 Samata E. Shetgaonkar
Samata E. Shetgaonkar Fateh V. Singh
Fateh V. Singh- Chemistry Division, School of Advanced Science, Vellore Institute of Technology, Chennai, India
Hypervalent iodine compounds are valuable and versatile reagents in synthetic organic chemistry, generating a diverse array of useful organic molecules. Owing to their non-toxic and environmentally friendly features, these reagents find potential applications in various oxidative functionalization reactions. In recent years, the use of hypervalent iodine reagents in palladium-catalyzed transformations has been widely studied as they are strong electrophiles and powerful oxidizing agents. For instance, extensive work has been carried out in the field of C–H bond functionalization via Pd-catalysis using hypervalent iodine reagents as oxidants. In addition, nowadays, iodine(III) reagents have been frequently employed as arylating agents in Pd-catalyzed C–H arylation or Heck-type cross-coupling reactions. In this review, recent advancements in the area of palladium-catalyzed oxidative cross-coupling reactions using hypervalent iodine reagents are summarized in detail.
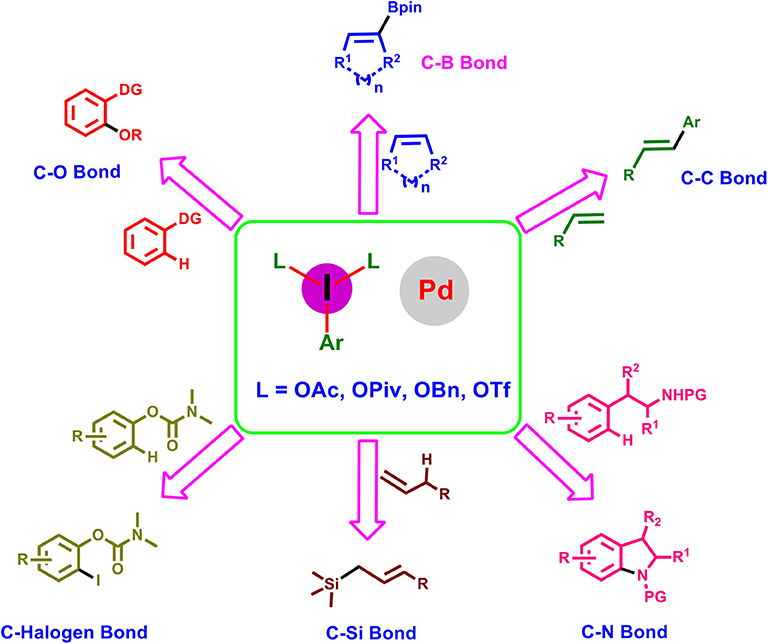
Graphical Abstract. Representative palladium-catalyzed reactions using hypervalent iodine reagents.
Introduction
Hypervalent iodine compounds are ubiquitous in organic synthesis since they have emerged as efficient and environmentally benign reagents for academic and industrial research (Zhdankin, 2014; Yoshimura and Zhdankin, 2016; Singh and Wirth, 2018). They are non-toxic, easily prepared, stable and alternative to metal-derived oxidants/catalysts in various oxidative transformations (Yusubov and Zhdankin, 2012; Singh and Wirth, 2014a,b, 2017; Mangaonkar et al., 2018; Singh et al., 2018a; Mangaonkar and Singh, 2019). Several research papers, book chapters and review articles have been published covering various aspects of hypervalent iodine compounds as reagents or as catalysts in α-functionalization of carbonyl compounds (Merritt and Olofsson, 2011; Dong et al., 2014), cyclizations (Singh and Wirth, 2011, 2012; Singh and Mangaonkar, 2018; Singh et al., 2018b), oxidative rearrangements (Singh et al., 2012; Singh and Wirth, 2013; Maertens and Canesi, 2015), alkene difunctionalizations (Li et al., 2018; Lee et al., 2019) and atom-transfer reactions (Li Y. et al., 2016). The inherent ability of hypervalent iodine reagents to act as oxidants as well as ligand transfer reagents is the key to the significant progress achieved in this area.
Representative examples of various hypervalent iodine(III)/(V) reagents commonly used as oxidants or atom transfer reagents are listed in Figure 1. Iodobenzenediacetate (PIDA) 1, [bis(trifluoroacetoxy)iodo] benzenes (PIFA) 2 and iodosobenzene dipivalate 3 are routinely used iodine(III) oxidants (Wirth, 2005; Zhdankin, 2009). Apart from this, other oxidizing agents such as iodosobenzene 4, Koser reagent 5, 2-iodosobenzoic acid (IBA) 6 and l-hydroxy-1H-1,2,3-benziodoxathiole 3,3-dioxide 7 were used in various oxidative transformations (Singh and Wirth, 2014a). Dess–Martin periodinane (DMP) 8 and 2-iodoxybenzoic acid (IBX) 9 are extensively used cyclic iodine(V) reagents (Tohma and Kita, 2004; Uyanik and Ishihara, 2009). 2,3,4,5-tetrafluoro-6-iodoxybenzoic acid (FIBX) 10 having higher reactivity than IBX 9 was synthesized by Richardson et al. (2007). Zhdankin's group prepared 2-iodoxybenzenesulfonic acid (IBS) 11 via oxone-mediated oxidation of 2-iodobenzenesulfonic acid (Koposov et al., 2006). Another important aspect of iodine(III)/(V) reagents that has received considerable attention is to act as electrophilic synthons for functional-group transfer reactions. For example, benziodoxol(on)e reagents 12–15 have been employed as a source of nucleophilic groups such as acetate, trifluoromethyl, fluorides, and alkynes in various transformations (Li Y. et al., 2016). Diaryliodonium salts 16 being electrophilic and a naturally good leaving group, are widely used as arylating reagents (Merritt and Olofsson, 2009).
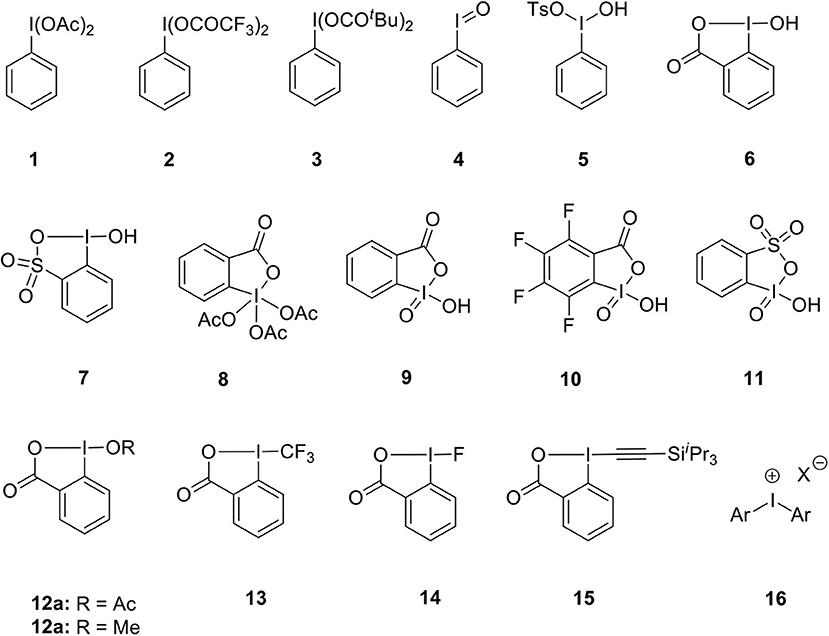
Figure 1. Examples of hypervalent iodine(III)/(V) reagents 1–16.
However, these reagents do have limitations such as stoichiometric generation of aryliodides as a byproduct, limited solubility in common organic solvents, and requirement of an excess amount of reagents thus lowering atom economy dramatically. For instance, IBA 6 is less explored due to its lower reactivity while DMP 8 is expensive and moisture sensitive. IBX 9 is associated with drawbacks such as an explosive nature, poor solubility in common organic solvents, and requirement of high reaction temperature which limits its synthetic applications to some extent (Singh and Wirth, 2014b). Also, IBS 11 is thermally unstable and highly reactive toward organic solvents, like acetonitrile, dimethyl sulfoxide, and methanol and readily decomposed into stable thia-analog of IBA 7 (Koposov et al., 2006). Although there are some drawbacks associated with these reagents, there is still great scope for these compounds as potential alternate candidates for toxic heavy-metal oxidants such as mercury, thallium, and lead based reagents (Silva L. F, 2002; Wirth et al., 2002).
On the other hand, palladium as a catalyst has become a fundamental part of various coupling reactions such as Suzuki-Miyaura, Heck, Buchwald–Hartwig, Sonagashira, and Negishi generating diverse array of useful molecules (Biffis et al., 2018). Hypervalent iodine reagents in palladium-catalyzed reactions constitute an interesting area of research in organic synthesis. Deprez and Sanford published the first review article explaining the unique reactivity of hypervalent iodine reagents in Pd-catalyzed reactions in 2007 (Deprez and Sanford, 2007). Later, the 2015 section of the review that describes the key role of polyvalent iodine reagents in high valent palladium chemistry was published by Wengryniuk's group (Silva et al., 2017). Owing to their excellent oxidizing and electrophilic nature, hypervalent iodine reagents reacts with palladium complexes and promotes reactions via Pd(0/II) or Pd(II/IV) redox cycles. For instance, iodine(III) reagents have been employed as a substitute for aryl halides in various Pd(0)/(II)-catalyzed cross-coupling reactions (Deprez and Sanford, 2007). Also, various Pd-catalyzed ligand transfer reactions employ few of these reagents as a source of aryl, alkynyl, and heteroatom ligands. Although substantial work has been carried out in Pd(0)/(II)-catalysis in the past, catalytic reactions via Pd(II)/(IV) pathway were less explored. However, with the aid of hypervalent iodine oxidants, Pd(II)/(IV)-catalysis have seen tremendous development in the last couple of decades enabling various synthetic transformations earlier inaccessible via traditional catalytic manifolds. For example, C–H functionalizations have become a powerful tool to construct new C–C and C–heteroatom bonds using Pd(II)/(IV)-catalysis.
Within this context, the present review focuses on the recent advancement accomplished in palladium-catalyzed transformations using hypervalent iodine reagents, highlighting its potential synthetic applications and mechanistic aspects. The article is classified on the basis of bonds formed such as C–O, C–N, C–C, C–Si, C–B, and C–halogen bonds and finally alkene difunctionalization reactions.
C–O Bond Formation
Palladium-catalyzed ligand assisted C–H functionalization has been proven to be one of the best, most atom economical, and effective tools in organic synthesis for the introduction of variable functional groups at the unactivated arene/alkane C–H bonds. Extensive research has been carried out by several groups in the field of C–O bond formation via palladium catalysis using hypervalent iodine reagents as oxidant or heteroatom ligand. Among them, Pd-catalyzed C–H oxidative cyclization, C–H acyloxylation, C–H alkoxylation and allylic oxidation constitutes important pathways in C–O bond formation reactions directed by directing functional groups such as oxime ether, oxazoline, amide, pyridine and pyrimidine, etc.
C–H Cyclization
Significant progress has been made in the field of Pd-catalyzed oxidative cyclization reactions using hypervalent iodine reagents, giving access to several oxygen-containing heterocycles. In 2010, Wang et al. developed a novel method for the construction of dihydrobenzofurans via Pd(OAc)2-catalyzed hydroxyl group-directed C–H activation/C–O cyclization reaction in the presence of (diacetoxyiodo)benzene 1 as a terminal oxidant and promoted by base Li2CO3. Moreover, the scope of the reaction was extended to the preparation of important scaffolds such as spirocyclic dihydrobenzofurans (Wang X. et al., 2010).
Later, Gevorgyan's group demonstrated intramolecular silanol group-directed C–H oxygenation of phenoxy silanols 17 employing Pd(OPiv)2 as catalyst and PhI(OAc)2 1 as an oxidant (Huang et al., 2011). This reaction involves the initial formation of cyclic silicon-protected catechols 18 which upon desilylation with TBAF/THF provides substituted catechols 19 (Scheme 1A). The plausible catalytic cycle involves initial coordination of Pd with silanols 17 to form palladacycle 20 accompanied by PIDA-mediated oxidation to forge Pd(IV)-intermediate 21. Next, intermediate 21 is transformed into acetoxylated intermediate 23 via reductive acetoxylation and regenerates back palladium catalyst. Finally, acid-catalyzed transesterification of 24 gives 25 which subsequently loose acetic acid to form cyclic silyl protected catechols 18 followed by subsequent desilylation with TBAF furnishes catechols 19. Further formation of products 22 through direct C–O reductive cyclization was eliminated based on 18O-labeling studies. Further reactions featured excellent site selectivity and broad substrates scope, particularly, electron-rich substrates reacted much faster and provided high yields. Another interesting work published by Gevorgyan's research group employing C–H oxygenation strategy is the convenient synthesis of oxasilacycles from substituted benzyl-silanols using combination of Pd(OAc)2 and PhI(OAc)2 1 in PhCF3 at 100°C (Huang et al., 2012).
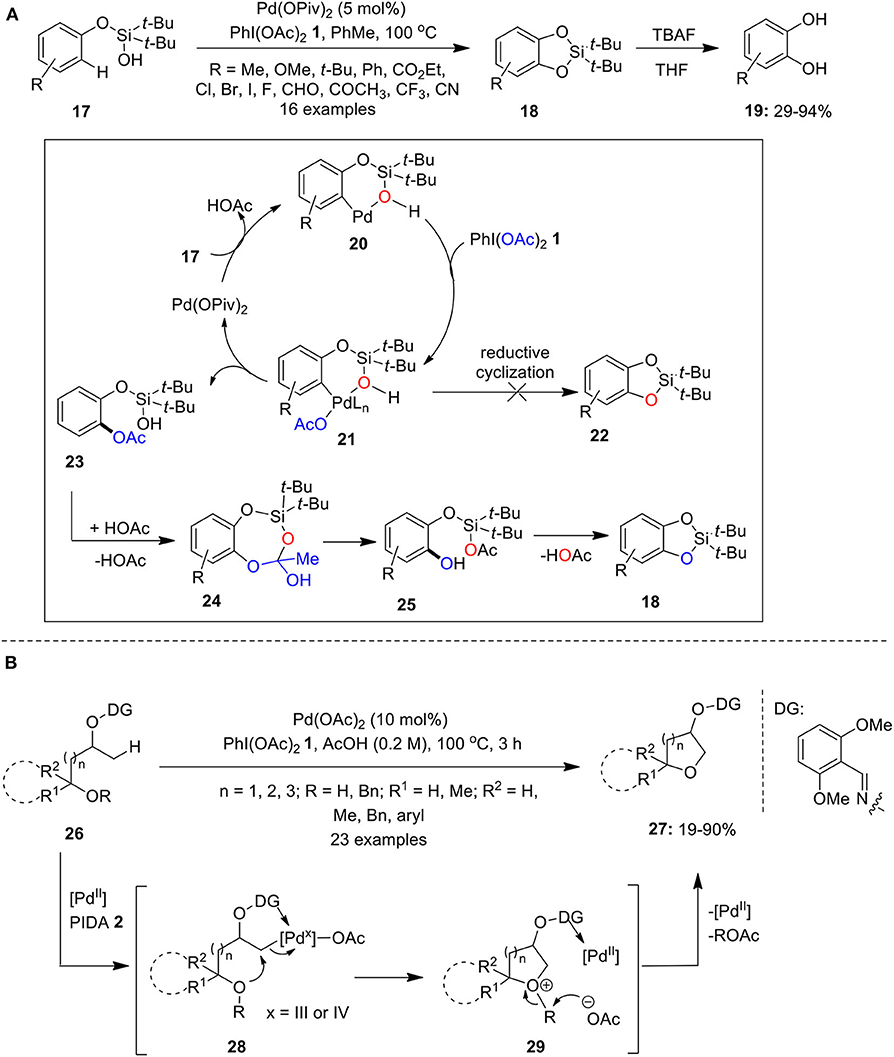
Scheme 1. (A) Pd(II)-catalyzed synthesis of substituted catechols 19 using PhI(OAc)2 1 as an oxidant and (B) Pd(II)-catalyzed synthesis of cyclic ethers 27 using PhI(OAc)2 1 as an oxidant.
Furthermore, Thompson et al. reported the preparation of cyclic ethers 27 via Pd-catalyzed oxime masked-alcohol directed dehydrogenative annulation of sp3 C–H bonds of substrates 26 using PhI(OAc)2 1 as an oxidant (Thompson et al., 2015). The reaction occurs selectively at the β-position and substrates 26 having primary, secondary, and tertiary hydroxyl group worked smoothly under the standard conditions (Scheme 1B). The reaction could proceed via C–H palladation, followed by oxidation of Pd to higher oxidation state species 28 and a subsequent intramolecular SN2 reaction to form oxonium intermediate 29. Finally, cyclic ethers 27 were obtained through deprotonation or debenzylation and regenerate Pd-catalyst.
Yang et al. reported Pd-catalyzed intramolecular lactonization of α, α-disubstituted arylacetic acids 30 in the presence of PhI(OAc)2 1 to furnish a series of α, α-disubstituted benzofuran-2-ones 31 in variable yields (Yang et al., 2013). The catalytic system composed of Pd(OAc)2 and the combination of NaOAc and CsOAc along with AgOAc as most effective bases (Scheme 2A). The authors proposed that C–H cleavage occurs via concerted metalation deprotonation to form six-membered palladacycle 32 which further undergoes oxidation and subsequent reductive elimination to afford 31. Similar catalytic C–H activation/C–O formation methods to construct functionalized benzofuranones (Cheng et al., 2013) and biaryl lactones (Li et al., 2013) were also developed employing acetyl-protected glycine (Ac-Gly-OH) as requisite ligand along with base KOAc in t-BuOH.
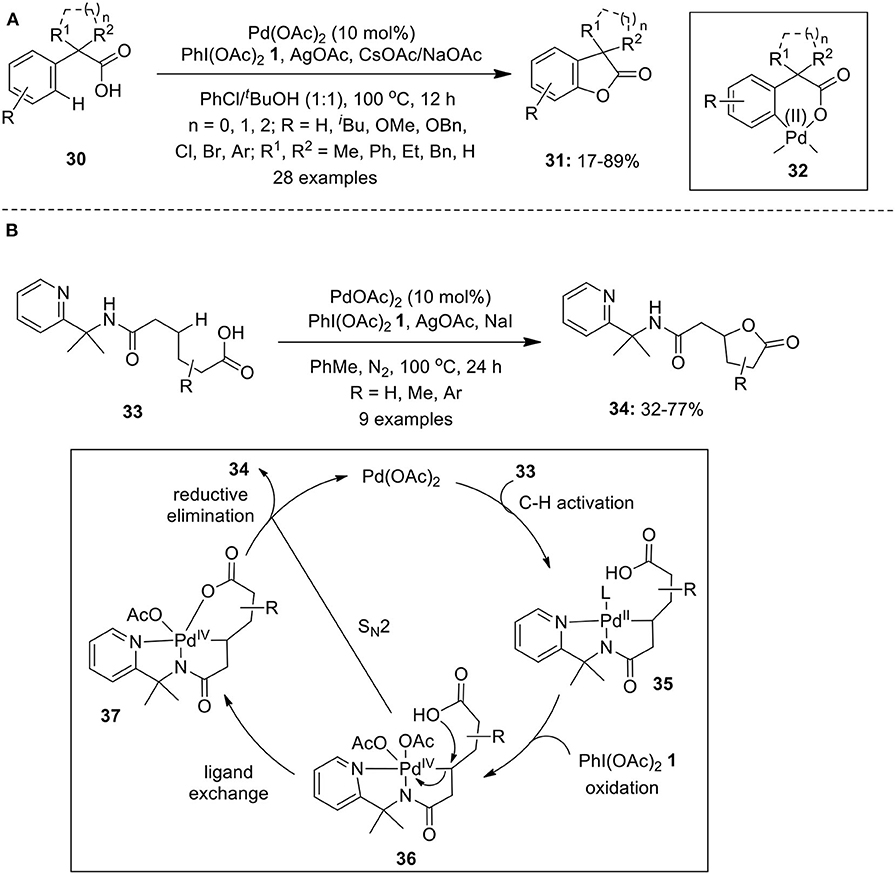
Scheme 2. (A) Pd(II)-catalyzed α,α-disubstituted benzofuran-2-ones 31 using PhI(OAc)2 1 and (B) Pd(II)-catalyzed synthesis of γ-lactones 34 using oxidant PhI(OAc)2 1.
In 2016, Shi's group described a concise route to access γ-lactones 34 featuring Pd(II)-catalyzed PIP auxiliary-directed lactonization of unactivated methylene C(sp3)–H bonds using oxidant PIDA 1 (Liu and Shi, 2016). The lactonization of aliphatic acids 33 bearing various substituents on the alkyl chain proceeded remarkably well to furnish anticipated γ-lactones 34 in 32–77% yields (Scheme 2B). The probable mechanism for the lactonization of aliphatic acids 32 involves formation of five-membered palladacycle 35 through Pd-catalyzed C–H activation assisted by bidentate auxiliary. In the presence of PhI(OAc)2 1, palladacycle 35 underwent oxidation to give Pd(IV) intermediate 36 followed by ligand exchange to form 37 which finally undergoes reductive elimination to release target product 34 along with Pd(II) catalyst to complete the catalytic cycle (Scheme 11). Another path to generate lactone 34 is via direct SN2-type attack by carboxylate group onto the Pd(IV)–C bond of 36 (Scheme 2B).
C(sp2/sp3)–H Acyloxylation
Over the years, C–H acyloxylation has made tremendous progress in transforming various unactivated sp2 and sp3 hybridized C–H bonds into valuable C–O bonds using palladium catalysts and iodine(III) reagents as oxidants. This method provides a concise route for the introduction of valuable ester functionality on to the aromatic or aliphatic substrates under mild conditions. Several C(sp2)–H acyloxylation protocols have been developed employing Pd(OAc)2/PhI(OAc)2 1 catalytic systems using a variety of directing groups such as pyrimidine (Gu et al., 2009), 8-aminoquinoline (Gou et al., 2009), oxime (Neufeldt and Sanford, 2010; Ren et al., 2015), pyridyldiisopropylsilyl (Chernyak et al., 2010; Gulevich et al., 2012), and 1,2,3-triazoles-pyridine (Ye et al., 2013).
Furthermore, a regioselective protocol featuring Pd(II)-catalyzed C–H benzoxylation of 2-arylpyridines 38 was performed by Zhang et al. affording mono-benzoxylation products 40 in moderate to excellent yields (Zhang et al., 2015). They employed easily accessible iodobenzene dibenzoate derivatives 39 as oxidants and sources of the benzoxyl group (Scheme 3). Furthermore, the present method was successfully applied for the benzoxylation of 2-thienyl pyridines to yield 3-benzoxylated thiophenes in high yields. The plausible mechanism proceeds via pyridyl-assisted C–H activation of substrates 38 with a palladium catalyst to form complex 41 which undergoes further oxidative addition with 39 to form high oxidation state complex 42. Finally, the reductive elimination of 42 gives desired product 40 and regenerates the palladium catalyst to complete the catalytic cycle.
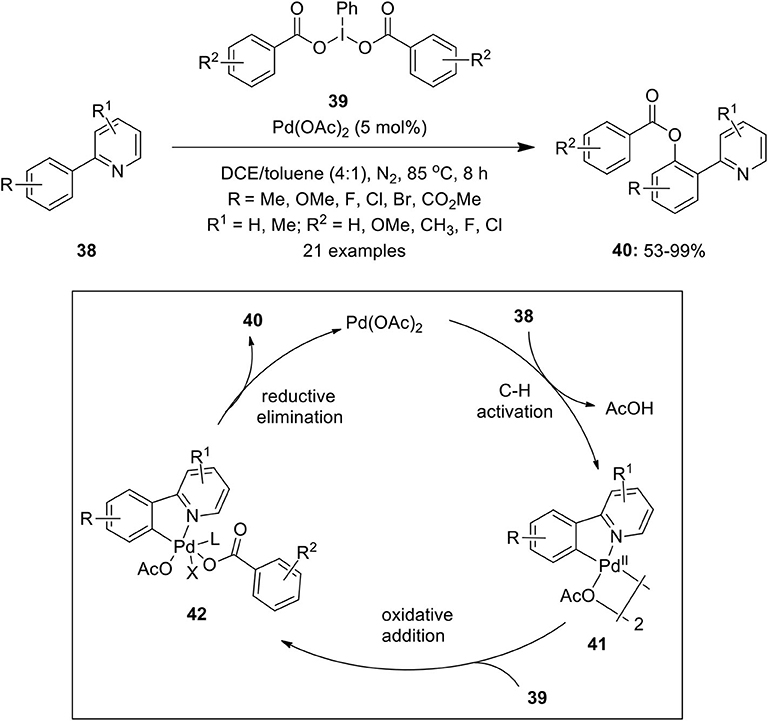
Scheme 3. Pd(II)-catalyzed ortho C–H benzoxylation of 2-arylpyridines 38 using substituted iodobenzene dibenzoates derivatives 39 as oxidant and source of benzoxyl group.
Although substantial development has been accomplished in the Pd-catalyzed ligand-directed C–H oxygenation reactions, the non-chelate assisted transformations are rarely explored. For instance, Pd-catalyzed C–H acetoxylation of arenes devoid of directing groups remains a challenge as it leads to the formation of mixtures of isomers. In this context, Emmert et al. developed non-chelate assisted palladium-catalyzed C–H acetoxylation of simple arenes using pyridine (Emmert et al., 2011) and acridine (Cook et al., 2013) as ancillary ligands in combination with hypervalent iodine reagents as oxidants.
Furthermore, the Pd-catalyzed allylic C–H acyloxylation represents one of the efficient methods in C–H functionalization reactions. Pilarski et al. have presented an excellent protocol for the preparation of allylic acetates or allylic benzoates via Pd-catalyzed allylic C–H acetoxylation/benzoyloxylation using PhI(OAc)2 1 or PhI(OBz)2 as oxidant and source of acyloxy group (Pilarski et al., 2009). Later, the same group described conversion of alkenes into allylic trifluoroacetates via Pd-catalyzed C–H trifluoroacetoxylation employing PhI(OCOCF3)2 2 as an oxidant and trifluoroacetoxy source (Alam et al., 2012).
Regioselective C–H functionalization of indoles has been found to be a straight forward route to access biologically important 3-acetoxyindoles. In this regard, Suna's and Kwong's groups independently reported the synthesis of 3-acetoxyindoles via direct C3-oxidation of indole derivatives catalyzed by Pd(OAc)2 (2–5 mol%) in the presence of terminal oxidant PhI(OAc)2 1 (Mutule et al., 2009; Choy et al., 2011). In continuation, Liu et al. developed a similar Pd-catalyzed approach for the selective C3-acetoxylation of substituted indoles 43 with reduced catalyst loading (1.5 mol%) using PhI(OAc)2 1 and KOH as a base (Scheme 4; Liu et al., 2011). Mechanistic insights revealed that the electrophilic palladation occurs at the C3 position of indole to generate Pd(II) species 45 which is oxidized to Pd(IV) intermediate 46 and subsequent reductive elimination affords corresponding C3-acetoxylated indoles 44.
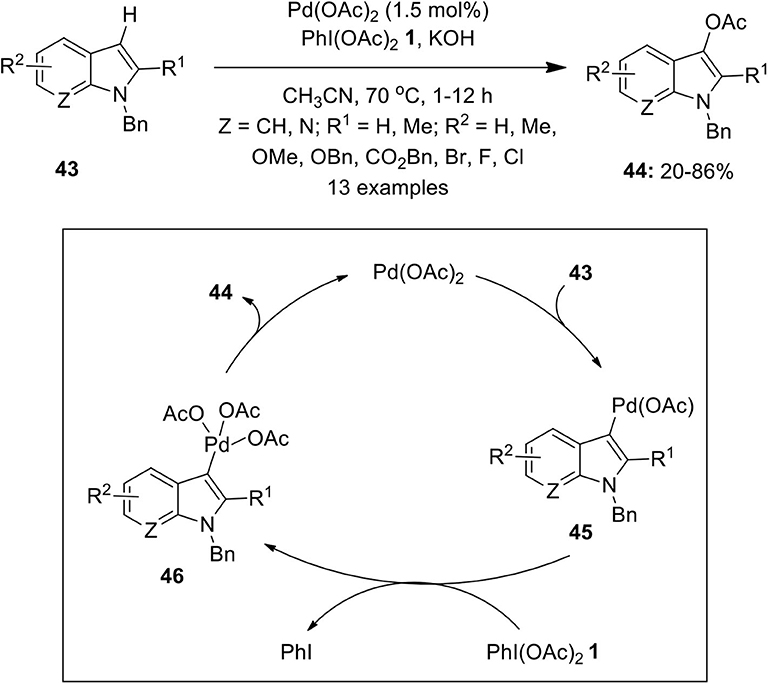
Scheme 4. Pd(II)-catalyzed C3-acetoxylation of substituted indoles 43 to afford C3-acetoxylated indoles 44 using PhI(OAc)2 1.
Acyloxylation of aliphatic sp3 C–H bond is another important method for regioselective C–O bond formation. Elegant protocols have been developed employing various nitrogen-based directing groups for the selective oxygenation of unactivated C(sp3)–H bond. In 2010, a novel chelation-assisted Pd-catalyzed C(sp3)–H acyloxylation of 8-methylquinolines in the presence of a stoichiometric amount of oxidant PhI(OAc)2 1 was developed employing inexpensive arene carboxylic acids as acyloxy source (Zhang et al., 2010). A similar method for the catalytic acetoxylation of unactivated primary β-C(sp3)–H bond of S-methyl-S-2-pyridylsulfoximine-N-amides at room temperature was described by Sahoo's group (Rit et al., 2012). This method employs S-methyl-S-2-pyridylsulfoximine (MPyS) as a bidentate directing group and β-C–H acetoxylated products were synthesized using different carboxylic acids as solvent and acetate sources.
Furthermore, a Pd-catalyzed oxime-directed β-C(sp3)–H acetoxylation of substrates 47 was performed employing PhI(OAc)2 1 as a terminal oxidant (Ren et al., 2012). The catalytic reaction was expected to generate five-membered exo-palladacycle 49 which on oxidation gives masked 1,2-diols 48 (Scheme 5A). Also, the selective acetoxylation of β-methylene (CH2) and β-methine (CH) groups in cyclopentanol and norbornyl-derived alcohols were also carried out under the same reaction conditions. Furthermore, the deprotection of DG and acetyl group was carried out by using Zn/AcOH & K2CO3/MeOH, respectively, to yield diols in excellent yield. Later, Zhang's group described an elegant work on Pd(II)-catalyzed benzylic C(sp3)–H acetoxylation of picolinoyl- or quinoline-2-carbonyl-protected toluidine derivatives using PhI(OAc)2 1 as an oxidant and acetate source (Ju et al., 2013).
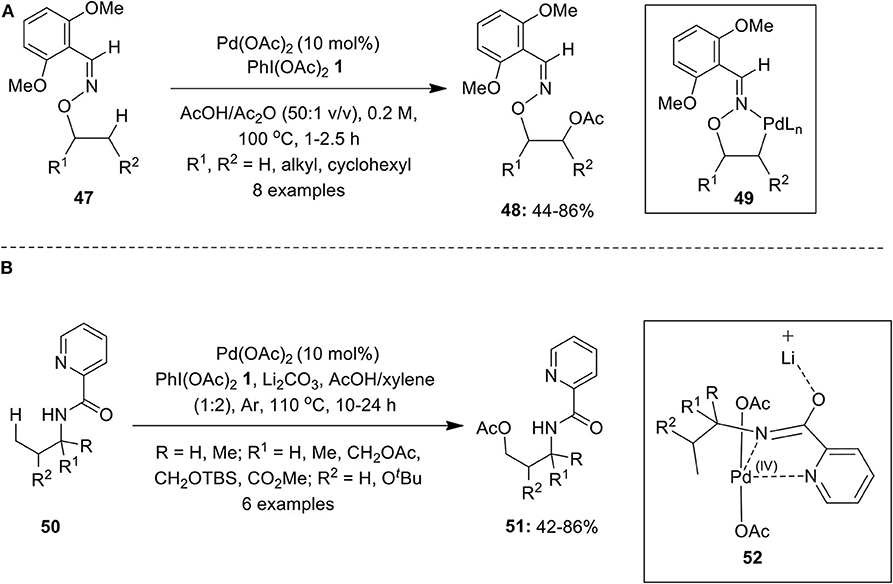
Scheme 5. (A) Pd-catalyzed PIDA-mediated C(sp3)–H acetoxylation of substrates 47 and (B) Pd-catalyzed C(sp3)–H acetoxylation of alkylamines 50 using oxidant PIDA 1.
In 2014, Li et al. performed a Pd(OAc)2-catalyzed acetoxylation of γ-C(sp3)–H bond of simple alkylamines 50 directed by picolinamide (PA) using oxidant PhI(OAc)2 1 under an argon atmosphere (Li et al., 2014). The process provides an easy access to γ-acetoxylated product 51 in good yields (Scheme 5B). Additionally, C–H acetoxylation of γ-methyl group of arylamines proceeded smoothly under this condition. Authors speculated that reaction likely to procced via C, N, N-pincer type Pd(IV) intermediate 52, in which Li2CO3 plays a crucial role as it interacts with O-imidate thereby suppressing the formation of cyclic azetidine through intramolecular C–H amination.
C(sp2/sp3)–H Alkoxylation
Another interesting Pd-catalyzed transformation enabling C–O bond formation is the C–H alkoxylation of sp2 and sp3 bonds using hypervalent iodine reagents as oxidant. Chen and Shi's group independently reported two methods for the palladium-catalyzed PhI(OAc)2-mediated alkoxylation of methylene and methyl C(sp3)–H bonds with a range of aliphatic alcohols as alkoxylation reagents, using picolinamide (Zhang et al., 2012) and pyridine (Chen et al., 2013) as easily removable directing groups. Later, azo group-directed selective C(sp2)–H alkoxylation of azobenzene compounds with alcohols as the alkoxylation reagents was reported by Yin et al. using palladium catalysis in the presence of oxidant PhI(OAc)2 1 (Yin et al., 2013). Additionally, Zhang and Sun demonstrated the regioselective alkoxylation of ortho-C(sp2)–H bond of 2-aryloxypyridines to provide ortho-alkoxylation products in the presence of catalytic amounts of Pd(OAc)2 and oxidant PhI(OAc)2 1 (Zhang and Sun, 2014).
Shan et al. for the first time employed cyclic hypervalent iodine(III) reagent 12 as an efficient oxidant in the Pd-catalyzed C(sp3)–H alkoxylation of unactivated methylene and methyl groups of 8-aminoquinoline-derived carboxylic acids 53 (Shan et al., 2013) (Scheme 6). The reaction worked perfectly well with DMP 8 (1.1 equivalents) at 110°C. Gratifyingly, C(sp3)-H alkoxylation of cyclic substrates such as cyclopentyl, cyclohexyl, and cycloheptyl were also perfomed under identical conditions. Furthermore, the synthetic applicability of present method was demonstrated for the alkoxylation of various analogs of Ibuprofen such as, Naproxen, Ketoprofen, and Flurbiprofen to afford alkoxylated products in variable yields. Based on preliminary mechanistic studies, authors proposed that either DMP 8 or 1-acetoxy-1,2-benziodoxole-3(1H)-one 12a serve as key precursors for the in situ generation of cyclic iodine(III) oxidant 56 which oxidizes cyclopalladium(II) intermediate 57 to Pd(IV) intermediate 58. Next, ArCO2 ligand of intermediate 58 could be easily displaced by alcohol 54 to give Pd(IV) intermediate 59 which finally undergoes C–O bond-forming reductive elimination to yield alkoxylated products 55 and regenerates palladium catalyst. Later, the same research group developed a similar method to prepare symmetrical and unsymmetric acetals via Pd-catalyzed regioselective double C(sp3)–H bond alkoxylation of 8-aminoquinoline-derived substrates using cyclic iodine(III) reagent 12 as an oxidant (Zong and Rao, 2014).
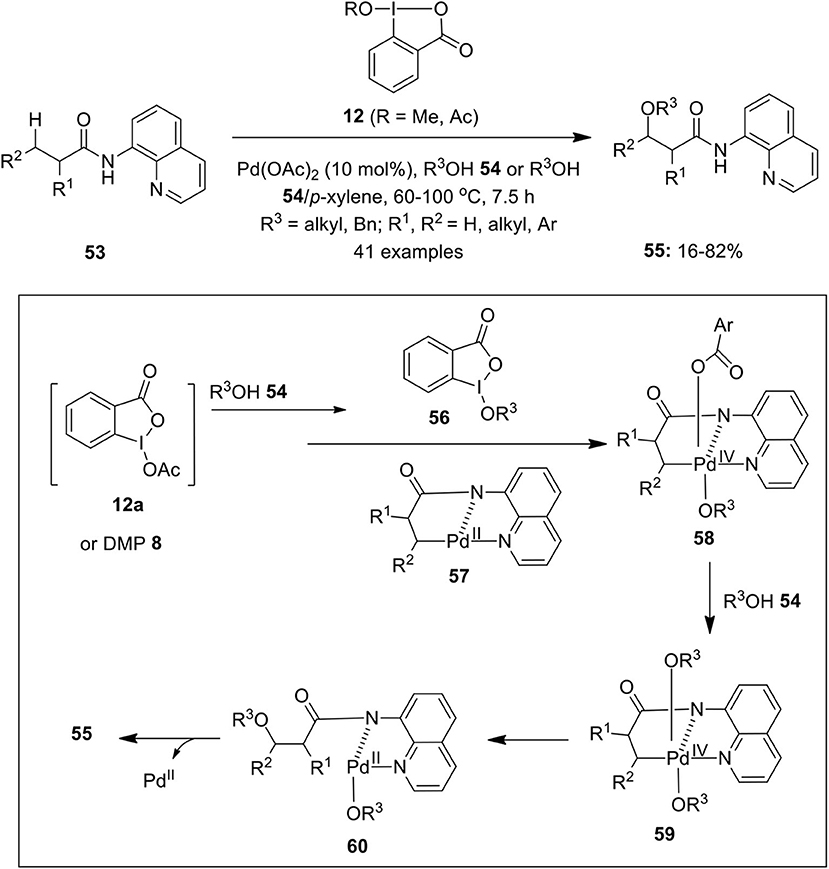
Scheme 6. Pd-catalyzed C(sp3)–H bond alkoxylation of aminoquinoline-derived substrates 53 to provide β-alkoxylated products 55 using cyclic hypervalent iodine(III) reagent 12.
C–H Oxidation and C–H Phosphorylation/Sulfonation
Chaudhari and Fernandes developed a Wacker–type oxidation protocol to convert various aliphatic and aromatic terminal alkenes 61 into functionally diversified methyl ketones 62 employing Dess-Martin Periodinane (DMP) 8 as an oxidant under nitrogen atmosphere (Chaudhari and Fernandes, 2016). Furthermore, the oxidation of a number of allylic or homoallylic compounds were also investigated under identical conditions to deliver methyl ketones in high yields. Excellent functional group compatibility, wide substrates scope and high isolated yields with complete Markovnikov selectivity are the key features associated with this methodology (Scheme 7A). A similar method for the conversion of terminal olefins into α,β-unsaturated ketones was developed by Bigi and White via a Wacker oxidation-dehydrogenation process employing Pd(CH3CN)4(BF4)2 (10 mol%) and PhI(OAc)2 1 (25 mol%) co-catalytic system in the presence of 1,4-benzoquinone as oxidant (Bigi and White, 2013). Interestingly, PhI(OAc)2 1 acts as a dehydrogenation catalyst and not as a terminal oxidant in this reaction.
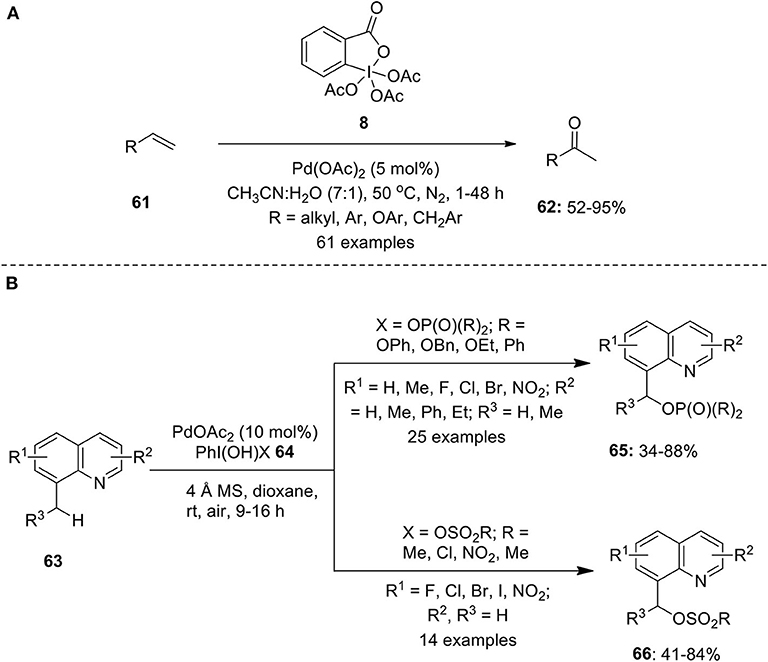
Scheme 7. (A) Pd(II)-catalyzed C–H oxidation of terminal alkenes 61 using DMP 8 as an oxidant and (B) Pd(II)-catalyzed C–H phosphorylation/sulfonation of methylquinolines 63 using hypervalent iodine reagent 64.
Very recently, He et al. reported Pd(II)-catalyzed phosphorylation and sulfonation of unactivated benzyl C(sp3)–H bond of 8-methylquinolines 63 employing organophosphorus or sulfonate hypervalent iodine(III) reagents 64 as an oxidant and as a functional group source (He et al., 2019). Using this protocol, desired products 65 or 66 were obtained in moderate to high yields with broad substrates scope (Scheme 7B). Additionally, the scope of reaction was extended for the C(sp2)–H hydroxylation and arylation of 2-phenylpyridines.
Furthermore, limited protocols are available for the direct C–O bond formation without involving C–H bond functionalization. For instance, Kitamura and his research group disclosed efficient conversion of (trimethylsilyl)arenes into acetoxyarenes via Pd(OAc)2-catalyzed desilylative acyloxylation strategy using PhI(OCOCF3)2 2 as an oxidant in AcOH (Gondo et al., 2015). Meanwhile, Cheng et al. reported Pd-catalyzed acetoxylative, hydroxylative and alkoxylative cycloisomerization of polysubstituted homoallenyl amides enabling preparation of functionalized 2-aminofurans using hypervalent iodine(III)reagents as oxidant (Cheng C. et al., 2015). Another striking example to prepare α-acetoxylated enones from propargylic substituted alkynes through Pd-catalyzed oxidative acetoxylation in the presence of terminal oxidant PhI(OAc)2 1 and additive benzoquinone (10 mol%) in DMSO was developed (Jiang et al., 2016).
C–C Bond Formation
Palladium-catalyzed C–H functionalization constitutes an important method in C–C bond formation reactions. A number of catalytic transformations have been developed using hypervalent iodine(III) reagents as oxidant.
via Oxidative Cyclization
An interesting domino process highlighting Pd-catalyzed C–H functionalization of N-arylpropiolamides 67 using iodine(III) reagent 68 as an aryl source was demonstrated by Tang et al. (2008a). This elegant protocol leads to the synthesis of 3-(1-arylmethylene)oxindoles 69 in useful yields (Scheme 8A). The effect of various electron-rich and electron-deficient substituents on the aryl ring as well as on the triple bond was investigated. The catalytic cycle for this reaction starts with the coordination of Pd(II)-catalyst with alkyne and nitrogen to form intermediate 70 followed by cis-addition with Pd and ArI(OAc)2 68 to give Pd(II) intermediate 71. Oxidation of intermediate 71 by ArI(OAc)2 68 forms Pd(IV) intermediate 72 which further gives intermediate 73 by activation of ortho C–H bond of 72. Finally, intermediate 73 underwent reductive elimination to provide anticipated products 69 and releases active Pd(II) species. The same group also reported the synthesis of (E)-(2-oxindolin-3-ylidene)phthalimides and (E)-(2-oxoindolin-3-ylidene)methyl acetates via Pd-catalyzed C–H functionalization of N-arylpropiolamides using phthalimide and carboxylic acids as nucleophiles, respectively (Tang et al., 2008b,c). Later, synthesis of CF3-substituted oxindoles was accomplished through palladium-catalyzed PhI(OAc)2-mediated intramolecular trifluoromethylation of N-alkyl-N-arylacrylamide derivatives using TMSCF3 as an efficient trifluoromethyl source in the presence of Lewis acid Yb(OTf)3 (Mu et al., 2012).
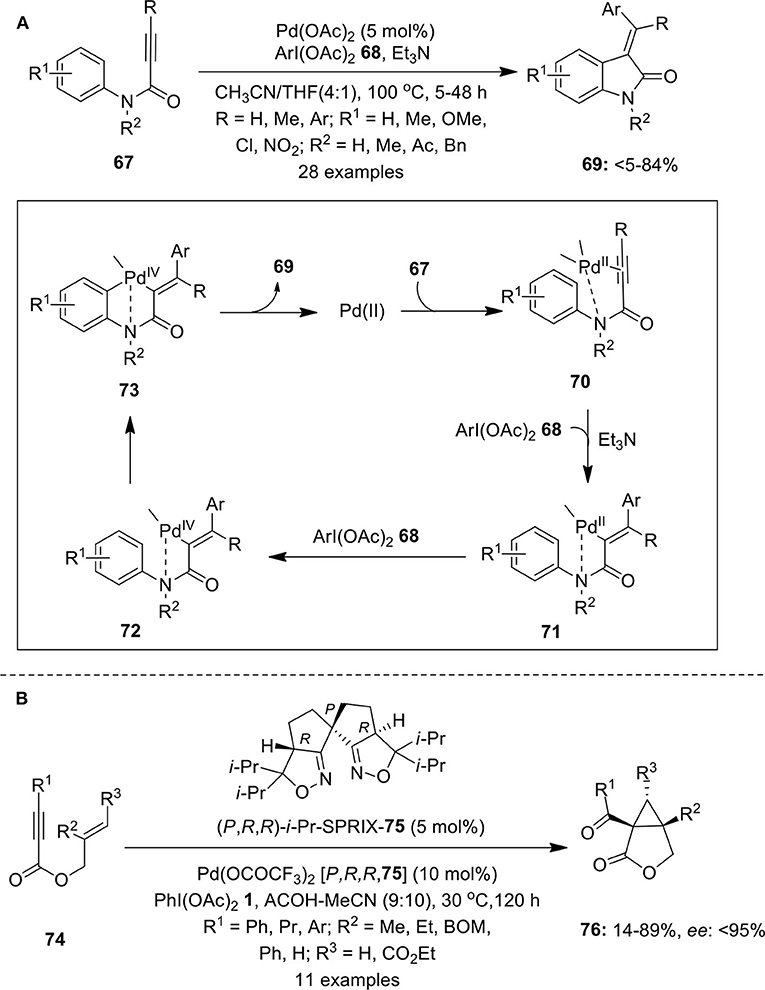
Scheme 8. (A) Pd(II)-catalyzed synthesis of 3-(1-arylmethylene)oxindoles 69 using iodine(III) reagent 68 and (B) Pd(II)-catalyzed enantioselective synthesis of bicyclic lactones 76 by employing terminal oxidant PhI(OAc)2 1.
Tong et al. coined the first Pd-catalyzed oxidative cyclization of 1,6-enynes into the corresponding bicyclo[3.1.0]hexane derivatives using oxidant PIDA 1 (Tong et al., 2007). Later, Sanford and Tong's research groups independently developed similar pathways toward the synthesis of multi-substituted bicyclo[3.1.0] ring systems using bipyridine as ligand (Welbes et al., 2007; Liu et al., 2008; Lyons and Sanford, 2009). Furthermore, Tsujihara et al. developed the first example featuring asymmetric Pd(II)/Pd(IV) catalysis for the enantioselective synthesis of bicyclic lactones 76 from 1,6-enynes 74 in variable yields with up to 95% enantiomeric excess (Scheme 8B) (Tsujihara et al., 2009). Reaction utilizes the spiro bis(isoxazoline) 75 (abbreviated as SPRIXs) as chiral ligand, Pd-SPRIX 75 complex as catalyst and PhI(OAc)2 1 as terminal oxidant.
Very recently, Xu et al. published an excellent example on the Pd(II/IV)-catalyzed intramolecular cycloaddition of the propargylic alcohol/amine and alkene of substrates 77 via acetoxylative (3 + 2) annulation strategy enabling the synthesis of bicyclic heterocycles 78 in significant yields (Xu et al., 2019) (Scheme 9). The possible catalytic cycle for this oxidative cycloaddition reaction initiates through the acetoxypalladation process generating alkenyl-Pd(II) intermediate 79 which is subsequently transform into alkyl-Pd(II) intermediate 81 via chair-like transition state (TS) 80. Next, PhI(OAc)2-mediated oxidation gives bicyclic Pd(IV) intermediate 82 which further gives 83 through loss of a molecule of AcOH. Finally, intermediate 83 undergoes direct C–O reductive elimination to deliver product 78 and regenerate palladium catalyst to continue the catalytic cycle (Scheme 9).
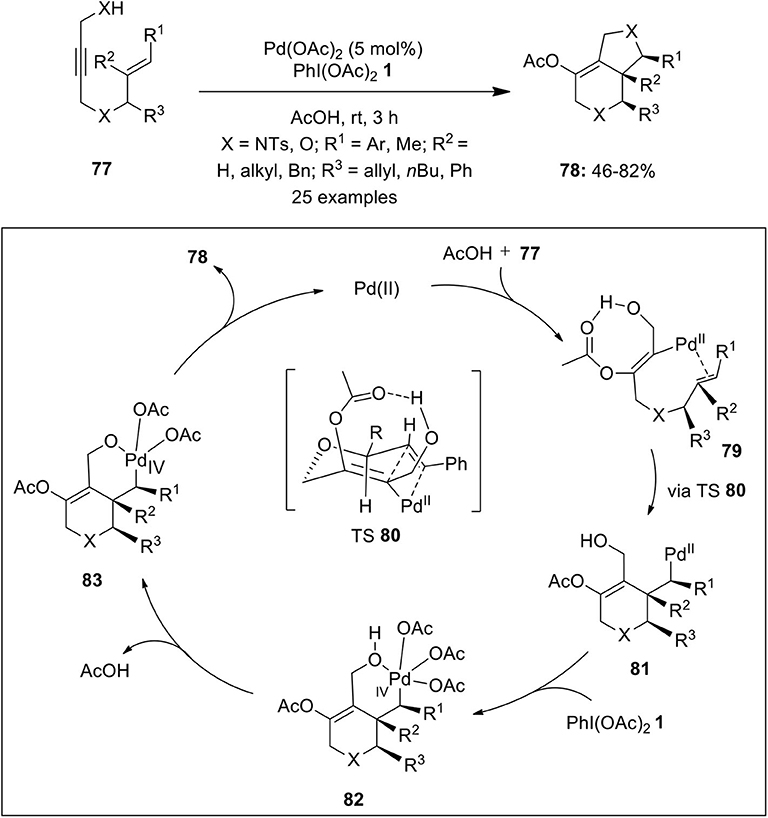
Scheme 9. Pd(II)-catalyzed synthesis of bicyclic heterocycles 78 from 1,6-enynes 77 using oxidant PhI(OAc)2 1.
via C–H Bond Arylation
In 2011, Qu et al. demonstrated a Heck-type coupling reaction between olefins 61 and iodobenzene generated in situ from hypervalent iodine reagents 84 (Qu et al., 2011). The coupling reaction was performed using Pd(OAc)2 (4 mol%), base K2CO3, and PEG-400 as solvent media under an open atmosphere at 40–60°C to afford coupling products 85 in 91–99% yields (Scheme 10). The catalytic system was free from any ligand or additive and possesses an excellent recyclability of catalyst (Scheme 10). A similar Pd-catalyzed Heck-type arylation of terminal alkenes with aryliodine(III) diacetates was developed by Evdokimov et al. (2011).
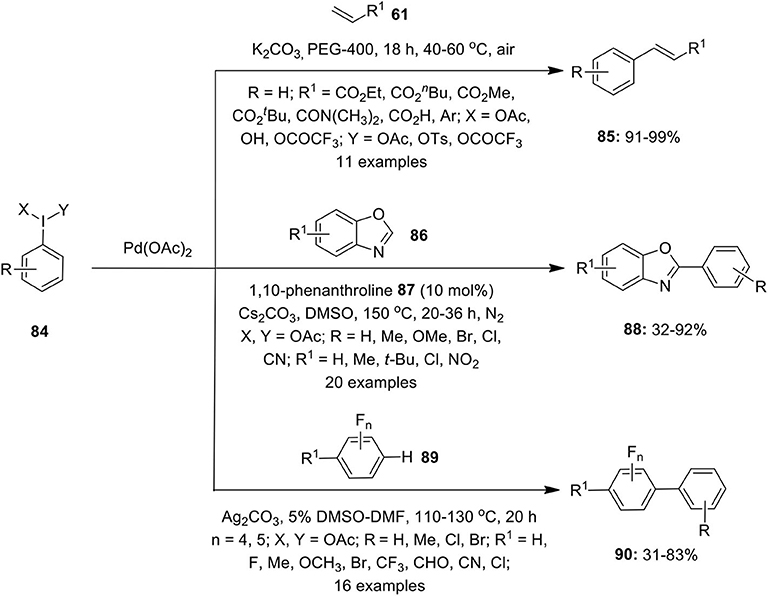
Scheme 10. Pd(II)-catalyzed Hseck-type coupling reactions using hypervalent iodine(III) reagents 84.
Later, Cheng's research group performed Pd(II)-catalyzed C–H functionalization of benzoxazoles 86 to furnish arylation products 88 using iodobenzene diacetates 84 as arylating reagent in the presence of ligand 1,10-phenanthroline 87 and base Cs2CO3 (Yu et al., 2012) (Scheme 10). Similarly, Fu et al. employed aryliodine(III) diacetates 84 as a coupling partner in the Pd-catalyzed C–H arylation of polyfluoroarenes 89 (Fu et al., 2015). Detailed mechanistic studies revealed in situ generation of aryliodides by the reduction of ArI(OAc)2 84 under basic conditions to furnish desired polyfluorobiaryls products 90 in moderate to good yields (Scheme 10).
Another interesting strategy to establish C–C bond formation is Pd-catalyzed oxidative coupling reactions of aromatic halides with different arylation reagents. Considering this, Xiong et al. illustrated the synthesis of useful symmetrical biaryls employing aryliodine(III) diacetates 68 as aryl precursor via Pd-catalyzed homocoupling reaction (Xiong et al., 2015). The reaction was carried out in DMF at 110°C in the presence of Pd(OAc)2 (10 mol %), K2CO3 (4 equiv.) under an air atmosphere. In 2016, Wang et al. reported ligand-free arylation of pyridines and quinolines via direct coupling of bromo-substituted pyridines or quinolines with hypervalent iodine(III) compounds as efficient arylating reagents in the presence of catalyst PdCl2 and base Cs2CO3 in DMF at 110°C (Wang et al., 2016).
via C–H Trifluoromethylation/Alkynylation
Mu et al. reported Pd-catalyzed C–H trifluoromethylation to prepare C2-trifluoromethylated indoles employing Ruppert–Prakash reagent, TMSCF3 as CF3 source in presence of oxidant PhI(OAc)2 1 and base CsF at room temperature (Mu et al., 2011). In 2013, Tolnai et al. employed 2 mol% of Pd(MeCN)4(BF4)2 catalyst in the regioselective C2-alkynylation of N-alkylated indoles using 1-[(Triisopropyllsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX) as an alkynylating reagent (Tolnai et al., 2013). This reaction gave direct access to substituted 1-alkylated-2-[(triisopropylsilyl)ethynyl]-1H-indole at ambient temperature under air atmosphere in significant yields. A number of substituents including Cl, Br, F, & I remained intact in the final products which could be used for further synthetic modifications.
C–N Bond Formation
Catalytic C–H activation/C–N bond formation has gained considerable attention in recent times as it represents the versatile approach for the construction of N-containing aliphatic/aromatic heterocycles. Several intra- and intermolecular protocols for the palladium-catalyzed amination of sp3 and sp2 C–H bonds have been developed using hypervalent iodine reagents as oxidant. Gaunt has disclosed an elegant work on intramolecular C–H amination of N-substituted biphenyls for the synthesis of carbazoles at room temperature using catalyst Pd(OAc)2 and oxidant PhI(OAc)2 1 in the presence of acetic acid in toluene (Jordan-Hore et al., 2008). Furthemore, Chen's research group synthesized azetidines 92 via intramolecular C–H amination of picolinamide-directed amine substrates 91 employing Pd(OAc)2 and PhI(OAc)2 1 in toluene under argon atmosphere (Scheme 11A; He et al., 2012a). Additionally, the synthesis of pyrrolidines was also accomplished under identical reaction conditions.
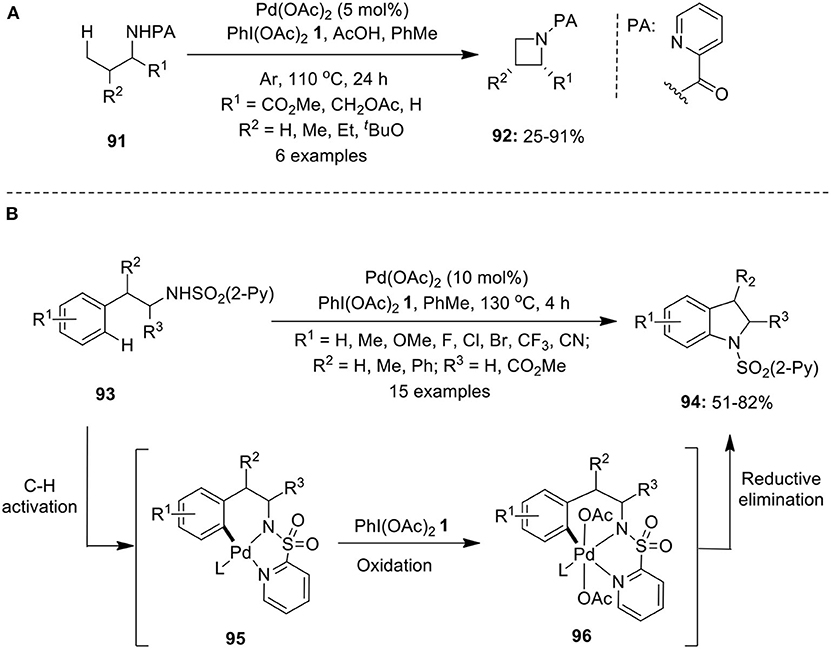
Scheme 11. (A) Pd(OAc)2-catalyzed synthesis of azetidines 92 using oxidant PhI(OAc)2 1 and (B) Pd(OAc)2-catalyzed synthesis of indolines 94 using oxidant PhI(OAc)2 1.
Mei et al. demonstrated the synthesis of indolines 94 via Pd(OAc)2-catalyzed intramolecular C–H activation/C–N cyclization of phenethylamine derivatives 93 using 2-pyridinesulfonyl as efficient directing group in the presence of oxidant PhI(OAc)2 1 (Mei et al., 2013). The intramolecular amination proposed to proceed via Pd-catalyzed C–H activation to generate organopalladium(II) complex 95, followed by oxidation to form palladium(IV) intermediate 96 and final C–N reductive elimination to provide anticipated products 94. Furthermore, the 2-pyridinesulfonyl moiety was easily removed under mild reaction conditions by treating with magnesium in MeOH at 0oC (Scheme 11B). Similar protocols to prepare indolines were independently reported by Daugulis, Chen, and Shi's research groups employing Pd(OAc)2/PhI(OAc)2 1 catalytic system (He et al., 2012b; Nadres and Daugulis, 2012; Ye et al., 2013).
On the other hand, Yin et al. disclosed an intermolecular allylic C–H amination of alkyl olefins to furnish linear (E)-allylimides using O-alkyl N-sulfonylcarbamates as nitrogen nucleophile. The catalytic system comprises of catalyst Pd(OAc)2 (5 mol%), terminal oxidant PhI(OPiv)2, additive 1,4-naphthoquinone (20 mol%) and base Bu4NOAc (Yin et al., 2010). Another interesting protocol featuring Pd-catalyzed regioselective intermolecular C–H amination of multi-substituted arenes using phthalimide as the nitrogen source was developed by Hartwig and co-workers (Shrestha et al., 2013).
C–B and C–SI Bond Formation
Sazabo and his research group demonstrated the first example of selective C–H borylation of simple alkenes 97 employing a palladium pincer complex 98 as an effective catalyst and PhI(TFA)2 2 as an essential oxidant (Selander et al., 2010). Reaction was performed in neat alkenes (Condition A) or diluted alkenes (Condition B) at ambient temperature (Scheme 12A). Simple cycloalkenes 97 such as cyclopentene and cyclohexene (R1: CH2; n = 1, 2) were readily borylated with B2pin2 99 to provide valuable organoboronates 100 with excellent vinylic selectivity. However, in the case that cycloheptane selectivity was reversed, the formation of allylic product was favored. Further borylation reaction of acyclic alkenes such as allylsilane and vinylboronate 98 (R1: CH2SiMe3, Bpin; n = 0) were also carried out under similar reaction conditions. Additionally, the course of reaction was also studied with Pd(OAc)2 as a catalyst, but products were obtained in albeit lower yields. The key step involves formation of Pd(IV) intermediate 101 via oxidation of catalyst using PhI(TFA)2 2, which further undergoes transmetalation with B2pin2 and subsequent reaction with alkenes to deliver desired products 100. Furthermore, the borylation reaction proceeded with excellent vinylic selectivity, except in the case of cycloheptane which yielded allylic products preferentially.
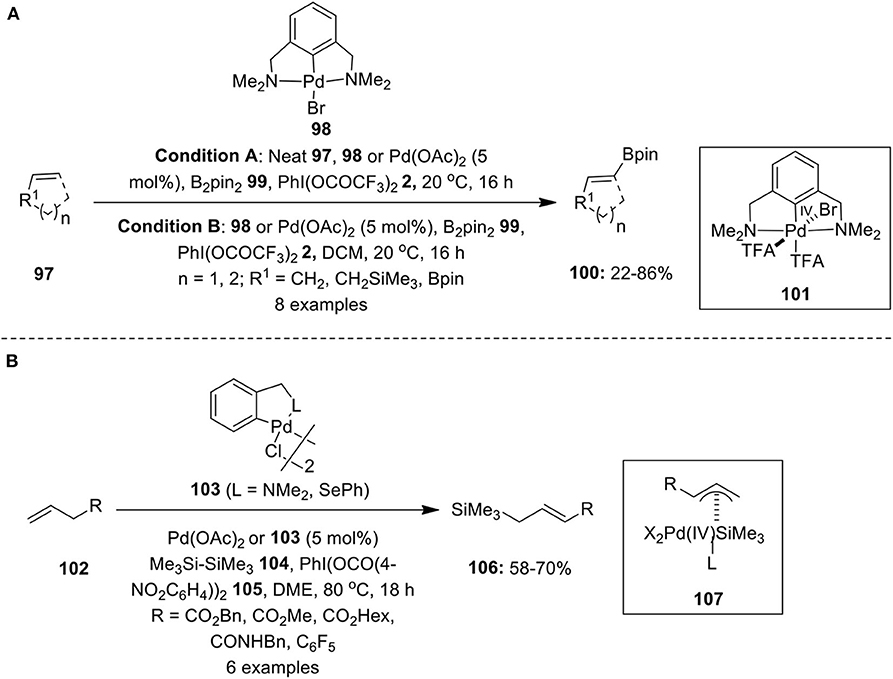
Scheme 12. (A) Palladium-catalyzed C–H borylation of olefins 97 mediated by PhI(TFA)2 2 and (B) Palladium-catalyzed C–H silylation of terminal olefins 102 using PhI(OCO(4-NO2C6H4))2 105 as an oxidant.
Later, the same research group developed the first Pd-catalyzed oxidative allylic C–H silylation of terminal olefins 102 using hexamethyldisilane 104 as the silyl source (Larsson et al., 2011). This catalytic process employs hypervalent iodine(III) reagent PhI(OCO(4-NO2C6H4))2 105 as an oxidant and Pd(OAc)2 or nitrogen and selenium-based palladium catalyst 103 (Scheme 12B). The reaction follows Pd(II)/Pd(IV) pathway involving formation of allylpalladium complex 107 which presumably undergoes reductive elimination to furnish allylsilanes 106. Moreover, the functional groups such as ester, benzyl, and amide were well-tolerated under the oxidizing conditions and anticipated products 106 were obtained with high regio- and stereoselectivity.
C–Halogen Bond Formation
Palladium-catalyzed conversion of C–H bond into C–Halogen bond is an attractive method to access valuable aryl halides. However, in the past limited studies have been carried out in C–H halogenation reactions via high valent palladium catalysis. McMurtrey et al. published the first report on Pd-catalyzed C–H fluorination employing AgF as a fluoride source in combination with iodosobenzene dipivalate, PhI(OPiv)2 3 as oxidant (McMurtrey et al., 2012). Although the detailed mechanism is not given, high valent Pd-alkyl fluoride complex 110 was hypothesized as the key intermediate in this process that would further undergo C–F bond-forming reductive elimination to yield fluorination products 109 in moderate to good yields (Scheme 13A). Furthermore, the substrates 108 bearing electron-withdrawing groups produced better results than those with electron-donating groups.
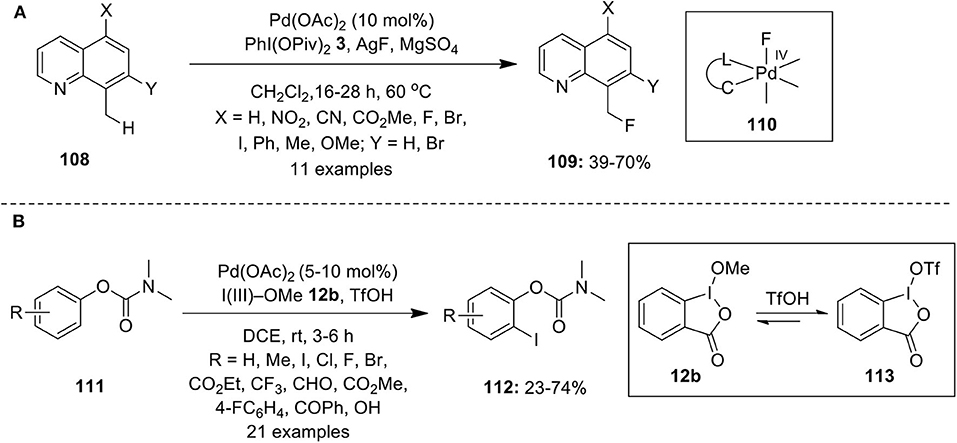
Scheme 13. (A) Pd(II)-catalyzed C–H fluorination of 8-methylquinoline analogs 108 using oxidant PhI(OPiv)2 3 and (B) Pd(II)-catalyzed C–H iodination of phenol carbamates 111 by employing Togni's reagent 12b.
In 2015, Rao's research group achieved ortho C–H iodination of phenol carbamates 111 through palladium catalysis employing Togni's reagent 12b as an iodine source and as oxidant in DCE/TfOH at room temperature (Sun et al., 2015). Both electron-rich and -deficient substituents were well-tolerated, leading to the preparation of o-iodinated phenol derivatives in fair to good yields (Scheme 13B). NMR studies revealed that some part of cyclic iodine(III)reagent on treatment with TfOH gets converted into 113 and probably both oxidants participates in this reaction.
Alkene Difunctionalization
Palladium-catalyzed difunctionalization of simple alkenes using hypervalent iodine reagents has emerged as a powerful method in organic synthesis. Various 1,1- and 1,2-difunctionalization protocols have been developed by several researchers for preparing a diverse array of useful molecules from alkenes. A general mechanism for the Pd-catalyzed oxidative functionalization of alkenes 61 is shown in Scheme 14. These transformations proposed to proceed through the formation of δ-alkyl PdII intermediate 114 obtained via olefin insertion into the aryl-Pd bond. Next, Heck intermediate 114 undergoes β-hydride elimination to form Heck product 116 and the resulting -HPdLn-species readds again to give benzylic Pd(II) intermediate 117 which can be intercepted with suitable nucleophile to furnish 1,1-difunctionalized product 118. Furthermore, Heck intermediates 114 could be oxidatively functionalized into 1,2-difunctionalized product 115 in the presence of suitable nucleophile or oxidant.
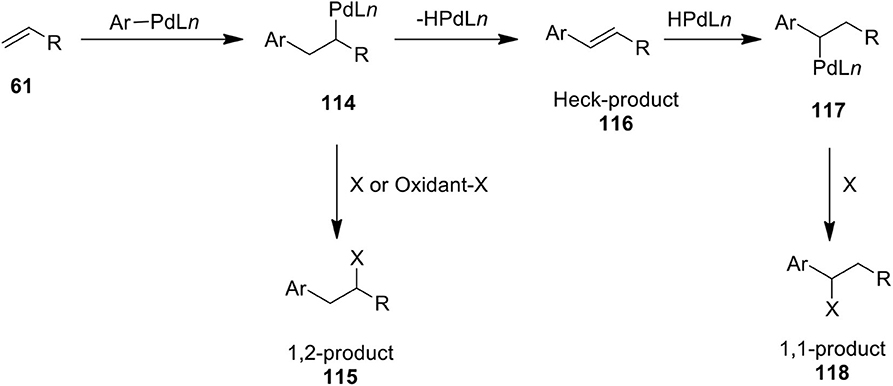
Scheme 14. Plausible mechanism for the Pd(II)-catalyzed difunctionalization of alkenes 61.
Pd(II)-Catalyzed 1,1-Difunctionalization of Alkenes
Pd-catalyzed hypervalent iodine mediated 1,1-difunctionalization of alkenes are rare and only few examples are available in the literature. Moran and co-author published an article highlighting 1,1-difuntionalization of acrylate derivatives 119 using palladium catalysis (Rodriguez and Moran, 2009). This reaction involves three component coupling of activated alkenes 119, substituted arenes 120 and hypervalent iodine(III) reagent 121 in acetic acid. Reaction possibly involves formation of Heck intermediate 123 via Heck-type addition of aryl-Pd complex to the alkene 119, followed by β-hydride elimination and subsequent readdition of Pd-H species 126 at benzylic site to form intermediate 128. Finally, oxidation of 128 to Pd(IV) intermediate using iodine(III) reagent 121 and subsequent functionalization with acetate ion lead to the formation of desired 1,1-aryloxygenated compounds 122 (Scheme 15A).
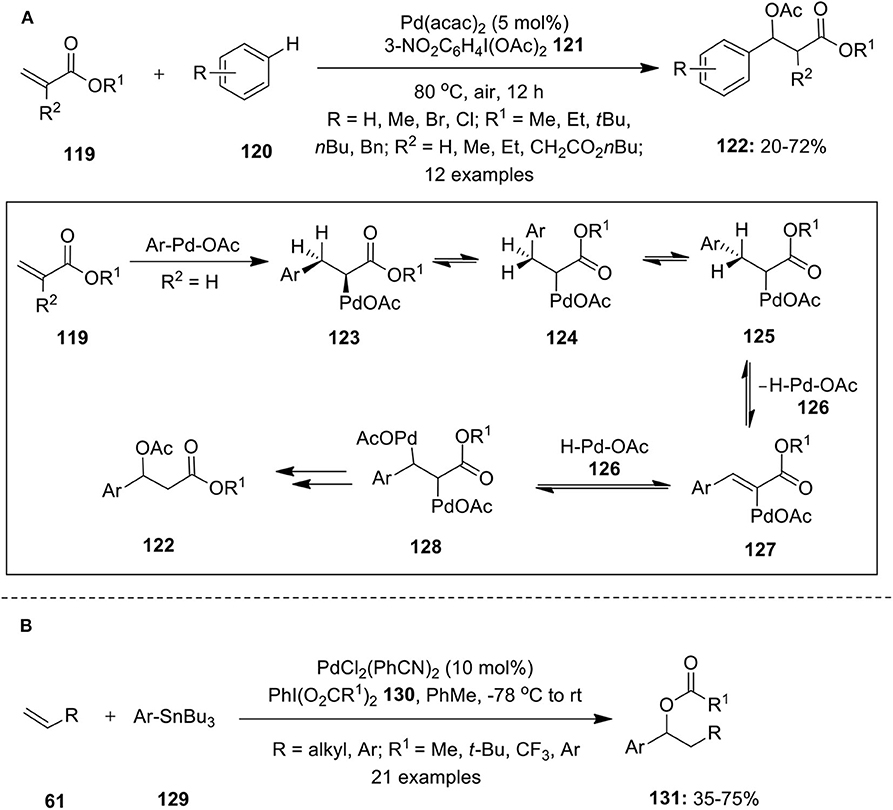
Scheme 15. (A) Pd(II)-catalyzed 1,1-difuntionalization of acrylate derivatives 119 using iodine(III) reagent 121 and (B) Pd(II)-catalyzed 1,1-difuntionalization of terminal olefins 61 using iodine(III) reagent 130 as oxidant.
Later, Satterfield et al. disclosed 1,1-aryloxygenation protocol wherein arylstannanes 129 were successfully coupled with terminal olefins 61 in the presence of hypervalent iodine reagents (PhI(O2CR1)2) 130 as an oxidant and palladium catalyst, PdCl2(PhCN)2 (Satterfield et al., 2011). This catalytic approach enabled simultaneous generation of C–C and C–O bond in a single step furnishing 1,1-arylacetoxylated products 131 in significant yields (Scheme 15B).
1,2-Difunctionalization of Alkenes
Over the years, great progress has been achieved in the field of Pd-catalyzed 1,2-difunctionalization of olefins using various hypervalent iodine reagents. Based on this strategy, a number intra- and intermolecular synthetic transformations such as diamination, dioxygenation, aminoacetoxylation, fluoroamination, and oxidative amination have been developed.
Intramolecular 1,2-Difunctionalization of Alkenes
Catalytic intramolecular difunctionalization of alkenes is one of the most powerful methods for the construction of heteroatom containing aromatic and aliphatic cyclic compounds. Sanford and co-workers reported Pd-catalyzed intramolecular oxidative amination strategy for the synthesis of tetrahydrofurans using oxidant PIDA 2 (Desai and Sanford, 2007). Subsequently, Muniz's research group developed an intramolecular alkene diamination protocol for the preparation of bisindoline and cyclic urea scaffolds using palladium catalysis (Streuff et al., 2005; Muniz, 2007; Muniz et al., 2008). Furthermore, Wu et al. performed Pd-catalyzed intramolecular aminofluorination of unactivated N-tosylamine alkenes with AgF as fluorinating reagent in the presence of PhI(OCOtBu)2 as terminal oxidant and MgSO4 as additive in acetonitrile at room temperature (Wu et al., 2009).
Zhu's research group reported domino carboacetoxylation of N-aryl acrylamides to synthesize 3,3′-disubstituted oxindoles and spiropyrrolidinyloxindoles using catalytic amount of Pd(OAc)2 or PdCl2 and oxidant PhI(OAc)2 1 in AcOH at 100°C (Jaegli et al., 2010). Furthermore, another intramolecular carboacetoxylation protocol for the preparation of acetoxymethyl-substituted cyclopentane derivatives via oxidative cyclization of 4-pentenyl-substituted malonate esters employing bis(acetonitrile)dichloropalladium catalyst and oxidant PhI(OAc)2 1 was developed (Fujino et al., 2010). The reaction was performed in the presence of base titanium tetraisopropoxide in DCE/Ac2O (1:1) solvent system at 50°C.
In 2010, Nicolai et al. reported the first intramolecular oxyalkynylation of non-activated terminal alkenes 132 using 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX) 15 as acetylene transfer reagent in DCM in the presence of 10 mol% of Pd(hfacac)2 as an efficient Pd species (Nicolai et al., 2010). The reaction was successful with different phenol substrates 132 giving cyclic ethers 133 via Pd(IV) intermediate 134 in variable yields (Scheme 16). Additionally, the scope of the reaction was examined with both aromatic and aliphatic carboxylic acids leading to the synthesis of γ-lactones under the same optimized reaction condition.
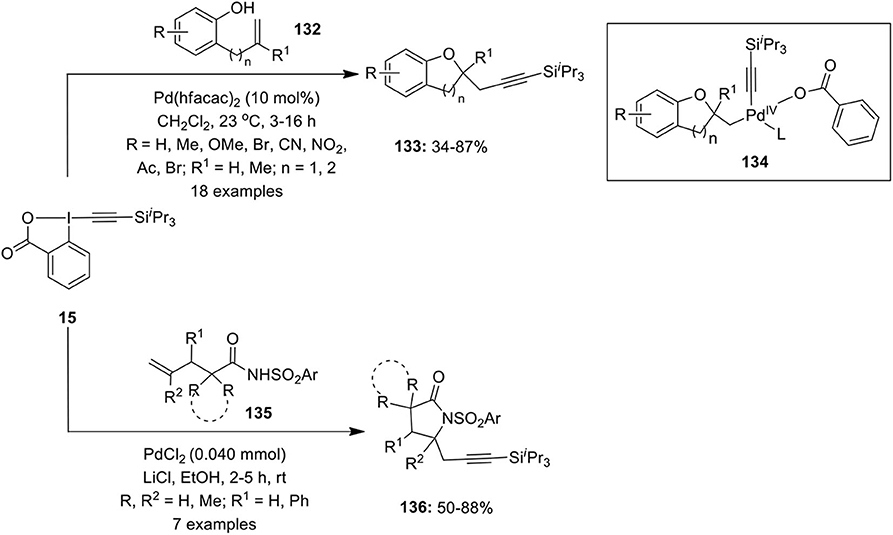
Scheme 16. Pd-catalyzed intramolecular oxyalkynylation of phenols 132 and aminoalkynylation of activated amides 135 by using TIPS-EBX 15 as acetylene transfer reagent.
In continuation with the difunctionalization reaction, the Waser's group described an intramolecular aminoalkynylation of activated olefins 135 to afford 4-propargyl lactams 136 employing TIPS-EBX 15 as alkynylating agent (Nicolai et al., 2011). In the presence of PdCl2 and LiCl, the active catalyst lithium palladate, Li2[PdCl4] generates in situ which catalyzes the carboamination reaction (Scheme 16). Furthermore, the present protocol was successfully utilized for the synthesis of 4-propargyl oxazolidinone and imidazolidinones through the cyclization of allyl carbamates and allyl ureas, respectively. Additionally, the synthesized γ- and δ-lactams were employed as precursors for the synthesis of bicyclic heterocycles pyrrolizidine and indolizidine and also in the total synthesis of natural product (±)-trachelanthamidine (Nicolai et al., 2011).
Intermolecular 1,2-Difunctionalization of Alkenes
Iglesias et al. developed the first example of Pd-catalyzed intermolecular diamination of terminal alkenes employing saccharin and bissulfonimides as the nitrogen sources in the presence of PhI(OPiv)2 3 as an oxidant (Iglesias et al., 2010). Further allylic ethers were subjected to similar catalytic intermolecular 1,2-diamination reaction using nitrogen sources, phthalimide, and N-fluoro-bis(phenylsulfonyl)imide (Muniz et al., 2011). Later, Martinez and Muniz successfully employed Pd/PhI(OPiv)2-catalytic system for the intermolecular vicinal diamination of internal alkenes 137 with phthalimide 138 and bissulfonimides 139 as the two nitrogen sources (Martinez and Muniz, 2012). This method employs alkene as the limiting reagent and desired diamination products 140 were isolated in moderate to high yields with complete regio- and diastereoselectivity (Scheme 17A). The mechanistic approach involves initial alkene 137 co-ordination with the Pd catalyst followed by subsequent aminopalladation involving nucleophilic addition of 138 via trans stereochemistry to form δ-alkylpalladium complex 141. Rapid oxidation of complex 141 gives Pd(IV) intermediate 142 which is attacked by bissulfonimide 139 to provide desired product 140 with net inversion of configuration at benzylic position.
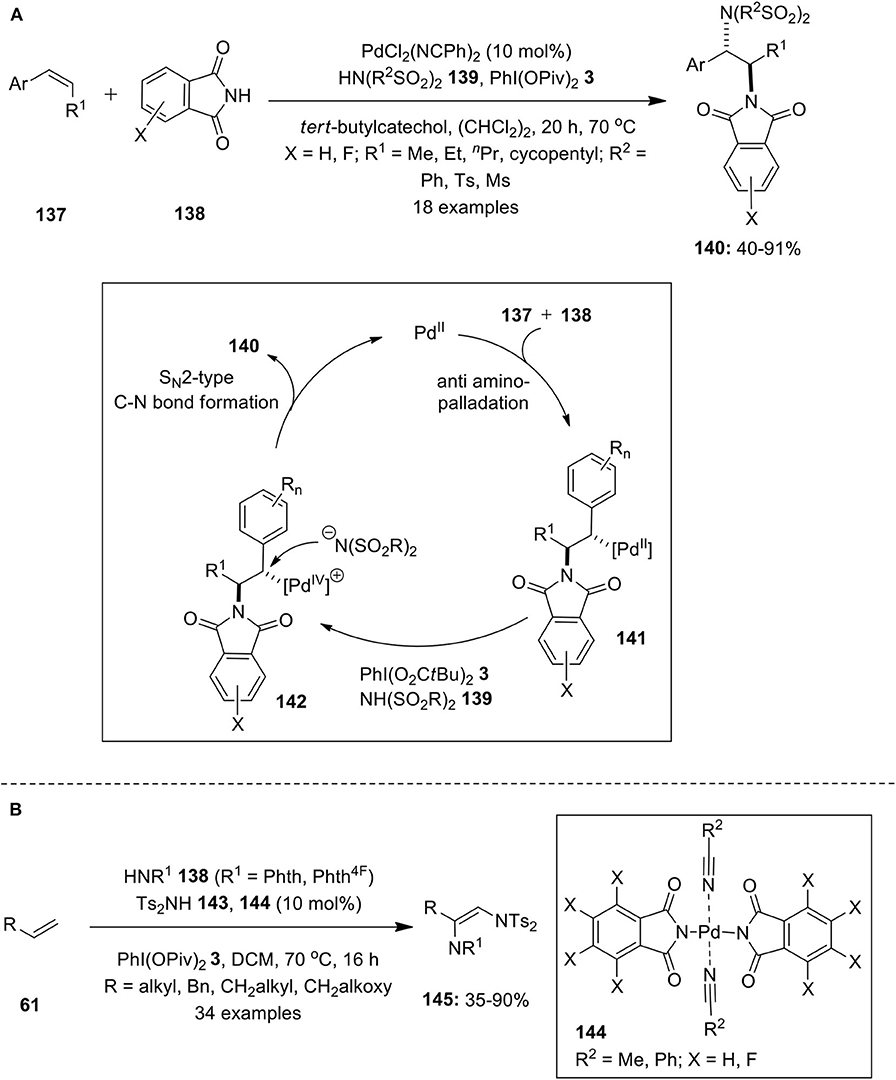
Scheme 17. (A) Intermolecular diamination of internal alkenes 137 using Pd/PhI(OPiv)2-catalytic system and (B) Palladium-phthalimidato complex-catalyzed diamination of alkenes 61 using PhI(OPiv)2 3 as oxidant.
In continuation, Martinez and Muniz reported the synthesis of new palladium-phthalimidato complexes 144 and demonstrated its broad applicability as catalysts in the vicinal diamination of alkenes 61 with phthalimide 143 and bissulfonimides 144 as nitrogen sources (Martinez and Muniz, 2016). The treatment of phthalimide 143 with Pd(OAc)2 in nitrile solution at room temperature resulted in the formation of palladium-phthalimidato complexes 144. The air-stable preformed phthalimidato complexes proved to be versatile catalysts for the present diamination reaction providing desired products 145 in useful yields (Scheme 17B). Moreover, the same research group synthesized other bissaccharido palladium(II) complexes and investigated their application in catalytic regioselective diaminations and aminooxygenation of alkenes (Martinez et al., 2016).
Pd-catalyzed aminoacetoxylation of alkenes were independently reported by Sorensen and Stahl's groups employing 2 equivalence of olefin with respect to nitrogen nucleophiles (Alexanian et al., 2005; Liu and Stahl, 2006). Later, Muniz with his co-workers reported a modified method for an intermolecular aminoacetoxylation of internal/terminal alkenes 137 using alkene substrates as limiting reagent (Martinez et al., 2013). A series of alkenes such as allyl ethers, allyl benzenes, (Z)-β-methylstyrene, etc. were oxidized and converted into aminoacetoxylated product 146 using phthalimide 138 as a nitrogen source (Scheme 18A). Based on the experimental evidences, it was concluded that PhI(OAc)2 1 influences the stereochemical aspect of the aminoacetoxylation reaction favoring trans-aminopalladation pathway.
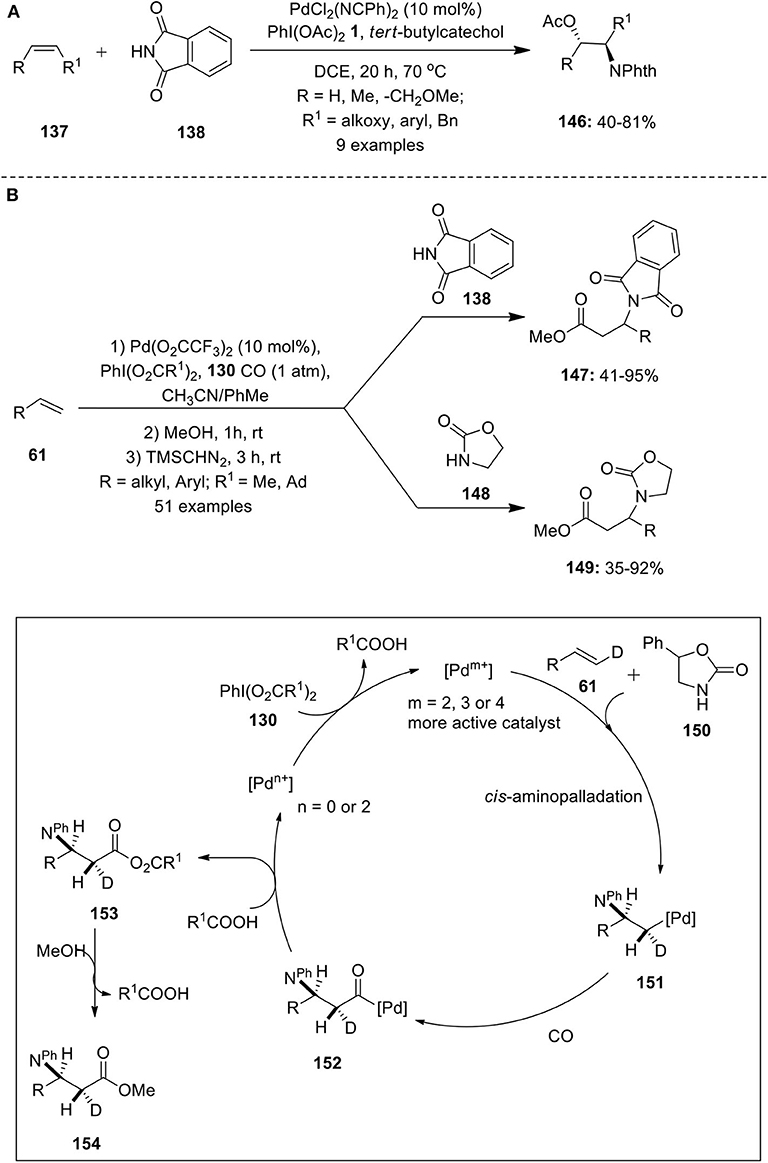
Scheme 18. (A) Pd(II)-catalyzed PIDA-mediated aminoacetoxylation of alkenes 137 and (B) Pd(II)-catalyzed aminocarbonylation of alkenes 61 induced by iodine(III) reagent 130.
In 2015, Cheng et al. have demonstrated an efficient and simple palladium-catalyzed protocol for the synthesis of β− amino acid derivatives 147 and 149 from alkenes 61 via intermolecular aminocarbonylation reaction (Cheng J. et al., 2015). An array of aliphatic or aromatic terminal alkenes 61 were reacted with either phthalimide 138 or with 2-oxazolidone 148 under a carbon monoxide atmosphere in the presence of PhI(O2CR1)2 130 as an oxidant (Scheme 18B). The reaction possessed excellent regioselectivity, broad substrates scope, and remarkable functional group tolerance. Reaction mechanism and stereochemistry was studied by taking deuterium labeled alkene 61 and substituted 2-oxazolidone 149 as an example. The reaction initiates with cis-aminopalladation of alkene 61 to form alkyl-Pd complex 151 and subsequent CO insertion gives intermediate 152. Next, nucleophilic attack of carboxylate on to the acyl-Pd species 152 afforded anhydride product 153 which upon alcoholysis yields carboxylic ester 154. Further the experimental evindence revealed that the iodine(III) reagent plays a crucial role in accelerating the intermolecular aminopalladation process.
The same research group developed a novel Pd-catalyzed intermolecular oxycarbonylation of terminal 155 and internal alkenes 157 under CO atmosphere using PIDA 1 as oxidant (Li M. et al., 2016). This difunctionalization reaction leads to the facile synthesis of various β-oxycarboxylic acids 156 and 158 in useful yields with excellent functional group compatibility, regioselectivity, and diastereoselectivity (Scheme 19). The potential scope of this method was extended toward the synthesis of natural product, (+)-honaucin C in 48% yield with ee up to 99% (Li M. et al., 2016).
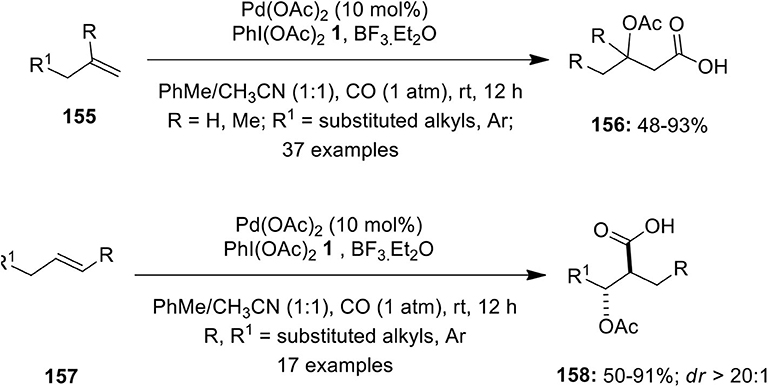
Scheme 19. Pd(II)-catalyzed oxycarbonylation of alkenes 154 and 156 using PhI(OAc)2 1 as an oxidant.
Vicinal alkene dioxygenation is an important method for the preparation of valuble 1,2-dioxygenated scaffolds. In this regard, Dong and Shi's research groups independently reported Pd-catalyzed vicinal dioxygenation of olefins employing hypervalent iodine reagent as terminal oxidant following distinct Pd(II)/Pd(IV) mechanism (Li et al., 2008; Wang W. et al., 2010). Subsequently, Sanford's group developed Pd(II)-catalyzed chiral oxime-directed asymmetric 1,2-dioxygenation of alkenes 159 using PhI(OBz)2 160 as an oxidant and benzoyloxy source (Neufeldt and Sanford, 2013). Various chiral allyl oxime ethers were screened and it was observed that menthone-derived substrates provided best reactivity and diastereoselectivity. This approach enabled efficient preparation of dibenzoylated products 161 with diasteriomeric ratio up to 9:1 (Scheme 20A). The proposed mechanism initiates with the coordination of oxime ether 159 with Pd catalyst to give intermediate 162 followed by subsequent oxypalladation and oxidation proceeding via either trans or cis geometry to deliver intermediate 163 or 164. Final, reductive elimination could occur by direct (path “a” and “c”) or by SN2-type mechanisms (path b and d) that would lead to the formation of syn or anti products. Further, based on the experimental data authors suggested that reaction proceeds via syn addition and primary operative mechanism could be either “b” or “c”.
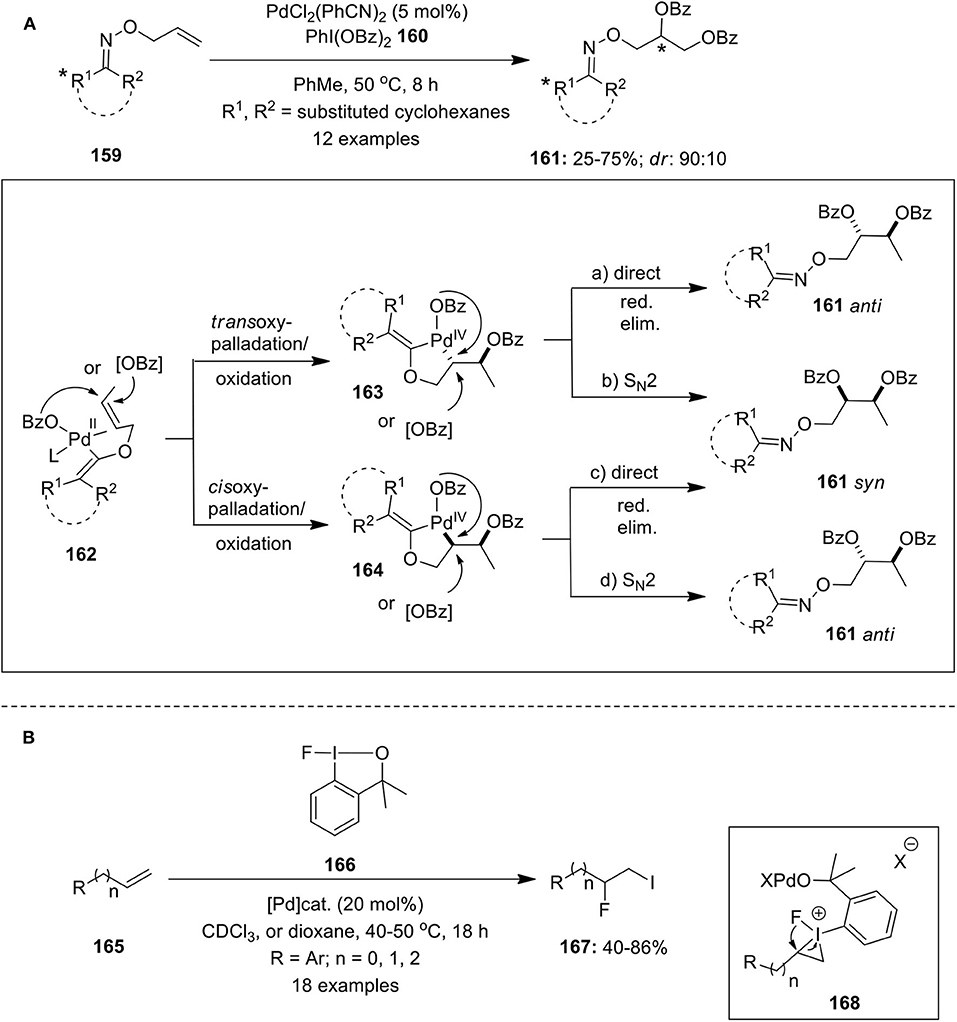
Scheme 20. (A) Pd(II)-catalyzed asymmetric 1,2-dioxygenation chiral oxime allyl ether 107 using PhI(OBz)2 160 and (B) Pd(II)-catalyzed iodofluorination of alkenes 110 employing fluoroiodane reagent 166.
Furthermore, the Pd-catalyzed iodofluorination of alkenes 165 was reported by Ilchenko et al. employing fluoroiodane reagent 166 as iodine and fluorine sources (Ilchenko et al., 2016). The reaction was carried out using either Pd(BF4)2(MeCN)4 or PdCl2(MeCN)2 or Pd(OAc)2 as palladium catalysts in CDCl3 (Scheme 20B). The reaction proceeds via three-membered iodonium intermediate 168 which upon ring opening and subsequent C(sp2)–I bond cleavage delivers the desired iodofluorinated products 167. Simple cycloalkenes such as cyclohexene and cycloheptene also undergoes iodofluorination reaction to give iodofluorinated products in modest yields.
Conclusion
In conclusion, this review described various achievements made in palladium-catalyzed reactions mediated by hypervalent iodine compounds. Hypervalent iodine reagents are versatile, non-toxic, environment friendly, and easy to handle reagents in organic synthesis. In recent years, the use of hypervalent iodine reagents in palladium-catalyzed transformations has been widely studied as they are strong electrophiles and powerful oxidizing agents. Most of the reactions proceed via Pd(II/IV) redox cycles involving hypervalent iodine reagents-induced oxidation to generate Pd(IV) center as the key step. The inherent oxidizing nature and unique reactivity of these reagents with palladium catalysts enabled efficient preparation of synthetically useful molecules through C–O, C–N, C–C, C–Si, C–B, and C–halogen bond formation reactions. In addition, a number of Pd-catalyzed alkene difunctionalization reactions have been developed in recent times employing hypervalent iodine reagents. More importantly, oxidation of an unactivated C(sp3)–H bond has been successfully accomplished using a palladium and hypervalent iodine catalytic system which is a challenging problem. From future perspectives, palladium catalysis is well-established with respect to achiral synthesis, however reactions concerning to asymmetric synthesis are limited. Thus, development of enantioselective Pd-catalyzed reaction with hypervalent iodine reagents would be a much-anticipated area of research in the near future. Apart from this, use of recyclable polymer-supported hypervalent iodine reagents in palladium catalyzed reactions would be another interesting field of research.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SS and FS are thankful to VIT Chennai for providing infrastructure for preparing the review article.
References
Alam, R., Pilarski, L. T., Pershagen, E., and Szabo, K. J. (2012). Stereoselective intermolecular allylic C–H trifluoroacetoxylation of functionalized alkenes. J. Am. Chem. Soc. 134, 8778–8781. doi: 10.1021/ja302457p
Alexanian, E. J., Lee, C., and Sorensen, E. J. (2005). Palladium-catalyzed ring-forming aminoacetoxylation of alkenes. J. Am. Chem. Soc. 127, 7690–7691. doi: 10.1021/ja051406k
Biffis, A., Centomo, P., Zotto, A. D., and Zecca, M. (2018). Pd metal catalysts for cross-couplings and related reactions in the 21st Century: a critical review. Chem. Rev. 118, 2249–2295. doi: 10.1021/acs.chemrev.7b00443
Bigi, M. A., and White, M. C. (2013). Terminal olefins to linear α,β-unsaturated ketones: Pd(II)/ hypervalent iodine co-catalyzed Wacker oxidation–dehydrogenation. J. Am. Chem. Soc. 135, 7831–7834. doi: 10.1021/ja402651q
Chaudhari, D. A., and Fernandes, R. A. (2016). Hypervalent iodine as a terminal oxidant in wacker-type oxidation of terminal olefins to methyl ketones. J. Org. Chem. 81, 2113–2121. doi: 10.1021/acs.joc.6b00137
Chen, F.-J., Zhao, S., Hu, F., Chen, K., Zhang, Q., Zhang, S.-Q., et al. (2013). Pd(II)-catalyzed alkoxylation of unactivated C(sp3)–H and C(sp2)–H bonds using a removable directing group: efficient synthesis of alkyl ethers. Chem. Sci. 4, 4187–4192. doi: 10.1039/c3sc51993g
Cheng, C., Liu, S., and Zhu, G. (2015). Divergent synthesis of 2-aminofurans via palladium-catalyzed acetoxylative, alkoxylative, and hydroxylative cycloisomerization of homoallenyl amides. J. Org. Chem. 80, 7604–7612. doi: 10.1021/acs.joc.5b01182
Cheng, J., Qi, X., Li, M., Chen, P., and Liu, G. (2015). Palladium-catalyzed intermolecular aminocarbonylation of alkenes: efficient access of β-amino acid derivatives. J. Am. Chem. Soc. 137, 2480–2483. doi: 10.1021/jacs.5b00719
Cheng, X.-F., Li, Y., Su, Y.-M., Yin, F., Wang, J.-Y., Sheng, J., et al. (2013). Pd(II)-catalyzed enantioselective C–H activation/C–O bond formation: synthesis of chiral benzofuranones. J. Am. Chem. Soc. 135, 1236–1239. doi: 10.1021/ja311259x
Chernyak, N., Dudnik, A. S., Huang, C., and Gevorgyan, V. (2010). PyDipSi: a general and easily modifiable/traceless Si-tethered directing group for C–H acyloxylation of arenes. J. Am. Chem. Soc. 132, 8270–8272. doi: 10.1021/ja1033167
Choy, P. Y., Lau, C. P., and Kwong, F. Y. (2011). Palladium-catalyzed direct and regioselective C–H bond functionalization/oxidative acetoxylation of indoles. J. Org. Chem. 76, 80–84. doi: 10.1021/jo101584k
Cook, A. K., Emmert, M. H., and Sanford, M. S. (2013). Steric control of site selectivity in the Pd-catalyzed C–H acetoxylation of simple arenes. Org. Lett. 15, 5428–5431. doi: 10.1021/ol4024248
Deprez, N. R., and Sanford, M. S. (2007). Reactions of hypervalent iodine reagents with palladium: Mechanisms and applications in organic synthesis. Inorg. Chem. 46, 1924–1935. doi: 10.1021/ic0620337
Desai, L. V., and Sanford, M. S. (2007). Construction of tetrahydrofurans by PdII/PdIV-catalyzed aminooxygenation of alkenes. Angew. Chem. Int. Ed. 46, 5737–5740. doi: 10.1002/anie.200701454
Dong, D.-Q., Hao, S.-H., Wang, Z.-L., and Chen, C. (2014). Hypervalent iodine: a powerful electrophile for asymmetric α-functionalization of carbonyl compounds. Org. Biomol. Chem. 12, 4278–4289. doi: 10.1039/c4ob00318g
Emmert, M. H., Cook, A. K., Xie, Y. J., and Sanford, M. S. (2011). Remarkably high reactivity of Pd(OAc)2/pyridine catalysts: nondirected C–H oxygenation of arenes. Angew. Chem. Int. Ed. 50, 9409–9412. doi: 10.1002/anie.201103327
Evdokimov, N. M., Kornienko, A., and Magedov, I. V. (2011). Aryliodine(III) diacetates as substrates for Pd–Ag catalyzed arylation of alkenes. Tetrahedron Lett. 52, 4327–4329. doi: 10.1016/j.tetlet.2011.06.051
Fu, Z., Xiong, Q., Zhang, W., Li, Z., and Cai, H. (2015). Pd-catalyzed direct arylation of electron-deficient polyfluoroarenes with aryliodine(III) diacetates. Tetrahedron Lett. 56, 123–126. doi: 10.1016/j.tetlet.2014.11.033
Fujino, D., Yorimitsu, H., and Oshima, K. (2010). Palladium-catalyzed intramolecular carboacetoxylation of 4-pentenyl-substituted malonate esters with iodobenzene diacetate. Chem. Asian J. 5, 1758–1760. doi: 10.1002/asia.201000194
Gondo, K., Oyamada, J., and Kitamura, T. (2015). Palladium-catalyzed desilylative acyloxylation of silicon–carbon bonds on (trimethylsilyl)arenes: synthesis of phenol derivatives from trimethylsilylarenes. Org. Lett. 17, 4778–4781. doi: 10.1021/acs.orglett.5b02336
Gou, F.-R., Wang, X.-C., Huo, P.-F., Bi, H.-P., Guan, Z.-H., and Liang, Y.-M. (2009). Palladium-catalyzed aryl C–H bonds activation/acetoxylation utilizing a bidentate system. Org. Lett. 11, 5726–5729. doi: 10.1021/ol902497k
Gu, S., Chen, C., and Chen, W. (2009). Ortho-functionalization of 2-phenoxypyrimidines via palladium-catalyzed C–H bond activation. J. Org. Chem. 74, 7203–7206. doi: 10.1021/jo901316b
Gulevich, A. V., Melkonyan, F. S., Sarkar, D., and Gevorgyan, V. (2012). Double-Fold C–H oxygenation of arenes using PyrDipSi: a general and efficient traceless/modifiable silicon-tethered directing group. J. Am. Chem. Soc. 134, 5528–5531. doi: 10.1021/ja3010545
He, G., Lu, C., Zhao, Y., Nack, W. A., and Chen, G. (2012b). Improved protocol for indoline synthesis via palladium-catalyzed intramolecular C(sp2)–H amination. Org. Lett. 14, 2944–2947. doi: 10.1021/ol301352v
He, G., Zhao, Y., Zhang, S., Lu, C., and Chen, G. (2012a). Highly efficient syntheses of azetidines, pyrrolidines, and indolines via palladium catalyzed intramolecular amination of C(sp3)–H and C(sp2)–H bonds at γ and δ positions. J. Am. Chem. Soc. 134, 3–6. doi: 10.1021/ja210660g
He, Y., Huang, L., Xie, L., Liu, P., Wei, Q., Mao, F., et al. (2019). Palladium-catalyzed C–H bond functionalization reactions using phosphate/sulfonate hypervalent iodine reagents. J. Org. Chem. 84, 10088–10101. doi: 10.1021/acs.joc.9b01278
Huang, C., Ghavtadze, N., Chattopadhyay, B., and Gevorgyan, V. (2011). Synthesis of catechols from phenols via Pd-catalyzed silanol-directed C–H oxygenation. J. Am. Chem. Soc. 133, 17630–17633. doi: 10.1021/ja208572v
Huang, C., Ghavtadze, N., Godoi, B., and Gevorgyan, V. (2012). Pd-catalyzed modifiable silanol-directed aromatic C–H oxygenation. Chem. Eur. J. 18, 9789–9792. doi: 10.1002/chem.201201616
Iglesias, A., Perez, E. G., and Muniz, K. (2010). An intermolecular palladium-catalyzed diamination of unactivated alkenes. Angew. Chem. Int. Ed. 49, 8109–8111. doi: 10.1002/anie.201003653
Ilchenko, N. O., Cortes, M. A., and Szabo, K. J. (2016). Palladium-catalyzed iodofluorination of alkenes using fluoro-iodoxole reagent. ACS Catal. 6, 447–450. doi: 10.1021/acscatal.5b02022
Jaegli, S., Dufour, J., Wei, H., Piou, T., Duan, X., Vors, J., et al. (2010). Palladium-catalyzed carbo-heterofunctionalization of alkenes for the synthesis of oxindoles and spirooxindoles. Org. Lett. 12, 4498–4501. doi: 10.1021/ol101778c
Jiang, T., Quan, X., Zhu, C., Andersson, P. G., and Backvall, J.-E. (2016). Palladium-catalyzed oxidative synthesis of α. Palladium-catnones from alkynes. Angew. Chem. Int. Ed. 55, 5824–5828. doi: 10.1002/anie.201600696
Jordan-Hore, J. A., Johansson, C. C. C., Gulias, M., Beck, E. M., and Gaunt, M. J. (2008). Oxidative Pd(II)-catalyzed C–H bond amination to carbazole at ambient temperature. J. Am. Chem. Soc. 130, 16184–16186. doi: 10.1021/ja806543s
Ju, L., Yao, J., Wu, Z., Liu, Z., and Zhang, Y. (2013). Palladium-catalyzed oxidative acetoxylation of benzylic C–H bond using bidentate auxiliary. J. Org. Chem. 78, 10821–10831. doi: 10.1021/jo401830k
Koposov, A. Y., Litvinov, D. N., Zhdankin, V. V., Ferguson, M. J., McDonald, R., Tykwinski, R. R., et al. (2006). Preparation and reductive decomposition of 2-iodoxybenzenesulfonic acid. X-ray crystal structure of 1-hydroxy-1H-1, 2, 3-benziodoxathiole 3, 3-dioxide. Eur. J. Org. Chem. 2006, 4791–4795. doi: 10.1002/ejoc.200600683
Larsson, J. M., Zhao, T. S. N., and Szabo, K. J. (2011). Palladium-catalyzed oxidative allylic C–H silylation. Org. Lett. 13, 1888–1891. doi: 10.1021/ol200445b
Lee, J. H., Choi, S., and Hong, K. B. (2019). Alkene difunctionalization using hypervalent iodine reagents: progress and developments in the past ten years. Molecules 24, 2634–2657. doi: 10.3390/molecules24142634
Li, M., Yu, F., Qi, X., Chen, P., and Liu, G. (2016). A cooperative strategy for the highly selective intermolecular oxycarbonylation reaction of alkenes using a palladium catalyst. Angew. Chem. Int. Ed. 55, 13843–13848. doi: 10.1002/anie.201607248
Li, Q., Zhang, S.-Y., He, G., Nack, W. A., and Chen, G. (2014). Palladium-catalyzed picolinamide-directed acetoxylation of unactivated γ. Pap3)–H bonds of alkylamines. Adv. Synth. Catal. 356, 1544–1548. doi: 10.1002/adsc.201400121
Li, X., Chen, P., and Liu, G. (2018). Recent advances in hypervalent iodine(III)-catalyzed functionalization of alkenes. Beilstein J. Org. Chem. 14, 1813–1825. doi: 10.3762/bjoc.14.154
Li, Y., Ding, Y.-J., Wang, J.-Y., Su, Y.-M., and Wang, X.-S. (2013). Pd-catalyzed C–H lactonization for expedient synthesis of biaryl lactones and total synthesis of cannabinol. Org. Lett. 15, 2574–2577. doi: 10.1021/ol400877q
Li, Y., Hari, D. P., Vita, M. V., and Waser, J. (2016). Cyclic hypervalent iodine reagents for atom-transfer reactions: beyond trifluoromethylation. Angew. Chem. Int. Ed. 55, 4436–4454. doi: 10.1002/anie.201509073
Li, Y., Song, D., and Dong, V. M. (2008). Palladium-catalyzed olefin dioxygenation. J. Am. Chem. Soc. 130, 2962–2964. doi: 10.1021/ja711029u
Liu, B., and Shi, B.-F. (2016). γ-Lactone synthesis via palladium(II)-catalyzed lactonization of unactivated methylene C(sp3)–H bonds. Synlett 27, 2396–2400. doi: 10.1055/s-0035-1562508
Liu, G., and Stahl, S. S. (2006). Highly regioselective Pd-catalyzed intermolecular aminoacetoxylation of alkenes and evidence for cis-aminopalladation and SN2 C–O bond formation. J. Am. Chem. Soc. 128, 7179–7181. doi: 10.1002/chin.200641044
Liu, H., Yu, J., Wang, L., and Tong, X. (2008). Cyclization–oxidation of 1,6-enyne derivatived from baylis–hillman adducts via Pd(II)/Pd(IV)-catalyzed reactions: stereoselective synthesis of multi-substituted bicyclo[3.1.0] hexanes and insight into reaction pathways. Tetrahedron Lett. 49, 6924–6928. doi: 10.1016/j.tetlet.2008.09.108
Liu, Q., Li, G., Yi, H., Wu, P., Liu, J., and Lei, A. (2011). Pd-catalyzed direct and selective C–H functionalization: C3-acetoxylation of indoles. Chem. Eur. J. 17, 2353–2357. doi: 10.1002/chem.201002547
Lyons, T. W., and Sanford, M. S. (2009). Palladium (II/IV) catalyzed cyclopropanation reactions: scope and mechanism. Tetrahedron 65, 3211–3221. doi: 10.1016/j.tet.2008.10.107
Maertens, G., and Canesi, S. (2015). 'Rearrangements induced by hypervalent iodine. Topics Curr. Chem. 373, 223–241. doi: 10.1007/128_2015_657
Mangaonkar, S. R., Kole, P. B., and Singh, F. V. (2018). Oxidation of organosulfides to organosulfones with trifluoromethyl 3-Oxo-1λ3,2-benziodoxole-1(3H)-carboxylate as an oxidant. Synlett 18, 199–202. doi: 10.1055/s-0036-1588575
Mangaonkar, S. R., and Singh, F. V. (2019). Hypervalent iodine(III)-catalyzed epoxidation of β-cyanostyrenes. Synthesis 51, 4473–4486. doi: 10.1055/s-0039-1690621
Martinez, C., and Muniz, K. (2012). Palladium-catalyzed vicinal difunctionalization of internal alkenes: diastereoselective synthesis of diamines. Angew. Chem. Int. Ed. 51, 7031–7034. doi: 10.1002/anie.201201719
Martinez, C., and Muniz, K. (2016). Defined palladium–phthalimidato catalysts for improved oxidative amination. Chem. Eur. J. 22, 7367–7370. doi: 10.1002/chem.201601128
Martinez, C., Perez, E. G., Iglesias, A., Escudero-Adan, E. C., and Muniz, K. (2016). Regioselective intermolecular diamination and aminooxygenation of alkenes with saccharin. Org. Lett. 18, 2998–3001. doi: 10.1021/acs.orglett.6b01368
Martinez, C., Wu, Y., Weinstein, A. B., Stahl, S. S., Liu, G., and Muniz, K. (2013). Palladium-catalyzed intermolecular aminoacetoxylation of alkenes and the influence of PhI(OAc)2 on aminopalladation stereoselectivity. J. Org. Chem. 78, 6309–6315. doi: 10.1021/jo400671q
McMurtrey, K. B., Racowski, J. M., and Sanford, M. S. (2012). Pd-catalyzed C–H fluorination with nucleophilic fluoride. Org. Lett. 14, 4094–4097. doi: 10.1021/ol301739f
Mei, T., Leow, D., Xiao, H., Laforteza, B. N., and Yu, J. (2013). Synthesis of indolines via Pd(II)-catalyzed amination of C–H bonds using PhI(OAc)2 as the bystanding oxidant. Org. Lett. 15, 3058–3061. doi: 10.1021/ol401246u
Merritt, E. A., and Olofsson, B. (2011). α-Functionalization of carbonyl compounds using hypervalent iodine reagents. Synthesis 4, 517–538. doi: 10.1055/s-0030-1258328
Merritt, E. A., and Olofsson, B. O. (2009). Diaryliodonium salts: journey from obscurity to fame. Angew. Chem. Int. Ed. 48, 9052–9070. doi: 10.1002/anie.200904689
Mu, X., Chen, S., Zhen, X., and Liu, G. (2011). Palladium-catalyzed oxidative trifluoromethylation of indoles at room temperature. Chem. Eur. J. 17, 6039–6042. doi: 10.1002/chem.201100283
Mu, X., Wu, T., Wang, H., Guo, Y.-L., and Liu, G. (2012). Palladium-catalyzed oxidative aryltrifluoromethylation of activated alkenes at room temperature. J. Am. Chem. Soc. 134, 878–881. doi: 10.1021/ja210614y
Muniz, K. (2007). Advancing palladium-catalyzed C–N bond formation: bisindoline construction from successive amide transfer to internal alkenes. J. Am. Chem. Soc. 129, 14542–14543. doi: 10.1021/ja075655f
Muniz, K., Hovelmann, C. H., and Streuff, J. (2008). Oxidative diamination of alkenes with ureas as nitrogen sources: mechanistic pathways in the presence of a high oxidation state palladium catalyst. J. Am. Chem. Soc. 130, 763–773. doi: 10.1021/ja075041a
Muniz, K., Kirsch, J., and Chavez, P. (2011). Intermolecular regioselective 1,2-diamination of allylic ethers. Adv. Synth. Catal. 353, 689–694. doi: 10.1002/adsc.201000813
Mutule, I., Suna, E., Olofsson, K., and Pelcman, B. (2009). Catalytic direct acetoxylation of indoles. J. Org. Chem. 74, 7195–7198. doi: 10.1021/jo901321b
Nadres, E. T., and Daugulis, O. (2012). Heterocycle Synthesis via direct C–H/N–H coupling. J. Am. Chem. Soc. 134, 7–10. doi: 10.1021/ja210959p
Neufeldt, S. R., and Sanford, M. S. (2010). O-Acetyl oximes as transformable directing groups for Pd-catalyzed C–H bond functionalization. Org. Lett. 12, 532–535. doi: 10.1021/ol902720d
Neufeldt, S. R., and Sanford, M. S. (2013). Asymmetric chiral ligand-directed alkene dioxygenation. Org. Lett. 15, 46–49. doi: 10.1021/ol303003g
Nicolai, S., Erard, S., Gonzalez, D. F., and Waser, J. (2010). Pd-catalyzed intramolecular oxyalkynylation of alkenes with hypervalent iodine. Org. Lett. 12, 384–387. doi: 10.1021/ol9027286
Nicolai, S., Piemontesi, C., and Waser, J. (2011). A palladium-catalyzed aminoalkynylation strategy towards bicyclic heterocycles: synthesis of (±)-trachelanthamidine. Angew. Chem. Int. Ed. 50, 4680–4683. doi: 10.1002/anie.201100718
Pilarski, L. T., Selander, N., Bose, D., and Szabo, K. J. (2009). Catalytic allylic C–H acetoxylation and benzoyloxylation via suggested (η3-allyl)palladium(IV) intermediates. Org. Lett. 11, 5518–5521. doi: 10.1021/ol9023369
Qu, X., Sun, P., Li, T., and Mao, J. (2011). Ligand-free reusable palladium-catalyzed Heck-type coupling reactions of hypervalent iodine reagents under mild conditions. Adv. Synth. Catal. 353, 1061–1066. doi: 10.1002/adsc.201100008
Ren, Z., Mo, F., and Dong, G. (2012). Catalytic functionalization of unactivated sp3 C–H bonds via exo-directing groups: synthesis of chemically differentiated 1,2-diols. J. Am. Chem. Soc. 134, 16991–16994. doi: 10.1021/ja3082186
Ren, Z., Schulz, J. E., and Dong, G. (2015). Catalytic ortho-acetoxylation of masked benzyl alcohols via an exo-directing mode. Org. Lett. 17, 2696–2699. doi: 10.1021/acs.orglett.5b01098
Richardson, R. D., Zayed, J. M., Altermann, S., Smith, D., and Wirth, T. (2007). Tetrafluoro-IBA and-IBX: hypervalent iodine reagents. Angew. Chem. Int. Ed. 46, 6529–6532. doi: 10.1002/anie.200702313
Rit, R. K., Yadav, M. R., and Sahoo, A. K. (2012). Pd(II)-catalyzed primary-C(sp3)–H acyloxylation at room temperature. Org. Lett. 14, 3724–3727. doi: 10.1021/ol301579q
Rodriguez, A., and Moran, W. J. (2009). Palladium-catalyzed three-component coupling reactions: 1,1-difunctionalization of activated alkenes. Eur. J. Org. Chem. 2009, 1313–1316. doi: 10.1002/ejoc.200801245
Satterfield, A. D., Kubota, A., and Sanford, M. S. (2011). Palladium-catalyzed 1,1-aryloxygenation of terminal olefins. Org. Lett. 13, 1076–1079. doi: 10.1021/ol103121r
Selander, N., Willy, B., and Szabo, K. J. (2010). Selective C–H borylation of alkenes by palladium pincer complex catalyzed oxidative functionalization. Angew. Chem. Int. Ed. 49, 4051–4053. doi: 10.1002/anie.201000690
Shan, G., Yang, X., Zong, Y., and Rao, Y. (2013). An efficient palladium-catalyzed C–H alkoxylation of unactivated methylene and methyl groups with cyclic hypervalent iodine (I3+) oxidants. Angew. Chem. Int. Ed. 52, 13606–13610. doi: 10.1002/anie.201307090
Shrestha, R., Mukherjee, P., Tan, Y., Litman, Z. C., and Hartwig, J. F. (2013). Sterically controlled, palladium-catalyzed intermolecular amination of arenes. J. Am. Chem. Soc. 135, 8480–8483. doi: 10.1021/ja4032677
Silva, L. F. Jr. (2002). Construction of cyclopentyl units by ring contraction reactions. Tetrahedron 58, 9137–9161. doi: 10.1016/S0040-4020(02)00990-0
Silva, F. C. S., Tierno, A. F., and Wengryniuk, S. E. (2017). Hypervalent iodine reagents in high valent transition metal chemistry. Molecules 22:780. doi: 10.3390/molecules22050780
Singh, F. V., Kole, P. B., Mangaonkar, S. R., and Shetgaonkar, S. E. (2018a). Synthesis of spirocyclic scaffolds using hypervalent iodine reagents. Beilstein J. Org. Chem. 14, 1778–1805. doi: 10.3762/bjoc.14.152
Singh, F. V., and Mangaonkar, S. R. (2018). Hypervalent iodine(III)-catalyzed synthesis of 2-arylbenzofurans. Synthesis 50, 4940–4948. doi: 10.1055/s-0037-1610650
Singh, F. V., Mangaonkar, S. R., and Kole, P. B. (2018b). Ultrasound-assisted rapid synthesis of β-cyanoepoxides using hypervalent iodine reagents. Synth. Commun. 48, 2169–2176. doi: 10.1080/00397911.2018.1479760
Singh, F. V., Rehbein, J., and Wirth, T. (2012). Facile oxidative rearrangements using hypervalent iodine reagents. ChemistryOpen 1, 245–250. doi: 10.1002/open.201200037
Singh, F. V., and Wirth, T. (2011). Selenium-catalyzed regioselective cyclization of unsaturated carboxylic acids using hypervalent iodine oxidants. Org. Lett. 13, 6504–6507. doi: 10.1021/ol202800k
Singh, F. V., and Wirth, T. (2012). Hypervalent iodine(III) mediated cyclization of ortho-stillbenes into benzofurans. Synthesis 44, 1171–1177. doi: 10.1055/s-0031-1290588
Singh, F. V., and Wirth, T. (2013). Oxidative rearrangements with hypervalent iodine reagents. Synthesis 45, 2499–2511. doi: 10.1055/s-0033-1339679
Singh, F. V., and Wirth, T. (2014a). “Oxidative functionalization with hypervalent halides,” in Comprehensive Organic Synthesis II, eds G. A. Molander and P. Knochel (Oxford: Elsevier), 880–933.
Singh, F. V., and Wirth, T. (2014b). Hypervalent iodine-catalyzed oxidative functionalizations including stereoselective reactions. Chem. Asian J. 9, 950–971. doi: 10.1002/asia.201301582
Singh, F. V., and Wirth, T. (2017). Catalytic oxidation with hypervalent iodine. Catal. Oxidat. Organic Synthes. 1, 29–62.
Singh, F. V., and Wirth, T. (2018). “Stereoselective reactions,” in Patai's Chemistry of Functional Groups, eds I. Marek, B. Olofsson, and Z. Rappoport (Chichester: John Wiley & Sons, Ltd).
Streuff, J., Hovelmann, C. H., Nieger, M., and Muniz, K. (2005). Palladium(II)-catalyzed intramolecular diamination of unfunctionalized alkenes. J. Am. Chem. Soc. 127, 14586. doi: 10.1021/ja055190y
Sun, X., Yao, X., Zhang, C., and Rao, Y. (2015). Pd(II) catalyzed ortho C–H iodination of phenylcarbamates at room temperature using cyclic hypervalent iodine reagents. Chem. Commun. 51, 10014–10017. doi: 10.1039/C5CC02533H
Tang, S., Peng, P., Pi, S., Liang, Y., Wang, N., and Li, J. (2008b). Sequential intermolecular aminopalladation/ortho-arene C-H activation reactions of N-phenylpropiolamides with phthalimide. Org. Lett. 10, 1179–1182. doi: 10.1021/ol800080w
Tang, S., Peng, P., Wang, Z., Tang, B., Deng, C., Li, J., et al. (2008c). Synthesis of (2-oxoindolin-3-ylidene)methyl acetates involving a C–H functionalization process. Org. Lett. 10, 1875–1878. doi: 10.1021/ol8006315
Tang, S., Peng, P., Zhong, P., and Li, J.-H. (2008a). Palladium-catalyzed C–H functionalization of N-arylpropiolamides with aryliodonium salts: selective synthesis of 3-(1-arylmethylene)oxindoles. J. Org. Chem. 73, 5476–5480. doi: 10.1021/jo8008808
Thompson, S. J., Thach, D. Q., and Dong, G. (2015). Cyclic ether synthesis via palladium-catalyzed directed dehydrogenative annulation at unactivated terminal positions. J. Am. Chem. Soc. 137, 11586–11589. doi: 10.1021/jacs.5b07384
Tohma, H., and Kita, Y. (2004). Hypervalent iodine reagents for the oxidation of alcohols and their application to complex molecule synthesis. Adv. Synth. Catal. 346, 111–124. doi: 10.1002/adsc.200303203
Tolnai, G. L., Ganss, S., Brand, J. P., and Waser, J. (2013). C2-selective direct alkynylation of indoles. Org. Lett. 15, 112–115. doi: 10.1021/ol3031389
Tong, X., Beller, M., and Tse, M. K. (2007). A Palladium-catalyzed cyclization-oxidation sequence: synthesis of bicyclo[3.1.0]hexanes and evidence for SN2 C–O bond formation. J. Am. Chem. Soc. 129, 4906–4907. doi: 10.1021/ja070919j
Tsujihara, T., Takenaka, K., Onitsuka, K., Hatanaka, M., and Sasai, H. (2009). PdII/PdIV Catalytic enantioselective synthesis of bicyclo[3.1.0]hexanes via oxidative cyclization of enynes. J. Am. Chem. Soc. 131, 3452–3453. doi: 10.1021/ja809965e
Uyanik, M., and Ishihara, K. (2009). Hypervalent iodine-mediated oxidation of alcohols. Chem. Commun. 2009, 2086–2099. doi: 10.1039/b823399c
Wang, W., Wang, F., and Shi, M. (2010). Bis(NHC)-palladium(II) complex-catalyzed dioxygenation of alkenes. Organometallics 29, 928–933. doi: 10.1021/om900975a
Wang, X., He, Y., Ren, M., Liu, S., Liu, H., and Huang, G. (2016). Pd-Catalyzed ligand-free synthesis of arylated heteroaromatics by coupling of N-heteroaromatic bromides with iodobenzene diacetate, iodosobenzene, or diphenyliodonium salts. J. Org. Chem. 81, 7958–7962. doi: 10.1021/acs.joc.6b01103
Wang, X., Lu, Y., Dai, H.-X., and Yu, J.-Q. (2010). Pd(II)-catalyzed hydroxyl-directed C-H activation/C-O cyclization: expedient construction of dihydrobenzofurans. J. Am. Chem. Soc. 132, 12203–12205. doi: 10.1021/ja105366u
Welbes, L. L., Lyons, T. W., Cychosz, K. A., and Sanford, M. S. (2007). Synthesis of cyclopropanes via Pd(II/IV)-catalyzed reactions of enynes. J. Am. Chem. Soc. 129, 5836–5837. doi: 10.1021/ja071204j
Wirth, T. (2005). Hypervalent iodine chemistry in synthesis: scope and new directions. Angew. Chem. Int. Ed. 44, 3656–3665. doi: 10.1002/anie.200500115
Wirth, T., Ochiai, M., Zhdankin, V. V., Koser, G. F., Tohma, H., Kita, Y., et al. (2002). Topics in Current Chemistry: Hypervalent Iodine Chemistry- Modern Developments in Organic Synthesis. Berlin: Spinger-Verlag.
Wu, T., Yin, G., and Liu, G. (2009). Palladium-catalyzed intramolecular aminofluorination of unactivated alkenes. J. Am. Chem. Soc. 131, 16354–16355. doi: 10.1021/ja9076588
Xiong, Q., Fu, Z., Li, Z., and Cai, H. (2015). Synthesis of symmetrical biaryls through palladium-catalyzed ligand-free homocoupling of aryliodine(III) diacetates. Synlett 26, 975–979. doi: 10.1055/s-0034-1380320
Xu, T., Wang, D., and Tong, X. (2019). Pd(II)-catalyzed intramolecular acetoxylative (3 + 2) annulation of propargylic alcohol and alkene: Polycyclic oxa-heterocycle synthesis and mechanistic insight. Org. Lett. 21, 5368–5372. doi: 10.1021/acs.orglett.9b02096
Yang, M., Jiang, X., Shi, W.-J., Zhu, Q.-L., and Shi, Z.-J. (2013). Direct lactonization of 2-arylacetic acids through Pd(II)-catalyzed C–H activation/C–O formation. Org. Lett. 15, 690–693. doi: 10.1021/ol303569b
Ye, X., He, Z., Ahmed, T., Weise, K., Akhmedov, N. G., Petersen, J. L., et al. (2013). 1,2,3-triazoles as versatile directing group for selective sp2 and sp3 C–H activation: cyclization vs substitution. Chem. Sci. 4, 3712–3716. doi: 10.1039/c3sc51211h
Yin, G., Wu, Y., and Liu, G. (2010). Scope and mechanism of allylic C–H amination of terminal alkenes by the palladium/PhI(OPiv)2 catalyst system: Insights into the effect of naphthoquinone. J. Am. Chem. Soc. 132, 11978–11987. doi: 10.1021/ja1030936
Yin, Z., Jiang, X., and Sun, P. (2013). Palladium-catalyzed direct ortho alkoxylation of aromatic azo compounds with alcohols. J. Org. Chem. 78, 10002–10007. doi: 10.1021/jo401623j
Yoshimura, A., and Zhdankin, V. V. (2016). Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 116, 3328–3435. doi: 10.1021/acs.chemrev.5b00547
Yu, P., Zhang, G., Chen, F., and Cheng, J. (2012). Direct arylation of benzoxazole C–H bonds with iodobenzene diacetates. Tetrahedron Lett. 53 4588–4590. doi: 10.1016/j.tetlet.2012.06.076
Yusubov, M. S., and Zhdankin, V. V. (2012). Hypervalent iodine reagents and green chemistry. Curr. Org. Synth. 9, 247–272. doi: 10.2174/157017912799829021
Zhang, C., and Sun, P. (2014). Palladium-catalyzed direct C(sp2)–H alkoxylation of 2-aryloxypyridines using 2-pyridyloxyl as the directing group. J. Org. Chem. 79, 8457–8461. doi: 10.1021/jo5014146
Zhang, Q., Wang, Y., Yang, T., Li, L., and Li, D. (2015). Palladium catalyzed ortho-C–H-benzoxylation of 2-arylpyridines using iodobenzene dibenzoates. Tetrahedron Lett. 56, 6136–6141. doi: 10.1016/j.tetlet.2015.09.097
Zhang, S., Luo, F., Wang, W., Jia, X., Hu, M., and Cheng, J. (2010). Chelation-assisted palladium-catalyzed acyloxylation of benzyl sp3 C–H bonds using PhI(OAc)2 as oxidant. Tetrahedron Lett. 51, 3317–3319. doi: 10.1016/j.tetlet.2010.04.075
Zhang, S.-Y., He, G., Zhao, Y., Wright, K., Nack, W. A., and Chen, G. (2012). Efficient alkyl ether synthesis via palladium-catalyzed, picolinamide-directed alkoxylation of unactivated C(sp3)–H and C(sp2)–H bonds at remote positions. J. Am. Chem. Soc. 134, 7313–7316. doi: 10.1021/ja3023972
Zhdankin, V. V. (2009). Hypervalent iodine(III) reagents in organic synthesis. ARKIVOC 2009, 1–62. doi: 10.3998/ark.5550190.0010.101
Zhdankin, V. V. (2014). Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Application of Polyvalent Iodine Compounds. Chichester: John Wiley & Sons Ltd. doi: 10.1002/9781118341155
Keywords: hypervalent iodine reagents, palladium, oxidant, catalyst, bond formation
Citation: Shetgaonkar SE and Singh FV (2020) Hypervalent Iodine Reagents in Palladium-Catalyzed Oxidative Cross-Coupling Reactions. Front. Chem. 8:705. doi: 10.3389/fchem.2020.00705
Received: 24 April 2020; Accepted: 09 July 2020;
Published: 29 September 2020.
Edited by:
Ravi Kumar, J.C. Bose University of Science and Technology, YMCA, IndiaReviewed by:
Michal Szostak, Rutgers University, Newark, United StatesEgle Maria Beccalli, University of Milan, Italy
Copyright © 2020 Shetgaonkar and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fateh V. Singh, ZmF0ZWh2ZWVyLnNpbmdoQHZpdC5hYy5pbg==