 Elisabetta Di Bello
Elisabetta Di Bello Clemens Zwergel
Clemens Zwergel Antonello Mai
Antonello Mai Sergio Valente
Sergio Valente
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Chem. , 18 June 2020
Sec. Medicinal and Pharmaceutical Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00516
This article is part of the Research Topic Chemical Innovative Approaches in Cancer Molecular Medicine and Translational Clinical Research View all 8 articles
Numerous and different types of cancers possess the dysregulation of the mevalonate pathway as a common feature. Statins, traditionally applied in cardiovascular diseases to reduce lipid levels, subsequently have been discovered to exhibit anti-cancer activities also. Indeed, statins influence proliferation, migration, and survival of cancer cells by regulating crucial signaling proteins, such as Rho, Ras, and Rac. Recently, several studies have demonstrated that simvastatin, fluvastatin, and lovastatin are implicated in different pathways that enhance the survival time of patients with cancer under treatment in combination with antineoplastic agents. In this minireview, we present an overview of the most important studies conducted regarding the use of statins in cancer therapy up to date.
To date, there are a lot of types of cancers described in the literature, with many associated treatments in constant evolution. In many forms of cancer, a dysregulation of the lipid metabolism (Fritz et al., 2013) and the mevalonate pathway (MVP) is observed (Freed-Pastor et al., 2012). Cholesterol is an indispensable component of cell membranes, and it is a precursor of bile acids, lipoproteins, and steroid hormones. Its biosynthesis is controlled by the MVP, which controls protein farnesylation and geranylation. These post-translational modifications are critical for the downstream signaling activity of Ras, Rho, or Rac proteins, that are part of the small GTPases superfamily (Takai et al., 2001), involved in tumorigenesis, progression (Buhaescu and Izzedine, 2007), proliferation, migration, and survival of tumor cells (Kidera et al., 2010). Like healthy cells, also cancer cells esterify fatty acids in phospholipids that are essential cell membrane components. These necessary lipids are obtained by the endogenous metabolites deriving from the MVP (Notarnicola et al., 2014). Inhibiting this vital process could be beneficial in cancer cells as they are usually rapidly proliferating without affecting too much healthy cells with a slower reproduction rate. Statins are able to decrease lipid levels in the plasma by inhibiting HMG-CoA reductase (HMGCR). Several studies have shown an intense correlation between the use of statins and cancer. First promising studies showed that statins are able to improve the outcome in cancer, i.e., prolonged the survival time (Gupta et al., 2019). In this minireview, we would like to illustrate new perspectives and innovative targets regarding the use of statins, such as simvastatin, fluvastatin, and lovastatin, in cancer alone or in combination with other drugs.
The first statin, mevastatin, was identified and isolated by Endo (2004), being the first cholesterol-lowering drug. Today statins are classified into two groups: type-I derivatives are derived from fermentation products, such as mevastatin, lovastatin, pravastatin, and simvastatin. Type-II statins, including fluvastatin, atorvastatin, cerivastatin, pitavastatin, and rosuvastatin, are drugs of synthetic origin (Oryan et al., 2015). Statins are characterized by a characteristic pharmacophore group either being a lactone ring as a prodrug or a long chain carboxylic acid as an active form (red moiety in Figure 1A), responsible for the inhibiting activity, and a ring system moiety that is different for each type (blue moiety in Figure 1A) (Gazzerro et al., 2012). Lovastatin and simvastatin are lactones, thus closed ring prodrugs, which are transformed into their active open forms in the body; other statins are administered orally as active, opened rings, forming hydroxyl acids (Corsini et al., 1995) as their inhibitory effect on HMGCR is heavily depending on their solubility. However, one of the major limitations restricting the application of statins lies, for some of them, in the low bioavailability often below 50%. To improve bioavailability, nanocarriers loaded with statins, such as micelles, nanocrystals, and lipid-based nanoparticles (NPs), allowing a better drug uptake in the gastrointestinal tract, have been studied with promising results (Korani et al., 2019).
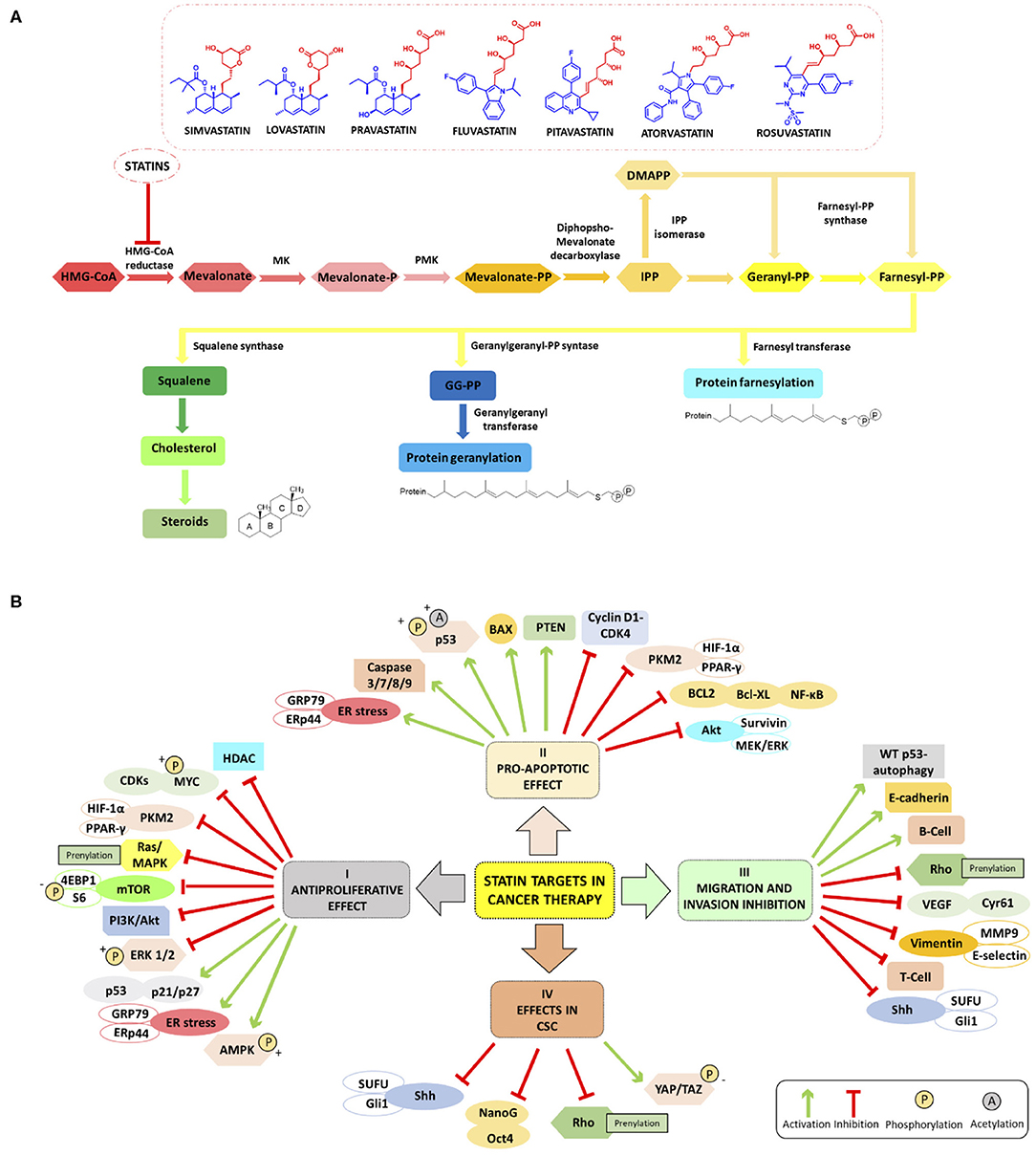
Figure 1. (A) Schematic summary of Mevalonate Pathway (MVP) and chemical structure of HMGCR inhibitors mainly studied in cancer (simvastatin, lovastatin, pravastatin, fluvastatin, pitavastatin, atorvastatin and rosuvastatin); the pharmacophore moiety in red and the ring system moiety in blue). (B) Overview of the main molecular targets of statins in cancer. I Antiproliferative effect: statins inhibit the proliferation of cancer cell increasing ER stress markers GRP78 and ERp44, phosphorylating S6 and 4EBP1 and inhibiting mTOR pathway (Okubo et al., 2020), and inhibiting HDACs (Lin et al., 2008; Chou et al., 2019). Statins involve LKB1-AMPK-p38MAPK-p53 survivin signaling cascade thanks to the phosphorylation and acetylation of p53 in cancer cells (Huang et al., 2020). II Pro-apoptotic effect: statins enhance apoptosis through inhibition of survivin protein expression (see above) (Huang et al., 2020), by inhibiting HIF-1α/PPAR-γ/PKM2-mediated glycolysis (Feng et al., 2020), induce caspase 3/7/8/9 protease, decrease the expression of cyclin D1 and CDK4 Bcl-XL (Okubo et al., 2020), and block the transcription factors NF-κB (Lee et al., 2014; Branvall et al., 2020). III Migration and invasion inhibition: statins induce T-cell suppression and B-cell survival (Branvall et al., 2020), and enhance the WT p53-dependent autophagy (Chou et al., 2019). IV Effects in CSC: statins can reduce the expression of Sonic Hedgehog (Shh) and consequently, the migration and invasion of cancer cells (Yin et al., 2018). Other cancerogenic pathways are reviewed in Iannelli et al. (2018).
As already mentioned, statins are traditionally used for the reduction of lipid profiles in cardiovascular diseases (Goldstein and Brown, 2015). Mechanistically, statins inhibit HMGCR, and the depletion of intracellular cholesterol levels induces the expression of LDL receptors that decrease circulating LDL cholesterol in plasma, resulting in a decreased biosynthesis and increased cholesterol catabolism (Schonewille et al., 2016). For dyslipidemia, red yeast rice is used as a treatment option in traditional Chinese medicine as it contains a variable percentage of monacolin K which possesses actually the same chemical structure of lovastatin. However, red yeast rice, in lay literature often entitled as a superfood, should not be considered especially in cancer, as the monacolin K levels are not standardized. Besides the dosage problem, often carcinogens or other harmful by-products are found in this modified fermented rice, which make this “food” a risky choice (Dujovne, 2017). Statins have gained much attention for the prevention and treatment of cancer as they are capable to inhibit inflammation, angiogenesis, and proliferation and to induce apoptosis (Demierre et al., 2005). The antitumor effect of statins in monotherapy or in combination with anti-cancer agents is associated with cancer-related survival (Pisanti et al., 2014; Vallianou et al., 2014). Figure 1B summarizes the main molecular targets of statins in cancer. In the following paragraphs, we will give an overview of the main studies reported to date in the literature and the most innovative drug combinations that include both statins (simvastatin, lovastatin, pravastatin, fluvastatin, pitavastatin, atorvastatin, and rosuvastatin) and many types of anti-cancer agents endowed with antiproliferative, proapoptotic effects also able to impair migration and invasion processes in several cancers including cancer stem cells (CSCs).
The rather lipophilic simvastatin has been shown in population-based cohort study to be more capable to reduct cancer-specific mortality than other hydrophilic statins. In a previous study, comparing atorvastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin, and simvastatin, the latter one gave the best results regarding patients' survival (Cardwell et al., 2015). Recently, Chou et al. have revealed the role of statins in lung cancer. First, they retrospectively analyzed patients diagnosed with lung cancer between 1998 and 2011, and found a high percentage of patients with p53 mutations observing a reduced 5-years mortality under simvastatin treatment in this population-based study (Chou et al., 2019). The p53 gene is well-known tumor suppressor modulating numerous vital cellular functions, such as apoptosis, autophagy, senescence, often correlated with metabolic reprogramming (Bieging et al., 2014). In a second step, they wanted to verify the impact of p53 mutations upon treatment with simvastatin in lung adenocarcinoma cells (Chou et al., 2019).
In more detail, different types of cell lines (such as p53 wild type or p53 null) were used to study the cytotoxic effects of simvastatin in cell growth inhibition and decrease of lipid rafts in lung cancer cells with p53 mutations. Statins are likely to exhibit their antitumor effects (such as proliferation and migration impairment) by influencing inflammatory and oxidative stress-related tumorigenesis. Moreover, simvastatin inhibits tumor metastasis by WT p53-dependent autophagy. Statins have very likely tumor-suppressive effects in both normal and mutant p53 cells by regulating different signaling pathways (Chou et al., 2019).
Pancreatic Ductal Adenocarcinoma (PDA) is a disease with high therapy resistance, early metastasis, and poor prognosis (Siegel et al., 2014). An essential role for pancreatic CSCs is playing by Shh, which is a fundamental part of the Hedgehog signaling pathway, and whose expression is often highly upregulated in CSCs (Pak and Segal, 2016) affecting the survival of malignant cells. Moreover, Hedgehog signaling is depending on cholesterol and its levels (Porter et al., 1996). Yin et al. (2018) investigated if statins, especially simvastatin, could stop the progression of PDA. In their hands, simvastatin increased the efficacy of the chemotherapeutic agent gemcitabine in PDA cancer cells as well as in a chicken-egg xenograft model resulting in inhibited tumor growth, and reduced pancreatic CSC features acting on Shh signaling. Thus, this cancer-specific effect of simvastatin might be Hedgehog-dependent (Yin et al., 2018). Staying on this type of tumor, McGregor and colleagues demonstrated in vitro and in vivo that PDA depends on MVP for de novo synthesizing Coenzyme Q (CoQ) (McGregor et al., 2020). They showed in vitro a synergistic interaction between simvastatin and the mitogen-activated protein kinase (MEK) inhibitor AZD6244. However, CoQ is available from the diet and might not be limited in the context of in vivo physiology. The authors further verified their results in a KPC PDA mouse model, and they observed that statin treatment induced elevated ROS production, and this increase was mitigated by cancer cells through upregulation of the antioxidant metabolic pathways. When the mice were treated with both statin and the MEK inhibitor AZD6244, there was a significant increase in apoptosis and cell death through elevated ROS production.
Hepatocellular carcinoma (HCC), a frequently occurring primary malignant tumors worldwide (Bray et al., 2018), is associated with delays in diagnosis and a high tendency to metastasis development (Niu et al., 2017). Sorafenib is used in cancer therapy since it possesses antiproliferative effects via the inhibition of kinases, such as CRAF, BRAF, and c-KIT and moreover is able to reduce angiogenesis through the inhibition of vascular endothelial growth factor 2/3 (VEGFR-2/3). Feng et al. (2020) have studied the problem of sorafenib resistance in HCC, demonstrating that simvastatin is able to counteract this resistance when administered in combination with sorafenib. They have used LM3, and sorafenib-resistant LM3 (LM3-SR) cell lines to study cell proliferation, apoptosis and glycolysis levels, and afterwards, they translated both cell lines into xenograft mouse models using oral formulations containing sorafenib and simvastatin alone or in combination. Their results have shown that the suppression of HIF-1α/PPAR-γ/PKM2 axis by simvastatin augments the sensitivity of LM3-SR cells to sorafenib when combined with simvastatin. Furthermore, this treatment decreased proliferation and increased apoptosis in HCC cells.
Moreover, about HCC, the cyclooxygenase-2 (COX-2) has been shown to be overexpressed in this type of cancer (Shiota et al., 1999). The primary metabolite of COX-2 is prostaglandin E2 and is associated with cell proliferation and resistance to apoptosis, thus the use of celecoxib, a COX-2 specific inhibitor, might result in the inhibition of HCC cell growth (Gao et al., 2010). Lee et al. supposed that the combination between simvastatin and NS398, a COX-2 inhibitor, could maximize pharmacological efficacy and may minimize adverse effects in HCC exhibiting a synergistic effect in Hep3B and Huh-7 cell lines. The aforementioned drug combination was shown to suppress the nuclear localization of nuclear factor-kappa-B (NF-κB), to inhibit the Akt pathway, to decrease procaspase 3 and Bcl-2 levels, and to augment caspase 9 levels, being more effective than the single treatments (Lee et al., 2014).
The NF-κB pathway is also upregulated in multiple myeloma (MM) cells, and it may be targeted in novel, innovative MM treatments (Annunziata et al., 2007). As we have seen in HCC, statins might lead to a downregulation of the NF-κB pathway, thus underpinning the probable positive outcomes in MM patients under statin treatment (Martin-Ventura et al., 2005). Brånvall et al. used a Swedish population-based national health register and performed a follow-up study on 4,315 MM cases. Various statins were followed up, but simvastatin was the most widely used. Some patients received no statins while others received them during the 6-months period preceding the MM diagnosis, and the last group during 6 months after diagnosis. The analysis revealed that the statin use before and after the diagnosis was linked to a reduced risk of mortality in both sexes (Branvall et al., 2020). The authors suppose in their discussion that the observed positive effects of statins are connected with a promotion of the host immune response by regulating mesenchymal stromal cell-induced T-cell suppression and B-lymphocyte survival as earlier described in vitro (Musso et al., 2011). The study mentioned above represents an interesting shred of evidence regarding the use of statins in MM, warranting further in vitro and in vivo experiments.
Simvastatin has also been tested to check its putative influence in Epithelial-Mesenchymal Transition (EMT). The transforming growth factor-β1 (TGF-β1) is a key inducer and driver of the EMT process. Yang et al. (2013) observed that simvastatin could attenuate TGF-β1-induced EMT in human alveolar A549 cells but failed to reverse the cell morphological changes. Despite the somewhat discouraging results, further research efforts are indispensable to dissect better the role of statins in lung cancer.
Epigenetics mechanisms, being up today strongly validated in cancer pathogenesis (see for details the following reviews) (Jones et al., 2016; Flavahan et al., 2017; Mai, 2020), have been recently studied even among the different biological effects of statins, and the role of the latter in epigenetics is more and more evolving.
Human tumors are frequently characterized by dysregulation between the activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Fraga et al., 2005). For instance, the acetylation of lysine residues on histone H3 and H4 increases the expression of p21, thus activating the tumor suppressor p53. Many pieces of evidence are showing that non-lipid effects by statins play a pivotal role in their anti-cancer activity (Wong et al., 2002). Lin et al. have compared the molecular structures of HDAC inhibitors (like vorinostat or TSA) and statins: both of them contain acidic moieties, such as hydroxamic and carboxylic acid functions, respectively, and they have discovered that statins probably induce hyperacetylation of H3 thanks to the inhibition of HDAC1/2. They also have performed molecular docking studies assessing that the carboxylic acid moiety of statins is able to enter the catalytic site of the human HDAC2. Next, the inhibition of HDAC by lovastatin, atorvastatin, pravastatin, and simvastatin was determined by using the Fluor-de-Lys HDAC activity assay kit. They have shown the dissociation of HDAC1/2 and the association of CREB binding protein (CBP) at the p21 promoter eliciting p21 expression and increased histone-H3 acetylation levels. The statins above mentioned were able to decrease the cell proliferation and tumor growth in several cellular models, such as lung cancer (A549, NCI-H292), colorectal cancer (HCT-116, HCT-116 p53), and AGS human gastric carcinoma cells, and in a xenograft female BALB/c nude mouse model of A549 lung cancer (Lin et al., 2008).
Recently, it has been shown by Okubo et al. (2020) that combinations of fluvastatin and vorinostat act cooperatively against renal cancer cells. In more detail, the anti-cancer activity of the HDAC inhibitor vorinostat was less evident because of the intrinsic activation of the mTOR pathway in a renal cancer cell model. The authors wanted to overcome this undesired activation through a co-treatment with fluvastatin, which is known to activate the AMP-activated protein kinase (AMPK) as a mTOR inhibitor. This is of particular interest because AMPK controls cellular metabolism pathways essential for cancer progression, thus being a vital regulator of cancer cell growth and proliferation (Faubert et al., 2015). The authors were able to demonstrate that the combination of vorinostat and fluvastatin enhanced the vorinostat-induced histone acetylation, suppressed the vorinostat-activated mTOR pathway via pivotal fluvastatin-related AMPK activation, thus resulting in renal anti-cancer effect in vitro (human renal cancer cells ACHN, A498, murine renal cancer Renca) and in vivo (Renca mouse model) via apoptosis induction. Noteworthy, the phosphorylation of downstream protein of the mTOR pathway 4EBP1 is decreased by vorinostat and fluvastatin co-treatment, and it reduced renal cancer viability. The results they have achieved indicate that the vorinostat-fluvastatin combination would be a safe and effective combination therapy warranting further examination.
These promising results might pave the way toward innovative cancer therapies and/or chemoprevention strategies. HDAC inhibitors and statins used alone do not give excellent results, and they are characterized by side effects or lower bioavailability. In 2013, Chen and colleagues designed some innovative dual-action inhibitors to target HDAC as well as HMGCR. Interestingly, the compounds showed potent inhibitory activities with IC50 values in the nanomolar range, via the presence of the two essential pharmacophoric elements: hydroxamic acid moiety responsible for the HDAC inhibition and dihydroxy-hydroxamate moiety responsible for the HMGCR inhibition. They used molecular docking to verify the affinity of their hybrid molecules to HDAC and HMGCR; then they performed in vitro studies to examine enzyme inhibition activity as well as cell growth inhibition in A549 human lung cancer cells, MEF normal mouse and HS68 human fibroblasts. Some of their dual-inhibitors possess a good selectivity for cancer cells when compared with healthy ones (Chen et al., 2013). The compounds mentioned above were studied further by the same research group in more detail. Wei et al. (2016a) tested the molecules in C57BL/6, BALB/c, NOD/SCID mice, and Apc(Min/+) colorectal cancer (CRC) mouse models obtaining inhibition of metastasis, angiogenesis, and cancer stemness; furthermore, such compounds boosted the effect of oxaliplatin in the aforementioned CRC mouse models. The potential benefits of the dual targeting HDAC/HMGCR inhibitor has been confirmed in a subsequent study of the same research team underlining the potential benefits of this promising lead compound able to prevent colitis-associated colorectal cancer (Wei et al., 2016b). Next, Weng et al. (2019) were able to demonstrate in a non-small lung cancer cell (NSCLC) model, the most common of lung cancers, that the hybrids are able to inhibit the EMT. This pivotal study underlines the feasibility of dual-targeting anti-cancer agents based on HDAC and statin moieties.
Otahal et al. demonstrated in vitro the effectiveness of the combination of fluvastatin or pitavastatin with erlotinib, an approved EGFR tyrosine kinase inhibitor (EGFR-TKI), in EGFR-TKI resistant human lung adenocarcinoma cell lines with mutated or overexpressed EGFR, such as A549, Calu6, and H1993, exhibiting promising cytotoxic effects. They proved that both statins combined with erlotinib led to apoptosis in NSCLC cell lines in vitro, particularly in K-Ras mutated cell lines (Otahal et al., 2020).
Regarding cancer-related deaths in women, breast cancer is one of the leading causes (Siegel et al., 2017). This cancer needs new strategies and/or new treatments due to its high mortality and aggressiveness. Survivin, the smallest member of the inhibitors of apoptosis protein (IAP) family, modulates numerous cellular processes for example mitosis, migration, angiogenesis or chemo-resistance. Recent studies demonstrated that the use of statins reduces breast cancer-specific mortality in patients (Cardwell et al., 2015; Liu et al., 2017). Huang et al. have highlighted that lovastatin could have a beneficial effect in various cell models, such as MCF-7, MDA-MB-231, and T47D. They showed that lovastatin inhibited cell proliferation and induced apoptosis in MCF-7 cells by causing p21 expression, thus leading to p53 level increase and survivin downregulation. The mechanism is not yet fully elucidated, but lovastatin exhibits its antitumor activities via the LKB1-AMPK-p38MAPK-p53 survivin cascade resulting in survivin reduction and ultimately cell death, thus suggesting an important role of this statin for breast cancer treatment (Huang et al., 2020). However, the precise mechanism by which lovastatin causes cell death needs to be studied more in-depth and probably depends on tumor subtypes and/or the patient's genetic background.
C-Myc, an important transcription factor regulating crucial biological cell functions, including cell cycle, differentiation and proliferation, is often deregulated in cancer through somatic mutation, chromosomal translocation, genomic amplification of defects in upstream regulators (Albihn et al., 2010). C-Myc is overexpressed in 31–64% of medulloblastoma types (Eberhart et al., 2004). MiR-33b is a specific inhibitor of c-Myc, which is frequently lost in medulloblastoma. Its overexpression leads to the down-regulation of c-Myc and its transactivation targets, such as cyclin E or ornithine decarboxylase (ODC). Takwi et al. (2012) have proven that lovastatin upregulated the expression of mi-R-33b, leading to reduced cell proliferation. Lovastatin treatment also impairs orthotopically xenografted cells tumor growth. This study could be important to purpose statins as a pharmacological modulator of c-Myc via miRNA-based therapeutics.
Jiao et al. (2020) discovered that the treatment with pitavastatin of human MCF10A exhibiting oncogenic defects, such as cells lacking PTEN or expressing K-RasG12V, led to geranylgeranyl diphosphate (GGPP) depletion of the MVP, to amino acid starvation and ultimately to cell death.
To sum up, statins have been used as lipid-lowering drugs to treat hyperlipidemia and to decrease cholesterol levels in the blood. It is now well-established that the antitumor effects of statins can be associated with MVP-mediated and non-MVP-mediated mechanisms (Figure 1). The MVP-pathway is interrupted at the first step through statin treatment that inhibits HMGCR halting the isoprenoid synthesis, such as GGPP and farnesyl diphosphate (FPP) for GTPase-proteins vital for cancer cells. Statins are regulators of the proliferation, migration, and survival of tumor cells by regulating Rho, Ras, and Rac proteins. Statins can also inhibit cancer cell growth by modulating specific other pathways. For example, very recently lovastatin was demonstrated to activate the LKB1-AMPK-p38MAPK-p53-survivin cascade causing MCF-7 breast cancer cell death (Huang et al., 2020). The simultaneous administration of fluvastatin and vorinostat effectively led to apoptosis and reduced renal cancer growth in vitro and in vivo through AMPK activation, histone acetylation, and ER stress induction (Okubo et al., 2020). Simvastatin is capable to inhibit the HIF-1α/PPAR-γ/PKM2 axis via the suppression of PKM2-mediated glycolysis, leading to a decreased proliferation and an increased apoptosis rate in HCC cells. Furthermore, HCC cell are resensitized to sorafenib treatment (Feng et al., 2020). Simvastatin treatment reduced CoQ synthesis and promoted oxidative stress and apoptosis in tumors when administered in combination with the MEK inhibitor AZD6244, highlighting a new mechanism through which statin treatment may impact PDA cancer growth (McGregor et al., 2020). Simvastatin led to mutant p53 protein degradation by activating a caspase-dependent apoptotic pathway and decreased motility in lung cancer cells possessing p53 missense mutations (Chou et al., 2019). The same drug hampered viability, stemness, tumor growth, and metastasis in pancreatic cells, via the inhibition of the Shh signaling leading to enhanced efficacy of gemcitabine treatment (Yin et al., 2018).
In this minireview, we have seen that statins are low cost drugs with few side effects that can be applied in numerous types of cancer. At present, in numerous clinical trials statins are investigated either alone or in combination with other anti-cancer agents, and an overview of the main ones is described in Table 1.
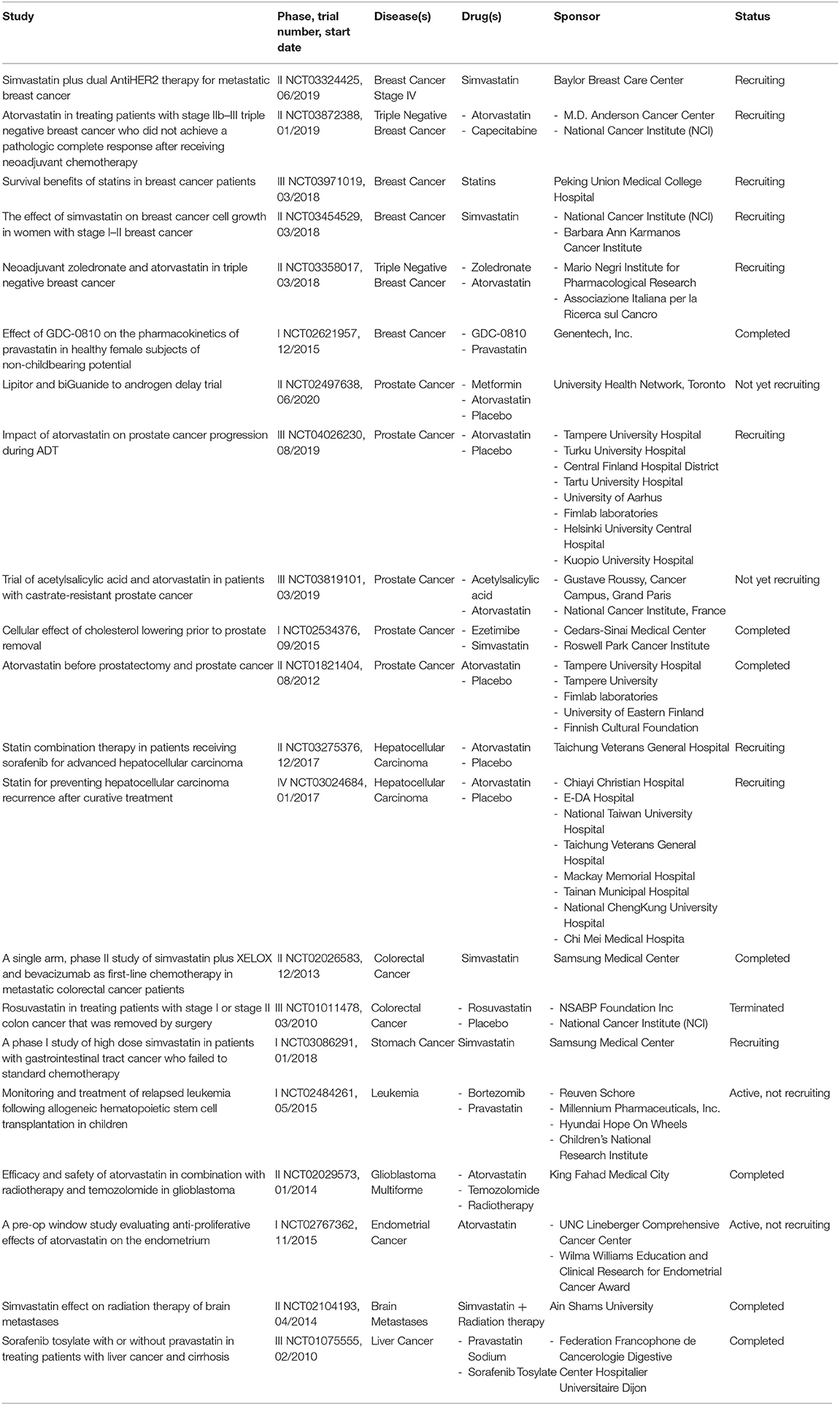
Table 1. Summary of the main clinical trials of statins investigated in cancer therapy.
These studies are in constant evolution, and we can expect new combination treatments to come in the near future. Scientists could probably step up and develop hybrid inhibitors, confirmed to be a feasible approach as in the pivotal study of Chen et al. (2013), to further enhance the anti-cancer activity and decrease the side effects. However, despite the promising results achieved by the treatment with statins alone or in combination with other anti-cancer agents, the precise mechanisms how they exert their antitumor effects often remain unclear. In the years to come, we can expect novel and promising approaches addressing the statins to fight cancer, probably also at clinical levels.
ED and CZ performed the literature research and drafted a first version of the manuscript. AM and SV supervised and coordinated the work as well as corrected the manuscript.
This research and publication costs were supported by the Italian Association for Cancer Research AIRC-19162 (AM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Albihn, A., Johnsen, J. I., and Henriksson, M. A. (2010). MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 107, 163–224. doi: 10.1016/S0065-230X(10)07006-5
Annunziata, C. M., Davis, R. E., Demchenko, Y., Bellamy, W., Gabrea, A., Zhan, F., et al. (2007). Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 12, 115–130. doi: 10.1016/j.ccr.2007.07.004
Bieging, K. T., Mello, S. S., and Attardi, L. D. (2014). Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370. doi: 10.1038/nrc3711
Branvall, E., Ekberg, S., Eloranta, S., Wasterlid, T., Birmann, B. M., and Smedby, K. E. (2020). Statin use is associated with improved survival in multiple myeloma: a Swedish population-based study of 4315 patients. Am. J. Hematol. 95, 652–661. doi: 10.1002/ajh.25778
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Buhaescu, I., and Izzedine, H. (2007). Mevalonate pathway: a review of clinical and therapeutical implications. Clin. Biochem. 40, 575–584. doi: 10.1016/j.clinbiochem.2007.03.016
Cardwell, C. R., Mc Menamin, U., Hughes, C. M., and Murray, L. J. (2015). Statin use and survival from lung cancer: a population-based cohort study. Cancer Epidemiol. Biomarkers Prev. 24, 833–841. doi: 10.1158/1055-9965.EPI-15-0052
Chen, J. B., Chern, T. R., Wei, T. T., Chen, C. C., Lin, J. H., and Fang, J. M. (2013). Design and synthesis of dual-action inhibitors targeting histone deacetylases and 3-hydroxy-3-methylglutaryl coenzyme a reductase for cancer treatment. J. Med. Chem. 56, 3645–3655. doi: 10.1021/jm400179b
Chou, C. W., Lin, C. H., Hsiao, T. H., Lo, C. C., Hsieh, C. Y., Huang, C. C., et al. (2019). Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 9:20403. doi: 10.1038/s41598-019-56532-6
Corsini, A., Maggi, F. M., and Catapano, A. L. (1995). Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol. Res. 31, 9–27. doi: 10.1016/1043-6618(95)80042-5
Demierre, M. F., Higgins, P. D., Gruber, S. B., Hawk, E., and Lippman, S. M. (2005). Statins and cancer prevention. Nat. Rev. Cancer 5, 930–942. doi: 10.1038/nrc1751
Dujovne, C. A. (2017). Red yeast rice preparations: are they suitable substitutions for statins? Am. J. Med. 130, 1148–1150. doi: 10.1016/j.amjmed.2017.05.013
Eberhart, C. G., Kratz, J., Wang, Y., Summers, K., Stearns, D., Cohen, K., et al. (2004). Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J. Neuropathol. Exp. Neurol. 63, 441–449. doi: 10.1093/jnen/63.5.441
Endo, A. (2004). The origin of the statins. Atheroscler. Suppl. 5, 125–130. doi: 10.1016/j.atherosclerosissup.2004.08.033
Faubert, B., Vincent, E. E., Poffenberger, M. C., and Jones, R. G. (2015). The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 356, 165–170. doi: 10.1016/j.canlet.2014.01.018
Feng, J., Dai, W., Mao, Y., Wu, L., Li, J., Chen, K., et al. (2020). Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J. Exp. Clin. Cancer Res. 39:24. doi: 10.1186/s13046-020-1528-x
Flavahan, W. A., Gaskell, E., and Bernstein, B. E. (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357:eaal2380. doi: 10.1126/science.aal2380
Fraga, M. F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., et al. (2005). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37, 391–400. doi: 10.1038/ng1531
Freed-Pastor, W. A., Mizuno, H., Zhao, X., Langerod, A., Moon, S. H., Rodriguez-Barrueco, R., et al. (2012). Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258. doi: 10.1016/j.cell.2011.12.017
Fritz, V., Benfodda, Z., Henriquet, C., Hure, S., Cristol, J. P., Michel, F., et al. (2013). Metabolic intervention on lipid synthesis converging pathways abrogates prostate cancer growth. Oncogene 32, 5101–5110. doi: 10.1038/onc.2012.523
Gao, J., Jia, W. D., Li, J. S., Wang, W., Xu, G. L., Ma, J. L., et al. (2010). Combined inhibitory effects of celecoxib and fluvastatin on the growth of human hepatocellular carcinoma xenografts in nude mice. J. Int. Med. Res. 38, 1413–1427. doi: 10.1177/147323001003800423
Gazzerro, P., Proto, M. C., Gangemi, G., Malfitano, A. M., Ciaglia, E., Pisanti, S., et al. (2012). Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol. Rev. 64, 102–146. doi: 10.1124/pr.111.004994
Goldstein, J. L., and Brown, M. S. (2015). A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161–172. doi: 10.1016/j.cell.2015.01.036
Gupta, A., Stokes, W., Eguchi, M., Hararah, M., Amini, A., Mueller, A., et al. (2019). Statin use associated with improved overall and cancer specific survival in patients with head and neck cancer. Oral Oncol. 90, 54–66. doi: 10.1016/j.oraloncology.2019.01.019
Huang, S. W., Chyuan, I. T., Shiue, C., Yu, M. C., Hsu, Y. F., and Hsu, M. J. (2020). Lovastatin-mediated MCF-7 cancer cell death involves LKB1-AMPK-p38MAPK-p53-survivin signalling cascade. J. Cell. Mol. Med. 24, 1822–1836. doi: 10.1111/jcmm.14879
Iannelli, F., Lombardi, R., Milone, M. R., Pucci, B., De Rienzo, S., Budillon, A, et al. (2018). Targeting mevalonate pathway in cancer treatment: repurposing of statins. Recent. Pat. Anticancer. Drug. Discov. 13, 184–200. doi: 10.2174/1574892812666171129141211.
Jiao, Z., Cai, H., Long, Y., Sirka, O. K., Padmanaban, V., Ewald, A. J., et al. (2020). Statin-induced GGPP depletion blocks macropinocytosis and starves cells with oncogenic defects. Proc. Natl. Acad. Sci. U.S.A. 117, 4158–4168. doi: 10.1073/pnas.1917938117
Jones, P. A., Issa, J. P., and Baylin, S. (2016). Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641. doi: 10.1038/nrg.2016.93
Kidera, Y., Tsubaki, M., Yamazoe, Y., Shoji, K., Nakamura, H., Ogaki, M., et al. (2010). Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J. Exp. Clin. Cancer Res. 29:127. doi: 10.1186/1756-9966-29-127
Korani, S., Korani, M., Bahrami, S., Johnston, T. P., Butler, A. E., Banach, M., et al. (2019). Application of nanotechnology to improve the therapeutic benefits of statins. Drug Discov. Today 24, 567–574. doi: 10.1016/j.drudis.2018.09.023
Lee, S. J., Hwang, J. W., Yim, H., Yim, H. J., Woo, S. U., Suh, S. J., et al. (2014). Synergistic effect of simvastatin plus NS398 on inhibition of proliferation and survival in hepatocellular carcinoma cell line. J. Gastroenterol. Hepatol. 29, 1299–1307. doi: 10.1111/jgh.12503
Lin, Y. C., Lin, J. H., Chou, C. W., Chang, Y. F., Yeh, S. H., and Chen, C. C. (2008). Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Res. 68, 2375–2383. doi: 10.1158/0008-5472.CAN-07-5807
Liu, B., Yi, Z., Guan, X., Zeng, Y. X., and Ma, F. (2017). The relationship between statins and breast cancer prognosis varies by statin type and exposure time: a meta-analysis. Breast Cancer Res. Treat 164, 1–11. doi: 10.1007/s10549-017-4246-0
Martin-Ventura, J. L., Blanco-Colio, L. M., Gomez-Hernandez, A., Munoz-Garcia, B., Vega, M., Serrano, J., et al. (2005). Intensive treatment with atorvastatin reduces inflammation in mononuclear cells and human atherosclerotic lesions in one month. Stroke 36, 1796–1800. doi: 10.1161/01.STR.0000174289.34110.b0
McGregor, G. H., Campbell, A. D., Fey, S. K., Tumanov, S., Sumpton, D., Blanco, G. R., et al. (2020). Targeting the metabolic response to statin-mediated oxidative stress produces a synergistic antitumor response. Cancer Res. 80, 175–188. doi: 10.1158/0008-5472.CAN-19-0644
Musso, A., Zocchi, M. R., and Poggi, A. (2011). Relevance of the mevalonate biosynthetic pathway in the regulation of bone marrow mesenchymal stromal cell-mediated effects on T-cell proliferation and B-cell survival. Haematologica 96, 16–23. doi: 10.3324/haematol.2010.031633
Niu, L., Liu, L., Yang, S., Ren, J., Lai, P. B. S., and Chen, G. G. (2017). New insights into sorafenib resistance in hepatocellular carcinoma: responsible mechanisms and promising strategies. Biochim. Biophys. Acta Rev. Cancer 1868, 564–570. doi: 10.1016/j.bbcan.2017.10.002
Notarnicola, M., Tutino, V., and Caruso, M. G. (2014). Tumor-induced alterations in lipid metabolism. Curr. Med. Chem. 21, 2729–2733. doi: 10.2174/0929867321666140303122426
Okubo, K., Isono, M., Miyai, K., Asano, T., and Sato, A. (2020). Fluvastatin potentiates anticancer activity of vorinostat in renal cancer cells. Cancer Sci. 111, 112–126. doi: 10.1111/cas.14225
Oryan, A., Kamali, A., and Moshiri, A. (2015). Potential mechanisms and applications of statins on osteogenesis: current modalities, conflicts and future directions. J. Control. Release 215, 12–24. doi: 10.1016/j.jconrel.2015.07.022
Otahal, A., Aydemir, D., Tomasich, E., and Minichsdorfer, C. (2020). Delineation of cell death mechanisms induced by synergistic effects of statins and erlotinib in non-small cell lung cancer cell (NSCLC) lines. Sci. Rep. 10:959. doi: 10.1038/s41598-020-57707-2
Pak, E., and Segal, R. A. (2016). Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy. Dev. Cell 38, 333–344. doi: 10.1016/j.devcel.2016.07.026
Pisanti, S., Picardi, P., Ciaglia, E., D'Alessandro, A., and Bifulco, M. (2014). Novel prospects of statins as therapeutic agents in cancer. Pharmacol. Res. 88, 84–98. doi: 10.1016/j.phrs.2014.06.013
Porter, J. A., Young, K. E., and Beachy, P. A. (1996). Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259. doi: 10.1126/science.274.5285.255
Schonewille, M., de Boer, J. F., Mele, L., Wolters, H., Bloks, V. W., Wolters, J. C., et al. (2016). Statins increase hepatic cholesterol synthesis and stimulate fecal cholesterol elimination in mice. J. Lipid Res. 57, 1455–1464. doi: 10.1194/jlr.M067488
Shiota, G., Okubo, M., Noumi, T., Noguchi, N., Oyama, K., Takano, Y., et al. (1999). Cyclooxygenase-2 expression in hepatocellular carcinoma. Hepatogastroenterology 46, 407–412.
Siegel, R., Ma, J., Zou, Z., and Jemal, A. (2014). Cancer statistics, 2014. CA Cancer J. Clin. 64, 9–29. doi: 10.3322/caac.21208
Siegel, R. L., Miller, K. D., and Jemal, A. (2017). Cancer statistic, 2017. CA Cancer J. Clin. 67, 7–30. doi: 10.3322/caac.21387
Takai, Y., Sasaki, T., and Matozaki, T. (2001). Small GTP-binding proteins. Physiol. Rev. 81, 153–208. doi: 10.1152/physrev.2001.81.1.153
Takwi, A. A., Li, Y., Becker Buscaglia, L. E., Zhang, J., Choudhury, S., Park, A. K., et al. (2012). A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol. Med. 4, 896–909. doi: 10.1002/emmm.201101045
Vallianou, N. G., Kostantinou, A., Kougias, M., and Kazazis, C. (2014). Statins and cancer. Anticancer. Agents Med. Chem. 14, 706–712. doi: 10.2174/1871520613666131129105035
Wei, T. T., Lin, Y. T., Chen, W. S., Luo, P., Lin, Y. C., Shun, C. T., et al. (2016a). Dual targeting of 3-hydroxy-3-methylglutaryl coenzyme A reductase and histone deacetylase as a therapy for colorectal cancer. EBioMedicine 10, 124–136. doi: 10.1016/j.ebiom.2016.07.019
Wei, T. T., Lin, Y. T., Tseng, R. Y., Shun, C. T., Lin, Y. C., Wu, M. S., et al. (2016b). Prevention of colitis and colitis-associated colorectal cancer by a novel polypharmacological histone deacetylase inhibitor. Clin. Cancer Res. 22, 4158–4169. doi: 10.1158/1078-0432.CCR-15-2379
Weng, C. H., Chen, L. Y., Lin, Y. C., Shih, J. Y., Lin, Y. C., Tseng, R. Y., et al. (2019). Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 38, 455–468. doi: 10.1038/s41388-018-0454-2
Wong, W. W., Dimitroulakos, J., Minden, M. D., and Penn, L. Z. (2002). HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 16, 508–519. doi: 10.1038/sj.leu.2402476
Yang, T., Chen, M., and Sun, T. (2013). Simvastatin attenuates TGF-beta1-induced epithelial-mesenchymal transition in human alveolar epithelial cells. Cell. Physiol. Biochem. 31, 863–874. doi: 10.1159/000350104
Keywords: cancer, statins, drug combination, target therapy, mevalonate pathway, HMG-CoA reductase
Citation: Di Bello E, Zwergel C, Mai A and Valente S (2020) The Innovative Potential of Statins in Cancer: New Targets for New Therapies. Front. Chem. 8:516. doi: 10.3389/fchem.2020.00516
Received: 19 April 2020; Accepted: 19 May 2020;
Published: 18 June 2020.
Edited by:
Alfonso Carotenuto, University of Naples Federico II, ItalyReviewed by:
Adriano Mollica, University “G. d'Annunzio” of Chieti-Pescara, ItalyCopyright © 2020 Di Bello, Zwergel, Mai and Valente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonello Mai, YW50b25lbGxvLm1haUB1bmlyb21hMS5pdA==; Sergio Valente, c2VyZ2lvLnZhbGVudGVAdW5pcm9tYTEuaXQ=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.