
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 01 March 2019
Sec. Inorganic Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00097
This article is part of the Research TopicAdvances in the Development of Artificial MetalloenzymesView all 13 articles
 Biswanath Das1Afnan Al-Hunaiti2†
Biswanath Das1Afnan Al-Hunaiti2† Brenda N. Sánchez-Eguía3Erica Zeglio1Serhiy Demeshko4
Brenda N. Sánchez-Eguía3Erica Zeglio1Serhiy Demeshko4 Sebastian Dechert4Steffen Braunger4Matti Haukka5Timo Repo2
Sebastian Dechert4Steffen Braunger4Matti Haukka5Timo Repo2 Ivan Castillo3*
Ivan Castillo3* Ebbe Nordlander1*
Ebbe Nordlander1*The new di- and tetranuclear Fe(III) μ-oxido complexes [Fe4(μ-O)4(PTEBIA)4](CF3SO3)4(CH3CN)2] (1a), [Fe2(μ-O)Cl2(PTEBIA)2](CF3SO3)2 (1b), and [Fe2(μ-O)(HCOO)2(PTEBIA)2](ClO4)2 (MeOH) (2) were prepared from the sulfur-containing ligand (2-((2,4-dimethylphenyl)thio)-N,N-bis ((1-methyl-benzimidazol-2-yl)methyl)ethanamine (PTEBIA). The tetrairon complex 1a features four μ-oxido bridges, while in dinuclear 1b, the sulfur moiety of the ligand occupies one of the six coordination sites of each Fe(III) ion with a long Fe-S distance of 2.814(6) Å. In 2, two Fe(III) centers are bridged by one oxido and two formate units, the latter likely formed by methanol oxidation. Complexes 1a and 1b show broad sulfur-to-iron charge transfer bands around 400–430 nm at room temperature, consistent with mononuclear structures featuring Fe-S interactions. In contrast, acetonitrile solutions of 2 display a sulfur-to-iron charge transfer band only at low temperature (228 K) upon addition of H2O2/CH3COOH, with an absorption maximum at 410 nm. Homogeneous oxidative catalytic activity was observed for 1a and 1b using H2O2 as oxidant, but with low product selectivity. High valent iron-oxo intermediates could not be detected by UV-vis spectroscopy or ESI mass spectrometry. Rather, evidence suggest preferential ligand oxidation, in line with the relatively low selectivity and catalytic activity observed in the reactions.
The interaction between iron and sulfur in metalloproteins and in biomimetic molecular systems has attracted increased attention in recent years (Beinert et al., 1997; Ohnishi, 1998; Rao and Holm, 2004; Ballmann et al., 2008a; Meyer, 2008; Lill, 2009). Among these, the majority of the Fe/S structural motifs are dominated by clusters where the iron center is in distorted tetrahedral environments (Beinert et al., 1997; Meyer, 2008); nonetheless, there are examples of molecular systems where the Fe centers display diverse coordination geometries (Ballmann et al., 2008b; Fuchs et al., 2010). In some typical molecular systems, the metal center has also been found to be involved in secondary bonding interactions with ether-O and thioether-S units (Ballmann et al., 2008b). Although in all these examples sulfur, being a soft donor, displays preference for low valent Fe(II), there are molecular systems where the sulfur atom has been found to be bonded to high valent (III and IV) iron centers with distorted octahedral geometry (Harrop and Mascharak, 2004; McDonald et al., 2010; Widger et al., 2014). Overall, the Fe-S interaction in various molecular systems having iron in different formal oxidation states is an interesting field of research.
On the other hand, the selective and environmentally benign oxidation of hydrocarbons using affordable and efficient catalysts is another important area of modern synthetic chemistry. In this regard, bio-inspired iron chemistry has received increasing attention in recent decades due to the natural iron abundance in the earth crust, and the highly selective catalytic hydrocarbon oxidations of iron-containing oxygenases such as cytochrome P450 (Hasemann et al., 1995), soluble methane monooxygenase (sMMO) (Tinberg and Lippard, 2011), and Rieske dioxygenases (Wackett, 2002). Among the (catalytic) properties of these iron-based enzymes, the selective activation of C-H bonds under mild conditions is one of the most striking aspects. Many mono- and diiron complexes with various multidentate N-based ligands have been investigated in order to mimic the structures as well as functions of non-heme iron enzymes, and to potentially develop sophisticated oxidation catalysts (Costas et al., 2000, 2004; Sun et al., 2011; Lindhorst et al., 2015; Gamba et al., 2017). Such studies indicate that fine tuning of the coordination environment of the iron centers plays a crucial role in the catalytic activity, which includes O2 binding, followed by electron-transfer from Fe to O2 to afford either iron–superoxo (FeIII-), iron–peroxo (FeIII-), or iron–oxo (FeIV/V = O) species after initial O-O bond cleavage (Kim et al., 1997; Hazell et al., 2002; Rohde et al., 2003; Ye and Neese, 2011; De Visser et al., 2013; Wang et al., 2013a; Mitra et al., 2014; Nam, 2015).
A number of dinuclear Fe(III)-μ-oxido complexes relevant to the aforementioned enzymes from both structural and functional perspectives, have been studied as oxidation catalysts (Romakh et al., 2007; Visvaganesan et al., 2009; Wang et al., 2013a). Depending on the specific ligand environment, catalytic oxidation reactions using such dinuclear μ-oxido Fe(III) complexes can proceed via a radical intermediate or a high valent metal oxo species, or a combination of both, and the nature of these intermediates/active oxidants exerts a profound influence on the product distribution (Costas et al., 2000, 2004). The experimental evidence in all cases indicate that there is a strong influence of the ligand system on the catalytic activity, as well as the choice of oxidant (i.e., ultimate oxygen donor), which can also affect the product distribution (Costas et al., 2000, 2004). In spite of the extensive oxidative catalytic activity studies of these dinuclear Fe(III)-μ-oxido complexes, to the best of our knowledge there are very few well-characterized complexes that have been used as oxidation catalysts where sulfur occupies one of the coordination sites (McQuilken and Goldberg, 2012). The easily oxidizable nature of sulfur argues against its use in these types of systems (Widger et al., 2014), despite the fact that studying small molecular catalysts with sulfur-containing ligands can be very useful in modeling key metal-sulfur interactions that play a significant role in non-heme enzymes. Studies on sulfur oxygenation in a number of biomimetic non-heme iron(III)-thiolate complexes indicate that a long Fe-S bond distance makes the sulfur unit susceptible to attack by O2 in a reaction where iron maintains the +3 oxidation state (McQuilken and Goldberg, 2012). The active site of the non-heme iron enzyme cysteine dioxygenase (McCoy et al., 2006) is believed to pass through a Fe(III)-superoxo intermediate. DFT and QM/MM computational studies predict that the formation of the superoxo intermediate is followed by formation of an energetically favorable cyclic four-membered Fe-O-O-S ring structure that undergoes O-O bond cleavage to form a Fe(IV)(Oxo)-sulfinate analog. This metal oxo unit transfers the second oxygen atom to generate the cysteine-sulfinic acid product (Kumar et al., 2011; McQuilken and Goldberg, 2012).
Goldberg and coworkers have reported Fe(II) complexes of interesting pentadentate ligand systems incorporating one sulfur donor in efforts to model the reactivity of the active site of cysteine dioxygenase. While a pentadentate ligand with one thiolate donor is oxidized to the corresponding sulfoxide upon reaction with dioxygen, an analogous thioether ligand permits formation of a non-heme Fe(IV)oxo species that can perform oxygen atom transfer (Widger et al., 2014). In this context, it is worth mentioning that one of the most realistic biomimetic model systems (both in terms of structure and reactivity) for cysteine dioxygenase, [TpMe,PhFeIICysOEt] (TpMe,Ph = hydridotris(3-phenyl-5-methylpyrazol-1-yl)borate), was reported by Limberg and coworkers (Sallmann et al., 2012), who used isotope (16O/18O) experiments to confirm that the treatment with dioxygen mainly leads to cysteine dioxygenase activity, i.e., deoxygenation of the bound cysteine ethyl ester.
Here, we describe new di- and tetranuclear Fe(III)-μ-oxido complexes with the thioether-containing PTEBIA ligand (Castillo et al., 2012), viz [Fe4(μ-O)4(PTEBIA)4](CF3SO3)4(CH3CN)2] (1a), and [Fe2(μ-O)Cl2(PTEBIA)2](CF3SO3)2 (1b) (Figure 1). The Fe-S interaction in these complexes and their efficiency as homogeneous oxidation catalyst precursors will be discussed. The UV-vis and mass spectrometric investigations of plausible active species in solution are also presented. In order to gain a better understanding of the spectroscopic features of the complexes, specifically regarding the Fe(III)-S interaction, we have also synthesized the dinuclear complex [Fe2(μ-O)(μ-HCOO)2(PTEBIA)2](ClO4)2(MeOH) (2) that features additional bridging formate ligands.
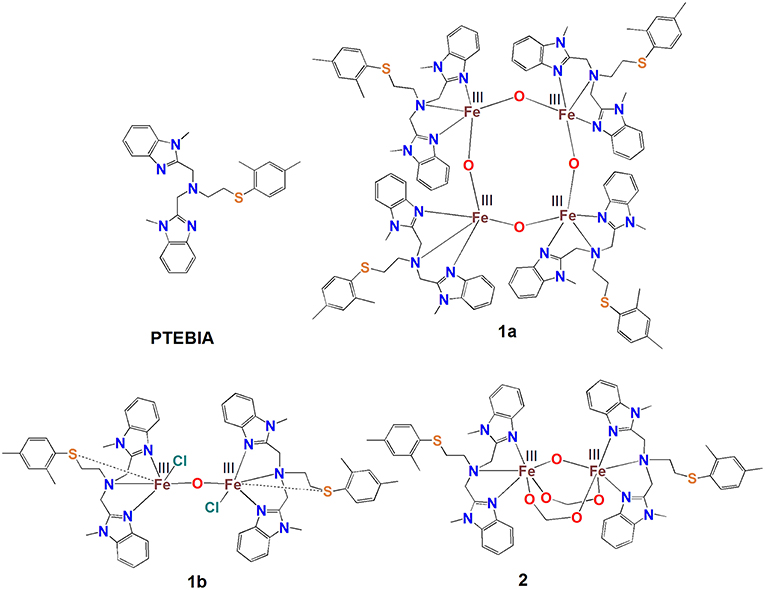
Figure 1. Schematic depiction of the structures of the PTEBIA ligand and complexes 1a, 1b, and 2.
The PTEBIA ligand was prepared following the procedure reported by Castillo and coworkers (Castillo et al., 2012). Addition of 1 equivalent of Fe(II)(OTf)2 (OTf = triflate, CF3) to a tetrahydrofuran solution of PTEBIA leads to an immediate change of the color of the ligand solution to green. Refluxing of this green solution for 4 h, followed by crystallization by slow diffusion of diethylether into an acetonitrile solution of the product leads to yellow, needle shaped crystals of 1a (Figure 2).
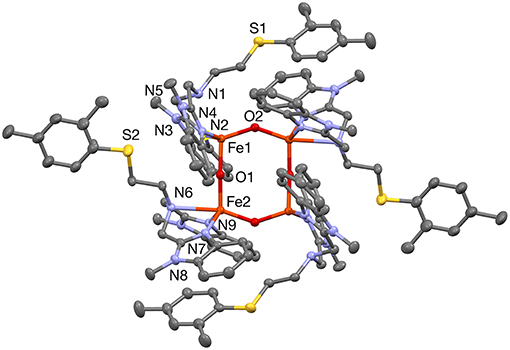
Figure 2. A plot (30% probability thermal ellipsoids) of the molecular structure of the cation of 1a, [Fe4(μ-O)4(PTEBIA)4]4+ (hydrogen atoms and disorder omitted for clarity). Symmetry transformation used to generate equivalent atoms: (') 1/2–x, 3/2–y, 1–z.
The solid-state structure of 1a consists of a tetranuclear unit formed by four PTEBIA ligands, four bridging oxido ligands and four Fe(III) ions. To the best of our knowledge, this represents the first example of a tetrairon, tetra-oxido cluster with a sulfur-containing ligand. All the Fe(III) units have a distorted trigonal bipyramidal geometry, with N3O2 coordination environments. Selected bond lengths and bond angles for 1a are listed in Tables 1, 2, respectively. There are two types of Fe-O-Fe bond angles and four different Fe-O bond distances present in the cluster. The angles are 142.36(15)° for Fe1-O2-Fe2′ and 171.64(18)° for Fe1-O1-Fe2 (Symmetry transformation used to generate equivalent atoms: (') 1/2–x, 3/2–y, 1–z). The corresponding bond lengths are 1.780(3) Å for Fe1-O2 and 1.778(6) Å for Fe2-O2,' and 1.811(3) Å for Fe1-O1, and 1.776(3) Å for Fe2-O1. The sulfur donor sites are > 5.2 Å away from the nearest Fe(III) unit, and thus there is no plausible intramolecular Fe(III)-S interaction.
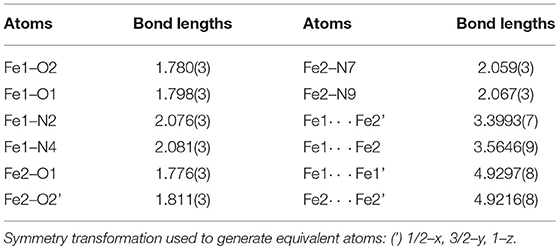
Table 1. Selected bond lengths [Å] for 1a.
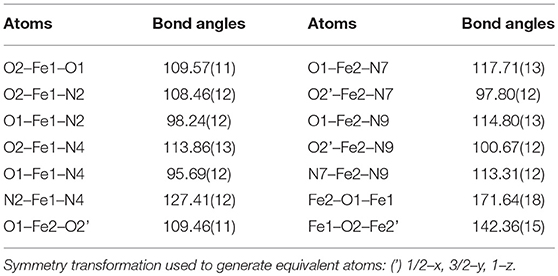
Table 2. Selected bond angles [°] for 1a.
When PTEBIA contained a sub-stoichiometric amount of HCl from incomplete neutralization during the last synthetic step in the PTEBIA synthesis (as evidenced by the immediate precipitation of AgCl upon addition of AgNO3 to a chloroform solution of PTEBIA•xHCl) (Castillo et al., 2012), formation of 1a was accompanied by brown crystals of 1b; the latter complex was also prepared independently from FeCl2. Both complexes appear to be stable over a period of days in acetonitrile solution, but evaporation of the solvent results in oily products. Nonetheless, X-ray quality crystals of 1b were isolated from cold acetonitrile solutions to acquire data at liquid N2 temperature. The molecular structure of the cation in [Fe2(μ-O)Cl2(PTEBIA)2](CF3SO3)2 (1b) is shown in Figure 3, and selected bond lengths and bond angles are collated in Tables 3, 4, respectively. In the solid state, 1b is a μ-oxido diiron(III) complex, with Fe(III) centers in a slightly distorted octahedral N3SOCl coordination environment. The thioether moiety coordinates weakly with Fe(III)-S bond lengths of Fe-S bonds of 2.8364(17) and 2.8147(16) Å for the two crystallographically independent molecules found in the asymmetric unit (symmetry transformations used to generate equivalent atoms: (') 1–x, 1–y, 1–z; (”) 2–x, 1–y, –z). This Fe-S distance is long in comparison to Fe-S distances reported by Widger et al. for mononuclear Fe(II) complexes (~2.3 Å) with the thioether containing ligand N3PyamideSR (R = -(CH2)2CN) (Widger et al., 2014), but it is comparable to the long Cu(I/II)-S(thioether) distance observed for coordination of methionine in plastocyanin (2.9 Å) (Sahoo and Ray, 2007). The Fe(III) centers are oxido-bridged and are 3.5821(11) and 3.5809(11)Å (two crystallographically independent molecules) apart from each other with an Fe-O-Fe angle of 180°, which is in accordance with similar dinuclear Fe(III)-μ-oxido complexes reported by McKenzie and coworkers (Vad et al., 2012) and Wang et al. (2003).
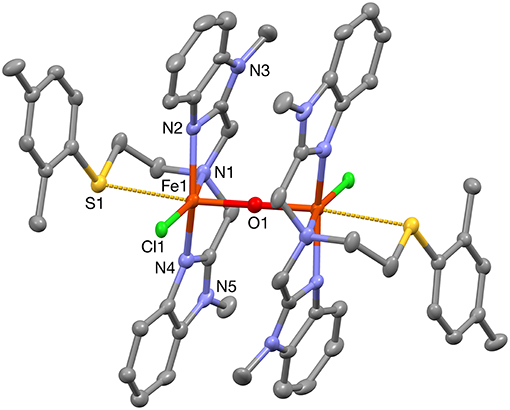
Figure 3. A plot (30% probability thermal ellipsoids) of the molecular structure of the cation of 1b, [Fe2(μ-O)Cl2(PTEBIA)2]2+ (hydrogen atoms omitted for clarity). Only one of the two crystallographically independent molecules is shown. Symmetry transformations used to generate equivalent atoms: (') 1–x, 1–y, 1–z.
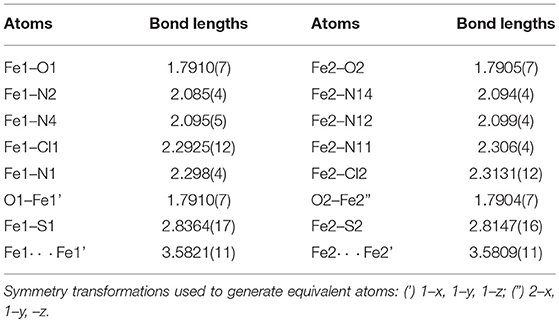
Table 3. Selected bond lengths [Å] for 1b.
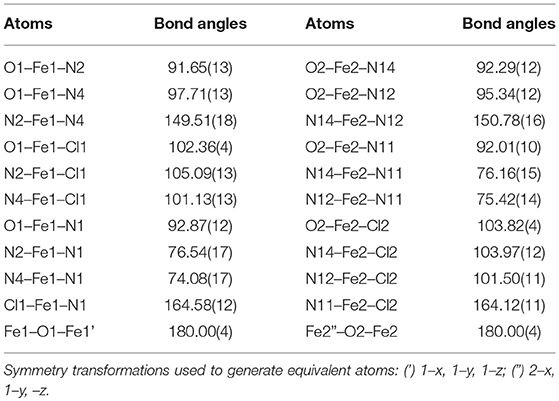
Table 4. Selected bond angles [°] for 1b.
Mass spectrometric measurements of 1a gave rise to two major peaks corresponding to the mononuclear complex, i.e. at 673.7 amu for [Fe(PTEBIA)(CF3SO3)]+, and at 469.8 corresponding to protonated [HPTEBIA]+ (Figure S1). A peak arising from 1b was also observed at 559.7 amu, assigned to [Fe(PTEBIA)(Cl)]+; no μ-oxido bridged species could be detected by either ESI or MALDI-TOF mass spectrometry. IR characterization revealed sharp resonances between 820 and 600 cm−1 assigned to asymmetric and symmetric Fe-O stretching modes of the Fe-O-Fe units in 1a and 1b (819, 749, 637 cm−1 in the former; 815, 780, 746, 633 cm−1 in the latter). Additional characterization of 1a and 1b was obtained by Mössbauer spectroscopy from 0.02 g of a batch of mixed crystals obtained from the reaction of Fe(OTf)2 with PTEBIA, revealing the presence of two high spin Fe(III) centers with different coordination environments in a 1:1 ratio of 1a and 1b (Figure 4). Since more ionic coordination environments with higher coordination numbers result in higher isomer shifts (δ), the subspectrum with δ = 0.46 mm·s−1 can be assigned to 1b, whereas the subspectrum with δ = 0.38 corresponds to 1a.
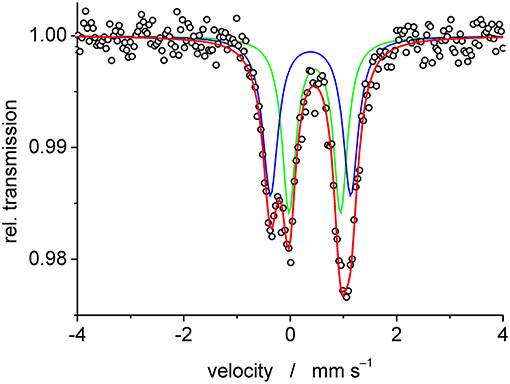
Figure 4. Experimental (open circles) and simulated (blue doublet: δ = 0.38 mm·s−1, ΔEQ = 1.51 mm·s−1 and green doublet: δ = 0.46 mm·s−1, ΔEQ = 0.97 mm·s−1) Mössbauer spectrum of complexes 1a and 1b recorded at 80 K.
Acetonitrile solutions of 1a (0.4 mM) exhibit broad peaks with absorption maxima at 330 and 430 nm (ε = 3,600 and 2,280 M−1 cm−1, respectively), while 1b features slightly narrower peaks with absorption maxima at 320 and 390 nm (ε = 2,400 and 1,990 M−1 cm−1, Figure 5). The peaks around 320–340 nm are in the so called “oxo dimer region” and are characteristic of Fe-O-Fe moieties, as has been observed for similar (μ-oxo)diiron(III) complexes (Reem et al., 1989; Kurtz, 1990; Do et al., 2012). The broad absorption bands around 390–430 nm can be assigned to weak sulfur to Fe(III) charge transfer bands (LMCT), similar to those observed in Cu(II) complexes with PTEBIA (Rodríguez Solano et al., 2011; Castillo et al., 2012).
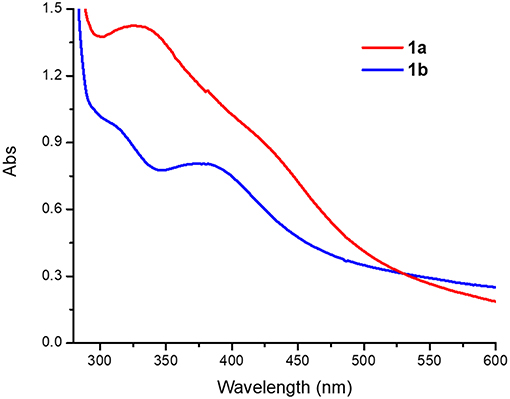
Figure 5. UV-Vis spectra of 1a and 1b in acetonitrile solution (0.4 mM).
Conversion of 1a to 1b was achieved in acetonitrile solution, as evidenced by UV-vis spectroscopy: addition of two equivalents of NBu4Cl as a source of chloride to 0.4 mM solutions of 1a resulted in spectra that are virtually identical to those of the chlorido-containing 1b, see Figure 5 and Figure S2.
To further probe the coordination mode of the PTEBIA ligand toward Fe(III) centers, equimolar amounts of Fe(II)(ClO4)2 and PTEBIA were heated to reflux in THF solution, followed by recrystallization from a methanolic solution by slow vapor diffusion of diethylether. Electrospray mass spectrometry on methanolic solutions of the green crystals obtained from the reaction show a major peak at 262.8 amu (see Figure S3), corresponding to the dicationic species [Fe(PTEBIA)]2+. The solid-state structure reveals that the complex consists of another μ-oxido diiron(III) species ([Fe2(μ-O)(HCOO)2(PTEBIA)2](ClO4)2(MeOH) (2), Figure 6), where both Fe(III) centers are in distorted octahedral environments with N3O3 donor sets. Selected bond distance and bond angles are collated in Tables 5, 6, respectively. The sulfur atoms are far apart from the Fe(III) centers (> 5.86 Å), ruling out any kind of direct interaction. The Fe(III) ions (Fe…Fe distance 3.121(9) Å) are coordinated by the N-donors of two PTEBIA ligands, as well as one bridging oxido and two formate ligands. The corresponding Fe-O-Fe angle is 121.046(1)°. The unexpected formate bridge is likely due to the aerobic oxidation of the methanol solvent in the presence of Fe(III) and PTEBIA, as has been observed by Que and co-workers in the related complex [Fe2(μ-O)(μ-HCOO)(TPA)2](ClO4)3 (TPA = tris(2-pyridylmethyl)amine) (Norman et al., 1998).
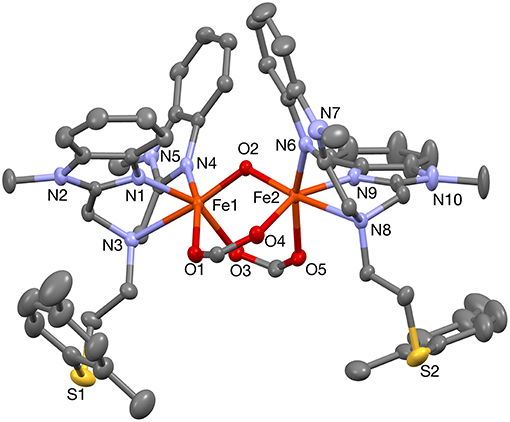
Figure 6. Molecular structure of [Fe2(μ-O)(HCOO)2(PTEBIA)2] (2). Thermal ellipsoids are plotted at 30% probability; all hydrogen atoms and perchlorate anions have been omitted for the sake of clarity.
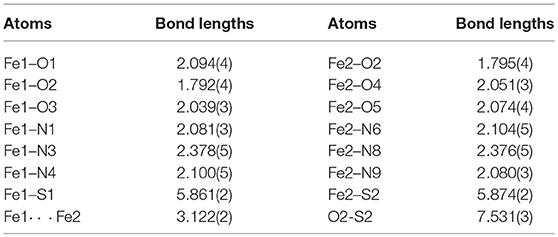
Table 5. Selected bond lengths [Å] for 2.
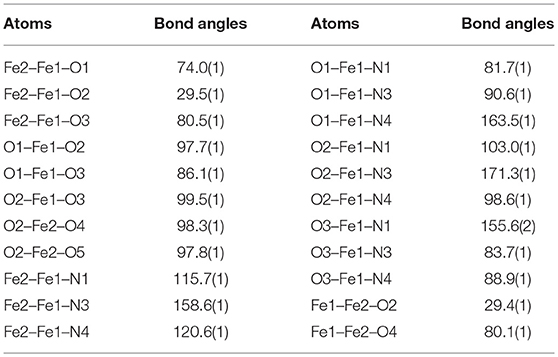
Table 6. Selected bond angles [°] for 2.
IR characterization of 2 shows characteristic asymmetric (νasym) and symmetric (νsym) C = O stretching bands as a broad peak at 1,614 cm−1 and two very sharp peaks for νsym at 1,490 and 1,453 cm−1, consistent with the presence of two bridging formates (Deacon and Phillips, 1980; Kurtz, 1990; Das et al., 2014a,b, 2018). In addition, the presence of a very sharp band at 1,080 cm−1 confirms the presence of perchlorate anions; the bands at 744 and 620 cm−1 correspond to the asymmetric and symmetric stretching modes of the Fe-O-Fe units (Kurtz, 1990; Norman et al., 1990). Acetonitrile solutions of 2 (0.5 mM) exhibit an absorption maximum at 342 nm (ε = 4,400 M−1 cm−1), characteristic of the Fe-O-Fe moiety (Kurtz, 1990; Norman et al., 1990; Do et al., 2012); unlike 1a and 1b, no lower energy bands were observed as the sulfur atom is not bound to the metal centers (Figure 7). Low temperature UV-vis experiments using a 1:1 (molar equivalent) of H2O2 (30 wt.% in H2O) and CH3COOH (> 99.7%) solution as oxidant and keeping overall complex:oxidant ratio at 1:2.5 reveal that the formate ligands of 2 may be protonated under these conditions, thus allowing the sulfur atom of PTEBIA to interact with the Fe(III) centers, as evidenced by the new absorption band at around 410 nm. This band is more prominent at low temperature (228 K) and appears only as a shoulder above 250 K (Figure 7) and was assigned to a S → Fe LMCT transition.
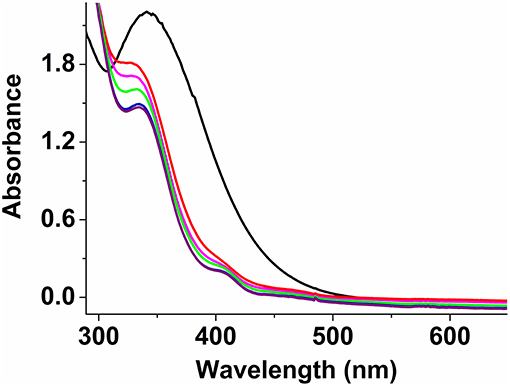
Figure 7. UV-Vis spectra of complex 2 in acetonitrile solution at 298 K (black), and after addition of H2O2/acetic acid mixture (1:1) at 228 K (purple), 238 K (blue), 248 K (green), 258 K (pink), and 268 K (red).
Jacobsen et al. (White et al., 2001) have effected efficient epoxidation of terminal long-chain alkenes using an iron complex based on the tetradentate N4-donor ligand mep (N,N'-dimethyl-N,N'-bis(2-pyridylmethyl)-ethane), H2O2 as oxidant and acetic acid as a promoter. These authors proposed that the active catalyst was the dinuclear ferric complex [Fe2(μ-O)(μ-OAc)(mep)2]+. Later studies by Fujita and Que indicated that the catalyst was rather the mononuclear complex [Fe(II)(mep)(solv)2]2+ (solv = solvent (NCMe)) (Fujita and Que, 2004). Similarly, Stack and coworkers (Dubois et al., 2003) used an Fe(III)-O-Fe(III) complex with aqua and nitrogen donor ligands, [Fe2(μ-O)(OH2)2(phen)2]4+ (phen = phenanthroline) as a catalyst/catalyst precursor for alkene epoxidation using peracetic acid as the oxidant. The combination of hydrogen peroxide with a suitable carboxylic acid (e.g., acetic acid) is believed to generate a peracid that in turn may generate a high valent metal oxo complex that functions as an active alkane/alkene oxidant (Fujita and Que, 2004), but it has also been suggested that the reaction of an Fe(II) complex with peracetic acid can lead to an Fe(III) κ2-peracetate complex with subsequent dissociation of the peracetate ligand to form a ferryl acyl radical species, i.e., an (OAc.)Fe(IV) = O species, that is an active oxidant (Wang et al., 2013b).
Palaniandavar and coworkers (Mayilmurugan et al., 2009) studied the use of Fe(III)2(μ-O) complexes, containing pentadentate Fe(III) ions chelated by tetradentate N2O2 salen-based ligands, as catalysts/precatalysts for the oxidation of alkanes and arenes using the peracid meta-chloroperbenzoic acid (m-CPBA) as the ultimate oxidant. With few exceptions, the obtained yields and alcohol/ketone ratios were relatively low, suggesting a mixture of metal-based and radical oxidation reactions. We have previously investigated the ability of Fe(III)2(μ-O) complexes with nitrogen and oxygen-based donor sets as active catalysts or precursors for the oxidation of alkanes and alkenes with hydrogen peroxide (Jarenmark et al., 2010; Das et al., 2015). A μ-oxo diiron(III) complex [{Fe(HIPCPMP)}2(μ-O)(Piv)]ClO4 (H2IPCPMP = 2-{N-isopropyl-N-[(2-pyridyl)methyl]aminomethyl}-6-{N(carboxymethyl)-N-[(2-pyridyl)methyl] aminomethyl}-4-methylphenol; Piv = Pivalate) showed moderate activity in cyclohexane oxidation, using H2O2 as the oxidant, and evidence suggests that both metal-based and radical mechanisms were involved in the process (Jarenmark et al., 2010). A greater contribution of the metal-based mechanism was found when the tetranuclear Fe2Li2 complex [O(LiDPCPMPP)2] [DPCPMPP = 3-[(3-{[bis(pyridin-2-ylmethyl)amino]methyl}-2-hydroxy-5-methylbenzyl)(pyridin-2-ylmethyl)amino]propanoate] was used as catalyst, based on the retention of configuration of the products observed in the oxidation of cis- or trans-1,2-dimethylcyclohexane (Das et al., 2015).
To assess the effect of the mixed N3S donor set of PTEBIA on the activity and selectivity of iron complexes in the oxygenation of alkenes, we investigated the oxidation of styrene and a number of prototypical cyclic alkene substrates, viz. cyclohexene, 3-ethylcyclohexene, cyclooctene, under mild conditions (Scheme 1) using a bulk sample containing an approximate 1:1 mixture of 1a and 1b as catalyst precursor(s). The oxidation conditions were optimized by using cyclohexene as the model compound (Table 7). Based on the maximum reactivity and selectivity, acetonitrile was used as solvent with CH3COOH as additive and H2O2 as oxidant. These optimized conditions were applied in all oxidation experiments involving the different substrates. Mass spectrometry (ESI-MS) indicated that mixtures of 1a and 1b convert to dimeric [(Fe2)(OAc)2(μ-O)(PTEBIA)2(CF3SO3)]+ in acetonitrile solution, as evidenced by the peak detected at 1332.8 amu (Figure S4, Supplementary Material). Cyclohexene, cyclooctene and 3-ethylcyclohexene were oxidized in moderate conversions (29, 44, and 15%, Table 8) to the overoxidation products of the epoxides, namely diols and diketones. Although styrene oxidation (63% yield) afforded benzylmethanol (by epoxide ring opening), the formation of benzaldehyde as the major product indicates that the main active species is likely a transient radical (single electron oxidation). In comparison to the previously reported μ-oxido di-iron(III) complexes with N and O-based ligands [e.g., 2,6-bis(N-methylbenzimidazol-2-yl)pyridine (Wang et al., 2003), DPCPMPP (Das et al., 2015)], the catalytic efficiency (% of product formation and TON) of 1a and 1b is comparatively low (Romakh et al., 2007; Das et al., 2015).
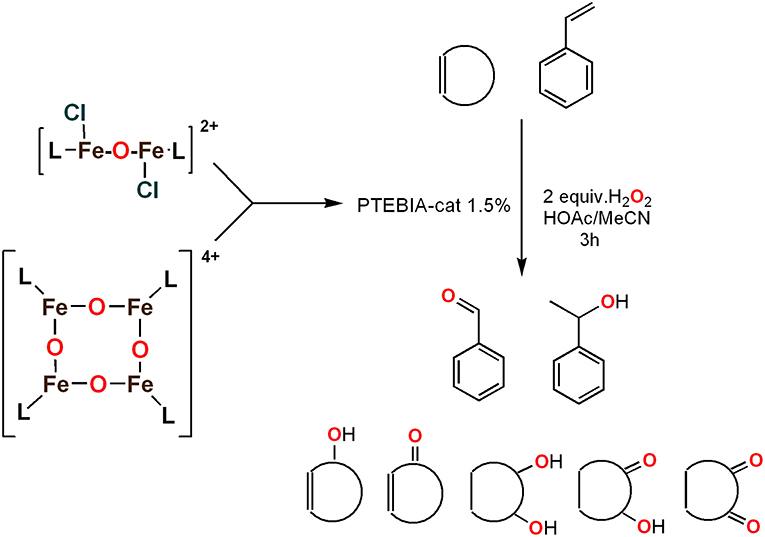
Scheme 1. Schematic depiction of the catalytic oxidation reactions effected by complexes 1a/1b; cf. text and Table 8.
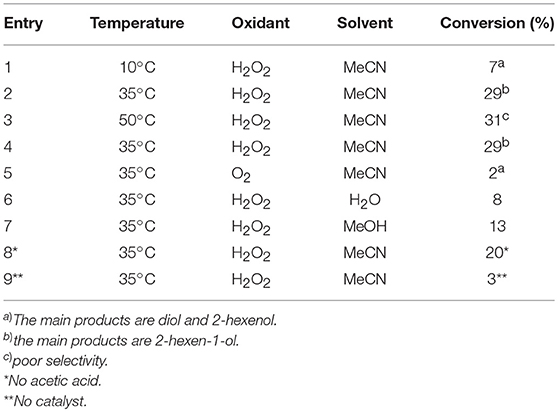
Table 7. Optimization conditions using cyclohexene as a model substrate; substrate:catalyst:oxidant:AcOH (100:1.5:200:80), solvent 3 ml, 3 h reaction time.
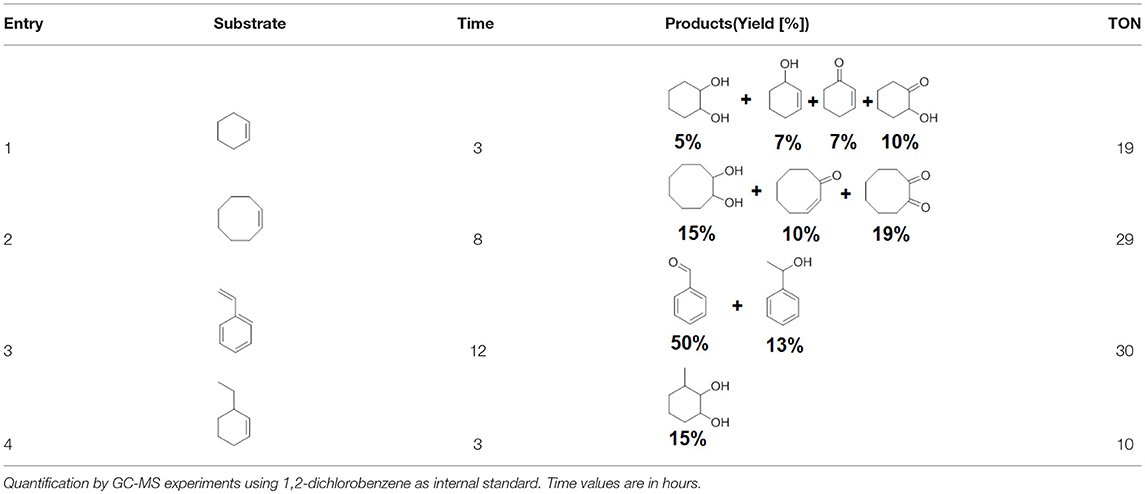
Table 8. Reaction conditions: Substrate:catalyst:H2O2:AcOH (100:1.5: 200:80), MeCN as solvent 3 mL, temperature 35°C.
In addition to [(Fe2)(OAc)2(μ-O)(PTEBIA)2(CF3SO3)]+ detected by ESI MS, the main peaks in the mass spectra, and their isotopic patterns are consistent with species formulated as monomeric [Fe(HPTEBIA)(OH)]+, [Fe(HPTEBIA)(O)(OH)]+, [Fe(PTEBIA)(O)(CH3COO)]+, and the protonated form of the oxidized ligand [(HPTEBIA)(O)]+ (see Figures S4–S6, Supplementary Material), irrespective of whether 1a and 1b were analyzed separately or as a 1:1 mixture. This indicates that the active species present during turnover conditions appear to be identical regardless of the precursor. Preferential oxidation of the thioether moiety (generating the corresponding sulfoxide) over the metal center may be occurring, as reported by Goldberg and coworkers with the [FeII(N3PyamideSR)](BF4)2 system in the presence of 5 equivalents of mCPBA (McQuilken and Goldberg, 2012). No evidence of the formation of Fe(IV) = O species was observed in analogous experiments with mCPBA or PhIO as oxo-transfer reagents. From the catalytic oxidation results induced by H2O2 and CH3COOH, we presume that catalysis with the current complex system does not involve heterolysis of the O-O bond to form a highly reactive Fe(V)O species, which should result in higher product selectivity (Prat et al., 2013). Instead, it may proceed via homolysis to give transient [LFe(IV)O] and hydroxyl radical (OH) intermediates, with participation of both species in the observed oxidations (Trettenhahn et al., 2006).
We have synthesized and characterized a new tetrairon tetraoxo cluster (1a) with a sulfur-containing ligand. In the UV-vis spectroscopic experiments, a prominent sulfur to iron charge transfer band (390–430 nm) was observed at room temperature for 1a and the corresponding dinuclear Fe(III)-O-Fe(III) complex 1b, whereas for the related dinuclear dicarboxylate-bridged Fe(III)-O-Fe(III) complex 2 it was only visible at low temperature (228 K) in the presence of H2O2/CH3COOH (presumably after initial protonation/dissociation of the oxo and formate groups of 2), and disappears at room temperature, likely due to loss of the Fe-S bond and/or sulfoxidation of PTEBIA. The catalytic efficiency of the μ-oxido iron(III) complex mixture of 1a and 1b for the oxidation of alkenes has been investigated, revealing that they act as moderate oxidation catalyst(s)/catalyst precursor(s). The reaction appears to proceed partially through a metal-centered process in tandem with free-radical oxidation by reactive oxygen species. Low temperature UV-vis spectroscopy and mass spectrometry were employed to gain insight into possible reactive intermediates/active oxidation catalysts, revealing that both H2O2/CH3COOH and PhIO preferably oxidize the thioether group of the ligand, in contrast with previous oxygenations with related copper-based PTEBIA systems, where the thioether functionality remains intact.
The ligand PTEBIA was prepared following the procedure reported by Castillo et al. (2012). All reagents and solvents were of analytical or spectroscopic grade purchased from Sigma Aldrich, Fisher chemicals or VWR, and were used without further purification. Cyclohexene (≥ 99.0%) contained ~0.01% of 2,6-di-tert-butyl-4-methylphenol as stabilizer and was used as received. Caution! Even though no problems were encountered in this work, caution should always be taken while using high concentrations of hydrogen peroxide (H2O2), as well as metal perchlorates.
Infrared spectra were recorded in the 4,000–400 cm−1 range on a Nicolet Avatar 360 FTIR spectrometer, as KBr pellets. Mass spectra were obtained on a JEOL JMS-SX-102A mass spectrometer at an accelerating voltage of 10 kV with a nitrobenzyl alcohol matrix and Xenon atoms at 6 keV (FAB+), a JEOL JMS-AX505HA spectrometer (Electron Ionization), or a Bruker Daltonics Esquire 6000 spectrometer with ion trap (Electrospray). Elemental analyses were performed at the microanalytical facility of the Instituto de Química, UNAM, Mexico. Analytical achiral GC was performed on an Agilent 6850 GC with FID detector using an Agilent DB-WAX (30.0 m × 0.25 mm) column at mL/min He carrier gas flow. Chiral GC was performed on an Agilent 6850 GC with FID detector. The 1H NMR spectra were recorded with a Varian Gemini 200 apparatus or a Varian Mercury 300 MHz spectrometer.
Substrate conversions in catalytic experiments were determined by GC-MS. The GC-MS analyses were performed with an Agilent 6890 N Network GC system equipped with a DB-1MS column (30 m × 0.25 mm) and an Agilent 5973 Network MS detector. Calibration curves were obtained from commercial products purchased from Aldrich or TCI when available or from pure isolated products obtained from a catalytic reaction using a FID-detector GC with a HP-INOWAX column (30 m × 0.25 mm) (1,2-dichlorobenzene used as an internal standard). The concentrations of each organic product were calibrated relative to that of an internal standard (1,2-dichlorobenzene) with a known concentration.
To 15 mL of a tetrahydrofuran solution of 0.13 g (0.26 mmol) of PTEBIA, 0.10 g (0.26 mmol) of Fe(II)(triflate)2 were added and the solution was refluxed for 4 h under vigorous stirring. Immediately after the addition of Fe(II)(triflate)2, the solution becomes turbid green, and on reflux it turns yellowish brown. The yellowish brown solution was filtered and the filtrate was collected in a 50 mL flask. Evaporation of the solvent under vacuum produces a tan oil. Overnight slow vapor diffusion of diethyl ether to a concentrated acetonitrile solution of the tan residue produces 0.12 g of 1a [Fe4(μ-O)4(PTEBIA)4](CF3SO3)4(CH3CN)2 as yellow microcrystals (65%). UV-vis (CH3CN): λmax = 335 nm (ε = 3,600 M−1 cm−1), 430 nm (ε~ 2,280 M−1 cm−1). 57Fe Mössbauer (80 K) δ = 0.55 mm/s; ΔEQ = 1.15 mm/s; ESI-MS in acetonitrile solution calculated for [Fe(PTEBIA)(CF3SO3)]+ (C29H31F3FeN5O3S2) (mononuclear species): 674.1; found 673.8. IR (KBr, cm−1): 2953, 2924, 2856, 1767, 1722, 1539, 1501, 1456, 1365, 1250, 1226, 1160, 1028, 967, 913, 819, 749, 636, 574, 517, 432. Anal. Calcd for C116H124F12Fe4N20O16S4: C, 50.44; H, 4.52; N, 10.14; S, 9.29; found: C, 50.19; H, 4.50; N, 9.88; S, 9.02.
The procedure is analogous to that for 1a, employing 33 mg (0.26 mmol) of FeCl2. The brown solution was filtered and the filtrate was collected in a 50 mL flask. Evaporation of the solvent under vacuum produces a brown solid. Overnight slow vapor diffusion of diethyl ether to a concentrated acetonitrile solution of the brown solid affords 0.11 g of 1b [Fe2(μ-O)Cl2(PTEBIA)2](CF3SO3)2 as brown microcystals [m.p. 195-197°C (dec)] (61%). UV-vis (CH3CN): λmax = 320 nm (ε = 2,400 M–1 cm–1), 390 nm (ε ~ 1,990 M–1 cm–1). 57Fe Mössbauer (80 K) δ = 0.28 mm/s; ΔEQ = 1.31 mm/s; ESI- MS in acetonitrile solution calculated for [Fe(PTEBIA)(Cl)]+ (C28H31ClFeN5S) (mononuclear species): 560.1; found 559.7. IR (KBr, cm–1): 2935, 1736, 1597, 1491, 1452, 1426, 1378, 1256, 1223, 1151, 1100, 1054, 1029, 959, 932, 815, 780, 746, 700, 633, 572, 545, 516, 430. Anal. Calcd for C58H62Cl2F6Fe2N10O7S4: C, 48.51; H, 4.35; N, 9.75; S, 8.93; found: C, 48.32; H, 4.39; N, 9.00; S, 8.40.
To a 20 mL tetrahydrofuran solution of 0.30 g (0.64 mmol) of PTEBIA, 0.16 g (0.64 mmol) of Fe(II)(ClO4)2 was added and the solution was refluxed for 3 h. The colorless solution of the ligand changes immediately to turbid green on addition of Fe(ClO4)2 and to yellowish green after 3 h of reflux. This yellowish green solution was filtered and the filtrate was dried under vacuum overnight in a 50 mL round bottom flask to get 0.64 g of 2 (74% yield). Slow vapor diffusion of diethyl ether for 4 days to the methanolic solution of 2 leads to needle-shaped yellowish green crystals of X-ray quality. UV-Vis (CH3CN): λmax = 335 nm (ε = 4,400 M–1 cm–1). ESI-MS in acetonitrile solution calculated for [Fe(PTEBIA)]2+ (C28H31FeN5S)2+ (mononuclear species): 262.6; found 262.8. IR (KBr, cm–1): 3063, 3031, 2945, 2917, 1614, 1490, 1453, 1359, 1328, 1289, 1269, 1238, 1080, 928, 896, 865, 814, 795, 744, 695, 620, 545, 522, 489, 432. Anal. Calcd for 2: C, 51.38; H, 4.76; N, 10.33; S, 4.73; found: C, 50.99; H, 4.85; N, 10.45; S, 4.75.
Each catalytic experiment was performed at least twice and the reported conversion is the average value. A general procedure for the oxidation experiments is as follows: magnetic stirring bar, catalyst complex (18 μmol), 2 mL of CH3CN, acetic acid (AcOH, 50 μL, 85 μmol), 230 μL of H2O2 (33% in water, 2.0 equivalents with respect to the substrate) and substrate (1 mmol) were placed in a Schlenk flask. The reaction mixture was stirred under argon at 35°C for the designated time. Sodium thiosulfate (ca. 400 mg, 2.5 mmol) was then added to the reaction mixture to quench further oxidation. 1,2-dicholorobenzene was added to the mixture followed by extraction with n-pentane and filtering through a silica gel column for analysis by GC-MS.
Crystal data and details of the data collections are given in Table 9. X-ray data for complexes 1a and 1b were collected on a STOE IPDS II diffractometer (graphite monochromated Mo-Kα radiation, λ = 0.71073 Å) by use of ω scans at −140°C. The structures were solved by direct methods (SHELXS-2014) and refined on F2 using all reflections with SHELXL-2014 (Sheldrick, 2008). Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and assigned to an isotropic displacement parameter of 1.2 / 1.5 Ueq(C). In each compound one of the two CF3 ions was found to be disordered [occupancy factors: 1a = 0.929(15)/0.071(15), 1b = 0.784(4)/0.216(4)]. SAME restraints and EADP constraints were used to model the respective disorders. In 1a one 2,4-dimethylphenyl moiety involving the carbon atoms C51(A/B/C) to C58(A/B/C) was found to be disordered about three positions A, B, and C along with acetonitrile (N21, C95, C96, belonging to B) and water (O31, belonging to C). After initial refinement the occupancy factors were set to 0.5 for A, 0.1 for B and 0.4 for C. RIGU, FLAT, SADI [d(1,3)C(ar)···C(Me)] and DFIX (dC(ar)−C(Me) = 1.5 Å) restraints and EADP constraints were applied to model the disorder. The AFIX 66 instruction was applied for the carbon atoms of the ring. Furthermore, acetonitrile disordered about a 2-fold rotation axis and about two positions [occupancy factors: 0.277(7)/ 0.223(7)] was refined using RIGU restraints. Face-indexed absorption corrections were performed numerically with the program X-RED (Stoe & Cie and X-RED, 2005). The crystal of 2 was immersed in cryo-oil, mounted in a MiTeGen loop, and measured at a temperature of 170 K on a Rigaku Oxford Diffraction Supernova diffractometer using Mo Kα radiation. The CrysAlisPro (Rigaku Oxford Diffraction, 2013) software was used for cell refinement and data reduction. Empirical absorption correction based on equivalent reflections was [CrysAlisPro (Rigaku Oxford Diffraction, 2013)] was applied to the intensities before structure solutions. The structure was solved by charge flipping method using the SUPERFLIP (Palatinus and Chapuis, 2007) and the structure refinement was carried out using SHELXL (Sheldrick, 2015) program. The crystal of 2 contained solvent accessible voids but no satisfactory solvent model could be found. The contribution of the missing solvent to the calculated structure factors was taken into account by using the SQUEEZE routine of PLATON (Spek, 2009). The missing solvent was not taken into account in the unit cell content. Hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C-H = 0.95–0.99 Å, O-H = 0.84 Å, and Uiso = 1.2–1.5 Ueq (parent atom). The highest peak is located 1.40 Å from atom S1 and the deepest hole is located 0.59 Å from atom S1.
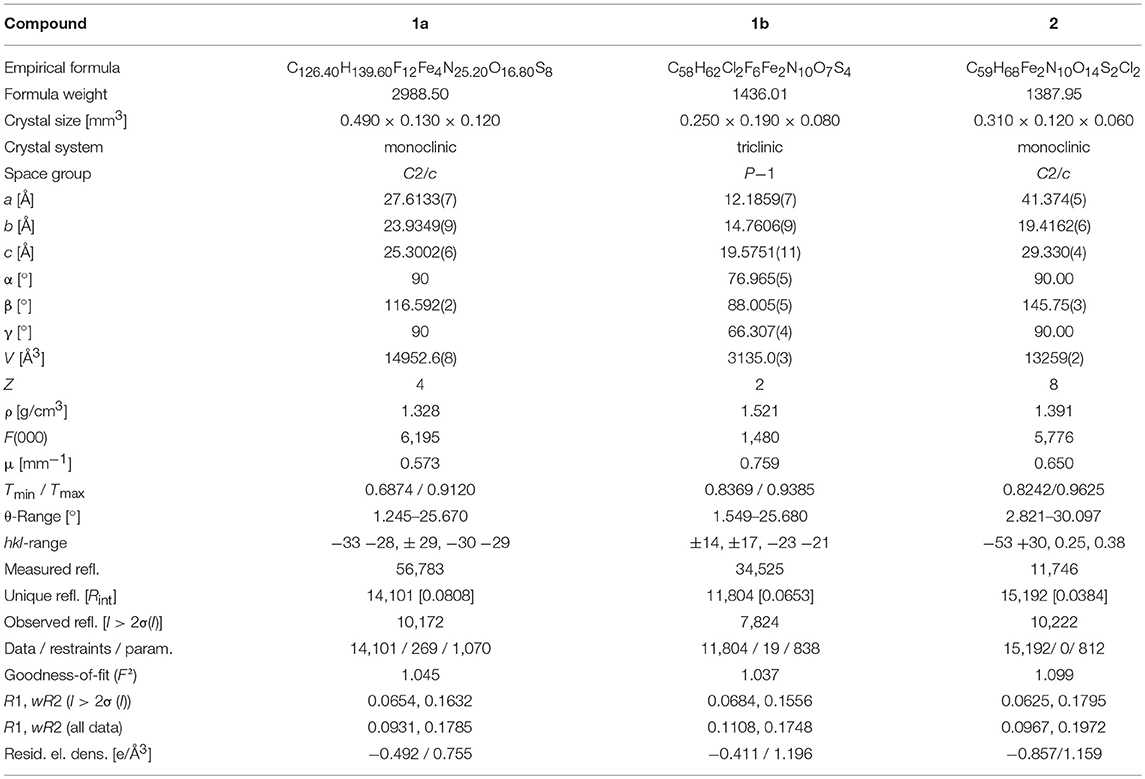
Table 9. Crystal data and refinement details for 1a, 1b, and 2.
The Mössbauer spectrum was recorded at 80 K with a 57Co source in a Rh matrix, using an alternating constant-acceleration Wissel Mössbauer spectrometer operated in the transmission mode and equipped with a Janis closed-cycle helium cryostat or with a Mössbauer-Spectromag cryostat. Isomer shifts (cf. caption, Figure 3) are given relative to iron metal at ambient temperature. Experimental data were simulated using the Mfit software (developed by E. Bill, Max-Planck Institute for Chemical Energy Conversion, Mülheim/Ruhr, Germany, 2008).
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher. Crystallographic data for the structures in this paper have been deposited with the Cambridge Crystallographic Data Center, CCDC, 12 Union Road, Cambridge CB21EZ, UK. Copies of the data can be obtained free of charge on quoting the depository number CCDC 1874712 (1a), 1874713 (1b) or 1874714 (2) (Fax:+44-1223-336-033; E-Mail: ZGVwb3NpdEBjY2RjLmNhbS5hYy51aw==, http://www.ccdc.cam.ac.uk).
BD performed all complex syntheses, performed low temperature studies in collaboration with BS-E, participated in the oxidation experiments, and contributed to the writing of the manuscript. AA-H performed all oxidation experiments and contributed to the writing of the manuscript. BS-E and EZ synthesized the PTEBIA ligand. SebD performed all Mössbauer measurements and analysis of the Mössbauer data. SerD, SB, and MH collected X-ray data and solved the crystal structures. TR supervised the oxidation studies and analyzed the results in collaboration with AA-H and BD, and supplied funding for the work. IC led and designed the study, contributed to the writing of the manuscript and supplied funding for the work. EN led and designed the study, contributed to the writing of the manuscript and supplied funding for the work.
This research has been carried out within the framework of the International Research Training Group, Metal Sites in Biomolecules: Structures, Regulation, and Mechanisms (www.biometals.eu) and supported by COST Action CM1003, Conacyt (151837, Beca 254496), the Swedish Research Council (2014-0429), and DGAPA-PAPIIT (IN210214).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BD thanks the European Commission for an Erasmus Mundus predoctoral fellowship. We thank Carmen Márquez for ESI-MS, María de la Paz Orta for combustion analysis, Rocío Patiño for IR measurements, and Dr. S. Maji and Prof. Franc Meyer for useful discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00097/full#supplementary-material
Ballmann, J., Albers, A., Demeshko, S., Dechert, S., Bill, E., Bothe, E., et al. (2008a). A synthetic analogue of rieske-type [2Fe-2S] clusters. Angew. Chem. Int. Ed. 47, 9537–9541. doi: 10.1002/anie.200803418
Ballmann, J., Dechert, S., Bill, E., Ryde, U., and Meyer, F. (2008b). Secondary bonding interactions in biomimetic [2Fe−2S] clusters. Inorg. Chem. 47, 1586–1596. doi: 10.1021/ic702095a
Beinert, H., Holm, R. H., and Münck, E. (1997). Iron-sulfur clusters: nature's modular, multipurpose structures. Science 277, 653–659.
Castillo, I., Ugalde-Saldívar, V. M., Rodríguez Solano, L. A., Sánchez Eguía, B. N., Zeglio, E., and Nordlander, E. (2012). Structural, spectroscopic, and electrochemical properties of tri- and tetradentate N3 and N3S copper complexes with mixed benzimidazole/thioether donors. Dalton Trans. 41, 9394–9404. doi: 10.1039/c2dt30756a
Costas, M., Chen, K., and Que, L. (2000). Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 200–202, 517–544. doi: 10.1016/S0010-8545(00)00320-9
Costas, M., Mehn, M. P., Jensen, M. P., and Lawrence Que, J. (2004). Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986. doi: 10.1021/cr020628n
Das, B., Al-Hunaiti, A., Haukka, M., Demeshko, S., Meyer, S., Shteinman, A. A., et al., Full list of authors (2015). Catalytic oxidation of alkanes and alkenes by H2O2 with a μ-oxido diiron(III) complex as catalyst/catalyst precursor. Eur. J. Inorg. Chem. 2015, 3590–3601. doi: 10.1002/ejic.201500576
Das, B., Daver, H., Pyrkosz-Bulska, M., Gumienna-Kontecka, E., Himo, F., and Nordlander, E. (2018). An unsymmetric ligand with a N5O2 donor set and its corresponding dizinc complex: a structural and functional phosphoesterase model. Eur. J. Inorg. Chem. 30, 4004–4013. doi: 10.1002/ejic.201701416
Das, B., Daver, H., Pyrkosz-Bulska, M., Persch, E., Barman, S. K., Mukherjee, R., et al. (2014a). A dinuclear zinc(II) complex of a new unsymmetric ligand with an N5O2 donor set: a structural and functional model for the active site of zinc phosphoesterases. J. Inorg. Biochem. 132, 6–17. doi: 10.1016/j.jinorgbio.2013.08.001
Das, B., Daver, H., Singh, A., Singh, R., Haukka, M., Demeshko, S., et al. (2014b). A heterobimetallic Fe III Mn II complex of an unsymmetrical dinucleating ligand: a structural and functional model complex for the active site of purple acid phosphatase of sweet potato. Eur. J. Inorg. Chem. 2014, 2204–2212. doi: 10.1002/ejic.201301375
De Visser, S. P., Rohde, J. U., Lee, Y. M., Cho, J., and Nam, W. (2013). Intrinsic properties and reactivities of mononuclear nonheme iron–oxygen complexes bearing the tetramethylcyclam ligand. Coord. Chem. Rev. 257, 381–393. doi: 10.1016/j.ccr.2012.06.002
Deacon, G. B., and Phillips, R. J. (1980). Relationships between the carbon-oxygen stretching frequencies of carboxylato complexes and the type of carboxylate coordination. Coord. Chem. Rev. 33, 227–250. doi: 10.1016/S0010-8545(00)80455-5
Do, L. H., Xue, G., Que, L., and Lippard, S. J. (2012). Evaluating the identity and diiron core transformations of a (μ-Oxo)diiron(III) complex supported by electron-rich tris(pyridyl-2-methyl)amine ligands. Inorg. Chem. 51, 2393–2402. doi: 10.1021/ic202379b
Dubois, G., Murphy, A., and Stack, T. D. P. (2003). Simple iron catalyst for terminal alkene epoxidation. Org. Lett. 5, 2469–2472. doi: 10.1021/ol0347085
Fuchs, M. G., Dechert, S., Demeshko, S., Ryde, U., and Meyer, F. (2010). A five-coordinate [2Fe−2S] cluster. Inorg. Chem. 49, 5853–5858. doi: 10.1021/ic902559n
Fujita, M., and Que, L. (2004). In situ formation of peracetic acid in iron-catalyzed epoxidations by hydrogen peroxide in the presence of acetic acid. Adv. Synth. Catal. 346, 190–194. doi: 10.1002/adsc.200303204
Gamba, I., Codolà, Z., Lloret-Fillol, J., and Costas, M. (2017). Making and breaking of the OO bond at iron complexes. Coord. Chem. Rev. 334, 2–24. doi: 10.1016/j.ccr.2016.11.007
Harrop, T. C., and Mascharak, P. K. (2004). Fe(III) and Co(III) centers with carboxamido nitrogen and modified sulfur coordination: lessons learned from nitrile hydratase. Acc. Chem. Res. 37, 253–260. doi: 10.1021/ar0301532
Hasemann, C. A., Kurumbail, R. G., Boddupalli, S. S., Peterson, J. A., and Deisenhofer, J. (1995). Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure 3, 41–62. doi: 10.1016/S0969-2126(01)00134-4
Hazell, A., McKenzie, C. J., Nielsen, L. P., Schindler, S., and Weitzer, M. (2002). Mononuclear non-heme iron(III) peroxide complexes: syntheses, characterisation, mass spectrometric and kinetic studies. Dalton Trans. 310–317. doi: 10.1039/b103844n
Jarenmark, M., Turitsyna, E. A., Haukka, M., Shteinman, A. A., and Nordlander, E. (2010). A monocarboxylate-bridged diiron(III) μ-oxido complex that catalyzes alkane oxidation by hydrogen peroxide. N. J. Chem. 34, 2118–2121. doi: 10.1039/c0nj00293c
Kim, C., Chen, K., Kim, J., and Que, L. (1997). Stereospecific alkane hydroxylation with H2O2 catalyzed by an iron(II)–tris(2-pyridylmethyl)amine complex. J. Am. Chem. Soc. 119, 5964–5965. doi: 10.1021/ja9642572
Kumar, D., Thiel, W., and de Visser, S. P. (2011). Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J. Am. Chem. Soc. 133, 3869–3882. doi: 10.1021/ja107514f
Kurtz, D. M. (1990). Oxo- and hydroxo-bridged diiron complexes: a chemical perspective on a biological unit. Chem. Rev. 90, 585–606. doi: 10.1021/cr00102a002
Lill, R. (2009). Function and biogenesis of iron–sulphur proteins. Nature 460, 831–838. doi: 10.1038/nature08301
Lindhorst, A. C., Haslinger, S., and Kühn, F. E. (2015). Molecular iron complexes as catalysts for selective C–H bond oxygenation reactions. Chem. Commun. 51, 17193–17212. doi: 10.1039/C5CC07146A
Mayilmurugan, R., Stoeckli-Evans, H., Suresh, E., and Palaniandavar, M. (2009). Chemoselective and biomimetic hydroxylation of hydrocarbons by non-heme μ-oxo-bridged diiron(iii) catalysts using m-CPBA as oxidant. Dalton Trans. 5101–5114. doi: 10.1039/b820771b
McCoy, J. G., Bailey, L. J., Bitto, E., Bingman, C. A., Aceti, D. J., Fox, B. G., et al. (2006). Structure and mechanism of mouse cysteine dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 3084–3089. doi: 10.1073/pnas.0509262103
McDonald, A. R., Bukowski, M. R., Farquhar, E. R., Jackson, T. A., Koehntop, K. D., Seo, M. S., et al. (2010). Sulfur versus iron oxidation in an iron–thiolate model complex. J. Am. Chem. Soc. 132, 17118–17129. doi: 10.1021/ja1045428
McQuilken, A. C., and Goldberg, D. P. (2012). Sulfur oxygenation in biomimetic non-heme iron–thiolate complexes. Dalton Trans. 41, 10883–10899. doi: 10.1039/c2dt30806a
Meyer, J. (2008). Iron–sulfur protein folds, iron–sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 13, 157–170. doi: 10.1007/s00775-007-0318-7
Mitra, M., Lloret-Fillol, J., Haukka, M., Costas, M., and Nordlander, E. (2014). Evidence that steric factors modulate reactivity of tautomeric iron–oxo species in stereospecific alkane C–H hydroxylation. Chem. Commun. 50, 1408–1410. doi: 10.1039/C3CC47830K
Nam, W. (2015). Synthetic mononuclear nonheme iron–oxygen intermediates. Acc. Chem. Res. 48, 2415–2423. doi: 10.1021/acs.accounts.5b00218
Norman, R. E., Leising, R. A., Yan, S., and Que, L. (1998). Unexpected assembly of a (μ-oxo)(μ-formato)diiron(III) complex from an aerobic methanolic solution of Fe(III) and the TPA ligand. Inorg. Chim. Acta 273, 393–396. doi: 10.1016/S0020-1693(97)06058-1
Norman, R. E., Yan, S., Que, L., Backes, G., Ling, J., Sanders-Loehr, J., et al. (1990). (μ.-Oxo)(μ.-carboxylato)diiron(III) complexes with distinct iron sites. Consequences of the inequivalence and its relevance to dinuclear iron-oxo proteins. J. Am. Chem. Soc. 112, 1554–1562. doi: 10.1021/ja00160a039
Ohnishi, T. (1998). Iron–sulfur clusters/semiquinones in Complex I. Biochim. Biophys. Acta Bioenerg. 1364, 186–206. doi: 10.1016/S0005-2728(98)00027-9
Palatinus, L., and Chapuis, G. (2007). SUPERFLIP – a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 40, 786–790. doi: 10.1107/S0021889807029238
Prat, I., Company, A., Postils, V., Ribas, X., Que, L., Luis, J. M., et al. (2013). The mechanism of stereospecific C–H oxidation by Fe(Pytacn) complexes: bioinspired non-heme iron catalysts containing cis-labile exchangeable sites. Chem. A Eur. J. 19, 6724–6738. doi: 10.1002/chem.201300110
Rao, P. V., and Holm, R. H. (2004). Synthetic analogues of the active sites of iron–sulfur proteins. Chem. Rev. 104, 527–560. doi: 10.1021/cr020615+
Reem, R. C., McCormick, J. M., Richardson, D. E., Devlin, F. J., Stephens, P. J., Musselman, R. L., et al. (1989). Spectroscopic studies of the coupled binuclear ferric active site in methemerythrins and oxyhemerythrin: the electronic structure of each iron center and the iron-oxo and iron-peroxide bonds. J. Am. Chem. Soc. 111, 4688–4704. doi: 10.1021/ja00195a024
Rodríguez Solano, L. A., Aguiñiga, I., López Ortiz, M., Tiburcio, R., Luviano, A., Regla, I., et al. (2011). Bis(2-methylbenzimidazolyl)amine-derived copper complexes and their antineoplastic activity. Eur. J. Inorg. Chem. 2011, 3454–3460. doi: 10.1002/ejic.201100301
Rohde, J. U., In, J. H., Lim, M. H., Brennessel, W. W., Bukowski, M. R., Stubna, A., et al. (2003). Crystallographic and spectroscopic characterization of a nonheme Fe(IV)-O complex. Science 299, 1037–1039. doi: 10.1126/science.299.5609.1037
Romakh, V. B., Therrien, B., Georg, S.-F., and Shul'pin, G. B. (2007). Dinuclear manganese complexes containing chiral 1,4,7-triazacyclononane-derived ligands and their catalytic potential for the oxidation of olefins, alkanes, and alcohols. Inorg. Chem. 46, 1315–1331. doi: 10.1021/ic061950+
Sahoo, S. C., and Ray, M. (2007). Ferrocene substitution in amino acids strengthens the axial binding in Cu(II) complexes and separates the hydrophobic and hydrophilic region in the crystals. Dalton Trans. 5148–5155. doi: 10.1039/b709662c
Sallmann, M., Siewert, I., Fohlmeister, L., Limberg, C., and Knispel, C. (2012). A Trispyrazolylborato iron cysteinato complex as a functional model for the cysteine dioxygenase. Angew. Chemie Int. Ed. 51, 2234–2237. doi: 10.1002/anie.201107345
Sheldrick, G. M. (2008). A short history of SHELX. Acta Crystallogr. Sect. A 64, 112–122. doi: 10.1107/S0108767307043930
Sheldrick, G. M. (2015). Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 71, 3–8. doi: 10.1107/S2053229614024218
Spek, A. L. (2009). Structure validation in chemical crystallography. Acta Crystallogr. Sect. D 65, 148–155. doi: 10.1107/S090744490804362X
Stoe & Cie and X-RED (2005). Program for Data Reduction and Absorption Correction. Darmstadt: Stoe & Cie GmbH.
Sun, C. L., Li, B. J., and Shi, Z. J. (2011). Direct C–H transformation via iron catalysis. Chem. Rev. 111, 1293–1314. doi: 10.1021/cr100198w
Tinberg, C. E., and Lippard, S. J. (2011). Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Res. 44, 280–288. doi: 10.1021/ar1001473
Trettenhahn, G., Nagl, M., Neuwirth, N., Arion, V. B., Jary, W., Pöchlauer, P., et al. (2006). A hexanuclear iron(III) carboxylate with an [Fe6(μ3-O)3(μ2-OH)]11+ core as an efficient catalyst for cycloalkane oxidation. Angew. Chemie Int. Ed. 45, 2794–2798. doi: 10.1002/anie.200504406
Vad, M. S., Lennartson, A., Nielsen, A., Harmer, J., McGrady, J. E., Frandsen, C., et al. (2012). An aqueous non-heme Fe(IV)oxo complex with a basic group in the second coordination sphere. Chem. Commun. 48, 10880–10882. doi: 10.1039/c2cc35746a
Visvaganesan, K., Suresh, E., and Palaniandavar, M. (2009). Highly selective hydroxylation of alkanes catalyzed by (μ-oxo)bis(μ-carboxylato)-bridged diiron(III) complexes: involvement of mononuclear iron(III) species in catalysis. Dalton Trans. 3814–3823. doi: 10.1039/b901508f
Wackett, L. P. (2002). Mechanism and applications of Rieske non-heme iron dioxygenases. Enzyme Microb. Technol. 31, 577–587. doi: 10.1016/S0141-0229(02)00129-1
Wang, D., Ray, K., Collins, M. J., Farquhar, E. R., Frisch, J. R., Gómez, L., et al. (2013a). Nonheme oxoiron(IV) complexes of pentadentate N5 ligands: spectroscopy, electrochemistry, and oxidative reactivity. Chem. Sci. 4, 282–291. doi: 10.1039/C2SC21318D
Wang, X., Wang, S., Li, L., Sundberg, E. B., and Gacho, G. P. (2003). Synthesis, structure, and catalytic activity of mononuclear iron and (μ-oxo)diiron complexes with the ligand 2,6-bis(n-methylbenzimidazol-2-yl)pyridine. Inorg. Chem. 42, 7799–7808. doi: 10.1021/ic0259437
Wang, Y., Janardanan, D., Usharani, D., Han, K., Que, L., and Shaik, S. (2013b). Nonheme iron oxidant formed in the presence of H2O2 and acetic acid is the cyclic ferric peracetate complex, not a perferryloxo complex. ACS Catal. 3, 1334–1341. doi: 10.1021/cs400134g
White, M. C., Doyle, A. G., and Jacobsen, E. N. (2001). A synthetically useful, self-assembling MMO mimic system for catalytic alkene epoxidation with aqueous H2O2. J. Am. Chem. Soc. 123, 7194–7195. doi: 10.1021/ja015884g
Widger, L. R., Davies, C. G., Yang, T., Siegler, M. A., Troeppner, O., Jameson, G. N. L., et al. (2014). Dramatically accelerated selective oxygen-atom transfer by a nonheme iron(IV)-oxo complex: tuning of the first and second coordination spheres. J. Am. Chem. Soc. 136, 2699–2702. doi: 10.1021/ja410240c
Keywords: Fe-S interaction, oxidation, homogeneous catalysis, thioether, iron-oxo complex
Citation: Das B, Al-Hunaiti A, Sánchez-Eguía BN, Zeglio E, Demeshko S, Dechert S, Braunger S, Haukka M, Repo T, Castillo I and Nordlander E (2019) Di- and Tetrairon(III) μ-Oxido Complexes of an N3S-Donor Ligand: Catalyst Precursors for Alkene Oxidations. Front. Chem. 7:97. doi: 10.3389/fchem.2019.00097
Received: 26 October 2018; Accepted: 04 February 2019;
Published: 01 March 2019.
Edited by:
Andrea Erxleben, National University of Ireland Galway, IrelandReviewed by:
Sam P. De Visser, University of Manchester, United KingdomCopyright © 2019 Das, Al-Hunaiti, Sánchez-Eguía, Zeglio, Demeshko, Dechert, Braunger, Haukka, Repo, Castillo and Nordlander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivan Castillo, am9zZWl2YW5AdW5hbS5teA==
Ebbe Nordlander, RWJiZS5Ob3JkbGFuZGVyQGNoZW1waHlzLmx1LnNl
†Present Address: Afnan Al-Hunaiti, Department of Chemistry, School of Science, University of Jordan, Amman, Jordan
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.