
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 28 April 2020
Sec. Theoretical and Computational Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00338
This article is part of the Research Topic Computational Modeling of Spintronic Materials View all 18 articles
 Meng Ju1,2
Meng Ju1,2 Jingjing Wang2*Jing Huang1Chuanzhao Zhang3*Yuanyuan Jin3Weiguo Sun4Shichang Li5Yunhong Chen2
Jingjing Wang2*Jing Huang1Chuanzhao Zhang3*Yuanyuan Jin3Weiguo Sun4Shichang Li5Yunhong Chen2Trivalent Cerium (Ce3+) doped Yttrium Oxide (Y2O3) host crystal has drawn considerable interest due to its popular optical 5d-4f transition. The outstanding optical properties of Y2O3:Ce system have been demonstrated by previous studies but the microstructures still remain unclear. The lacks of Y2O3:Ce microstructures could constitute a problem to further exploit its potential applications. In this sense, we have comprehensively investigated the structural evolutions of Y2O3:Ce crystals based on the CALYPSO structure search method in conjunction with density functional theory calculations. Our result uncovers a new rhombohedral phase of Y2O3:Ce with R-3 group symmetry. In the host crystal, the Y3+ ion at central site can be naturally replaced by the doped Ce3+, resulting in a perfect cage-like configuration. We find an interesting phase transition that the crystallographic symmetry of Y2O3 changes from cubic to rhombohedral when the impurity Ce3+ is doped into the host crystal. With the nominal concentration of Ce3+ at 3.125%, many metastable structures are also identified due to the different occupying points in the host crystal. The X-ray diffraction patterns of Y2O3:Ce are simulated and the theoretical result is comparable to experimental data, thus demonstrating the validity of the lowest energy structure. The result of phonon dispersions shows that the ground state structure is dynamically stable. The analysis of electronic properties indicate that the Y2O3:Ce possesses a band gap of 4.20 eV which suggests that the incorporation of impurity Ce3+ ion into Y2O3 host crystal leads to an insulator to semiconductor transition. Meanwhile, the strong covalent bonds of O atoms in the crystal, which may greatly contribute to the stability of ground state structure, are evidenced by electron localization function. These obtained results elucidate the structural and bonding characters of Y2O3:Ce and could also provide useful insights for understanding the experimental phenomena.
The rare-earth Cerium ions doped crystals constitute an attractive class of materials that have been extensively used in many kinds of fields including scintillation phosphors, laser medium, and white light emitting diode phosphors (Han et al., 2019; Lin et al., 2019; Masanori et al., 2020). The outstanding optical behaviors of trivalent Cerium ion (Ce3+) has drawn considerable interest due to its popular optical 5d-4f transition. Among various host materials, Yttrium Oxide (Y2O3) crystal is considered to be the most promising sesquioxide host because of its unique chemical and thermal stability. The Y2O3 host crystal is also one of the multifunctional materials that can give rise to many application areas owing to its fabulous capacity of incorporating the activated laser ions (Ming et al., 2018; Wang et al., 2018; Ju et al., 2020). The latest study has indicated that the Ce3+ doped Y2O3 crystals (Y2O3:Ce) exhibit dominant emission bands at around 380 nm and relatively low intensive band at 560 nm (Gieszczyk et al., 2019). The results further demonstrate the ideal applications of energy storage phosphors for Y2O3:Ce. The excellent advantages of Y2O3:Ce can also be evidenced by the effective use as various laser ceramics (Lupei et al., 2017).
It is well-known that the laser actions can be generally identified in the absorption and emission spectra of rare-earth doped materials. In order to explore the luminescent properties of Y2O3:Ce, Jia et al. (2001) had synthesized the Y2O3:Ce nanoparticles in experiments and measured the photoluminescence spectra at room temperature. Their results revealed that the strong emissions cover the ultraviolet band from 240 nm to 380 nm. To explain the emission lines of the spectra, Loitongbam et al. (2013) measured the luminescence intensities of Y2O3:Ce and found that the characteristic blue color emissions at 424 and 486 nm are originated from Ce3+ ion 5d (spectra terms) → 4f (spectra terms). An unexpected optical activity, including up and down conversions, for Y2O3:Ce crystal was firstly observed by Marin et al. (2013). Although the laser actions were established by a few studies, many researchers were motivated to probe the structural properties of Y2O3:Ce. The effect of doping Ce3+ ion into Y2O3 fibers was investigated by Zhu et al. (2008). They found that the obvious quenching of the luminescence occurred at Ce3+ concentration of 5%. By using the solid-state-reactive method, Liu et al. (2020) carried out a study on the structures of a series of Ce3+ doped Y2O3 ternary ceramics. The results demonstrated that the solubility of Ce3+ concentration at 4% could broaden emission spectra and lead to a large red-shift, which is attractive for the white light emitting. A recent research on the structural properties of Y2O3:Ce was conducted by Krutikova et al. (2020). The nanopowders were obtained by laser ablation and the X-ray diffraction (XRD) patterns of Y2O3:Ce crystal were reported. By looking at the investigations concerning Y2O3:Ce in the literatures, it can be concluded that the systematic electronic structures have not yet been explored, especially for the theoretical insights. Furthermore, the lacks of Y2O3:Ce microstructures constitute a problem to exploit its potential prospects in many applications.
In this paper, we have performed a systematic study on the stable structures and electronic properties for Y2O3 doped with Ce3+ system. By using the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) structure search method (Wang et al., 2010, 2012; Li et al., 2014) combined with first-principle calculation, the low-lying energy structures of Y2O3:Ce are extensively searched. A large number of candidate structures are obtained and the ground state structure together with the first four metastable structures is analyzed in detail. Based on the obtained lowest energy structure of Y2O3:Ce, we thoroughly conduct a calculation of the electronic properties, which could provide powerful guidance for further experimental and theoretical studies.
We have carried out an unbiased structure search for Y2O3 doped with Ce3+ system based on the CALYPSO method (Wang et al., 2010, 2012; Li et al., 2014). The CALYPSO is able to successfully predict the stable structures only with given chemical composition of the system (Lu et al., 2013, 2017, 2018; Lu and Chen, 2018). The detailed method of CALYPSO has been reported in many papers (Ju et al., 2016, 2017, 2019a,b). In this work, the structure searches are performed for Y2O3 doped with Ce system at 80 atoms in one unit cell. The obtained low-lying energy structures are used to perform further geometric optimizations. We conduct the ab initio structural relaxations and electronic properties calculations in the framework of density functional theory (DFT) by using the local density approximation (LDA) exchange correlation functional, as implemented in the Vienna Ab Initio Simulation Package (VASP) (Kresse and Hafner, 1993; Kresse and Furthmuller, 1996; Perdew et al., 1996). Considering the strong f-electrons correlations within the heavy Ce3+ ion, an onsite Coulomb repulsion U = 5.0 eV is employed in the calculations (Herbst and Waston, 1978). We use the projector-augmented wave method to simulate the valence electron space of Ce, Y, and O atoms. The used electrons are 4f15s25p65d16s2, 4s24p64d15s2, and 2s22p4, respectively. Sufficiently fine Monkhorst-Pack k meshes and 500 eV cutoff energy have been chosen to make sure that the calculated enthalpy of each atom is <1 meV. By using a super cell approach, the phonon dispersion spectra are calculated in PHONOPY code (Atsushi et al., 1993). The electron localization function (ELF) (Becke and Edgecombe, 1990; Savin et al., 1992) analysis of Y2O3:Ce is performed and the results are depicted in the VESTA software (Momma and Izumi, 2011). The projected Crystal Orbital Hamilton Population (COHP) (Richard and Peter, 1993) are calculated by the LOBSTER code (Volker et al., 2011; Stefan et al., 2016).
The stable structures for Y2O3:Ce system are favorably identified by using the method described in section Computational Details. On the basis of total energies from low to high, we have plotted the lowest-energy structure of Y2O3:Ce in Figure 1, together with the local [CeO6]9− complex ligand. Noticeably, the ground state structure of Y2O3:Ce possesses a novel structure with R-3 (No. 148) space group. To the best of our knowledge, the rhombohedral phase of Y2O3:Ce crystal is uncovered for the first time. This result indicates an interesting phase transition that the crystallographic symmetry of Y2O3 changes from cubic (Ia-3) to rhombohedral (R-3) when the impurity Ce3+ is doped into the host crystal. It is clearly seen from Figure 1 that the host Y3+ ion can be naturally occupied by the impurity Ce3+ ion. Interestingly, the Wyckoff position of Ce3+ is 1b (0.5, 0.5, 0.5), suggesting that the ground state Y2O3:Ce is a standard cage-like structure. This result is different from that of Y2O3:Nd system (Ju et al., 2020). For reference, the coordinates of all atoms for the ground state Y2O3:Ce are summarized in Table 1. The estimated unit cell parameters and volume for Y2O3:Ce are a = b = c = 10.541 Å and 1171.371 Å (Han et al., 2019), respectively. These values are slightly smaller than those of pure Y2O3 but are comparable to the results reported by Kumar et al. (2017). As regard to the local structure, the Ce3+ ion is calculated to be 6-fold coordinated by O2−, forming the [CeO6]9− complex ligand. The cationic site symmetry of Ce3+ is C3i with six equal Ce–O bonds of 2.369 Å. This bond length is similar with that of Y–O bonds because the effective radius of Ce3+ (1.03 Å) is very close to Y3+ (0.90 Å).
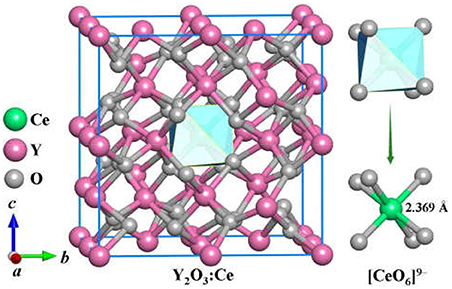
Figure 1. The ground state structure of Y2O3:Ce.
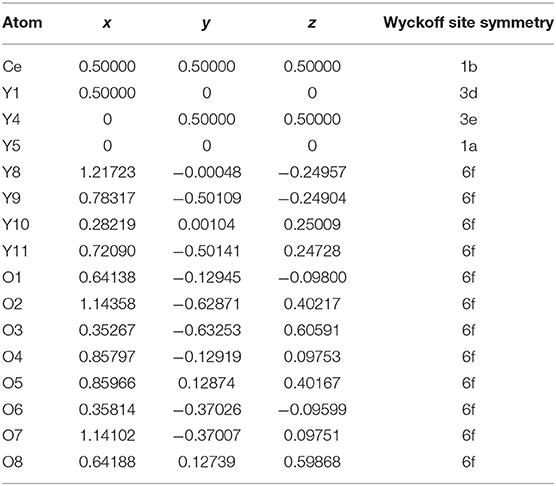
Table 1. Coordinates of all atoms for the ground state Y2O3:Ce.
In the structure prediction, we adopt the chemical composition of Ce:Y: O = 1: 31: 48 to obtain the stable structures with nominal concentration of Ce3+. In this sense, the impurity Ce3+ in Y2O3 crystal is equal to 3.125 at %. Apart from the ground state structure, the CALYPSO also identifies a large number of candidate isomers that can be useful to study the structural evolution of the Y2O3:Ce. Figure 2 illustrates the first four metastable structures of Y2O3:Ce. The isomer (a) has the same R-3 space group as the lowest energy structure while the impurity Ce3+ ions are likely to substitute the Y3+ at the lattice vertexes. The Ce3+ ion of isomer (a) takes the 1a (0, 0, 0) position. It is evidenced that the calculated crystal lattice parameters (10.543 Å) are nearly same as those of lowest-energy structure. The group symmetry of isomer (b) is predicted to be P1 with a triclinic phase. The Wyckoff position of Ce3+ is predicted to be 1a (0.25, 0.75, 0.25). Calculated result reveals that the isomer (c) exhibits a monoclinic structure which belongs to P2 symmetry. The impurity Ce3+ ion occupies the 1b (0, 0.47157, 05) position. For the configuration of isomer (d), it is seen that the Ce3+ ions appear at the center sites of bottom and top in the crystal lattice. The isomer (d) is assigned to P1 group symmetry and is 0.27 eV energetically higher than ground state structure.
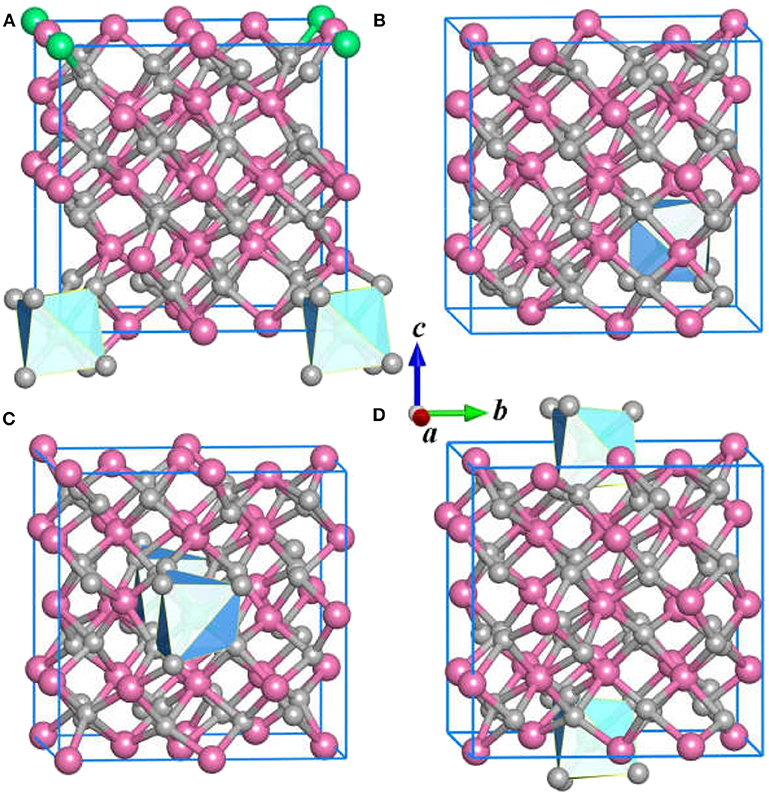
Figure 2. The (A–D) isomers for Y2O3:Ce.
Although the X-ray powder diffraction (XRD) patterns of Y2O3:Ce crystals have been extensively studied, there appears to be inconsistencies of the spectra (Chien and Yu, 2008; Taibeche et al., 2016; Kumar et al., 2017). In order to clarify the crystal characters of the lowest-energy structure, we simulate the XRD patterns of Y2O3:Ce in the 2θ range of 15–65°. The result compared with experimental data is presented in Figure 3. It is evident that the calculated spectrum is in perfect agreement with the values measured by Kumar et al. (2017), demonstrating the validity of the lowest energy structure as well as the accuracy of our theoretical calculations. It should be pointed out that the simulated diffraction peak at 34° is ascribed to the (400) plane direction. This is accord with the result obtained by Taibeche et al. (2016) but different from the measured value proposed by Chien and Yu (2008). For comparison, the XRD patterns of the four isomers (a), (b), (c), and (d) are also provided in Figure 3. Although the overall distribution of the peaks in isomers is closely similar with each other, there are minor differences in the relative intensities. To evaluate the dynamical stability of Y2O3:Ce, the phonon spectrum within the Brillouin zone of ground state structure are calculated. Figure 4 illustrates the phonon dispersion curves along the high-symmetry directions including F, Γ, and Z. Clearly, the overall values in Figure 4 are positive and no virtual frequencies are observed in the full Brillouin zone. It is concluded that the rhombohedral phase structure of Y2O3:Ce crystal is dynamically stable.
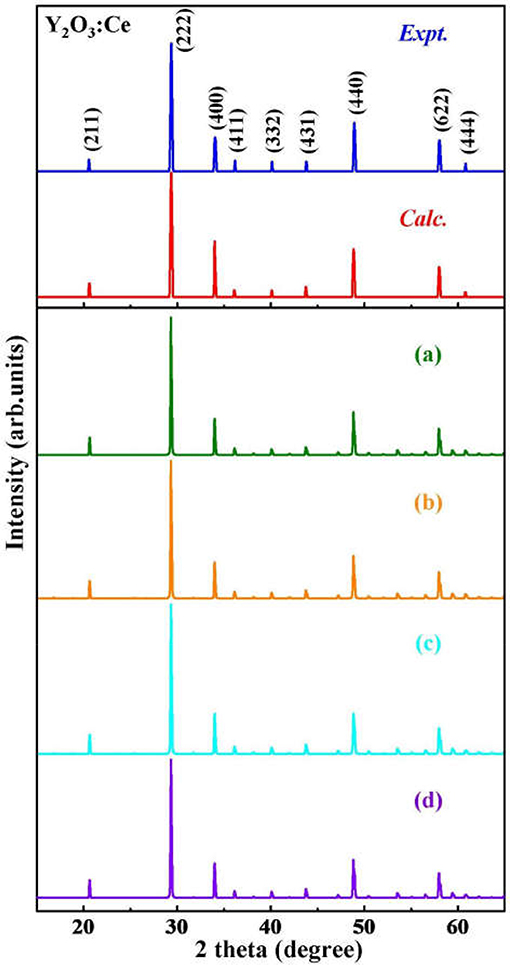
Figure 3. The simulated XRD patterns for ground state Y2O3:Ce and isomers (a–d), compared with measured spectra.
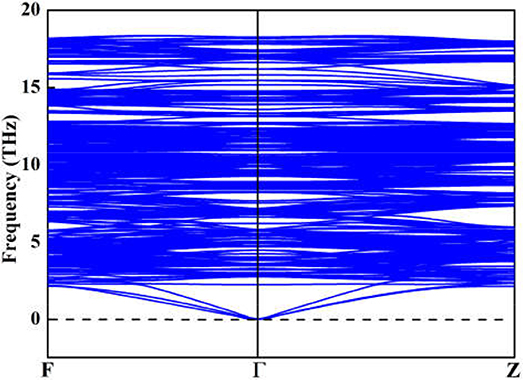
Figure 4. Simulated phonon dispersions of the lowest energy structure for Y2O3:Ce.
To further elucidate the electronic properties of Y2O3:Ce crystal, we have performed a series of ab initio calculations including the electronic band structures, total and partial electronic density of states and electron localization functions. The calculated band structure and density of states (DOS) are plotted together in Figure 5. Our calculated results show that both of the conduction band minimum and valence band maximum are identified at Γ site. The band structure is considered to be a typical of semiconductor with relatively flat top of the valence bands. According to the calculations, the band gap value of ground state Y2O3:Ce is equal to 4.20 eV directly at Γ point. This result is very close to the energy gap of Y2O3:Nd system (Ju et al., 2020) but significantly smaller than that of pure Y2O3 crystal (Wilk and Wallace, 2002). The direct band gap of 4.20 eV suggests a semiconductor character of the Y2O3:Ce. In addition to the electronic band gap, the electronic calculations of high-symmetry directions are in accordance with the above analysis based on phonon spectrum. In Figure 5A, we can clearly see that the band structures can be divided into three parts. The high conduction band is above 4.20 eV while the low valence band is below −0.17 eV. Interestingly, an extremely narrow valence band is observed just below the Fermi level. This result is greatly different with the band structures of pure Y2O3. The calculations show that the narrow valence band is caused by the electronic Alpha states. In contrast, the Beta electrons are not identified near the Fermi level. In order to explore the origins of the electronic bands, we further calculate the partial DOS including s, p, d and f states. The calculated DOS are depicted in Figure 5B. It can be clearly seen that the high conduction bands are mainly formed by d and p states. The p electrons are calculated to be the strongest state in the low valence bands. Moreover, the partial DOS of Y2O3:Ce reveals that the extremely narrow valence band near Fermi level is ascribed to the f orbital, which suggests that the impurity Ce3+ ion leads to a dramatic reduction of the band gap. In other words, it is concluded that the incorporation of the doped Ce3+ ion into Y2O3 host crystal results in an insulator to semiconductor transition.
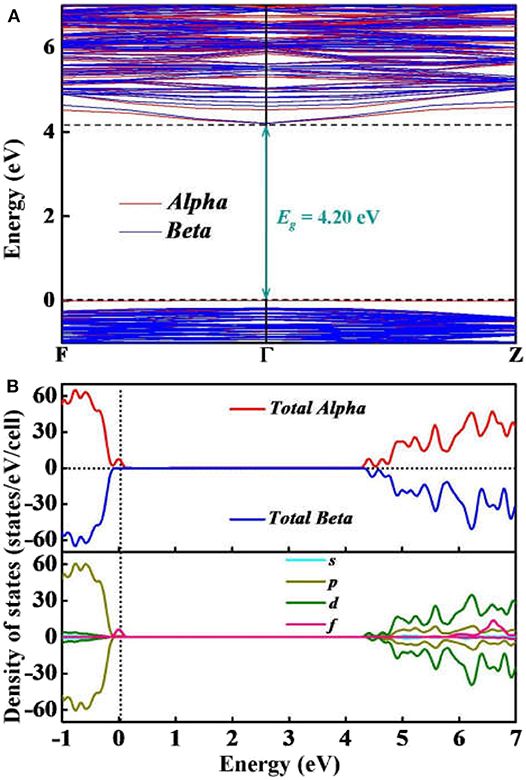
Figure 5. Simulated (A) electronic bands and (B) total and partial DOS of Y2O3:Ce.
To achieve foundational understanding of the bonding character and distribution of electrons of Y2O3:Ce crystal, we have carried out a calculation on the electron localization functions based on the ground state structure. The visually ELF of the structure and (100) plane are presented together in Figure 6. Obviously, the electrons near the cationic atoms are greatly localized with ELF values at ~0.9 while the ELF values in the crystal lattice are nearly zero. This result indicates that the electrons localization on Ce and Y atoms broadens toward O atoms, forming a complete charge delocalization in the vicinity of O atoms. The strong ionic bonds are identified between Ce-O and Y-O. Furthermore, our calculations also show that the value of ELF at Ce atom is relatively larger than the ELF of Y atoms. This phenomenon can be explained as the remaining 4f (Masanori et al., 2020) electron of Ce3+ ion. It should be pointed out that there are strong charge localizations between O-O atoms, demonstrating the covalent bond of O atoms. To further quantitatively estimate the contribution of bonds between O atoms, we have presented the projected Crystal Orbital Hamilton Population (-pCOHP) curves for the O-O bonds in Y2O3:Ce. As shown in Figure 7, the strong bonding contributions of O-O bonds are evidenced. The bond features near the Fermi level can be ascribed to covalent. It is convinced that the excellent stability of Y2O3:Ce crystal is owing to the strong covalent bonds of O atoms.
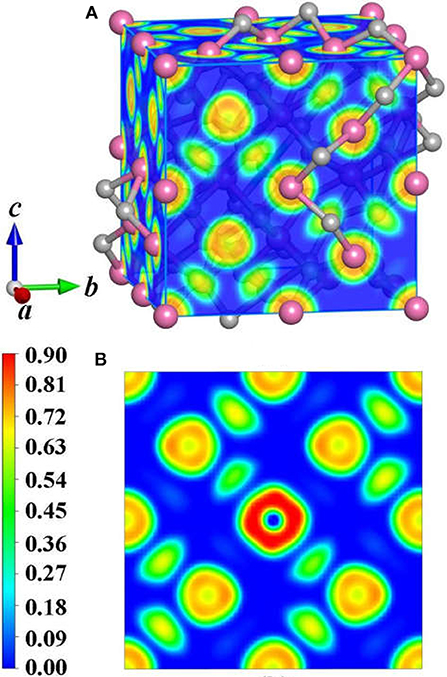
Figure 6. ELF of the (A) structure and (B) <100> plane for Y2O3:Ce.
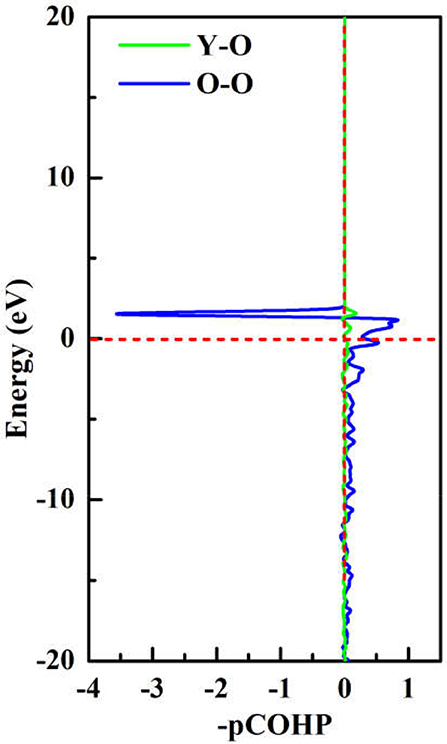
Figure 7. The projected Crystal Orbital Hamilton Population (-pCOHP) curves for the O-O bonds in Y2O3:Ce.
To summarize, we have systematically reported the structural evolutions, doping site locations and electronic properties of Y2O3 crystal doped with Ce3+ ions. By using the CALYPSO method in conjunction with first-principles calculations, a novel stable phase with R-3 space group is identified for the first time. For the ground state structure, the doped Ce3+ can naturally occupy the central Y3+ site in the crystal lattice of Y2O3, forming a standard cage-like structure. The cationic site symmetry of Ce3+ is calculated to be C3i with six equal Ce–O bonds. The first four candidate isomers present different doping sites for Ce3+, which is helpful to investigate the structural evolution of Y2O3:Ce. By comparing the simulated XRD patterns with experimental data, we demonstrate the validity of the lowest energy structure. The dynamically stability of Y2O3:Ce crystal is carefully examined through the calculation of phonon dispersions. Our results of electronic band structures reveal that both of the conduction band minimum and valence band maximum are located at Γ site, leading to a band gap value of 4.20 eV. This band gap suggests a semiconductor character of Y2O3:Ce system. Interestingly, an extremely narrow valence band near Fermi level is observed in the band structure and the contribution of this band is assigned to f orbital. In addition, the calculated results of visually ELF show that the charge localizations between O-O atoms are dramatically strong, suggesting the covalent bond character of O atoms in the Y2O3:Ce crystal. These findings could provide important information of the microstructures of rare-earth doped laser materials.
All datasets generated for this study are included in the article/supplementary material.
MJ, JW, and CZ conceived the idea. MJ, JH, YJ, YC, WS, and SL performed the calculations. MJ, JH, and WS wrote the manuscript. All authors reviewed the manuscript.
This work was financial supported in part by National Natural Science Foundation of China (Nos. 11904297, 11747139, and 11804031) and the Fundamental Research Funds for the Central Universities (SWU118055), the Scientific Research Project of Education Department of Hubei Province (No. Q20191301), the Open Research Projects of Wuhan Huada (No. NERCEL-OP2015001) and the Provincial Teaching Research Project in Hubei province of China (Grant No. 2017369).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Atsushi, T., Fumiyasu, O., and Isao, T. (1993). First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 78:134106.
Becke, A. D., and Edgecombe, K. E. (1990). A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 92, 5397–5403. doi: 10.1063/1.458517
Chien, W. C., and Yu, Y. Y. (2008). Preparation of Y2O3:Ce3+ phosphors by homogeneous precipitation inside bicontinuous cubic phase. Mater. Lett. 62, 4217–4219. doi: 10.1016/j.matlet.2008.06.041
Gieszczyk, W., Bilski, P., Kłosowski, M., Mrozik, A., Zorenko, Yu., Zorenko, T., et al. (2019). Luminescent properties of undoped and Ce3+ doped crystals in Y2O3-Lu2O3-Al2O3 triple oxide system grown by micro-pulling-down method. Opt. Mater. 89, 408–413. doi: 10.1016/j.optmat.2019.01.023
Han, Z. K., Zhang, L., Liu, M., Maria, V. G. P., and Gao, Y. (2019). The structure of oxygen vacancies in the near-surface of reduced CeO2 (111) under strain. Front. Chem. 7:436. doi: 10.3389/fchem.2019.00795
Herbst, J. F., and Waston, R. E. (1978). Relativistic calculations of 4f excitation energies in the rare-earth metals: further results. Phys. Rev. B 17, 3089–3098. doi: 10.1103/PhysRevB.17.3089
Jia, W., Wang, Y., Fernandez, F., Wang, X., Huang, S., and Yen, W. M. (2001). Photoluminescence of Ce3+,Tb3+:Y2O3 nanoclusters embedded in SiO2 sol-gel glasses. Mater. Sci. Eng. C 16, 55–58. doi: 10.1016/S0928-4931(01)00298-3
Ju, M., Lu, C., Yeung, Y., Kuang, X., Wang, J., and Zhu, Y. (2016). Structural evolutions and crystal field characterizations of Tm-doped YAlO3: new theoretical insights. ACS Appl. Mater. Interfaces 8, 30422–30429. doi: 10.1021/acsami.6b09079
Ju, M., Sun, G., Kuang, X., Lu, C., Zhu, Y., and Yeung, Y. (2017). Theoretical investigation of the electronic structure and luminescence properties for NdxY1–xAl3(BO3)4 non-linear laser crystal. J. Mater. Chem. C 5, 7174–7181. doi: 10.1039/C7TC01911D
Ju, M., Xiao, Y., Sun, W., Lu, C., and Yeung, Y. (2020). In-depth determination of the microstructure and energy transition mechanism for Nd3+-doped yttrium oxide laser crystals. J. Phys. Chem. C 124, 2113–2119. doi: 10.1021/acs.jpcc.9b10482
Ju, M., Xiao, Y., Zhong, M., Sun, W., Xia, X., Yeung, Y., et al. (2019b). New theoretical insights into the crystal-field splitting and transition mechanism for Nd3+-doped Y3Al5O12. ACS Appl. Mater. Interfaces 11, 10745–10750. doi: 10.1021/acsami.9b00973
Ju, M., Zhong, M., Lu, C., and Yeung, Y. (2019a). Deciphering the microstructure and energy-level splitting of Tm3+-doped yttrium aluminum garnet. Inorg. Chem. 58, 1058–1066. doi: 10.1021/acs.inorgchem.8b02009
Kresse, G., and Furthmuller, J. (1996). Efficient iterative schemes for ab initio totalenergy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186. doi: 10.1103/PhysRevB.54.11169
Kresse, G., and Hafner, J. (1993). Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561. doi: 10.1103/PhysRevB.47.558
Krutikova, I., Ivanov, M., Murzakaev, A., and Nefedova, K. (2020). Laser-synthesized Ce3+ and Pr3+ doped Y2O3 nanoparticles and their characteristics. Mater. Lett. 265:127435. doi: 10.1016/j.matlet.2020.127435
Kumar, P., Nagpal, K., and Gupta, B. K. (2017). Unclonable security codes designed from multicolour luminescent lanthanide doped Y2O3 nanorods for anti-counterfeiting. ACS Appl. Mater. Interfaces 9, 14301–14308. doi: 10.1021/acsami.7b03353
Li, L., Hao, J., Liu, H., Li, Y., and Ma, Y. (2014). The metallization and superconductivity of dense hydrogen sulfide. J. Chem. Phys. 140:174712. doi: 10.1063/1.4874158
Lin, Y. C., Bettinelli, M., and Karlsson, M. (2019). Unraveling the mechanisms of thermal quenching of luminescence in Ce3+−doped garnet phosphors. Chem. Mater. 31, 3851–3862. doi: 10.1021/acs.chemmater.8b05300
Liu, Y., Hu, S., Zhang, Y., Wang, Z., Zhou, G., and Wang, S. (2020). Crystal structure evolution and luminescence property of Ce3+-doped Y2O3-Al2O3-Sc2O3 ternary ceramics. J. Eur. Ceram. Soc. 40, 840–846. doi: 10.1016/j.jeurceramsoc.2019.10.022
Loitongbam, R. S., Singh, W. R., Phaomei, G., and Singh, N. S. (2013). Blue and green emission from Ce3+ and Tb3+ co-doped Y2O3 nanoparticles. J. Lumin. 140, 95–102. doi: 10.1016/j.jlumin.2013.02.049
Lu, C., Amsler, M., and Chen, C. (2018). Unraveling the structure and bonding evolution of the newly discovered iron oxide FeO2. Phys. Rev. B 98:054102. doi: 10.1103/PhysRevB.98.054102
Lu, C., and Chen, C. (2018). High-Pressure evolution of crystal bonding structures and properties of FeOOH. J. Phys. Chem. Lett. 9, 2181–2185. doi: 10.1021/acs.jpclett.8b00947
Lu, C., Li, Q., Ma, Y., and Chen, C. (2017). Extraordinary indentation strain stiffening produces superhard tungsten nitrides. Phys. Rev. Lett. 119:11550. doi: 10.1103/PhysRevLett.119.115503
Lu, C., Miao, M., and Ma, Y. (2013). Structural evolution of carbon dioxide under high pressure. J. Am. Chem. Soc. 135, 14167–14171. doi: 10.1021/ja404854x
Lupei, A., Lupei, V., and Hau, S. (2017). Vibronics in optical spectra of Yb3+ and Ce3+ in YAG and Y2O3 ceramics. Opt. Mater. 63, 143–152. doi: 10.1016/j.optmat.2016.06.024
Marin, R., Back, M., Mazzucco, N., Enrichi, F., Frattini, R., Benedetti, A., et al. (2013). Unexpected optical activity of cerium in Y2O3:Ce3+,Yb3+, Er3+ up and down-conversion system. Dalton Trans. 42, 16837–16845. doi: 10.1039/C3DT51297E
Masanori, N., Akira, M., Daisuke, U., Yuki, M., Yosuke, G., Yoshikazu, M., et al. (2020). Flux growth and superconducting properties of (Ce,Pr)OBiS2 single crystals. Front. Chem. 8:44. doi: 10.3389/fchem.2020.00044
Ming, C., Pei, M., Ren, X., Xie, N., Cai, Y., Qin, Y., et al. (2018). Improving luminescent penetrability by Tm3+/Ce3+ doped Y2O3 nanocrystals for optical imaging. Mater. Lett. 218, 154–156. doi: 10.1016/j.matlet.2018.01.169
Momma, K., and Izumi, F. (2011). VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276. doi: 10.1107/S0021889811038970
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Richard, D., and Peter, E. B. (1993). Crystal Orbital Hamilton Populations (COHP). Energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617–8624. doi: 10.1021/j100135a014
Savin, A., Jepsen, O., Flad, J., Andersen, O. K., Preuss, H., and von Schnering, H. G. (1992). Electron localization in solid-state structures of the elements: the diamond structure. Angew. Chem. Int. Ed. Engl. 31, 187–188. doi: 10.1002/anie.199201871
Stefan, M., Volker, L. D., Andrei, L. T., and Richard, D. (2016). LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035. doi: 10.1002/jcc.24300
Taibeche, M., Guerbous, L., Boukerika, A., Kechouane, M., Nedjar, R., and Zergoug, T. (2016). Ab-initio simulations at the atomic scale of an exceptional experimental photoluminescence signal observed in Ce3+- doped Y2O3 sesquioxide system. Opt. 127, 10561–10568. doi: 10.1016/j.ijleo.2016.08.092
Volker, L. D., Andrei, L. T., and Richard, D. (2011). Crystal Orbital Hamilton Population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 115, 5461–5466. doi: 10.1021/jp202489s
Wang, N., He, J., Ye, K., Song, X., and L, T. (2018). Sol-gel synthesis and enhanced 1.54 μm emission in Y2O3:Yb3+, Er3+ nanophosphors co-doped with Ce3+ ions. Infrared Phys. Technol. 93, 77–80. doi: 10.1016/j.infrared.2018.07.023
Wang, Y., Lv, J., Zhu, L., and Ma, Y. (2010). Crystal structure prediction via particleswarm optimization. Phys. Rev. B 82:094116. doi: 10.1103/PhysRevB.82.094116
Wang, Y., Lv, J., Zhu, L., and Ma, Y. (2012). CALYPSO: a method for crystal structure prediction. Comput. Phys. Commun. 183, 2063–2070. doi: 10.1016/j.cpc.2012.05.008
Wilk, G., and Wallace, R. M. (2002). Alternative gate dielectrics for microelectronics. Mater. Res. Soc. Bull. 27:3. doi: 10.1557/mrs2002.70
Keywords: crystal structure prediction (CSP), electronic propercies, structural evolution, rare earth element, theoretical calculation DFT
Citation: Ju M, Wang J, Huang J, Zhang C, Jin Y, Sun W, Li S and Chen Y (2020) The Microstructure and Electronic Properties of Yttrium Oxide Doped With Cerium: A Theoretical Insight. Front. Chem. 8:338. doi: 10.3389/fchem.2020.00338
Received: 05 March 2020; Accepted: 01 April 2020;
Published: 28 April 2020.
Edited by:
Zhenxiang Cheng, University of Wollongong, AustraliaReviewed by:
Li Zhu, Carnegie Institution for Science (CIS), United StatesCopyright © 2020 Ju, Wang, Huang, Zhang, Jin, Sun, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingjing Wang, c2N1X3dqakAxNjMuY29t; Chuanzhao Zhang, emN6MTk4NzA1MTdAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.