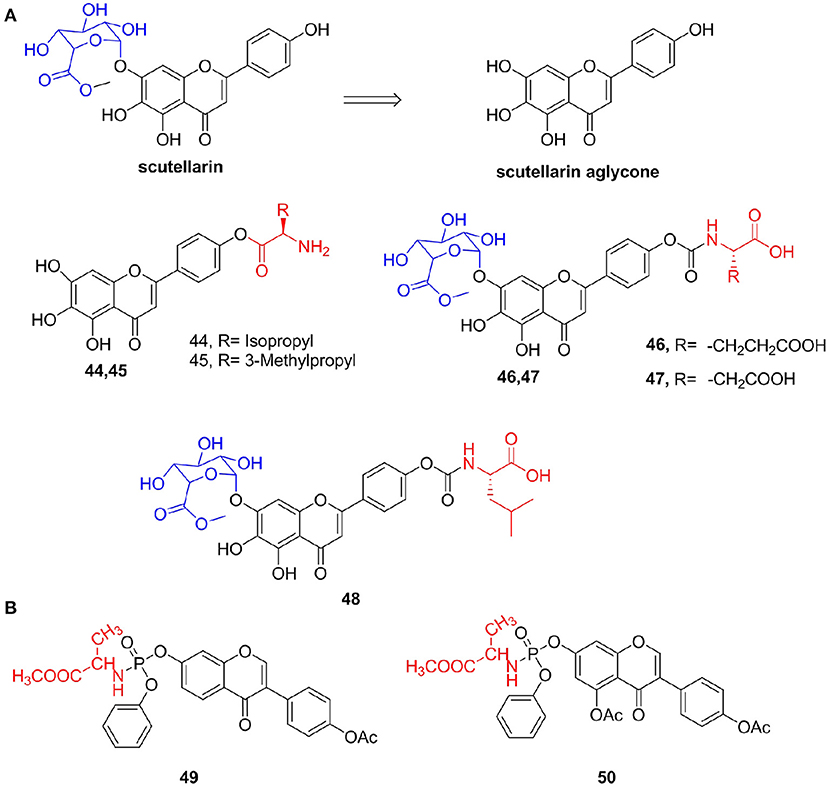
Figure 6. (A) Scutellarin amino acid derivatives. (B) Isoflavone amino acid derivatives.
Li et al. (2016) synthesized a series of isoflavone-7-phosphoramidate derivatives and determined their antiproliferative activity in vitro against the HepG2 cell line and L02 cell line. Compounds 49 (IC50 = 5.5 μM) and 50 (IC50 = 6.6 μM), which incorporated the methyl alanine, exhibited high inhibitory activities against the HepG2 cell line. Compound 49 can significantly induce G2/M arrest and lead to early apoptosis in HepG2 cells (Figure 6B).
Chalcones, with α,β-unsaturated ketone structures, are derived from natural compounds and chemical synthesis, possessing a variety of biological properties (Zhuang et al., 2017). Jin et al. (2012) synthesized a chalcone derivative containing L-phenylalanine and evaluated its antibacterial activity. Among which compound 51 (MIC = 2 mg/mL) exhibited the best antimicrobial activity against Staphylococcus aureus RN4220. This compound showed the most potent activity against all the MDR clinical isolates tested. Based on compound 51, Song M. X. et al. (2014) changed the types of amino acids and synthesized another series of derivatives and found that, among all, leucine (52, MIC = 1 mg/mL) was the most potent against two methicillin-resistant S. aureus (Figure 7A).
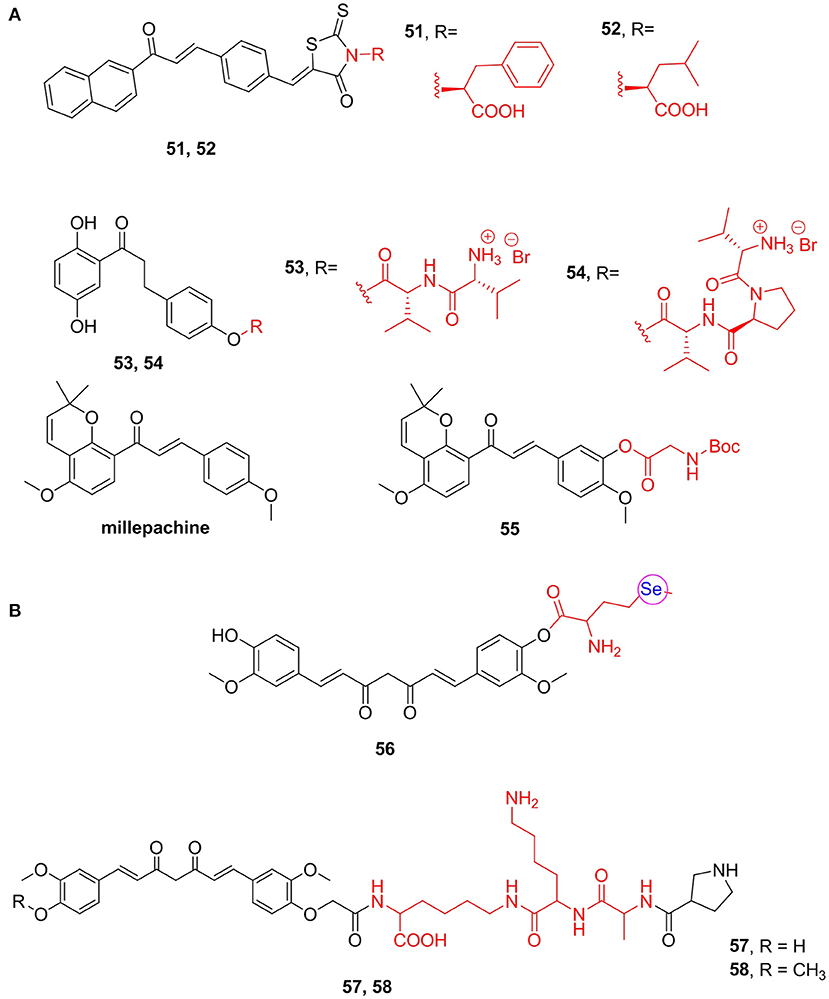
Figure 7. (A) Chalcones amino acid derivatives. (B) Curcumin amino acid derivatives.
1,3-Diphenylpropanones is a positive allosteric modulator of α7 nAChRs, which have good analgesic activity but poor pharmacokinetics. To improve its biological activity in vivo, Criado et al. (2016) have prepared amino acid and peptide derivatives as prodrugs of these modulators. Compounds 53 (inhibition of α7 nAChR currents = 14.7%) and 54 (inhibition of α7 nAChR currents = 24.7), prodrugs of 1,3-diphenylpropanones, showed obviously antinociceptive activity in vivo. Compound 54 shows roughly the same analgesic effect as the parent compound but long-lasting effects (Figure 7A).
Furthermore, millepachine is a natural pyranochalcone isolated from Millettia pachycarpa (Zhou Z. Y. et al., 2019). This compound has a benzopyran structure, which has good cell membrane permeability (Nicolaou et al., 2000); however, its water solubility is not optimistic. Thus, Wu et al. (2015) prepared a series of amino acid derivatives of millepachine and tested their solubility and antitumor activity. They found that compound 55, a glycine derivative, with an IC50 value for MDAMB-231 cells of 0.13 μM, showed the best potency and long-term inhibition of cell viability and induced apoptosis. The solubility of compound 55 in human plasma is 2.98 mg/mL, which is significantly stronger than the lead compound millepachine (0.15 mg/mL) (Figure 7A).
Additionally, curcumin has been reported to have a variety of pharmacological activities, such as anticancer, antioxidant, and anti-inflammatory activities. However, low bioavailability, poor water solubility, and poor pharmacokinetics hinder the clinical application of curcumin (Kotha and Luthria, 2019). Thus, Cao et al. (2014) synthesized curcumin derivatives with enhanced bioactivity and reported that compounds containing one selenomethionine molecule (56) attached to the hydroxy group of curcuminoids showed high antimicrobial activity. Moreover, they exhibited strong DPPH scavenging ability, which was about 10 times stronger than that of curcumin and has good antiproliferative activity on a variety of cancer cells, with an IC50 value of <1 μM. Furthermore, Bi et al. (2016) got a new class of derivatives that can associate apoptosis induced by ischemia/reperfusion injury and reverse mitochondrial oxidative stress. The results suggest that selected curcumin polypeptide analogs 57 (EC50/.OH = 25.6 μM) and 58 (EC50/.OH = 31.4 μM) exhibited good hydroxyl radical scavenging activity and can facilitate the recovery of mitochondrial reticular networks and cell rescue (Figure 7B).
Gambogic acid (GA), the most important compound of Garcinia, has been reported to be a hopeful antitumor agent. Chemical modification of GA C-37 position by introducing hydrophilic amines can increase activity (Li et al., 2012). Notably, the antiproliferative activity of compound 59 was almost 2-fold more active than that of GA against HCT 116 cell lines with an IC50 value of 1.35 μM (Figure 8A).
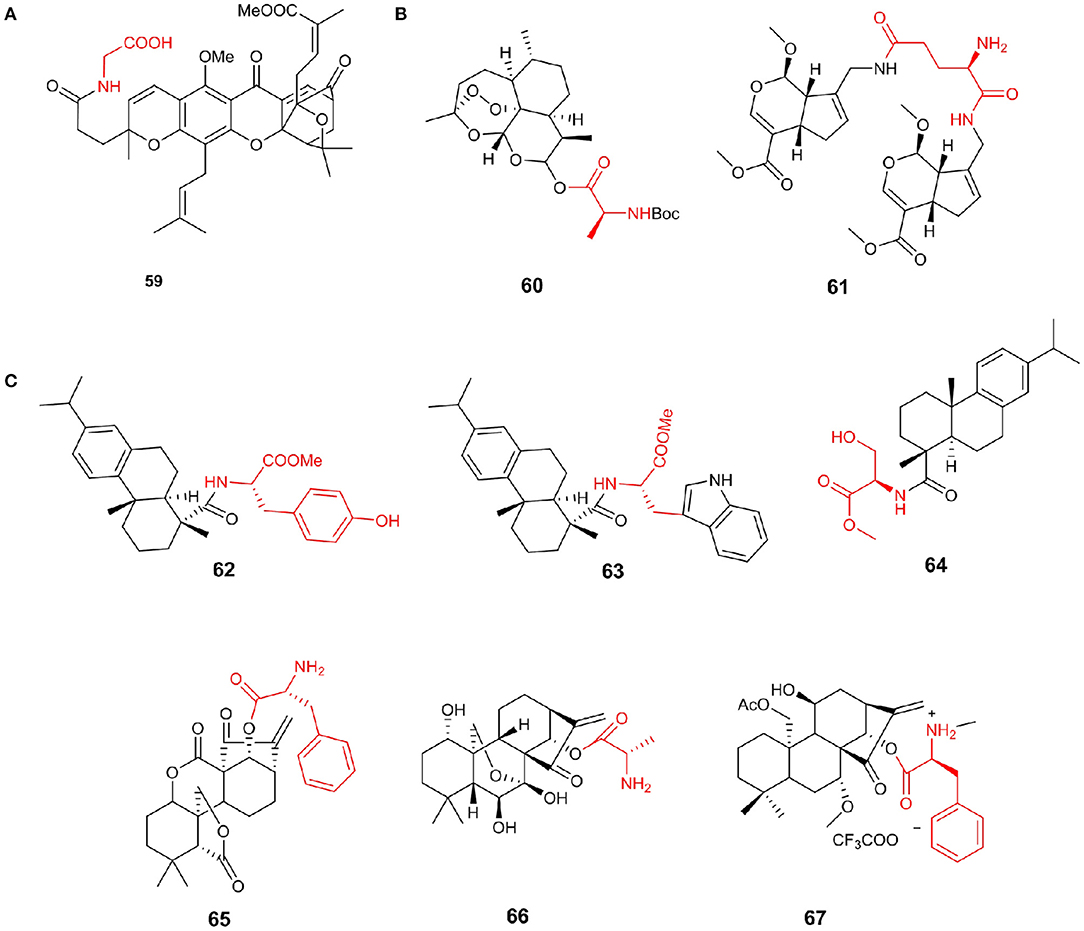
Figure 8. (A) Gambogic acid amino acid derivatives. (B) Sesquiterpenes amino acid derivative. (C) Diterpenes amino acid derivative.
Terpenoids are the general term for polymers and their derivatives of isoprene or isopentane structural units, with general structural formula (C5H8)n and a very wide range of biological activities. There are many natural products from terpenes used in clinical applications, such as the antimalarial drug artemisinin (Sowunmi et al., 2016), the antitumor drug paclitaxel (Shi et al., 2018), and the anti-infective drug andrographolide (Wen et al., 2015).
It has been reported that artemisinin has anti-Toxoplasma activity; however, the activity is not strong (Sarciron et al., 2000). Deng et al. (2020) discovered that compound 60 (selectivity index = 6.44), showed the most effective anti-T. gondii activity and low cytotoxicity. Furthermore, compound 60 had better growth inhibitory activity on T. gondii than positive control drug and remarkably reduced the number of tachyzoites in vivo (P < 0.01). The mouse Toxoplasma infection model proved the effectiveness of compound 60 anti-T. gondii property in vivo and its role in the reduction of liver damage caused by T. gondii. Additionally, genipin is the product of geniposide hydrolysis by β-glucosidase and has anticancer activity. Fang et al. (2018) reported genipin analogs obtained by a biomimetic synthesis strategy and evaluated their cytotoxic activity. Compound 61 was reported to have a significantly stronger inhibitory effect on tumor cell proliferation than that of cisplatin, with IC50 values of 0.43–1.18 μM (Figure 8B).
Abietic and dehydroabietic acids are a class of diterpenoids that show moderate cytotoxicity. Ukiya et al. (2013) prepared amino acid conjugated derivatives from these acids to evaluate their cytotoxicities in a variety of cells. Among the derivatives, compound 62 showed potential cytotoxic activities against multiple cancer cell lines with IC50 values of 2.3–8.1 μM, and the cytotoxic activity of leukemia cells has the highest selectivity. It is estimated that more than 65% of all bacterial infections resistant to chemotherapy are related to biofilms (Lebeaux et al., 2013). Dehydroabietic acid was reported to prevent the formation of S. aureus biofilms in the low micromolar range and only 2–4-fold higher concentrations to significantly reduce the viability and biomass of existing S. aureus biofilms (Fallarero et al., 2013). The combination of dehydroabietic acid scaffolds and different amino acids can simultaneously target planktonic bacteria and biofilm bacteria in S. aureus strains and has stronger anti-biofilm agents than conventional antibiotics (Manner et al., 2015). Compound 63 was the most potent abietane-type anti-biofilm agent and exhibits strong activity on pre-formed biofilms at a concentration that is threefold higher than the concentration required to inhibit biofilm formation. Abietic acid and dehydroabietic acid, in a shortage of antibiotics and antimicrobial resistance problems looming against the growing respect, showing potential value. Helfenstein et al. (2017) discovered a dehydroabietic acid derivative 64 containing a serine side chain; its MIC90 against Staphylococcus aureus ATCC 25923 was 60 μg/mL, and the IC50 value on Balb/c 3T3L cells was 45 μg /mL (Figure 8C).
Oridonin was isolated from isodon with a special structural scaffold and has excellent bioactivity, including anticancer activity (Wang L. et al., 2012). However, poor solubility and moderate bioavailability of oridonin hindered its clinical research on cancer treatment. Many researchers have modified the structure of oridonin, hoping to obtain good anticancer drugs, including the candidate drug HAO472, which is undergoing phase I clinical trials. Hu et al. (2019) designed and synthesized a series of enmein-type diterpenoid amino acid ester derivatives according to HAO472 and tested their antiproliferative activity on various cancer cell lines and L-02 normal hepatocytes. The results show that compound 65 (IC50/Bel−7402 = 0.07 μM) has the strongest cytotoxicity against human liver cancer Bel-7402 and chronic myeloid leukemia K562 cells at the submicromolar level more potent than L-alanine-(14-oridonin) ester 66(IC50/Bel−7402 = 9.56 μM). Simultaneously, it shows low cytotoxicity (IC50 for L02 cells > 50 μM) to normal cells and high selectivity. At the same time, Ke et al. synthesized a series of Flexicaulin A amino acid derivatives and evaluated their antiproliferative activity against four kinds of cancer cells. Compared with Flexicaulin A, the anticancer activity and solubility of most derivatives were remarkably improved. Compound 67 showed the most excellent activity, and its IC50 value for TE-1 cells was 0.75 μM. It could inhibit the proliferation of cancer cells and the formation of cell colonies and increase the level of ROS in TE1 cells (Figure 8C).
Panax ginseng is widely used as a healthy functional food due to its obvious health benefits. Ginsenosides are the main bioactive constituents of Panax ginseng. Panaxadiol (PD), protopanaxadiol (PPD), 25-methoxyprotopanaxadiol (AD1), and 25-hydroxyprotopanaxadiol (AD2) are obtained by hydrolysis of ginsenosides and have a variety of biological activities, including anticancer (Zhang et al., 2007; Salvador et al., 2017), antiliver fibrosis (Han et al., 2018), and antibacterial (Amoussa et al., 2016) activities. Many researchers have introduced amino acids into ginsenosides and obtained ginsenoside amino acid derivatives with enhanced activity. Liu et al. (2011) synthesized a series of PD amino acid derivatives by modifying the 3-hydroxyl group of panaxadiol and evaluated their activities against a panel of human tumor cell lines. All amino acid derivatives showed stronger antiproliferative activities than PD on the growth of different cancer cells in vitro. From the results of the antitumor tests in HepG2, compound 68 (IC50, 5.03 μM) exhibited 7.5 times the strongest activities than that of PD (IC50, 37.91 μM). For the A549 cell line, compound 69 (IC50, 7.63 μM) showed the strongest activity, which was 5.6 times stronger than PD (43.13 μM). Ocotillol enantiopure and its derivatives showed good antibacterial activity anti-gram-positive bacteria (Bi et al., 2015; Ren et al., 2019), and they can be obtained by the oxidation of PPD with m-CPBA. Wang et al. (2018) introduced free amino groups at the C-3 position to confirm whether the group can enhance the antibacterial activity. The results showed that compound 70 showed excellent antibacterial activity with MIC of 2 μg/mL against MRSA USA300 and 4 μg/mL against B. subtilis. Derivatives of AD2 were measured on six distinct human tumor cell lines (Wang et al., 2013). Compound 71 exhibited the best antitumor activity with IC50 values ranging from 2.77 to 6.83 μM for six cell lines compared to the parent AD2. Qu et al. (2015) continued to synthesize AD-2 amino acid derivatives and evaluated the in vitro antitumor activities of five human tumor cell lines. Compounds 72 (IC50 = 4.2–9.9 μM) and 73 (IC50 = 4.2–7.1 μM) displayed the most potent anticancer activities. The two derivatives could remarkably induce the apoptosis of human prostatic carcinoma DU145 cells and have low toxicity in IOSE144 cells and HOSEpiC cells. Yuan et al. (2018) modified AD1 and AD2 with non-protein amino acids and obtained two series of derivatives of AD1 and AD2. Compounds 74 and 75 exhibited strong biological activity against HCT-116 and BGC-823 cell lines, with_IC50 values in the range of 4.76–9.03 μM, and exhibited higher cytotoxic activity than AD-2 and AD-1. The relative plasma stability of esters is closely related to the hydrolytic enzymes in plasma. It is reported that the increase in steric hindrance near the hydrolyzable group will reduce the affinity between the compound and the hydrolase, thereby improving the plasma stability of the compound (Borthwick et al., 2003; Carroux et al., 2013). Therefore, among these ginsenoside compounds, the plasma stability of compound 70 should be the strongest.
Research has demonstrated that AD-1 could regulate c-FLIP pathway-mediated activation of NF-κB, thereby showing liver protection and reversal of activated HSC (Wu et al., 2011). At the same time, AD-1 improved hepatic injury, fibrosis, and inflammation induced by thioacetamide in C57BL/6 mice (Han et al., 2018). The present study aimed to introduce amino acids to AD-1 and obtained safer, more effective, and developable derivatives, which could be used to prevent or improve liver fibrosis, and simultaneously provide amino acid dietary supplements. Conjugate 76 has a significant inhibitory effect on cell proliferation in activated t-HSC/Cl-6 cells (IC50 = 3.8 μM) and appeared to be non-toxic to L02 cells (Figure 9).
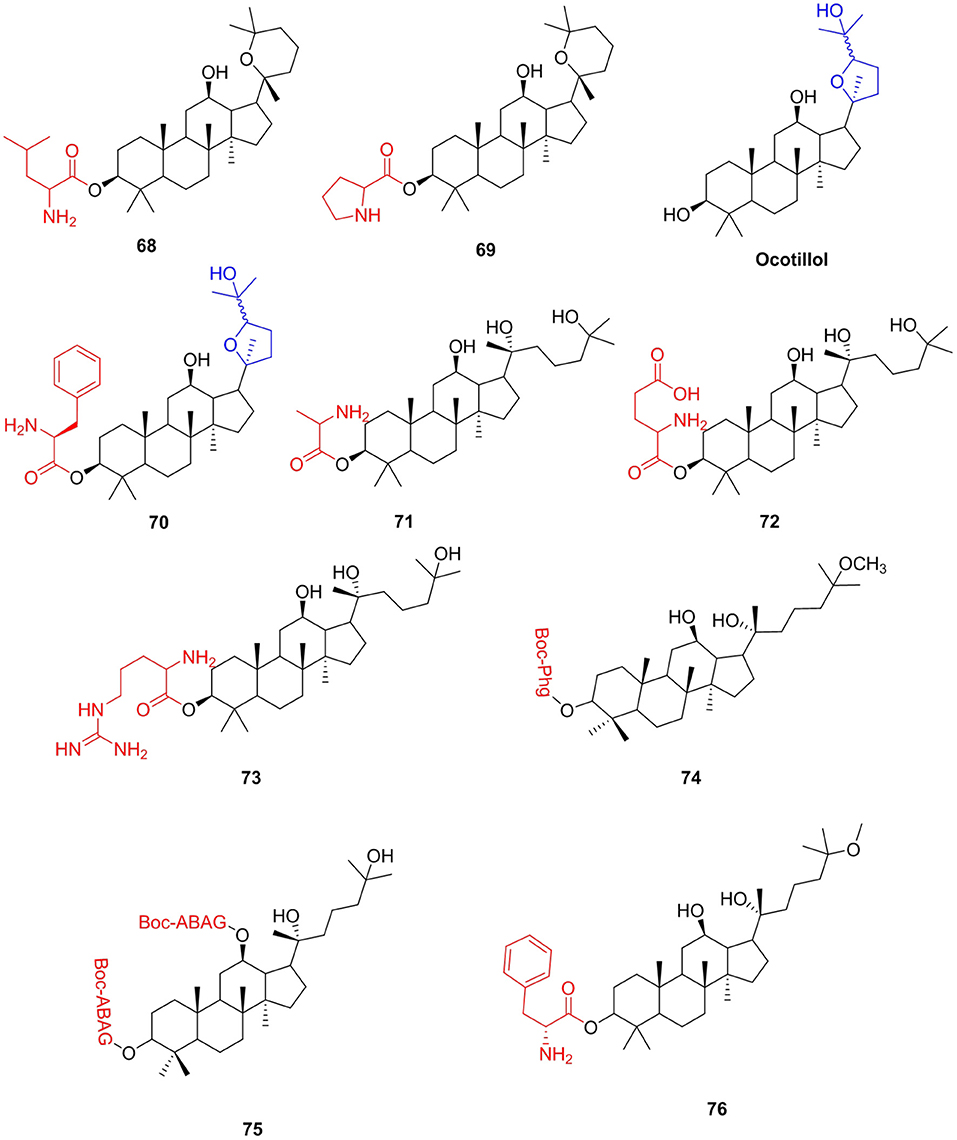
Figure 9. Ginsenosides amino acid derivatives.
23-Hydroxybetulinic acid existed in the root of Pulsatilla chinensis, which has anti-HIV and antitumor activities similar to betulinic acid and botulin (Bi et al., 2007; Zhu et al., 2009). Unfortunately, both betulinic acid and botulin are very poorly soluble in aqueous buffers; thus, their bioavailability and biodistribution are inadequate in terms of medical applications (Drag-Zalesinska et al., 2009). Suitable modification at the right positions of natural products may yield analogs with significantly improved potency. A set of 17-carboxylic acid-modified 23-hydroxybetulinic acid derivatives that displayed slightly improved antiproliferative potencies were reported previously (Bi et al., 2007). To increase the anticancer activity and structural diversity of 23-hydroxybetulinic acid, Lan et al. (2011) prepared a series of new compounds and screened these compounds for in vitro antiproliferative activity against various cancer cell lines. The amino acid derivatives 77 and 78 with IC50 <10 μM in all tested cell lines were considered to be the most hopeful compounds.
Betulinic acid exhibits anticancer and anti-HIV biological activities, and its derivative Bevirimat (79) is undergoing phase II clinical trials against HIV (Smith et al., 2007). Bevirimat exhibited promising pharmacokinetic profiles in clinical trials, but its effectuality was compromised by the high baseline resistance of HIV-1 variants with polymorphism. A series of betulinic acid (BA) derivatives were measured for their inhibition of the chymotrypsin-like activity of the 20S proteasome (Qian et al., 2011). Compounds 80 and 81, comprising leucine, illustrated the best proteasome inhibition activity with IC50 values of 1.56 and 1.80 μM, respectively, which are 3–4-fold more potent than the proteasome inhibition controls LLM-F and lactacystin. To determine whether the viruses with bevirimat-resistant polymorphism also altered their sensitivity to betulinic acid derivatives that inhibit HIV-1 entry, a series of new betulinic acid inhibitors were tested for their activities against HIV-1 NL4-3 and NL4-3 variants resistant to bevirimat (Dang et al., 2012). The results show that the bevirimat-resistant viruses were ~5–10-fold more sensitive to three new glycine ester derivatives 82, 83, and 84, and showed potent anti-HIV activity with EC50 ranging from 0.04 to 0.12 μM. In contrast, wild-type NL4-3 and bevirimat-resistant variants were equally sensitive to the HIV-1 RT inhibitor AZT. Compound A43D exhibited the most potent anti-HIV-1 entry activity (Huang et al., 2006). Compared with A43D (CLint = 0.37 ml/min/mg), these three new compounds 82 (CLint = 0.23 ml/min/mg), 83 (CLint = 0.17 ml/min/mg), and 84 (CLint = 0.22 ml/min/mg) showed significantly improved microsomal stability (Figure 10).
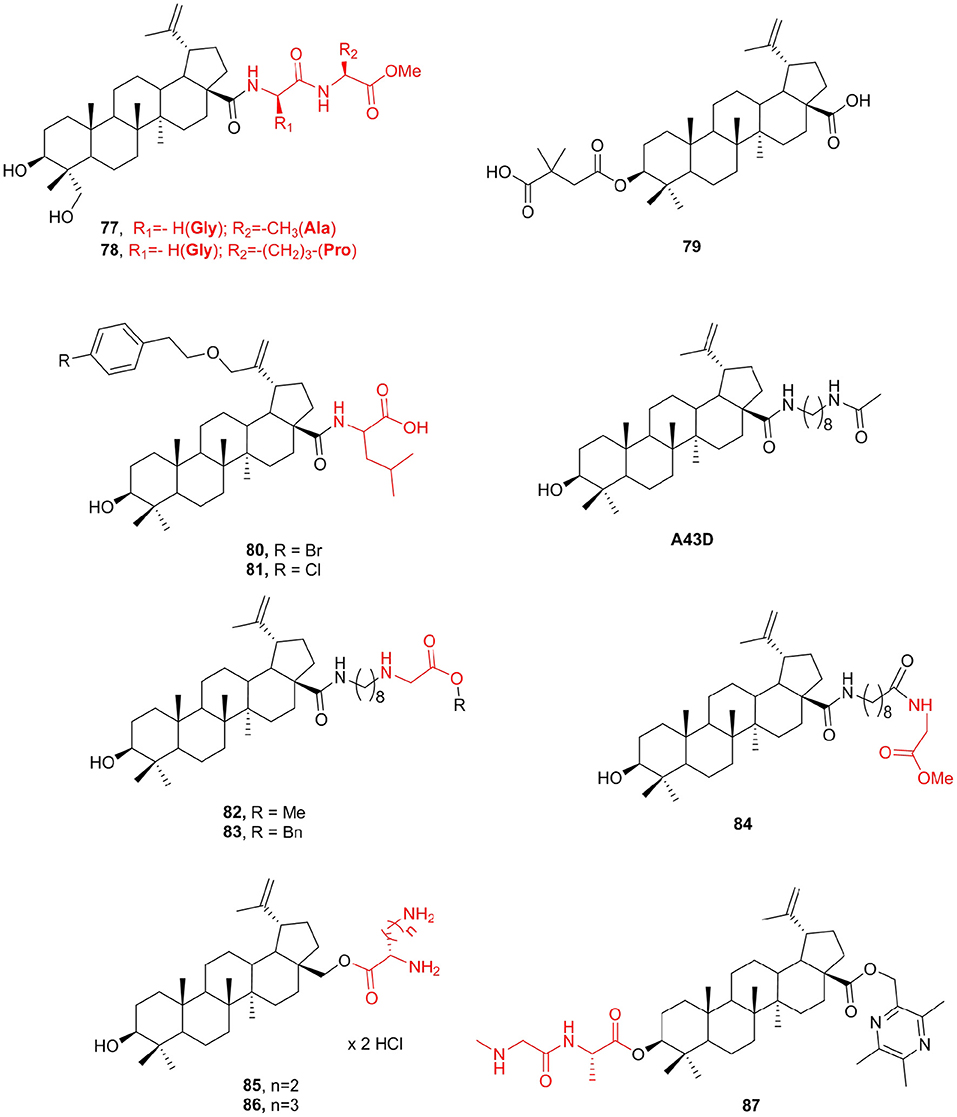
Figure 10. Betulinic acid derivatives.
The new amino acid derivatives of betulinic acid and betulin were synthesized, and their anticancer activity was evaluated (Drag-Zalesinska et al., 2015). In particular, compounds containing a lysine side chain (85, IC50 = 7 μM) and ornithine (86, IC50 = 10 μM) are stronger than the parent compounds, and the biggest advantage was that new compounds are safe for normal human keratinocytes. In addition, the literature reports that the introduction of amino acids into triterpenes could enhance selective cytotoxicity and water solubility (Schwarz et al., 2014). Based on the above, researchers attempted the synthesis of several ligustrazine-betulinic acid (TBA) amino acid derivatives BAX by introducing several amino acids to the C3 of TBA to enhance its antitumor activities and tumor targeting. These compounds were screened for selective cytotoxic activity against five cancer cell lines and the non-malignant MDCK cell line. Most of the tested TBA-amino acid derivatives showed better antiproliferative activity against all tumor cell lines than TBA. Among them, compound 87 showed the best cytotoxic activity on tumor cell lines (IC50 = 2.31 μM), which was 2-fold higher than that of the positive drug cisplatin (DDP), while it showed lower cytotoxicity in MDCK cells than DDP cells (Figure 10).
Glycyrrhetinic acid (GA) is the main component of licorice root (Baltina, 2003; Schwarz and Csuk, 2010); its antitumor activity is low compared with other members of the triterpenoid family (Tatsuzaki et al., 2007). GA is easily inexpensive and shows apoptotic effects on tumor cells. These facts make GA and its derivatives the focus of our scientific interest. In addition, GA demonstrated only poor selectivity (Schwarz et al., 2010). Here, we tried to enhance the poor cytotoxicity of GA via simple derivatization. Csuk et al. (2011a) make use of various glutamyl and aspartyl substituents for the synthesis of C esters of GA methyl ester. Compound 88 (IC50 = 1.27–2.33 μM) demonstrated 67-fold higher cytotoxicity and an up to 140-fold better selectivity toward tumor cells than that of GA. To improve the cytotoxicity of GA, Csuk et al. (2011b) synthesized new derivatives of GA, differing in structure and lipophilicity. The most active compound 89, a benzyl glycyrrhizinate with an extra 3-N-(3aminopropyl)glycyl substituent, showed an IC50 between 1.96 and 5.14 μM for five human cancer cell lines and triggers apoptosis in 80% of the cells. Use various amino acids, including methionine (90), threonine (91), valine (92), and phenylalanine (93), to modify glycyrrhetinic acid to obtain more effective derivatives (Wang J. et al., 2012). These compounds could protect the growth of E. coli, Bacillus subtilis, and yeast against high concentrations of DMF. To improve the cytotoxicity of GA and explore the effect of the bonding mode on antitumor activity, Zhou F. et al. (2019) synthesized a series of amino acid derivatives and evaluated their antitumor activity. All the deprotected (without Boc group) derivatives displayed much stronger cytotoxic activity than GA, and surprisingly enough, all the GA-NH2 series of the compounds were more effective than GA-OH series against different tumor cells. Among them, compound 94 showed better antitumor activity against A549 (IC50 = 2 μM) than cisplatin (IC50 = 9 μM). Further studies indicated that compound 94 could induce A549 apoptosis by nuclei fragmentation and prevented the transition from S to G2 phase (Figure 11).
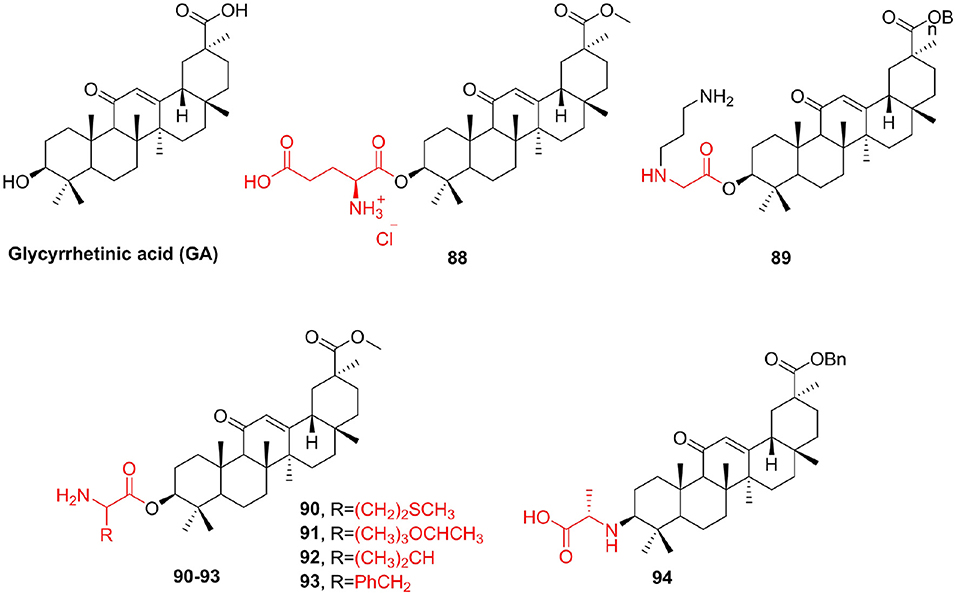
Figure 11. Glycyrrhetinic acid derivatives.
In addition to those listed above, other pentacyclic triterpene compounds often react with amino acids to improve their unfavorable properties. Ursolic acid is abundant in the plant kingdom. Ursolic acid and its derivatives have been reported to have interesting bioactivities (Choi et al., 2000; Chattopadhyay et al., 2002). Shao et al. (2011) synthesized some ursolic acid derivatives and determined their cytotoxic activity on various cancer cells. Among them, the derivative 95 introduced into serine showed greater anti-A549 cell activity than ursolic acid, with an IC50 of 35.3 μM. Fu H. J. et al. (2014) prepared a series of new ursolic acid derivatives under the guidance of virtual screening and used RBL2H3 cells and ovariectomized (OVX) rats for biological evaluation. Compound 96, which contains a glycine structure, showed potent inhibitory activity on serotonin biosynthesis, which inhibited the protein and mRNA expression of Tph-1 and lowered serotonin contents in the serum and gut without influencing brain serotonin. Due to their strong hemolytic activity, the clinical development of Pulsatilla saponins A (PSA) and D (PSD) is limited (HD50 = 10 μM) (Wang and Fang, 2009). To solve the problem, Chen et al. (2015) prepared C-28 position derivatives of PSA/PSD and evaluated the hemolytic activity and cytotoxicity of these compounds. Compared with PSA, the acute toxicity of compound 97 to mice is greatly reduced. At the same time, compound 97 (HD50 > 500 μM) is a potential antitumor agent, thereby preventing hemolysis (Figure 12).
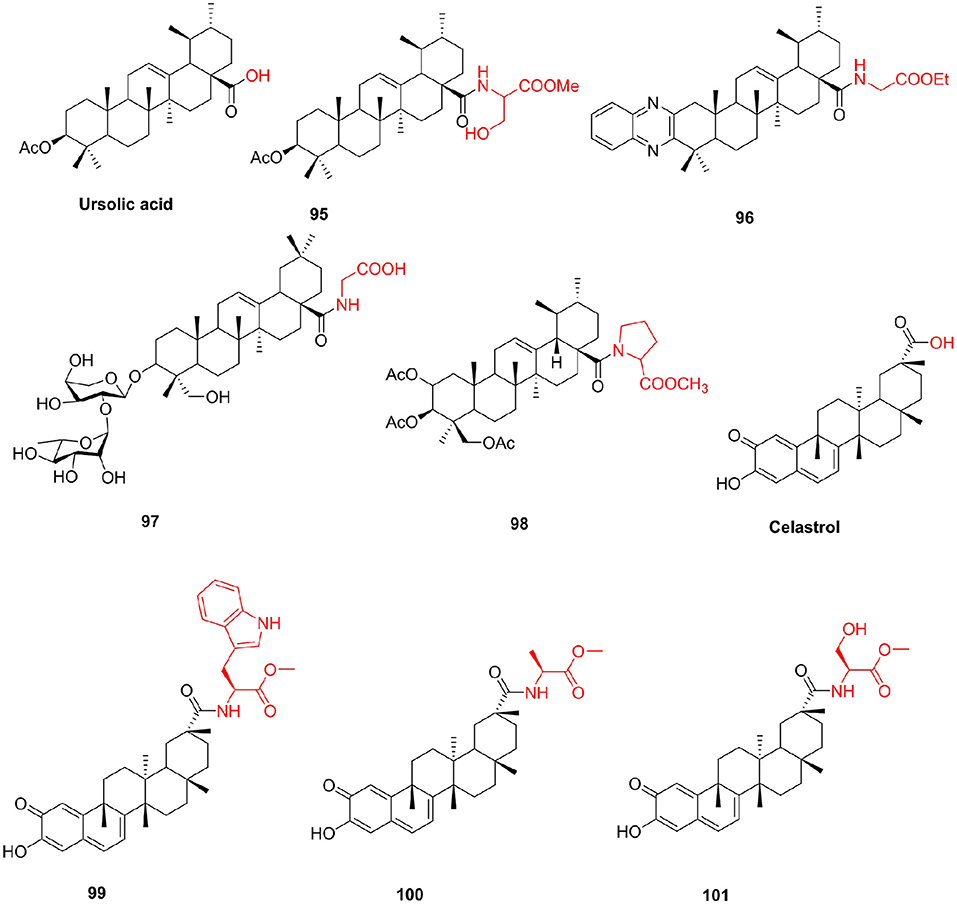
Figure 12. Other triterpenoids derivatives.
Asiatic acid, an active pentacyclic triterpenoid found in Centella asiatica, can be easily prepared by the hydrolysis of asiaticoside. Asiatic acid also has many biological effects (Mook-Jung et al., 1999; Park et al., 2005), but the efficacy of the original asiatic acid is relatively poor. Jing et al. (2015) synthesized the derivatives of Asiatic acid and evaluated their biological activities. The derivative 98, containing the proline structure, had markedly better antitumor activity (IC50 < 10 μM) than both Asiatic acid and other derivatives, with stability similar to that of its parent compound Asiatic acid. Celastrol has many important pharmacological activities, including anticancer (Zheng et al., 2014), anti-inflammatory (Jia et al., 2015), and resistance to neurodegenerative disease activity (Paris et al., 2010), but it has several drawbacks such as poor water solubility, high toxicity, and poor stability, which restrict its clinical application. Zhang et al. (2017) designed and synthesized new celastrol derivatives and evaluated their anticancer activities. In the series of amino acid derivatives, compound 99 showed the highest anticancer activity against AGS cell lines (IC50 = 0.44 μM) and showed better anticancer activity against HCT-116, BEL-7402, and HepG-2 cell lines than the other compounds. Eight novel celastrol amino acid derivatives were synthesized, which displayed strong cytotoxic activities against A549, HeLa, and HepG-2 cell lines (Pang et al., 2018). In particular, the cytotoxicities of 100 and 101 against HeLa and A549 cells with IC50 values ranging from 0.109 to 0.371 μM, the effect of cytotoxic activity was remarkably stronger than that of celastrol and cisplatin. The most potent compound 101 showed 10- and 55-fold better antiproliferative activity of anti-A549 cell than celastrol and cisplatin, respectively. Compounds 100 and 101 can induce apoptosis in A549 cells at the concentration of 0.2 μM (Figure 12).
Alkaloids are a class of nitrogen-containing natural compounds with physiological effects. Most of them have complex nitrogen heterocyclic structures, and a few are non-nitrogen heterocyclic organic amines. Alkaloids are widely distributed in nature, especially in the plant kingdom, but also in the animal kingdom, such as bufotenine from toads (Kostakis and Byard, 2009) and hypoxanthine from earthworms (Huang et al., 2016). Camptothecin (CPT) is a plant anticancer drug extracted from Camptotheca acuminata (Pu et al., 2020), which plays an important role in clinical cancer treatment, and its derivatives are a favorite of pharmaceutical chemists. CPT mainly forms a ternary complex by interacting with DNA and topoisomerase I, blocking DNA replication, leading to cell death, and thus showing significant antitumor activity (Fan et al., 1998; Cincinelli et al., 2013). The shortcomings of CPT, such as poor solubility and large side effects, have seriously affected its clinical application (Ye et al., 2012). Many researchers have synthesized a large number of CPT derivatives, hoping to find targets with fewer side effects. 10-Hydoxycamptothecin (HCPT) is a natural CPT derivative that was approved for marketing in 1988 (Guerrant et al., 2013). Meng et al. (2014) synthesized a series of amino acid derivatives to improve the antitumor activity and reduce the side effects of irinotecan. Among them, the derivative 102 (IC50 for KB cell line = 1.39 nM, LogP = 0.74) of valine and HCPT not only maintained the tumor proliferation inhibitory activity equivalent to HCPT (IC50 for KB cell line = 1.48 nM), but also its water solubility was significantly improved compared with LogP of HCPT of 1.53 (Figure 13).
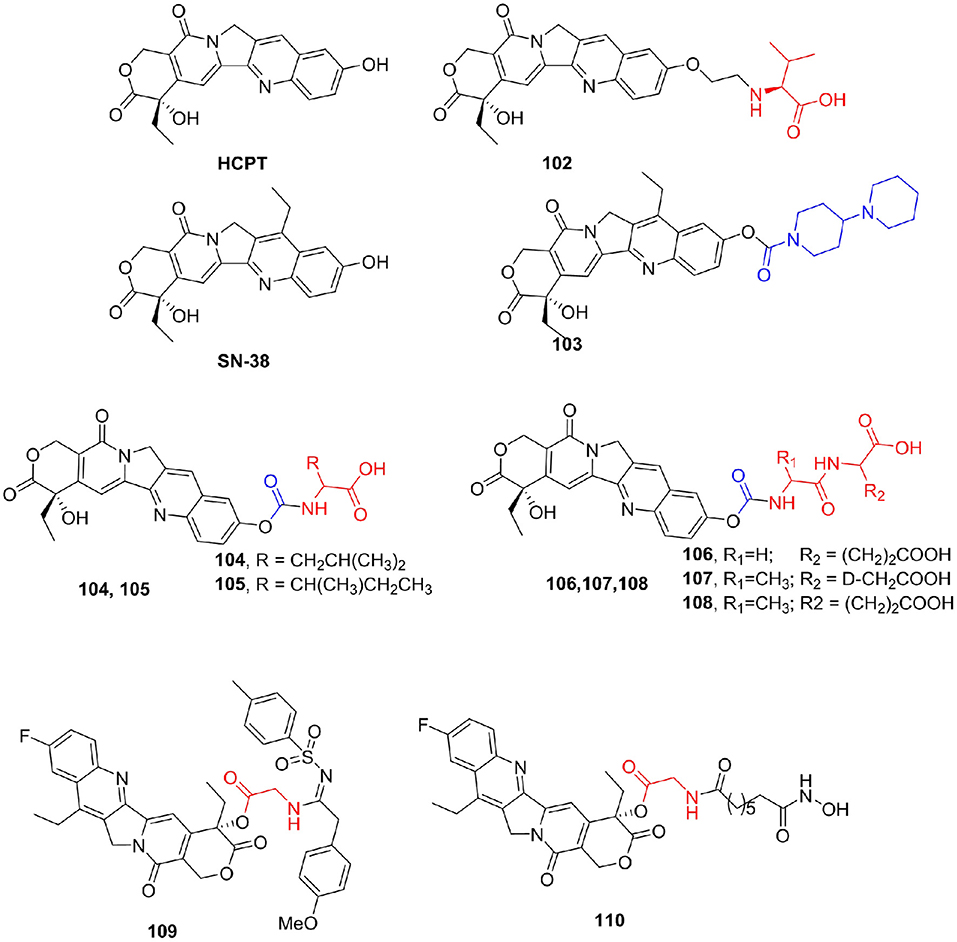
Figure 13. Camptothecin derivatives.
Irinotecan (103) is a water-soluble prodrug of the CPT derivative SN-38. However, high individual variation in efficacy, toxicity, and side effects of irinotecan preclude its clinical use (Dodds and Rivory, 1999). Novel derivatives of SN-38 were synthesized by conjugating amino acids to the 10-hydroxyl group via a carbamate linkage (Zhou et al., 2014). All prodrug compounds showed much greater in vitro antitumor activities against SGC-7901 cells and HeLa cells than irinotecan. The most active compounds 104–107 exhibited IC50 values that were 1,000 times lower against HeLa cells and 30 times lower against SGC-7901 cells than that of irinotecan, and the inhibitory activities of these prodrugs against AchE were remarkably reduced, with IC50 values more than 6.8 times better than that of irinotecan. In addition, compound 108 showed the same level of tumor growth inhibitory activity (Inhibitory Rate = 51%) as irinotecan (Inhibitory Rate = 51%) in vivo. In an attempt to improve the antitumor activity of camptothecins (CPTs), novel 10-fluoro-CPT derivatives were designed, synthesized, and evaluated for cytotoxicity against various human cancer cell lines (Yang et al., 2017). Remarkably, compound 109 (IC50 = 99.2 nM) displayed the strongest cytotoxicity against the MDR KB-VIN cell line. Yu et al. (2012) prepared a series of CPT prodrugs that exhibit histone deacetylase (HDAC) inhibition activity based on the synergistic effect between camptothecin derivatives and HDAC inhibitors. Compound 110 exhibited strong antiproliferative activity in A549 (IC50 = 137.7 nM) and HCT-116 (IC50 = 30.5 nM) cell lines (Figure 13).
Based on the reported anti-Leishmanial activity of the natural alkaloid piperine (Kapil, 1993), the combination of piperine is synthesized by hydrolyzing piperine to piperonic acid and then reacting with amino acid methyl ester (Singh et al., 2010). All the compounds showed better activity than piperine. Piperoyl-valine methyl ester (111) was the strongest active compound with an IC50 of 0.075 mM against the amastigotes.
Liguzinediol (Ligu) is a para-xylene derivative of ligustrazine, an alkaloid compound, which is a potential treatment for heart failure, but its safety is low (Liu et al., 2009). To determine whether Ligu metabolites have positive inotropic effects, Zhu et al. (2018) synthesized two amino acid-linked metabolite derivatives (112, 113). Through pharmacological evaluation, Ligu's positive inotropic activity is mainly since the parent drug and its metabolites have no adverse reactions such as arrhythmia. Through pharmacokinetics, it was found that Ligu is eliminated faster in vivo and has a shorter half-life. After studying the structure–activity relationship between Ligu metabolites in vivo, Li et al. (2017) synthesized Ligu valine ester (114) prodrugs and determined the solubility of the compound (224 mg/mL) and the ester–water partition coefficient (−0.47). The experimental results show that the amino acid ester prodrug retains the solubility characteristics of the original drug, and the in vivo and in vitro experiments have obtained good experimental results, which significantly prolong the in vivo action time of Ligu; but the fat solubility of the compound was not improved (Figure 14).
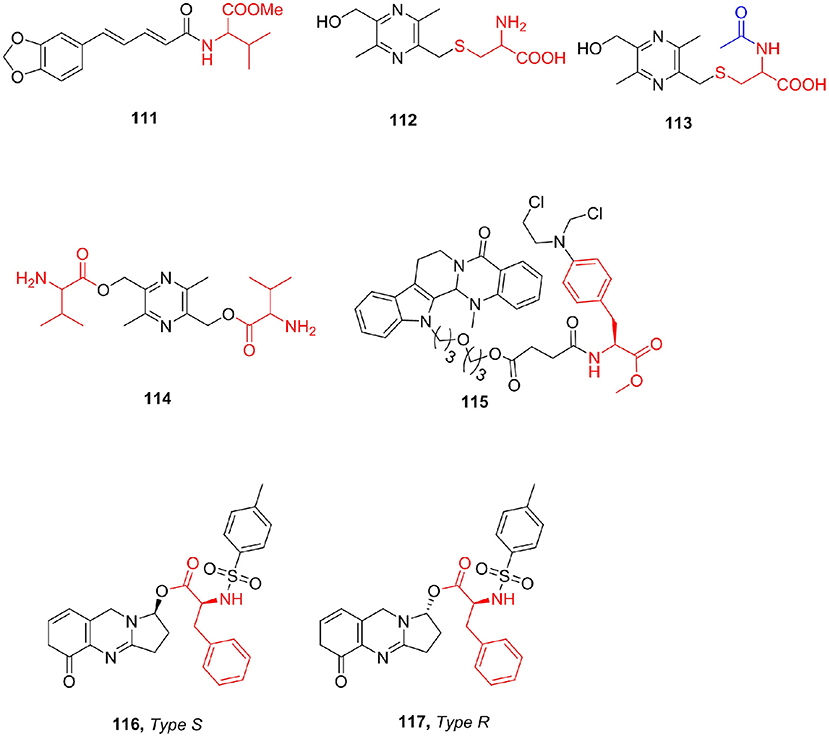
Figure 14. Alkaloids amino acid derivatives.
Evodiamine is an indole alkaloid that exists in the fruits of the traditional Chinese herb Evodiae fructus, which is widely used to treat a variety of human diseases (Yu et al., 2013). Although evodiamine showed good antitumor activity, the development of evodiamine for clinical cancer treatment was stunted by its relatively moderate potency. Hu et al. (2017) reported a series of novel evodiamine with antiproliferative properties. All derivatives showed antiproliferative activities against tested human tumor cell to some extent and almost non-toxic (>200 μM) to human normal PBMCs. Compound 115 showed the strongest cytotoxicity against two tumor cell lines (THP-1 and HL-60) with IC50 values of 4.05 and 0.50 μM, respectively, and selected for further mechanism study in HL-60 cells, and compound 115 could induce HL-60 cell apoptosis and arrest at the G2 phase at low concentrations. (±)-Vasicinone was isolated from the seeds of P. harmala. Filali et al. (2019) reported the synthesis of a new series of α-amino acid ester derivatives of (±)-vasicinone and tested for their in vitro AChE, anti-5-LOX, and cytotoxic activities (MCF-7, OVCAR-3, and HCT-116 cell lines). Most of the derivatives showed cytotoxic activity against the three cell lines used. Only two diastereoisomers, 116 and 117, exhibited activity against the 5- LOX enzyme with IC50 values of 63 and 79 μM, respectively (Figure 14).
Danshensu is one of the main water-soluble active ingredients of Salvia miltiorrhiza Bunge (Danshen), which has been applied in clinical practice for centuries in Asia (Zhang et al., 2010). After the hydroxyl group of Danshensu is esterified or etherified, Danshensu is not easily oxidized, fat solubility is improved, and toxicity is reduced. Jia et al. (2012) designed and synthesized a series of novel amide and thioester conjugates between Danshensu and cysteine derivatives under the guidance of “medicinal chemical hybridization” (MCH). The amide conjugates 118–120 displayed widely protective effects against H2O2-HUVECs. Pretreatment with these derivatives could increase GSH levels and decrease MDA levels. Salvianolic acid C, a polyphenolic benzofuran derivative that exists in Danshen, is reported to have a potent XO inhibitory activity (Fu Y. et al., 2014). Nevertheless, few have attempted to study salvianolic acid C except for the total synthesis and pharmacokinetic study of rat plasma (Shen et al., 2012). A series of 2-arylbenzo[b]furan derivatives were prepared based on salvianolic acid C and tested for XO inhibitory and antioxidant activities (Tang et al., 2016). Compounds 121–124 showed effective XO inhibitory activities with IC50 values ranging from 4.0 to 6.4 mM, which were comparable with that of allopurinol. The representative derivative 123 could bind to either XO or the xanthine oxidase–xanthine complex, showing a mixed-type competitive mechanism (Figure 15).
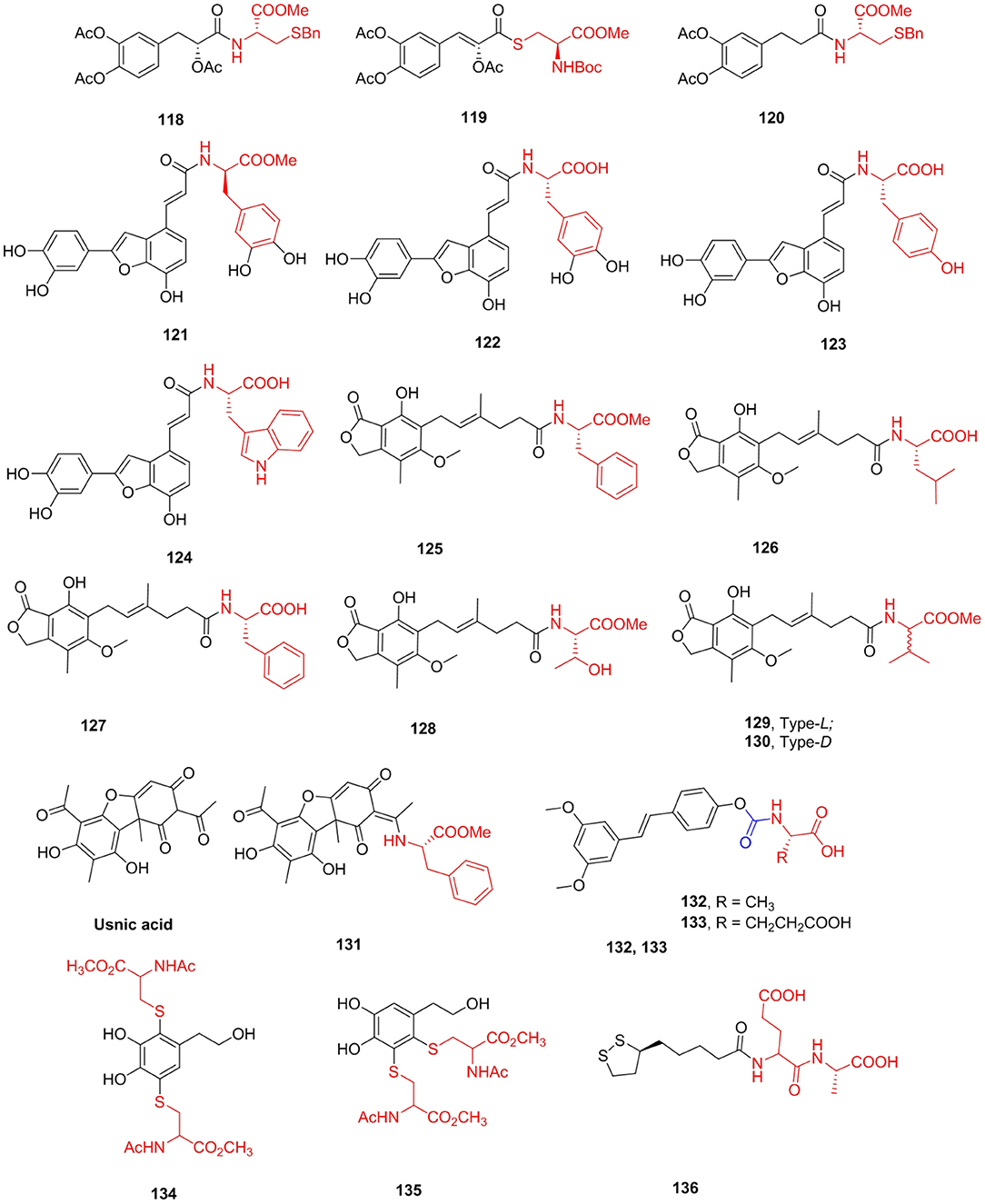
Figure 15. Polyphenols amino acid derivatives.
Mycophenolic acid (MPA), first isolated in 1896 by Gosio from Pennicillium stoloniferum and was probably the first antibiotic (Bentley, 2000), is one of the most effective inhibitors of hIMPDH (human IMPDH; Ki = 7 nM) (Hedstrom, 2009). MPA reduces the availability of guanine nucleotides, especially GTP. Mycophenolate mofetil (MMF, CellCept) and mycophenolate sodium (MPS, Myfortic) are used clinically as immunosuppressants. Many structural modifications were made to MPA, but only a few showed similar or better immunosuppressive activity. Iwaszkiewicz-Grzes et al. (2013) synthesized 11 amino acid derivatives of MPA and evaluated new analogs as growth inhibitors of Jurkat cells and hPBMCs cells from healthy donors. The cytotoxicity depends on the substituent and configuration of the chiral center in the amino acid unit. Compounds 125 (SI = 2871.4), 126 (SI = 33,787.9), and 127 (SI = 7166.7) exhibited higher potency than MPA in vitro. In addition, Siebert et al. (2018) obtained novel amino acid MPA derivatives as potential antibacterial agents. Peptide derivatives proved to be the most versatile, and their strains were relative to most MIC values lower than MPA. Among them, the MIC value of compound 128 against S. aureus MSSA ATCC 25923 was 8 μg/mL, which was almost equivalent to kanamycin (MIC = 7.8 μg/mL) and significantly stronger than that of MPA (MIC > 178 μg/mL). As can be seen, the activity of amino acid derivatives depends on the configuration at the chiral center in the amino acid unit, and methyl esters revealed better antimicrobial activity than derivatives with free carboxylic groups. Felczak et al. (2014) prepared mycophenolic adenine dinucleotide analogs linked by amino acids. Mycophenolic-(L)- and (D)-valine adenine diamide derivatives 129 (Ki = 9 nM) and 130 (Ki = 3 nM) were found to be very potent enzymatically but did not inhibit the proliferation of cancer cells (Figure 15).
Usnic acid (UA), isolated from lichen, has a variety of biological activities (Galanty et al., 2017), including antiparasitic activity. However, the toxicity of usnic acid, especially liver failure (Piska et al., 2018) and cultured mouse hepatocyte necrosis (Foti et al., 2008), has also been reported. Nevertheless, poor solubility in water (Erba et al., 1998) limits the application of usnic acid to some extent. Guo et al. (2019) synthesized some (+)-usnic acid derivatives containing amino acids and evaluated their anti-Toxoplasma activity. Compared with positive control drugs sulfadiazine (selectivity 1.15), pyrimethamine (selectivity index = 0.89), spiramycin (selectivity index = 0.72), and the lead compound (+)-usnic acid (selectivity index = 0.96), compound 131 showed the most effective anti-T. gondii activity (selectivity index > 2.77). Furthermore, compound 131 had better inhibitory effects on T. gondii (inhibition rates 76.0%) in vivo than spiramycin (inhibition rate 55.2%). The number of tachyzoites was significantly reduced (p < 0.001) and reduced hepatotoxicity and significantly increased antioxidative effects in comparison to the normal group (Figure 15).
The peroxisome proliferator-activated receptor subtype α (PPARα) has been established as a target in drug discovery studies of new lipid-lowering drugs. Pterostilbene is a naturally occurring PPARα agonist that has been shown to decrease plasma lipid concentrations through activation of PPARα (Kim and Chong, 2013). Of the conjugates investigated, 4-OMe-pterostilbene had a lower activating effect than pterostilbene, but the pterostilbenes with either amino acids 132 (Fl = 18.4) and 133 (Fl = 18.9) demonstrated a small but remarkable increase in PPARα activity compared with pterostilbene (Fl = 17.1) (Figure 15).
Polyphenols are one of the biologically active substances in many plant-derived foods. They have effective reducing properties, which can inhibit the oxidative deterioration of food. Previous attempts have been made to use this polyphenol to avoid oxidative browning of MbO2. However, the effective antioxidant polyphenols accelerated the oxidation of MbO2 to MetMb (Masuda et al., 2013). Honda et al. (2016) synthesized some polyphenols linked to amino acid compounds. Dydroxytyrosol substituted with dicysteine 134 and 135 showed the most effective reduction effect on MetMb and no oxidation effect on MbO2 (Figure 15).
Lipoic acid (LA) is a pharmacophore with unique antioxidant (Whiteman et al., 1996) and cytoprotective properties (Pugazhenthi et al., 2007). Based on its chemical and biological properties, LA is an attractive candidate for modification. Kates et al. (2014) prepared and screened a selection of LA analogs for cytoprotective activity. Compound 136 was selected based on these attributes as well as a favorable ADMET and receptor screen profile for further preclinical and clinical investigation, which showed statistically remarkable protection against myocardial damage associated with percutaneous coronary intervention (Figure 15).
Natural products have long been considered an important source of effective therapeutic drugs. About 60% of the world's population relies on herbal medicines and natural medicines to treat diseases. With natural products as lead compounds, the relationship between structure and biological activity and metabolic research can be further carried out for structural modification and analog synthesis, which can improve drug efficacy and reduce toxicity. Amino acids are an important class of bioorganic molecules. They have the characteristics of structural diversity, ease of availability, and easy connection with other amino acids and biologically active molecules. They can effectively change the low solubility and biological activity of natural products. The introduction of amino acids into natural compounds can often improve the solubility and increase the pharmacological activities of natural products, and a few amino acids are used to increase the selectivity of these products. Certain compounds, such as anthraquinones and flavonoids, are difficult to dissolve in water. The introduction of amino acids into their structures and the preparation of amino acid hydrochlorides can greatly improve their solubility. Some compounds have strong activity. For example, PPT lacks favorable selectivity to normal cells, which leads to its increased toxicity and side effects. After the introduction of amino acids, its selectivity increases and cytotoxicity to normal cells decreases. Regarding the amino acid-protecting group, N-Boc, there is no clear conclusion about whether it needs to be deprotected at the end when the structure is modified.
According to previous studies, natural products have diverse structure types and pharmacological activities. For compounds with weak biological activity and low water solubility, we can first consider amino acids. For compounds with strong activity and high toxicity, we can consider adding amino acids to prepared prodrugs to reduce toxicity. When we modify the structure of a natural compound, we should first fully understand the various properties of the natural product, such as solubility, reported pharmacological activity, and structural modification. Combined with computer-simulated drug design and rapid determination of activity, researchers can quickly and efficiently modify the directional structure of natural compounds. In the structural modification of natural products, the application of amino acids can improve many properties. Therefore, amino acids have great application prospects in the structural modification of natural products.
QX and HD conceptualized the project. QX, HD, and XL contributed to the development and writing of the manuscript. XL and Z-SQ contributed in validating, reviewing, and supervising the project. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No. 81960626).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Thanks for the language editing service provided by the Cactus Communications (Shanghai) Co., Ltd. for the manuscript.
Amoussa, A. M., Lagnika, L., Bourjot, M., Vonthron-Senecheau, C., and Sanni, A. (2016). Triterpenoids from Acacia ataxacantha DC: antimicrobial and antioxidant activities. BMC Complement Altern. Med. 16:284. doi: 10.1186/s12906-016-1266-y
Anbharasi, V., Cao, N., and Feng, S. S. (2010). Doxorubicin conjugated to D-alpha-tocopheryl polyethylene glycol succinate and folic acid as a prodrug for targeted chemotherapy. J. Biomed. Mater Res. A 94, 730–743. doi: 10.1002/jbm.a.32734
Baloch, S. K., Ma, L., Wang, X. L., Shi, J., Zhu, Y., Wu, F. Y., et al. (2015). Design, synthesis and mechanism of novel shikonin derivatives as potent anticancer agents. Rsc. Adv. 5, 31759–31767. doi: 10.1039/C5RA01872B
Baltina, L. (2003). Chemical modification of glycyrrhizic acid as a route to new bioactive compounds for medicine. Curr. Med. Chem. 10, 155–171. doi: 10.2174/0929867033368538
Barnham, K. J., Djuran, M. I., Murdoch, P. S., Ranford, J. D., and Sadler, P. J. (1996). Ring-opened adducts of the anticancer drug carboplatin with sulfur amino acids. Inorg. Chem. 35, 1065–1072. doi: 10.1021/ic950973d
Bentley, R. (2000). Mycophenolic Acid: a one hundred year odyssey from antibiotic to immunosuppressant. Chem. Rev. 100, 3801–3826. doi: 10.1021/cr990097b
Bi, W., Bi, Y., Gao, X., Yan, X., Zhang, Y., Harris, J., et al. (2016). Pharmacological protection of mitochondrial function mitigates acute limb ischemia/reperfusion injury. Bioorg. Med. Chem. Lett. 26, 4042–4051. doi: 10.1016/j.bmcl.2016.06.079
Bi, Y., Xu, J., Wu, X., Ye, W., Yuan, S., and Zhang, L. (2007). Synthesis and cytotoxic activity of 17-carboxylic acid modified 23-hydroxy betulinic acid ester derivatives. Bioorg. Med. Chem. Lett. 17, 1475–1478. doi: 10.1016/j.bmcl.2006.09.096
Bi, Y., Yang, X., Zhang, T., Liu, Z., Zhang, X., Lu, J., et al. (2015). Design, synthesis, nitric oxide release and antibacterial evaluation of novel nitrated ocotillol-type derivatives. Eur. J. Med. Chem. 101, 71–80. doi: 10.1016/j.ejmech.2015.06.021
Borthwick, A. D., Davies, D. E., Ertl, P. F., Exall, A. M., Haley, T. M., Hart, G. J., et al. (2003). Design and synthesis of pyrrolidine-5,5'-trans-lactams (5-oxo-hexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 4. Antiviral activity and plasma stability. J. Med. Chem. 46, 4428–4449. doi: 10.1021/jm030810w
Brady, S. F., Pawluczyk, J. M., Lumma, P. K., Feng, D. M., Wai, J. M., Jones, R., et al. (2002). Design and synthesis of a pro-drug of vinblastine targeted at treatment of prostate cancer with enhanced efficacy and reduced systemic toxicity. J. Med. Chem. 45, 4706–4715. doi: 10.1021/jm020139f
Cai, E., Guo, S., Yang, L., Han, M., Xia, J., Zhao, Y., et al. (2018). Synthesis and antitumour activity of arctigenin amino acid ester derivatives against H22 hepatocellular carcinoma. Nat. Prod. Res. 32, 406–411. doi: 10.1080/14786419.2017.1314279
Cai, E. B., Yang, L. M., Jia, C. X., Zhang, W. Y., Zhao, Y., Li, W., et al. (2016). The synthesis and evaluation of arctigenin amino acid ester derivatives. Chem. Pharm. Bull. 64, 1466–1473. doi: 10.1248/cpb.c16-0429
Cao, Y. K., Li, H. J., Song, Z. F., Li, Y., and Huai, Q. Y. (2014). Synthesis and biological evaluation of novel curcuminoid derivatives. Molecules 19, 16349–16372. doi: 10.3390/molecules191016349
Carroux, C. J., Moeker, J., Motte, J., Lopez, M., Bornaghi, L. F., Katneni, K., et al. (2013). Synthesis of acylated glycoconjugates as templates to investigate in vitro biopharmaceutical properties. Bioorg. Med. Chem. Lett. 23, 455–459 doi: 10.1016/j.bmcl.2012.11.056
Carubelli, V., Castrini, A. I., Lazzarini, V., Gheorghiade, M., Metra, M., and Lombardi, C. (2015). Amino acids and derivatives, new treatment of chronicheart failure? Heart Fail Rev. 20, 39–51. doi: 10.1007/s10741-014-9436-9
Cha, Y. F., Zhang, S., Su, H., Ou, Y., Fu, X. Z., Jiang, F. J., et al. (2014). L-Amino acid carbamate prodrugs of scutellarin: synthesis, physiochemical property, Caco-2 cell permeability, and in vitro anti-oxidative activity. Med. Chem. Res. 24, 2238–2246. doi: 10.1007/s00044-014-1286-4
Chattopadhyay, D., Arunachalam, G., Mandal, A. B., Sur, T. K., Mandal, S. C., and Bhattacharya, S. K. (2002). Antimicrobial and anti-inflammatory activity of folklore: Mallotus peltatus leaf extract. J. Ethnopharmacol. 82, 229–237. doi: 10.1016/S0378-8741(02)00165-4
Chen, A., Weber, I. T., Harrison, R. W., and Leis, J. (2006). Identification of amino acids in HIV-1 and avian sarcoma virus integrase subsites required for specific recognition of the long terminal repeat Ends. J. Biol. Chem. 281, 4173–4182. doi: 10.1074/jbc.M510628200
Chen, S. H., Wang, X. L., Xie, X. H., Zheng, L. Z., Yao, D., Wang, D. P., et al. (2012). Comparative study of osteogenic potential of a composite scaffold incorporating either endogenous bone morphogenetic protein-2 or exogenous phytomolecule icaritin: an in vitro efficacy study. Acta Biomater. 8, 3128–3137. doi: 10.1016/j.actbio.2012.04.030
Chen, X., Cui, L., Duan, X., Ma, B., and Zhong, D. (2006). Pharmacokinetics and metabolism of the flavonoid scutellarin in humans after a single oral administration. Drug Metab. Dispos. 34, 1345–1352. doi: 10.1124/dmd.106.009779
Chen, X., Yang, L., Oppenheim, J. J., and Howard, M. Z. (2002). Cellular pharmacology studies of shikonin derivatives. Phytother. Res. 16, 199–209. doi: 10.1002/ptr.1100
Chen, Z., Duan, H., Wang, M., Han, L., Liu, Y., Zhu, Y., et al. (2015). Synthesis, cytotoxicity and haemolytic activity of Pulsatilla saponin A, D derivatives. Bioorg. Med. Chem. Lett. 25, 2550–2554. doi: 10.1016/j.bmcl.2015.04.049
Choi, Y. H., Baek, J. H., Yoo, M. A., Chung, H. Y., Kim, N. D., and Kim, K. W. (2000). Induction of apoptosis by ursolic acid through activation of caspases and down-regulation of c-IAPs in human prostate epithelial cells. Int. J. Oncol. 17, 565–571. doi: 10.3892/ijo.17.3.565
Cincinelli, R., Musso, L., Dallavalle, S., Artali, R., Tinelli, S., Colangelo, D., et al. (2013). Design, modeling, synthesis and biological activity evaluation of camptothecin-linked platinum anticancer agents. Eur. J. Med. Chem. 63, 387–400. doi: 10.1016/j.ejmech.2013.02.022
Criado, M., Balsera, B., Mulet, J., Sala, S., Sala, F., de la Torre-Martinez, R., et al. (2016). 1,3-diphenylpropan-1-ones as allosteric modulators of alpha7 nACh receptors with analgesic and antioxidant properties. Future Med. Chem. 8, 731–749. doi: 10.4155/fmc-2015-0001
Csuk, R., Schwarz, S., Kluge, R., and Strohl, D. (2011a). Improvement of the cytotoxicity and tumor selectivity of glycyrrhetinic acid by derivatization with bifunctional amino acids. Arch Pharm. 344, 505–513. doi: 10.1002/ardp.201100030
Csuk, R., Schwarz, S., Siewert, B., Kluge, R., and Strohl, D. (2011b). Synthesis and antitumor activity of ring A modified glycyrrhetinic acid derivatives. Eur. J. Med. Chem. 46, 5356–5369. doi: 10.1016/j.ejmech.2011.08.038
Cui, X. R., Tsukada, M., Suzuki, N., Shimamura, T., Gao, L., Koyanagi, J., et al. (2008). Comparison of the cytotoxic activities of naturally occurring hydroxyanthraquinones and hydroxynaphthoquinones. Eur. J. Med. Chem. 43, 1206–1215. doi: 10.1016/j.ejmech.2007.08.009
Dang, Z., Qian, K., Ho, P., Zhu, L., Lee, K. H., Huang, L., et al. (2012). Synthesis of betulinic acid derivatives as entry inhibitors against HIV-1 and bevirimat-resistant HIV-1 variants. Bioorg. Med. Chem. Lett. 22, 5190–5194. doi: 10.1016/j.bmcl.2012.06.080
Deng, H., Huang, X., Jin, C., Jin, C. M., and Quan, Z. S. (2020). Synthesis, in vitro and in vivo biological evaluation of dihydroartemisinin derivatives with potential anti-Toxoplasma gondii agents. Bioorg. Chem. 94:103467. doi: 10.1016/j.bioorg.2019.103467
Dodds, H. M., and Rivory, L. P. (1999). The mechanism for the inhibition of acetylcholinesterases by irinotecan (CPT-11). Mol. Pharmacol. 56, 1346–1353. doi: 10.1124/mol.56.6.1346
Drag-Zalesinska, M., Kulbacka, J., Saczko, J., Wysocka, T., Zabel, M., Surowiak, P., et al. (2009). Esters of betulin and betulinic acid with amino acids have improved water solubility and are selectively cytotoxic toward cancer cells. Bioorg. Med. Chem. Lett. 19, 4814–4817. doi: 10.1016/j.bmcl.2009.06.046
Drag-Zalesinska, M., Wysocka, T., Borska, S., Drag, M., Poreba, M., Choromanska, A., et al. (2015). The new esters derivatives of betulin and betulinic acid in epidermoid squamous carcinoma treatment - in vitro studies. Biomed. Pharmacother. 72, 91–97. doi: 10.1016/j.biopha.2015.04.003
Erba, E., Pocar, D., and Rossi, L. M. (1998). New esters of R-(+)-usnic acid. Farmaco 53, 718–720. doi: 10.1016/S0014-827X(98)00113-X
Fallarero, A., Skogman, M., Kujala, J., Rajaratnam, M., Moreira, V. M., Yli-Kauhaluoma, J., et al. (2013). (+)-Dehydroabietic acid, an abietane-type diterpene, inhibits Staphylococcus aureus biofilms in vitro. Int. J. Mol. Sci. 14, 12054–12072. doi: 10.3390/ijms140612054
Fan, Y., Weinstein, J. N., Kohn, K. W., Shi, L. M., and Pommier, Y. (1998). Molecular modeling studies of the DNA-topoisomerase I ternary cleavable complex with camptothecin. J. Med. Chem. 41, 2216–2226. doi: 10.1021/jm9605445
Fang, J., Huang, T., Xia, M., Deng, L., Hao, X., Wang, Y., et al. (2018). Design and synthesis of novel monoterpenoid indole alkaloid-like analogues and their antitumour activities in vitro. Org. Biomol. Chem. 16, 3026–3037. doi: 10.1039/C8OB00677F
Felczak, K., Vince, R., and Pankiewicz, K. W. (2014). NAD-based inhibitors with anticancer potential. Bioorg. Med. Chem. Lett. 24, 332–336. doi: 10.1016/j.bmcl.2013.11.005
Filali, I., Jelassi, A., and Jannet, H. B. (2019). New bioactive esters and phosphonates semisynthesized from (±)-vasicinone: an alkaloid isolated from peganum harmala. Nat. Prod. Comm.14, 1–9. doi: 10.1177/1934578X19893544
Foti, R. S., Dickmann, L. J., Davis, J. A., Greene, R. J., Hill, J. J., Howard, M. L., et al. (2008). Metabolism and related human risk factors for hepatic damage by usnic acid containing nutritional supplements. Xenobiotica 38, 264–280. doi: 10.1080/00498250701802514
Francisco, C. S., Rodrigues, L. R., Cerqueira, N. M., Oliveira-Campos, A. M., and Rodrigues, L. M. (2012). Synthesis of novel benzofurocoumarin analogues and their anti-proliferative effect on human cancer cell lines. Eur. J. Med. Chem. 47, 370–376. doi: 10.1016/j.ejmech.2011.11.005
Fu, H. J., Zhou, Y. R., Bao, B. H., Jia, M. X., Zhao, Y., Zhang, L., et al. (2014). Tryptophan hydroxylase 1 (Tph-1)-targeted bone anabolic agents for osteoporosis. J. Med. Chem. 57, 4692–4709. doi: 10.1021/jm5002293
Fu, X. Z., Zhang, W., Wang, Y. L., Lan, Y. Y., Wang, A. M., Zhou, W., et al. (2011). Design, synthesis and anti-oxidative evaluation of L-amino acid prodrugs of scutellarein. Yao Xue Xue Bao 46, 548–555. doi: 10.1007/s11606-010-1517-4
Fu, Y., Mo, H. Y., Gao, W., Hong, J. Y., Lu, J., Li, P., et al. (2014). Affinity selection-based two-dimensional chromatography coupled with high-performance liquid chromatography-mass spectrometry for discovering xanthine oxidase inhibitors from Radix Salviae Miltiorrhizae. Anal. Bioanal. Chem. 406, 4987–4995. doi: 10.1007/s00216-014-7902-9
Galanty, A., Koczurkiewicz, P., Wnuk, D., Paw, M., Karnas, E., Podolak, I., et al. (2017). Usnic acid and atranorin exert selective cytostatic and anti-invasive effects on human prostate and melanoma cancer cells. Toxicol. in Vitro 40, 161–169. doi: 10.1016/j.tiv.2017.01.008
Garcia, E. S., and Azambuja, P. (2004). Lignoids in insects: chemical probes for the study of ecdysis, excretion and Trypanosoma cruzi-triatomine interactions. Toxicon 44, 431–440. doi: 10.1016/j.toxicon.2004.05.007
Gobec, M., Tomasic, T., Stimac, A., Frkanec, R., Trontelj, J., Anderluh, M., et al. (2018). Discovery of nanomolar desmuramylpeptide agonists of the innate immune receptor Nucleotide-Binding Oligomerization Domain-Containing Protein 2 (NOD2) possessing immunostimulatory properties. J. Med. Chem. 61, 2707–2724. doi: 10.1021/acs.jmedchem.7b01052
Guerrant, W., Patil, V., Canzoneri, J. C., Yao, L. P., Hood, R., and Oyelere, A. K. (2013). Dual-acting histone deacetylase-topoisomerase I inhibitors. Bioorg. Med. Chem. Lett. 23, 3283–3287. doi: 10.1016/j.bmcl.2013.03.108
Guo, H. Y., Jin, C. M., Zhang, H. M., Jin, C. M., Shen, Q. K., and Quan, Z. S. (2019). Synthesis and biological evaluation of (+)-usnic acid derivatives as potential anti-Toxoplasma gondii agents. J. Agric. Food Chem. 67, 9630–9642. doi: 10.1021/acs.jafc.9b02173
Hamed, A., Frese, M., Elgaafary, M., Syrovets, T., Sewald, N., Simmet, T., et al. (2020). Synthesis of novel feruloyl dipeptides with proapoptotic potential against different cancer cell lines. Bioorg. Chem. 97:103678. doi: 10.1016/j.bioorg.2020.103678
Han, H. W., Qiu, H. Y., Hu, C., Sun, W. X., Yang, R. W., Qi, J. L., et al. (2016). Design, synthesis and anti-cancer activity evaluation of podophyllotoxin-norcantharidin hybrid drugs. Bioorg. Med. Chem. Lett. 26, 3237–3242. doi: 10.1016/j.bmcl.2016.05.063
Han, X., Song, J., Lian, L. H., Yao, Y. L., Shao, D. Y., Fan, Y., et al. (2018). Ginsenoside 25-OCH3-PPD promotes activity of LXRs to ameliorate P2X7R-Mediated NLRP3 inflammasome in the development of hepatic fibrosis. J. Agric. Food Chem. 66, 7023–7035. doi: 10.1021/acs.jafc.8b01982
Hedstrom, L. (2009). IMP dehydrogenase: structure, mechanism, and inhibition. Chem. Rev. 109, 2903–2928. doi: 10.1021/cr900021w
Helfenstein, A., Vahermo, M., Nawrot, D. A., Demirci, F., Iscan, G., Krogerus, S., et al. (2017). Antibacterial profiling of abietane-type diterpenoids. Bioorg. Med. Chem. 25, 132–137. doi: 10.1016/j.bmc.2016.10.019
Honda, S., Miura, Y., Masuda, T., and Masuda, A. (2016). Effective conversion of metmyoglobin to oxymyoglobin by cysteine-substituted polyphenols. J. Agric. Food Chem. 64, 806–811. doi: 10.1021/acs.jafc.5b05511
Hu, X., Bai, Z., Qiao, J., Li, H., Xu, S., Wang, X., et al. (2019). Effective enmein-type mimics of clinical candidate HAO472: design, synthesis and biological evaluation. Eur. J. Med. Chem. 171, 169–179. doi: 10.1016/j.ejmech.2019.03.046
Hu, X., Gao, C., Tan, C., Zhang, C., Zhang, H., Li, S., et al. (2011). Design and synthesis of N-phosphoryl peptide modified podophyllotoxin derivatives as potent anticancer agents. Protein Pept. Lett. 18, 1258–1264. doi: 10.2174/092986611797642652
Hu, X., Wang, Y., Xue, J., Han, T., Jiao, R., Li, Z., et al. (2017). Design and synthesis of novel nitrogen mustard-evodiamine hybrids with selective antiproliferative activity. Bioorg. Med. Chem. Lett. 27, 4989–4993. doi: 10.1016/j.bmcl.2017.10.014
Huang, C. Q., Li, W., Wu, B., Chen, W. M., Chen, L. H., Mo, G. W., et al. (2016). Pheretima aspergillum decoction suppresses inflammation and relieves asthma in a mouse model of bronchial asthma by NF-kappaB inhibition. J. Ethnopharmacol. 189, 22–30. doi: 10.1016/j.jep.2016.05.028
Huang, L., Ho, P., Lee, K. H., and Chen, C. H. (2006). Synthesis and anti-HIV activity of bi-functional betulinic acid derivatives. Bioorg. Med. Chem. 14, 2279–2289. doi: 10.1016/j.bmc.2005.11.016
Huang, R. Z., Jin, L., Yao, G. Y., Dai, W. L., Huang, X. C., Liao, Z. X., et al. (2017). Synthesis and molecular docking study of novel alizarin derivatives containing phosphoryl amino acid moiety as potential antitumor agents. Med. Chem. Res. 26, 2363–2374. doi: 10.1007/s00044-017-1938-2
Ikeda, M. (2003). Amino acid production processes. Adv. Biochem. Eng. Biotechnol. 79, 1–35. doi: 10.1007/3-540-45989-8_1
Imbert, T. F. (1998). Discovery of podophyllotoxins. Biochimie 80, 207–222. doi: 10.1016/S0300-9084(98)80004-7
Iwaszkiewicz-Grzes, D., Cholewinski, G., Kot-Wasik, A., Trzonkowski, P., and Dzierzbicka, K. (2013). Synthesis and biological activity of mycophenolic acid-amino acid derivatives. Eur. J. Med. Chem. 69, 863–871. doi: 10.1016/j.ejmech.2013.09.026
Jeong, Y. H., Chung, S. Y., Han, A. R., Sung, M. K., Jang, D. S., Lee, J., et al. (2007). P-glycoprotein inhibitory activity of two phenolic compounds, (-)-syringaresinol and tricin from Sasa borealis. Chem. Biodivers. 4, 12–16. doi: 10.1002/cbdv.200790001
Ji, Q., Ge, Z., Ge, Z., Chen, K., Wu, H., Liu, X., et al. (2016). Synthesis and biological evaluation of novel phosphoramidate derivatives of coumarin as chitin synthase inhibitors and antifungal agents. Eur. J. Med. Chem. 108, 166–176. doi: 10.1016/j.ejmech.2015.11.027
Jia, Y., Dong, X., Zhou, P., Liu, X., Pan, L., Xin, H., et al. (2012). The synthesis and biological evaluation of novel Danshensu-cysteine analog conjugates as cardiovascular-protective agents. Eur. J. Med. Chem. 55, 176–187. doi: 10.1016/j.ejmech.2012.07.016
Jia, Z., Xu, C., Shen, J., Xia, T., Yang, J., and He, Y. (2015). The natural compound celastrol inhibits necroptosis and alleviates ulcerative colitis in mice. Int. Immunopharmacol. 29, 552–559. doi: 10.1016/j.intimp.2015.09.029
Jiang, F. J., Fu, X. Z., Wang, S. W., Huang, Y., Zhou, W., Wang, A. M., et al. (2013). Synthesis and physiochemical property evaluation of carbamate derivatives of scutellarin methyl ester. Chin. Chem. Lett. 24, 338–340. doi: 10.1016/j.cclet.2013.02.003
Jin, X., Zheng, C. J., Song, M. X., Wu, Y., Sun, L. P., Li, Y. J., et al. (2012). Synthesis and antimicrobial evaluation of L-phenylalanine-derived C5-substituted rhodanine and chalcone derivatives containing thiobarbituric acid or 2-thioxo-4-thiazolidinone. Eur. J. Med. Chem. 56, 203–209. doi: 10.1016/j.ejmech.2012.08.026
Jing, Y., Wang, G., Ge, Y., Xu, M., and Gong, Z. (2015). Synthesis, anti-tumor and anti-angiogenic activity evaluations of asiatic Acid amino Acid derivatives. Molecules 20, 7309–7324. doi: 10.3390/molecules20047309
Kapil, A. (1993). Piperine: a potent inhibitor of Leishmania donovani promastigotes in vitro. Planta Med. 59, 474. doi: 10.1055/s-2006-959737
Kates, S. A., Casale, R. A., Baguisi, A., and Beeuwkes, R. (2014). Lipoic acid analogs with enhanced pharmacological activity. Bioorg. Med. Chem. 22, 505–512. doi: 10.1016/j.bmc.2013.10.057
Kawaguchi, T., Izumi, N., Charlton, M. R., and Sata, M. (2011). Branched-chain amino acids as pharmacological nutrients in chronic liver disease. Hepatology 54, 1063–1070. doi: 10.1002/hep.24412
Kim, M. K., and Chong, Y. (2013). Design, synthesis, and biological evaluation of resveratrol derivatives as PPAR alpha agonists. J. Korean Soc. Applied Biol. Chem. 56, 353–356. doi: 10.1007/s13765-013-3086-9
Kim, M. K., Choo, H., and Chong, Y. (2014). Water-soluble and cleavable quercetin-amino acid conjugates as safe modulators for P-glycoprotein-based multidrug resistance. J. Med. Chem. 57, 7216–7233. doi: 10.1021/jm500290c
Kim, M. K., Kim, Y., Choo, H., and Chong, Y. (2017). Quercetin-glutamic acid conjugate with a non-hydrolysable linker; a novel scaffold for multidrug resistance reversal agents through inhibition of P-glycoprotein. Bioorg. Med. Chem. 25, 1219–1226. doi: 10.1016/j.bmc.2016.12.034
Kim, M. K., Park, K. S., Yeo, W. S., Choo, H., and Chong, Y. (2009). In vitro solubility, stability and permeability of novel quercetin-amino acid conjugates. Bioorg. Med. Chem. 17, 1164–1171. doi: 10.1016/j.bmc.2008.12.043
Kostakis, C., and Byard, R. W. (2009). Sudden death associated with intravenous injection of toad extract. Forensic. Sci. Int. 188, 1–5. doi: 10.1016/j.forsciint.2009.02.006
Kotha, R. R., and Luthria, D. L. (2019). Curcumin: biological, pharmaceutical, nutraceutical, and analytical aspects. Molecules 24::2930. doi: 10.3390/molecules24162930
Lan, P., Wang, J., Zhang, D. M., Shu, C., Cao, H. H., Sun, P. H., et al. (2011). Synthesis and antiproliferative evaluation of 23-hydroxybetulinic acid derivatives. Eur. J. Med. Chem. 46, 2490–2502. doi: 10.1016/j.ejmech.2011.03.038
LaVail, J. H., Tauscher, A. N., Aghaian, E., Harrabi, O., and Sidhu, S. S. (2003). Axonal transport and sorting of herpes simplex virus components in a mature mouse visual system. J. Virol. 77, 6117–6126. doi: 10.1128/JVI.77.11.6117-6126.2003
Lebeaux, D., Chauhan, A., Rendueles, O., and Beloin, C. (2013). From in vitro to in vivo models of bacterial biofilm-related infections. Pathogens. 2, 288–356. doi: 10.3390/pathogens2020288
Lee, C. C., Huang, K. F., Chang, D. M., Hsu, J. J., Huang, F. C., Shih, K. N., et al. (2012). Design, synthesis and evaluation of telomerase inhibitory, hTERT repressing, and anti-proliferation activities of symmetrical 1,8-disubstituted amidoanthraquinones. Eur. J. Med. Chem. 50, 102–112. doi: 10.1016/j.ejmech.2012.01.044
Lee, K. H. (2004). Current developments in the discovery and design of new drug candidates from plant natural product leads. J. Nat. Prod. 67, 273–283. doi: 10.1021/np030373o
Li, L., Liu, W. Y., Feng, F., Wu, C. Y., and Xie, N. (2013). Synthesis and in vitro cytotoxicity evaluation of baicalein amino acid derivatives. Chin. J. Nat. Med. 11, 284–288. doi: 10.3724/SP.J.1009.2013.00284
Li, X., Zhang, X., Sun, H., Zhang, L., Gao, Y., Wang, J., et al. (2012). Synthesis and anti-tumor evaluation of novel C-37 modified derivatives of gambogic acid. Chin. J. Chem. 30, 1083–1091. doi: 10.1002/cjoc.201100693
Li, Y. Q., Yang, F., Wang, L., Cao, Z., Han, T. J., Duan, Z. A., et al. (2016). Phosphoramidate protides of five flavones and their antiproliferative activity against HepG2 and L-O2 cell lines. Eur. J. Med. Chem. 112, 196–208. doi: 10.1016/j.ejmech.2016.02.012
Li, Z., Shen, M. Z., and Li, W. (2017). Design, synthesis and druggability evaluation of liguzinediol valine ester prodrug as promissing inotropic agent. J. Nanjing Univ. Tradit Chin. Med. 33, 412–416. doi: 10.14148/j.issn.1672-0482.2017.0412
Lin, H. Y., Chen, W., Shi, J., Kong, W. Y., Qi, J. L., Wang, X. M., et al. (2013). Design, synthesis and biological evaluation of cinnamic acyl shikonin derivatives. Chem. Biol. Drug Des. 81, 275–283. doi: 10.1111/cbdd.12077
Lin, H. Y., Li, Z. K., Bai, L. F., Baloch, S. K., Wang, F., Qiu, H. Y., et al. (2015). Synthesis of aryl dihydrothiazol acyl shikonin ester derivatives as anticancer agents through microtubule stabilization. Biochem. Pharmacol. 96, 93–106. doi: 10.1016/j.bcp.2015.04.021
Lin, L. L., Liu, A. J., Liu, J. G., Yu, X. H., Qin, L. P., and Su, D. F. (2007). Protective effects of scutellarin and breviscapine on brain and heart ischemia in rats. J. Cardiovasc. Pharmacol. 50, 327–332. doi: 10.1097/FJC.0b013e3180cbd0e7
Lin, S., Koh, J. J., Aung, T. T., Sin, W. L. W., Lim, F., Wang, L., et al. (2017). Semisynthetic flavone-derived antimicrobials with therapeutic potential against Methicillin-Resistant Staphylococcus aureus (MRSA). J. Med. Chem. 60, 6152–6165. doi: 10.1021/acs.jmedchem.7b00380
Liu, X., Niu, H., Li, Q., and Gu, P. (2019). Metabolic engineering for the production of l-phenylalanine in Escherichia coli. 3 Biotech 9:85. doi: 10.1007/s13205-019-1619-6
Liu, X. K., Ye, B. J., Wu, Y., Lin, Z. H., Zhao, Y. Q., and Piao, H. R. (2011). Synthesis and anti-tumor evaluation of panaxadiol derivatives. Eur. J. Med. Chem. 46, 1997–2002. doi: 10.1016/j.ejmech.2011.02.022
Liu, Z., Xiao, M., Waters, J., and Velazquez, O. C. (2009). QS248. a novel approach in regulating neovascularization: targeting stromal fibroblasts by modulation of Notch1 signaling. J. Surg. Res. 151, 293–293. doi: 10.1016/j.jss.2008.11.551
Lo, Y. L., Ho, C. T., and Tsai, F. L. (2008). Inhibit multidrug resistance and induce apoptosis by using glycocholic acid and epirubicin. Eur. J. Pharm. Sci. 35, 52–67. doi: 10.1016/j.ejps.2008.06.003
Ma, C. M., Kully, M., Khan, J. K., Hattori, M., and Daneshtalab, M. (2007). Synthesis of chlorogenic acid derivatives with promising antifungal activity. Bioorg. Med. Chem. 15, 6830–6833. doi: 10.1016/j.bmc.2007.07.038
Ma, C. M., Nakamura, N., Hattori, M., Kakuda, H., Qiao, J. C., and Yu, H. L. (2000). Inhibitory effects on HIV-1 protease of constituents from the wood of Xanthoceras sorbifolia. J. Nat. Prod. 63, 238–242. doi: 10.1021/np9902441
Malysheva, Y. B., Voitovich, Y. V., Sharonova, E. A., Combes, S., Svirshchevskaya, E. V., Vodovozova, E. L., et al. (2014). Novel water-soluble anticancer agents derived from 4-arylcoumarins. Russ. Chem. Bull. 62, 1103–1110. doi: 10.1007/s11172-013-0149-3
Manner, S., Vahermo, M., Skogman, M. E., Krogerus, S., Vuorela, P. M., Yli-Kauhaluoma, J., et al. (2015). New derivatives of dehydroabietic acid target planktonic and biofilm bacteria in Staphylococcus aureus and effectively disrupt bacterial membrane integrity. Eur. J. Med. Chem. 102, 68–79. doi: 10.1016/j.ejmech.2015.07.038
Masuda, T., Inai, M., Miura, Y., Masuda, A., and Yamauchi, S. (2013). Effect of polyphenols on oxymyoglobin oxidation: prooxidant activity of polyphenols in vitro and inhibition by amino acids. J. Agric. Food Chem. 61, 1097–1104. doi: 10.1021/jf304775x
Meng, G., Li, J., Wang, G., Dong, M., and Zhang, Q. (2014). Synthesis and cytotoxic activities of the amino acid-conjugates of 10-Hydroxycamptothecin. Chin. J. Org. Chem. 34, 155–160. doi: 10.6023/cjoc201307049Z
Mook-Jung, I., Shin, J. E., Yun, S. H., Huh, K., Koh, J. Y., Park, H. K., et al. (1999). Protective effects of asiaticoside derivatives against beta-amyloid neurotoxicity. J. Neurosci. Res. 58, 417–425. doi: 10.1002/(SICI)1097-4547(19991101)58:3<417::AID-JNR7>3.0.CO;2-G
Moscatelli, V., Hnatyszyn, O., Acevedo, C., Megias, J., Alcaraz, M. J., and Ferraro, G. (2006). Flavonoids from Artemisia copa with anti-inflammatory activity. Planta Med. 72, 72–74. doi: 10.1055/s-2005-873177
Nicolaou, K. C., Pfefferkorn, J. A., Roecker, A. J., Cao, G. Q., Barluenga, S., and Mitchell, H. J. (2000). Natural product-like combinatorial libraries based on privileged structures. 1. general principles and solid-phase synthesis of benzopyrans. J. Amer. Chem. Soc. 122, 9939–9953. doi: 10.1021/ja002033k
Nitz, K., Lacy, M., and Atzler, D. (2019). Amino acids and their metabolism in atherosclerosis. Arterioscler. Thromb. Vasc. Bio. 39, 319–330. doi: 10.1161/ATVBAHA.118.311572
Pang, C., Luo, J., Liu, C., Wu, X., and Wang, D. (2018). Synthesis and biological evaluation of a series of novel celastrol derivatives with amino acid chain. Chem. Biodivers. 15, 1–11. doi: 10.1002/cbdv.201800059
Paris, D., Ganey, N. J., Laporte, V., Patel, N. S., Beaulieu-Abdelahad, D., Bachmeier, C., et al. (2010). Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer's disease. J. Neuroinflam. 7:17. doi: 10.1186/1742-2094-7-17
Park, B. C., Bosire, K. O., Lee, E. S., Lee, Y. S., and Kim, J. A. (2005). Asiatic acid induces apoptosis in SK-MEL-2 human melanoma cells. Cancer Lett. 218, 81–90. doi: 10.1016/j.canlet.2004.06.039
Patagar, D., Kusanur, R., Sitwala, N. D., Ghate, M. D., Saravanakumar, S., Nembenna, S., et al. (2019). Synthesis of novel 4-substituted coumarins, docking studies, and DHODH inhibitory activity. J. Heterocyclic Chem. 56, 2761–2771. doi: 10.1002/jhet.3644
Pecere, T., Gazzola, M. V., Mucignat, C., Parolin, C., and Palu, G. (2000). Aloe-emodin is a new type of anticancer agent with selective activity against neuroectodermal tumors1. Cancer Res. 60, 2800–2804.
Piska, K., Galanty, A., Koczurkiewicz, P., Zmudzki, P., Potaczek, J., Podolak, I., et al. (2018). Usnic acid reactive metabolites formation in human, rat, and mice microsomes. implication for hepatotoxicity. Food Chem. Toxicol. 120, 112–118. doi: 10.1016/j.fct.2018.07.005
Pu, X., Zhang, C. R., Gao, H. C., Gao, Y. J., Huang, L., Zhu, L., et al. (2020). Biosynthesis-inspired mining and identification of untapped alkaloids in Camptotheca acuminate for enzyme discovery using ultra-high performance liquid chromatography coupled with quadrupole-time of flight-mass spectrometry. J. Chromatogr. A 1620:461036. doi: 10.1016/j.chroma.2020.461036
Pugazhenthi, S., Akhov, L., Selvaraj, G., Wang, M., and Alam, J. (2007). Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by a PI3-kinase/Akt-mediated pathway in mouse beta-cells. Am. J. Physiol. Endocrinol. Metab. 293, 645–655. doi: 10.1152/ajpendo.00111.2007
Qian, K., Kim, S. Y., Hung, H. Y., Huang, L., Chen, C. H., and Lee, K. H. (2011). New betulinic acid derivatives as potent proteasome inhibitors. Bioorg. Med. Chem. Lett. 21, 5944–5947. doi: 10.1016/j.bmcl.2011.07.072
Qu, F., Zhao, C., Liu, Y., Cao, J., Li, W., and Zhao, Y. (2015). Semi-synthesis and anti-tumor evaluation of novel 25-hydroxyprotopanaxadiol derivatives as apoptosis inducing agents. Med. Chem. Comm. 6, 2004–2011. doi: 10.1039/C5MD00382B
Ren, Q., Yang, G., Guo, M., Guo, J., Li, Y., Lu, J., et al. (2019). Design, synthesis, and discovery of ocotillol-type amide derivatives as orally available modulators of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 161, 118–130. doi: 10.1016/j.ejmech.2018.10.038
Rodrigues, T., Reker, D., Schneider, P., and Schneider, G. (2016). Counting on natural products for drug design. Nat. Chem. 8, 531–541. doi: 10.1038/nchem.2479
Salvador, J. A. R., Leal, A. S., Valdeira, A. S., Goncalves, B. M. F., Alho, D. P. S., Figueiredo, S. A. C., et al. (2017). Oleanane-, ursane-, and quinone methide friedelane-type triterpenoid derivatives: recent advances in cancer treatment. Eur. J. Med. Chem. 142, 95–130. doi: 10.1016/j.ejmech.2017.07.013
Sarciron, M. E., Saccharin, C., Petavy, A. F., and Peyron, F. (2000). Effects of artesunate, dihydroartemisinin, and an artesunate-dihydroartemisinin combination against Toxoplasma gondii. Am. J. Trop Med. Hyg. 62, 73–76. doi: 10.4269/ajtmh.2000.62.73
Schwarz, S., and Csuk, R. (2010). Synthesis and antitumour activity of glycyrrhetinic acid derivatives. Bioorg. Med. Chem. 18, 7458–7474. doi: 10.1016/j.bmc.2010.08.054
Schwarz, S., Siewert, B., Xavier, N. M., Jesus, A. R., Rauter, A. P., and Csuk, R. (2014). A “natural” approach: synthesis and cytoxicity of monodesmosidic glycyrrhetinic acid glycosides. Eur. J. Med. Chem. 72, 78–83. doi: 10.1016/j.ejmech.2013.11.024
Shao, J. W., Dai, Y. C., Xue, J. P., Wang, J. C., Lin, F. P., and Guo, Y. H. (2011). In vitro and in vivo anticancer activity evaluation of ursolic acid derivatives. Eur. J. Med. Chem. 46, 2652–2661. doi: 10.1016/j.ejmech.2011.03.050
Shen, S. D., Zhang, G. P., Lei, M., and Hua, L. H. (2012). First total synthesis of salvianolic acid C, tournefolic acid A, and tournefolal. Arkivoc 6, 204–213. doi: 10.3998/ark.5550190.0013.619
Shi, M., Sun, J., Zhou, J., Yu, H., Yu, S., Xia, G., et al. (2018). Phase I dose escalation and pharmacokinetic study on the nanoparticle formulation of polymeric micellar paclitaxel for injection in patients with advanced solid malignancies. Invest. New Drugs 36, 269–277. doi: 10.1007/s10637-017-0506-4
Shi, Q. W., Li, L. G., Huo, C. H., Zhang, M. L., and Wang, Y. F. (2010). Study on natural medicinal chemistry and new drug development. Chin. Trad. Herbal Drugs 41, 1583–1589.
Siebert, A., Wysocka, M., Krawczyk, B., Cholewinski, G., and Rachon, J. (2018). Synthesis and antimicrobial activity of amino acid and peptide derivatives of mycophenolic acid. Eur. J. Med. Chem. 143, 646–655. doi: 10.1016/j.ejmech.2017.11.094
Singh, I. P., Jain, S. K., Kaur, A., Singh, S., Kumar, R., Garg, P., et al. (2010). Synthesis and antileishmanial activity of piperoyl-amino acid conjugates. Eur. J. Med. Chem. 45, 3439–3445. doi: 10.1016/j.ejmech.2010.04.033
Smith, P. F., Ogundele, A., Forrest, A., Wilton, J., Salzwedel, K., Doto, J., et al. (2007). Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3',3'-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob. Agents Chemother. 51, 3574–3581. doi: 10.1128/AAC.00152-07
Song, M. X., Deng, X. Q., Li, Y. R., Zheng, C. J., Hong, L., and Piao, H. R. (2014). Synthesis and biological evaluation of (E)-1-(substituted)-3-phenylprop-2-en-1-ones bearing rhodanines as potent anti-microbial agents. J. Enzyme Inhib. Med. Chem. 29, 647–653. doi: 10.3109/14756366.2013.837899
Song, X., Liu, Y., Ma, J., He, J., Zheng, X., Lei, X., et al. (2014). Synthesis of novel amino acid derivatives containing chrysin as anti-tumor agents against human gastric carcinoma MGC-803 cells. Med. Chem. Res. 24, 1789–1798. doi: 10.1007/s00044-014-1267-7
Sowunmi, A., Akano, K., Ayede, A. I., Ntadom, G., Aderoyeje, T., Adewoye, E. O., et al. (2016). Clinical illness and outcomes in Nigerian children with late-appearing anaemia after artemisinin-based combination treatments of uncomplicated falciparum malaria. BMC Infect. Dis. 16:240. doi: 10.1186/s12879-016-1565-4
Sun, P., Wu, G., Qiu, Z., and Chen, Y. (2014). Preparation of L-alanine-(14-oridonin) Ester Trifluoroacetate for Treatment of Cancer. Jiangsu Hengrui Medicine Co., Ltd., Peop. Rep. China, 10.
Tang, H. J., Zhang, X. W., Yang, L., Li, W., Li, J. H., Wang, J. X., et al. (2016). Synthesis and evaluation of xanthine oxidase inhibitory and antioxidant activities of 2-arylbenzo[b]furan derivatives based on salvianolic acid C. Eur. J. Med. Chem. 124, 637–648. doi: 10.1016/j.ejmech.2016.08.019
Tatsuzaki, J., Taniguchi, M., Bastow, K. F., Nakagawa-Goto, K., Morris-Natschke, S. L., Itokawa, H., et al. (2007). Anti-tumor agents 255: novel glycyrrhetinic acid-dehydrozingerone conjugates as cytotoxic agents. Bioorg. Med. Chem. 15, 6193–6199. doi: 10.1016/j.bmc.2007.06.027
Thimmegowda, N. R., Park, C., Shwetha, B., Sakchaisri, K., Liu, K., Hwang, J., et al. (2015). Synthesis and antitumor activity of natural compound aloe emodin derivatives. Chem. Biol. Drug Des. 85, 638–644. doi: 10.1111/cbdd.12448
Ukiya, M., Kawaguchi, T., Ishii, K., Ogihara, E., Tachi, Y., Kurita, M., et al. (2013). Cytotoxic activities of amino acid-conjugate derivatives of abietane-type diterpenoids against human cancer cell lines. Chem. Biodivers. 10, 1260–1268. doi: 10.1002/cbdv.201300043
Verghese, J., Nguyen, T., Oppegard, L. M., Seivert, L. M., Hiasa, H., and Ellis, K. C. (2013). Flavone-based analogues inspired by the natural product simocyclinone D8 as DNA gyrase inhibitors. Bioorg. Med. Chem. Lett. 23, 5874–5877. doi: 10.1016/j.bmcl.2013.08.094
Wang, J., Hu, X. L., Wen, W. H., Yang, L. L., and Zhu, Y. L. (2012). Synthesis and activity of 3-Amino acid derivatives of glycyrrhetinic acid. Chin. J. Appl. Chem. 29, 873–877. doi: 10.3724/SP.J.1095.2012.00371
Wang, K. Y., Zhou, Z. W., Zhang, H. Y., Cao, Y. C., Xu, J. Y., Ma, C., et al. (2018). Design, Synthesis and Antibacterial Evaluation of 3-Substituted Ocotillol-Type Derivatives. Molecules 23, 87–101. doi: 10.3390/molecules23123320
Wang, L., Li, D., Xu, S., Cai, H., Yao, H., Zhang, Y., et al. (2012). The conversion of oridonin to spirolactone-type or enmein-type diterpenoid: synthesis and biological evaluation of ent-6,7-seco-oridonin derivatives as novel potential anticancer agents. Eur. J. Med. Chem. 52, 242–250. doi: 10.1016/j.ejmech.2012.03.024
Wang, P., Bi, X. L., Xu, J., Yuan, H. N., Piao, H. R., and Zhao, Y. Q. (2013). Synthesis and anti-tumor evaluation of novel 25-hydroxyprotopanaxadiol analogs incorporating natural amino acids. Steroids 78, 203–209. doi: 10.1016/j.steroids.2012.09.012
Wang, S. R., and Fang, W. S. (2009). Pentacyclic triterpenoids and their saponins with apoptosis-inducing activity. Curr. Topics Med. Chem. 9, 1581–1596. doi: 10.2174/156802609789909821
Wen, T., Xu, W., Liang, L., Li, J., Ding, X., Chen, X., et al. (2015). Clinical efficacy of andrographolide sulfonate in the treatment of severe Hand, Foot, and Mouth Disease (HFMD) is dependent upon inhibition of neutrophil activation. Phytoth. Res. 29, 1161–1167. doi: 10.1002/ptr.5361
Whiteman, M., Tritschler, H., and Halliwell, B. (1996). Protection against peroxynitrite-dependent tyrosine nitration and α1-antiproteinase inactivation by oxidized and reduced lipoic acid. FEBS Lett. 379, 74–76. doi: 10.1016/0014-5793(95)01489-6
Wu, G. (2009). Amino acids: metabolism, functions, and nutrition. Amino Acids 37, 1–17. doi: 10.1007/s00726-009-0269-0
Wu, G. R., Xu, B., Yang, Y. Q., Zhang, X. Y., Fang, K., Ma, T., et al. (2018). Synthesis and biological evaluation of podophyllotoxin derivatives as selective antitumor agents. Eur. J. Med. Chem. 155, 183–196. doi: 10.1016/j.ejmech.2018.05.052
Wu, Y. L., Wan, Y., Jin, X. J., OuYang, B. Q., Bai, T., Zhao, Y. Q., et al. (2011). 25-OCH3-PPD induces the apoptosis of activated t-HSC/Cl-6 cells via c-FLIP-mediated NF-kappaB activation. Chem. Biol. Interact. 194, 106–112. doi: 10.1016/j.cbi.2011.08.010
Wu, Y. Z., Cao, D., Wang, F., Ma, L., Gao, G., and Chen, L. J. (2015). Synthesis and evaluation of millepachine amino acid prodrugs with enhanced solubility as antitumor agents. Chem. Bio. Drug Des. 86, 559–567. doi: 10.1111/cbdd.12507
Yang, C. J., Song, Z. L., Goto, M., Hsu, P. L., Zhang, X. S., Yang, Q. R., et al. (2017). Design, semisynthesis and potent cytotoxic activity of novel 10-fluorocamptothecin derivatives. Bioorg. Med. Chem. Lett. 27, 4694–4697. doi: 10.1016/j.bmcl.2017.09.012
Ye, D., Shi, Q., Leung, C. H., Kim, S. W., Park, S. Y., Gullen, E. A., et al. (2012). Antitumor agents 294. novel E-ring-modified camptothecin-4beta-anilino-4'-O-demethyl-epipodophyllotoxin conjugates as DNA topoisomerase I inhibitors and cytotoxic agents. Bioorg. Med. Chem. 20, 4489–4494. doi: 10.1016/j.bmc.2012.05.030
You, L., Zhao, M., Regenstein, J. M., and Ren, J. (2011). In vitro antioxidant activity and in vivo anti-fatigue effect of loach (Misgurnus anguillicaudatus) peptides prepared by papain digestion. Food Chem. 124, 188–194. doi: 10.1016/j.foodchem.2010.06.007
Yu, H., Jin, H., Gong, W., Wang, Z., and Liang, H. (2013). Pharmacological actions of multi-target-directed evodiamine. Molecules 18, 1826–1843. doi: 10.3390/molecules18021826
Yu, X. M., Ramiandrasoa, F., Guetzoyan, L., Pradines, B., Quintino, E., Gadelle, D., et al. (2012). Synthesis and biological evaluation of acridine derivatives as antimalarial agents. Chem. Med. Chem. 7, 587–605. doi: 10.1002/cmdc.201100554
Yuan, W., Guo, J., Wang, X., Su, G., and Zhao, Y. (2018). Non-protein amino acid derivatives of 25-methoxylprotopanaxadiol/25-hydroxyprotopanaxadioland their anti-tumour activity evaluation. Steroids 129, 1–8. doi: 10.1016/j.steroids.2017.11.003
Zhang, C., Li, X., Gao, Y., Zhang, L., and Fu, X. (2007). Synthesis and primary research on antitumor activity of three new panaxadiol fatty acid esters. Chem. Res. Chin. Univ. 23, 176–182. doi: 10.1016/S1005-9040(07)60037-3
Zhang, G., Ye, X., Ji, D., Zhang, H., Sun, F., Shang, C., et al. (2014). Inhibition of lung tumor growth by targeting EGFR/VEGFR-Akt/NF-kappaB pathways with novel theanine derivatives. Oncotarget 5, 8528–8543. doi: 10.18632/oncotarget.2336
Zhang, H. B., Shen, Q. K., Wang, H., Jin, C. M., Jin, C. M., and Quan, Z. S. (2018). Synthesis and evaluation of novel arctigenin derivatives as potential anti-Toxoplasma gondii agents. Eur. J. Med. Chem. 158, 414–427. doi: 10.1016/j.ejmech.2018.08.087
Zhang, H. J., Zhang, G. R., Piao, H. R., and Quan, Z. S. (2017). Synthesis and characterisation of celastrol derivatives as potential anticancer agents. J. Enzyme Inhibit. Med. Chem. 33, 190–198. doi: 10.1080/14756366.2017.1404590
Zhang, L. J., Chen, L., Lu, Y., Wu, J. M., Xu, B., Sun, Z. G., et al. (2010). Danshensu has anti-tumor activity in B16F10 melanoma by inhibiting angiogenesis and tumor cell invasion. Eur. J. Pharmacol. 643, 195–201. doi: 10.1016/j.ejphar.2010.06.045
Zhang, T. J., Li, S. Y., Yuan, W. Y., Zhang, Y., and Meng, F. H. (2018). Design, synthesis, and molecular docking studies of N-(9,10-anthraquinone-2-carbonyl)amino acid derivatives as xanthine oxidase inhibitors. Chem. Biol. Drug Des. 91, 893–901. doi: 10.1111/cbdd.13156
Zhang, Y. N., Zhang, W., Hong, D., Shi, L., Shen, Q., Li, J. Y., et al. (2008). Oleanolic acid and its derivatives: new inhibitor of protein tyrosine phosphatase 1B with cellular activities. Bioorg. Med. Chem. 16, 8697–8705. doi: 10.1016/j.bmc.2008.07.080
Zhao, H., Guo, Y., Li, S., Han, R., Ying, J., Zhu, H., et al. (2015). A novel anti-cancer agent Icaritin suppresses hepatocellular carcinoma initiation and malignant growth through the IL-6/Jak2/Stat3 pathway. Oncotarget 6, 31927–31943. doi: 10.18632/oncotarget.5578
Zheng, L., Fu, Y., Zhuang, L., Gai, R., Ma, J., Lou, J., et al. (2014). Simultaneous NF-kappaB inhibition and E-cadherin upregulation mediate mutually synergistic anticancer activity of celastrol and SAHA in vitro and in vivo. Int. J. Cancer 135, 1721–1732. doi: 10.1002/ijc.28810
Zhou, F., Wu, G. R., Cai, D. S., Xu, B., Yan, M. M., Ma, T., et al. (2019). Synthesis and biological activity of glycyrrhetinic acid derivatives as antitumor agents. Eur. J. Med. Chem. 178, 623–635. doi: 10.1016/j.ejmech.2019.06.029
Zhou, M., Liu, M., He, X., Yu, H., Wu, D., Yao, Y., et al. (2014). Synthesis and biological evaluation of novel 10-substituted-7-ethyl-10-hydroxycamptothecin (SN-38) prodrugs. Molecules 19, 19718–19731. doi: 10.3390/molecules191219718
Zhou, Z. Y., Zhao, W. R., Shi, W. T., Xiao, Y., Ma, Z. L., Xue, J. G., et al. (2019). Endothelial-dependent and independent vascular relaxation effect of tetrahydropalmatine on rat aorta. Front. Pharmacol. 10:336. doi: 10.3389/fphar.2019.00336
Zhu, P., Bi, Y., Xu, J., Li, Z., Liu, J., Zhang, L., et al. (2009). Terpenoids. III: synthesis and biological evaluation of 23-hydroxybetulinic acid derivatives as novel inhibitors of glycogen phosphorylase. Bioorg. Med. Chem. Lett. 19, 6966–6969. doi: 10.1016/j.bmcl.2009.10.055
Zhu, Q., Yu, X., Shen, Q., Zhang, Q., Su, M., Zhou, Y., et al. (2018). A series of camptothecin prodrugs exhibit HDAC inhibition activity. Bioorg. Med. Chem. 26, 4706–4715. doi: 10.1016/j.bmc.2018.08.008
Keywords: derivatives, research progress, structure modification, natural products, amino acids
Citation: Xu Q, Deng H, Li X and Quan Z-S (2021) Application of Amino Acids in the Structural Modification of Natural Products: A Review. Front. Chem. 9:650569. doi: 10.3389/fchem.2021.650569
Received: 07 January 2021; Accepted: 02 March 2021;
Published: 29 April 2021.
Edited by:
Tara Louise Pukala, University of Adelaide, AustraliaReviewed by:
Assem Barakat, King Saud University, Saudi ArabiaCopyright © 2021 Xu, Deng, Li and Quan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoting Li, c3lwdWxpeGlhb3RpbmdAMTYzLmNvbQ==; Zhe-Shan Quan, enNxdWFuQHlidS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.