
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 30 June 2021
Sec. Organic Chemistry
Volume 9 - 2021 | https://doi.org/10.3389/fchem.2021.642614
This article is part of the Research TopicOrganic Chemistry Editor’s Pick 2022View all 11 articles
 Satya Kumar Avula1
Satya Kumar Avula1 Majid Khan1,2
Majid Khan1,2 Sobia Ahsan Halim1
Sobia Ahsan Halim1 Ajmal Khan1
Ajmal Khan1 Samia Ahmed Al-Riyami1
Samia Ahmed Al-Riyami1 Rene Csuk3Biswanath Das1
Rene Csuk3Biswanath Das1 Ahmed Al-Harrasi1*
Ahmed Al-Harrasi1*A series of novel 1H-1,2,3-triazole analogs (9a–j) were synthesized via “Click” chemistry and Suzuki–Miyaura cross-coupling reaction in aqueous medium. The compounds were evaluated for their carbonic anhydrase-II enzyme inhibitory activity in vitro. The synthesis of triazole 7a was accomplished using (S)-(-) ethyl lactate as a starting material. This compound (7a) underwent Suzuki–Miyaura cross-coupling reaction with different arylboronic acids in aqueous medium to afford the target molecules, 9a–j in good yields. All newly synthesized compounds were characterized by 1H NMR, 13C NMR, FT-IR, HRMS, and where applicable 19F NMR spectroscopy (9b, 9e, 9h, and 9j). The new compounds have shown moderate inhibition potential against carbonic anhydrase-II enzyme. A preliminary structure-activity relationship suggested that the presence of polar group at the 1H-1,2,3-triazole substituted phenyl ring in these derivatives (9a–j) has contributed to the overall activity of these compounds. Furthermore, via molecular docking, it was deduced that the compounds exhibit inhibitory potential through direct binding with the active site residues of carbonic anhydrase-II enzyme. This study has unraveled a new series of triazole derivatives as good inhibitors against carbonic anhydrase-II.
1H-1,2,3-Triazole molecules play a vital role in pharmaceuticals and agrochemicals (Abdel-Wahab et al., 2012). The triazole moiety is very important in organic chemistry due to its broad range of applications in biomedicinal, biochemical, and material sciences (Singh et al., 2010). The chemistry of the compounds containing this moiety underwent substantial growth over the past decades (Thirumurugan et al., 2013). These compounds are widely used in industrial applications such as dyes, photographic materials, photostabilizers, agrochemicals, and corrosion inhibitors (copper alloys) (Fan et al., 1996).
Recent literature studies (Kumar et al., 2018; Sharma et al., 2019) demonstrated that 1H-1,2,3-triazole ring containing heterocycles was showing superior carbonic anhydrase inhibitors (Figure 1A).
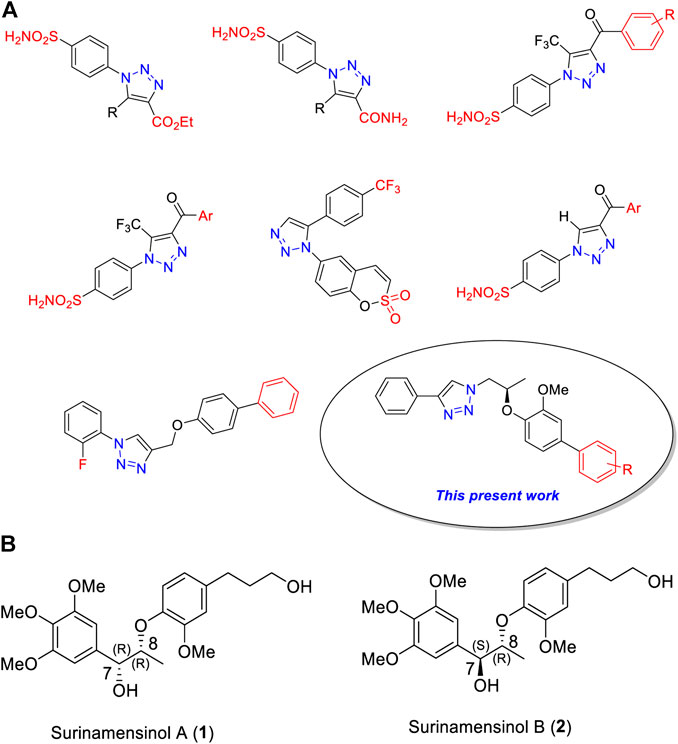
FIGURE 1. (A) Chemical structures of some clinically used 1H-1,2,3-triazole analogs as potential carbonic anhydrase-II inhibitors. (B) Chemical structures of surinamensinols A and B.
Carbonic anhydrase (CAs, EC 4.2.1.1), a Zn+2 containing metallic enzyme, catalyzes the reversible reaction of carbon dioxide into bicarbonate ions (Supuran and De Simone, 2015; Ozensoy and Guler et al., 2016). There are fifteen isoforms of CA which have been identified so far (Lindskog, 1997; Aggarwal et al., 2013). They possess a difference in their organ distributions, levels of gene expression, molecular sequence features, and kinetic parameters (Krishnamurthy et al., 2008). CAs are key contributors to various physiological and pathological processes. Thus, they are considered the prime therapeutic target for the treatment of several chronic diseases.
In continuation of our research work on 1H-1,2,3-triazole derivatives (Avula et al., 2018; Avula et al., 2019), we herein report a new series of 1H-1,2,3-triazole analogs (9a–j) as carbonic anhydrase-II inhibitors (Huisgen, 1963; Pham Thi et al., 2016). We have selected the 8th triazole structure (Figure 1A) for our present synthesis because it contains a chiral dioxyaryl moiety which is present in some bioactive natural products such as surinamensinols A and B (Figure 1B) (add two references). A structural-activity relationship is discussed to demonstrate the influence of structural moieties on the triazole derivatives.
The first step employed Mitsunobu reaction between (S)-(-) ethyl lactate 1 and 4-bromo-2-methoxy phenol using diisopropylazodicarboxylate (DIAD). The reaction afforded the expected compound 2 in 86% yield. In this step, a chiral center has been successfully introduced. Reduction of compound 2 to compound 3 was achieved by using DIBAL-H which furnished compound 3 in 90% yield. The hydroxyl group of 3 was converted into a tosyl moiety to provide compounds 4 in high yield (95%). The latter was treated with NaN3 and afforded the corresponding azide derivative 5 in 78% yield.
Azide 5 underwent 1,3-dipolar cycloaddition with the alkyne derivative 6a in the presence of CuI and Hunig’s base. The reaction furnished the desired product 1H-1,2,3-triazole derivative 7a as a colorless amorphous solid in 76% yield. The 1H NMR spectrum of compound 7a showed singlet at δ 8.05 for triazole proton (–CH–N3). The eight aromatic protons appeared in the region of δ 7.78–6.63 ppm. One multiplet and doublet of doublet signals at δ 4.61–4.56 and δ 4.48 correspond to the–CH–O– and –CH–N–, respectively. A singlet peak at δ 3.69 is due to methoxy protons on the phenyl ring and a doublet at δ 1.24 is attributed to methyl protons of –CH–O–Ph. The high-resolution mass spectrometric data at 388.0661 (M+) support the structure of compound 7a. Similarly, using the same reaction conditions described for the synthesis of compound 7a, compound 7b was obtained in 82% yield using different alkyne derivative (6b). The synthesis of compounds 7a and 7b is summarized in Scheme 1.
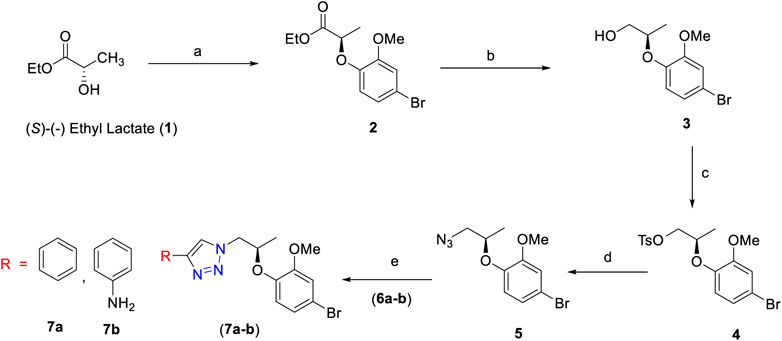
SCHEME 1. Reagents and conditions: (A) 4-bromo-2-methoxyphenol, PPh3, DIAD, dry THF, 0°C to room temperature, 3 h, 86%; (B) DIBAL-H, dry DCM, 0°C to room temperature, 3 h, 90%; (C) TsCl, Et3N, dry DCM, DMAP, 0°C to room temperature, 5 h, 95%; (D) NaN3, DMF, 70°C, 3 h, 78%; (E) alkyne derivative (6a–b), CuI, Et3N, MeCN, room temperature, 3 h, 7a (76%) and 7b (82%).
The final step of this series was Suzuki–Miyaura cross-coupling reaction (Shaulis et al., 2002; Polshettiwar et al., 2010) of compound 7a with different arylboronic acids (8a–j) to afford a novel series of 1H-1,2,3-triazole analogs (9a–j) in good yields (82–91%) (Scheme 2, Table 1). Here, both electron-withdrawing and electron-donating arylboronic acids were successfully employed. The best yields were obtained in THF : H2O (3 : 1) at 85–90°C for 10–12 h using Pd(OAc)2 as a catalyst (5 mol%) and K2CO3 (3.0 equiv) as a base. The structures of all the new synthesized compounds (9a–j) were confirmed by spectroscopic techniques (1H NMR, 13C NMR, FT-IR, HRMS, and where applicable 19F NMR). The synthesis of a novel series of 1H-1,2,3-triazole analogs (9a–j) is summarized in Scheme 2.
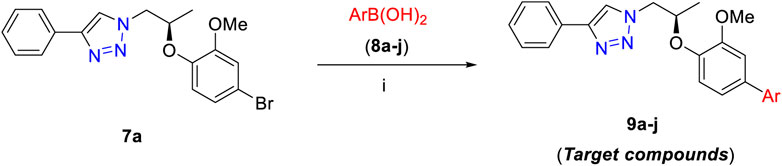
SCHEME 2. Synthesis of 9a–j. Reagents and conditions: (i) 7a (1.0 equiv), 8a–j (1.2–1.5 equiv), Pd(OAc)2 (5 mol%), K2CO3 (3.0 equiv), THF:H2O (3 : 1), 80–85°C 10–12 h (82–91%).
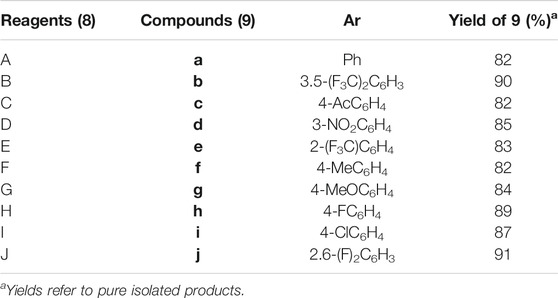
TABLE 1. Synthesis of cross-coupled 1H-1,2,3-triazole analogs (9a–j).
The general structural features of the synthesized molecules (9) are presented in Figure 2. Two pharmacophoric elements (triazole and chiral dioxyaryl) were considered as a rigid motif with an aryl group attached to the chiral dioxyaryl moiety which is mainly determining the varying degree of activity. A diverse array of functional groups (both electropositive and electronegative) influencing the activity toward carbonic anhydrase-II inhibition has been utilized.
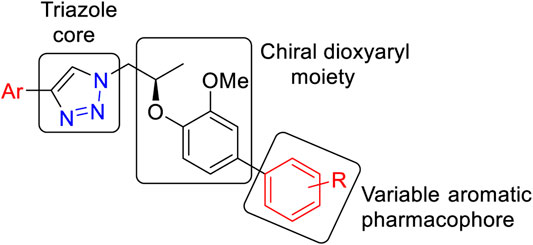
FIGURE 2. General structural feature of the synthesized molecules (9).
All synthesized 1H-1,2,3-triazole analogs (9a–j) were evaluated against bovine carbonic anhydrase-II enzyme to know their therapeutic potential. All compounds showed potent to significant activities with IC50 values in the range of 13.8–35.7 µM, as compared to standard acetazolamide (18.2 ± 0.23 µM). In in vitro carbonic anhydrase-II assay, compounds 7b (13.8 ± 0.63 µM) and 9e (18.1 ± 1.31 µM) showed potent activities, followed by 9d (20.7 ± 1.13 µM), 9c (21.5 ± 0.21 µM), 9b (25.1 ± 1.04 µM), and 9f (26.6 ± 0.80 µM), while compounds 9h (32.3 ± 0.77 µM) and 9j (35.7 ± 0.57 µM) demonstrated lower activities. The detailed predictive SAR was deduced from molecular docking studies. The in vitro and docking results are tabularized in Table 2.
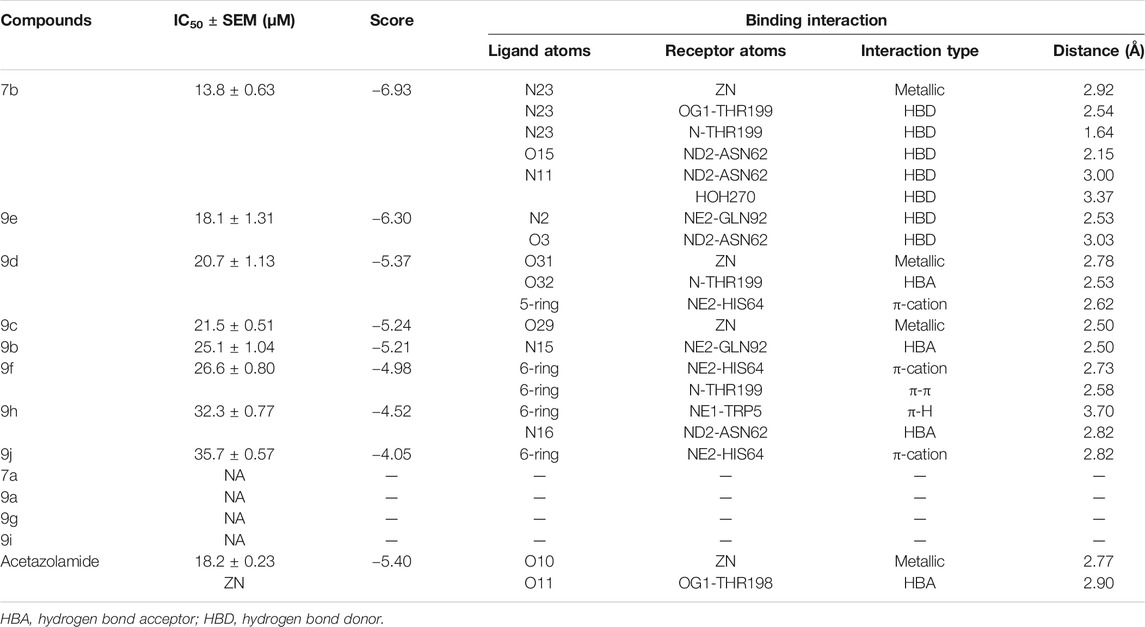
TABLE 2. In vitro and in silico results of the compounds against carbonic anhydrase-II.
Molecular docking studies of all active triazole derivatives were performed using molecular operating software (MOE) (Chemical Computing Group, 2014), in order to determine the best plausible binding modes of the ligands in the active site of the enzyme. The active site of CA-II is depicted in Figure 3A. Compound 7b (IC50 = 13.8 ± 0.63 µM) was found to be the most active compound of the series and its best predicted binding pose is presented in Figure 3B. The binding interaction of 7b demonstrated that its aniline moiety formed a metallic bond with Zn+2 ion and hydrogen bonding with the amino and –OH groups of Thr199. Furthermore, the dioxyaryl group of 7b exhibited H-bonding with the amide group of Asn62. Additionally, a water molecule (HOH270) also offered an H-bond to the triazole moiety of 7b. This multiple bonding of the compound with the active site residues is responsible for the enhanced biological activity of 7b as compared to the rest of the compounds. Similarly, the triazole nitrogen and methoxy oxygen of the 9e (IC50 = 18.1 ± 1.31 µM) interacted with the side chains of Gln92 and Asn62, respectively. The loss of hydrogen bond donor/acceptor group at triazole substituted aryl group of 9e makes the molecule less active than 7b; moreover, water molecule does not contribute to protein-ligand bridging for 9e. The nitro group of 9d mediated H-bonding and metallic interaction with the amide group of Thr199 and Zn ion, respectively. Additionally, the side chain of His64 provided π-cation interaction to the triazole ring of 9d. The dioxyaryl group does not interact with the surrounding residues including Asn62, Asn67, Gln92, and water molecules; this may be the reason for lower activity of 9d than 7b and 9e. The docked view of 9c showed that the carbonyl oxygen interacted with the Zn ion via metallic bond; however, the other polar groups do not interact with the active site residues and the docked orientation of 9c is surface exposed. This is the reason for the further reduced activity of 9c. Similarly, the triazole substituted aryl group of 9b does not possess any polar moiety to interact with Zn ion or Thr199 and His94. However, the triazole nitrogen of 9b formed H-bond with the side chain of Gln92. The dioxyaryl moiety and its substituted R group of 9b remained surface exposed, which further decreased the inhibitory activity of 9b. Likewise, the triazole substituted aryl group and dioxyaryl substituted aryl group of 9f mediated hydrophobic interactions with the side chains of Thr199 and His64, respectively, while the triazole ring and the dioxyaryl group lost interaction with Gln92 and Asn62, respectively. Due to the loss of these interactions, the compound exhibited less biological activity than 9b. The binding mode of 9h was similar to the docked view of 9f; however, the triazole nitrogen and the dioxyaryl substituted aryl group of 9h formed H-bonding and hydrophobic interaction with Asn62 and Trp5, respectively. Similarly, the least active compound, 9j, exhibited only π-cation interaction with the side chain of His64, while its triazole ring and the dioxyaryl group do not interact with the surrounding residues; due to the loss of major H-bonding or metallic interactions, the compound exhibited the least inhibitory activity against CA-II. The best-docked poses of the active compounds are depicted in Figure 3C. The docking scores and the binding interactions are tabulated in Table 2. Acetazolamide was used as a positive control in docking which exhibited biological activity with IC50 value of 18.2 µM. The sulfate group of acetazolamide interacted with the Zn ion and the side chain of Thr198 through metallic interaction and H-bonding, respectively. The docking score of acetazolamide is −5.40, which is lesser than the docking scores of 7b and 9e, while being greater than the docking scores of 9d, 9c, 9b, 9f, 9h, and 9j. The docked view of acetazolamide is shown in Figure 4 in both 3D and 2D format. The docking scores and binding interactions of the compounds are well correlated with the inhibitory activities of the compounds. The docked orientation of compounds showed that Thr199 and Zn ions play important role in the stabilization of the triazole substituted aryl group, while the dioxyaryl group interacts with Asn62 or Gln92. Additionally, His64 provides hydrophobic interactions to the compounds.
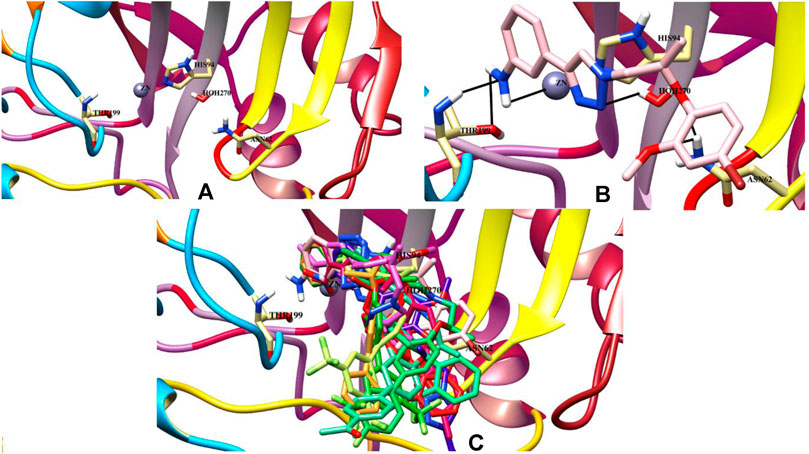
FIGURE 3. (A) The active site of the carbonic anhydrase is shown. (B) The binding mode of the most active compound (7b) within the active site of carbonic anhydrase. Ligand is presented as a light pink stick model and hydrogen bonds are shown in black lines. (C) The docked poses of all active ligands in the active site of the bovine carbonic anhydrase.
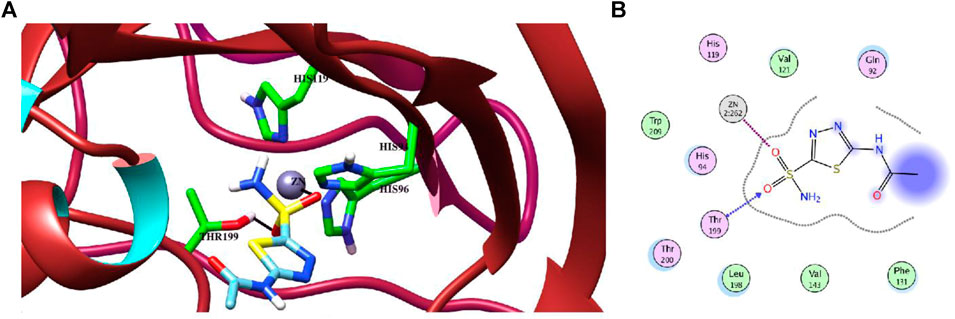
FIGURE 4. (A) The binding mode of the standard inhibitor “acetazolamide (AZM)” in the active site of CA-II in the 3D form. H-bond and metal-ligand interaction are shown in black lines. (B) The protein-ligand interaction is shown in 2D form. The H-bond and metal-ligand interaction are displayed in blue and magenta dotted lines, respectively.
In summary, a series of novel 1H-1,2,3-triazole analogs were synthesized (9a–j) and evaluated for their carbonic anhydrase-II inhibitory activity in vitro. (S)-(-) Ethyl lactate was used as a starting material to introduce the chirality of the target molecules. The triazole moiety was prepared via “Click” chemistry and the aryl derivatives by utilizing Suzuki–Miyaura cross-coupling reaction in aqueous medium. All the compounds have shown moderate inhibition potential against carbonic anhydrase-II enzyme reported for the first time. The molecular docking studies showed that all the active compounds well accommodate to the active site of the CA enzyme.
Reagents were obtained from Sigma-Aldrich, Germany. Silica gel for column chromatography was of 100–200 mesh. Solvents were purified by following standard procedures. Thin-layer chromatography (TLC) was carried using silica gel F254 precoated plates. UV-light and I2 stain were used to visualize the spots. The 1H and 13C NMR spectra were recorded on NMR spectrometer (Bruker: 600 MHz for 1H, 150 MHz for 13C, and 564 MHz for 19F) using CDCl3 as a solvent. The high-resolution electrospray ionization mass spectra (HR-ESI-MS) were recorded on Agilent 6530 LC Q-TOF instrument. Organic extracts and solutions of pure compounds were dried over anhydrous MgSO4.
The key intermediate azide compound 5 was prepared according to our earlier literature method (Avula et al., 2018).
CuI (2.0 equiv) and triethylamine (3.0 equiv) were added to a solution of azide compound 5 (1.0 equiv) and alkyne derivatives 6a–b (1.2 equiv) in acetonitrile (10 ml) at room temperature, and the mixture was stirred for 3 h. The reaction mixture was diluted with EtOAc (20 ml), 10 ml of aqueous NH4Cl was added, the aqueous layer was extracted with EtOAc (3 × 15 ml), and the combined organic layer was washed with brine solution, dried over anhydrous Na2SO4, and concentrated in vacuo to obtain a crude residue that was purified by flash chromatography to obtain desired 1H-1,2,3-triazole derivatives (7a) 76% and (7b) 82%.
Colorless amorphous solid; Yield = 76%; IR (solid): 1498, 1265, 1128, 732 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.05 (s, 1H), 7.78 (d, J = 7.7 Hz, 2H), 7.35 (t, J = 7.6 Hz, 2H), 7.25 (d, J = 7.6 Hz, 1H), 6.92–6.87 (m, 2H), 6.63 (d, J = 8.4 Hz, 1H), 4.61–4.56 (m, 2H), 4.48 (dd, J = 14.8, 7.5 Hz, 1H), 3.69 (s, 3H), 1.24 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 151.5, 147.4, 130.8, 128.8, 128.0, 125.5, 123.5, 121.5, 119.2, 115.6, 115.0, 75.1, 55.9, 54.8, 17.1; HRMS (ESI+): Found (M+H+): 388.0661 C18H1979BrN3O2 required 388.0664. Found (M+H+): 390.0642 C18H1981BrN3O2 required 390.0639.
Brown color solid; Yield = 82%; IR (solid): 3380, 1586, 1496, 1268, 1130, 842, 736 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.90 (s, 1H), 7.43 (d, J = 8.0 Hz, 2H), 6.88–6.83 (m, 2H), 6.65 (d, J = 8.5 Hz, 1H), 6.59 (d, J = 8.1 Hz, 2H), 4.55 (ddd, J = 29.5, 12.1, 4.9 Hz, 2H), 4.46 (dd, J = 14.2, 6.8 Hz, 1H), 4.23 (brs, 2H), 3.67 (s, 3H), 1.20 (d, J = 6.5 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 151.5, 145.5, 126.5, 123.4, 120.1, 119.1, 115.7, 114.9, 114.7, 75.0, 56.0, 54.6, 17.2; HRMS (ESI+): Found (M+H+): 404.2002 C24H2479BrN3O2 required 404.2004. Found (M+H+): 406.0714 C24H2481BrN3O2 required 406.0716.
Different substituted boronic acids 8a–j (1.2–1.5 equiv per crossing coupling step), K2CO3 (3.0 equiv), and Pd(OAc)2 (5 mol%) were added to a mixture of phenyl 1H-1,2,3-triazole 7a (1.0 equiv), THF : H2O (3 : 1, 5 ml). The reaction was heated at 80–85°C with stirring under a nitrogen atmosphere for 10–12 h, cooled to room temperature, and diluted with water (5 ml). The resulting reaction mixture was extracted with Et2O. The organic layers were combined and washed with brine solution, dried over anhydrous MgSO4, and then filtered off. The solvent was removed under vacuum, and the crude product was purified by silica gel column chromatography (eluting with EtOAc/hexane, 9 : 1) to give pure cross-coupled products 9a–j.
White solid; Yield = 82%; IR (solid): 1498, 1214, 774, 746, 667 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.16 (s, 1H), 7.83–7.79 (m, 2H), 7.52–7.46 (m, 2H), 7.39 (td, J = 7.7, 2.8 Hz, 4H), 7.29 (t, J = 7.4 Hz, 2H), 7.10–7.01 (m, 2H), 6.88 (d, J = 8.1 Hz, 1H), 4.77–4.69 (m, 2H), 4.59 (dd, J = 14.4, 7.2 Hz, 1H), 3.85 (s, 3H), 1.37 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 150.6, 147.4, 145.4, 140.5, 136.3, 130.6, 128.6, 127.8, 126.9, 126.7, 125.4, 121.4, 119.4, 117.7, 111.1, 75.0, 55.6, 54.8, 17.1; HRMS (ESI+): Found (M+H+): 386.1856 C24H24N3O2 required 386.1854.
Colorless amorphous solid; Yield = 90%; IR (solid): 1496, 1328, 1267, 1168, 1219, 774 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.14 (s, 1H), 7.90 (s, 2H), 7.81 (d, J = 8.3 Hz, 3H), 7.41 (t, J = 7.6 Hz, 2H), 7.34–7.28 (m, 1H), 7.09–7.02 (m, 2H), 6.93 (d, J = 8.2 Hz, 1H), 4.86–4.72 (m, 2H), 4.62 (dd, J = 14.3, 7.3 Hz, 1H), 3.90 (s, 3H), 1.41 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 151.1, 147.5, 147.0, 142.8, 133.1, 132.1, 131.9, 130.6, 128.7, 128.0, 126.8, 125.5, 124.2, 122.4, 121.5, 120.5, 119.9, 117.6, 111.1, 75.0, 56.0, 55.0, 17.2; 19F NMR (564 MHz, chloroform-d): δ −62.82; HRMS (ESI+): Found (M+H+): 522.1627 C26H22F6N3O2 required 522.1630.
Pale yellow amorphous solid; Yield = 82%; IR (solid): 1680, 1498, 1217, 842, 754, 671 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.11 (s, 1H), 7.87 (s, 2H), 7.83 (d, J = 8.3 Hz, 2H), 7.79 (d, J = 8.6 Hz, 3H), 7.39 (t, J = 7.7 Hz, 2H), 7.05 (d, J = 8.6 Hz, 1H), 6.85 (d, J = 8.3 Hz, 2H), 4.81–4.70 (m, 2H), 4.60 (dd, J = 14.3, 7.2 Hz, 1H), 3.88 (s, 3H), 2.50 (s, 3H), 1.39 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 195.3, 152.3, 145.9, 144.2, 137.4, 132.5, 130.7, 128.8, 128.0, 127.5, 126.9, 125.6, 125.1, 121.4, 120.6, 119.9, 115.6, 115.3, 111.1, 75.1, 56.0, 55.0, 17.3; HRMS (ESI+): Found (M +H+): 428.1973 C26H26N3O3 required 428.1971.
Light brown amorphous solid; Yield = 85%; IR (solid): 1598, 1496, 1343, 1216, 842, 771, 747, 669 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.09 (s, 1H), 7.63 (dd, J = 12.4, 7.7 Hz, 2H), 7.54 (d, J = 7.8 Hz, 1H), 7.44 (t, J = 8.0 Hz, 2H), 7.39 (t, J = 7.6 Hz, 3H), 7.29 (s, 1H), 6.93 (d, J = 8.4 Hz, 2H), 6.67 (d, J = 8.2 Hz, 1H), 4.65 (td, J = 13.7, 4.7 Hz, 2H), 4.54 (dd, J = 13.8, 6.7 Hz, 1H), 3.74 (s, 3H), 1.30 (d, J = 6.1 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 151.5, 147.5, 145.3, 132.8, 132.1, 132.0, 130.6, 129.6, 128.8, 128.6, 128.5, 128.1, 125.6, 123.6, 121.6, 119.2, 115.6, 115.2, 75.3, 55.9, 54.9, 17.1; HRMS (ESI+): Found (M+H+): 431.1732 C24H23N4O4 required 431.1735.
White amorphous solid; Yield = 83%; IR (solid): 1498, 1329, 1263, 1164, 1219, 845, 772, 667 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.08 (s, 1H), 7.79 (d, J = 7.8 Hz, 2H), 7.60 (dd, J = 12.7, 7.5 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 7.7 Hz, 2H), 7.31 (d, J = 7.5 Hz, 1H), 6.95 (d, J = 7.3 Hz, 2H), 6.68 (d, J = 8.5 Hz, 1H), 4.69–4.62 (m, 2H), 4.54 (dd, J = 13.9, 6.9 Hz, 1H), 3.74 (s, 3H), 1.30 (d, J = 6.1 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 151.5, 147.6, 145.3, 133.7, 132.1, 132.0, 130.9, 130.5, 129.2, 128.8, 128.6, 128.5, 128.1, 125.6, 123.6, 121.6, 119.2, 115.6, 115.2, 75.3, 55.9, 54.9, 17.1; 19F NMR (564 MHz, chloroform-d): δ −59.28; HRMS (ESI+): Found (M+H+): 454.1744 C25H23F3N3O2 required 454.1742.
White solid; Yield = 82%; IR (solid): 1496, 1217, 845, 753, 669 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.16 (s, 1H), 7.81 (d, J = 7.6 Hz, 2H), 7.39 (t, J = 7.4 Hz, 4H), 7.29 (t, J = 7.4 Hz, 1H), 7.18 (s, 2H), 7.05–7.01 (m, 2H), 6.86 (d, J = 8.1 Hz, 1H), 4.74–4.68 (m, 2H), 4.58 (dd, J = 14.7, 7.4 Hz, 1H), 3.84 (s, 3H), 2.35 (s, 3H), 1.36 (d, J = 6.1 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 150.8, 147.5, 145.4, 137.8, 136.9, 136.5, 130.8, 129.4, 128.7, 127.9, 126.7, 125.6, 121.6, 119.3, 118.0, 111.1, 75.1, 55.7, 55.0, 21.0, 17.3; HRMS (ESI+): Found (M+H+): 400.2017 C25H26N3O2 required 400.2020.
Pale yellow amorphous solid; Yield = 84%; IR (solid): 1497, 1239, 1214, 770, 744, 668 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.18 (d, J = 1.1 Hz, 1H), 7.84–7.80 (m, 2H), 7.45–7.39 (m, 4H), 7.33–7.29 (m, 1H), 7.04–7.00 (m, 2H), 6.96–6.92 (m, 2H), 6.88 (d, J = 8.1 Hz, 1H), 4.76–4.71 (m, 2H), 4.60 (dd, J = 14.7, 7.4 Hz, 1H), 3.85 (d, J = 1.1 Hz, 3H), 3.83 (d, J = 1.1 Hz, 3H), 1.38 (d, J = 6.1 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 158.9, 150.8, 147.5, 145.1, 136.2, 133.3, 130.8, 128.7, 127.9, 125.6, 121.5, 119.0, 118.0, 114.1, 110.9, 75.1, 55.7, 55.3, 54.9, 17.3; HRMS (ESI+): Found (M+H+): 416.1977 C25H26N3O3 required 416.1974.
White amorphous solid; Yield = 89%; IR (solid): 1496, 1217, 773, 745, 669 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.20 (s, 1H), 7.86 (d, J = 7.6 Hz, 2H), 7.48 (dd, J = 8.3, 5.3 Hz, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.11 (t, J = 8.5 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.0 Hz, 1H), 4.83–4.74 (m, 2H), 4.64 (dd, J = 14.5, 7.4 Hz, 1H), 3.89 (s, 3H), 1.42 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 163.1, 161.4, 150.8, 147.5, 145.6, 136.8, 135.6, 130.6, 128.8, 128.4, 128.0, 125.6, 121.6, 119.4, 118.0, 115.6, 115.5, 111.1, 75.1, 55.8, 55.1, 17.3; 19F NMR (564 MHz, chloroform-d): δ −115.90; HRMS (ESI+): Found (M +H+): 404.1777 C24H23FN3O2 required 404.1779.
White solid; Yield = 87%; IR (solid): 1217, 772, 743, 668 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.15 (s, 1H), 7.81 (d, J = 7.6 Hz, 2H), 7.43–7.38 (m, 4H), 7.35 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 14.9 Hz, 1H), 7.00 (dd, J = 4.2, 2.4 Hz, 2H), 6.87 (d, J = 8.7 Hz, 1H), 4.78–4.70 (m, 2H), 4.59 (dd, J = 14.4, 7.3 Hz, 1H), 3.85 (s, 3H), 1.38 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 150.8, 147.6, 145.9, 139.1, 135.2, 133.1, 130.7, 128.8, 128.1, 128.0, 125.6, 121.5, 119.4, 117.8, 111.0, 75.1, 55.8, 55.0, 17.3; HRMS (ESI+): Found (M +H+): 420.1478 C24H23ClN3O2 required 420.1475.
Colorless amorphous solid; Yield = 91%; IR (solid): 1497, 1216, 756, 666 cm−1; 1H NMR (600 MHz, chloroform-d) δ 8.14 (s, 1H), 7.82–7.78 (m, 2H), 7.39 (t, J = 7.6 Hz, 2H), 7.29 (d, J = 7.5 Hz, 1H), 7.09–6.98 (m, 4H), 6.94 (dq, J = 8.7, 3.9 Hz, 1H), 6.88 (d, J = 8.2 Hz, 1H), 4.78–4.69 (m, 2H), 4.59 (dd, J = 14.2, 7.1 Hz, 1H), 3.83 (s, 3H), 1.38 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, chloroform-d) δ 159.5, 157.8, 156.4, 154.7, 150.4, 147.6, 146.2, 130.7, 129.5, 128.8, 128.0, 125.6, 121.5, 117.2, 116.5, 115.0, 112.9, 75.0, 55.8, 54.9, 17.3; 19F NMR (564 MHz, chloroform-d): δ −119.02, 124.00; HRMS (ESI+): Found (M+H+): 422.2606 C24H22F2N3O2 required 422.2608.
In vitro experiment of bovine erythrocyte CA-II was conducted in HEPES-Tris buffer (20 mM) to maintain the pH 7.4. The purified was dissolved in HEPES-Tris buffer (0.1 mg/ml), and the reaction mixture was comprised of 140 μL of the HEPES-Tris buffer, 20 μL of the enzyme, and 20 μL of test compound (prepared in DMSO) and was incubated for 15 min at 25°C. After completion of the preincubation, the reaction was started by adding instantly the substrate p-nitrophenylacetate (P-NPA) at a concentration of 0.7 mM and prepared in methanol. To initiate the reaction, the 96-well plate was placed in a microplate reader and continuous product formation was monitored with a one-minute time interval for 30 min at 400 nm. The assay temperature was strictly controlled and kept at 25°C. All the reaction was conducted in triplicate, and the results are presented as mean.
The molecular docking experiment was performed on Molecular Operating Environment (MOE, 2014.14). The structures of ligands were prepared by MOE and minimized with MMFF94x force field until an RMSD gradient of 0.1 kcal mol−1Å−1 was attained, and partial charges were automatically calculated. The crystal structure of bovine carbonic anhydrase-II (PDB ID: 1V9E) was downloaded from the Protein Data Bank (https://www.rcsb.org/). Water molecules at an active site within the vicinity of 3 Å were retained and the rest of them were removed. The enzyme structure was then prepared for docking simulation using Protonate 3D option in MOE. Triangle Matcher placement method and London dG scoring function were used for docking using Zn2+ metal ion as a constrain for molecular docking. The best-docked pose of each compound was selected based on the binding interactions and docking score. The docking protocol was first validated by redocking the standard acetazolamide in the active site of the enzyme. The best-docked pose of the known inhibitor showed a highly negative docking score (S = −5.40 kcal/mol). The calculated RMSD between the docked and the native confirmation of sulfonamide was 2.31 Å. The docked view and binding interactions of the known active inhibitor are shown in Figure 4. The validated molecular docking protocol was then used to predict the binding pattern of the newly synthesized triazole derivatives in the active site of CA-II and to elucidate their structure-activity relationship.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
SA designed the study and wrote the manuscript; MK and AK were involved in biological activity; SH and SA-R performed molecular docking studies; RC and BD were involved in manuscript corrections and writing part.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the University of Nizwa for the generous support of this project. The project was supported by grant from The Oman Research Council (TRC) through the funded project (BFP/RGP/HSS/19/198).
Abdel-Wahab, B. F., Abdel-Latif, E., Mohamed, H. A., and Awad, G. E. A. (2012). Design and synthesis of new 4-pyrazolin-3-yl-1,2,3-triazoles and 1,2,3-triazol-4-yl-pyrazolin-1-ylthiazoles as potential antimicrobial agents. Eur. J. Med. Chem. 52, 263–268. doi:10.1016/j.ejmech.2012.03.023
Aggarwal, M., Boone, C. D., Kondeti, B., and McKenna, R. (2013). Structural annotation of human carbonic anhydrases. J. Enzyme Inhib. Med. Chem. 28, 267–277. doi:10.3109/14756366.2012.737323
Avula, S. K., Khan, A., Halim, S. A., Al-Abri, Z., Anwar, M. U., Al-Rawahi, A., et al. (2019). Synthesis of novel (R)-4-fluorophenyl-1H-1,2,3-triazoles: a new class of α-glucosidase inhibitors. Bioorg. Chem. 91, 103182. doi:10.1016/j.bioorg.2019.103182
Avula, S. K., Khan, A., Rehman, N. U., Anwar, M. U., Al-Abri, Z., Wadood, A., et al. (2018). Synthesis of 1H-1,2,3-triazole derivatives as new α-glucosidase inhibitors and their molecular docking studies. Bioorg. Chem. 81, 98–106. doi:10.1016/j.bioorg.2018.08.008
Chemical Computing Group ULC. (2014). 1010 Sherbooke St. West, Suite 910. Montreal, QC: Canada. Available at: www.chemcomp.com.
Huisgen, R. (1963). 1,3-dipolar cycloadditions. Past and Future. Angew. Chem. Int. Ed. Engl. 2, 565–598. doi:10.1002/anie.196305651
Fan, W. Q., Katritzky, A. R., Rees, C. W., and Scriven, E. F. V. (Editors) (1996). Comprehensive heterocyclic chemistry II. (Oxford, Newyork: Elsevier Science) 4, 1–126.
Krishnamurthy, V. M., Kaufman, G. K., Urbach, A. R., Gitlin, I., Gudiksen, K. L., Weibel, D. B., et al. (2008). Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem. Rev. 108, 946–1051. doi:10.1021/cr050262p
Kumar, R., Vats, L., Bua, S., Supuran, C. T., and Sharma, P. K. (2018). Design and synthesis of novel benzenesulfonamide containing 1,2,3-triazoles as potent human carbonic anhydrase isoforms I, II, IV and IX inhibitors. Eur. J. Med. Chem. 155, 545–551. doi:10.1016/j.ejmech.2018.06.021
Lindskog, S. (1997). Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 74, 1–20. doi:10.1016/S0163-7258(96)00198-2
Nguyen, V., Decottignies, A., Len, C., and Fihri, A. (2010). Suzuki-Miyaura cross-coupling reactions in aqueous media: green and sustainable syntheses of biaryls. ChemSusChem 3, 502–522. doi:10.1002/cssc.200900221
Ozensoy Guler, O., Capasso, C., and Supuran, C. T. (2016). A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J. Enzyme Inhib. Med. Chem. 31, 689–694. doi:10.3109/14756366.2015.1059333
Pham Thi, T., Le Nhat, T. G., Ngo Hanh, T., Luc Quang, T., Pham The, C., Dang Thi, T. A., et al. (2016). Synthesis and cytotoxic evaluation of novel indenoisoquinoline-substituted triazole hybrids. Bioorg. Med. Chem. Lett. 26, 3652–3657. doi:10.1016/j.bmcl.2016.05.092
Sharma, V., Kumar, R., Bua, S., Supuran, C. T., and Sharma, P. K. (2019). Synthesis of novel benzenesulfonamide bearing 1,2,3-triazole linked hydroxy-trifluoromethylpyrazolines and hydrazones as selective carbonic anhydrase isoforms IX and XII inhibitors. Bioorg. Chem. 85, 198–208. doi:10.1016/j.bioorg.2019.01.002
Shaulis, K. M., Hoskin, B. L., Townsend, J. R., Goodson, F. E., Incarvito, C. D., and Rheingold, A. L. (2002). Tandem Suzuki coupling−norbornadiene insertion reactions. a convenient route to 5,6-diarylnorbornene compounds. J. Org. Chem. 67, 5860–5863. doi:10.1021/jo016212b
Singh, N., Pandey, S. K., and Tripathi, R. P. (2010). Regioselective [3+2] cycloaddition of chalcones with a sugar azide: easy access to 1-(5-deoxy-d-xylofuranos-5-yl)-4,5-disubstituted-1H-1,2,3-triazolesH-1,2,3-triazoles. Carbohydr. Res. 345, 1641–1648. doi:10.1016/j.carres.2010.04.019
C. T. Supuran, and G. De Simone (Editors) (2015). Carbonic anhydrases as biocatalysts – from theory to medical and industrial applications, carbonic anhydrases: an overview. Amsterdam, Netherlands: Elsevier.
Keywords: synthesis, 1H-1, 2, 3-triazole analogs, click chemistry, aqueous medium, carbonic anhydrase-II inhibitory activity, molecular docking studies
Citation: Avula SK, Khan M, Halim SA, Khan A, Al-Riyami SA, Csuk R, Das B and Al-Harrasi A (2021) Synthesis of New 1H-1,2,3-Triazole Analogs in Aqueous Medium via “Click” Chemistry: A Novel Class of Potential Carbonic Anhydrase-II Inhibitors. Front. Chem. 9:642614. doi: 10.3389/fchem.2021.642614
Received: 16 December 2020; Accepted: 28 January 2021;
Published: 30 June 2021.
Edited by:
Andrea Basso, University of Genoa, ItalyReviewed by:
Cheng Zhang, National Renewable Energy Laboratory (DOE), United StatesCopyright © 2021 Avula, Khan, Halim, Khan, Al-Riyami, Csuk, Das and Al-Harrasi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Al-Harrasi, YWhhcnJhc2lAdW5pendhLmVkdS5vbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.