
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 18 February 2020
Sec. Organic Chemistry
Volume 8 - 2020 | https://doi.org/10.3389/fchem.2020.00095
This article is part of the Research Topic Bioactive Compounds from Microorganisms View all 8 articles
 Zhiyin Yu1,2
Zhiyin Yu1,2 Hao Jiang1
Hao Jiang1 Li Wang2
Li Wang2 Feng-Xian Yang2
Feng-Xian Yang2 Jian-Ping Huang2
Jian-Ping Huang2 Chongxi Liu1,2
Chongxi Liu1,2 Xiaowei Guo1,2
Xiaowei Guo1,2 Wensheng Xiang1*
Wensheng Xiang1* Sheng-Xiong Huang2*
Sheng-Xiong Huang2*Six new pimprinine alkaloids (1–6), including four dimers, dipimprinines A–D (1–4), and two monomers, (±)-Pimprinol D (5), and pimprinone A (6), along with six known congeners (7–12), were isolated from a soil-derived actinomycete Streptomyces sp. NEAU-C99. Structures of the new compounds were elucidated by extensive spectroscopic analyses, single-crystal X-ray diffractions, and ECD calculations. Dipimprinines A–D (1–4) showed weak cytotoxic activities against five tumor cell lines, including HL-60, SMMC-7721, A-549, MCF-7, and SW-480, with IC50 values ranging from 12.7 to 30.7 μM.
Natural products, in particular secondary metabolites derived from actinomycetes, Gram-positive bacteria (Hoshino et al., 2018; Yang et al., 2018), such as antibiotics, enzymes, enzyme inhibitors, and other pharmacologically active agents (Sripreechasak et al., 2013), have contributed substantially to modern medical care (Onaka, 2017). These microbial natural products are still an attractive and indispensable resources for drug discovery due to their potential productivity of unique core skeletons, such as the antiparasitic drug ivermectin (Cragg and Newman, 2013) and the anticancer agent eribulin (Yu et al., 2011). Pimprinine is an indole alkaloid, which was first isolated from the filtrates of Streptomyces pimprina cultures in 1963 (Joshi et al., 1963). Members of this family display a range of biological activities, such as antiepileptic (Naik et al., 2001; Roy et al., 2006), platelet-aggregation-inhibitory (Miao et al., 2004), antitumor (Pettit et al., 2002), fungicidal (Zhang et al., 2012), and anti-plant-viral activities (Liu et al., 2019).
In the continuation of our chemical and biological screenings of the extracts libraries from endophytes (mainly actinomycetes) in traditional Chinese medicinal (TCM) plants and extremophiles from un- and underexplored ecological niches (Yu et al., 2016; Yang et al., 2017; He et al., 2019), the extract of Streptomyces sp. NEAU-C99, isolated from a soil sample collected in Mount Song, Henan province, China, in 2016, indicated distinct UV absorptions compared with the extracts of other strains. As a result, six new pimprinine alkaloids (1–6), along with six known congeners (7–12) including pimprinol C (7) (Raju et al., 2012), pimprinol A (8) (Raju et al., 2012), (5-(1H-indol-3-yl)oxazol-2-yl)methanol (9) (Liu et al., 2019), pimprinine (10) (Noltemeyer et al., 1982), pimprinethine (11) (Pettit et al., 2002), and WS-30581 A (12) (Wei et al., 2014), were isolated from Streptomyces sp. NEAU-C99 (Figure 1). Herein, we describe the isolation and structure elucidation of six new pimprinine alkaloids analogs (1–6), as well as their cytotoxic activities against HL-60, SMMC-7721, A-549, MCF-7, and SW-480 cell lines.
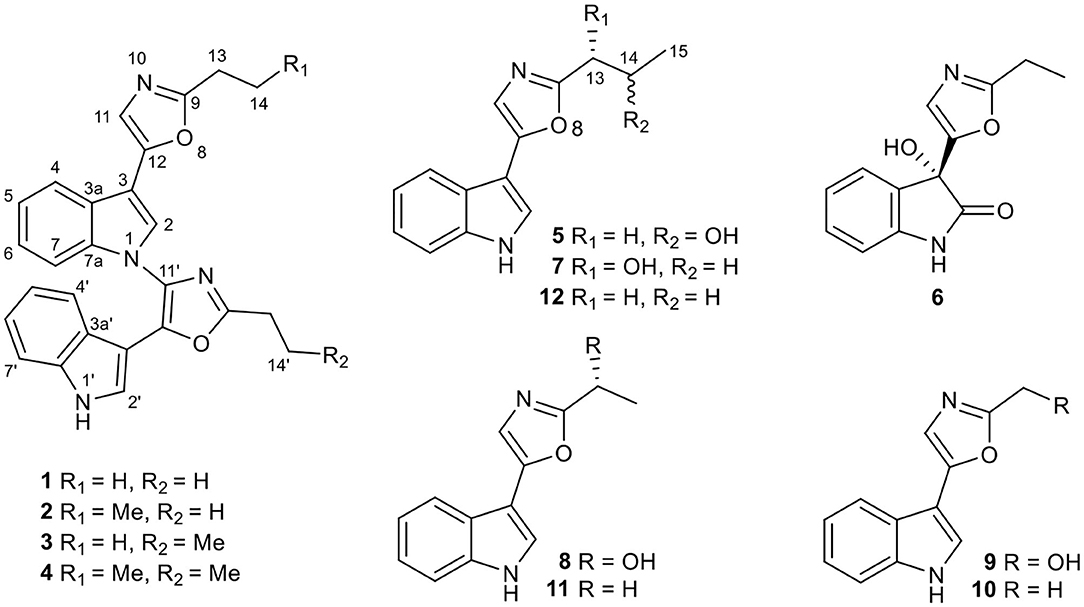
Figure 1. Chemical structures of compounds 1–12.
NMR spectra were recorded in methanol-d4 or CDCl3 using a Bruker AVANCE III-600 or AVANCE III-400 spectrometer (Bruker Corp., Switzerland), and tetramethylsilane (TMS) was used as internal standard. HRESIMS data were obtained using an Agilent G6230 Q-TOF mass instrument (Agilent Corp., USA) or a Shimadzu UPLC-IT-TOF mass instrument (Shimadzu Corp., Japan). Optical rotation data were determined in MeOH on an Autopol VI S2&Plus polarimeter (Rudolph Research Analytical, Hackettstown, USA). CD spectra were recorded on an Applied Photophysics digital circular dichroism chiroptical spectrometer (Applied Photophysics Limited, Surrey, United Kingdom). IR spectra were measured on a Nicolet™ iS™ 10 FT-IR spectrometer with KBr disks (Thermo Fisher Scientific, Waltham, USA). X-ray crystallographic analysis was carried out with a Bruker APEX DUO single crystal X-ray diffractometer (Bruker Corp., Switzerland). Thin-layer chromatography (TLC) was performed using precoated silica gel GF254 plates (0.25 mm in thickness, Qingdao Marine Chemical Inc., China), and spots were visualized by UV light (254 nm) and colored by spraying heated silica gel plates with 10% H2SO4 in ethanol. Semipreparative HPLC was conducted on a HITACHI Chromaster system (Hitachi Corp., Japan) equipped with a DAD detector, an YMC-Hydrosphere C18 column (250 × 10 mm i.d., 5 μm) at a flow rate of 3.0 mL/min and a column temperature of 25°C.
The strain Streptomyces sp. NEAU-C99 was isolated from a soil sample collected in Mount Song, Henan Province, China, in 2016. It was identified as Streptomyces sp. on the basis of the morphological characteristics and 16S rRNA gene sequence (GenBank: MN647558) with closest homology to that of Streptomyces netropsis strain SXYM16 (100% similarity, GenBank: JN999913.1).
The strain Streptomyces sp. NEAU-C99 was grown on ISP3 agar plates (Oatmeal 20 g, KNO3 0.2 g, MgSO4·7H2O 0.2 g, K2HPO4·3H2O 0.5 g, and Agar 20 g in 1 L of water, pH 7.2) for 7 days at 28°C. Then it was inoculated into 250 mL baffled erlenmeyer flasks containing 50 mL of sterile seed medium (Tryptone Soy Broth, 30 g/L) and cultivated for 2 days at 30°C on a rotary shaker (200 rpm). After that, aliquots (12.5 mL) of the seed culture were transferred into 1,000 mL baffled Erlenmeyer flasks filled with 250 mL of production medium consisting of 2% soluble starch (w/v), 2% tryptone (w/v), 1% glycerol (w/v), 0.05% NaCl (w/v), 0.05% K2HPO4·3H2O (w/v), 0.05% MgSO4·7H2O (w/v), 0.05% FeSO4·7H2O (w/v), and 0.1% KNO3 (w/v), and cultured on a rotary shaker (200 rpm) at 30°C for a week.
The fermentation broth (25 L) was centrifuged (4,000 rpm, 20 min), and the supernatant was extracted with EtOAc for three times. The EtOAc extract was subsequently evaporated in vacuo to afford 10.0 g of oily crude extract. The mycelia were extracted with methanol (1 L × 3) and then concentrated in vacuo to remove the methanol to yield the aqueous concentrate. This aqueous concentrate was finally extracted with EtOAc (1 L × 3) to give 1.0 g of oily crude extract after removing the EtOAc. Both extracts revealed an identical set of metabolites based on HPLC and TLC analyses, and therefore, they were combined for further purification.
The crude extract in total (11.0 g) was subjected to silica gel column chromatography (CC) using a successive elution of petroleum ether/EtOAc (1:0, 10:1, 5:1, 3:1, 1:1, and 0:1, v/v) to yield fractions A–F. Fr.A (petroleum ether/EtOAc, 10:1, v/v) was subjected to semipreparative HPLC (0–20.0 min, 45% CH3CN in H2O; 20.1–48.0 min, 69% CH3CN in H2O; 48.1–52.0 min, 100% CH3CN) directly to afford compounds 1 (tR = 36.4 min, 2.2 mg), 2 (tR = 41.4 min, 2.6 mg), 3 (tR = 42.4 min, 2.3 mg), and 4 (tR = 49.2 min, 2.0 mg). Fr.B (petroleum ether/EtOAc, 5:1, v/v) was further purified by semipreparative HPLC (0–20.0 min, 45% CH3CN in H2O; 20.1–48.0 min, 69% CH3CN in H2O; 48.1–52.0 min, 100% CH3CN) to give 10 (tR = 17.5 min, 30.7 mg) and 11 (tR = 24.3 min, 5.7 mg). Fr.C (petroleum ether/EtOAc, 3:1, v/v) was applied to semipreparative HPLC (0–20.0 min, 40% CH3OH in H2O; 20.1–35.0 min, 62% CH3OH in H2O; 35.1–40 min, 100% CH3OH) to obtain 12 (tR = 35.1 min, 5.0 mg). Compounds 9 (tR = 18.9 min,1.4 mg), 8 (tR = 25.3 min, 22.6 mg), 5 (tR = 32.2 min, 4.8 mg), 6 (tR = 33.2 min, 5.7 mg), and 7 (tR = 40.0 min, 8.1 mg) were obtained from fraction D (Petroleum ether/EtOAc, 1:1, v/v) by semipreparative HPLC (0–33.0 min, 48% CH3OH in H2O; 33.1–43.0 min, 56% CH3OH in H2O; 43.1–58.0 min, 78% CH3OH in H2O).
Dipimprinine A (1): yellow powder (MeOH), UV (MeOH) λmax (log ε): 227 (4.75), 266 (4.60) nm; IR (KBr) νmax 3,399, 2,962, 2,925, 2,854, 1,644, 1,572, 1,541, 1,461, 1,261, 1,098, 1,016, 802, 743 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data (see Table 1); HRESIMS m/z 421.1670 [M-H]− (calcd for C26H21N4O2, 421.1670).
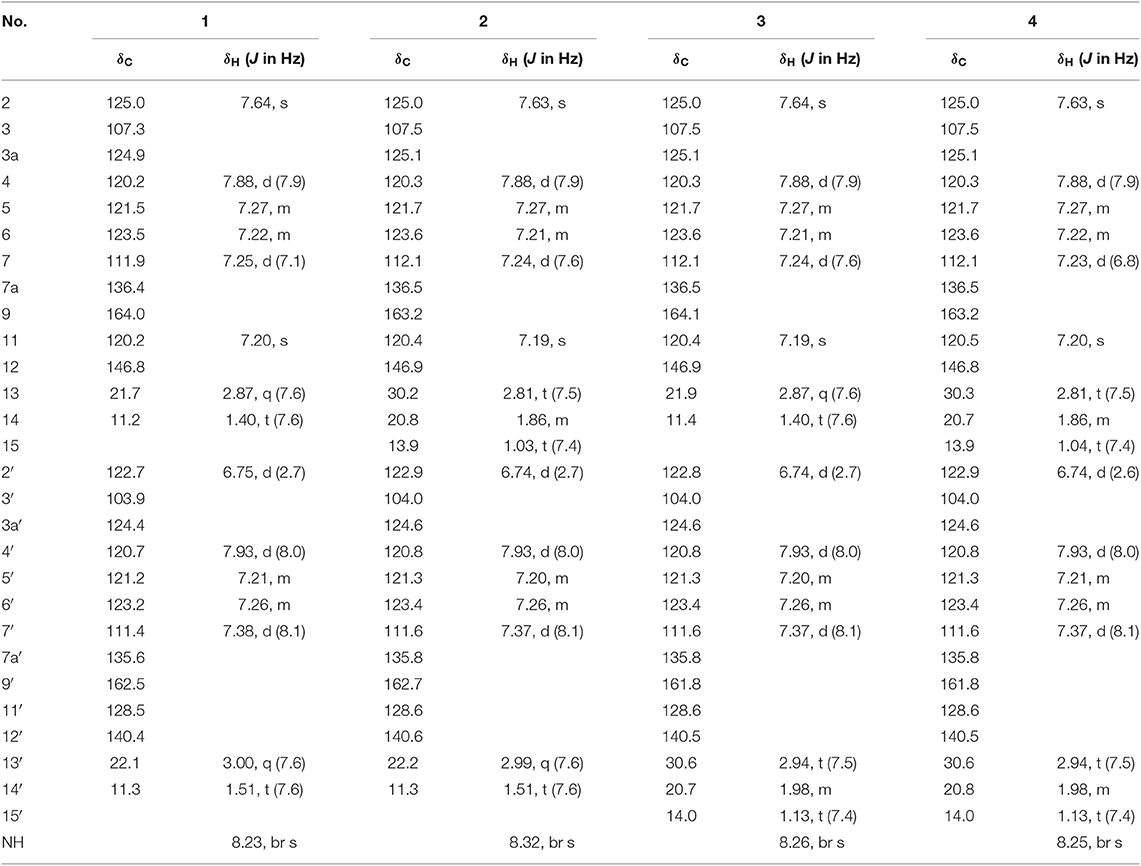
Table 1. 1H (600 MHz) and 13C (150 MHz) NMR Data of Compounds 1–4 in CDCl3.
Dipimprinine B (2): yellow powder (MeOH), UV (MeOH) λmax (log ε): 224 (4.56), 266 (4.32) nm; IR (KBr) νmax 3,411, 2,963, 2,930, 2,874, 1,642, 1,572, 1,542, 1,463, 1,236, 1,193, 1,128, 1,014, 8,01, 743 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data (see Table 1); HRESIMS m/z 435.1839 [M-H]− (calcd for C27H23N4O2, 435.1826).
Dipimprinine C (3): yellow powder (MeOH), UV (MeOH) λmax (log ε): 224 (4.56), 266 (4.31) nm; IR (KBr) νmax 3,403, 2,962, 2,927, 1,643, 1,572, 1,541, 1,462, 1,261, 1,193, 1,127, 1,099, 1,013, 803, 743 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data (see Table 1); HRESIMS m/z 435.1840 [M-H]− (calcd for C27H23N4O2, 435.1826).
Dipimprinine D (4): yellow powder (MeOH), UV (MeOH) λmax (log ε): 224 (4.74), 266 (4.52) nm; IR (KBr) νmax 3,412, 2,962, 2,929, 2,873, 1,641, 1,571, 1,542, 1,462, 1,260, 1,192, 1,097, 1,013, 803, 742 cm−1; 1H (600 MHz, CDCl3) and 13C (150 MHz, CDCl3) NMR data (see Table 1); HRESIMS m/z 449.1991 [M-H]− (calcd for C28H25N4O2, 449.1983).
(±)-Pimprinol D (5): white block crystals (CHCl3:MeOH:H2O 10:5:1), UV (MeOH) λmax (log ε): 225 (4.40), 267 (4.26) nm; IR (KBr) νmax 3,244, 2,968, 1,638, 1,581, 1,442, 1,354, 1,247, 1,133, 1,120, 1,079, 733 cm−1; 1H (600 MHz, CD3OD) and 13C (150 MHz, CD3OD) NMR data (see Table 2); HRESIMS m/z 243.1128 [M+H]+ (calcd for C14H15N2O2, 243.1128).

Table 2. 1H (600 MHz) and 13C (150 MHz) NMR Data of Compounds 5 and 6 in CD3OD.
Crystal data for 5: C14H14N2O2, M = 242.27, a = 16.7789(4) Å, b = 7.4526(2) Å, c = 19.6259(4) Å, α = 90°, β = 90°, γ = 90°, V = 2454.15(10) Å3, T = 100. (2) K, space group Pbca, Z = 8, μ(Cu Kα) = 0.724 mm−1, 25,660 reflections measured, 2,428 independent reflections (Rint = 0.0470). The final R1 values were 0.0566 [I > 2σ(I)]. The final wR(F2) values were 0.1354 [I > 2σ(I)]. The final R1 values were 0.0578 (all data). The final wR (F2) values were 0.1362 (all data). The goodness of fit on F2 was 1.126. Original crystallographic data of 5 has been deposited in the Cambridge Crystallographic Data Center (CCDC), with deposition number of CCDC1964253. Copies of the data can be obtained from the website of CCDC free of charge.
Pimprinone A (6): yellow oil (MeOH), −20.92 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 209 (4.78), 215 (4.76), 294 (3.49) nm; ECD (MeOH) λ(ε) 292 (−0.21), 278 (−0.04), 265 (−0.18), 227 (+1.06), 207 (−1.02); IR (KBr) νmax 3,212, 2,984, 1,728, 1,621, 1,561, 1,473, 1,384, 1,327, 1,225, 1,185, 1,110, 1,062, 1,001, 911, 756, 689 cm−1; 1H (600 MHz, CD3OD) and 13C (150 MHz, CD3OD) NMR data (see Table 2); HRESIMS m/z 267.0731 [M+Na]+ (calcd for C13H12N2O3Na, 267.0740).
Five tested human tumor cell lines, human leukemia (HL-60), hepatocellular carcinoma (SMMC-7721), lung cancer (A-549), breast adenocarcinoma (MCF-7), and colon carcinoma (SW-480), were purchased from ATCC (Manassas, VA, USA). Each of these cell lines was incubated in medium DMEM or RPMI-1640 containing 10% fetal bovine serum at 37°C under humidified atmosphere with 5% CO2. Cytotoxicity of the isolates toward these tumor cell lines was assessed via the 3-(4, 5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfopheny)-2H tetrazolium (MTS) (Promega, Madison, WI, USA) method (Cory et al., 1991), and cisplatin (Sigma) was used as a positive control. The cell lines were inoculated into each well of the normal 96-well plates and incubated for 12 h before addition of the test isolates. Different concentrations of each compound were added and exposed to the cells for a continuous cultivation of 48 h. The isolates with inhibition rates ≥50% against the cell lines were further assessed in triplicate at different concentrations (0.064, 0.32, 1.6, 8, and 40 μM). The IC50 values were measured based on Reed and Muench's method (Reed and Muench, 1938). All the experiments were carried out in triplicate.
Compound 1 was obtained as yellow amorphous powder, and its molecular formula C26H22N4O2 was determined by high resolution electrospray ionization mass spectrometry (HRESIMS) data (m/z 421.1670 [M-H]−, calcd for 421.1670), corresponding to 18 degrees of unsaturation (Figure S8). The 1H NMR and 1H-1H COSY data (Table 1, Figures S3, S5) indicated a 1,3-substituted indole ring with signals at δH 7.88 (1H, d, J = 7.9 Hz, H-4), 7.64 (1H, s, H-2), 7.27(m, H-5), 7.25 (1H, d, J = 7.1 Hz, H-7), and 7.22 (m, H-6) and a 3-substituted indole ring with signals at δH 7.93 (1H, d, J = 8.0 Hz, H-4′), 7.38 (1H, d, J = 8.1 Hz, H-7′), 7.26 (m, H-6′), 7.21 (m, H-5′), and 6.75 (1H, d, J = 2.7 Hz, H-2′), along with the active amine-hydrogen signal (δH 8.23, H-1′). The 13C and DEPT spectra of 1 suggested the presence of 26 carbons, which were classified into two methyls, two methylenes, 11 aromatic nonprotonated carbons, and 11 aromatic methine carbons (Table 1, Figure S4). These signals appeared in pairs in the 13C NMR spectrum, which were very similar to those of pimprinethine (Pettit et al., 2002). The aforementioned spectroscopic evidences suggested that compound 1 was likely a dimeric pimprinine alkaloid.
In unit A, the 1H–1H COSY and HSQC spectra of 1 showed two spin-coupling systems, H-14/H-13 and H-4/H-5/H-6/H-7 (Figure 2, Figures S5, S6). The HMBC cross-peaks from H-2 and H-4 to C-3/C-3a/C-7a, from H-7 to C3a/C-7a, further revealed the presence of an indole moiety. The HMBC cross-peaks from H-11 to C-9/C-12/C-3, from H2-13 to C-9/C-11/C-12, and from H-2 to C-12 were observed in the HMBC spectrum (Figure 2, Figure S7), which suggested a 2-ethyl-oxazole was connected to C-3 of an indole moiety. The above data resembled those of pimprinethine. Similarly, unit B in 1 was constructed by the following signals, correlations of H-4′ to H-7′, NH-1′/H-2′, and H2-13′/H3-14′ observed in the 1H–1H COSY spectrum, and cross-peaks of H-4′ with C-3′/C-3a′/C-7a′, H-7′ with C-3a′, NH-1′ with C-2′/C-3′/C-3a′/C-7a′, H-2′ with C-3′/C-3a′/C-7a′/C-12′, and H2-13′ with C-9′/C-11′/C-12′ observed in the HMBC spectrum. Units A and B were finally established as being bridged via the N-1–C-11′ bond based on the key HMBC correlation from H-2 to C-11′ (Figure 2). Thus, the structure of compound 1 was determined as shown in Figure 1, and named as dipimprinine A.
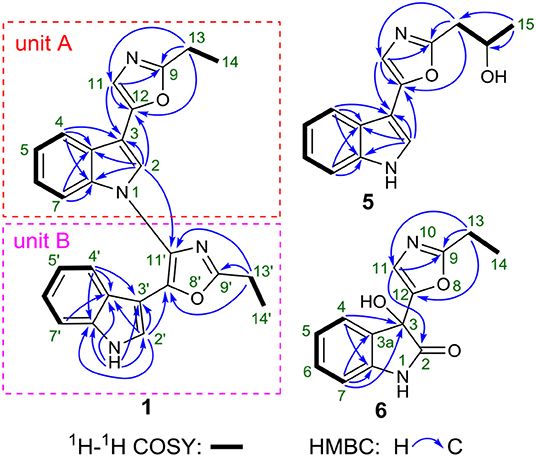
Figure 2. 2D NMR correlations of 1, 5, and 6.
Dipimprinine B (2) has a molecular formula of C27H24N4O2 as established by HRESIMS (m/z 435.1839 [M-H]−, calcd for 435.1826), which showed 14 mass units more than that of compound 1 (Figure S16). The 1H and 13C NMR spectra (Table 1, Figures S11, S12) of 2 showed high similarity to those of compound 1, except for the presence of an additional methylene at δC 30.2 (C-13) and δH 2.81 (2H, t, J = 7.5 Hz, H-13), suggesting that compound 2 was a derivative of 1. This deduction was further confirmed by the 1H–1H COSY coupling fragment of H2-13/H2-14/H3-15 and HMBC correlations from H3-15 to C-14/C-13 (Figures S2, S13, S15). Consequently, the structure of compound 2 was thus elucidated as shown (Figure 1).
Dipimprinine C (3) shared the same molecular formula C28H26N4O2 with 2 as determined by the HRESIMS ion peak at m/z 435.1840 [M-H]− (calcd for 435.1826) (Figure S24), suggesting 3 is an isomer of 2. Indeed, the 1H and 13C-NMR chemical shifts of 3 were almost the same as those of 2 (Table 1, Figures S19, S20), but differed in the 1H-NMR splitting pattern of the proton signals at δH 2.99 and δH 2.81, the signal at δH 2.99 was a quartet in 2 but a triplet in 3, while the other signal at δH 2.81 was a triplet in 2 but a quartet in 3. Based on the in-depth interpretation of its 1D NMR data (Table 1) and 2D NMR data (Figures S21–S23), particularly the 1H–1H COSY and HMBC correlations, 3 was further revealed as a structural analog of 2 with the obvious HMBC correlations from H2-13 to C-14 and from H2-13′ to C-14′/C-15′ and the 1H–1H COSY cross-peaks of H2-13/H3-14 and H2-13′/H2-14′/H3-15′ (Figure S2). Therefore, the structure of compound 3 was identified as shown in Figure 1.
Dipimprinine D (4) was isolated as yellow powder and its molecular formula was assigned as C28H26N4O2 based on HRESIMS analysis (m/z 449.1991 [M-H]−, calcd for 449.1983), with 18 degrees of unsaturation (Figure S32). Its 1H and 13C NMR data closely resembled those of 1, apart from two additional sp3 methylene resonances at δH 1.86 (2H, m, H-14), δC 20.7 (C-14) and δH 1.98 (m, 2H, H-14′), δC 20.8 (C-14′) (Table 1, Figures S27, S28). It can be inferred that the two ethyl moieties at C-9 and C-9′ in 1 were replaced by two propyl groups in 4, which was further supported by 1H–1H COSY cross-peaks of H2-13/H2-14/H3-15, and H2-13′/H2-14′/H3-15′ (Figures S2, S29). Hence, the structure of compound 4 was established.
(±)-Pimprinol D (5) possesses a molecular formula of C14H14N2O2 from its HRESIMS data (m/z 243.1128 [M+H]+, calcd for 243.1128) (Figure S40). The 13C NMR spectrum of 5 showed a total of 14 carbon resonances (Table 2). Detailed analyses of its 1D NMR and HSQC data enabled the classification of these carbons as one methyl, one methylene, one sp3 methine, six sp2 methines and five sp2 quaternary carbons (Figures S35, S36, S38). The 1H and 13C NMR spectra of 5 had similar features to those of pimprinol C (7) (Raju et al., 2012). The major difference was that the C-14 was replaced by a hydroxy group in 5, which was confirmed by the 1H–1H COSY correlations of H2-13/H-14/H3-15 (Figure 2, Figure S37). To assign the absolute configuration of 5, its X-ray diffraction data was obtained using Cu Kα radiation. Its X-ray crystallographic data showed a space group of Pbca (Yesilyurt et al., 2018; Cai et al., 2019). Detailed X-ray crystallographic analysis showed that compound 5 was a racemate, and the indole ring and the oxazole ring were almost coplanar in 5 (Figure 3). Hence, the structure of 5 was elucidated as shown in Figure 1, and it was named as (±)-Pimprinol D (5).

Figure 3. X–ray crystallographic structures of 5.
The molecular formula of pimprinone A (6) was determined as C13H12N2O3 on the basis of HRESIMS (m/z 267.0731 [M+Na]+, calcd for 267.0740), accounting for nine degrees of unsaturation (Figure S47). The 1H NMR spectrum exhibited nine signals, including one methyl signals at δH 1.28 (3H, t, J = 7.6 Hz, H-14), one methylene signal at δH 2.77 (2H, q, J = 7.6 Hz, H-13), one single olefin proton signal at δH 6.88 (s, H-11), and four mutually coupled signals of aryl protons at δH 7.44 (1H, d, J = 7.4 Hz, H-4), 7.32 (1H, td, J = 7.7, 0.9 Hz, H-6), 7.09 (1H, t, J = 7.4 Hz, H-5), and 6.94 (1H, d, J = 7.8 Hz, H-7), indicating the presence of an ortho-disubstituted benzene (Table 2, Figure S42). The 13C NMR spectrum revealed a carbonyl carbon (δC 178.0), six aromatic carbons (δC 142.9, 131.5, 130.6, 126.3, 124.0, 111.6), and an oxygenated tertiary carbon (δC 73.9) (Figure S43). These spectroscopic data implied the presence of 3-hydroxy-oxindole (Park et al., 2018) moiety in 6. The 1H and 13C NMR data of 6 were similar to those of pimprinethine (11) (Pettit et al., 2002), except that the olefinic bond at C-2/C-3 was substituted by a carbonyl (δC 178.0, C-2) and a sp3 non-protonated carbon (δC 73.9, C-3). The assumption was confirmed by the HMBC correlations from H-7 (δH 6.94) to C-3 (δC 73.9), from H-4 (δH 7.44) to C-3 (δC 73.9), and from H-11 (δH 6.88) to C-2 (δC 178.0) (Figure 2), and evidenced from the molecular formula, respectively. Analysis of the 2D NMR data confirmed that the other parts of 6 were the same as those of pimprinethine (11) (Figures S44–S46). Therefore, the planar structure of 6 was elucidated as depicted in Figure 1. To confirm the absolute configuration of 6, we then performed electronic circular dichroism (ECD) calculations of (3R)-6 using time-dependent density functional theory (TDDFT) (Supplementary Material, p. S5). The calculated ECD spectrum of 6 was in good agreement with the experimental one (Figure 4). Ultimately, the absolute configuration of the only chiral carbon C-3 in 6 was identified as R.
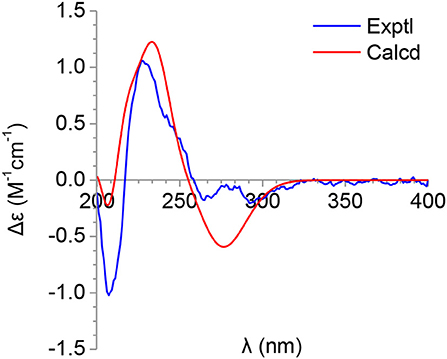
Figure 4. Calculated and experimental ECDs of 6.
As mentioned above, the stereochemistry of rings in the monomeric pimprinines turns out to be planar according to the result of X-ray crystallographic analysis (Figure 3). To investigate the possible potential axial chirality in dimeric pimprinine molecules, CD spectra for dipimprinines A–D (1–4) were acquired (Figures S10, S18, S26, S34). Unlike the reported natural dimeric atropisomers (Wang et al., 2013; Tshitenge et al., 2019), no Cotton effects can be found in any CD spectra of dipimprinines A–D (1–4). Consequently, either dipimprinines A–D (1–4) have no atropisomeric stereochemistry (that's to say a plane structure) or they were all racemates. Actually, it's more likely that compounds 1–4 have no atropisomeric stereochemistry. The carbon-nitrogen bond (N-1–C-11′) in compounds 1–4 can rotate in a circle without any steric hindrance for the reason that no substituents can be found at neither C-2 nor N-10′.
All the new compounds were evaluated for their cytotoxic activities against five human tumor cell lines, human leukemia (HL-60), hepatocellular carcinoma (SMMC-7721), lung cancer (A-549), breast adenocarcinoma (MCF-7), and colon carcinoma (SW-480), and cisplatin was used as a positive control. As shown in Table 3, compounds 1, 2, and 4 showed antiproliferative activity against breast adenocarcinoma cell line MCF-7 with IC50 values ranging from 13.8 to 18.2 μM, while the same treatment on cisplatin turned out to be an IC50 value of 26.8 μM. Compound 3 showed weak inhibitory activity against hepatocellular carcinoma cell line SMMC-7721 with an IC50 value of 25.2 μM.
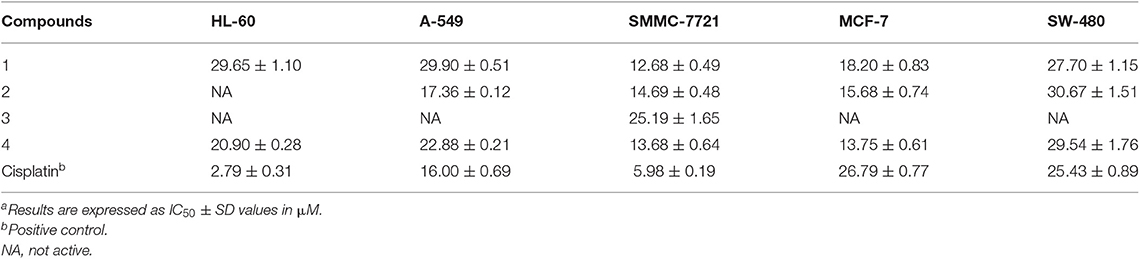
Table 3. Cytotoxicity of compounds 1–4 against five human tumor cell linesa.
In summary, this work describes the isolation and characterization of six new pimprinine alkaloids (1–6) from a soil-derived actinomycete Streptomyces sp. NEAU-C99. Their structures including absolute configurations were determined by extensive spectroscopic data, single-crystal X-ray diffraction analysis, and ECD calculations. Cytotoxicity assays showed that compounds 1, 2, and 4 displayed moderate antitumor activity against breast adenocarcinoma MCF-7. Compounds 1–4 were represented as the first examples of dimeric pimprinine alkaloids, which could further enrich the structure diversities of pimprinine alkaloids.
All datasets for this study are included in the article/Supplementary Material.
ZY performed the experiments, identified the structures, and prepared the original manuscript. HJ isolated and identified the strain, and conducted the cytotoxicity assay. LW collected the spectrographic data, assisted with the structure elucidation and manuscript revision. F-XY, J-PH, CL and XG revised the manuscript. WX and S-XH designed and supervised the research and revised the manuscript.
This research was financially supported by the National Natural Science Foundation of China to S-XH (U1702285 and 81522044), and to WX (31672092), Key Research Program of Frontier Sciences and the Strategic Priority Research Program, CAS (QYZDB-SSW-SMC051 and XDB27020205), Yunnan Innovative Research Team for Discovery and Biosynthesis of Bioactive Natural Products (2018HC012), and the Youth Innovation Promotion Association of CAS (2018424).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thanked Dr. Xiao-Nian Li at Analysis and Testing Center, Kunming Institute of Botany, Chinese Academy of Sciences, for the measurement and analysis of the single-crystal X-ray diffraction data, and Dr. Jing Yang, associate professor in State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, for the help of theoretical ECD calculations.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00095/full#supplementary-material HRESIMS, IR, CD, 1D and 2D NMR spectra of compounds 1–6, detailed ECD calculations of compound 6, as well as the NMR data of compounds 7–12 (PDF).
Cai, Z.-Q., Zhao, C.-K., Li, M.-Y., Shuai, X.-M., Ding, H.-G., Wang, Q.-L., et al. (2019). Synthesis, crystal structure and biological activity of 6-(3-chloropropoxy)-4-(2-fluorophenylamino)-7-methoxyquinazoline. J. Chem. Res. 43, 97–100. doi: 10.1177/1747519819841831
Cory, A. H., Owen, T. C., Barltrop, J. A., and Cory, J. G. (1991). Use of an aqueous soluble tetrazolium/ formazan assay for cell-growth assays in culture. Cancer Commun. 3, 207–212. doi: 10.3727/095535491820873191
Cragg, G. M., and Newman, D. J. (2013). Natural products: a continuing source of novel drug leads. Biochim. Biophys. Acta 1830, 3670–3695. doi: 10.1016/j.bbagen.2013.02.008
He, X., Wang, Y., Luo, R.-H., Yang, L.-M., Wang, L., Guo, D., et al. (2019). Dimeric pyranonaphthoquinone glycosides with anti-HIV and cytotoxic activities from a soil-derived Streptomyces. J. Nat. Prod. 82, 1813–1819. doi: 10.1021/acs.jnatprod.9b00022
Hoshino, S., Ozeki, M., Awakawa, T., Morita, H., Onaka, H., and Abe, I. (2018). Catenulobactins A and B, heterocyclic peptides from culturing Catenuloplanes sp. with a mycolic acid-containing bacterium. J. Nat. Prod. 81, 2106–2110. doi: 10.1021/acs.jnatprod.8b00261
Joshi, B. S., Taylor, W. I., Bhate, D. S., and Karmarkar, S. S. (1963). The structure and synthesis of pimprinine. Tetrahedron 19, 1437–1439. doi: 10.1016/S0040-4020(01)98569-2
Liu, B., Li, R., Li, Y., Li, S., Yu, J., Zhao, B., et al. (2019). Discovery of pimprinine alkaloids as novel agents against a plant virus. J. Agric. Food. Chem. 67, 1795–1806. doi: 10.1021/acs.jafc.8b06175
Miao, Y. P., Wen, R., Aoshima, H., and Zhou, P. G. (2004). Synthesis and antioxidative activity of 2-substituted phenyl-5-(3'-indolyl)-oxazole derivatives. Acta Pharm. Sin. 39, 37–40. doi: 10.16438/j.0513-4870.2004.01.009
Naik, S. R., Harindran, J., and Varde, A. B. (2001). Pimprinine, an extracellular alkaloid produced by Streptomyces CDRIL-312: fermentation, isolation and pharmacological activity. J. Biotechnol. 88, 1–10. doi: 10.1016/S0168-1656(01)00244-9
Noltemeyer, M., Sheldrick, G. M., Hoppe, H. U., and Zeeck, A. (1982). 2-Ethyl-5-(3-indolyl)oxazole from Streptomyces cinnamomeus discovered by chemical screening. Characterization and structure elucidation by X-ray analysis. J. Antibiot. 35, 549–555. doi: 10.7164/antibiotics.35.549
Onaka, H. (2017). Novel antibiotic screening methods to awaken silent or cryptic secondary metabolic pathways in actinomycetes. J. Antibiot. 70, 865–870. doi: 10.1038/ja.2017.51
Park, K. J., Kim, D. H., Kim, C. S., Oh, J., Subedi, L., Son, M. W., et al. (2018). Isolation of indole alkaloid and anthranilic acid derivatives from Indigo Pulverata levis. Tetrahedron Lett. 59, 4380–4383. doi: 10.1016/j.tetlet.2018.10.057
Pettit, G. R., Knight, J. C., Herald, D. L., Davenport, R., Pettit, R. K., Tucker, B. E., et al. (2002). Isolation of labradorins 1 and 2 from Pseudomonas syringae pv. coronafaciens. J. Nat. Prod. 65, 1793–1797. doi: 10.1021/np020173x
Raju, R., Gromyko, O., Fedorenko, V., Luzhetskyy, A., and Muller, R. (2012). Pimprinols A-C, from the terrestrial actinomycete, Streptomyces sp. Tetrahedron Lett. 53, 3009–3011. doi: 10.1016/j.tetlet.2012.03.134
Reed, L. J., and Muench, H. (1938). A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 27, 493–497. doi: 10.1093/oxfordjournals.aje.a118408
Roy, S., Haque, S., and Gribble, G. W. (2006). Synthesis of novel oxazolyl-indoles. Synthesis 23, 3948–3954. doi: 10.1055/s-2006-950318
Sripreechasak, P., Matsumoto, A., Suwanborirux, K., Inahashi, Y., Shiomi, K., Tanasupawat, S., et al. (2013). Streptomyces siamensis sp. nov., and Streptomyces similanensis sp. nov., isolated from Thai soils. J. Antibiot. 66, 633–640. doi: 10.1038/ja.2013.60
Tshitenge, D. T., Bruhn, T., Feineis, D., Schmidt, D., Mudogo, V., Kaiser, M., et al. (2019). Ealamines A–H, a series of naphthylisoquinolines with the rare 7,8-coupling site, from the Congolese Liana Ancistrocladus ealaensis, targeting pancreatic cancer cells. J. Nat. Prod. 82, 3150–3164. doi: 10.1021/acs.jnatprod.9b00755
Wang, L.-Y., Wu, J., Yang, Z., Wang, X.-J., Fu, Y., Liu, S.-Z., et al. (2013). (M)-and (P)-bicelaphanol A, dimeric trinorditerpenes with promising neuroprotective activity from Celastrus orbiculatus. J. Nat. Prod. 76, 745–749. doi: 10.1021/np3008182
Wei, Y., Fang, W., Wan, Z., Wang, K., Yang, Q., Cai, X., et al. (2014). Antiviral effects against EV71 of pimprinine and its derivatives isolated from Streptomyces sp. Virol. J. 11, 195–208. doi: 10.1186/s12985-014-0195-y
Yang, G.-X., Ma, G.-L., Li, H., Huang, T., Xiong, J., and Hu, J.-F. (2018). Advanced natural products chemistry research in China between 2015 and 2017. Chin. J. Nat. Med. 16, 881–906. doi: 10.1016/S1875-5364(18)30131-6
Yang, R., Yang, J., Wang, L., Huang, J.-P., Xiong, Z., Luo, J., et al. (2017). Lorneic acid aanalogues from an endophytic actinomycete. J. Nat. Prod. 80, 2615–2619. doi: 10.1021/acs.jnatprod.7b00056
Yesilyurt, F., Aydin, A., Gul, H. I., Akkurt, M., and Ozcelik, N. D. (2018). Crystal structure of (2E)-1-(4-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one. Eur. J. Chem. 9, 147–150. doi: 10.5155/eurjchem.9.2.147-150.1733
Yu, M. J., Zheng, W., Seletsky, B. M., Littlefield, B. A., and Kishi, Y. (2011). Case history: discovery of eribulin (HALAVENTM), a halichondrin B analogue that prolongs overall survival in patients with metastatic breast cancer. Annu. Rep. Med. Chem. 46, 227–241. doi: 10.1016/B978-0-12-386009-5.00013-8
Yu, Z., Wang, L., Yang, J., Zhang, F., Sun, Y., Yu, M., et al. (2016). A new antifungal macrolide from Streptomyces sp. KIB-H869 and structure revision of halichomycin. Tetrahedron Lett. 57, 1375–1378. doi: 10.1016/j.tetlet.2016.02.061
Keywords: Streptomyces, pimprinine, alkaloids, structural characterization, cytotoxicity
Citation: Yu Z, Jiang H, Wang L, Yang F-X, Huang J-P, Liu C, Guo X, Xiang W and Huang S-X (2020) Dimeric Pimprinine Alkaloids From Soil-Derived Streptomyces sp. NEAU-C99. Front. Chem. 8:95. doi: 10.3389/fchem.2020.00095
Received: 21 November 2019; Accepted: 31 January 2020;
Published: 18 February 2020.
Edited by:
Huiming Ge, Nanjing University, ChinaReviewed by:
Yongbo Xue, Huazhong University of Science and Technology, ChinaCopyright © 2020 Yu, Jiang, Wang, Yang, Huang, Liu, Guo, Xiang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wensheng Xiang, eGlhbmd3ZW5zaGVuZ0BuZWF1LmVkdS5jbg==; Sheng-Xiong Huang, c3hodWFuZ0BtYWlsLmtpYi5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.