
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 01 September 2020
Sec. Medicinal and Pharmaceutical Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00795
This article is part of the Research Topic Medicinal and Pharmaceutical Chemistry Editor’s Pick 2021 View all 21 articles
 Letizia Crocetti1
Letizia Crocetti1 Maria Paola Giovannoni1*
Maria Paola Giovannoni1* Niccolò Cantini1
Niccolò Cantini1 Gabriella Guerrini1
Gabriella Guerrini1 Claudia Vergelli1
Claudia Vergelli1 Igor A. Schepetkin2
Igor A. Schepetkin2 Andrei I. Khlebnikov3
Andrei I. Khlebnikov3 Mark T. Quinn2
Mark T. Quinn2Human neutrophil elastase (HNE) is involved in a number of essential physiological processes and has been identified as a potential therapeutic target for treating acute and chronic lung injury. Nevertheless, only one drug, Sivelestat, has been approved for clinical use and just in Japan and the Republic of Korea. Thus, there is an urgent need for the development of low-molecular-weight synthetic HNE inhibitors, and we have developed a wide variety of HNE inhibitors with various chemical scaffolds. We hypothesized that substitution of the active fragment of Sivelestat into these HNE inhibitor scaffolds could modulate their inhibitory activity, potentially resulting in higher efficacy and/or improved chemical stability. Here, we report the synthesis, biological evaluation, and molecular modeling studies of novel compounds substituted with the 4-(sulfamoyl)phenyl pivalate fragment necessary for Sivelestat activity. Many of these compounds were potent HNE inhibitors with activity in the nanomolar range (IC50 = 19–30 nM for compounds 3a, 3b, 3f, 3g, and 9a), confirming that the 4-(sulfamoyl)phenyl pivalate fragment could substitute for the N-CO group at position 1 and offer a different point of attack for Ser195. Results of molecular docking of the these pivaloyl-containing compounds into the HNE binding site supported the mechanism of inhibitory activity involving a nucleophilic attack of Ser195 from the catalytic triad onto the pivaloyl carbonyl group. Furthermore, some compounds (e.g., 3a and 3f) had a relatively good stability in aqueous buffer (t1/2 > 9 h). Thus, this novel approach led to the identification of a number of potent HNE inhibitors that could be used as leads for the further development of new therapeutics.
Human neutrophil elastase (HNE) is a multifunctional enzyme involved in the killing of pathogens, regulation of inflammatory processes, and tissue homeostasis. HNE is also involved in chemotaxis and the release of inflammatory mediators through the cleavage of adhesion molecules in cellular junctions (Pham, 2006; Korkmaz et al., 2010). Under physiological conditions, HNE is regulated by a group of endogenous protease inhibitors called “serpins” (Silverman et al., 2001; Heutinck et al., 2010). However, when this balance fails in favor of the proteolytic enzyme, excessive HNE activity can cause tissue damage. Among the pathologies associated with increased HNE activity are acute respiratory distress syndrome (ARDS) and acute lung injury (ALI) (Polverino et al., 2017), chronic obstructive pulmonary disease (Pandey et al., 2017), cystic fibrosis (Kelly et al., 2008; Dittrich et al., 2018), and other disorders with an inflammatory component, such as rheumatoid arthritis (Hilbert et al., 2002; Di Cesare Mannelli et al., 2016), atherosclerosis (Henriksen and Sallenave, 2008; Wen et al., 2018), psoriasis, and dermatitis (Marto et al., 2018). HNE has also been implicated in the progression of non-small cell lung cancer (Lerman and Hammes, 2017; Lerman et al., 2017).
The development of new and selective HNE inhibitors is of great interest, both in the academic and industrial world, for therapeutic development. Despite the discovery of numerous classes of potent HNE inhibitors (Groutas et al., 2011; Von Nussbaum and Li, 2015; Crocetti et al., 2019), only two drugs are currently available on the market: the peptide inhibitor Prolastin® (purified α1-antitrypsin, from Alpha Therapeutic Corp, 2003) and the small molecule Sivelestat. Sivelestat (ONO-5046, Figure 1) developed by Ono Pharmaceutical and marketed in Japan and Korea exclusively as a sodium salt in the injectable formulation Elaspol® 100 for the treatment of ARDS and ALI associated with systemic inflammatory response syndrome and in pediatric surgery to alleviate the inflammatory response induced by cardiopulmonary bypass (Kawabata et al., 1991; Fujii et al., 2010; Inoue et al., 2013). Sivelestat acts as acyl-enzyme inhibitor (IC50 = 44 nM) (Ohbayashi, 2005), as demonstrated by electrospray ionization mass spectrometry (ESI), which highlighted the formation of a HNE-Sivelestat complex after 0–10 min incubation of the drug with HNE (Figure 2) (Nakayama et al., 2002). We have been working for some time on the design and synthesis of HNE inhibitors with different nitrogen monocyclic and bicyclic scaffolds (Crocetti et al., 2011, 2013, 2016, 2017, 2018; Giovannoni et al., 2016, 2018, 2019; Vergelli et al., 2017). Figure 3 illustrates the compounds already investigated (A-F) and the range of activity for each series. Many of these compounds have very potent HNE inhibitory activity (IC50 values in the low nanomolar range), and kinetic experiments have characterized our compounds as competitive and pseudo-irreversible acyl-enzyme inhibitors. Furthermore, molecular modeling studies on compound interactions with HNE indicated that Ser195-OH of the catalytic triad attacks the carbonyl group of the N-CO group at position 1 in the bicyclic compounds (A-E) and the CO at position 5 in the monocyclic derivatives F (Vergelli et al., 2017; Giovannoni et al., 2018).
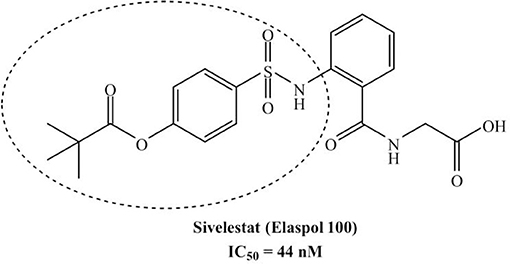
Figure 1. Sivelestat (ONO-5046) claimed by Ono Pharmaceutical (Kawabata et al., 1991).
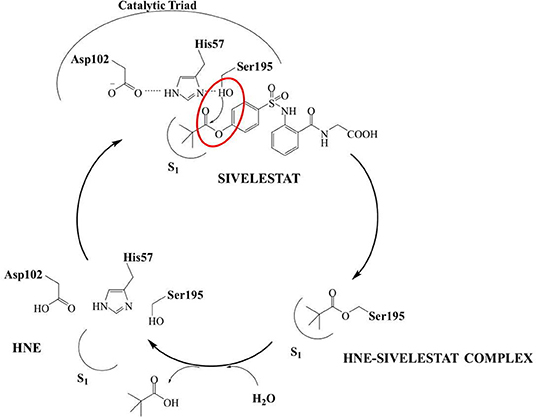
Figure 2. Mechanism of HNE inhibition by Sivelestat.
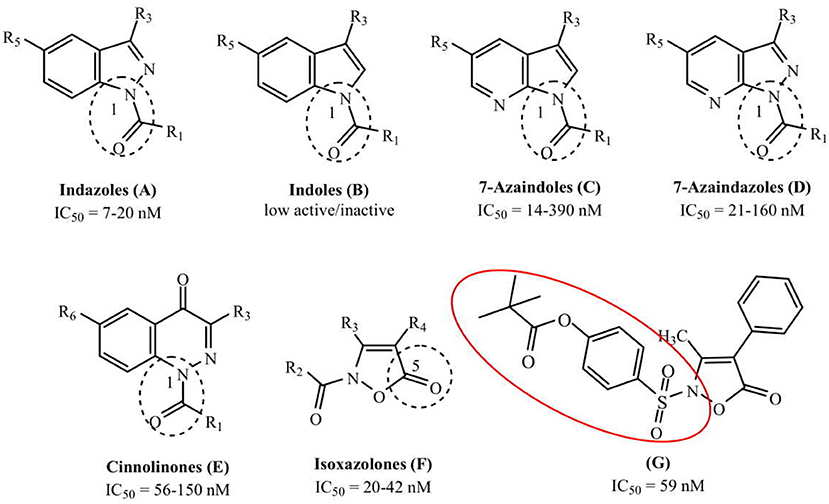
Figure 3. The investigated scaffolds of HNE inhibitors and range of their inhibitory activity. The segmented circle underlines the point of attack by OH group of Ser195 (G: 3b; Giovannoni et al., 2018).
Since the first patent in which Sivelestat was reported (Ono Pharmaceutical Co. and LTD., 1989), followed by the publication of Kawabata and coworkers (Kawabata et al., 1991), few articles have appeared in the literature describing Sivelestat analogs and (or) derivatives. The patent filed in 2002 (Macias, 2002) reports the introduction of substituents on the benzoyl moiety of Sivelestat, whereas Hwang et al. (2015) performed structural modification of Sivelestat by replacing the glycine moiety with an oxime group and obtained a slightly more potent compound. Recently, we synthesized the isoxazolone derivative G (Giovannoni et al., 2018) (Figure 3) containing the 4-(sulfonyl)phenyl pivalate fragment bound to the aniline nitrogen of Sivelestat. This compound exhibited high HNE inhibitory activity (IC50 = 59 nM) and excellent chemical stability in aqueous buffer (data not shown), which was improved over that of our previously published HNE inhibitors.
In the present studies, we expanded our strategy to evaluate how addition of the 4-(sulfonyl)phenyl pivalate fragment could modulate inhibitory activity of other HNE inhibitor scaffolds to evaluate if an additive effect would occur or whether these modifications could improve chemical stability of these new compounds. Therefore, we selected a number of compounds from our HNE inhibitor library belonging to the series A-E shown in Figure 3 and inserted the active 4-(sulfamoyl)phenyl pivalate fragment of Sivelestat into our scaffolds while leaving/maintaining the best substituents at positions 3 and 5/6. In most of these new bicyclic compounds, the carbonyl group of the N-CO function at position 1 was replaced with the Sivelestat pivalate fragment. In a few compounds, the N-CO function of our original compounds was left unchanged, and the active fragment of Sivelestat was inserted into a different position in order to produce two possible points of interaction with Ser195.
All melting points were determined on a Büchi apparatus (New Castle, DE) and are uncorrected. Extracts were dried over Na2SO4, and the solvents were removed under vacuum. Merck F-254 commercial plates (Merck, Durham, NC) were used for analytical TLC to follow the course of reactions. Silica gel 60 (Merck 70–230 mesh, Merck, Durham, NC) was used for column chromatography. 1H NMR and 13C NMR spectra were recorded on an Avance 400 instrument (Bruker Biospin Version 002 with SGU, Bruker Inc., Billerica, MA). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using the solvent as an internal standard. Coupling constants (J values) are given in Hz and were calculated using TopSpin 1.3 software (Nicolet Instrument Corp., Madison, WI) and are rounded to the nearest 0.1 Hz. Mass spectra (m/z) were recorded on an ESI-TOF mass spectrometer (Brucker Micro TOF, Bruker Inc., Billerica, MA), and reported mass values are within the error limits of ±5 ppm mass units. Microanalyses indicated by the symbols of the elements or functions were performed with a PerkinElmer 260 elemental analyzer (PerkinElmer, Inc., Waltham, MA) for C, H, and N, and the results were within ±0.4% of the theoretical values, unless otherwise stated. Reagents and starting material were commercially available.
To a suspension of the substrate 1a-c (1a: Shahidul et al., 2006; 1b: DeGraw and Goodman, 1964; 1c: Yuen et al., 2013) (0.43 mmol) in 10 mL of anhydrous THF, 0.86 mmol of sodium hydride (60% dispersion in mineral oil) was added while stirring. After 30 min, 0.56 mmol of 4-(chlorosulfonyl)phenyl pivalate 2 (Hwang et al., 2015) was added, and the mixture was stirred at room temperature overnight. After evaporation of the solvent in vacuo, the residue was diluted with ice-cold water (10 mL), neutralized with HCl 6N, and extracted with ethyl acetate (3 × 15 mL). The organic phase was dried over sodium sulfate, and the solvent was evaporated in vacuo to obtain the final compounds 3a-c, which were purified by flash column chromatography using cyclohexane/ethyl acetate 4:1 (for 3a) and 6:1 (for 3c) or hexane/ethyl acetate 5:2 for 3b as eluents.
Yield = 15%; mp = 118–121°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, 1H, Ar), 8.16 (dd, 1H, Ar, J = 2.2 Hz and J = 6.6 Hz), 7.99 (m, 3H, Ar), 7.38 (m, 2H, Ar), 7.22 (d, 2H, Ar, J = 8.8 Hz), 4.41 (q, 2H, CH2CH3, J = 7.1 Hz), 1.45 (t, 3H, CH2CH3, J = 7.1 Hz), 1.33 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 175.95 (C), 163.59 (C), 155.74 (C), 134.83 (C), 134.48 (C), 131.80 (CH), 128.82 (CH), 127.88 (C), 125.52 (CH), 124.56 (CH), 122.77 (CH), 122.35 (CH), 114.26 (C), 113.23 (CH), 60.61 (CH2), 39.27 (C), 26.94 (CH3), 14.42 (CH3). IR (ν) = 1,753 cm−1 (CO), 1,718 cm−1 (CO). ESI-MS calcd. for C22H23NO6S, 429.49; found: m/z 430.13 [M + H]+. Anal. C22H23NO6S (C, H, N).
Yield = 25%; mp = 170-173°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 9.04 (d, 1H, Ar, J = 2.4 Hz), 8.39 (s, 1H, Ar), 8.27 (dd, 1H, Ar, J = 2.4 Hz, and J = 9.2 Hz), 8.09 (d, 1H, Ar, J = 9.2 Hz), 8.01 (d, 2H, Ar, J = 8.8 Hz), 7.27 (d, 2H, Ar, J = 8.8 Hz), 4.46 (q, 2H, CH2CH3, J = 7.2 Hz), 1.46 (t, 3H, CH2CH3, J = 7.2 Hz); 1.33 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 175.88 (C), 162.53 (C), 156.32 (C), 145.15 (C), 137.49 (C), 134.16 (CH), 133.76 (C), 128.97 (CH), 127.91 (C), 123.16 (C), 120.69 (CH), 118.89 (CH), 114.90 (C), 113.67 (CH), 61.19 (CH2), 29.70 (C), 26.91 (CH3), 14.38 (CH3). IR (ν) = 1,747 cm−1 (CO), 1,712 cm−1 (CO), 1,517, and 1,377 cm−1 (NO2). ESI-MS calcd. for C22H22N2O8S, 474.48; found: m/z 475.11 [M + H]+. Anal. C22H22N2O8S (C, H, N).
Yield = 14%; mp = 133–136°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.11 (s, 1H, Ar), 8.0 (m, 3H, Ar), 7.72 (d, 1H, Ar, J = 7.6 Hz), 7.47 (t, 1H, Ar, J = 7.6 Hz), 7.41 (t, 1H, Ar, J = 7.6 Hz), 7.26 (d, 2H, Ar, J = 8.8 Hz), 1.34 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 156.13 (C), 133.87 (C), 133.64 (C), 132.98 (CH), 128.94 (CH), 128.38 (C), 126.73 (CH), 125.0 (CH), 123.01 (CH), 120.45 (CH), 113.69 (CH), 113.31 (C), 94.16 (C), 39.31 (C), 26.93 (CH3). IR (ν) = 2,231 cm−1 (CN), 1,743 cm−1 (CO). ESI-MS calcd. for C20H18N2O4S, 382.43; found: m/z 383.10 [M + H]+. Anal. C20H18N2O4S (C, H, N).
To a cooled (0°C) suspension of the appropriate substrate 1d-f (1d: Alaime et al., 2018; 1e: Crocetti et al., 2013; 1f: Crocetti et al., 2011) (0.32 mmol) in anhydrous CH2Cl2 (1–2 mL), 0.64 mmol of Et3N and 0.96 mmol of 4-(chlorosulfonyl)phenyl pivalate 2 (Hwang et al., 2015) were added. The solution was stirred at 0°C for 2 h and then for 2 h at room temperature. The organic solvent was evaporated under vacuum. After dilution with ice-cold water, the precipitate was filtered and washed with water (10–20 mL) to obtain the final compounds 3d-f, which were purified by crystallization from ethanol.
Yield = 69%; mp = 155–158°C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 8.28 (d, 1H, Ar, J = 7.2 Hz), 8.17 (d, 2H, Ar, J = 6.4 Hz), 7.99 (d, 1H, Ar, J = 6.4 Hz), 7.84 (m, 1H, Ar), 7.61 (m, 1H, Ar), 7.44 (d, 2H, Ar, J = 6.4 Hz), 1.25 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, DMSO-d6) δ 176.02 (C), 156.71 (C), 139.79 (C), 132.74 (C), 131.98 (CH), 130.37 (CH), 126.99 (CH), 125.95 (C), 125.33 (C), 124.27 (CH), 120.92 (CH), 113.62 (CH), 112.17 (C), 39.35 (C), 26.95 (CH3). IR (ν) = 2,247 cm−1 (CN), 1,753 cm−1 (CO). ESI-MS calcd. for C19H17N3O4S, 383.42; found: m/z 384.10 [M + H]+. Anal. C19H17N3O4S (C, H, N).
Yield = 44%; mp = 183–186°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.81 (d, 1H, Ar, J = 2.0 Hz), 8.57 (dd, 1H, Ar, J = 2.2 Hz and J = 9.4 Hz), 8.45 (d, 1H, Ar, J = 9.2 Hz), 8.14 (d, 2H, Ar, J = 8.8 Hz), 7.33 (d, 2H, Ar, J = 8.8 Hz), 1.36 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.02 (C), 157.08 (C), 145.73 (C), 141.85 (C), 132.30 (C), 130.28 (CH), 126. 99 (C), 125.40 (CH), 125.06 (C), 123.29 (CH), 116.95 (CH), 114.61 (CH), 110.38 (C), 39.36 (C), 26.93 (CH3). IR (ν) = 2,245 cm−1 (CN), 1,751 cm−1 (CO), 1,531 and 1,377 cm−1 (NO2). ESI-MS calcd. for C19H17N4O6S, 428.42; found: m/z 429.08 [M + H]+. Anal. C19H17N4O6S (C, H, N).
Yield = 44%; mp = 132–135°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.23 (m, 2H, Ar), 8.10 (d, 2H, Ar, J = 8.8 Hz), 7.62 (t, 1H, Ar, J = 7.6 Hz), 7.45 (t, 1H, Ar, J = 7.4 Hz), 7.22 (d, 2H, Ar, J = 8.8 Hz), 4.52 (q, 2H, CH2CH3, J = 7.1 Hz), 1.48 (t, 3H, CH2CH3, J = 7.1 Hz), 1.33 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.03 (C), 161.31 (C), 155.95 (C), 142.26 (C), 141.18 (C), 133.94 (C), 129.69 (CH), 129.65 (CH), 125.46 (CH), 124.43 (C), 122.79 (CH), 122.68 (CH), 112.99 (CH), 61.84 (CH2), 39.26 (C), 26.94 (CH3), 14.32 (CH3). IR (ν) = 1,745 cm−1 (CO), 1,732 cm−1 (CO). ESI-MS calcd. for C21H22N2O6S, 430.48; found: m/z 431.12 [M + H]+. Anal. C21H22N2O6S (C, H, N).
To a suspension of intermediate 1g (Crocetti et al., 2013) (0.64 mmol) in 5 mL of anhydrous pyridine, 1.92 mmol of 4-(chlorosulfonyl)phenyl pivalate 2 (Hwang et al., 2015) was added. The mixture was stirred at room temperature for 4 h. The solvent was concentrated in vacuo to obtain the final compound 3g, which was purified by crystallization from ethanol. Yield = 72%; mp = 185–188°C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 8.87 (s, 1H, Ar), 8.54 (d, 1H, Ar, J = 9.2 Hz), 8.43 (d, 1H, Ar, J = 9.2 Hz), 8.18 (d, 2H, Ar, J = 8.4 Hz), 7.45 (d, 2H, Ar, J = 8.4 Hz), 4.46 (q, 2H, CH2CH3, J = 7.0 Hz), 1.38 (t, 3H, CH2CH3, J = 7.0 Hz), 1.25 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, DMSO-d6) δ 176.08 (C), 160.26 (C), 156.80 (C), 145.67 (C), 143.29 (C), 142.85 (C), 132.84 (C), 130.44 (CH), 125.45 (CH), 124.37 (CH), 123.97 (C), 119.46 (C), 114.65 (CH), 62.62 (CH2), 39.18 (C), 26.98 (CH3), 14.49 (CH3). IR (ν) = 1,736 cm−1 (CO), 1,735 cm−1 (CO), 1,523 and 1,377 cm−1 (NO2). ESI-MS calcd. for C21H21N3O8S, 475.47; found: m/z 476.11 [M + H]+. Anal. C21H21N3O8S (C, H, N).
1H-Indazol-3-amine 1h (Lefebvre et al., 2010) (2.03 mmol) was dissolved in 13 mL of dry DMF and triethylamine (20.28 mmol). 4-(chlorosulfonyl)phenyl pivalate 2 (Hwang et al., 2015) (2.13 mmol) dissolved in 4 mL of dry 1,4-dioxane was added to the reaction mixture dropwise at 10°C, and the mixture was stirred at 50°C for 4 h. Ice-cold water was added to the reaction mixture, and the suspension was extracted with ethyl acetate (3 × 15 mL). The organic phase was dried over sodium sulfate, and the solvent was evaporated in vacuo to obtain the desired compound 3h, which was purified by flash column chromatography using cyclohexane/ethyl acetate 2:1 as eluent. Yield = 5%; mp = 189-192°C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 7.96 (d, 1H, Ar, J = 8.2 Hz), 7.79 (d, 1H, Ar, J = 8.2 Hz), 7.75 (d, 2H, Ar, J = 8.8 Hz), 7.59 (t, 1H, Ar, J = 7.6 Hz), 7.35 (t, 1H, Ar, J = 7.6 Hz), 7.26 (d, 2H, Ar, J = 8.8 Hz), 6.55 (exch br s, 2H, NH2), 1.24 (s, 9H, C(CH3)3). ESI-MS calcd. for C18H19N3O4S, 373.43; found: m/z 374.11 [M + H]+. Anal. C18H19N3O4S (C, H, N).
Compounds 3i-l and 4 were obtained starting from intermediates 1i-l (1i: Bahekar et al., 2007; 1j: Crocetti et al., 2018; 1k,l: Schirok et al., 2015) and 3h, respectively, following the same procedure described for 3d-f and using the appropriate sulfonyl chloride 2 (Hwang et al., 2015) (for 3i-l) or m-toluoyl chloride (for 4) as reagents. After evaporation of organic solvent, ice-cold water was added (20 mL). Compounds 3i,j and 4 were recovered by extraction with CH2Cl2 (3 × 15 mL), while compounds 3k, l were recovered by vacuum filtration. The final compounds 3i,j and 4 were purified by flash column chromatography using cyclohexane/ethyl acetate 4:1 (for 3i), 5:1 (for 3j) or toluene/ethyl acetate 95:5 (for 4) as eluents or by crystallization from ethanol (compounds 3k and 3l).
Yield = 22%; mp = 176–179°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.56 (dd, 1H, Ar, J = 1.6 Hz and J = 4.8 Hz), 8.33 (d, 2H, Ar, J = 7.2 Hz), 8.30 (s, 1H, Ar), 8.07 (dd, 1H, Ar, J = 1.6 Hz and J = 8.0 Hz), 7.38 (m, 1H, Ar), 7.28 (d, 2H, Ar, J = 7.2 Hz), 1.35 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.08 (C), 156.32 (C), 147.09 (CH), 145.64 (C), 133.66 (C), 133.17 (CH), 130.59 (CH), 129.11 (CH), 122.64 (CH), 121.01 (C), 120.46 (CH), 112.90 (C), 90.67 (C), 39.31 (C), 26.96 (CH3). IR (ν) = 2,233 cm−1 (CN), 1,755 cm−1 (CO). ESI-MS calcd. for C19H17N3O4S, 383.42; found: m/z 384.10 [M + H]+. Anal. C19H17N3O4S (C, H, N).
Yield = 8%; mp = 164–167°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.55 (m, 3H, Ar), 8.34 (d, 2H, Ar, J = 8.8 Hz), 7.39 (m, 1H, Ar), 7.26 (d, 2H, Ar, J = 8.8 Hz), 2.51 (s, 3H, CH3), 1.34 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.05 (C), 170.65 (C), 167.57 (C), 156.08 (C), 146.86 (C), 146.47 (CH), 134.18 (C), 130.66 (CH), 130.43 (CH), 128.78 (CH), 122.49 (CH), 120.38 (CH), 119.45 (C), 104.59 (C), 39.28 (C), 26.95 (CH3), 11.65 (CH3). IR (ν) = 1,757 cm−1 (CO). ESI-MS calcd. for C21H20N4O5S, 440.47; found: m/z 441.12 [M + H]+. Anal. C21H20N4O5S (C, H, N).
Yield = 24%; mp = 135–138°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.83 (d, 1H, Ar, J = 3.6 Hz), 8.32 (d, 2H, Ar, J = 8.8 Hz), 8.21 (d, 1H, Ar, J = 8.0 Hz), 7.46 (m, 1H, Ar), 7.27 (d, 2H, Ar, J = 8.8 Hz), 1.34 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.08 (C), 156.33 (C), 151.86 (CH), 151.10 (C), 133.81 (C), 131.90 (C), 130.42 (CH), 130.12 (CH), 122.74 (CH), 121.80 (C), 120.95 (CH), 113.95 (C), 39.27 (C), 26.92 (CH3). IR (ν) = 1,759 cm−1 (CO). ESI-MS calcd. for C18H16F3N3O4S, 427.40; found: m/z 428.08 [M + H]+. Anal. C18H16F3N3O4S (C, H, N).
Yield = 24%; mp = 166–169°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.86 (d, 1H, Ar, J = 4.4 Hz), 8.32 (d, 2H, Ar, J = 8.8 Hz), 8.24 (d, 1H, Ar, J = 8.0 Hz), 7.52 (m, 1H, Ar), 7.28 (d, 2H, Ar, J = 8.8 Hz), 1.34 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.06 (C), 156.62 (C), 152.34 (CH), 150.59 (C), 133.22 (C), 130.60 (CH), 129.65 (CH), 122.94 (CH), 121.50 (CH), 117.96 (C), 111.09 (C), 39.31 (C), 26.94 (CH3). IR (ν) = 2,250 cm−1 (CN), 1,762 cm−1 (CO). ESI-MS calcd. for C18H16N4O4S, 384.41; found: m/z 385.09 [M + H]+. Anal. C18H16N4O4S (C, H, N).
Yield = 37%; oil. 1H-NMR (400 MHz, DMSO-d6) δ 11.33 (exch br s, 1H, NH), 8.16 (d, 1H, Ar, J = 8.4 Hz), 8.01 (d, 2H, Ar, J = 8.8 Hz), 7.87 (m, 3H, Ar), 7.70 (t, 1H, Ar, J = 7.8 Hz), 7.43 (m, 3H, Ar), 7.38 (d, 2H, Ar, J = 8.8 Hz), 2.39 (s, 3H, m-CH3-Ph), 1.26 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, DMSO-d6) δ 176.13 (C), 166.51 (C), 155.87 (C), 148.72 (C), 141.66 (C), 138.32 (C), 133.67 (C), 133.49 (CH), 133.27 (C), 130.73 (CH), 129.58 (CH), 129.26 (CH), 128.87 (CH), 125.83 (CH), 124.77 (CH), 124.28 (CH), 123.81 (CH), 121.22 (C), 113.38 (CH), 39.36 (C), 26.99 (CH3), 21.36 (CH3). IR (ν) = 3,263 cm−1 (NH), 1,747 cm−1 (CO), 1,645 cm−1 (CO amide). ESI-MS calcd. for C26H25N3O5S, 491.56; found: m/z 492.15 [M + H]+. Anal. C26H25N3O5S (C, H, N).
A mixture of intermediate 1b (DeGraw and Goodman, 1964) or 1g (Bistocchi et al., 1981) (0.93 mmol), 1.40 mmol of Na2CO3, and 4.68 mmol of CH3I in 4 mL of anhydrous acetonitrile was stirred at 80°C for 6 h. After cooling, the organic solvent was evaporated under vacuum, and ice-cold water was added (10–15 mL). The precipitate was filtered and washed with water to obtain compounds 5a and 5d, which were purified by flash column chromatography using toluene/ethyl acetate 7:3 (for 5a) or 8:2 (for 5d) as eluents.
Yield = 22%; mp = 153–155°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 9.11 (d, 1H, Ar, J = 2.4 Hz), 8.22 (dd, 1H, Ar, J = 2.4 Hz and J = 9.2 Hz), 7.94 (s, 1H, Ar), 7.42 (d, 1H, Ar, J = 9.2 Hz), 4.45 (q, 2H, CH2CH3, J = 7.2 Hz), 3.93 (s, 3H, N-CH3); 1.47 (t, 3H, CH2CH3, J = 7.2 Hz). ESI-MS calcd. for C12H12N2O4, 248.24; found: m/z 249.08 [M + H]+. Anal. C12H12N2O4 (C, H, N).
Yield = 64%; mp = 178–181°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 9.18 (s, 1H, Ar), 8.36 (dd, 1H, Ar, J = 2.0 Hz and J = 9.2 Hz), 7.57 (d, 1H, Ar, J = 9.2 Hz), 4.59 (q, 2H, CH2CH3, J = 7.0 Hz), 4.26 (s, 3H, N-CH3), 1.53 (t, 3H, CH2CH3, J = 7.0 Hz). ESI-MS calcd. for C11H11N3O4, 249.23; found: m/z 250.08 [M + H]+. Anal. C11H11N3O4 (C, H, N).
Compounds 5a,b,d (5b: Crocetti et al., 2016) (0.60 mmol) were subjected to catalytic reduction in 96% EtOH (8 mL) for 2 h with a Parr instrument using 128 mg of 10% Pd/C as catalyst and hydrogen (30 psi). The catalyst was filtered, and the solvent was evaporated under vacuum, resulting in the desired products 6a, 6b, and 6d. Compounds 6b and 6d were purified by crystallization from ethanol.
Yield = 98%; oil. 1H-NMR (400 MHz, CDCl3) δ 7.67 (s, 1H, Ar), 7.50 (s, 1H, Ar), 7.13 (d, 1H, Ar, J = 8.2 Hz), 6.75 (d, 1H, Ar, J = 8.2 Hz), 4.37 (q, 2H, CH2CH3, J = 7.2 Hz), 3.75 (s, 3H, N-CH3), 1.41 (t, 3H, CH2CH3, J = 7.2 Hz). ESI-MS calcd. for C12H14N2O2, 218.26; found: m/z 219.11 [M + H]+. Anal. C12H14N2O2 (C, H, N).
Yield = 44%; mp = 154–157°C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 7.97 (d, 1H, Ar, J = 8.8 Hz), 7.66 (s, 1H, Ar), 7.59 (s, 1H, Ar), 7.53 (m, 3H, Ar), 7.27 (d, 1H, Ar, J = 2.2 Hz), 6.72 (dd, 1H, Ar, J = 2.2 Hz and J = 8.8 Hz), 5.23 (exch br s, 2H, NH2), 4.27 (q, 2H, CH2CH3, J = 7.1 Hz), 2.42 (s, 3H, m-CH3-Ph), 1.29 (t, 3H, CH2CH3, J = 7.1 Hz). IR (ν) = 3,414–3,336 cm−1 (NH2), 1,681 cm−1 (CO), 1,585 cm−1 (CO amide). ESI-MS calcd. for C19H18N2O3, 322.36; found: m/z 323.14 [M + H]+. Anal. C19H18N2O3 (C, H, N).
Yield = 46%; mp = 151-154°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 7.43 (s, 1H, Ar), 7.29 (d, 1H, Ar, J = 8.8 Hz), 6.92 (dd, 1H, Ar, J = 2.0 Hz and J = 8.8 Hz), 4.51 (q, 2H, CH2CH3, J = 7.1 Hz), 4.12 (s, 3H, N-CH3), 1.48 (t, 3H, CH2CH3, J = 7.1 Hz). ESI-MS calcd. for C11H13N3O2, 219.24; found: m/z 220.10 [M + H]+. Anal. C11H13N3O2 (C, H, N).
Compounds 7a,b were obtained starting from intermediates 6a,b following the same procedure described for 3d-f and 3i-l using 4-(chlorosulfonyl)phenyl pivalate 2 (Hwang et al., 2015) as reagent. After evaporation of the organic solvent, ice-cold water was added (20 mL). Compounds 7a,b were recovered by extraction with CH2Cl2 (3 × 15 mL) and purified by flash column chromatography using toluene/ethyl acetate 8:2 (for 7a) or cyclohexane/ethyl acetate 2:1 (for 7b) as eluents.
Yield = 64%; oil. 1H-NMR (400 MHz, CDCl3) δ 7.76 (d, 1H, Ar, J = 4.8 Hz), 7.75 (m, 3H, Ar), 7.18 (m, 2H, Ar), 7.11 exch br s, 1H, NH), 7.07 (d, 2H Ar, J = 7.2 Hz), 4.35 (q, 2H, CH2CH3, J = 7.2 Hz), 3.78 (s, 3H, N-CH3), 1.38 (t, 3H, CH2CH3, J = 7.2 Hz), 1.33 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.08 (C), 163.88 (C), 154.35 (C), 136.31 (C), 136.13 (CH), 135.45 (C), 130.70 (C), 128.91 (CH), 126.91 (C), 121.97 (CH), 119.20 (CH), 116.35 (CH), 110.51 (CH), 59.81 (CH2), 39.18 (C), 33.53 (CH3), 26.98 (CH3), 14.49 (CH3). IR (ν) = 3,242 cm−1 (NH), 1,747 cm−1 (CO), 1,714 cm−1 (CO). ESI-MS calcd. for C23H26N2O6S, 458.53; found: m/z 459.15 [M + H]+. Anal. C23H26N2O6S (C, H, N).
Yield = 38%; mp = 172-175°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.27 (d, 1H, Ar, J = 8.8 Hz), 8.0 (s, 1H, Ar), 7.90 (d, 1H, Ar, J = 2.2 Hz), 7.81 (dd, 2H, Ar, J = 2.2 Hz and J = 8.4 Hz), 7.56 (s, 1H, Ar), 7.48 (m, 3H, Ar), 7.21 (dd, 1H, Ar, J = 2.2 Hz and J = 8.8 Hz), 7.16 (dd, 2H, Ar, J = 2.2 Hz and J = 8.4 Hz), 6.64 (exch br s, 1H, NH), 4.39 (q, 2H, CH2CH3, J = 7.1 Hz), 2.47 (s, 3H, m-CH3-Ph), 1.41 (t, 3H, CH2CH3, J = 7.1 Hz), 1.35 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.08 (C), 163.88 (C), 154.56 (C), 139.07 (C), 136.06 (C), 134.40 (CH), 134.17 (C), 133.62 (CH), 133.35 (C), 133.0 (C), 129.89 (CH), 128.94 (CH), 128.75 (CH), 128.43 (C), 126.52 (CH), 122.17 (CH), 120.23 (CH), 117.02 (CH), 115.03 (CH), 113.33 (C), 60.75 (CH2), 39.22 (C), 26.99 (CH3), 21.36 (CH3), 14.37 (CH3). IR (ν) = 3,261 cm−1 (NH), 1,749 cm−1 (CO), 1,715 cm−1 (CO), 1,697 cm−1 (CO amide). ESI-MS calcd. for C30H30N2O7S, 562.64; found: m/z 563.18 [M + H]+. Anal. C30H30N2O7S (C, H, N).
Compounds 7c-e were obtained starting from intermediates 6c-e (6c: Purandare et al., 2014; 6e: Crocetti et al., 2013), respectively, following the same procedure described for 3g. The solvent was concentrated in vacuo to obtain the final compounds 7c-e, which were first purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent and then by crystallization from ethanol.
Yield = 63%; mp = 162–165°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 7.79 (d, 2H, Ar, J = 8.8 Hz), 7.49 (d, 1H, Ar, J = 1.2 Hz), 7.39 (m, 3H, 2H Ar and 1H NH), 7.16 (d, 2H, Ar, J = 8.8 Hz), 4.13 (s, 3H, N-CH3), 1.35 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.47 (C), 154.74 (C), 138.01 (C), 135.73 (C), 132.64 (C), 128.87 (CH), 125.57 (C), 124.05 (CH), 122.43 (CH), 117.15 (C), 113.32 (C), 111.89 (CH), 111.30 (CH), 39.25 (C), 36.90 (CH3), 26.98 (CH3). IR (ν) = 3,300 cm−1 (NH), 2,233 cm−1 (CN), 1,753 cm−1 (CO). ESI-MS calcd. for C20H20N4O4S, 412.46; found: m/z 413.12 [M + H]+. Anal. C20H20N4O4S (C, H, N).
Yield = 89%; mp = 130–133°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 7.85 (d, 2H, Ar, J = 6.8 Hz), 7.76 (d, 2H, Ar, J = 8.8 Hz), 7.30 (s, 1H, Ar), 7.08 (d, 2H, Ar, J = 8.8 Hz), 4.43 (q, 2H, CH2CH3, J = 7.1 Hz), 4.06 (s, 3H, N-CH3), 1.39 (t, 3H, CH2CH3, J = 7.1 Hz), 1.29 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 176.32 (C), 162.37 (C), 154.51 (C), 139.12 (C), 136.02 (C), 134.61 (C), 132.11 (C), 130.11 (C), 128.92 (CH), 123.75 (C), 123.40 (CH), 122.19 (CH), 115.20 (CH), 110.54 (CH), 61.10 (CH2), 39.18 (C), 36.50 (CH3), 26.96 (C), 14.38 (CH3). IR (ν) = 3,228 cm−1 (NH), 1,755 cm−1 (CO), 1,720 cm−1 (CO). ESI-MS calcd. for C22H25N3O6S, 459.52; found: m/z 460.15 [M + H]+. Anal. C22H25N3O6S (C, H, N).
Yield = 64%; mp = 180–182°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.46 (d, 1H, Ar, J = 8.8 Hz), 7.98 (s, 1H, Ar), 7.92 (s, 2H, Ar), 7.82 (d, 2H, Ar, J = 8.8 Hz), 7.43 (m, 3H, Ar), 7.18 (d, 2H, Ar, J = 8.8 Hz), 6.68 (exch br s, 1H, NH), 4.53 (q, 2H, CH2CH3, J = 7.1 Hz), 2.47 (s, 3H, m-CH3-Ph), 1.48 (t, 3H, CH2CH3, J = 7.1 Hz), 1.34 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 168.15 (C), 161.51 (C), 154.79 (C), 140.66 (C), 139.26 (C), 138.04 (C), 135.83 (C), 134.30 (C), 133.92 (CH), 132.05 (CH), 131.70 (C), 128.92 (CH), 128.08 (CH), 125.09 (C), 124.62 (CH), 122.38 (CH), 116.76 (CH), 114.44 (CH), 61.93 (CH2), 39.23 (C), 26.99 (CH3), 21.36 (CH3), 14.25 (CH3). IR (ν) = 3,250 cm−1 (NH), 1,745 cm−1 (CO), 1,712 cm−1 (CO), 1,698 cm−1 (CO amide). ESI-MS calcd. for C29H29N3O7S, 563.63; found: m/z 564.18 [M + H]+. Anal. C29H29N3O7S (C, H, N).
Compounds 9a,b were obtained starting from intermediates 8a,b (8a: Muthupplaniappan et al., 2009; 8b: Giovannoni et al., 2016) following the same procedure described for 3d-f, 3i-l, and 7a,b. Compounds 9a,b were recovered by extraction with CH2Cl2 (3 × 15 mL) and purified by flash column chromatography using cyclohexane/ethyl acetate 2:1 (for 9a) or petroleum ether/ethyl acetate 5:1 (for 9b) as eluents.
Yield = 8%; mp = 116–119°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.63 (d, 1H, Ar, J = 8.6 Hz), 8.27 (d, 1H, Ar, J = 8.6 Hz), 8.08 (d, 2H, Ar, J = 8.8 Hz), 7.83 (s, 1H, Ar), 7.81 (t, 1H, Ar, J = 7.6 Hz), 7.51 (t, 1H, Ar, J = 7.6 Hz), 7.30 (d, 2H, Ar, J = 8.8 Hz), 1.36 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 175.99 (C), 170.78 (C), 156.26 (C), 141.52 (CH), 139.39 (C), 134.93 (CH), 133.27 (C), 130.16 (CH), 126.70 (CH), 123.79 (C), 122.82 (CH), 117.05 (CH), 39.33 (C), 26.96 (CH3). IR (ν) = 1,745 cm−1 (CO), 1,716 cm−1 (CO). ESI-MS calcd. for C19H18N2O5S, 386.42; found: m/z 387.10 [M + H]+. Anal. C19H18N2O5S (C, H, N).
Yield = 25%; mp = 121–123°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.62 (d, 1H, Ar, J = 8.8 Hz), 8.29 (d, 1H, Ar, J = 8.8 Hz), 8.02 (d, 2H, Ar, J = 8.8 Hz), 7.74 (t, 1H, Ar, J = 7.6 Hz), 7.46 (t, 1H, Ar, J = 7.6 Hz), 7.28 (d, 2H, Ar, J = 8.8 Hz), 2.54 (m, 1H, CH C3H5), 1.36 (s, 9H, C(CH3)3), 0.99 (m, 2H, CH2 C3H5), 0.85 (m, 2H, CH2 C3H5). 13C-NMR (100 MHz, CDCl3) δ 176.0 (C), 170.55 (C), 156.04 (C), 153.20 (C), 139.71 (C), 134.36 (CH), 133.70 (C), 130.15 (CH), 126.42 (CH), 125.93 (CH), 122.53 (CH), 122.0 (C), 116.86 (CH), 39.31 (C), 26.97 (CH3), 9.90 (CH2), 9.09 (CH). IR (ν) = 1,745 cm−1 (CO), 1,732 cm−1 (CO). ESI-MS calcd. for C22H22N2O5S, 426.49; found: m/z 427.13 [M + H]+. Anal. C22H22N2O5S (C, H, N).
Compound 11 was obtained starting from intermediate 10 (Becalli et al., 1984) by reaction with 4-(chlorosulfonyl)phenyl pivalate 2 (Hwang et al., 2015) following the general procedure described for 3g and 7c-e. The pyridine was concentrated in vacuo to obtain the final compound 11, which was purified by flash column chromatography using hexane/acetone 1:1 as eluent. Yield = 18%; mp = 166–168°C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 8.35 (s, 1H, CH), 7.93 (d, 2H, Ar, J = 8.8 Hz), 7.66 (d, 2H, Ar, J = 8.0 Hz), 7.37 (d, 3H, Ar, J = 6.4 Hz), 7.31 (d, 2H, Ar, J = 8.8 Hz), 1.34 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ 165.85 (C), 156.91 (C), 143.93 (CH), 131.16 (CH), 129.56 (CH), 128.99 (CH), 128.13 (C), 126.37 (C), 126.24 (CH), 122.89 (CH), 115.15 (C), 39.36 (C), 26.96 (CH3). IR (ν) = 3,099 cm−1 (CH isox), 1,759 cm−1 (CO), 1,745 cm−1 (CO), 1,597 cm−1 (C3=C4). ESI-MS calcd. for C20H19NO6S, 401.43; found: m/z 402.10 [M+H]+. Anal. C20H19NO6S (C, H, N).
Compounds were dissolved in 100% DMSO at 5 mM stock concentrations. The final concentration of DMSO in the reactions was 1%, and this level of DMSO had no effect on enzyme activity. HNE inhibition assays were performed in black flat-bottom 96-well microtiter plates. Briefly, a solution containing 200 mM Tris–HCl, pH 7.5, 0.01% bovine serum albumin, 0.05% Tween®-20, and 20 mU/mL of HNE (Calbiochem) was added to wells containing different concentrations of each compound. The reaction was initiated by addition of 25 μM elastase substrate (N-methylsuccinyl-Ala-Ala-Pro-Val-7-amino-4-methylcoumarin, Calbiochem) in a final reaction volume of 100 μL/well. Kinetic measurements were obtained every 30 s for 10 min at 25°C using a Fluoroskan Ascent FL fluorescence microplate reader (Thermo Electron, MA) with excitation and emission wavelengths of 355 and 460 nm, respectively. For all compounds tested, the concentration of inhibitor that caused 50% inhibition of the enzymatic reaction (IC50) was calculated by plotting % inhibition vs. logarithm of inhibitor concentration (at least six points). The data are presented as the mean values of at least three independent experiments with relative standard deviations of <15%.
Spontaneous hydrolysis of selected derivatives was evaluated at 25°C in 0.05 M phosphate buffer, pH 7.3. Kinetics of compound hydrolysis was monitored by measuring changes in absorbance spectra over time using a SpectraMax ABS Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). Absorbance (At) at the characteristic absorption maxima of each compound was monitored over time until no further absorbance decreases occurred (A∞). Using these measurements, we created semilogarithmic plots of log(At-A∞) vs. time, and k′ values were determined from the slopes of these plots. Half-conversion times were calculated using t1/2 = 0.693/k′.
Structures of Sivelestat (in the form of a carboxylate anion) and compounds 3a, 7b, 7d, and 7e, were created using ChemOffice 2016 software, pre-optimized with the MM2 force field and saved in Tripos MOL2 format. The ligand structures were then imported into the Molegro Virtual Docker 6.0 program (MVD). The structure of HNE complexed with 1-{3-methyl-2-[4-(morpholine-4-carbonyl)-benzoylamino]-butyryl}-pyrrolidine-2-carboxylic acid (3,3,4,4,4-pentafluoro-1-isopropyl-2-oxo-butyl)-amide (SEI) ligand was downloaded from the Protein Data Bank (PDB code 1B0F) and also imported into MVD. The co-crystallized water molecules were removed from the 1B0F structure on importing. A search space for docking was defined in the HNE binding site as a sphere of radius 12 Å positioned at the geometric center of gravity of the SEI ligand, and the investigated compounds were docked into the binding site. MolDock score functions (Thomsen and Christensen, 2006) were applied with a 0.3 Å grid resolution. Ligand flexibility was accounted for with respect to torsion angles auto-detected in MVD. Structure of the protein was considered rigid. The “Internal HBond” and “sp2-sp2 torsions” options were activated in the “Ligand evaluation” menu of the MVD Docking Wizard. Three hundred docking runs were performed for each molecule. Our attempts to enhance number of docking runs up to 600 did not lead to better scored docking poses. The option “Return multiple poses for each run” was enabled, and the post-processing options “Energy minimization” and “Optimize H-bonds” were applied after docking. Similar poses were clustered at a RMSD threshold of 1 Å.
All final compounds were synthesized as reported in Schemes 1–3, and the structures were confirmed on the basis of analytical and spectral data. The 4-(chlorosulfonyl)phenyl pivalate fragment 2 representing the active portion of Sivelestat that was incorporated into all new compounds was synthesized as reported previously (Hwang et al., 2015).
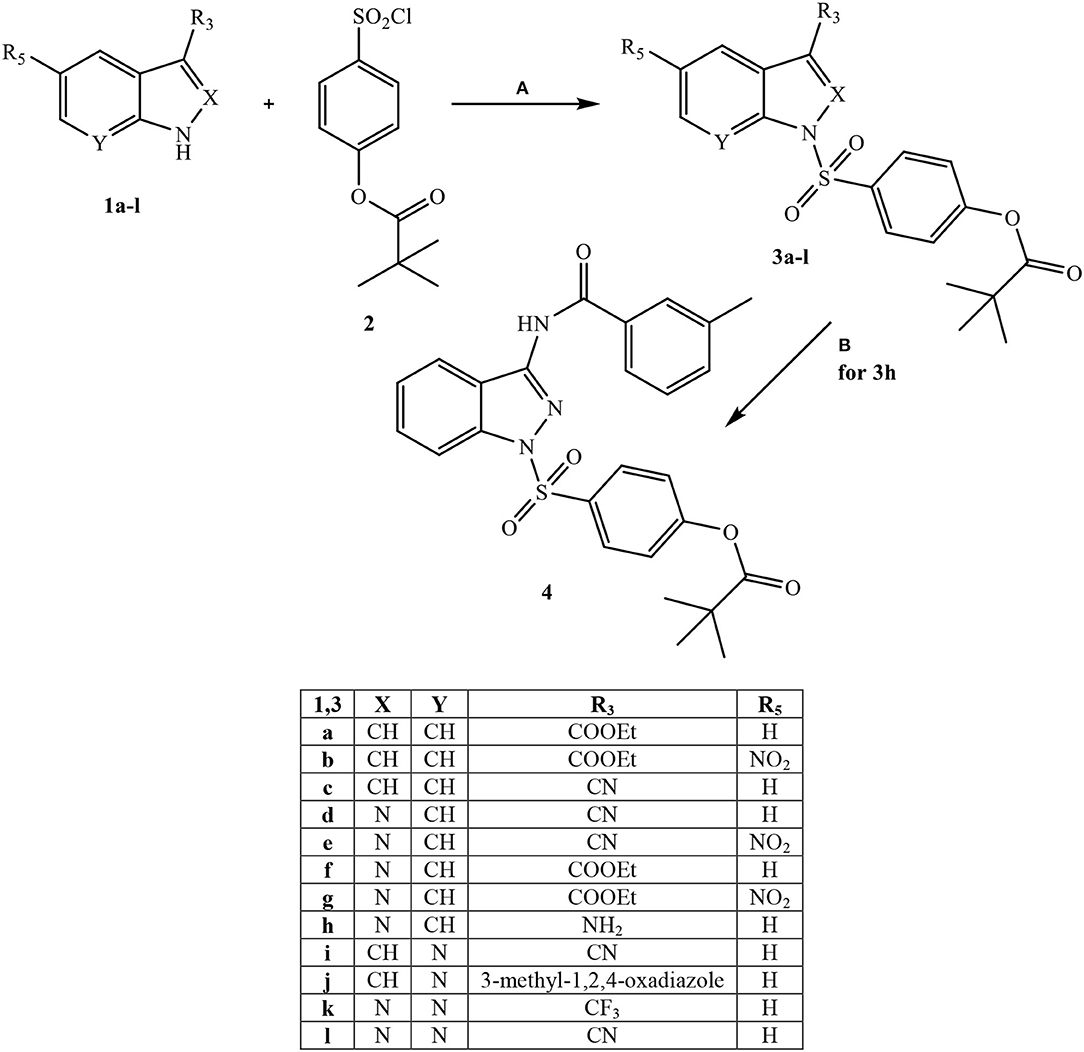
Scheme 1. aReagents and conditions. (A) for 3a-c: NaH (60% dispersion in mineral oil), anhydrous THF, r.t., 24 h; for 3d-f and 3i-l: Et3N, anhydrous CH2Cl2, 0°C, 2 h then r.t., 2 h; for 3g: dry pyridine, r.t., 4 h; for 3h: Et3N, dry DMF, 1,4-dioxane, 50 °C, 4h; (B) m-Toluoyl chloride, Et3N, anhydrous CH2Cl2, 0°C, 2 h then r.t., 2 h.
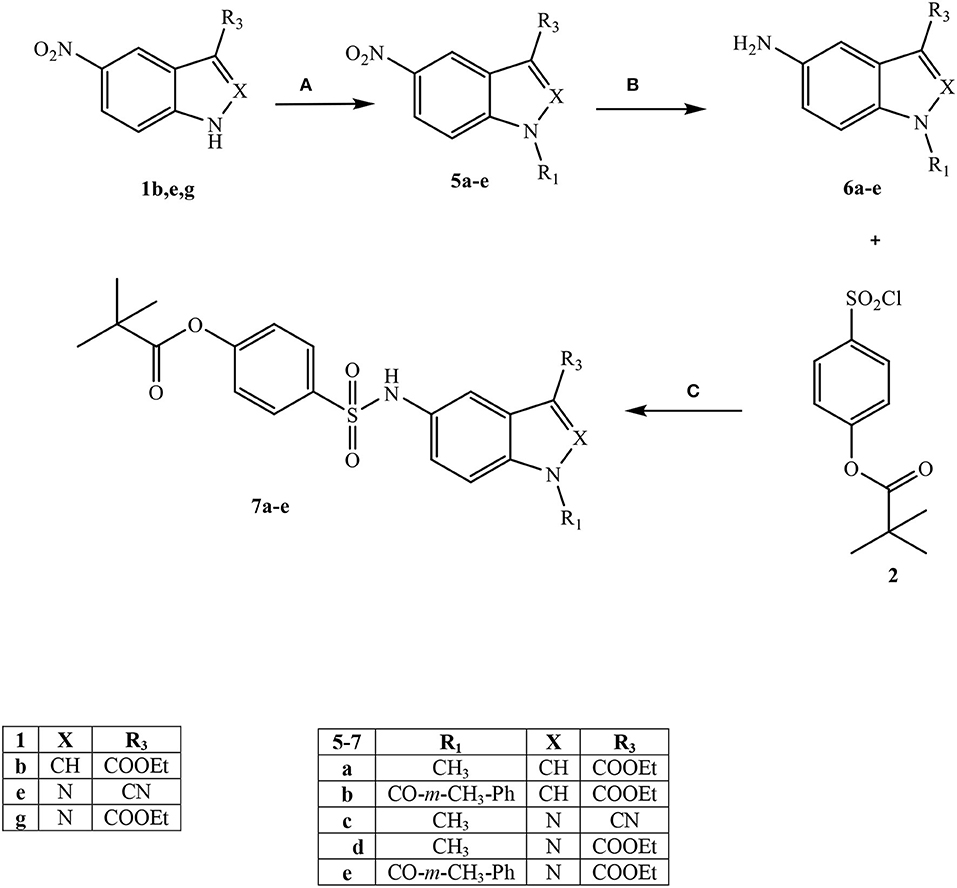
Scheme 2. aReagents and conditions. (A) for 5a,c,d: CH3I, Na2CO3, anhydrous CH3CN, 80 C, 6 h; for 5b: m-Toluoyl chloride, NaH (60% dispersion in mineral oil), anhydrous THF, r.t., 24 h; for 5e: m-Toluoyl chloride, Et3N, anhydrous CH2Cl2, 0°C, 2 h then r.t., 2 h (B) H2 (Parr), Pd/C, EtOH 96%, 30 min (for 6c) and 2 h (for 6a,b,d,e); (C) for 7a,b: Et3N, anhydrous CH2Cl2, 0°C, 2 h then r.t., 2 h; for 7c-e: dry pyridine, r.t., 4 h.
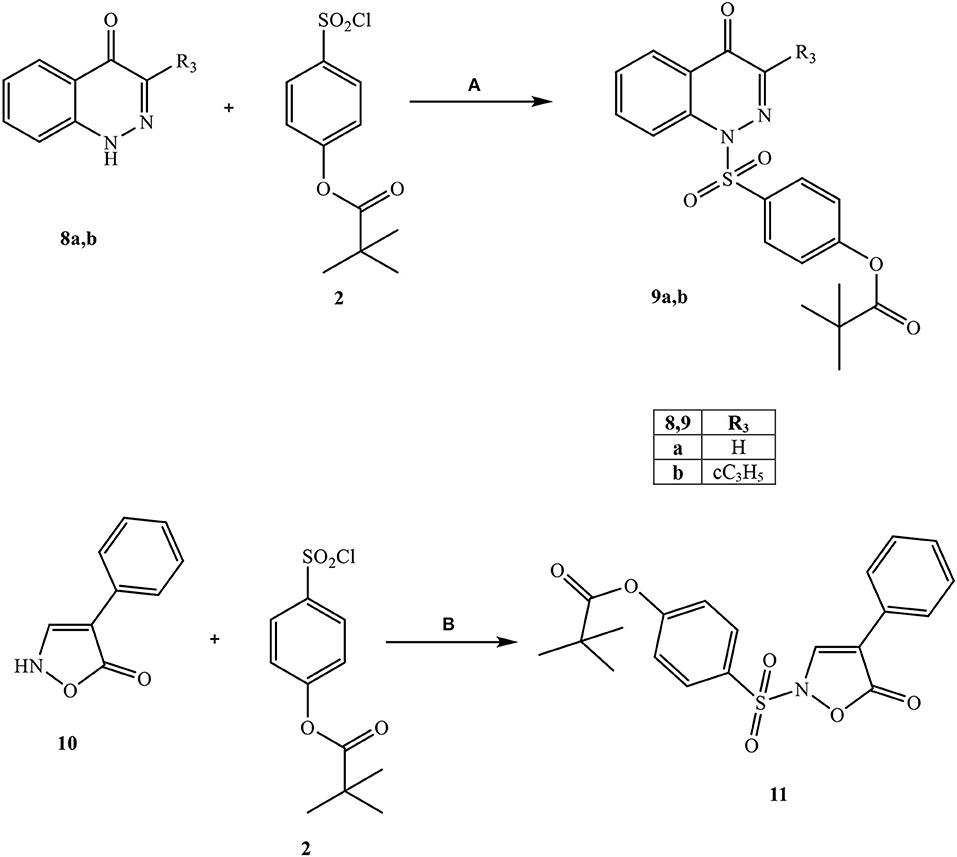
Scheme 3. aReagents and conditions. (A) Et3N, anhydrous CH2Cl2, 0°C, 2 h then r.t., 2 h; (B) dry pyridine, r.t., 4 h.
Starting from Scheme 1, fragment 2 was inserted at the N-1 position of four different bi-heterocycles that were previously investigated by our research group as N(1)-CO- aryl(alkyl) derivatives: indoles (Crocetti et al., 2016), indazoles (Crocetti et al., 2011, 2013), 7-azaindoles (Crocetti et al., 2018; Giovannoni et al., 2019) and 7-azaindazoles (data not shown). Indole derivatives 3a-c were obtained starting from the precursors 1a-c (1a: Shahidul et al., 2006; 1b: DeGraw and Goodman, 1964; 1c: Yuen et al., 2013) by treatment with 4-(chlorosulfonyl)phenyl pivalate 2 and sodium hydride in anhydrous tetrahydrofuran (THF) at room temperature. To obtain the final indazoles 3d-g two different procedures were followed: treatment of the appropriate intermediate 1d-g (1d: Alaime et al., 2018; 1e,g: Crocetti et al., 2013; 1f: Crocetti et al., 2011) with the sulfonyl chloride 2 in anhydrous CH2Cl2 and Et3N (compounds 3d-f) or in dry pyridine at room temperature (3g). The 3-aminoindazole 1h (Lefebvre et al., 2010) was reacted with 4-(chlorosulfonyl)phenyl pivalate 2 in dry 1,4-dioxane/DMF, Et3N at 50 °C to obtain the intermediate 3h, which was further elaborated by treatment with m-toluoyl chloride in anhydrous CH2Cl2 and Et3N to obtain the final compound 4, containing a benzamido moiety at position 3. Lastly, synthesis of the 7-azaindoles 3i, j and 7-azaindazoles 3k, l was performed starting from precursors 1i,j and 1-k,l (1i: Bahekar et al., 2007; 1j: Crocetti et al., 2018; 1k,l: Schirok et al., 2015), respectively, under the same conditions described for compounds 3d-f.
Scheme 2 shows the synthetic route followed to obtain compounds 7a-e bearing the active fragment of Sivelestat at position 5 of the indazole or indole scaffolds. Treatment of precursors 1b, 1e, and 1g with iodomethane, sodium carbonate in anhydrous acetonitrile at reflux afforded intermediates 5a, c, d (5c: Purandare et al., 2014), while compounds 5b and 5e were obtained by treatment of 1b with m-toluoyl chloride in anhydrous dichloromethane and Et3N and 1g with m-toluoyl chloride and sodium hydride in tetrahydrofuran, respectively (5b: Crocetti et al., 2016; 5e: Crocetti et al., 2013). The 5-NO2 derivatives 5a-e were transformed into the corresponding 5-amino compounds 6a-e (6b: Crocetti et al., 2016; 6c: Purandare et al., 2014) through catalytic reduction with a Parr instrument and a subsequent reaction with 4-(chlorosulfonyl)phenyl pivalate 2 (Hwang et al., 2015) under the same conditions reported in Scheme 1, leading to compounds 7a-e. Finally, Scheme 3 shows the synthesis of the cinnolinone derivatives of type 9 and isoxazolone 11. In all precursors 8a, b (8a: Muthupplaniappan et al., 2009; 8b: Giovannoni et al., 2016) and 10 (Becalli et al., 1984), the insertion of fragment 2 was carried out under the same conditions as described in Scheme 1.
All new products were evaluated for HNE inhibitory activity, and the results are reported in Tables 1, 2 in comparison with Sivelestat. Table 1 presents the results of compounds lacking the 1-N-CO function responsible for activity in our original compounds (Figure 3). These include 3a-g, 3i-l, 4, and 9a, b, which contain the sulfamoyl fragment of Sivelestat at N-1 of the bicyclic nucleus, and compounds 7a, 7c, and 7d, which have the (sulfonyl)phenyl pivalate chain at position five of the nucleus and a methyl group at N-1 (N-methyl derivatives). Many of the new derivatives exhibited very potent HNE inhibitory activity, with IC50 values between 15 and 78 nM for most compounds, which is comparable to or better that that of Sivelestat (IC50 = 44 nM). The most potent compounds were the indazole 3f and the cinnoline 9a, which had IC50 values of 15 and 19 nM, respectively. These results clearly demonstrated that replacement of the NCO function at N-1 with the active fragment of Sivelestat did not affect HNE inhibitory activity (Table 1). Previously, we found that indoles of type B (Figure 3), which were designed as 2-deaza analogs of highly active N-benzoylindazole compounds, were inactive or low activity HNE inhibitors due to the lack of the nitrogen at position two, which forms an important interaction with Gly193 of the catalytic site (Crocetti et al., 2016). Here, we report that introduction of the (sulfonyl)phenyl pivalate chain at N-1 of the indole nucleus into these compounds (i.e., 3a-c) resulted in very potent HNE inhibitors, with IC50 values of 30, 25, and 49 nM, respectively, suggesting that inclusion of the active fragment of Sivelestat leads to a different interaction of the indole scaffold with HNE. Finally, the N-1 methyl derivatives 7a, c, d bearing the (sulfonyl)phenyl pivalate chain at position five of the nucleus retained some inhibitory activity, although they were not as active as the other compounds described above.
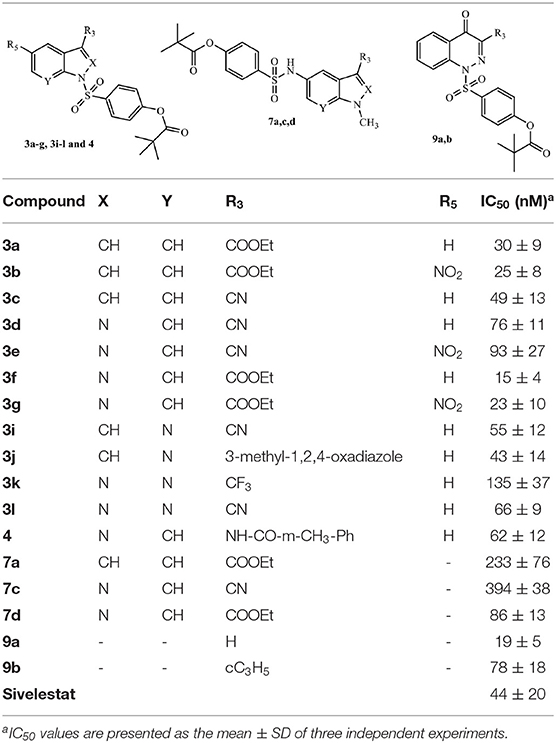
Table 1. HNE inhibitory activity of compounds 3a-g, 3i-l, 4, 7a,c,d, and 9a,b.
The results reported in the Table 1 indicate that in practice, the pivaloyl fragment of Sivelestat can “replace” the role of the N-CO group at position 1 and offer a different point of attack for Ser195. It is also clear that the selected scaffolds with adequate substitutions are appropriate carriers for the Sivelestat pharmacophore. On the other hand, compounds maintaining the NCO function at position 1 (compounds 7b and 7e) or CO at position five (compound 11) (Table 2) as the point of attack for Ser195, and simultaneously bearing the active pivaloyl fragment of Sivelestat, only exhibited moderate HNE inhibitory activity (IC50 = 0.29–5.2 μM), with the exception of the previously published compound G (IC50 = 59 nM), which has activity comparable to its analogs lacking the Sivelestat fragment (Giovannoni et al., 2018). However, these data also clearly indicate that this strategy does not produce the expected additive effect, probably due to the increased hindrance of the molecules.
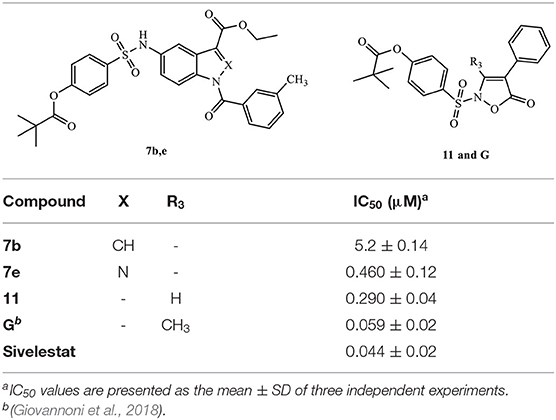
Table 2. HNE inhibitory activity of compounds 7b, e, 11 in comparison with compound G and Sivelestat.
A set of the most potent HNE inhibitors, as well as low activity compound 7b, were evaluated for their chemical stability in aqueous buffer. Spontaneous hydrolysis rates of the inhibitors were measured in phosphate buffer at pH 7.3 and 25°C. As shown in Table 3, compounds 3a, 3c, 3f, and 7d had a relatively good stability (t1/2 > 9 h) with high HNE inhibitory activity (IC50 <100 nM).
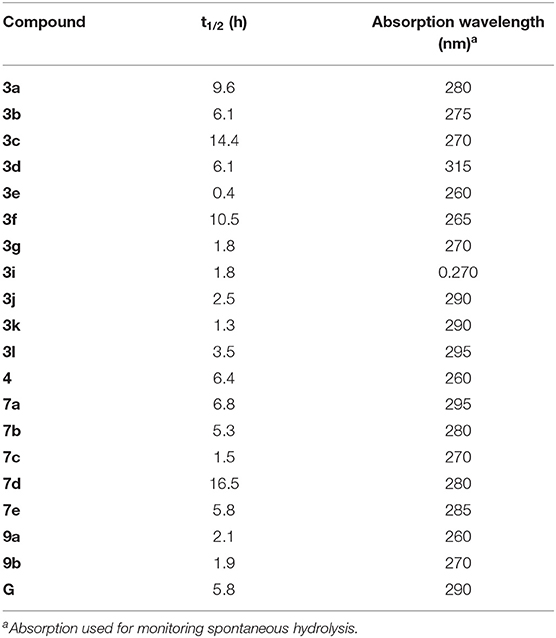
Table 3. Half-life (t1/2) for the spontaneous hydrolysis of selected derivatives.
The relatively high enzymatic stability of HNE allowed us to evaluate reversibility of the enzyme inhibition over time. As an example, Figure 4 shows kinetic curves monitoring substrate cleavage catalyzed by HNE over a 10-h period in the presence of selected sulfonamide derivatives (5 μM) and compared to Sivelestat. Persistence of selected HNE inhibitors over an extended period of time (16 h) was also evaluated and showed that the most effective HNE inhibitors over time were 3c, 3d, 3i, 3l, and 9a (Table 4). Inhibitory activities of these compounds were comparable or better than Sivelestat in this assay.
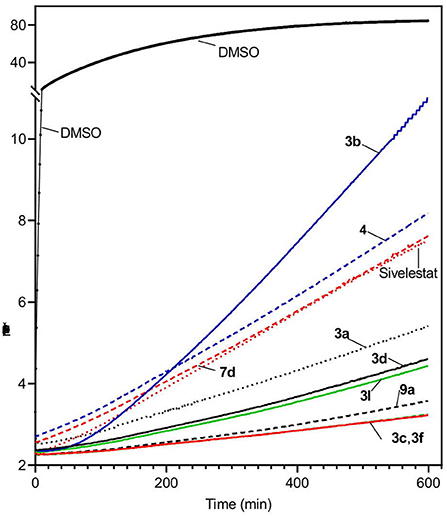
Figure 4. Evaluation of HNE inhibition by representative sulfonamides and Sivelestat over time. HNE was incubated with the indicated selected compounds (5 μM), and kinetic curves monitoring substrate cleavage catalyzed by HNE over time are shown. Representative curves are from two independent experiments.

Table 4. Potency of selected HNE inhibitors (with IC50 <100 nM) over extended period of time (16 h).
Molecular docking studies of some HNE inhibitors, including Sivelestat and triterpenes, were previously made (Feng et al., 2012, 2013) based on 1B0F structure from PDB. Hence, we also used this structure in our docking calculations. The MVD program was validated on HNE by confirming the ability of the program to reproduce the position of the co-crystallized SEI ligand contained in the 1B0F structure taken from the Protein Data Bank (Cregge et al., 1998). Independent docking of SEI into the HNE binding site was performed, and comparison of the resulting pose with the experimental ligand location demonstrated that the MVD program accurately reproduced the location of the SEI ligand in the HNE binding site (RMSD of non-hydrogen atom positions between the two structures is 1.24 Å) (Figure S1, see Supplementary Material).
One of the goals of our molecular modeling study consisted in clarifying the possibilities for Nakayama's mechanism for inhibitory action of pivaloyl-containing compounds (Nakayama et al., 2002) via an attack of Ser195 hydroxyl oxygen atom at the carbon center of C=O group in the inhibitor molecule. Docking of Sivelestat in its anionic form into HNE using MVD software gave a ligand position (Figure 5A) similar to the pose obtained by Feng and co-authors with the free docking program AutoDock (Feng et al., 2012). In this docking pose, Sivelestat forms H-bonds between the oxygen atom of the sulfonamide group and Ser195 and Gly193, as well as an H-bond between the carboxyl group and Ser214. In addition, the Sivelestat pose is near to hydrophobic residues Leu99B, Phe192, His57, Val216, Cys191, and Phe41. All of these features of the ligand location are consistent with the docking results previously reported for Sivelestat (Feng et al., 2012). According to our data and the results obtained by Feng et al. (2012) on the binding of Sivelestat to HNE, Ser195 forms an H-bond with the sulfonamide oxygen atom, hence Ser195 is far from the carbonyl carbon atom of the pivaloyl group, which is a potential reaction center in the reported mechanism (Nakayama et al., 2002). Thus, for the docking pose of Sivelestat, we calculated the distance O(Ser195)····C=O(pivaloyl) to be 9.4 Å. In this regard, the experimental results of Nakayama and co-authors (Nakayama et al., 2002) can be explained by the presence of other possibilities for binding of Sivelestat to HNE using conformations other than the optimal docking pose that we obtained. Indeed, we found another pose for Sivelestat in which the pivaloyl group is located close to the elastase catalytic triad, forming H-bonds with Ser195 and Asp194. In addition, H-bonds were formed between the amide nitrogen atom of the ligand and Val216 and between the carboxyl group and Gly218 and Gly219 (Figure 5B). This alternative docking pose of Sivelestat is favorable for nucleophilic attack of the Ser195 oxygen atom on the carbonyl carbon of the pivaloyl group, resulting the distance O(Ser195)····C=O(pivaloyl) of 3.04 Å, which is consistent with the reported mechanism (Nakayama et al., 2002). It should be noted that the MolDock score for this pose is only 1.4 units higher than that for the optimal pose shown in Figure 5A.
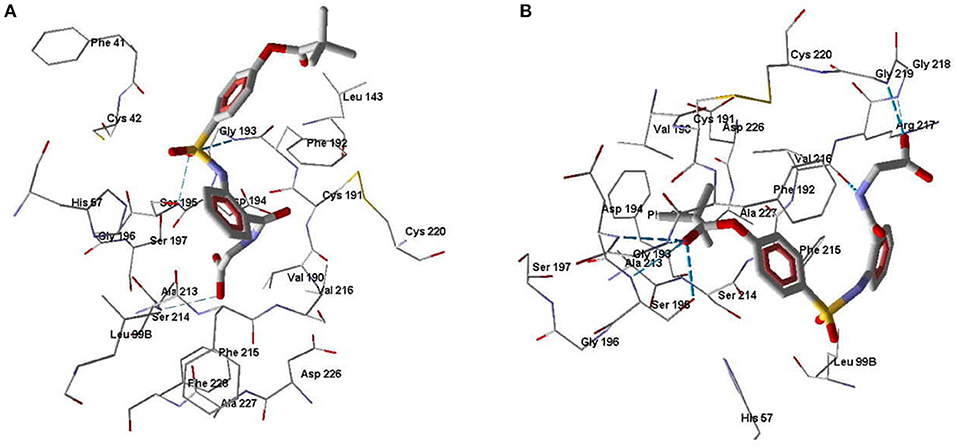
Figure 5. Docking poses of Sivelestat. (A) The lowest-score docking pose of Sivelestat. (B) Alternative docking pose of Sivelestat. Residues within 5 Å from the pose are visible. H-bonds are shown by blue dashed lines.
Molecule 7d forms H-bonds with Ser195 via participation of both oxygen atoms of the ethoxycarbonyl group. In addition, the carbonyl oxygen of the ethoxycarbonyl substituent forms Hbonds with Gly193 and Asp194, while the sulfonamide nitrogen atom is H-bonded to Ser214 (Figure 6). The docking pose of molecule 7d in the HNE binding site is characterized by a distance of 5.40 Å between the oxygen atom of Ser195 and the carbonyl carbon of the pivaloyl group. This does not exclude the possibility of nucleophilic addition of Ser195 to the C=O group according to the reported mechanism (Nakayama et al., 2002), because as a result of thermal movements of the ligand and receptor, the carbonyl oxygen may be available for nucleophilic attack.
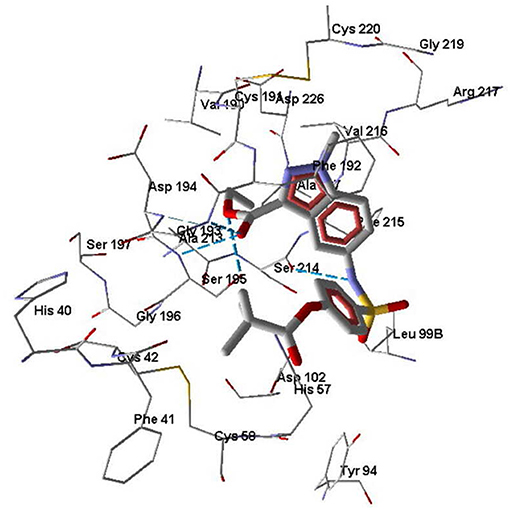
Figure 6. Docking pose of compound 7d. Residues within 5 Å from the pose are visible. H-bonds are shown by blue dashed lines.
According to our docking results, compound 3a forms strong H-bonds with Ser195 (two possible H-bonds), Asp194, and Gly193 with participation of the carbonyl oxygen atom of the pivaloyl group (Figure 7A). With this position in the binding site, 3a is quite accessible for attack by Ser195 at the carbonyl carbon atom, i.e., according to the direction of metabolism proposed by Nakayama et al. (2002). The distance O(Ser195)····C=O(pivaloyl) in this case is 3.05 Å, which is comparable to the corresponding distance for Sivelestat (see above). Compound 7e in its docking pose forms several H-bonds with HNE, one of them being a bond between the pyrazole nitrogen and Ser195 (Figure 7B). Additionally, the amide oxygen atom forms a strong Hbond with Gly193, while the sulfonamide nitrogen H-bonds with Val216. The distance O(Ser195)····C=O(pivaloyl) for the pose of compound 7e is 5.96 Å. Thus, compound 7e is anchored significantly within the binding site. Compound 7b differs from 7e by the presence of a CH group in the 5-membered ring. This reduces opportunities for the formation of H-bonds involving participation of the heterocycle. Accordingly, 7b is slightly shifted away from Ser195 and neighboring residues (Figure 8A). Molecule 7b is H-bonded to Val216 with participation of the sulfonamide nitrogen atom and also with Asp194 and Gly193 via participation of oxygen atom in the m-methylbenzoyl substituent. In Figure 8B, the poses of 7e and 7b are shown together. Visible amino acid residues lie within 5 Å of the co-crystallized ligand SEI. The distance O(Ser195)····C=O(pivaloyl) for the pose of 7b in the binding site is 6.74 Å, which is the largest value of the investigated compounds (Table 5). Perhaps, due to the remoteness of the pivaloyl group from the key residue Ser195 of the catalytic triad, compound 7b is the least active among the sulfonamides investigated.
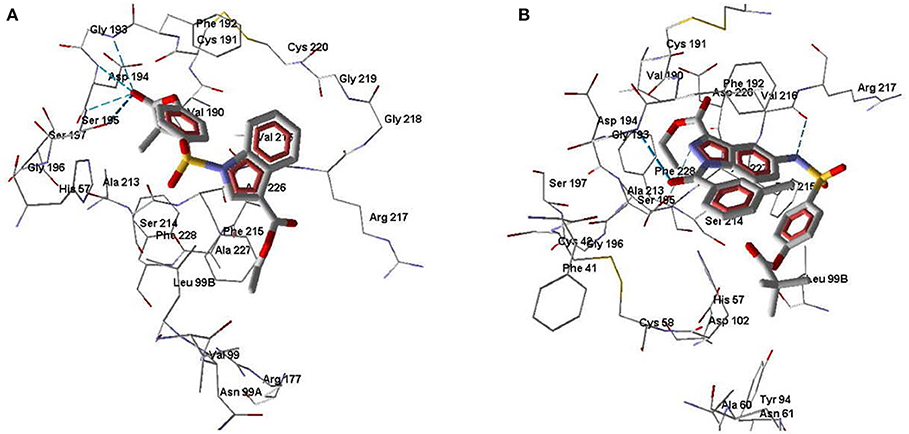
Figure 7. Docking pose of compounds 3a (A) and 7e (B). Residues within 5 Å from the pose are visible. H-bonds are shown by blue dashed lines.
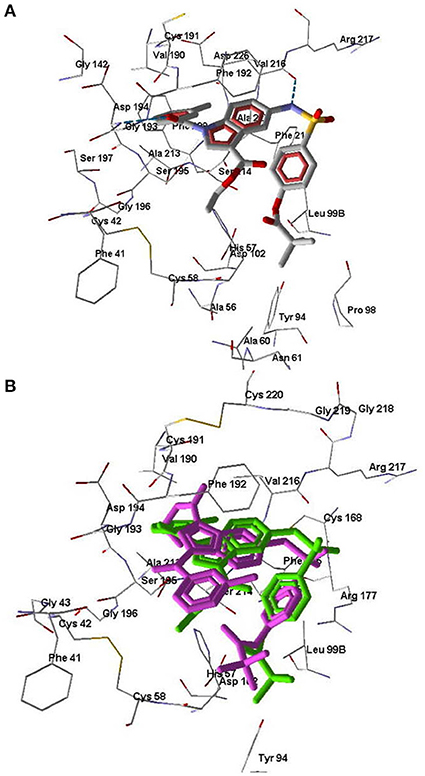
Figure 8. Docking poses of compounds 7b and 7e. (A) Docking pose of compounds 7b. H-bonds are shown by blue dashed lines. (B) Superimposed docking poses of compounds 7e (purple) and 7b (green). Amino acid residues within 5 Å from the co-crystallized SEI ligand (not shown) are visible.
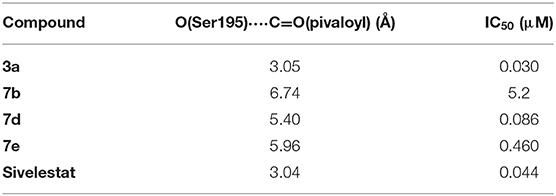
Table 5. Geometric parameters of the docking poses along with biological activities of sulfonamides.
Docking scores for the obtained poses of compounds 3a, 7b, 7d, 7e, and Sivelestat anion are equal to −127.45, −108.94, −114.37, −133.75, and −125.04 MolDock units, respectively. It should be noted that these values did not show any significant correlation with IC50 indicating that specific protein-ligand interactions rather than total complementarity play role in appearing the inhibitory activity. Indeed, according to our results, the specific mechanism of HNE inhibition proposed by Nakayama et al. (2002), which includes the Ser195 attack on the carbonyl carbon of the pivaloyl group, can be easily achieved for compounds 3a and Sivelestat (Table 5). The IC50 values obtained for compounds 7b, 7d, and 7e are also in agreement with the geometric characteristics of their docking poses (Table 5).
Previously, we demonstrated that the isoxazolone derivative G had high HNE inhibitory activity (Giovannoni et al., 2018) and excellent chemical stability in aqueous buffer (data not shown). Since this compound contains the 4-(sulfamoyl)phenyl pivalate fragment that is necessary for Sivelestat activity, we hypothesized that substitution of this active fragment onto other HNE inhibitor scaffolds could modulate their inhibitory activity, potentially resulting in higher efficacy and/or improved chemical stability of these new compounds. Based on this novel approach, we synthesized and characterized a number of new derivatives and demonstrated that the 4-(sulfamoyl)phenyl pivalate fragment could “replace” the role of the N-CO group at position 1 and offer a different point of attack for Ser195. Indeed, results of molecular docking of the these pivaloyl-containing compounds into the HNE binding site supported the mechanism of inhibitory activity involving a nucleophilic attack of Ser195 from the catalytic triad onto the carbonyl group of the pivaloyl moiety. Clearly, the selected scaffolds with adequate substituents can be appropriate carriers for the Sivelestat pharmacophore since many of the new compounds had high inhibitory activity in the nanomolar range, with the most potent inhibitors being 3a, 3b, 3f, 3g, and 9a (IC50= 19–30 nM). However, these data also indicate that this strategy does not produce an expected additive effect of inhibitor potency, probably due to increased steric hindrance of the pivaloyl substituent.
All datasets presented in this study are included in the article/Supplementary Material.
LC and MG designed the compounds and wrote the manuscript. NC, GG, and CV synthesized the compounds and checked the final version of the manuscript. IS and MQ performed in vitro studies (inhibition essay), analyzed the stability of compounds and wrote the pharmacological section. AK performed molecular modeling studies. All of the authors have given approval to the final version of the manuscript.
This research was supported in part by National Institutes of Health IDeA Program Grants GM110732, GM115371, and GM103474; USDA National Institute of Food and Agriculture Hatch project 1009546; the Montana State University Agricultural Experiment Station; and the Tomsk Polytechnic University Competitiveness Enhancement Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00795/full#supplementary-material
Alaime, T., Daniel, M., Hiebel, M.-A., Pasquinet, E., Suzenet, F., and Guillaumet, G. (2018). Access to 1H-indazoles, 1H-benzoindazoles and 1H-azaindazoles from (het)aryl azides: a one-pot Staudinger-aza-Wittig reaction leading to N–N bond formation? Chem. Comm. 54, 8411–8414. doi: 10.1039/C8CC03612H
Alpha Therapeutic Corp (2003). Alpha Therapeutic Corporation Receives FDA Approval for Aralast (TM) Alpha-1 Proteinase Inhibitor. Los Angeles, CA: Release.
Bahekar, R. H., Jain, M. R., Jadav, P. A., Prajapati, V. M., Patel, D. N., Gupta, A. A., et al. (2007). Synthesis and antidiabetic activity of 2,5-disubstituted-3-imidazol-2-yl-pyrrolo[2,3-b]pyridines and thieno[2,3-b]pyridines. Bioorg. Med. Chem. 15, 6782–6795. doi: 10.1016/j.bmc.2007.08.005
Becalli, E. M., La Rosa, C., and Marchesini, A. (1984). Oxidation of 4-aryl-substituted isoxazoline-5-ones. A new synthesis of 2,5-diaryl-1,3-oxazin-6-ones. J. Org. Chem. 49, 4287–4290. doi: 10.1021/jo00196a034
Bistocchi, G. A., De Meo, G., Pedini, M., Ricci, A., Brouilhet, H., and Boucherie, S. (1981) N-substituted ethyl-1H-indazole-3-carboxylate 1H-indazole-3-hydroxamic acids. Farmaco Edizione Sci. 36, 315–333.
Cregge, R. J., Durham, S. L., Farr, R. A., Gallion, S. L., Hare, C. M., Hoffman, R. V., et al. (1998). Inhibition of Human Neutrophil Elastase. 4. Design, synthesis, X-ray crystallographic analysis, and structure-activity relationships for a series of P2-modified, orally active peptidyl pentafluoroethyl ketones. J. Med. Chem. 41, 2461–2480. doi: 10.1021/jm970812e
Crocetti, L., Bartolucci, G., Cilibrizzi, A., Giovannoni, M. P., Guerrini, G., Iacovone, A., et al. (2017). Synthesis and analytical characterization of new thiazol-2-(3H)-ones as human neutrophil elastase (HNE) inhibitors. Chem. Cent. J. 11:127. doi: 10.1186/s13065-017-0358-1
Crocetti, L., Giovannoni, M. P., Schepetkin, I. A., Quinn, M. T., Khlebnikov, A. I., Cantini, N., et al. (2018). 1H-pyrrolo[2,3-b]pyridine: a new scaffold for human neutrophil elastase (HNE) inhibitor. Bioorg. Med. Chem. 26, 5583–5595. doi: 10.1016/j.bmc.2018.09.034
Crocetti, L., Giovannoni, M. P., Schepetkin, I. A., Quinn, M. T., Khlebnikov, A. I., Cilibrizzi, A., et al. (2011). Design, synthesis and evaluation of N-benzoylindazole derivatives and analogues as inhibitors of human neutrophil elastase. Bioorg. Med. Chem. 19, 4460–4472. doi: 10.1016/j.bmc.2011.06.036
Crocetti, L., Quinn, M. T., Schepetkin, I. A., and Giovannoni, M. P. (2019). A patenting perspective on human neutrophil elastase (HNE) inhibitors (2014-2018) and their therapeutic applications. Expert Opin. Ther. Pat. 29, 555–578. doi: 10.1080/13543776.2019.1630379
Crocetti, L., Schepetkin, I. A., Ciciani, G., Giovannoni, M. P., Guerrini, G., Iacovone, A., et al. (2016). Synthesis and pharmacological evaluation of indole derivatives as deaza analogues of potent human neutrophil elastase inhibitors. Drug. Dev. Res. 77, 285–299. doi: 10.1002/ddr.21323
Crocetti, L., Schepetkin, I. A., Cilibrizzi, A., Graziano, A., Vergelli, C., Giomi, D., et al. (2013). Optimization of N-benzoylindazole derivatives as inhibitors of human neutrophil elastase. J. Med. Chem. 56, 6259–6272. doi: 10.1021/jm400742j
DeGraw, J. I., and Goodman, L. (1964). Alkylating agents derived from indole-IV-synthesis of the 5-nitrogen mustard of indole-3-carboxylic acid. J. Med. Chem. 7, 213–215. doi: 10.1021/jm00332a019
Di Cesare Mannelli, L., Micheli, L., Cinci, L., et al. (2016). Effects of the neutrophil elastase inhibitor EL-17 in rat adjuvant-induced arthritis. Rheumatology (Oxford) 55, 1285–1294. doi: 10.1093/rheumatology/kew055
Dittrich, A. S., Kühbandner, I., Gehrig, S., Rickert-Zacharias, V., Twigg, M., Wege, S., et al. (2018). Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur. Respir. J. 51:1701910. doi: 10.1183/13993003.01910-2017
Feng, L., Zhu, W., Huang, C., and Li, Y. (2012). Direct interaction of ONO-5046 with human neutrophil elastase through 1H NMR and molecular docking. Int. J. Biol. Macromol. 51, 196–200. doi: 10.1016/j.ijbiomac.2012.04.023
Feng, L.i., Liu, X., Zhu, W., Guo, F., Wu, Y., Wang, R., et al. (2013). Inhibition of human neutrophil elastase by pentacyclic triterpenes. PLoS ONE 8:e82794. doi: 10.1371/journal.pone.0082794
Fujii, M., Miyagi, Y., Bessho, R., Nitta, T., Ochi, M., and Shimizu, K. (2010). Effect of a neutrophil elastase inhibitor on acute lung injury after cardiopulmonary bypass. Interact. Cardiov. Thor. Surg. 10, 859–862. doi: 10.1510/icvts.2009.225243
Giovannoni, M. P., Cantini, N., Crocetti, L., Guerrini, G., Iacovone, A., Schepetkin, I. A., et al. (2019). Further modifications of 1H-pyrrolo[2,3-b]pyridine derivatives as inhibitors of human neutrophil elastase. Drug Dev. Res. 80, 617–628. doi: 10.1002/ddr.21539
Giovannoni, M. P., Schepetkin, I. A., Crocetti, L., Ciciani, G., Cilibrizzi, A., Guerrini, G., et al. (2016). Cinnoline derivatives as human neutrophil elastase inhibitors. J. Enzyme Inhib. Med. Chem. 31, 628–639. doi: 10.3109/14756366.2015.1057718
Giovannoni, M. P., Schepetkin, I. A., Quinn, M. T., Cantini, N., Crocetti, L., Guerrini, G., et al. (2018). Synthesis, biological evaluation, and molecular modeling studies of potent human neutrophil elastase (HNE) inhibitors. J. Enz. Inhib. Med Chem. 33, 1108–1124. doi: 10.1080/14756366.2018.1480615
Groutas, W. C., Dou, D., and Alliston, K. R. (2011). Neutrophil elastase inhibitors. Expert Opin. Ther. Pat. 21, 339–354. doi: 10.1517/13543776.2011.551115
Henriksen, P. A., and Sallenave, J.-M. (2008). Human neutrophil elastase: mediator and therapeutic target in atherosclerosis. Int. J. Biochem. Cell. Biol. 40, 1095–1100. doi: 10.1016/j.biocel.2008.01.004
Heutinck, K. M., Ten Berge, I. J., Hack, C. E., Hamann, J., and Rowshani, A. T. (2010). Serine proteases of the human immune system in health and disease. Mol. Immunol. 47, 1943–1955. doi: 10.1016/j.molimm.2010.04.020
Hilbert, N., Schiller, J., Arnhold, J., and Arnold, K. (2002). Cartilage degradation by stimulated human neutrophils: elastase is mainly responsible for cartilage damage. Bioorg. Chem. 30, 119–132. doi: 10.1006/bioo.2002.1242
Hwang, T. L., Wang, W. H., Wang, T. Y., Yu, H. P., and Hsieh, P. W. (2015). Synthesis and pharmacological characterization of 2-aminobenzaldehyde oxime analogs as dual inhibitors of neutrophil elastase and proteinase 3. Bioorg. Med. Chem. 23, 1123–1134. doi: 10.1016/j.bmc.2014.12.056
Inoue, N., Oka, N., Kitamura, T., Shibata, K., Itatani, K., Tomoyasu, T., et al. (2013). Neutrophil elastase inhibitor Sivelestat attenuates perioperative inflammatory response in pediatric heart surgery with cardiopulmonary bypass. A prospective randomized study. Int. Heart J. 54, 149–153. doi: 10.1536/ihj.54.149
Kawabata, K., Suzuki, M., Sugitani, M., Imaki, K., Toda, M., and Miyamoto, T. (1991). ONO-5046, a novel inhibitor of human neutrophil elastase. Biochem. Biophys. Res. Commun. 177, 814–820. doi: 10.1016/0006-291X(91)91862-7
Kelly, E., Greene, C. M., and McElvaney, N. G. (2008). Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 12, 145–157. doi: 10.1517/14728222.12.2.145
Korkmaz, B., Horwitz, M. S., Jenne, D. E., and Gauthier, F. (2010). Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 62, 726–759. doi: 10.1124/pr.110.002733
Lefebvre, V., Cailly, T., Fabis, F., and Rault, S. (2010). Two-step synthesis of substituted 3-aminoindazoles from 2-bromobenzonitriles. J. Org. Chem. 75, 2730–2732. doi: 10.1021/jo100243c
Lerman, I., Garcia-Hernandez, M. L., Rangel-Moreno, J., Chiriboga, L., Pan, C., Nastiuk, K. L., et al. (2017). Infiltrating myeloid cells exert protumorigenic actions via neutrophil elastase. Mol. Cancer Res. 15, 1138–1152. doi: 10.1158/1541-7786.MCR-17-0003
Lerman, I., and Hammes, S. R. (2017). Neutrophil elastase in the tumor microenvironment. Steroids 133, 96–101. doi: 10.1016/j.steroids.2017.11.006
Macias, W. L. (2002). Combination Therapy for the Treatment of Inflammatory and Respiratory Diseases. US2002/198138 A1. Indianapolis, IN.
Marto, J., Ruivo, E., Lucas, S. D., Gonçalves, L. M., Simões, S., Gouveia, L. F., et al. (2018). Starch nanocapsules containing a novel neutrophil elastase inhibitor with improved pharmaceutical performance. Eur. J. Pharm. Biopharm. 127, 1–11. doi: 10.1016/j.ejpb.2018.01.011
Muthupplaniappan, M., Margal, S., Sukeerthi, K., Khairatkar-Joshi, N., and Karnik, P. (2009). Novel Cannabinoid Receptor Ligands, Pharmaceutical Compositions Containing Them, and Process for Their Preparation. PCT Int. Appl. WO 2009053799 A1 20090430. Navi Mumbai.
Nakayama, Y., Odagaki, Y., Fujita, S., Matsuoka, S., Hamanaka, N., Nakai, H., et al. (2002). Clarification of mechanism of human sputum elastase inhibition by a new inhibitor, ONO-5046, using electrospray ionization mass spectrometry. Bioorg. Med. Chem. Lett. 12, 2349–2353. doi: 10.1016/S0960-894X(02)00393-1
Ohbayashi, H. (2005). Current synthetic inhibitors of human neutrophil elastase in 2005. Expert. Opin. Ther. Pat. 15, 759–771. doi: 10.1517/13543776.15.7.759
Ono Pharmaceutical Co. and LTD. (1989). Derivatives of p-Substituted Phenyl Ester of Pivalic Acid. EP 0347168 A1. Osaka.
Pandey, K. C., De, S., and Mishra, P. K. (2017). Role of proteases in chronic obstructive pulmonary disease. Front. Pharmacol. 8, 512–519. doi: 10.3389/fphar.2017.00512
Pham, C. T. (2006). Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol. 6, 541–550. doi: 10.1038/nri1841
Polverino, E., Rosales-Mayor, E., Dale, G. E., Dembowsky, K., and Torres, A. (2017). The role of neutrophil elastase inhibitors in lung diseases. Chest. 152, 249–262. doi: 10.1016/j.chest.2017.03.056
Purandare, A. V., Fink, B. E., Johnson, W. L., Hart, A. C., He, L., Huynh, T. N., et al. (2014). Imidazotriazinecarbonitriles Useful as Kinase Inhibitors. PCT Int. Appl. WO 2014011974 A120140116. Princeton, NJ.
Schirok, H., Griebenow, N., Füstner, C., and Dilmac, A. M. (2015). Use of 3-(trifluoromethyl)-1H-pyrazolo-[3,4-b]pyridine as a versatile building block. Tetrahedron 71, 5597–5601. doi: 10.1016/j.tet.2015.06.050
Shahidul, I. M., Courtney, B., Qinwei, W., and Mahmun, M. H. (2006). Convenient method of synthesizing 3-ethoxycarbonyl indoles. J. Org. Chem. 71, 4675–4677. doi: 10.1021/jo0601821
Silverman, G. A., Bird, P. I., Carrell, W., et al. (2001). The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 276, 33293–33296. doi: 10.1074/jbc.R100016200
Thomsen, R., and Christensen, M. H. (2006). MolDock: a new technique for high-accuracy molecular docking. J. Med. Chem. 49, 3315–3321. doi: 10.1021/jm051197e
Vergelli, C., Schepetkin, I. A., Crocetti, L., Iacovone, A., Giovannoni, M. P., Guerrini, G., et al. (2017). Isoxazol-5(2H)-one: a new scaffold for potent human neutrophil elastase (HNE) inhibitors. J. Enz. Inhib. Med. Chem. 32, 821–831. doi: 10.1080/14756366.2017.1326915
Von Nussbaum, F., and Li, V. M. J. (2015). Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Bioorg. Med. Chem. 25, 4370–4381. doi: 10.1016/j.bmcl.2015.08.049
Wen, G., An, W., Chen, J., Maguire, E. M., Chen, Q., Yang, F., et al. (2018). Genetic and pharmacologic inhibition of the neutrophil elastase inhibits experimental atherosclerosis. J. Am. Heart. Assoc. 7:e008187. doi: 10.1161/JAHA.117.008187
Keywords: human neutrophil elastase, inhibitor, Sivelestat, stability, molecular modeling
Citation: Crocetti L, Giovannoni MP, Cantini N, Guerrini G, Vergelli C, Schepetkin IA, Khlebnikov AI and Quinn MT (2020) Novel Sulfonamide Analogs of Sivelestat as Potent Human Neutrophil Elastase Inhibitors. Front. Chem. 8:795. doi: 10.3389/fchem.2020.00795
Received: 28 May 2020; Accepted: 29 July 2020;
Published: 01 September 2020.
Edited by:
Simona Rapposelli, University of Pisa, ItalyReviewed by:
Eddy Sotelo, Universidad de Santiago de Compostela, SpainCopyright © 2020 Crocetti, Giovannoni, Cantini, Guerrini, Vergelli, Schepetkin, Khlebnikov and Quinn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Paola Giovannoni, bWFyaWFwYW9sYS5naW92YW5ub25pQHVuaWZpLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.