 Fang Ying
Fang Ying Yutong Zhang1
Yutong Zhang1 Hujun Xie
Hujun Xie- 1Department of Applied Chemistry, Zhejiang Gongshang University, Hangzhou, China
- 2Hangzhou Environmental Monitoring Center Station, Hangzhou, China
- 3Department of Chemistry, Zhejiang University, Hangzhou, China
DFT calculations were performed to elucidate mechanistic details of an unusual palladium-catalyzed methylcyclopropanation from [2 + 1] cycloadditions of (Z)-2-bromovinylbenzene and endo-N-(p-tolyl)-norbornenesuccinimide. The reaction proceeds via oxidative addition (OA), intermolecular alkene insertion, deprotonation/protonation, intramolecular alkene insertion, β-H elimination and reductive elimination (RE). Protonation is the rate-limiting step and requires an overall barrier of 28.5 kcal/mol. The sources of two protons for protonation and exchange have also been clarified and the calculations agree with experimental observations.
Introduction
Cyclopropane skeleton has attracted tremendous attention from organic chemists and can be found in many important biomolecules and pharmaceutical drugs (Hofmann et al., 1954; Crowley et al., 1961; Wiberg, 1996; de Meijere, 2003; Fedorynski, 2003; Lebel et al., 2003; Pietruszka, 2003; Reissig and Zimmer, 2003; Wessjohann et al., 2003; Hata et al., 2011; Chen et al., 2014; Hiratsuka et al., 2014). Many methods have been used to construct the cyclopropane scaffold, including transition metal mediated C–C and C–H bond activations (Satake and Nakata, 1998; Goudreau and Charette, 2010; Oonishi et al., 2012; Masutomi et al., 2014; Du et al., 2015), carbene/carbenoid cycloadditions (Miki et al., 2002; Biswas et al., 2012; Lindsay et al., 2013), Simmons–Smith reactions (Simmons and Smith, 1958; Beaulieu et al., 2013), Michael-initiated ring closure (MIRC) (Xie et al., 2007; Xuan et al., 2009), cycloisomerizations (Bruneau, 2005; Miege et al., 2011), and the coupling of norbornenes with organoboron reagents or alkynes (Bigeault et al., 2005; Miura et al., 2006).
However, the cyclopropanation of halohydrocarbon with alkenes catalyzed by transition metal catalysts by a non-carbene mechanism is still underdeveloped (Mao and Bao, 2014a; Mao et al., 2014). Recently, we firstly reported the palladium-catalyzed methylcyclopropanation of bromostyrenes with norbornenes via [2 + 1] cycloaddition, and the reactions proceed by a methylene protonation and a H/D exchange with CD3OD (Mao et al., 2015). A methylcyclopropane group was constructed through a three-fold domino method including an important protonation process. The experimental results demonstrated that a norbornenylpalladium intermediate could capture one proton from research systems (Palucki et al., 1997; Torraca et al., 2000; Kuwabe et al., 2001; Matsukawa et al., 2005; Tseng et al., 2006; Dash and Janni, 2012; Mao and Bao, 2014b). The mechanistic studies revealed that the methylcyclopropanation step proceeds via a protonation and a H/D exchange with CD3OD. As shown in Scheme 1, two different deuterium atoms from CD3OD were chemoselectively added into the two positions of methylcyclopropane derivatives. Herein, quantum chemistry (QC) calculations have been used to elucidate the reaction mechanisms, and the protonation step and a H/D exchange process from CD3OD have also been explored and discussed.
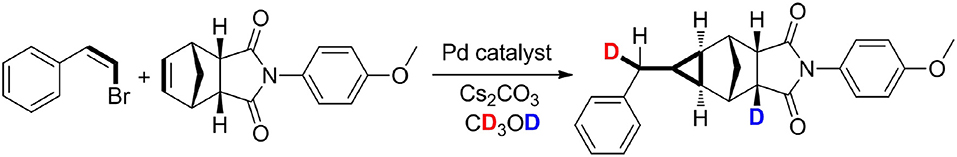
Graphical Abstract. Palladium-Catalyzed Methylcyclopropanation of Norbornenes With Vinyl Bromides.
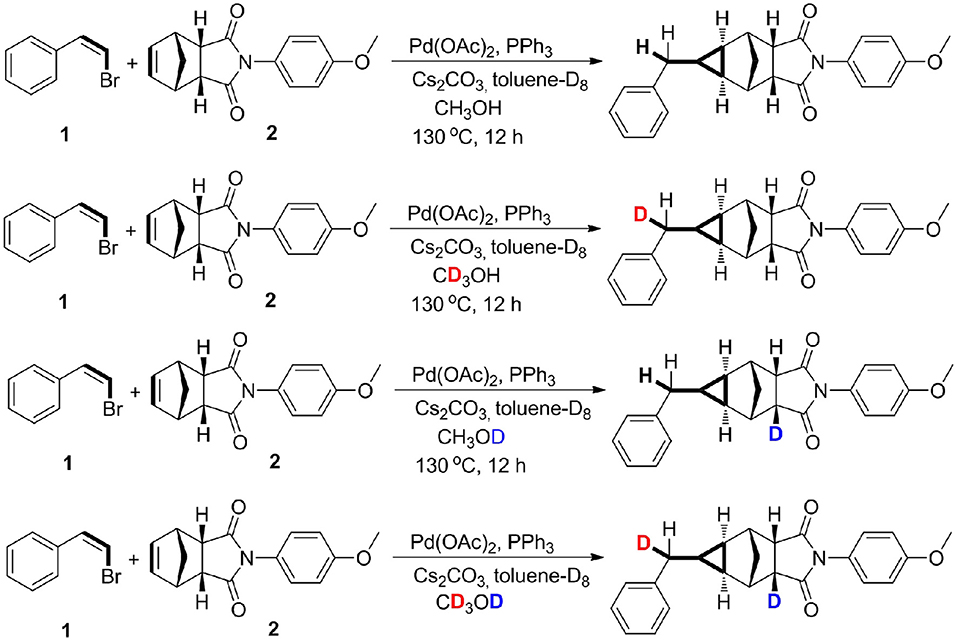
Scheme 1. Deuterium-labeling studies.
Computational Methods
All of species were optimized through M06 functional (Zhao and Truhlar, 2006a,b, 2008) in combination with 6-31G(d,p) basis set for H, C, O and N atoms. The Pd, P, Br, and Cs atoms were described by LANL2DZ basis set (Ehlers et al., 1993; Check et al., 2001). The polarization functions involving Pd(ζf) = 1.472 (Huzinaga, 1984), Br(ζd) = 0.389, P(ζd) = 0.340, and Cs(ζf) = 0.306 were also added (Amatore et al., 1992). The structural parameters of complex 1 from calculations are consistent with the measured parameters from experiments (Figure 1; Mao et al., 2015) suggesting that the computational method in our calculations is right. Frequency analyses have been used to obtain the zero-point energies (ZPE), and then confirmed the transition states with only one imaginary frequency and the intermediates with zero imaginary frequency. Each transition state was also validated through intrinsic reaction coordinate calculations to connect the reactant and product (Fukui, 1970, 1981). Natural bond orbital (NBO) was carried out to obtain atomic charge distribution (Reed and Weinhold, 1985; Reed et al., 1985, 1988). In order to reduce the costs for computation, the triphenylphosphine (PPh3) ligand used in experiments was replaced by trimethylphosphine (PMe3), and the reliability of this models has been validated by previous calculations (Xie et al., 2013a,b). All calculations were performed by Gaussian09 software (Frisch et al., 2009).
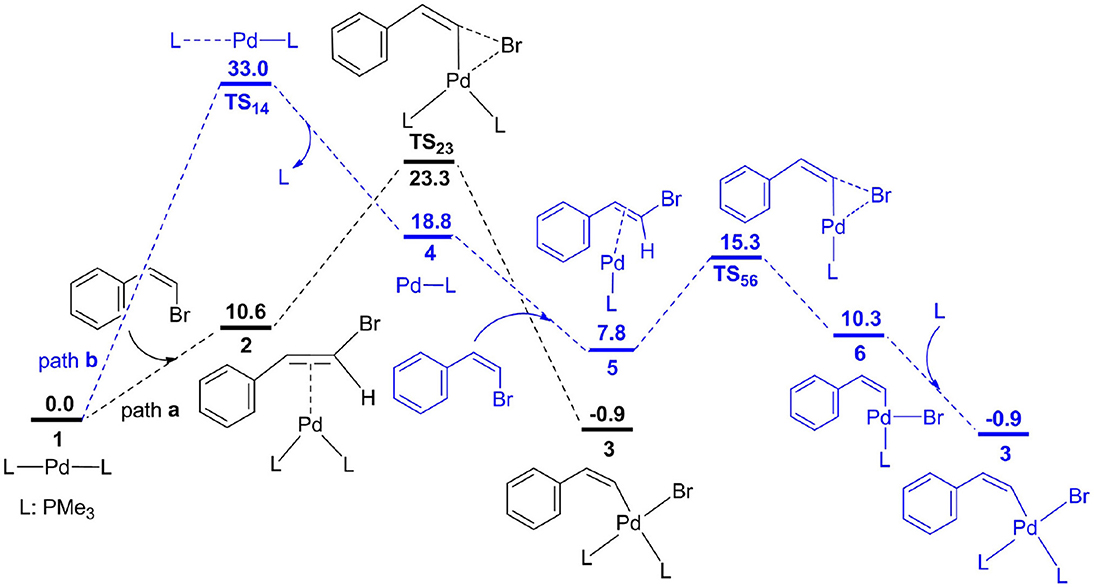
Figure 1. Free energy profiles for two possible oxidative addition pathways.
A continuum medium strategy based on the optimized species in gas-phase was performed to obtain single point energy in solvent. We selected the conductor-like polarizable continuum model (CPCM) involving an UAHF radii method (Barone and Cossi, 1998; Cossi et al., 2003). Toluene was utilized as solvent based on reaction conditions.
The entropy change was taken into consideration in a bimolecular process, and the corrections were added to the free energies based on the free volume theory (Benson, 1982). For 2 to 1 (or 1 to 2) change, a correction of −2.6 (or 2.6) kcal/mol was necessary. The corrections have been validated by previous calculations (Okuno, 1997; Ardura et al., 2005; Liu et al., 2009, 2012; Schoenebeck and Houk, 2010; Wang et al., 2012a,b). The relative Gibbs free energies from solvent were adopted to analyze the reaction mechanisms in this manuscript.
Results and Discussion
Oxidative addition is expected to be the initial step for Pd-catalyzed methylcyclopropanation of norbornene with vinyl bromide, and the corresponding free energy profiles are shown in Figure 1, and optimized geometries for different transition states are described in Figure 2. From palladium bisphosphine complex 1, two possible pathways for the formation of complex 3 are proposed. Path a (black) is related to the bisphosphine pathway and path b (blue) involves the monodentate phosphine pathway. The calculation results showed that path a is preferred. In path a, the double bond of substrate (Z)-2-bromovinylbenzene is coordinated to the Pd center to produce complex 2, and the process is endergonic via 10.6 kcal/mol. Subsequently, the three-membered ring oxidative addition transition state has been located with an overall barrier of 23.3 kcal/mol from 1 to TS23, and generates a square-planar complex 3. In path b, one phosphine ligand of complex 1 is dissociated to give complex 4, and the barrier is predicted to be 33.0 kcal/mol for dissociation process based on the method proposed by Hall and coworkers (Hartwig et al., 2005). From 4, the substrate enters into reaction system to yield complex 5, followed by oxidative addition with a barrier (TS56) of 7.5 kcal/mol to afford a three-coordinate complex 6. Finally, complex 3 is produced via the coordination of phosphine ligand.
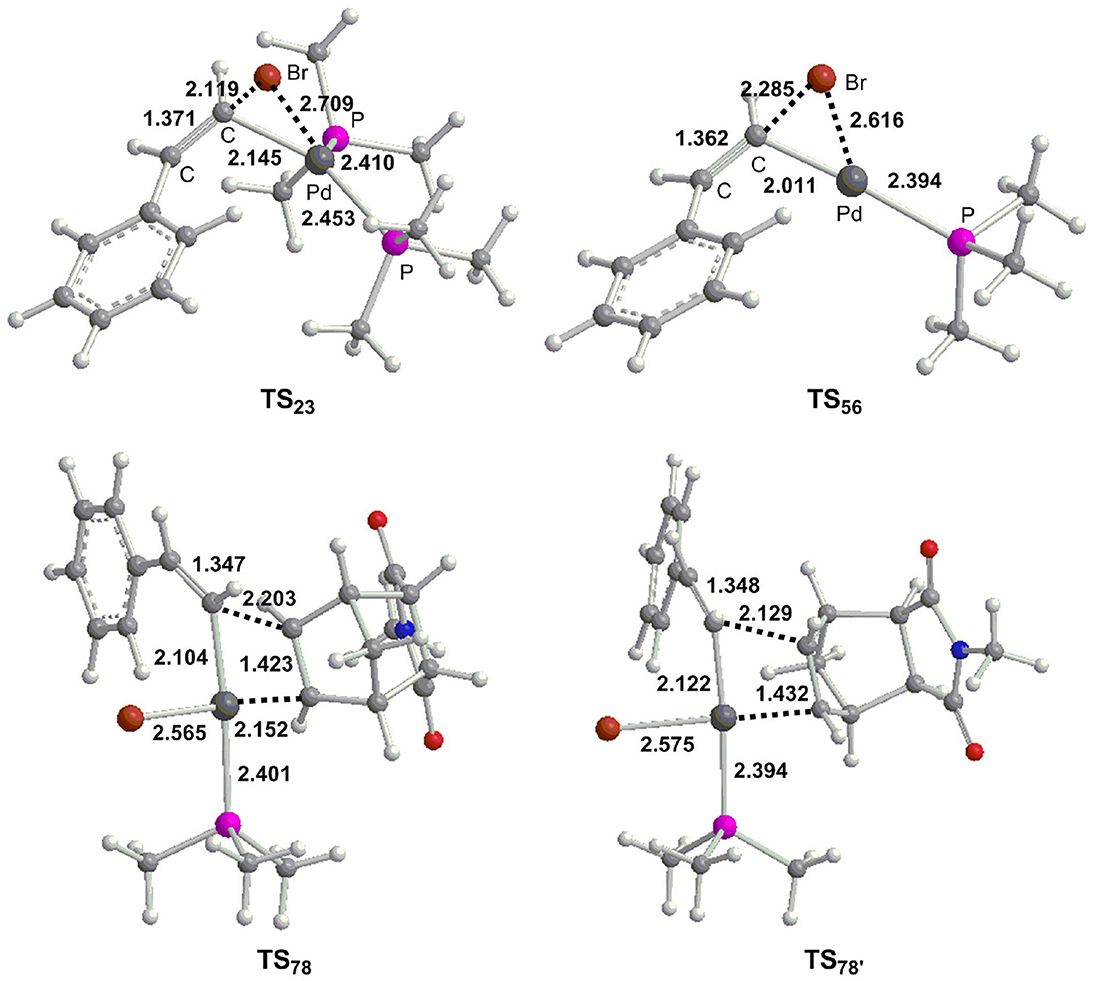
Figure 2. Optimized geometries (Å) for selected transition states.
From 3, the reaction proceeds by intermolecular alkene insertion step, and two possible pathways are presented considering different coordination directions of endo-N-(p-tolyl)-norbornenesuccinimide (Figure 3). In path c, two bridge-hydrogen atoms and the bridge-carbon atom of norbornene moieties are outside of the plane. While in path d, two bridge-hydrogen atoms and the bridge-carbon atom of norbornene moieties locate inside of the plane. According to the calculations, path c (12.2 kcal/mol for TS78) is more favorable than path d (17.3 kcal/mol for ) by 5.1 kcal/mol, then a stable four-coordinate intermediate 8 is formed and this process is obviously exergonic by 17.7 kcal/mol.
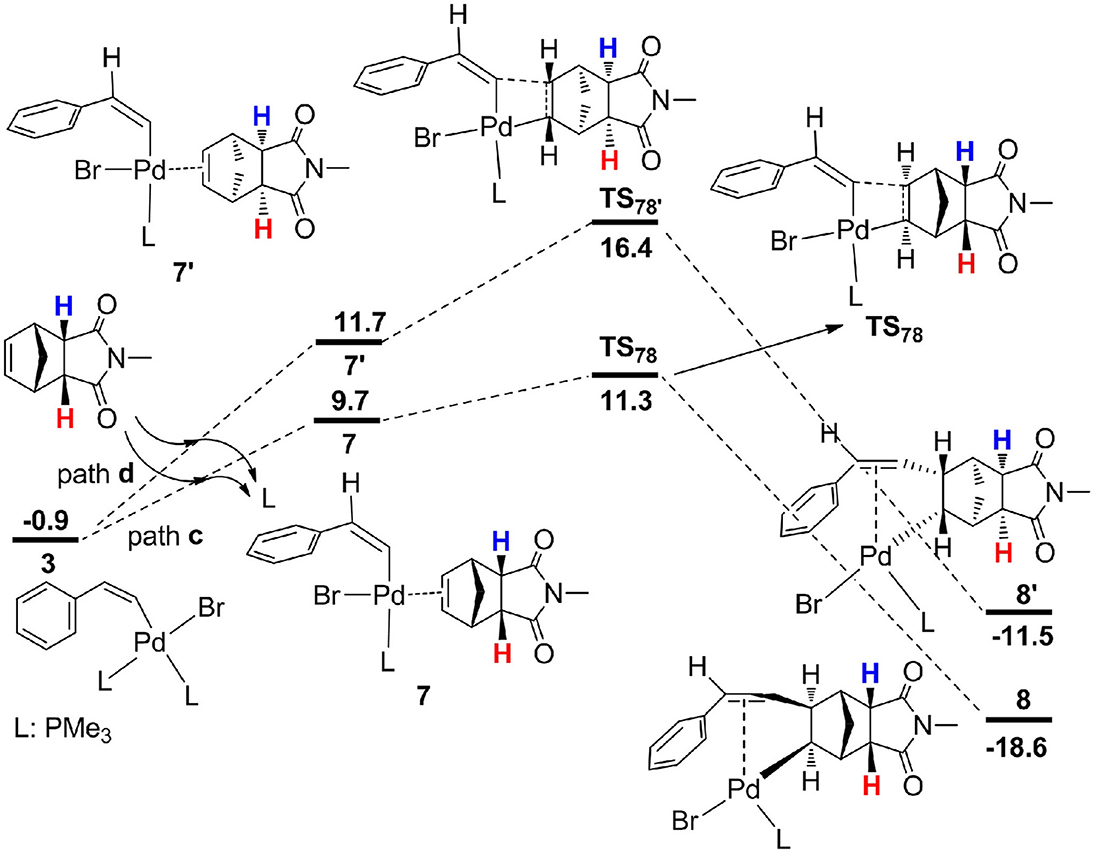
Figure 3. Free energy profiles for two possible intermolecular alkene insertion pathways.
From 8, we consider the possibility for the formation of ion pair complex 9' as described in previous experiments (Mao et al., 2015). The calculations showed the relative Gibbs free energy of this complex is very high with a value of 68.1 kcal/mol (Figure 4), thus we exclude this possibility. Alternatively, we investigate the key role of base in deprotonation, which has been confirmed in previous experiments (Wasa et al., 2009; Liang et al., 2012) and calculations (Biswas et al., 2000; Davies et al., 2005; Lafrance et al., 2007; Ess et al., 2008; Kefalidis et al., 2010; Figg et al., 2013; Xie et al., 2013c, 2016). However, it is interesting to note that the γ-H1 in complex 8 is far away from palladium center with the Pd–H1 distance of 5.268 Å (Figure 4), therefore, it is very difficult to activate this C–H1 bond. The γ-C–H activation has been previously accomplished by Yu et al. (Li et al., 2014; Jiang et al., 2016; Wu et al., 2016; Shao et al., 2017, 2018; Zhu et al., 2018), and they developed a weakly coordinating directing group to help the C–H bond activation. From 8, the ligand substitution of Cs2CO3 and for Br− occurs to give a stable complex 9, where the γ-H1 generates weak hydrogen bond interaction with the oxygen atom of . The γ-C–H1 distance is 1.110 Å in complex 9 (Figure 5), indicating that this bond has been activated. Subsequently, the deprotonation is easy to take place to give complex 10 with a barrier (TS9−10) of only 8.9 kcal/mol. The C–H1 and O–H1 bond length in TS9−10 are 1.430 Å and 1.221 Å, respectively (Figure 5). For comparison, the α-H and β-H on the same side of Pd center can be activated by palladium center, and the barriers for α-H (26.8 kcal/mol) and β-H (14.2 kcal/mol) are much higher than that of γ-H. From 10, the ligand substitution of five CH3OH molecules for Cs2CO3 and CsHCO3 takes place to generate an unstable complex 10, and this process is significantly endergonic by 25.7 kcal/mol, accompanied by protonation via TS11−12 to yield complex 12. It is worth noting that the proton comes from hydroxyl of methanol. An overall barrier of protonation step is 28.5 kcal/mol from 10 to TS11−12, which is the rate-limiting step of catalytic cycle. We have used several density functionals including B3LYP-D3 (Becke, 1993; Stephens et al., 1994), TPSS (Tao et al., 2003), M06-2X (Zhao and Truhlar, 2008), WB97X-D (Chai and Head-Gordon, 2008) to evaluate the functional dependency of this transition metal system. The calculations demonstrated that different functionals have slight effect on the rate-determining state. The barriers (TS11−12) for B3LYP-D3, TPSS, M06-2X, and WB97X-D are 26.9, 31.2, 29.3, and 27.8 kcal/mol, respectively. From 12, intramolecular alkene insertion occurs to give a cyclopropanepalladium complex 13 and it requires a barrier (TS12−13) of only 3.0 kcal/mol. Then complex 14 is generated via the release of four methanol molecules. We know that the γ-H1 in complex 8 is far away from palladium center, thus five CH3OH molecules are necessary to form the hydrogen bonding network between γ-H1 and Pd center for proton transfer in TS11−12. In addition, we also considered the influence of methanol number on the barriers for proton transfer, and the calculations showed that it has only slight effect. The barriers are 28.5 kcal/mol (TS11−12) for five methanol molecules, 31.4 kcal/mol (TS11−12_A) for six methanol molecules, 30.9 kcal/mol (TS11−12_B) for seven methanol molecules, and 30.3 kcal/mol (TS11−12_C) for eight methanol molecules, respectively (see Supporting Information). We also consider the other possible pathway for proton exchange with CH3OH and intramolecular alkene insertion, where the intramolecular alkene insertion occurs first (see Figure S1). The calculations illustrated that the protonation by methanol molecule is the rate-determining step for catalytic cycle, and needs much higher overall barrier (35.8 kcal/mol from 11′ to ) than the barrier mentioned above (28.5 kcal/mol from 10 to TS11−12).
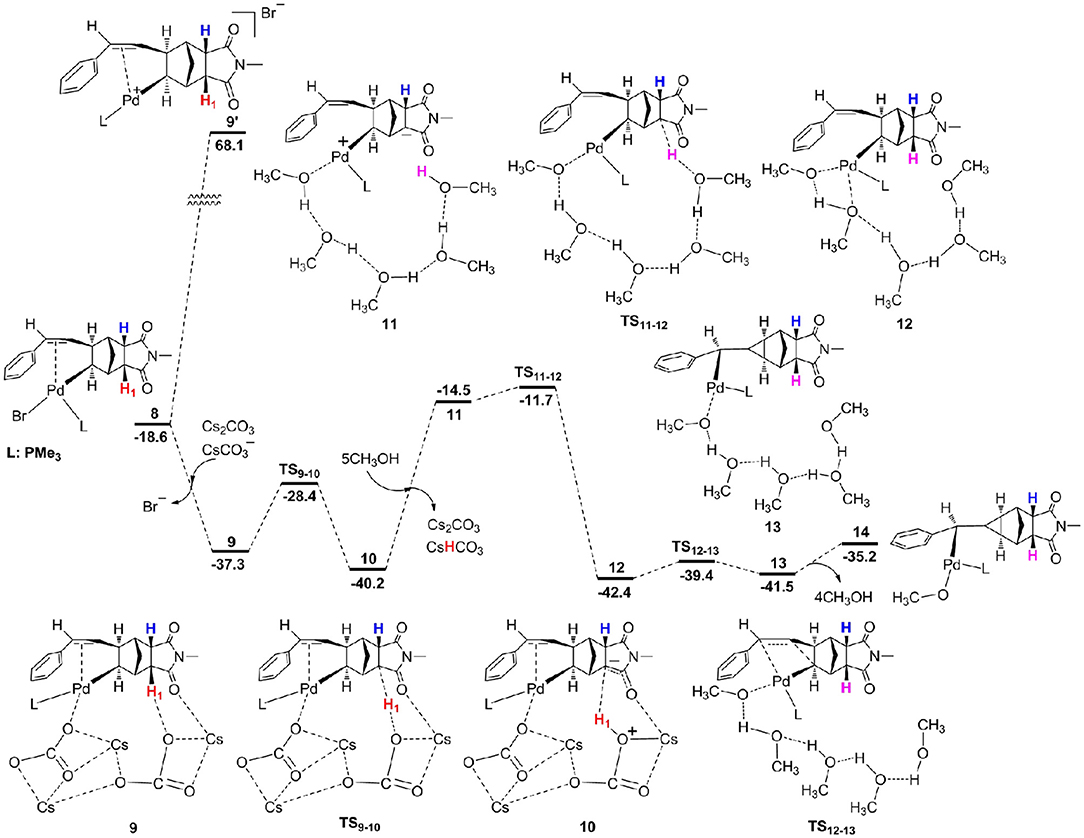
Figure 4. Free energy profiles for proton exchange with CH3OH and intramolecular alkene insertion.
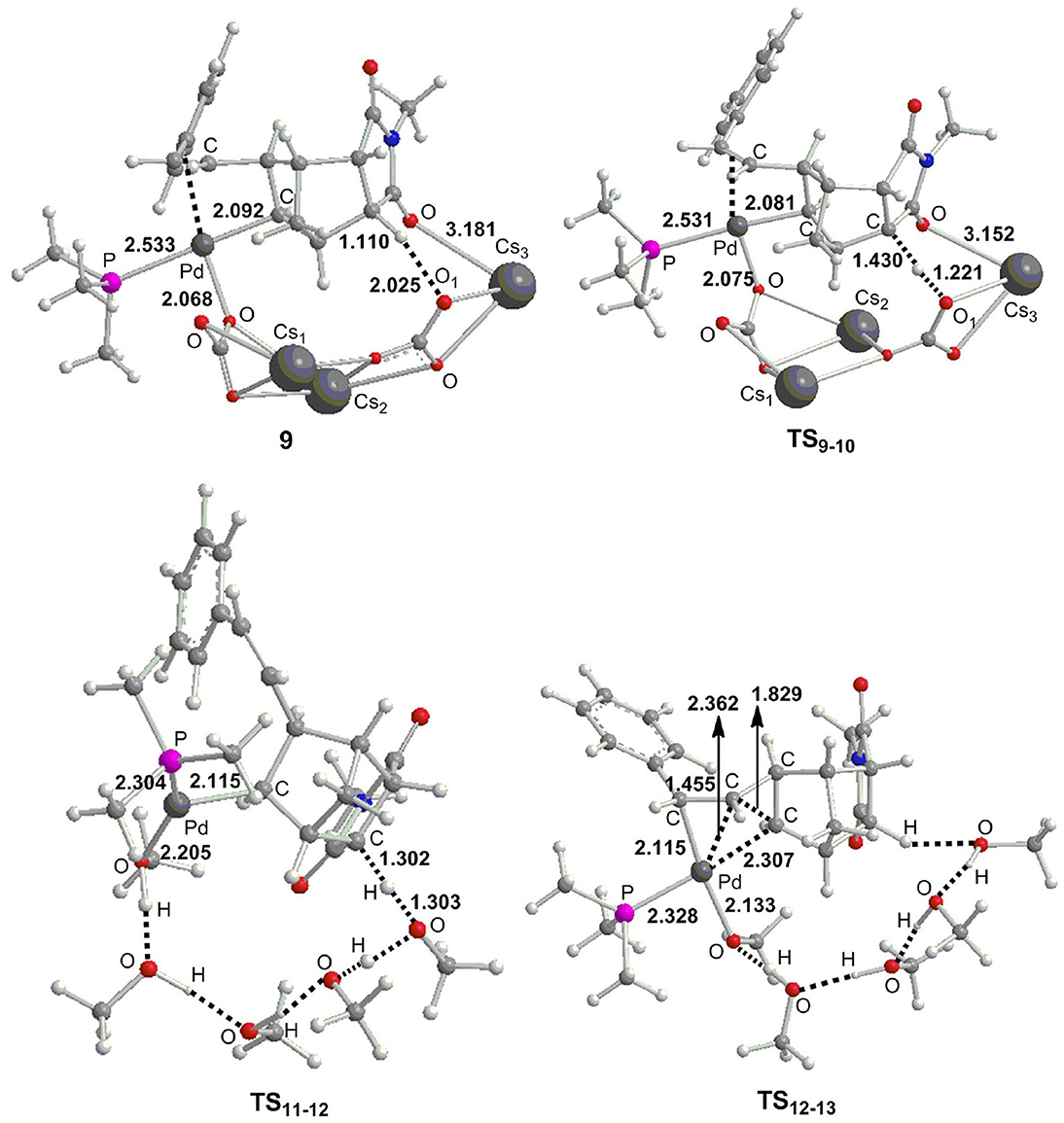
Figure 5. Optimized important geometries (Å) as presented in Figure 4.
From 14, the reaction can proceed via β-H elimination and two possible pathways are proposed due to the existence of two β-H atom for Pd center (Figure 6). One is from methoxyl group (path e) and the other is from the cyclopropane carbon-bonded hydrogen atom (path f). The calculations demonstrated that path e (16.7 kcal/mol for TS14−15) is more favorable than path d (23.0 kcal/mol for TS14−16), and optimized geometries of two transition states are described in Figure 7. Subsequently, a square-planar complex 15 is generated, followed by the release of methanal to produce complex 17. A methylcyclopropane product is then formed via the C–H bond reductive elimination, and it needs a barrier (TS17−4) of 9.5 kcal/mol. Finally, one phosphine ligand is coordinated to the Pd center to regenerate the catalyst. It is clearly to see that the proton for the protonation of a methylcyclopropane subunit comes from the methyl of CH3OH, which is consistent with the deuterium-labeling experiments (Fedorynski, 2003).
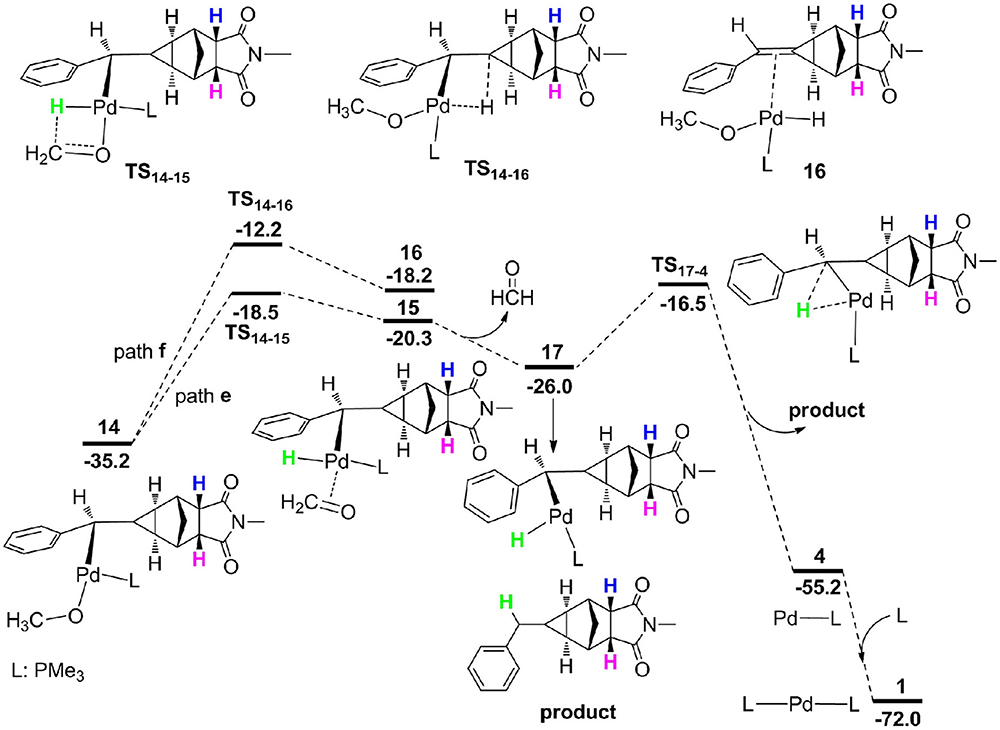
Figure 6. Free energy profiles for β-H elimination and C–H bond reductive elimination.
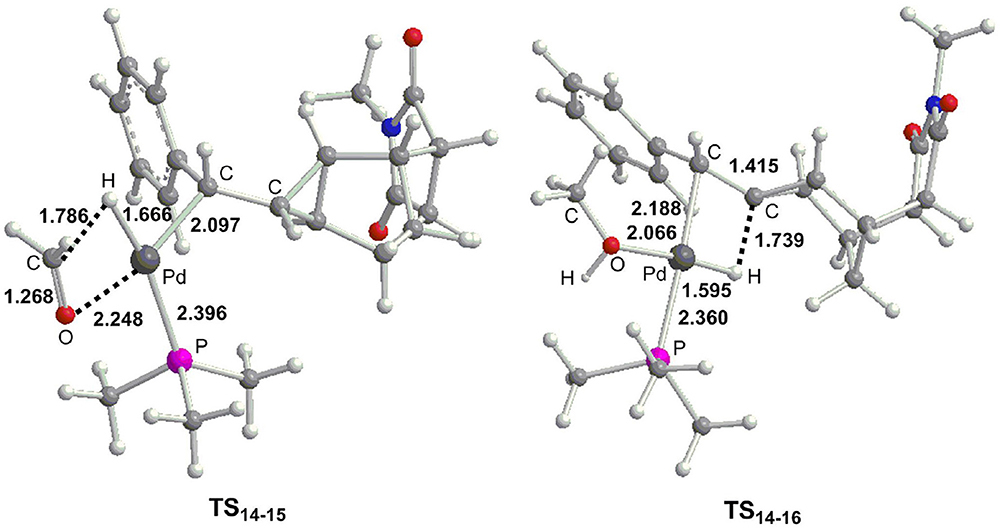
Figure 7. Optimized geometries (Å) for selected transition states as presented in Figure 6.
As described in Figure 8, the catalytic cycle for the reaction of (Z)-2-bromovinylbenzene with endo-N-(p-tolyl)-norbornenesuccinimide undergoes six steps, consist of oxidative addition (OA), intermolecular olefin insertion, deprotonation/protonation, intramolecular olefin insertion, β-H elimination and reductive elimination (RE), and protonation is the rate-determining step and requires an overall barrier of 28.5 kcal/mol from 10 to TS11−12.
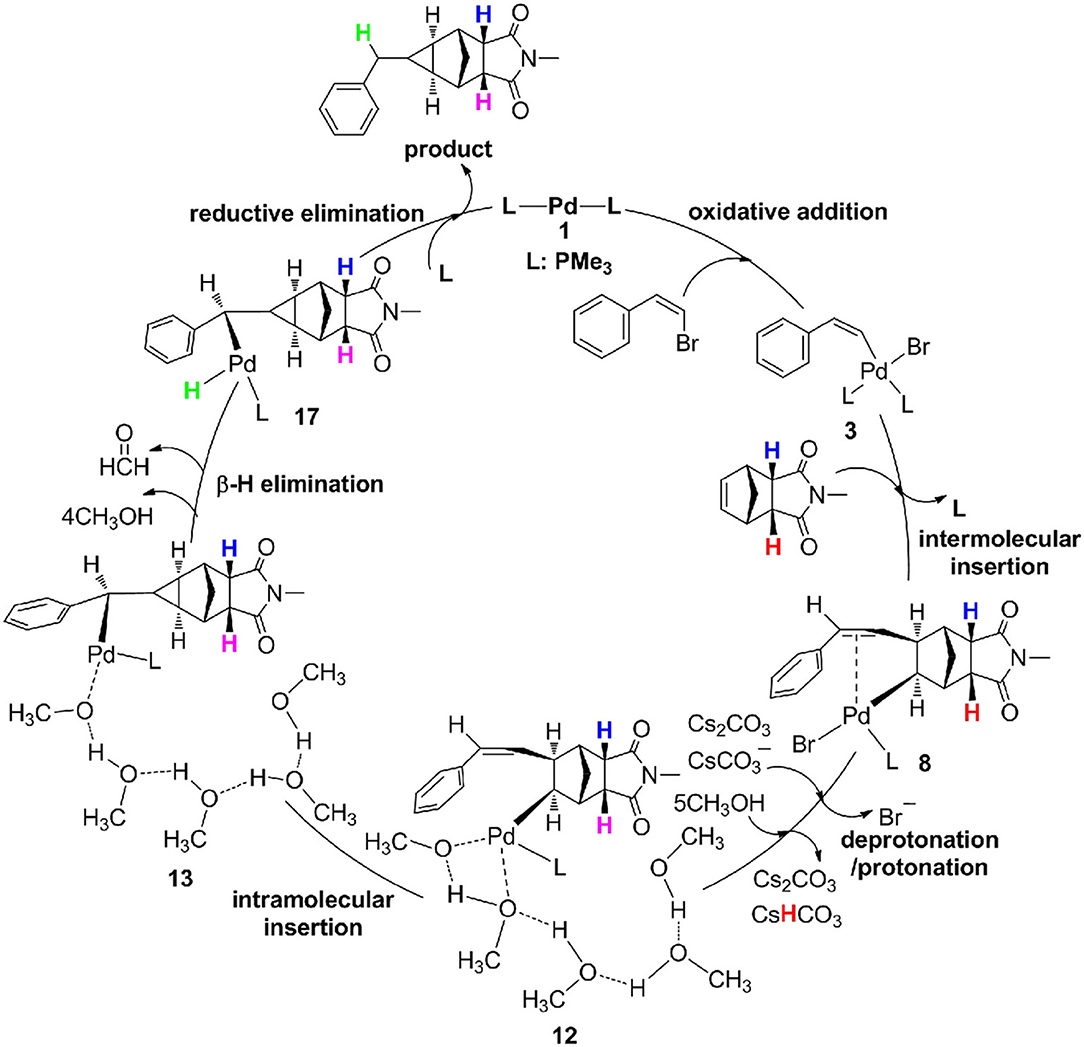
Figure 8. Catalytic cycle for palladium-catalyzed methylcyclopropanation between (Z)-2-bromovinylbenzene and endo-N-(p-tolyl)-norbornenesuccinimide.
Conclusions
In conclusion, Pd-catalyzed [2 + 1] cycloaddition domino reaction mechanisms of (Z)-2-bromovinylbenzene and endo-N-(p-tolyl)-norbornenesuccinimide have been studied by DFT calculations. The results revealed that the methylcyclopropanation process underwent six steps, including oxidative addition, intermolecular alkene insertion, deprotonation/protonation, intramolecular alkene insertion, β-H elimination and reductive elimination, and protonation by methanol is the rate-limiting step with an overall barrier of 28.5 kcal/mol. In addition, the hydrogen atoms for protonation and exchange are both from the methanol, and the former comes from the methyl of methanol, and the latter comes from the hydroxyl of methanol. These calculation results are consistent with the deuterium-labeling experiments.
Author Contributions
The work was completed by cooperation of all authors. HX and WB were responsible for the study of concept and design of the project. FY, YZ, CX, and ZS searched the intermediates and transition states and analyzed the data and drew energy profiles. FY, YZ, HX, and WB drafted and revised the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (21203166), the Natural Science Foundation of Zhejiang Province (LY17B050001).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00169/full#supplementary-material
References
Amatore, C., Jutand, A., and M'Barki, M.A (1992). Evidence of the formation of zerovalent palladium from Pd(OAc)2 and triphenylphosphine. Organometallics 11, 3009–3013. doi: 10.1021/om00045a012
Ardura, D., López, R., and Sordo, T. L. (2005). Relative gibbs energies in solution through continuum models: effect of the loss of translational degrees of freedom in bimolecular reactions on gibbs energy barriers. J. Phys. Chem. B 109, 23618–23623. doi: 10.1021/jp0540499
Barone, V., and Cossi, M. (1998). Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001. doi: 10.1021/jp9716997
Beaulieu, L. P., Schneider, J. F., and Charette, A. B. (2013). Highly enantioselective simmons–smith fluorocyclopropanation of allylic alcohols via the halogen ccrambling strategy of zinc carbenoids. J. Am. Chem. Soc. 135, 7819–7822. doi: 10.1021/ja402393w
Becke, A. D. (1993). Density-functional thermochemistry. III. the role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Bigeault, J., Giordano, L., and Buono, G. (2005). [2+1] Cycloadditions of terminal alkynes to norbornene derivatives catalyzed by palladium complexes with phosphinous acid ligands. Angew. Chem. Int. Ed. 44, 4753–4757. doi: 10.1002/anie.200500879
Biswas, A., De Sarkar, S., Tebben, L., and Studer, A. (2012). Enantioselective cyclopropanation of enals by oxidative N-heterocyclic carbene catalysis. Chem. Commu. 48, 5190–5192. doi: 10.1039/c2cc31501g
Biswas, B., Sugimoto, M., and Sakaki, S. (2000). C–H bond activation of benzene and methane by M(η2-O2CH)2 (M = Pd or Pt). a theoretical study. Organometallics 19, 3895–3908. doi: 10.1021/om000002s
Bruneau, C. (2005). Electrophilic activation and cycloisomerization of enynes: a new route to functional cyclopropanes. Angew. Chem., Int. Ed 44, 2328–2334. doi: 10.1002/anie.200462568
Chai, J. D., and Head-Gordon, M. (2008). Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 128:084106. doi: 10.1063/1.2834918
Check, C. E., Faust, T. O., Bailey, J. M., Wright, B. J., Gilbert, T. M., and Sunderlin, L. S. (2001). Addition of polarization and diffuse functions to the LANL2DZ basis set for P-block elements. J. Phys. Chem. A 105, 8111–8116. doi: 10.1021/jp011945l
Chen, J., Levant, B., Jiang, C., Keck, T. M., Newman, A. H., and Wang, S. (2014). Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D3 receptor antagonists. J. Med. Chem. 57, 4962–4968. doi: 10.1021/jm401798r
Cossi, M., Rega, N., Scalmani, G., and Barone, V. (2003). Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681. doi: 10.1002/jcc.10189
Crowley, M. P., Inglis, H. S., Snarey, M., and Thain, E. M. (1961). Biosynthesis of the pyrethrins. Nature 191, 281–282. doi: 10.1038/191281a0
Dash, P., and Janni, M. S. (2012). Perunchera-lathan, trideuteriomethoxylation of aryl and heteroaryl halides. Eur. J. Org. Chem. 2012, 4914–4917. doi: 10.1002/ejoc.201200753
Davies, D. L., Donald, M. A., and Macgregor, S. A. (2005). Computational study of the mechanism of cyclometalation by palladium acetate. J. Am. Chem. Soc. 127, 13754–13755. doi: 10.1021/ja052047w
de Meijere, A. (2003). Introduction: cyclopropanes and related rings. Chem. Rev. 103, 931–932. doi: 10.1021/cr0100289
Du, W., Gu, Q., Li, Z., and Yang, D. (2015). Palladium(II)-catalyzed intramolecular tandem aminoalkylation via divergent C(sp3)–H functionalization. J. Am. Chem. Soc. 137, 1130–1135. doi: 10.1021/ja5102739
Ehlers, A. W., Bohme, M., Dapprich, S., Gobbi, A., Hollwarth, A., Jonas, V., et al. (1993). A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au. Chem. Phys. Lett. 208, 111–114. doi: 10.1016/0009-2614(93)80086-5
Ess, D. H., Bischof, S. M., Oxgaard, J., Periana, R. A., and Goddard, W. A. (2008). Transition state energy decomposition study of acetate-assisted and internal electrophilic substitution C–H bond activation by (acac-O,O)2Ir(X) complexes (X = CH3COO, OH). Organometallics 27, 6440–6445. doi: 10.1021/om8006568
Fedorynski, M. (2003). Syntheses of gem-dihalocyclopropanes and their use in organic synthesis. Chem. Rev. 103, 1099–1132. doi: 10.1021/cr0100087
Figg, T. M., Wasa, M., Yu, J. Q., and Musaev, D. G. (2013). Understanding the reactivity of Pd(0)/PR3-catalyzed intermolecular C(sp3)-H bond arylation. J. Am. Chem. Soc. 135, 14206–14214. doi: 10.1021/ja4053416
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09, Revision A.1. Wallingford CT: Gaussian, Inc.
Fukui, K. (1970). Formulation of the reaction coordinate. J. Phys. Chem. 74, 4161–4163. doi: 10.1021/j100717a029
Fukui, K. (1981). The path of chemical reactions-the IRC approach. Acc. Chem. Res. 14, 363–368. doi: 10.1021/ar00072a001
Goudreau, S. R., and Charette, A. B. (2010). Defying ring strain: new approaches to cyclopropanes. Angew. Chem. Int. Ed. 49, 486–488. doi: 10.1002/anie.200905109
Hartwig, J. F., Cook, K. S., Hapke, M., Incarvito, C. D., Fan, Y. B., Webster, C. D., et al. (2005). Rhodium boryl complexes in the catalytic, terminal functionalization of alkanes. J. Am. Chem. Soc. 127, 2538–2552. doi: 10.1021/ja045090c
Hata, Y., Zimmermann, S., Quitschau, M., Kaiser, M., Hamburger, M., and Adams, M. (2011). Antiplasmodial and antitrypanosomal activity of pyrethrins and pyrethroids. J. Agric. Food Chem. 59, 9172–9176. doi: 10.1021/jf201776z
Hiratsuka, T., Suzuki, H., Kariya, R., Seo, T., Minami, A., and Oikawa, H. (2014). Enantioselective synthesis of α-alkylidene-γ-butyrolactones: intramolecular rauhut-currier reaction promoted by acid/base organocatalysts. Angew. Chem. Int. Ed. 53, 5423–5426. doi: 10.1002/anie.201402623
Hofmann, K., Jucker, O., Miller, W. R., Young, A. C., and Tausig, F. (1954). On the structure of lactobacillic acid. J. Am. Chem. Soc. 76, 1799–1804. doi: 10.1021/ja01636a020
Huzinaga, S. (1984). Gaussian Basis Sets for Molecular Calculations; Elsevier Science. Amsterdam: Pub. Co.
Jiang, H., He, J., Liu, T., and Yu, J. Q. (2016). Ligand-enabled γ-C(sp3)–H olefination of amines: en route to pyrrolidines. J. Am. Chem. Soc. 138, 2055–2059. doi: 10.1021/jacs.5b13462
Kefalidis, C. E., Baudoin, O., and Clot, E. (2010). DFT study of the mechanism of benzocyclobutene formation by palladium-catalysed C(sp3)–H activation: role of the nature of the base and the phosphine. Dalton Trans. 39, 10528–10535. doi: 10.1039/c0dt00578a
Kuwabe, S. I., Torraca, K. E., and Buchwald, S. L. (2001). Palladium-catalyzed intramolecular C–O bond formation. J. Am. Chem. Soc. 123, 12202–12206. doi: 10.1021/ja012046d
Lafrance, M., Gorelsky, S. I., and Fagnou, K. (2007). High-yielding palladium-catalyzed intramolecular alkane arylation: reaction development and mechanistic studies. J. Am. Chem. Soc. 129, 14570–14571. doi: 10.1021/ja076588s
Lebel, H., Marcoux, J. F., Molinaro, C., and Charette, A. B. (2003). Stereoselective cyclopropanation reactions. Chem. Rev. 103, 977–1050. doi: 10.1021/cr010007e
Li, S. H., Chen, G., Feng, C. G., Gong, W., and Yu, J. Q. (2014). Ligand-enabled γ-C–H olefination and carbonylation: construction of β-quaternary carbon centers. J. Am. Chem. Soc. 136, 5267–5270. doi: 10.1021/ja501689j
Liang, Y., Geng, W. Z., Wei, J. N., Ouyang, K. B., and Xi, Z. F. (2012). Palladium-catalyzed silyl C(sp3)–H bond activation. Org. Biomol. Chem. 10, 1537–1542. doi: 10.1039/c2ob06941e
Lindsay, V. N., Fiset, D., Gritsch, P. J., Azzi, S., and Charette, A. B. (2013). Stereoselective Rh2(S-IBAZ)4-catalyzed cyclopropanation of alkenes, alkynes, and allenes: asymmetric synthesis of diacceptor cyclopropylphosphonates and alkylidenecyclopropanes. J. Am. Chem. Soc. 135, 1463–1470. doi: 10.1021/ja3099728
Liu, B. W., Gao, M., Dang, L., Zhao, H. T., Marder, T. B., and Lin, Z. Y. (2012). DFT studies on the mechanisms of the platinum-catalyzed diboration of acyclic α,β-unsaturated carbonyl compounds. Organometallics 31, 3410–3425. doi: 10.1021/om3002153
Liu, Q., Lan, Y., Liu, J., Li, G., Wu, Y. D., and Lei, A. (2009). Revealing a second transmetalation step in the Negishi coupling and its competition with reductive elimination: improvement in the interpretation of the mechanism of biaryl syntheses. J. Am. Chem. Soc. 131, 10201–10210. doi: 10.1021/ja903277d
Mao, J., and Bao, W. L. (2014a). Palladium(0)-catalyzed methylenecyclopropanation of norbornenes with vinyl bromides. Org. Lett. 16, 2646–2649. doi: 10.1021/ol500829t
Mao, J., and Bao, W. L. (2014b). Palladium-catalyzed [2+1+1] annulation of norbornenes with (z)-bromostyrenes: synthesis of bismethylenecyclobutanes via twofold C(sp2)–H bond activation. Chem. Commun. 50, 15726–15729. doi: 10.1039/C4CC06545J
Mao, J., Xie, H. J., and Bao, W. L. (2015). Palladium(0)-catalyzed methylcyclopropanation of norbornenes with vinyl bromides and mechanism study. Org. Lett. 17, 3678–3681. doi: 10.1021/acs.orglett.5b01603
Mao, J. G., Zhang, S. Q., Shi, B. F., and Bao, W. L. (2014). Palladium(0)-catalyzed cyclopropanation of benzyl bromides via C(sp3)–H bond activation. Chem. Commun. 50, 3692–3694. doi: 10.1039/C3CC49231A
Masutomi, K., Oguchi, K., and Tanaka, K. (2014). Enantioselective cycloisomerization of 1,6-enynes to bicyclo[3.1.0]hexanes catalyzed by rhodium and benzoic acid. J. Am. Chem. Soc. 136, 7627–7630. doi: 10.1021/ja504048u
Matsukawa, Y., Mizukado, J., Quan, H., Tamura, M., and Sekiya, A. (2005). Palladium(0)-catalyzed hydroalkoxylation of hexafluoropropene: synthesis of hydrofluoroethers under neutral conditions. Angew. Chem. Int. Ed. 44, 1128–1130. doi: 10.1002/anie.200462200
Miege, F., Meyer, C., and Cossy, J. (2011). Rhodium-catalyzed cycloisomerization involving cyclopropenes: efficient stereoselective synthesis of medium-sized heterocyclic scaffolds. Angew. Chem. Int. Ed. 50, 5932–5937. doi: 10.1002/anie.201101220
Miki, K., Nishino, F., Ohe, K., and Uemura, S. (2002). Novel approach for catalytic cyclopropanation of akenes via (2-furyl)carbene complexes from 1-benzoyl-cis-1-buten-3-yne. J. Am. Chem. Soc. 124, 5260–5261. doi: 10.1021/ja025776+
Miura, T., Sasaki, T., Harumashi, T., and Murakami, M. (2006). Vinylcyclopropanation of olefins via 3-methoxy-1-propenylrhodium(I). J. Am. Chem. Soc. 128, 2516–2517. doi: 10.1021/ja0575326
Okuno, Y. (1997). Theoretical investigation of the mechanism of the baeyer-villiger reaction in nonpolar solvents. Chem. Eur. J. 3, 212–218. doi: 10.1002/chem.19970030208
Oonishi, Y., Kitano, Y., and Sato, Y. (2012). Palladium-catalyzed asymmetric synthesis of silicon-stereogenic dibenzosiloles via enantioselective C–H bond functionalization. Angew. Chem. Int. Ed. 51, 7305–7308. doi: 10.1002/anie.201203772
Palucki, M., Wolfe, J. P., and Buchwald, S. L. (1997). Palladium-catalyzed intermolecular carbon–oxygen bond formation: a new synthesis of aryl ethers. J. Am. Chem. Soc. 119, 3395–3396. doi: 10.1021/ja9640152
Pietruszka, J. (2003). Synthesis and properties of oligocyclopropyl-containing natural products and model compounds. Chem. Rev 103, 1051–1070. doi: 10.1021/cr010027g
Reed, A. E., Curtiss, L. A., and Weinhold, F. (1988). Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899–926. doi: 10.1021/cr00088a005
Reed, A. E., and Weinhold, F. (1985). Natural localized molecular orbitals. J. Chem. Phys. 83, 1736–1740. doi: 10.1063/1.449360
Reed, A. E., Weinstock, R. B., and Weinhold, F. (1985). Natural population analysis. J. Chem. Phys. 83, 735–746. doi: 10.1063/1.449486
Reissig, H. U., and Zimmer, R. (2003). Donor–acceptor-substituted cyclopropane derivatives and their application in organic synthesis. Chem. Rev. 103, 1151–1196. doi: 10.1021/cr010016n
Satake, A., and Nakata, T. (1998). Novel η3-allylpalladium–pyridinylpyrazole complex: synthesis, reactivity, and catalytic activity for cyclopropanation of ketene silyl acetal with allylic acetates. J. Am. Chem. Soc. 120, 10391–10396. doi: 10.1021/ja982269c
Schoenebeck, F., and Houk, K. N. (2010). Ligand-controlled regioselectivity in palladium-catalyzed cross coupling reactions. J. Am. Chem. Soc. 132, 2496–2497. doi: 10.1021/ja9077528
Shao, Q., He, J., Wu, Q. F., and Yu, J. Q. (2017). Ligand-enabled γ-C(sp3)–H cross-coupling of nosyl-protected amines with aryl- and alkylboron reagents. ACS Catal. 7, 7777–7782. doi: 10.1021/acscatal.7b02721
Shao, Q., Wu, Q. F., He, J., and Yu, J. Q. (2018). Enantioselective γ-C(sp3)–H activation of alkyl amines via Pd(II)/Pd(0) catalysis. J. Am. Chem. Soc. 140, 5322–5325. doi: 10.1021/jacs.8b01094
Simmons, H. E., and Smith, R. D. (1958). A new synthesis of cyclopropanes from oledins. J. Am. Chem. Soc. 80, 5323–5324. doi: 10.1021/ja01552a080
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627. doi: 10.1021/j100096a001
Tao, J., Perdew, J. P., Staroverov, V. N., and Scuseria, G. E. (2003). Climbing the density functional ladder: nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 91:146401. doi: 10.1103/PhysRevLett.91.146401
Torraca, K. E., Kuwabe, S. I., and Buchwald, S. L. (2000). A high-yield, general method for the catalytic frmation of oxygen heterocycles. J. Am. Chem. Soc. 122, 12907–12908. doi: 10.1021/ja005698v
Tseng, N. W., Mancuso, J., and Lautens, M. (2006). Rhodium-catalyzed tandem vinylcyclopropanation of strained alkenes. J. Am. Chem. Soc. 128, 5338–5339. doi: 10.1021/ja060877j
Wang, M. Y., Fan, T., and Lin, Z. Y. (2012a). DFT studies on copper-catalyzed arylation of aromatic C–H bonds. Organometallics 31, 560–569. doi: 10.1021/om2007612
Wang, M. Y., Fan, T., and Lin, Z. Y. (2012b). DFT studies on the reaction of CO2 with allyl-bridged dinuclear palladium(I) complexes. Polyhedron 32, 35–40. doi: 10.1016/j.poly.2011.05.016
Wasa, M., Engle, K. M., and Yu, J. Q. (2009). Pd(0)/PR3-catalyzed intermolecular arylation of sp3 C–H bonds. J. Am. Chem. Soc. 131, 9886–9887. doi: 10.1021/ja903573p
Wessjohann, L. A., Brandt, W., and Thiemann, T. (2003). Biosynthesis and metabolism of cyclopropane rings in natural compounds. Chem. Rev. 103, 1625–1648. doi: 10.1021/cr0100188
Wiberg, K. B. (1996). Bent bonds in organic compounds. Acc. Chem. Res. 29, 229–234. doi: 10.1021/ar950207a
Wu, Y. W., Chen, Y. Q., Liu, T., Eastgate, M. D., and Yu, J. Q. (2016). Pd-catalyzed γ-C(sp3)–H arylation of free amines using a transient directing group. J. Am. Chem. Soc. 138, 14554–14557. doi: 10.1021/jacs.6b09653
Xie, H., Zu, L., Li, H., Wang, J., and Wang, W. J. (2007). Organocatalytic enantioselective cascade michael-alkylation reactions: synthesis of chiral cyclopropanes and investigation of unexpected organocatalyzed stereoselective ring opening of cyclopropanes. J. Am. Chem. Soc. 129, 10886–10894. doi: 10.1021/ja073262a
Xie, H. J., Fan, T., Lei, Q. F., and Fang, W.,J. (2016). New progress in theoretical studies on palladium-catalyzed C–C bond-forming reaction mechanisms. Sci. China Chem. 59, 1432–1447. doi: 10.1007/s11426-016-0018-2
Xie, H. J., Lin, F. R., Lei, Q. F., and Fang, W. J. (2013b). Mechanism and substrate-dependent rate-determining step in palladium-catalyzed intramolecular decarboxylative coupling of arenecarboxylic acids with aryl bromides: a DFT study. Organometallics 32, 6957–6968. doi: 10.1021/om400503x
Xie, H. J., Zhang, H., and Lin, Z. Y. (2013a). DFT studies on the palladium-catalyzed dearomatization reaction between chloromethylnaphthalene and the cyclic amine morpholine. Organometallics 32, 2336–2343. doi: 10.1021/om301215a
Xie, H. J., Zhang, H., and Lin, Z. Y. (2013c). DFT studies on the mechanisms of palladium-catalyzed intramolecular arylation of a silyl C(sp3)–H bond. N. J. Chem. 37, 2856–2861. doi: 10.1039/c3nj00531c
Xuan, Y., Nie, S., Dong, L., Zhang, J., and Yan, M. (2009). Highly enantioselective synthesis of nitrocyclopropanes via organocatalytic conjugate addition of bromomalonate to α,β-unsaturated nitroalkenes. Org. Lett. 11, 1583–1586. doi: 10.1021/ol900227j
Zhao, Y., and Truhlar, D. G. (2006a). Comparative DFT study of van der waals complexes: rare-gas dimers, alkaline-earth dimers, zinc dimer, and zinc-rare-gas dimers. J. Phys. Chem. A 110, 5121–5129. doi: 10.1021/jp060231d
Zhao, Y., and Truhlar, D. G. (2006b). Density functional for spectroscopy: no long-range self-interaction error, good performance for rydberg and charge-transfer states, and better performance on average than B3LYP for ground states. J. Phys. Chem. A 110, 13126–13130. doi: 10.1021/jp066479k
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi: 10.1007/s00214-007-0310-x
Keywords: Pd catalysis, DFT calculation, cycloaddition, mechanism, protonation
Citation: Ying F, Zhang Y, Xiang C, Song Z, Xie H and Bao W (2019) Key Mechanistic Features in Palladium-Catalyzed Methylcyclopropanation of Norbornenes With Vinyl Bromides: Insights From DFT Calculations. Front. Chem. 7:169. doi: 10.3389/fchem.2019.00169
Received: 11 January 2019; Accepted: 05 March 2019;
Published: 27 March 2019.
Edited by:
Zexing Cao, Xiamen University, ChinaReviewed by:
Chunsen Li, Fujian Institute of Research on the Structure of Matter (CAS), ChinaXinzheng Yang, Institute of Chemistry (CAS), China
Wei Guan, Northeast Normal University, China
Copyright © 2019 Ying, Zhang, Xiang, Song, Xie and Bao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hujun Xie, aHVqdW54aWVAZ21haWwuY29t