
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 30 September 2020
Sec. Organic Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.577834
This article is part of the Research Topic Design, Synthesis, and Application of Novel π-Conjugated Materials View all 13 articles
 Ruifa Jin1,2*
Ruifa Jin1,2* Jingfan Xin1,2
Jingfan Xin1,2A series of donor–acceptor (D–A) tricoordinated organoboron derivatives (1–10) have been systematically investigated for thermally activated delayed fluorescent (TADF)-based organic light-emitting diode (OLED) materials. The calculated results show that the designed molecules exhibit small singlet-triplet energy gap (ΔEST) values. Density functional theory (DFT) analysis indicated that the designed molecules display an efficient separation between donor and acceptor fragments because of a small overlap between donor and acceptor fragments on HOMOs and LUMOs. Furthermore, the delayed fluorescence emission color can be tuned effectively by introduction of different polycyclic aromatic fragments in parent molecule 1. The calculated results show that molecules 2, 3, and 4 possess more significant Stokes shifts and red emission with small ΔEST values. Nevertheless, other molecules exhibit green (1, 7, and 8), light green (6 and 10), and blue (5 and 9) emissions. Meanwhile, they are potential ambipolar charge transport materials except that 4 and 10 can serve as electron and hole transport materials only, respectively. Therefore, we proposed a rational way for the design of efficient TADF materials as well as charge transport materials for OLEDs simultaneously.
Organic light-emitting diodes (OLEDs) have drawn considerable attention for applications in displaying and lighting fields owing to their outstanding advantages nowadays (Choy et al., 2014; Zhang et al., 2015; Im et al., 2017a; Liu et al., 2017b, 2018; Pal et al., 2018; Zhu et al., 2018). Unfortunately, their commercialization applications are still limited by the low device performance at high luminance and low external quantum efficiency (EQE) of the emitters. It is noteworthy that charge recombination results in 25% singlet and 75% triplet excitons in the traditional fluorescence process (Tang and Vanslyke, 1987; Burroughes et al., 1990). The triplet excitons cannot be directly utilized by emitters because they are spin forbidden and decay is non-emissive in producing undesired heat. As a consequence, the internal quantum efficiency (IQE) of OLEDs does not exceed 25% using singlet excitons for traditional fluorescent materials. Remarkably, it is challenging to develop highly efficient emitters utilizing triplet excitons. The phosphorescent emitters incorporating heavy metals can reach an IQE of nearly 100% by enhancing electron spin–orbit coupling (Baldo et al., 1998; Adachi et al., 2001; Xia et al., 2014). However, the applications of phosphorescent materials containing heavy metals are limited owing to their high cost and environmental contamination (Li et al., 2013; Liu et al., 2017a; Wang et al., 2018). Accordingly, it is critically important to develop novel metal-free materials, which can be functionalized as efficient emitters and exhibit environment friendliness. To address this issue, thermally activated delayed fluorescence (TADF) materials have been considered recently as promising candidates because of their potential in achieving 100% IQEs by harvesting all the triplet excitons (Uoyama et al., 2012; Wu et al., 2017; Yang et al., 2017; Chatterjee and Wong, 2019; Wang et al., 2019). As for TADF materials, a small energy gap (ΔEST) between the lowest singlet (S1) and triplet (T1) states promotes efficient spin conversion from the triplet to singlet manifold by thermal activation through reverse intersystem crossing (RISC), subsequently resulting in fluorescence from the converted S1 to the ground (S0) states (Evans et al., 2018; Li et al., 2018b; Zhu et al., 2019). It is noteworthy that a small ΔEST is favorable for the RISC process and may obtain TADF. Furthermore, it is noticeable that the interplay between theory and experiment is capable of providing a deeper insight into the understanding of the optical and electronic properties of molecules in ground as well as excited states. Many theoretical research efforts have been made in this regard. It is critically important to get the relationship between topologic structure and optical as well as electronic properties for designing novel materials with improved properties. Density functional theory (DFT) and time-dependent DFT (TD-DFT) approaches have been remarkably successful in accurately evaluating a variety of ground and excited-state properties, in particular for TADF materials (Lu et al., 2015a,b; Wang et al., 2017; Hussain et al., 2019). In this regard, the rational design of a twisted donor–acceptor (D–A)-type structure can endow the molecules with TADF characteristics. These systems can efficiently reduce ΔEST in virtue of their separating the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) (Geng et al., 2017; Li et al., 2018a). Additionally, D–A architectures are beneficial for the intramolecular charge transfer (ICT) in the excited states (Santos et al., 2016; Deng et al., 2019; Zhang et al., 2020). In addition, TADF materials with different donor moieties such as 9,9-dimethyl-9,10-dihydroacridine, 10H-phenoxazine, 5,10-dihydrophenazine, 10H-phenothiazine, 9H-carbazole, 3,6-di-tert-butyl-9H-carbazole, 9,9-diphenyl-9,10-dihydroacridine 9H-spiro[4,5]fluorene-9,10-dihydroacridine, 9-(3-(9H-carbazol-9-yl)phenyl)-9H-carbazole, and diphenylamine possess excellent electroluminescence performance (Bezvikonnyi et al., 2019; Hosokai et al., 2019; Jin et al., 2019; Joo et al., 2019; Kim et al., 2019; Sharma et al., 2019; Zhong et al., 2019). Meanwhile, there are many TADF materials with different acceptor moieties such as benzonitrile, triazines, sulfones, benzophenone, quinoxaline, and naphthalimide reported in literature (Li et al., 2016; Nobuyasu et al., 2016; Tsujimoto et al., 2017; Sommer et al., 2018; Wu et al., 2018; Yu et al., 2018). The HOMO/LUMO distribution and energy levels of the typical donor and acceptor moieties have been found in the relevant literature (Im et al., 2017b). Among the numerous reported TADF materials, organoboron compounds have drawn significant interest recently owing to their appealing optical properties in OLEDs. Because of the lack of electrons in the Pz orbital, the central boron atoms in the organoboron compounds exhibit strong electron-accepting ability through p–π conjugations, which is favorable for the ICT (Ji et al., 2017; Giustra and Liu, 2018; Liang et al., 2018; Meng et al., 2019).
Considering the merits and characteristics mentioned above, in this work, we design a series of novel D–A tricoordinated organoboron compounds with 5,9-dioxa-13b-boranaphtho[3,2,1-de]anthracene (DOBA) as electron acceptors and different polycyclic aromatic fragments as electron donors for TADF molecules (Scheme 1). Applying DFT and TD-DFT approaches, optoelectronic properties including frontier molecular orbital (FMO) energies (EHOMO and ELUMO), HOMO–LUMO gaps (Eg), adiabatic ionization potentials (AIP), adiabatic electron affinities (AEA), reorganization energy (λ), and ΔEST were systematically investigated. The absorption, fluorescence, and phosphorescence spectra of the designed molecules were predicted using the TD-DFT method. This provides a useful insight for designing novel TADF materials as well as charge transport materials for OLEDs.
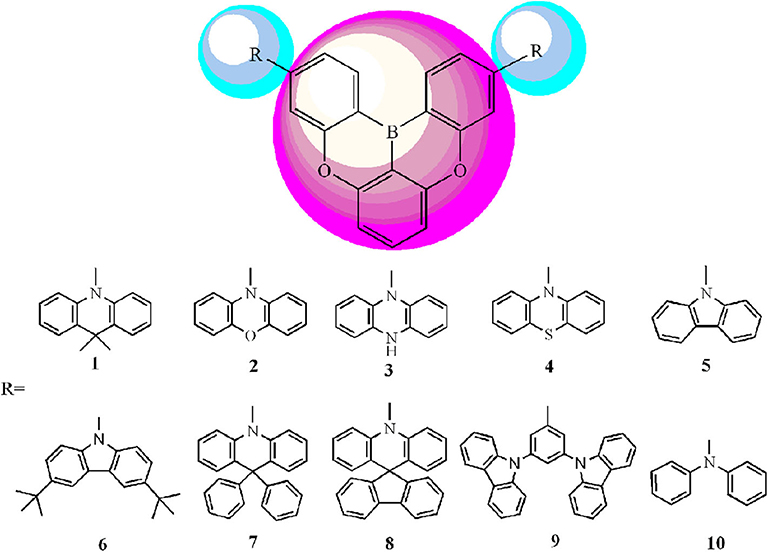
Scheme 1. Molecule models of 1–10 investigated in this work.
Geometry optimizations for all the designed molecules in the S0 states were accomplished using the DFT method in the Gaussian 09 package (Frisch et al., 2009). The corresponding frequency calculations were carried out at the same level to prove the nature of each stationary point. Using the TD-DFT approach, the structures in S1 and T1 states were optimized. Under the optimized structures in S0, S1, and T1 states, their absorption, fluorescence, and phosphorescence spectra were predicted by using the TD-DFT method. Meanwhile, the ΔEST values were evaluated by adiabatic excitation energy. The 6-31G (d,p) basis set was selected for all the calculations in this work. With the aim to select reasonable exchange correlation functionals, six functionals including B3LYP (Becke, 1993), PBE0 (Adamo and Barone, 1999), ωB97XD (Chai and Head-Gordon, 2008), M062X (Zhao and Truhlar, 2008), and CAM-B3LYP (Yanai et al., 2004) were employed to optimize the geometries of molecule 1 in S0, S1, and T1 states. On the basis of the optimized geometries in S0 and S1 states, the absorption and fluorescence spectra were predicted using the corresponding TD-DFT method. The ΔEST values were also calculated using the different methods. The corresponding results as well as the experimental data are shown in Figure 1 and Supplementary Table 1. As visualized in Figure 1, obviously, the fluorescence emission wavelength (λfl) obtained using the B3LYP functional (567 nm) is quite close to the experimental value (557 nm) (Meng et al., 2019), with the deviation of 10 nm. Furthermore, both the ΔEST values of molecule 1 obtained using B3LYP and PBE0 functionals (0.0068 and 0.0107 eV) are close to the experimental value (0.0091 eV). Additionally, comparing the optimized geometries of 1 (for the atom numbering, see Supplementary Figure 1) in S0 states with its crystal structure data [CCDC 1887610], one can find that the main geometrical parameters obtained using the B3LYP/6-31G(d,p) method are in better agreement with crystal structure data than those obtained with other methods (Supplementary Table 2). Therefore, geometry optimizations in S0, S1, and T1 states, band gaps Eg, ΔEST, absorption, fluorescence, and phosphorescence spectra of molecules under investigation were performed by the B3LYP/6-31G(d,p) and TD-B3LYP/6-31G(d,p) methods.
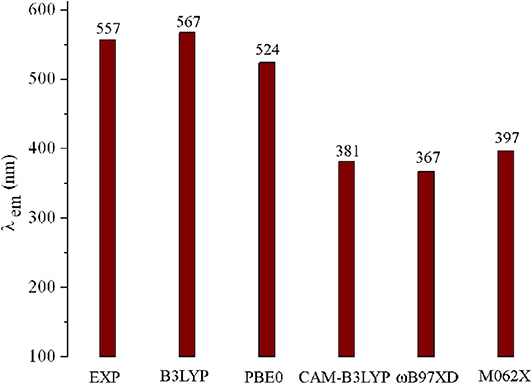
Figure 1. Calculated fluorescence wavelengths (λfl) of 1 using various functionals, along with available experimental data.
The reorganization energy λ is composed of external and internal reorganization energies (λext and λint). We focus only on the λint because λext is quite complicated to predict at this stage. λint can be expressed as (Köse et al., 2007; Sancho-García, 2007):
Here, E± and E stand for the energies of the charged and neutral states in the ground states, respectively; corresponds to the energy of the charged state with the optimized neutral molecule structure. E* represents the energy of neutral molecule with the optimized charged geometry. Furthermore, AIP and AEA were calculated by the adiabatic potential-energy surfaces of neutral/charged species. AIP can be obtained with the energy difference between cation and neutral specie. AEA was determined by the energy difference between neutral and anion specie. The AIP, AEA, and λ for the electron (λe) and hole (λh) of the designed molecules were predicted at the B3LYP/6-31G(d,p) level.
The main geometrical parameters of the designed molecules in S0, S1, and T1 states are given in Supplementary Table 3. The results displayed in Supplementary Table 3 reveal that there are no significant changes in the bond lengths of acceptor DOBA fragments for the designed molecules in S0 states. Compared with the parent molecule 1, the bond length change mainly appears on the bonds which connect the DOBA acceptor and different donor fragments. Obviously, the lengths of C5-N22 bonds for molecules 2, 3, 5–8, and 10 have been decreased by 0.005, 0.003, 0.02, 0.022, 0.001, and 0.024 Å, while the corresponding values of C5-N22 for 4 and C5-C22 for 9 have lengthened by 0.002 and 0.05 Å with respect to that of molecules 1, respectively. Similar phenomena are found for C18-N24 bonds for 2–8 and 10 and C18-C24 bond for 9. Furthermore, the designed molecules display a large twist angle (β) between their acceptor and donor fragments in the S0 states owing to their large steric hindrance. The β values of 1–4, 7, and 8 are about 81–100°, while the corresponding values of 5, 6, 9, and 10 are about 34–51°, respectively. The large β values facilitate for disrupting the electronic communication between D and A fragments. Comparing the geometrical parameters of the designed molecules in S1 states with those in the S0 states, the bond lengths of B1−C2, C4−C5, C6−C7, C9−C10, C13−C14, B1−C15, C17−C18, and C19−C20 (0.002–0.025 Å) are shortened, while other bond lengths are enlarged (0.001–0.025 Å), respectively. The more obvious bond length variations are found for the connecting bonds between acceptor and donor fragments for the designed molecules. The C5−N12 and C18−N24 bonds for 1–8 and 10 are stretched (0.005–0.038 Å and 0.005–0.038 Å) in the S1 states as compared to those in the S0 states, respectively. However, compared with the C5−C12 and C18−C24 bonds for 9 in the S0 state, obviously, they are shortened by 0.036 and 0.003 Å in the S1 state, respectively. Additionally, it can be seen that the twist angle β is closer to 90° in the S1 state as compared to those in the S0 states for the designed molecules except for 9, whose two β values in the S1 state are smaller than those in S0 states, respectively. Apparently, inspection of Supplementary Table 3 reveals that the geometrical parameters in T1 states are similar to those in S1 states for 1–4, 7, and 8. On the contrary, the molecular geometries in T1 states for 5, 6, 9, and 10 are closer to those in S0 states, respectively.
It is worth noting that the FMO energies (EHOMO and ELUMO) and Eg play dominant roles for the optical and electronic properties. The molecular geometry is affected by electron density redistribution that is caused by an electronic excitation (Forés et al., 1999; Helal et al., 2011). The distributions of HOMO and LUMO in S0 states for the designed molecules are plotted in Supplementary Figure 2. Additionally, we investigated the contributions of A and D fragments to the FMOs and the overlap value (ρ) between D and A fragments on HOMOs and LUMOs, as displayed in Supplementary Table 4. An inspection in Supplementary Figure 2 reveals clearly that the HOMOs of 1–10 are mainly localized on the D fragments, whereas the corresponding LUMOs predominantly reside at the A fragments. The contributions of D fragments for HOMOs in 1–4 and 7–9 are 93.1, 94.4, 95.6, 95.4, 93.1, 92.8, and 99.4%, while the corresponding contributions for 5, 6, and 10 are 83.1, 85.8, and 66.5%, respectively. For the LUMOs, the contributions of A fragments in 1–8 are larger than 90%, while the corresponding contributions for 9 and 10 are 75.9 and 84.5%, respectively. Apparently, the HOMO to LUMO transitions lead the electronic density to flow from D to A fragments. The percentages of charge transfer are in the order of 4 (89.3%) > 3 (88.9%) > 2 (87.5%) > 7 (86.4%) > 8 (85.9%) > 1 (85.7%) > 6 (76.9%)> 9 (75.3%)> 5 (74.8%) > 10 (51%). Moreover, the overlap (ρ) between D and A fragments on HOMOs is in the range of 0.001–0.040, while the corresponding ρ values for LUMOs are 0.013–0.026, for the designed molecules except for 10, whose ρ values on its HOMO and LUMO are 0.082 and 0.037, respectively. It indicates that the HOMO to LUMO transitions exhibit a strong charge transfer nature and slight electron exchange energy, resulting in small ΔEST values.
The qualitative HOMOs and LUMOs in S1 states for 1–10 are shown in Figure 2. The calculated EHOMO, ELUMO, and Eg, the overlap (ρ) between D and A fragments, and the contributions of A and D fragments (in %) to the FMOs of 1–10 are summarized in Table 1. Comparing the HOMOs and LUMOs in S1 states with those in S0 states, we find that the electron density plots of both HOMOs and LUMOs in S1 states are similar to those in S0 states for all the investigated molecules, respectively. The HOMOs are mainly centralized on the D fragments, whereas the corresponding LUMOs predominantly distributed on the A fragments. The contributions of D fragments for HOMOs are about 91.2–99.9%. The contributions of A fragments for the LUMOs are about 90.5–93.8% except that the contribution of A fragment for 9 is 68.7%. The percentages of charge transfer from D to A fragments are in the order of 3 (89.5%) > 4 (89.1%) > 2 (88.6%) > 6 (88.1%) > 5 (87.8%) > 7 (87.3%) > 8 (86.6%)> 1 (86.3%)> 10 (81.7%) > 9 (68.6%). Clearly, the charge transfer character in S1 states is more significant than that in S0 states. Furthermore, the ρ values for HOMOs are in the range of 0.001–0.019, while the corresponding ρ values for LUMOs are about 0.014–0.037. It suggests that the HOMOs and LUMOs are also separated efficiently in the S1 state. This implies the efficient separation between D and A fragments, which display the potential TADF features. The sequence of EHOMO and ELUMO is 3 > 4 > 2 > 1> 8 > 10 > 7 > 6 > 5 > 9 and 10 > 3 > 6 > 1> 7 > 8 > 5 > 2 > 4 > 9, respectively. Thus, the Eg values are in the order of 5 > 10 > 9 > 6> 7 > 8 > 1 > 2 > 4 > 3. It suggests that the Eg values of 2–4 decrease, while the corresponding value of 5–10 increases compared with that of 1. Therefore, one can conclude that 2–4 may possess a red shift, whereas 5–10 could exhibit a blue shift in their emission spectra in comparison with the parent molecule 1. It implies that the introduction of different aromatic heterocyclic donors can tune effectively the EHOMO, ELUMO, and Eg values for the designed molecules. Furthermore, we investigated the qualitative HOMO and LUMO plots in T1 states for 1–10 (Supplementary Figure 3). One can find that the qualitative HOMOs and LUMOs in T1 states are similar to those in S1 states for all the investigated molecules. It is also favorable for the RISC process from T1 to S1 states.

Figure 2. The distributions of HOMOs and LUMOs in S1 states for the designed molecules.
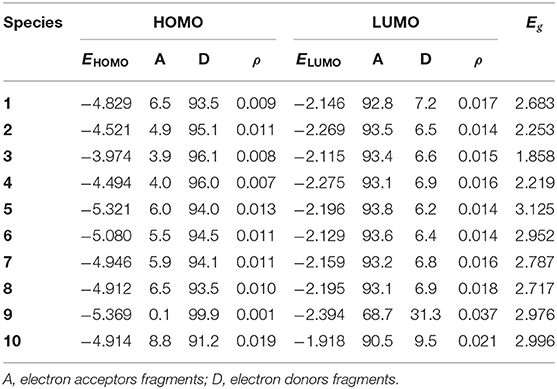
Table 1. The FMO energies EHOMO and ELUMO, HOMO–LUMO gaps Eg (all in eV), HOMO and LUMO contributions (%), and the overlap between D and A fragments on HOMOs and LUMOs (ρ) of 1–10 in S1 states.
Table 2 collected the vertical excitation energy (ES1 and ET1) in S1 and T1 states and ΔEST of 1–10 at the TD-B3LYP/6-31G (d,p) level. It is noticeable that the lower the ΔEST values, the easier the RISC process from the T1 to S1 states. The ES1 and ET1 values are in the orders 5 > 9 > 10 > 6 > 7 > 8 > 1 > 2 > 4 > 3 and 5 > 9 > 6 > 10 > 7 > 8 > 1 > 2 > 4 > 3, respectively. As expected, the order of ES1 values is similar to those Eg values except that a sequential interchange is found between 9 and 10. Moreover, the sequence of ET1 values is similar to ES1 values except that a sequential interchange is found between 6 and 10. One can find that all the designed molecules possess small ΔEST values. The prediction of ΔEST values is in the sequence 10 (0.2659) > 9 (0.2600) > 5 (0.1257) > 6 (0.0618) > 7 (0.0078) > 8 (0.0069) > 1 (0.0068) > 2 (0.0065) > 4 (0.0062) > 3 (0.0057). The results displayed in Table 2 reveal that the ΔEST values of 2–4, 7, and 8 (0.0057–0.0078 eV) are similar to that for the parent molecule 1 (0.0068 eV). On the contrary, molecules 5, 6, 9, and 10 have larger ΔEST values than the parent molecule 1, especially for molecules 9 and 10, whose ΔEST values are 0.2600 and 0.2659 eV, respectively. The reason might be that 2–4, 7, and 8 own the more effective separation between their donor/acceptor compositions (%) of the FMOs in the S1 state than those larger than 90% (Table 1 and Figure 2). Additionally, β is closer to 90° in the S1 state as compared to those of 5, 6, 9, and 10. For molecules 5 and 6, among their two β, β values are both close to 90°, while another β values are −60.6 and 59.1°, respectively. However, molecule 6 has two electron donor tert-butyl groups in donor fragments, which is more favorable for charge transfer and in turn decreases the ΔEST value. As a consequence, the ΔEST value of 6 is smaller than that of 5. For molecules 9 and 10, their smaller two β values may decrease the effective separation between their donor and acceptor fragments. Two β values are −12.7 and 32.4° for 9 and −51.7 and 77.2° for 10 in S1 states. It indicates that the introduction of 10H-phenoxazine (2) 5,10-dihydrophenazine (3), 10H-phenothiazine (4), 9,9-diphenyl-9,10-dihydroacridine (7), and 9H-spiro[4,5]fluorene-9,10-dihydroacridine (8) donor fragments does not significantly affect the ΔEST values compared with the parent molecule with 9,9-dimethyl-9,10-dihydroacridine donor fragments (1). On the contrary, the introduction of 9H-carbazole (5), 3,6-di-tert-butyl-9H-carbazole (6), 9-(3-(9H-carbazol-9-yl)phenyl)-9H-carbazole (9), diphenylamine (10), and donor fragments increase the ΔEST values compared with parent molecule 1. As a consequence, the RISC rate constant (kRISC) of 1–4, 7, and 8 should be higher than those of molecules 5, 6, 9, and 10 because a small ΔEST value is beneficial for the high kRISC value.
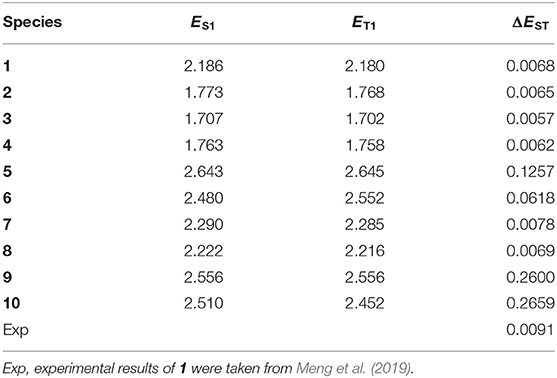
Table 2. The vertical excitation energy (ES1 and ET1) and singlet triplet energy gap (ΔEST) of 1–10 at the TD-B3LYP/6-31G(d,p) level (in eV).
The calculated wavelength of delayed fluorescence emission (λTADF), phosphorescence emission (λph), absorption (λabs), and Stokes shift of 1–10 are listed in Table 3. As visualized in Figure 2 and Supplementary Figures 2, 3, the FMO distributions in S0, S1, and T1 states possess π characteristics. The results presented in Table 3 show that the λabs values of 2–10 have slight bathochromic shifts compared with that of the parent compound 1, respectively. The λTADF values of 1–10 follow the tendency 3 > 4 > 2 > 1 > 8 > 7 > 6 > 10 > 9 > 5, which is similar to the reverse order of Eg values. By comparing with parent molecule 1, the λTADF values of 2–4 exhibit bathochromic shifts, 132.2, 159.5, and 136.9 nm, respectively, due to the increased electron-donating ability of the donor fragments. Conversely, it can be noted that the λTADF values of 5–10 have hypsochromic shifts, 97.9, 67.2, 25.7, 9.1, 82, and 73.1 nm compared with that of 1, respectively. Obviously, 2–4 possess more significant Stokes shifts (326.6, 320.0, and 330.5 nm) and red emission with small ΔEST values (0.0065, 0.0057, and 0.0062 eV). Nevertheless, 1, 7, and 8 exhibit green emissions with Stokes shifts, 196.7, 169.8, and 183.1 nm, respectively. In addition, 6 and 10 show light green emissions, while 5 and 9 display blue emissions with small Stokes shifts. The Stokes shifts of 5, 6, 9, and 10 are 60.0, 75.0, 68.1, and 92.2 nm, respectively. Accordingly, the delayed fluorescence emission color can be tuned effectively by introduction of different polycyclic aromatic fragments as electron donors in the parent molecule. This implies that the designed molecules are expected to be the promising candidates for TADF materials, particularly for 2–4, 7, and 8. Furthermore, the λph values are similar to the λTADF values. The small difference between the λTADF and λph values should facilitate for achieving the TADF phenomenon.
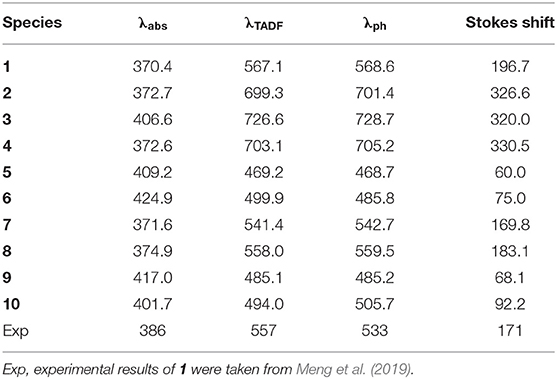
Table 3. The delayed fluorescence emission wavelength (λTADF) and phosphorescence emission wavelength (λph), corresponding to the absorption wavelength (λabs), and Stokes shift of 1–10 at the TD-B3LYP/6-31G (d,p) level.
It is quite clear that the charge injection and charge transfer play dominant roles in the device performance of OLEDs. λ can be used to estimate the charge transfer rate. The AIP and AEA are used to evaluate the energy barrier for the injection of holes and electrons. Generally, the low λ value corresponds to the big charge transfer rate (Marcus, 1964, 1993). The smaller the AIP value and the larger the AEA value, the easier the injection of holes and electrons, respectively. The calculated λe, λh, AIP, and AEA of 1–10 are listed in Table 4. As presented in Table 4, we find that 3 and 5 have the smallest and largest AIP values (5.151 and 6.387 eV), respectively. The AIP values of 1–10 are observed in the following: 5 > 9 > 6 > 7 > 10 > 8 > 1 > 4 > 2 > 3, suggesting that the hole injection and transportations of 2–4 are expected to be easier than the others with respect to parent molecule 1. On the other hand, the AEA values of 3, 5, 6, and 10 are lower, while the corresponding values of 2, 4, and 7–9 are higher than that of 1 (0.878 eV). The order of the AEA of 1–10 is as follows: 9 > 4 > 2 > 8 > 7 > 1 > 5 > 3 > 6 > 10; this implies that the abilities to accept electrons in 2, 4, and 7–9 are improved with respect to parent molecule 1. So, we can deduce that the introduction of 10H-phenoxazine (2) and 10H-phenothiazine (4) donor fragments will enhance both the hole and electron injection abilities, whereas the introduction of 9,9-diphenyl-9,10-dihydroacridine (7), 9H-spiro[4,5]fluorene-9,10-dihydroacridine (8), and 9-(3-(9H-carbazol-9-yl)phenyl)-9H-carbazole (9) donor fragments can improve the electron injection ability only compared with that of parent molecule 1.
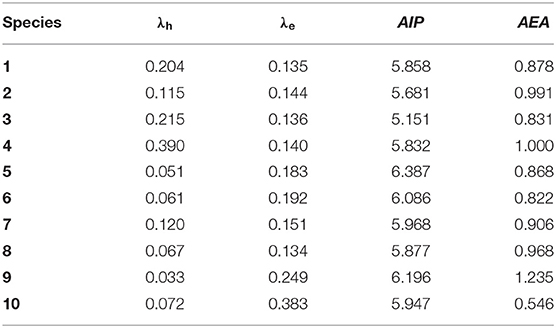
Table 4. Calculated molecular λe, λh, AIP, and AEA (all in eV) of 1–10 at the B3LYP/6-31G(d,p) level.
For charge transport materials in OLEDs, generally, TPD (λh = 0.290 eV) and Alq3 (λe = 0.276 eV) are used as typical hole and electron transport materials, respectively (Gruhn et al., 2002; Lin et al., 2005). According to Table 4, the calculated λh values of 1–3 and 5–10 are smaller than those of TPD. It suggests that the hole transfer rates of the designed molecules except for 4 may be higher than that of TPD. The λh values are in the order 4 > 3 > 1 > 2 > 7 > 10 > 8 > 6 > 5 > 9. On the other hand, the λe values of 1–9 are smaller than that of Alq3. It implies that the electron transfer rates of 1–9 may be higher than that of Alq3, suggesting that their electron transfer rates might be lower than that of Alq3. The order of the predicted λe is as follows: 10 > 9 > 6 > 5 > 7 > 2 > 3 > 6 > 1 > 8. Remarkably, the difference between λh and λe for 1–3, 5–9 (0.029–0.216 eV) is small enough, suggesting that they are potential ambipolar charge transport materials. Nevertheless, 4 and 10 can serve as electron and hole transport materials only, respectively.
In summary, a series of novel D–A tricoordinated organoboron derivatives have been systematically investigated for TADF-based OLED materials. The calculated results show that the designed molecules exhibit small ΔEST values, suggesting that they can be used as excellent TADF candidates. DFT analysis indicated that the designed molecules display efficient separation between donor and acceptor fragments because of the small overlap between donor and acceptor fragments (ρ) on HOMOs and LUMOs. Furthermore, the delayed fluorescence emission color can be tuned effectively by introduction of different polycyclic aromatic fragments in the parent molecule. Molecules 2, 3, and 4 possess more significant Stokes shifts (326.6, 320.0, and 330.5 nm) and red emission with small ΔEST values (0.0065, 0.0057, and 0.0062 eV). Nevertheless, other molecules exhibit green (1, 7, and 8), light green (6 and 10), and blue (5 and 9) emissions. Meanwhile, they are potential good ambipolar charge transport materials except that 4 and 10 can serve as electron and hole transport materials only, respectively. Therefore, we proposed a rational way for the design of efficient TADF materials as well as charge transport materials for OLEDs simultaneously.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
RJ conceived and designed the research, and headed, wrote, and revised the manuscript. JX contributed to the performance and analysis of the frontier molecular orbitals, absorption fluorescence, and phosphorescence spectra, and the reorganization energies. Both authors contributed to the manuscript revision and read and approved the submitted version.
This work was supported by NSFC (No. 21563002) and Natural Science Foundation of Inner Mongolia Autonomous Region (No. 2015MS0201).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.577834/full#supplementary-material
Adachi, C., Baldo, M. A., Thompson, M. E., and Forrest, S. R. (2001). Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 90, 5048–5051. doi: 10.1063/1.1409582
Adamo, C., and Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170. doi: 10.1063/1.478522
Baldo, M. A., O'Brien, D. F., You, Y., Shoustikov, A., Sibley, S., Thompson, M. E., et al. (1998). Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 395, 151–154. doi: 10.1038/25954
Becke, A. D. (1993). Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Bezvikonnyi, O., Gudeika, D., Volyniuk, D., Mimaite, V., Sebastine, B. R., and Grazulevicius, J. V. (2019). Effect of donor substituents on thermally activated delayed fluorescence of diphenylsulfone derivatives. J. Lumin. 206, 250–259. doi: 10.1016/j.jlumin.2018.10.018
Burroughes, J. H., Bradley, D. D. C., Brown, A. R., Marks, R. N., MacKay, K., Friend, R. H., et al. (1990). Light-emitting diodes based on conjugated polymers. Nature 347, 539–541. doi: 10.1038/348352a0
Chai, J. D., and Head-Gordon, M. (2008). Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. doi: 10.1039/B810189B
Chatterjee, T., and Wong, K.-T. (2019). Perspective on host materials for thermally activated delayed fluorescence organic light emitting diodes. Adv. Opt. Mater. 7:1800565. doi: 10.1002/adom.201800565
Choy, W. C. H., Chan, W. K., and Yuan, Y. (2014). Recent advances in transition metal complexes and light-management engineering in organic optoelectronic devices. Adv. Mater. 26, 5368–5399. doi: 10.1002/adma.201306133
Deng, Z., Ai, T., Li, R., Yuan, W., Zhang, K., Du, H., et al. (2019). Conjugated polymers containing building blocks 1,3,4,6-tetraarylpyrrolo[3,2-b]pyrrole-2,5-dione (isoDPP), benzodipyrrolidone (BDP) or naphthodipyrrolidone (NDP): a review. Polymers 11:1683. doi: 10.3390/polym11101683
Evans, E. W., Olivier, Y., Puttisong, Y., Myers, W. K., Hele, T. J. H., Menke, S. M., et al. (2018). Vibrationally assisted intersystem crossing in benchmark thermally activated delayed fluorescence molecules. J. Phys. Chem. Lett. 9, 4053–4058. doi: 10.1021/acs.jpclett.8b01556
Forés, M., Duran, M., Solà, M., and Adamowicz, L. (1999). Excited-state intramolecular proton transfer and rotamerism of 2-(2'-hydroxyvinyl)benzimidazole and 2-(2'-hydroxyphenyl)imidazole. J. Phys. Chem. A 103, 4413–4420. doi: 10.1021/jp9844765
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. Wallingford, CT: Gaussian, Inc.
Geng, Y., D'Aleo, A., Inada, K., Cui, L. S., Kim, J. U., Nakanotani, H., and Adachi, C. (2017). Donor-σ-acceptor motifs: thermally activated delayed fluorescence emitters with dual upconversion. Angew.Chem. Int. Ed. 56, 16536–16540. doi: 10.1002/anie.201708876
Giustra, Z. X., and Liu, S. Y. (2018). The state of the art in azaborine chemistry: new synthetic methods and applications. J. Am. Chem. Soc. 140, 1184–1194. doi: 10.1021/jacs.7b09446
Gruhn, N. E., da Silva Filho, D. A., Bill, T. G., Malagoli, M., Coropceanu, V., Kahn, A., et al. (2002). The vibrational reorganization energy in pentacene: molecular influences on charge transport. J. Am. Chem. Soc. 124, 7918–7919. doi: 10.1021/ja0175892
Helal, A., Rashid, M. H. O., Choi, C. H., and Kim, H. S. (2011). Chromogenic and fluorogenic sensing of Cu2+ based on coumarin. Tetrahedron 67, 2794–2802. doi: 10.1016/j.tet.2011.01.093
Hosokai, T., Nakanotani, H., Santou, S., Noda, H., Nakayama, Y., and Adachi, C. (2019). TADF activation by solvent freezing: the role of nonradiative triplet decay and spin-orbit coupling in carbazole benzonitrile derivatives. Synthet. Met. 252, 62–68. doi: 10.1016/j.synthmet.2019.04.005
Hussain, A., Yuan, H., Li, W., and Zhang, J. (2019). Theoretical investigations of the realization of sky-blue to blue TADF materials via CH/N and H/CN substitution at the diphenylsulphone acceptor. J. Mater. Chem. C 7, 6685–6691. doi: 10.1039/C9TC01449G
Im, Y., Byun, S. Y., Kim, J. H., Lee, D. R., Oh, C. S., Yook, K. S., et al. (2017a). Recent progress in high-efficiency blue-light-emitting materials for organic light-emitting diodes. Adv. Funct. Mater. 27:1603007. doi: 10.1002/adfm.201603007
Im, Y., Kim, M., Cho, Y. J., Seo, J.-A., Yook, K. S., and Lee, J. Y. (2017b). Molecular design strategy of organic thermally activated delayed fluorescence emitters. Chem. Mater. 29, 1946–1963. doi: 10.1021/acs.chemmater.6b05324
Ji, L., Griesbeck, S., and Marder, T. B. (2017). Recent developments in and perspectives on three-coordinate boron materials: a bright future. Chem. Sci. 8, 846–863. doi: 10.1039/C6SC04245G
Jin, J., Ying Gao, Y., and Geng, Y. (2019). A theoretical investigation on the thermally activated delayed fluorescence characteristics of the isomers of DTCB Py. J. Mol. Graph. Model. 86, 125–131. doi: 10.1016/j.jmgm.2018.10.002
Joo, C. W., Huseynova, G., Yifei, J., Yoo, J. M., Kim, Y. H., Cho, N. S., et al. (2019). Highly efficient solution-processed blue organic light-emitting diodes based on thermally activated delayed fluorescence emitters with spiroacridine donor. J. Ind. Eng. Chem. 78, 265–270. doi: 10.1016/j.jiec.2019.06.003
Kim, J. H., Lee, K. H., and Lee, J. Y. (2019). Design of thermally activated delayed fluorescent assistant dopants to suppress the nonradiative component in red fluorescent organic light-emitting diodes. Chem. Eur. J. 25, 9060–9070. doi: 10.1002/chem.201901135
Köse, M. E., Mitchell, W. J., Kopidakis, N., Chang, C. H., Shaheen, S. E., Kim, K., et al. (2007). Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 129, 14257–14270. doi: 10.1021/ja073455y
Li, C., Duan, C., Han, C., and Xu, H. (2018a). Secondary acceptor optimization for full-exciton radiation: toward sky-blue thermally activated delayed fluorescence diodes with external quantum efficiency of ≈30%. Adv. Mater. 30:1804228. doi: 10.1002/adma.201804228
Li, C., Wang, Y., Sun, D., Li, H., Sun, X., Ma, D., et al. (2018b). Thermally activated delayed fluorescence pendant copolymers with electron- and hole-transporting spacers. ACS Appl. Mater. Interfaces 10, 5731–5739. doi: 10.1021/acsami.8b00136
Li, J., Nakagawa, T., MacDonald, J., Zhang, Q., Nomura, H., Miyazaki, H., et al. (2013). Highly efficient organic light-emitting diode based on a hidden thermally activated delayed fluorescence channel in a heptazine derivative. Adv. Mater. 25, 3319–3323. doi: 10.1002/adma.201300575
Li, Y., Xie, G., Gong, S., Wu, K., and Yang, C. (2016). Dendronized delayed fluorescence emitters for non-doped, solution-processed organic light-emitting diodes with high efficiency and low efficiency roll-off simultaneously: two parallel emissive channels. Chem. Sci. 7, 5441–5447. doi: 10.1039/C6SC00943C
Liang, X., Yan, Z. P., Han, H. B., Wu, Z. G., Zheng, Y. X., Meng, H., et al. (2018). Peripheral amplification of multi-resonance induced thermally activated delayed fluorescence for highly efficient OLEDs. Angew.Chem. Int. Ed. 57, 11316–11320. doi: 10.1002/anie.201806323
Lin, B. C., Cheng, C. P., You, Z. Q., and Hsu, C. P. (2005). Charge transport properties of tris(8-hydroxyquinolinato)aluminum(III): why it is an electron transporter. J. Am. Chem. Soc. 127, 66–67. doi: 10.1021/ja045087t
Liu, B., Dang, F., Feng, Z., Tian, Z., Zhao, J., Wu, Y., et al. (2017a). Novel Iridium(III) complexes bearing dimesitylboron groups with nearly 100% phosphorescent quantum yields for highly efficient organic light-emitting diodes. J. Mater. Chem. C 5, 7871–7883. doi: 10.1039/C7TC02369C
Liu, B., Dang, F., Tian, Z., Feng, Z., Jin, D., Dang, W., et al. (2017b). High triplet energy level achieved by tuning the arrangement of building blocks in phosphorescent polymer backbones for furnishing high electroluminescent performances in both blue and white organic light-emitting devices. ACS Appl. Mater. Interfaces 9, 16360–16374. doi: 10.1021/acsami.7b04509
Liu, Y., Li, C., Ren, Z., Yan, S., and Bryce, M. R. (2018). All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 3:18020. doi: 10.1038/natrevmats.2018.20
Lu, J., Zheng, Y., and Zhang, J. (2015a). Rational design of phenoxazine-based donor–acceptor–donor thermally activated delayed fluorescent molecules with high performance. Phys. Chem. Chem. Phys. 17, 20014–20020. doi: 10.1039/c5cp02810h
Lu, J., Zheng, Y., and Zhang, J. (2015b). Tuning the color of thermally activated delayed fluorescent properties for spiro-acridine derivatives by structural modification of the acceptor fragment: a DFT study. RSC Adv. 5, 18588–18592. doi: 10.1039/c4ra15155k
Marcus, R. A. (1964). Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 15, 155–196.
Marcus, R. A. (1993). Electron transfer reactions in chemistry.theory and experiment. Rev. Mod. Phys. 65, 599–610.
Meng, G., Chen, X., Wang, X., Wang, N., Peng, T., and Wang, S. (2019). Isomeric bright sky-blue TADF emitters based on bisacridine decorated DBNA: impact of donor locations on luminescent and electroluminescent properties. Adv. Optical. Mater. 7:1900130. doi: 10.1002/adom.201900130
Nobuyasu, R. S., Ren, Z., Griffiths, G. C., Batsanov, A. S., Data, P., Yan, S., et al. (2016). Rational design of TADF polymers using a donor–acceptor monomer with enhanced TADF efficiency induced by the energy alignment of charge transfer and local triplet excited states. Adv. Opt. Mater. 4, 597–607. doi: 10.1002/adom.201500689
Pal, A. K., Krotkus, S., Fontani, M., Mackenzie, C. F. R., Cordes, D. B., Slawin, A. M. Z., et al. (2018). High-efficiency deep-blue-emitting organic light-emitting diodes based on Iridium(III) carbene complexes. Adv. Mater. 30:1804231. doi: 10.1002/adma.201804231
Sancho-García, J. C. (2007). Assessment of density-functional models for organic molecular semiconductors: the role of hartree–fock exchange in charge-transfer processes. Chem. Phys. 331, 321–331. doi: 10.1016/j.chemphys.2006.11.002
Santos, P. L., Ward, J. S., Data, P., Batsanov, A. S., Bryce, M. R., Dias, F. B., and Monkman, A. P. (2016). Engineering the singlet–triplet energy splitting in a TADF molecule. J. Mater. Chem. C 4, 3815–3824. doi: 10.1039/c5tc03849a
Sharma, N., Spuling, E., Mattern, C. M., Li, W., Fuhr, O., Tsuchiya, Y., et al. (2019). Turn on of sky-blue thermally activated delayed fluorescence and circularly polarized luminescence (CPL) via increased torsion by a bulky carbazolophane donor. Chem. Sci. 10, 6689–6696. doi: 10.1039/c9sc01821b
Sommer, G. A., Mataranga-Popa, L. N., Czerwieniec, R., Hofbeck, T., Homeier, H. H. H., Müller, T. J. J., et al. (2018). Design of conformationally distorted donor–acceptor dyads showing efficient thermally activated delayed fluorescence. J. Phys. Chem. Lett. 9, 3692–3697. doi: 10.1021/acs.jpclett.8b01511
Tang, C. W., and Vanslyke, S. A. (1987). Organic electroluminescent diodes. Appl. Phys. Lett. 51, 913–915. doi: 10.1063/1.98799
Tsujimoto, H., Ha, D. G., Markopoulos, G., Chae, H. S., Baldo, M. A., and Swager, T. M. (2017). Thermally activated delayed fluorescence and aggregation induced emission with through-space charge transfer. J. Am. Chem. Soc. 139, 4894–4900. doi: 10.1021/jacs.7b00873
Uoyama, H., Goushi, K., Shizu, K., Nomura, H., and Adachi, C. (2012). Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492:234. doi: 10.1038/nature11687
Wang, J., Lu, J., and Zhang, J. (2017). Tuning the electronic and optical properties of diphenylsulphone based thermally activated delayed fluorescent materials via structural modification: a theoretical study. Dyes. Pigments. 143, 42–47. doi: 10.1016/j.dyepig.2017.03.064
Wang, Q., Zhang, Y. X., Yuan, Y., Hu, Y., Tian, Q. S., Jiang, Z. Q., and Liao, L. S. (2019). Alleviating efficiency roll-off of hybrid single-emitting layer WOLED utilizing bipolar TADF material as host and emitter. ACS Appl. Mater. Interfa Interfaces 11, 2197–2204. doi: 10.1021/acsami.8b18665
Wang, S., Zhao, L., Zhang, B., Ding, J., Xie, Z., Wang, L., and Wong, W. Y. (2018). High-energy-level blue phosphor for solution-processed white organic light-emitting diodes with efficiency comparable to fluorescent tubes. Science 6, 128–137. doi: 10.1016/j.isci.2018.07.016
Wu, K., Wang, Z., Zhan, L., Zhong, C., Gong, S., Xie, G., et al. (2018). Realizing highly efficient solution-processed homojunction-like sky-blue OLEDs by using thermally activated delayed fluorescent emitters featuring an aggregation-induced emission property. J. Phys. Chem. Lett. 9, 1547–1553. doi: 10.1021/acs.jpclett.8b00344
Wu, S. F., Li, S. H., Wang, Y. K., Huang, C. C., Sun, Q., Liang, J. J., et al. (2017). White organic LED with a luminous efficacy exceeding 100 lm w−1 without light out-coupling enhancement techniques. Adv. Funct. Mater. 27:1701314. doi: 10.1002/adfm.201701314
Xia, D., Wang, B., Chen, B., Wang, S., Zhang, B., Ding, J., et al. (2014). Self-host blue-emitting iridium dendrimer with carbazoledendrons: nondoped phosphorescent organic light-emitting diodes. Angew. Chem. Int. Ed. 53, 1048–1052. doi: 10.1002/ange.201307311
Yanai, T., Tew, D. P., and Handy, N. C. (2004). A new hybrid exchange–correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57. doi: 10.1016/j.cplett.2004.06.011
Yang, Z., Mao, Z., Xie, Z., Zhang, Y., Liu, S., Zhao, J., et al. (2017). Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 46, 915–1016. doi: 10.1039/c6cs00368k
Yu, L., Wu, Z., Xie, G., Zeng, W., Ma, D., and Yang, C. (2018). Molecular design to regulate the photophysical properties of multifunctional TADF emitters towards high-performance TADF-based OLEDs with EQEs up to 22.4% and small efficiency roll-offs. Chem. Sci. 9, 1385–1391. doi: 10.1039/C7SC04669C
Zhang, H., Li, R., Deng, Z., Cui, S., Wang, Y., Zheng, M., et al. (2020). π-Conjugated oligomers based on aminobenzodifuranone and diketopyrrolopyrrole. Dyes. Pigments. 181:108552. doi: 10.1016/j.dyepig.2020.108552
Zhang, S., Yao, L., Peng, Q., Li, W., Pan, Y., Xiao, R., et al. (2015). Achieving a significantly increased efficiency in nondoped pure blue fluorescent OLED: a quasi-equivalent hybridized excited state. Adv. Funct. Mater. 25, 1755–1762. doi: 10.1002/adfm.201404260
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor.Chem.Acc.120, 215–241. doi: 10.1007/s00214-007-0401-8
Zhong, D., Yu, Y, Song, D., Yang, X., Zhang, Y., Chen, X., et al. (2019). Organic emitters with a rigid 9-phenyl-9-phosphafluorene oxide moiety as the acceptor and their thermally activated delayed fluorescence behavior. ACS Appl. Mater. Interfaces 11, 27112–27124. doi: 10.1021/acsami.9b05950
Zhu, X. D., Tian, Q. S., Zheng, Q., Tao, X. C., Yuan, Y., Yu, Y. J., et al. (2019). A sky-blue thermally activated delayed fluorescence emitter based on multimodifiedcarbazole donor for efficient organic light-emitting diodes. Org. Electron. 68, 113–120. doi: 10.1016/j.orgel.2019.02.001
Keywords: tricoordinated organoboron derivatives, thermally activated delayed fluorescent (TADF), photophysical properties, reorganization energy, organic light-emitting diodes (OLEDs)
Citation: Jin R and Xin J (2020) Rational Design of π-Conjugated Tricoordinated Organoboron Derivatives With Thermally Activated Delayed Fluorescent Properties for Application in Organic Light-Emitting Diodes. Front. Chem. 8:577834. doi: 10.3389/fchem.2020.577834
Received: 30 June 2020; Accepted: 26 August 2020;
Published: 30 September 2020.
Edited by:
Qixin Zhou, University of Akron, United StatesReviewed by:
Yang Zhou, Teknor Apex Company, United StatesCopyright © 2020 Jin and Xin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruifa Jin, UnVpZmFqaW5AMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.