 Silvia Pomella
Silvia Pomella Rossella Rota
Rossella Rota
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Chem. , 13 March 2020
Sec. Medicinal and Pharmaceutical Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00178
This article is part of the Research Topic Chemical Innovative Approaches in Cancer Molecular Medicine and Translational Clinical Research View all 8 articles
The RNA-guided clustered regularly interspaced palindromic repeats (CRISPR)/associated nuclease 9 (Cas9)-based genome editing technology has increasingly become a recognized method for translational research. In oncology, the ease and versatility of CRISPR/Cas9 has made it possible to obtain many results in the identification of new target genes and in unravel mechanisms of resistance to therapy. The majority of the studies have been made on adult tumors so far. In this mini review we present an overview on the major aspects of CRISPR/Cas9 technology with a focus on a group of rare pediatric malignancies, soft tissue sarcomas, on which this approach is having promising results.
Starting from the works of Jinek et al. (2012, 2013) and Qi et al. (2013), the prokaryotic adaptive system for the defense against exogenous viruses called clustered regularly interspaced palindromic repeats (CRISPR) paired with the CRISPR-associated endonuclease 9 (Cas9) has been more and more recognized as a powerful and efficient tool for genome editing. The RNA-guided CRISPR/Cas9 consists of a small guide RNA (sgRNA) in complex with Cas9 and whose pairing with the target DNA induces a single Cas9-dependent double-strand breaks (DSBs) (as reviewed by Lino et al., 2018). The resulting editing includes deletions or insertions of specific sequences or changes of pre-existing DNA sequences in specific target regions of the genome of living cells (Figure 1). More recently, the technology has been enriched to label DNA regions (Banito et al., 2018), modulate endogenous gene expression (La Russa and Qi, 2015) or change the epigenetic status (Vojta et al., 2016). Compared to transcription activator-like effector nucleases (TALENs) and zinc-finger nucleases (ZFNs) technologies, CRISPR/Cas9 allows multiplexed analyses, is of rapid construction and easier to deliver, has a higher editing efficiency, even if off-target cleavages are more frequent (Gupta and Musunuru, 2014).
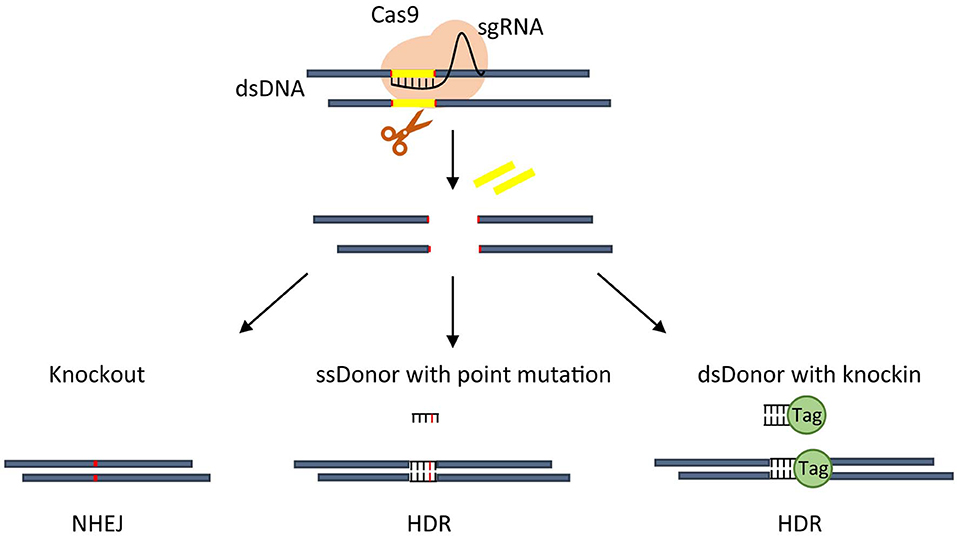
Figure 1. Overview of CRISPR-based approaches. An active CRISPR nuclease Cas9 is directed by sgRNA to make a cut on DNA that, through NHEJ repair, usually create a small indel thus allowing gene knockout (left). CRISPR-Cas9 guided cut, through HDR, can insert point mutation mediated by single strand (ss) donor with point mutation (middle), or generate an in frame knockin by using double strand (ds) donor carrying knockin sequence (right).
In the last decade, this approach has entered the basic and preclinical oncologic research on cancer cell survival, metastasis, and drug resistance.
In this minireview, we will give an overview of the CRISPR/Cas9 technology with a particular focus on its use to unravel tumorigenic mechanisms and identify pathways potentially targetable in a group of translocation-positive pediatric soft tissue sarcomas (STSs).
The repair of Cas9-created blunt-ended DSBs can be responsible for undesired effects including translocations (Kosicki et al., 2018) and/or activation of p53 (Haapaniemi et al., 2018; Ihry et al., 2018). Moreover, it often produces a mutational reproducible profile correlated to the target sequence (Brinkman et al., 2018).
The DSBs repair mainly uses the non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ is the usually active pathway in human cells, works faster than HDR but is error-prone randomly giving rise to insertions or deletions. It is able to specifically knockout a gene by creating frameshift mutations or translocations, even if with low efficiency, disrupting the coding sequence or the gene regulatory regions (as reviewed by Wang et al., 2020). Conversely, HDR is slower and more precise, operates only during certain cell cycle phases but needs a double-stranded donor to guide the repair. It is useful to introduce specific sequences (knockin) or mutations into a target genomic region or efficiently produce chromosomal translocations (as reviewed by Vanoli and Jasin, 2017).
Cas9 is a very efficient endonuclease and nucleotide binding does not require a complete identical sequence potentially allowing the annealing of mismatched sequences. Therefore, cleavages outside the on-target sites can lead to off-target genomic modifications causing unintentional impairment of normal genes expression or activation of silent genes (Fu et al., 2013; Anderson et al., 2018). Prediction and detection of off-targets events is still a key challenge for the field. Online tools have been developed to predict off-targets sites based on the specificity of sgRNA sequence (Haeussler, 2019; Li et al., 2019). In addition, biased methods based on the experimental detection of predicted off-target sites or unbiased off-target analysis to discover unintended cleavages can be applied for detecting off-targets events (Haeussler, 2019; Li et al., 2019).
Moreover, to increase the precision of DSB repair, base editing (Rees and Liu, 2018) or the very recently primed editing (Anzalone et al., 2019) have been suggested as alternative technologies, which can operate with no requirement of donor DNA templates. Primed edits can induce all the 12 possible base-to-base conversions directly into the genomic DNA decreasing off-targets. However, additional studies are needed to exploit all its potential.
Pediatric cancers affect children and adolescents and, even if considered rare due to the low number of patients, they are the leading cause of death in the young population (as reviewed by Kattner et al., 2019). They are different from the adult ones in term of pathogenic mechanisms and therapeutic approaches including the long-term side effects of therapy that highly impact the life of cancer survivors (Forrest et al., 2018).
Differently from adult cancers, characterized by a number of mutations and chromosomal alterations that accumulate over time (Siegel et al., 2020), pediatric cancers have a low mutational burden and about 10% of all pediatric tumors showed no mutation at all (Gröbner et al., 2018). This feature makes it difficult to find altered factors targetable with current therapies (Shern et al., 2014; Tirode et al., 2014; Pishas and Lessnick, 2016). This evidence also suggests that epigenetic rather than purely genetic abnormalities are involved in the malignant process (Monje, 2018).
We work in a Children's Hospital and Research Institute and we are particularly interested in solid tumors of childhood, and more specifically in rhabdomyosarcoma (RMS).
RMS is a myogenic STS of childhood, which includes translocation-positive and -negative subgroups. The most frequent recurrent chromosomal translocation t(2;13) in RMS results in the expression of the fusion oncoprotein PAX3-FOXO1, necessary for tumor cell survival (Gryder et al., 2017). Fusion-negative RMS are frequently RAS-mutated (Skapek et al., 2019). Despite currently available multimodal therapies, the prognosis for high risk patients with metastatic disease, including those harboring PAX3-FOXO1, remains dismal with a 5-year overall survival <30%, highlighting the need for novel therapeutic approaches (Missiaglia et al., 2012).
A number of STSs are dependent from fusion proteins that act as or in complexes with transcription factors (TFs) such as the afore-mentioned PAX3-FOXO1 in RMS, EWS-FLI1 in Ewing sarcomas (ES), an STS that affects also bones, and SS18-SSX in synovial sarcomas (SS) (Riggi et al., 2014; Gryder et al., 2017; McBride et al., 2018). These oncoproteins work in complexes of core regulatory (CR) TFs, specific for each type of tumor, regulating an oncogenic gene transcriptional program through binding to enhancer regions (Gryder et al., 2019a,b).
In addition, loss of specific gene components of the SWI/SNF chromatin-remodeling complex such as SMARCB1 or SMARCA4 characterizes rhabdoid sarcomas, another type of pediatric STS (Roberts et al., 2000; Lawrence et al., 2013). The SWI/SNF complex is considered a tumor suppressor and drives differentiation maintaining proper physiological enhancer functions (Alver et al., 2017; Wang et al., 2017). Overall, these TFs-dependent cancers can be considered “enhancers-driven” diseases, an aspect that has been unveiled only recently.
Unfortunately, TFs and nuclear DNA-bound complexes are difficult to target directly, and even more in the case of loss of function of a single gene. However, several new approaches are giving promising results (as reviewed by Bushweller, 2019). In this view, the identification of the bound enhancer regions and the composition and functioning of such transcriptional complexes has high relevance for the discovery of novel targetable molecules in pediatric STSs.
Enhancers are distally located short regions of non-coding DNA that “facilitate/enhance” the expression of a cis-encoded gene (Moreau et al., 1981). They act over a few to tens of kilobases, bear histone marks associated with open chromatin (H3K27ac, H3K4me1), are bound by cell-type specific TFs and then loop in 3D space to interact with their related promoters (Spitz and Furlong, 2012). In recent years, different methodologies have been applied to correctly identify, validate and characterize enhancers, among which (i) DNA sequence to map TFs binding motifs (ii) biochemical annotations to describe chromatin status (ChIP-seq, CUT&RUN, DNase-seq, MNase-seq, and ATAC-seq, DNA methylation, PRO-seq, RNA-seq), (iii) 3D conformation mapping to visualize enhancer-promoter communication (ChIA-PET, HiChIP, PLAC-seq, DNase Hi-C, 3C, 4C), (iv) massively parallel reporter assays (MPRAs) to test the functional activity of many candidate enhancers using barcodes (as reviewed by Gasperini et al., 2020). The emergent CRISPR/Cas9 technology is fully applied to perturb enhancer activity complementing the previous methodologies. CRISPR-based approaches are, indeed, used to: disrupt a TF binding site, delete the entire enhancer, engage a repressive or activating domain through a nuclease inactive dead (d)Cas9 (Qi et al., 2013) on a specific enhancer site to disrupt or activate its activity. CRISPR enables to specifically target short DNA sequences to precisely define the site necessary for TF transcriptional activity and the 3D fold looping.
In RMS we recently discovered CR TFs including PAX3-FOXO1 and the cell lineage type-specific master TFs MYOD and MYOG (Gryder et al., 2019a). We showed that these CR TFs co-regulate transcriptionally each other and activate the transcription of oncogenic genes in RMS by binding to SEs forming a tumor core regulatory circuitry (Gryder et al., 2019a). We were able to confirm the essential role of these CR TFs for cell survival and proliferation applying a CRISPR/Cas9 pooled library targeting each DNA-binding domain of all TFs expressed in one PAX3-FOXO1 RMS cell line, including the CR TFs, and comparing the results to those of all non-CR TFs (Gryder et al., 2019a).
Interestingly, EWS-FLI1 in ES binds to microsatellite chromatin regions at SEs to activate the transcription of target genes (Riggi et al., 2014). With the aim to identify mediators of EWS/FLI1 function, the component of chromatin-activating machinery BRD4 was pharmacologically inhibited in ES leading to the identification of PHF19, a protein associated with PRC2, as a novel EWS-FLI1-regulated gene (Gollavilli et al., 2018). Targeting of PHF19 with the CRISPR/Cas9 approach highlighted the contribution of this gene to the oncogenic program in ES.
In SS, the fusion oncoprotein SS18-SSX incorporates into SWI/SNF (BAF) complexes causing the eviction of the BAF47 tumor suppressor subunit, encoded by SMARCB1 (Kadoch and Crabtree, 2013). A subsequent work of the Kadoch's group demonstrated that the incorporation of SS18-SSX into the BAF complexes hijacks them from enhancers to PRC2-controlled bivalent promoters activating these latter and blocking, in the meantime, the accessibility of distal (enhancer) sites (McBride et al., 2018). This phenomenon was sufficient to foster a tumorigenic gene signature. SS18-SSX depletion led to the retargeting of BAF complexes to enhancers sites only in cells in which SMARCB1 has not been knocked out by CRISPR/Cas9, suggesting BAF47 as necessary for this step. Conversely, BAF47 appeared not needed for the proliferative arrest, as reported in rhabdoid tumors with loss of SMARCB1 (as reviewed by Kohashi and Oda, 2017), observed after SS18-SSX depletion (McBride et al., 2018). These results indicate that the roles for SSX18-SSX knock down and BAF47 rescue are functionally decoupled in SS.
Genome-scale CRISPR-Cas9 gene-knockout screens alone or in combination with small drug screenings are becoming increasingly employed in the study of pediatric cancers (Aguirre et al., 2016; Oberlick et al., 2019).
Actually, these types of large-scale perturbational screening in tumor cell lines can unveil novel therapeutic vulnerabilities in genomes lacking mutations or genomic rearrangements, which can be useful for preclinical investigations.
Through this approach, Oberlick et al. discovered a dependency of rhabdoid cells on different receptor tyrosine kinases (RTKs) among which PDGFRA and MET (Oberlick et al., 2019). They demonstrated that these kinases are up-regulated following SMARCB1 loss in cells lines and primary samples providing a proliferative advantage (Oberlick et al., 2019).
Stolte et al. (2018) used the data of their published CRISPR/Cas9 genome wide screening (Aguirre et al., 2016) to identify among others ES cell lines behaving as p53 wild-type, i.e., dependent on p53, better representing primary ESs genomic status conversely to the available p53m ES cell lines. Then, gene anti-correlated with the p53-dependency score such as MDM2, MDM4, PPM1D, and USP7 were identified as necessary for cell survival of TP53 wild-type ESs through pharmacological targeting with inhibitors or stapled peptides suggesting future clinical interventions.
Platforms for forward genetic screening using CRISPR/Cas9 have been used as a model to unravel mechanisms of drug actions and drug resistance in various cancer types. In addition, treatment of tumor cells with drugs and/or small molecules in combination with CRISPR/Cas9-induced specific deletions can reveal synthetic lethal interactions that can be potentially valuable for future treatments. It is known that tumor cells with defective DNA mismatch repair (dMMR) develop drug resistance due to mutations leading to drug-resistant alleles that can be identified by transcriptome or whole-exome sequencing (Han et al., 2017). Intriguingly, Povedano et al. (2019) exploited CRISPR/Cas9 genome editing to delete MSH2, a mismatch repair gene, in ES cells. Through this approach, they circumvent the paucity of mutations of this pediatric tumor inducing microsatellite instability and the emergence of hypermutated compound-resistant clones, which ultimately could facilitate the discovery of drug resistance mechanisms (Povedano et al., 2019).
Deletions of portions of a gene by CRISPR/Cas9 to study the contribution and requirement of specific domains for nuclear proteins has been applied to the histone demethylase KDM2B in SS (Banito et al., 2018). Banito et al. (2018) indeed, using this strategy discovered that the DNA binding domain of KDM2B, and not its enzymatic activity, is needed in SS cells to mediate the SS18-SSX binding to chromatin, which in turn leads to the de-repression of proliferative genes (Banito et al., 2018). Moreover, thanks to a tag inserted by CRISPR/Cas9 at the N-terminal of SS18-SSX, which is challenging to immunoprecipitate with usual antibodies, interactions between the fusion protein and almost all the components of the non-canonical Polycomb Repressor Complex (PRC) 1.1, among which KDM2B, were identified. Overall, a dependency of SS18-SSX on an epigenetic player like KDM2B has been thus unveiled.
Through CRISPR/Cas9 it has also been possible to model chromosomal translocations specific of ES (EWS-FLI1) and desmoplastic small round cell (DSRC) tumors (EWS-WT1) in HEK293 and human Mesenchymal cells (Torres et al., 2014; Spraggon et al., 2017) and RMS (Pax3-Foxo1) in murine myoblasts and mice in vitro (Lagutina et al., 2015).
To enhance the efficiency of rearrangements, the group of Ladanyi (Spraggon et al., 2017) combined CRISPR/Cas9 with HDR. The clear advantage of obtaining the expression of chromosomal translocations products is multiple because the role of specific fusion factors as drivers of the tumor can allow the discovery of the permissive cell context and can be exploited for drug testing.
The emerging need to study chromatin regulation in a more physiological context, led to the development of the inactive endonuclease dCas9 (Qi et al., 2013) fused to transcriptional transactivation domains of a specific factor. This complex is able to recruit chromatin effectors to a target gene by sgRNAs to repress (Gilbert et al., 2013) or activate its expression (Hilton et al., 2015) (Figure 2).
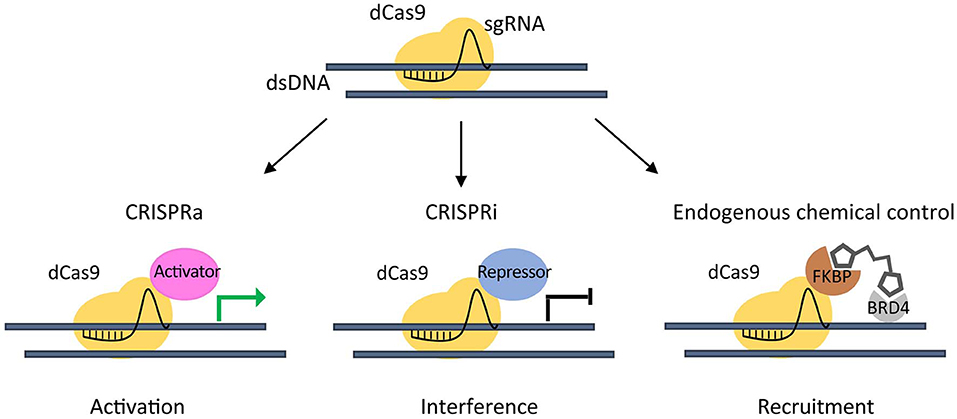
Figure 2. CRISPR/dCas9 chromatin engaging approaches. Inactive (dead) dCas9 is tethered to an activating domain (CRISPR activation- CRISPRa) that can potentially induce activation of a target gene (green arrow) (left), or to a repressive domain (CRISPR interference - CRISPRi) that can interfere gene expression (black arrow) (middle). A dCas9-FKBP fusion protein is used to recruit Chemical Epigenetic Modifiers (CEM) small molecules, in order to engage the endogenous BRD4 to a target gene (right).
Taking advantage of a dCas9 fused to truncated FKBP protein that can recruit Chemical Epigenetic Modifiers (CEM) small molecules, we were able to engage the endogenous BRD4 to target super-enhancer (SE) elements thus inducing an at least 20-fold increase in the activation of the already expressed MYOD1 gene in PAX3-FOXO1 RMS cells (Chiarella et al., 2020). This method enabled the activation of endogenous genes avoiding the need of exogenous transcriptional activators.
Genetically engineered mouse models (GEMM) of adult cancers have been created or used for the study of gene drivers by CRISPR/Cas9 (Heckl et al., 2014; Platt et al., 2014; Sanchez-Rivera et al., 2014; Chiou et al., 2015) or to generate actionable gene fusions such as EML4-ALK typical of lung cancers (Maddalo et al., 2014).
Albers et al. (2015) developed a lentiviral vector inducing the co-expression of Cas9, H-RasG12V and multiple sgRNAs against different genes such as Trp53, Cdkn2a and Pten (MuLE). By injecting MuLE vectors into the skeletal muscles of mice, they were able to show genetic cooperation between oncogenes and tumor-suppressor genes in generating models of undifferentiated sarcoma with pleomorphic (UPS) and fusion-negative RMS characteristics (Albers et al., 2015).
To obtain a fusion-positive model of RMS, a translocation of the murine Pax3-Foxo1 has been produced in mice firstly using an engineering strategy to invert Foxo1 orientation, which is in an opposite orientation to Pax3, in murine ES cells (Lagutina et al., 2015). Then the authors isolated and cultured myoblasts and fibroblasts (MEFs) from these Foxo1inv+/+ mice to virally deliver Cas9 and sgRNAs in vitro and produced a t(1;3) translocation, correspondent to the human t(2;13), and a functional Pax3-Foxo1 protein. Thus, these mice can be useful for future investigations on the tumorigenic steps of this chromosomal translocation (Lagutina et al., 2015).
Recently, a combined Cre-loxP and CRISPR/Cas9 approach in the production of GEMM of adult and pediatric sarcomas revealed the potentiality in generating UPS and malignant peripheral nerve sheath tumor (MPNST) sarcomas (Huang et al., 2017). UPS spontaneous tumors were generated in a Cre-loxP mouse model conditionally expressing oncogenic Kras and Cas9 after an intramuscular injection of an adenoviral Cre-recombinase and an sgRNA for Trp53 (the homologous of the human TP53). More interestingly, a single injection into the sciatic nerve of wild-type mice of an adenoviral vector co-expressing Cas9 and single sgRNAs individually targeting neurofibromin 1 (Nf1) and Trp53 induced the development of MPNSTs (Huang et al., 2017). These two types of sarcomas developed in vivo with a similar tumor onset, resembling their human counterparts, with similar characteristics of sarcomas obtained with the Cre-loxP system. Overall, the above CRISPR-based sarcoma models pave the way for future investigations by a growing number of researchers decreasing both the expenses and the time to obtain them.
Pediatric sarcomas are rare tumors molecularly highly heterogeneous and genetically diverse. The potentiality offered by the CRISPR/Cas9 has just begun to be exploited in this field and there is still much to study. In this “Gene Therapy Era” in which diseased people can be cured by CRISPR/Cas9-induced gene replacement, this technology has a lot to offer and new CRISPR approaches are continuously in development such as that using Cas13 nuclease able to cleave RNA molecules (Gulei et al., 2019). Several clinical studies based on CRISPR/Cas9 are underway for solid and hematological malignancies, infections (HIV and gastrointestinal), Sickle Cell Disease and Thalassemia (www.clinicaltrials.gov), which are expected to become ever more numerous.
In the study of pediatric STSs, the CRISPR/Cas9 can help in the understanding of mechanisms of malignancy development unveiling the cell(s) of origin of each subtype, which is still unknown for the majority of them, by manipulating normal cells/tissues and/or GEM models. In this regard, this technology, together or not with Cre-recombinase delivery in vivo, can be used to generate GEMM with mutations in multiple driver genes to obtain models of cancers bearing complex and heterogeneous genomic alterations.
Moreover, the huge effort in obtaining patient-derived models of rare cancers including pediatric STSs, can define a platform to combine CRISPR/Cas9 functional genomic screening with drug screenings to assist in the identification of novel targets in the “Precision Medicine” age.
It is however noteworthy that for preclinical studies aimed at identifying targetable genes, the combination of two techniques such as CRISPR/Cas9 and RNAi more robustly can confirm the results in the studied context. Indeed, the former induces gene deletions while the latter leads to suppression of gene expression, but both have specific off-target effects. In addition, in both cases tumor cells can show adaptation to long-term gene disruption/suppression, an effect that should be taken into account.
Oncological research and therapy have recently been revolutionized by the advent of immunotherapy giving hope even for forms of cancer considered incurable. A number of clinical trials are under evaluation for adoptive therapy including chimeric antigen receptor CAR-T cells in adult and pediatric sarcomas (www.clinicaltrials.gov). However, the majority of STSs show low if not a lack of T cells infiltration and inflammation and are so-called “cold tumors” (Bonaventura et al., 2019) and, consequently, not responsive to immunotherapeutic approaches requiring T cells homing. With respect to this latter aspect, the emerging data on the modulation of immune-checkpoints by CRISPR/Cas9 (Zhang et al., 2018; Afolabi et al., 2019) pave the way to a future of hope in switching the tumor microenvironment from “cold” to “hot” also in STSs.
RR conceived the paper and selected the literature. SP and RR wrote the paper, read, and approved the final manuscript.
This present work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC Project # 15312), Italian Ministry of Health Ricerca Finalizzata (Project # PE-2013-02355271), and Ricerca Corrente 2020 to RR. SP was a recipient of a Fondazione Veronesi fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Afolabi, L. O., Adeshakin, A. O., Sani, M. M., Bi, J., and Wan, X. (2019). Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology 158, 63–69. doi: 10.1111/imm.13094
Aguirre, A. J., Meyers, R. M., Weir, B. A., Vazquez, F., Zhang, C. Z., Ben-David, U., et al. (2016). Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 6, 914–929. doi: 10.1158/2159-8290.CD-16-0154
Albers, J., Danzer, C., Rechsteiner, M., Lehmann, H., Brandt, L. P., Hejhal, T., et al. (2015). A versatile modular vector system for rapid combinatorial mammalian genetics. J. Clin. Invest. 125, 1603–1619. doi: 10.1172/JCI79743
Alver, B. H., Kim, K. H., Lu, P., Wang, X., Manchester, H. E., Wang, W., et al. (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8:14648. doi: 10.1038/ncomms14648
Anderson, K. R., Haeussler, M., Watanabe, C., Janakiraman, V., Lund, J., Modrusan, Z., et al. (2018). CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods 15, 512–514. doi: 10.1038/s41592-018-0011-5
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. doi: 10.1038/s41586-019-1711-4
Banito, A., Li, X., Laporte, A. N., Roe, J. S., Sanchez-Vega, F., Huang, C. H., et al. (2018). The SS18-SSX oncoprotein hijacks KDM2B-PRC1.1 to drive synovial sarcoma. Cancer Cell 34, 346–348. doi: 10.1016/j.ccell.2018.07.006
Bonaventura, P., Shekarian, T., Alcazer, V., Valladeau-Guilemond, J., Valsesia-Wittmann, S., Amigorena, S., et al. (2019). Cold tumors: a therapeutic challenge for immunotherapy. Front. Immunol. 10:168. doi: 10.3389/fimmu.2019.00168
Brinkman, E. K., Chen, T., de Haas, M., Holland, H. A., Akhtar, W., and van Steensel, B. (2018). Kinetics and fidelity of the repair of Cas9-induced double-strand DNA breaks. Mol. Cell. 70, 801–813.e6. doi: 10.1016/j.molcel.2018.04.016
Bushweller, J. H. (2019). Targeting transcription factors in cancer - from undruggable to reality. Nat. Rev. Cancer 19, 611–624. doi: 10.1038/s41568-019-0196-7
Chiarella, A. M., Butler, K. V., Gryder, B. E., Lu, D., Wang, T. A., Yu, X., et al. (2020). Dose-dependent activation of gene expression is achieved using CRISPR and small molecules that recruit endogenous chromatin machinery. Nat. Biotechnol. 38, 50–55. doi: 10.1038/s41587-019-0296-7
Chiou, S. H., Winters, I. P., Wang, J., Naranjo, S., Dudgeon, C., Tamburini, F. B., et al. (2015). Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 29, 1576–1585. doi: 10.1101/gad.264861.115
Forrest, S. J., Geoerger, B., and Janeway, K. A. (2018). Precision medicine in pediatric oncology. Curr. Opin. Pediatr. 30, 17–24. doi: 10.1097/MOP.0000000000000570
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. doi: 10.1038/nbt.2623
Gasperini, M., Tome, J. M., and Shendure, J. (2020). Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. doi: 10.1038/s41576-019-0209-0. [Epub ahead of print].
Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. doi: 10.1016/j.cell.2013.06.044
Gollavilli, P. N., Pawar, A., Wilder-Romans, K., Natesan, R., Engelke, C. G., Dommeti, V. L., et al. (2018). EWS/ETS-driven ewing sarcoma requires BET bromodomain proteins. Cancer Res. 78, 4760–4773. doi: 10.1158/0008-5472.CAN-18-0484
Gröbner, S. N., Worst, B. C., Weischenfeldt, J., Buchhalter, I., Kleinheinz, K., Rudneva, V. A., Johann, P. D., et al. (2018). The landscape of genomic alterations across childhood cancers. Nature 555, 321–327. doi: 10.1038/nature25480
Gryder, B. E., Pomella, S., Sayers, C., Wu, X. S., Song, Y., Chiarella, A. M., et al. (2019a). Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 51, 1714–1722. doi: 10.1038/s41588-019-0534-4
Gryder, B. E., Wu, L., Woldemichael, G. M., Pomella, S., Quinn, T. R., Park, P. M. C., et al. (2019b). Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 10:3004. doi: 10.1038/s41467-019-11046-7
Gryder, B. E., Yohe, M. E., Chou, H.-C., Zhang, X., Marques, J., Wachtel, M., et al. (2017). PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 7, 884–899. doi: 10.1158/2159-8290.CD-16-1297
Gulei, D., Raduly, L., Berindan-Neagoe, I., and Calin, G. A. (2019). CRISPR-based RNA editing: diagnostic applications and therapeutic options. Expert Rev. Mol. Diagn. 19, 83–88. doi: 10.1080/14737159.2019.1568242
Gupta, R. M., and Musunuru, K. (2014). Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Invest. 124, 4154–4161. doi: 10.1172/JCI72992
Haapaniemi, E., Botla, S., Persson, J., Schmierer, B., and Taipale, J. (2018). CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930. doi: 10.1038/s41591-018-0049-z
Haeussler, M. (2019). CRISPR off-targets: a question of context. Cell Biol. Toxicol. 47:W171–W174. doi: 10.1007/s10565-019-09497-1
Han, T., Goralski, M., Gaskill, N., Capota, E., Kim, J., Ting, T. C., et al. (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 356:eaal3755. doi: 10.1126/science.aal3755
Heckl, D., Kowalczyk, M. S., Yudovich, D., Belizaire, R., Puram, R. V., McConkey, M. E., et al. (2014). Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 32, 941–946. doi: 10.1038/nbt.2951
Hilton, I. B., D'Ippolito, A. M., Vockley, C. M., Thakore, P. I., Crawford, G. E., Reddy, T. E., et al. (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517. doi: 10.1038/nbt.3199
Huang, J., Chen, M., Whitley, M. J., Kuo, H. C., Xu, E. S., Walens, A., et al. (2017). Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat. Commun. 8:15999. doi: 10.1038/ncomms15999
Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D., Theriault, K., et al. (2018). P53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946. doi: 10.1038/s41591-018-0050-6
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Jinek, M., East, A., Cheng, A., Lin, S., Ma, E., and Doudna, J. (2013). RNA-programmed genome editing in human cells. Elife 2:e00471. doi: 10.7554/eLife.00471
Kadoch, C., and Crabtree, G. R. (2013). Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85. doi: 10.1016/j.cell.2013.02.036
Kattner, P., Strobel, H., Khoshnevis, N., Grunert, M., Bartholomae, S., Pruss, M., et al. (2019). Compare and contrast: pediatric cancer versus adult malignancies. Cancer Metastasis Rev. 38, 673–682. doi: 10.1007/s10555-019-09836-y
Kohashi, K., and Oda, Y. (2017). Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 108:547–552. doi: 10.1111/cas.13173
Kosicki, M., Tomberg, K., and Bradley, A. (2018). Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771. doi: 10.1038/nbt.4192
La Russa, M. F., and Qi, L. S. (2015). The new state of the art: cas9 for gene activation and repression. Mol. Cell. Biol. 35, 3800–3809. doi: 10.1128/MCB.00512-15
Lagutina, I. V., Valentine, V., Picchione, F., Harwood, F., Valentine, M. B., Villarejo-Balcells, B., et al. (2015). Modeling of the human alveolar rhabdomyosarcoma Pax3-Foxo1 chromosome translocation in mouse myoblasts using CRISPR-Cas9 nuclease. PLoS Genet. 11:e1004951. doi: 10.1371/journal.pgen.1004951
Lawrence, M. S., Stojanov, P., Polak, P., Kryukov, G. V., Cibulskis, K., Sivachenko, A., et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. doi: 10.1038/nature12213
Li, J., Hong, S., Chen, W., Zuo, E., and Yang, H. (2019). Advances in detecting and reducing off-target effects generated by CRISPR-mediated genome editing. J. Genet. Genomics 46, 513–521. doi: 10.1016/j.jgg.2019.11.002
Lino, C. A., Harper, J. C., Carney, J. P., and Timlin, J. A. (2018). Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 25, 1234–1257. doi: 10.1080/10717544.2018.1474964
Maddalo, D., Manchado, E., Concepcion, C. P., Bonetti, C., Vidigal, J. A., Han, Y. C., et al. (2014). In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427. doi: 10.1038/nature13902
McBride, M. J., Pulice, J. L., Beird, H. C., Ingram, D. R., D'Avino, A. R., Shern, J. F., et al. (2018). The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell 33, 1128–41.e7. doi: 10.1016/j.ccell.2018.05.002
Missiaglia, E., Williamson, D., Chisholm, J., Wirapati, P., Pierron, G., Petel, F., et al. (2012). PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 30, 1670–1677. doi: 10.1200/JCO.2011.38.5591
Monje, M. (2018). Open questions: why are babies rarely born with cancer? BMC Biol. 16:129. doi: 10.1186/s12915-018-0601-9
Moreau, P., Hen, R., Wasylyk, B., Everett, R., Gaub, M. P., and Chambon, P. (1981). The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res. 9, 6047–6056. doi: 10.1093/nar/9.22.6047
Oberlick, E. M., Rees, M. G., Seashore-Ludlow, B., Vazquez, F., Nelson, G. M., Dharia, N. V., et al. (2019). Small-molecule and CRISPR screening converge to reveal receptor tyrosine kinase dependencies in pediatric rhabdoid tumors. Cell Rep. 28, 2331–2344. doi: 10.1016/j.celrep.2019.07.021
Pishas, K. I., and Lessnick, S. L. (2016). Recent advances in targeted therapy for Ewing sarcoma. F1000Res. 5:F1000 Faculty Rev-2077. doi: 10.12688/f1000research.8631.1
Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. doi: 10.1016/j.cell.2014.09.014
Povedano, J. M., Liou, J., Wei, D., Srivatsav, A., Kim, J., Xie, Y., et al. (2019). Engineering forward genetics into cultured cancer cells for chemical target identification. Cell Chem. Biol. 26, 1315–1321.e3. doi: 10.1016/j.chembiol.2019.06.006
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi: 10.1016/j.cell.2013.02.022
Rees, H. A., and Liu, D. R. (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770–788. doi: 10.1038/s41576-018-0059-1
Riggi, N., Knoechel, B., Gillespie, S. M., Rheinbay, E., Boulay, G., Suvà, M. L., et al. (2014). EWS-FLI1Utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in ewing sarcoma. Cancer Cell 26, 668–681. doi: 10.1016/j.ccell.2014.10.004
Roberts, C. W. M., Galusha, S. A., McMenamin, M. E., Fletcher, C. D. M., and Orkin, S. H. (2000). Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. U.S.A. 97, 13796–13800. doi: 10.1073/pnas.250492697
Sanchez-Rivera, F. J., Papagiannakopoulos, T., Romero, R., Tammela, T., Bauer, M. R., Bhutkar, A., et al. (2014). Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431. doi: 10.1038/nature13906
Shern, J. F., Chen, L., Chmielecki, J., Wei, J. S., Patidar, R., Rosenberg, M., et al. (2014). Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 4, 216–231. doi: 10.1158/2159-8290.CD-13-0639
Siegel, R. L., Miller, K. D., and Jemal, A. (2020). Cancer statistics, 2020. CA Cancer J Clin. 70, 7–30. doi: 10.3322/caac.21590
Skapek, S. X., Ferrari, A., Gupta, A. A., Lupo, P. J., Butler, E., Shipley, J., et al. (2019). Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 5:1. doi: 10.1038/s41572-018-0051-2
Spitz, F., and Furlong, E. E. M. (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. doi: 10.1038/nrg3207
Spraggon, L., Martelotto, L. G., Hmeljak, J., Hitchman, T. D., Wang, J., Wang, L., et al. (2017). Generation of conditional oncogenic chromosomal translocations using CRISPR–Cas9 genomic editing and homology-directed repair. J. Pathol. 242, 102–112. doi: 10.1002/path.4883
Stolte, B., Iniguez, A. B., Dharia, N. V., Robichaud, A. L., Conway, A. S., Morgan, A. M., et al. (2018). Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J. Exp. Med. 215, 2137–2155. doi: 10.1084/jem.20171066
Tirode, F., Surdez, D., Ma, X., Parker, M., Le Deley, M. C., Bahrami, A., et al. (2014). Genomic landscape of ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 4, 1342–1353. doi: 10.1158/2159-8290.CD-14-0622
Torres, R., Martin, M. C., Garcia, A., Cigudosa, J. C., Ramirez, J. C., and Rodriguez-Perales, S. (2014). Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 5:3964. doi: 10.1038/ncomms4964
Vanoli, F., and Jasin, M. (2017). Generation of chromosomal translocations that lead to conditional fusion protein expression using CRISPR-Cas9 and homology-directed repair. Methods 121–122:138–145. doi: 10.1016/j.ymeth.2017.05.006
Vojta, A., Dobrinic, P., Tadic, V., Bockor, L., Korac, P., Julg, B., et al. (2016). Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 44, 5615–5628. doi: 10.1093/nar/gkw159
Wang, J., Zhang, C., and Feng, B. (2020). The rapidly advancing Class 2 CRISPR-Cas technologies: a customizable toolbox for molecular manipulations. J Cell Mol Med. 23, 6797–6804. doi: 10.1111/jcmm.15039
Wang, X., Lee, R. S., Alver, B. H., Haswell, J. R., Wang, S., Mieczkowski, J., et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295. doi: 10.1038/ng.3746
Keywords: CRISPR/Cas9, sarcoma, enhancers, chromosomal translocation, mice models
Citation: Pomella S and Rota R (2020) The CRISP(Y) Future of Pediatric Soft Tissue Sarcomas. Front. Chem. 8:178. doi: 10.3389/fchem.2020.00178
Received: 19 February 2020; Accepted: 26 February 2020;
Published: 13 March 2020.
Edited by:
Clemens Zwergel, Sapienza University of Rome, ItalyReviewed by:
Piia Kokkonen, University of Eastern Finland, FinlandCopyright © 2020 Pomella and Rota. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rossella Rota, cm9zc2VsbGEucm90YUBvcGJnLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.