 Stephen Rhatigan
Stephen Rhatigan Michael Nolan
Michael Nolan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 12 February 2019
Sec. Chemical Physics and Physical Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00067
This article is part of the Research TopicThe Role of Non-Stoichiometry in the Functional Properties of Oxide MaterialsView all 14 articles
Surface modification of titania surfaces with dispersed metal oxide nanoclusters has the potential to enhance photocatalytic activity. These modifications can induce visible light absorption and suppress charge carrier recombination which are vital in improving the efficiency. We have studied heterostructures of Mn4O6 nanoclusters modifying the TiO2 rutile (110) and anatase (101) surfaces using density functional theory (DFT) corrected for on-site Coulomb interactions (DFT + U). Such studies typically focus on the pristine surface, free of the point defects and surface hydroxyls present in real surfaces. In our study we have considered partial hydroxylation of the rutile and anatase surfaces and the role of cation reduction, via oxygen vacancy formation, and how this impacts on a variety of properties governing the photocatalytic performance such as nanocluster adsorption, light absorption, charge separation, and reducibility. Our results indicate that the modifiers adsorb strongly at the surface and that modification extends light absorption into the visible range. MnOx-modified titania can show an off-stoichiometric ground state, through oxygen vacancy formation and cation reduction spontaneously, and both modified rutile and anatase are highly reducible with moderate energy costs. Manganese ions are therefore present in a mixture of oxidation states. Photoexcited electrons and holes localize at cluster metal and oxygen sites, respectively. The interaction of water at the modified surfaces depends on the stoichiometry and spontaneous dissociation to surface bound hydroxyls is favored in the presence of oxygen vacancies and reduced metal cations. Comparisons with bare TiO2 and other TiO2-based photocatalyst materials are presented throughout.
Photocatalysts are semiconductor materials which absorb photons of energies in excess of the bandgap to produce electron-hole pairs. These charge carriers separate and migrate to the surface of the catalyst where they drive chemical reactions via reduction and oxidation of adsorbed species. Photocatalysis has a variety of applications, including, but not limited to, the solar production of hydrogen from water splitting (Ni et al., 2007; Fujishima et al., 2008; Maeda and Domen, 2010; Jiang et al., 2017).
A practical photocatalyst must meet a number of criteria, such as visible light absorption, efficient charge carrier separation, stability, and active surface sites for the adsorption of feedstock species. Reducible metal cations in the catalyst can be important for enhancing the activity of the catalyst toward the difficult step of water dissociation. The development of metal oxide photocatalysts is of interest as these materials are cheap, earth abundant and, in many instances, non-toxic. Indeed, the most widely studied photocatalyst is titanium dioxide (TiO2) (Ni et al., 2007; Fujishima et al., 2008; Dimitrijevic et al., 2011; Pelaez et al., 2012; Habisreutinger et al., 2013; Tada et al., 2014; Etacheri et al., 2015) which was first demonstrated as a photoanode for water splitting by Fujishima and Honda (1972). The large bandgap (>3 eV) means that photoactivity is restricted to the UV and has limited the real-world application of TiO2-based photocatalyst technologies. As a result, significant scientific effort has focused on extending the light absorption edge of TiO2 to longer wavelengths.
Substitutional doping of TiO2 with cations and/or anions is a widely studied approach to inducing visible light absorption through the emergence of impurity-derived energy levels in the TiO2 bandgap (Di Valentin et al., 2007; Czoska et al., 2008; Haowei et al., 2008; Ikeda et al., 2008; Gai et al., 2009; Nie et al., 2009; Valentin et al., 2009; Xu et al., 2009; Yang et al., 2009; Yu et al., 2009; Zhu et al., 2009; Long and English, 2010a,b; Zhang et al., 2010; Zheng et al., 2010; Herrmann, 2012; Etacheri et al., 2015; Li, 2015; Na Phattalung et al., 2017). First principles studies of such doped systems typically focus on bandgap reduction (Cui et al., 2008; Yang et al., 2009; Zhu et al., 2009; Long and English, 2010a,b; Zhang et al., 2010, 2014; Chand et al., 2011; Guo and Du, 2012; Na Phattalung et al., 2017) and questions of charge localization and surface reactivity are often overlooked. These are important considerations as dopant-derived defect states have been shown to act as recombination centers (Herrmann, 2012; Etacheri et al., 2015; Li, 2015) and photocatalysis is generally a surface mediated phenomenon.
Studies of the chemistry and electronic properties of surfaces and interfaces are key to understanding and screening materials for photocatalysis. The enhanced performance of the benchmark material, P25, which consists of chemically interfaced rutile and anatase phases, has been attributed to the favorable alignment of the conduction and valence bands at the interface which facilitates charge transfer between phases and the suppression of charge carrier recombination (Scanlon et al., 2013). In addition, the interface can promote the formation of active catalytic sites. This effect can be tuned by considering heterostructures with metal oxides of different compositions. Such heterostructures have been realized experimentally and shown to exhibit enhanced photocatalytic activity (Boppana and Lobo, 2011; Boppana et al., 2013; Chae et al., 2017; Sotelo-Vazquez et al., 2017; Wang et al., 2017). Nanostructuring of metal oxides has been investigated as an approach to enhancing charge transfer kinetics and increasing surface area while providing low-coordinated metal and oxygen sites for the adsorption of feedstock species (Gordon et al., 2012; Bhatia and Verma, 2017; Zhang et al., 2017a; Ong et al., 2018). Further, nanostructuring can also facilitate the reduction of metal cations.
Surface modification of metal oxide surfaces with dispersed metal oxide nanoclusters combines the properties of hetero- and nano-structuring. Sub-nm nanoclusters of iron oxide were deposited on TiO2 surfaces via chemisorption-calcination cycle (CCC) (Jin et al., 2011; Tada et al., 2014) and atomic layer deposition (ALD) (Libera et al., 2010). FeOx-modified TiO2 exhibited bandgap reduction and enhanced visible light photocatalytic activity. The modification was shown to suppress carrier recombination as indicated by photoluminescence spectroscopy (Jin et al., 2011). The red-shift in light absorption was attributed to cluster-derived states above the valence band maximum (VBM) which were identified by X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) simulations (Jin et al., 2011; Nolan, 2011a; Tada et al., 2014).
These studies, and the subsequent development of similar systems, (Jin et al., 2012; Boppana et al., 2013; Iwaszuk et al., 2013; Bhachu et al., 2014; Fronzi et al., 2016a) mean that a multitude of nanocluster-surface composites can be investigated. Considerations for tuning these systems for optimal performance include composition, surface termination, nanocluster size, and stoichiometry; all of which contribute to the light absorption properties, charge carrier mobility and surface reactivity. DFT simulations can be used to illuminate the properties underpinning experimental observations (Nolan, 2011a; Jin et al., 2012; Nolan et al., 2012; Iwaszuk et al., 2013) and to screen candidate materials worthy of further investigation (Park et al., 2009; Graciani et al., 2010; Nolan, 2011b, 2012, 2018; Iwaszuk and Nolan, 2013; Lucid et al., 2014; Nolan et al., 2014; Fronzi et al., 2016b; Rhatigan and Nolan, 2018a,b).
In the present study we use first principles DFT calculations to examine the photocatalytic properties of manganese oxide modified TiO2, using model systems of Mn4O6-nanoclusters modifying the rutile (110) and anatase (101) surfaces and consider the role of partial surface hydroxylation in the interfacial chemistry. Our analysis includes an assessment of the stability of the composite surfaces, their ground state stoichiometry and reducibility via oxygen vacancy formation. Point defects, such as oxygen vacancies, are active sites at metal oxide surfaces and can be produced thermally. A more reducible surface will lose oxygen more readily and be more active in solar thermal (Muhich et al., 2016) or Mars and van Krevelen processes 1954, (Ganduglia-Pirovano et al., 2007). Computed density of states plots elucidate the impact of modification on the light absorption properties and a model for the photoexcited state (Di Valentin and Selloni, 2011) is used to examine charge separation and localization. Finally, we study the interaction of water with the modified surfaces and focus particularly on the role of oxygen vacancies and reduced cations on water adsorption. We identify the characteristics of activation, such as dissociation, geometry distortions and charge transfer to the adsorbed species. The importance of oxygen vacancies as active sites for water dissociation at the rutile (110) surface (Schaub et al., 2001; Henderson et al., 2003) and ceria surfaces (Mullins et al., 2012) has been widely discussed and reduced Ti3+ ions have been shown to be active in the chemistry at titania surfaces (Lira et al., 2011; Xiong et al., 2012). For anatase TiO2, oxygen vacancies have been shown to be more stable at subsurface and bulk sites than on the surface (He et al., 2009; Scheiber et al., 2012). However the surface can be reduced by electron bombardment (Scheiber et al., 2012; Setvin et al., 2016) and the reaction of these vacancy sites with water and O2 results in water dissociation. These studies highlight the necessity of engineering photocatalytic surfaces for which vacancies can be produced with moderate energy costs.
MnOx is an interesting modifier as manganese is a multi-valent, reducible element which crystallizes in oxides with a variety of oxidation states; (Franchini et al., 2007) this will have implications for the light absorption properties and reducibility of sub-nm nanoclusters of MnOx dispersed at the titania surfaces. We have previously studied similar systems of MnOx-modified TiO2, in collaboration with experiment, to interrogate their activity for CO2 capture and reduction (Schwartzenberg et al., 2017). In the present study, we focus on the potential for these catalysts to be active toward water activation. Furthermore, we investigate the impact of surface hydroxylation on the reduction of the heterostructures via oxygen vacancy formation and apply a model for photoexcitation to examine the associated energetics and charge localization. In reference Schwartzenberg et al. (2017), the Mn4O6-TiO2 composites were found to be stoichiometric in the ground state for both modified rutile and anatase, albeit with moderate costs to produce reducing oxygen vacancies (+0.59 eV for rutile and +1.1 eV for anatase). However, the impact of surface hydroxyls on the formation of oxygen vacancies was not investigated; in this paper we show that vacancy formation is in fact promoted with hydroxyls already present at the TiO2 surfaces. The photoexcited state model, which examines localization of electrons and holes at nanocluster metal and oxygen sites, sheds light on experimental observations which suggest that the MnOx-modifiers may facilitate recombination (Schwartzenberg et al., 2017). In addition, active oxygen vacancy sites play a crucial role in the subsequent interaction of water molecules and their adsorption modes. In particular, dissociation is favored for the reduced systems; this is an important step in the water oxidation reaction.
Periodic plane wave DFT calculations are performed using the VASP5.4 code (Kresse and Hafner, 1994; Furthmüller et al., 1996) with an energy cut-off of 400 eV. The core-valence interaction is described with projector augmented wave (PAW) potentials, (Blöchl, 1994; Kresse and Joubert, 1999) with 4 valence electrons for Ti, 6 for O, 13 for Mn and 1 for H species. The Perdew-Wang (PW91) approximation to the exchange-correlation functional is used (Perdew et al., 1996).
The TiO2 rutile (110) and anatase (101) substrates are modeled as 18 and 12 atomic layer slabs, respectively. The bulk lattice parameters for rutile were computed as a = 4.639 Å and c = 2.974 Å, and the rutile (110) surface was modeled with a (2×4) surface expansion. For anatase the bulk lattice parameters are a = 3.814 Å and c = 9.581 Å and a (1 × 4) expansion was used for the anatase (101) surface. These parameters correspond to surface areas per supercell of 13.120 × 11.896 Å and 10.312 × 15.255 Å for rutile (110) and anatase (101), respectively. The surfaces are separated from their periodic images by vacuum gaps of 20 Å, as used in our previous studies (Fronzi and Nolan, 2017; Schwartzenberg et al., 2017; Nolan, 2018; Rhatigan and Nolan, 2018a,b). Γ-point sampling is used and the convergence criteria for the energy and forces are 10−4 eV and 0.02 eV−2, respectively. All calculations are spin polarized.
A Hubbard U correction is implemented to consistently describe the partially filled Mn 3d states and reduced Ti3+ states (Anisimov et al., 1991; Dudarev et al., 1998). The values of U used are U(Ti) = 4.5 eV and U(Mn) = 4 eV and these have been chosen based on previous work on TiO2 (Morgan and Watson, 2007; Nolan et al., 2008; Iwaszuk and Nolan, 2011; Nolan, 2011a; Fronzi et al., 2016a; Fronzi and Nolan, 2017; Rhatigan and Nolan, 2018a) and manganese oxides (Franchini et al., 2007; Kitchaev et al., 2016).
To model surface hydroxylation (before the nanoclusters are adsorbed) and the impact on the heterostructure chemistry, four water molecules are dissociatively adsorbed at the clean rutile (110) and anatase (101) surfaces which gives a partial coverage of 50%. The computed energy gain when the TiO2 surfaces are hydroxylated at half coverage is −1.03 eV per water molecule for rutile (110) and −0.8 eV for anatase (101), referenced to the total energy of four gas phase water molecules. These indicate favorable water adsorption and surface hydroxylation and these models have been used in our previous studies (Fronzi et al., 2016a; Fronzi and Nolan, 2017; Schwartzenberg et al., 2017; Rhatigan and Nolan, 2018a). The nature of water molecules adsorbed at metal oxide surfaces, and in particular TiO2 surfaces, is widely investigated both experimentally and computationally (Valdés et al., 2008; Fronzi and Nolan, 2017; Rhatigan and Nolan, 2018a) and readers are referred to reference (Mu et al., 2017) for a review of the state of the art. These models are representative of hydroxylated rutile and anatase surfaces, while we are not attempting to describe the most stable solutions for water or dissociative water adsorption at these titania surfaces (Fronzi et al., 2016a; Fronzi and Nolan, 2017; Rhatigan and Nolan, 2018a). The hydroxylated surfaces are denoted by OH-r110 and OH-a101. For the O2− ions of the pristine titania surfaces, computed Bader charges are in the range of 7.3–7.4 electrons and this is our reference. After hydroxylation, Bader charges for those oxygen atoms of the surface to which H atoms are adsorbed increase to values in the range 7.6–7.7 electrons, with similar values for oxygen ions of the water-derived hydroxyls.
The Mn4O6 nanocluster (see Supporting Information) was adsorbed in different configurations at the hydroxylated rutile (110) and anatase (101) surfaces and the adsorption energies are computed using:
where Esurf+A, Esurf and EA are the energies of the adsorbate-surface composite system, the hydroxylated titania surface and the gas phase nanocluster, respectively.
For the reduction of the composite surface, each of the six O sites of the supported nanocluster is considered for the formation of an oxygen vacancy, OV. One oxygen ion is removed from the Mn4Ox cluster and the vacancy formation energy is calculated as:
where the first and third terms on the right hand side are the total energy of the cluster-surface composite with and without an oxygen vacancy and the energy is referenced to half the total energy for molecular O2. Having identified the most stable structure with a single OV, the calculation is repeated for each of the five remaining O sites to determine the most stable structure with two OV. Oxidation states are investigated with Bader charge analysis and computed spin magnetizations.
We apply a model for photoexcitation to the ground state configuration of each modified surface and to the unmodified OH-r110 and OH-a101 surfaces for comparison. This model involves imposing a triplet electronic state on the system (Di Valentin and Selloni, 2011) to promote an electron to the conduction band, with a corresponding hole in the valence band. The analysis of the energies and charge localization is discussed in more detail in the Supporting Information.
For the interaction of water with the modified surfaces, H2O molecules are adsorbed in various configurations at the oxygen deficient systems and the adsorption energies are calculated as:
where Esurf+H2O, Esurf and EH2O refer to the energies of the H2O molecule and modified surface in interaction, the modified surface, and the gas phase H2O, respectively.
Oxygen atoms of the surface, cluster and surface-bound hydroxyls are denoted OS, OC and OOH, respectively, and similar notation is adopted for OH groups. For the interaction of water with the modified surfaces, water-derived oxygen and hydroxyls are denoted OW and OHW.
Figures 1A,B show the adsorption energies and relaxed atomic structures of the stoichiometric Mn4O6-nanocluster modifying the OH-r110 and OH-a101 surfaces. The large, negative adsorption energies indicate that the nanocluster-surface interaction is favorable and that the nanoclusters will be stable against desorption and aggregation (Fronzi et al., 2016b; Nolan et al., 2016; Fronzi and Nolan, 2017; Nolan, 2018; Rhatigan and Nolan, 2018a,b). For Mn4O6-OH-r110 (Figure 1A), three Mn ions are 4-fold coordinated and to each of these is bound a terminal OH. Of these OH groups, one has migrated from a Ti site in the rutile surface to an Mn ion of the cluster (OHOH) and two OH groups result from the migration of hydrogen from surface hydroxyls to OC atoms (OHC). The fourth Mn ion is 5-fold coordinated and is bound to three OC and two OS ions (one bridging OS and one in-plane OS). Five O ions of the OH-r110 surface bind with Mn of the nanocluster (three OS and two OOH) and two OC ions bind to Ti of the surface. Mn-O distances are in the range 1.8–2.1 Å; the shorter distances involve 2-fold coordinated O ions and for Mn bound to the in-plane OS ion the Mn-O distance is 2.2 Å. Ti ions which bind to the nanocluster migrate out from the surface by 0.1 Å, however, distortions to the geometry of the rutile (110) surface are minimal.
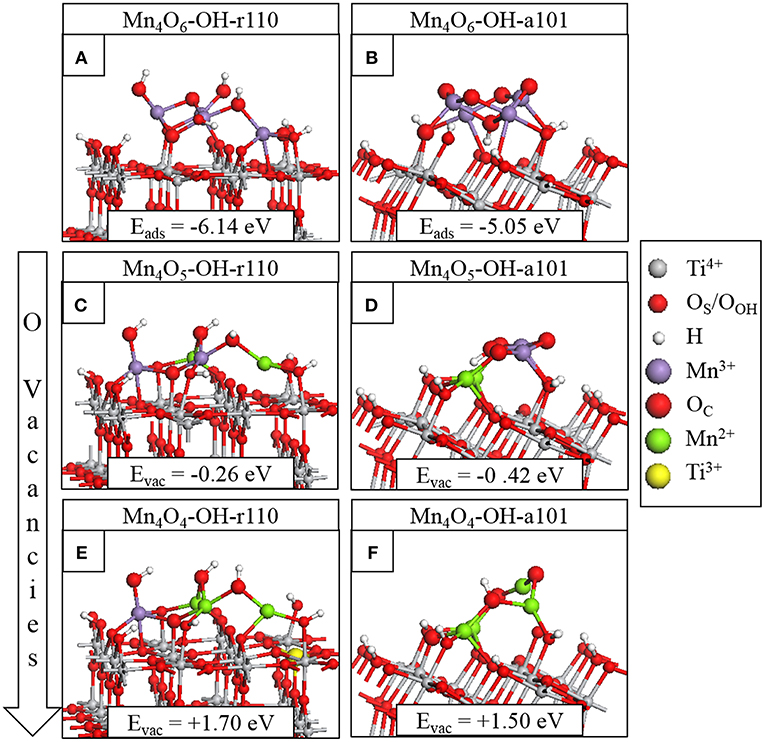
Figure 1. Relaxed atomic structures of Mn4Ox modifying the hydroxylated titania surfaces. The stoichiometric composites are shown in (A) for Mn4O6-OH-r110 and (B) for Mn4O6-OH-a101; the nanocluster adsorption energies are included in the inset. The atomic structures after formation of the most stable single OV are shown in panels (C) for Mn4O5-OH-r110 and (D) for Mn4O5-OH-a101. The atomic structures of the most stable composites with two OV are shown in panels (E) for Mn4O4-OH-r110 and (F) for Mn4O4-OH-a101. The energy costs to produce OV are included and computed relative to the structure with one less OV. Atomic species and oxidation states are indicated by the colors in the legend on the right hand side.
For Mn4O6-OH-a101 (Figure 1B), three Mn ions are 4-fold coordinated and one Mn is 5-fold coordinated. Five OC sites are 2-fold coordinated with one OC ion binding to three Mn ions and a H atom which has migrated from a bridging OS site. Of the six interfacial bonds between the Mn4O6 nanocluster and OH-a101, three involve Mn and OHOH groups; two involve Mn and bridging OS sites and the sixth is a Ti-OC bond. Mn-O distances are in the range 1.7–2.1 Å.
For Mn4O6 adsorbed at OH-r110 and OH-a101, the computed Bader charge for each of the Mn ions is 11.3 electrons, which are typical of Mn3+ ions (see Table 1).(Schwartzenberg et al., 2017) The spin magnetizations for these sites are each 3.9 μB, which reflects the 3d4 configuration of the Mn3+ ion.
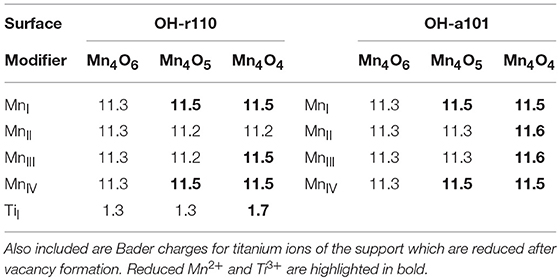
Table 1. Computed Bader charges for the manganese ions in the supported nanoclusters before and after formation of one or more OV.
For the Mn4O6 nanocluster adsorbed at OH-a101, there is an accumulation of positive charge at those OC sites which are doubly-coordinated to Mn ions of the nanocluster. Computed Bader charges of 7.0 electrons for these OC sites compare with 7.3–7.7 electrons computed for O2− anions of the OH-a101 surface. The nanocluster-surface interaction is not as strong at the OH-a101 surface as indicated by the smaller adsorption energy. The consequence of this is that the supported nanocluster retains characteristics of the gas phase, for which the OC ions have computed Bader charges in the range 7.0–7.1 electrons.
The most stable modified surfaces with a single OV are shown in Figure 1C for Mn4O5-OH-r110 and Figure 1D for Mn4O5-OH-a101. For the modified OH-r110 surface the formation energy of a single OV is −0.26 eV and this formation energy indicates that OV will form spontaneously. The next three most stable vacancy sites have formation energies in the range 0.60–0.82 eV. After formation of the most stable vacancy, two Mn ions are 3-fold coordinated and the third and fourth Mn cations are 4- and 5-fold coordinated. Two bridging and one in-plane surface oxygen are bound to Mn ions of the nanocluster. Two OC ions bind to surface Ti sites while three OC ions are bound only to Mn and H ions.
The formation of the neutral oxygen vacancy releases two electrons. Bader charge analysis reveals that the electrons localize at the 3-fold coordinated Mn sites of the nanocluster. The computed Bader charges on these sites increase from 11.3 to 11.5 electrons; see (Table 1) for computed Bader charges of reduced Ti and all Mn sites. The computed spin magnetizations are 4.6 μB for these Mn sites; this is typical of the formation of reduced Mn2+ ions which has an electronic configuration of 3d5.
The most favorable structure with one OV is more stable than the second most favorable by 0.9 eV. However, the relaxed atomic structures of these configurations are very similar (compare Figure 1C with Figure S3A). The difference in energy arises from the distibution of excess charge. For the OV structure shown in Figure S3A, one excess charge localizes at a 5-fold coordinated surface Ti site for which the Bader charge increases from 1.3 to 1.7 electrons. A computed spin magnetization of 1.0 μB reflects the 3d1 configuration of reduced Ti3+.
For the modified OH-a101 surface, the most stable OV has a formation energy of −0.42 eV which indicates that it will spontaneously form, so that the ground state is off-stoichiometric (vacancy formation energies for other sites of the nanocluster were in the range 0.5–1.3 eV). This compares with Mn4O6 modifying bare anatase (101) which was found to be stoichiometric in the ground state (Schwartzenberg et al., 2017). After the formation of this OV, two Mn ions relax toward the titania surface and bind with bridging OS sites so that, in this configuration, each Mn ion is 4-fold coordinated. Bader charge analysis and computed spin densities indicate that two Mn ions are reduced to Mn2+, having computed Bader charges of 11.5 electrons and computed spin magnetisations of 4.6 μB. The next most stable structure with one OV is shown in Figure S3B; in this configuration three Mn ions are reduced to Mn2+ and this is accompanied by an accumulation of postive charge on 2-fold coordinated OC ions for which the Bader charges were computed as 7.0 electrons.
The formation of the second OV has a moderate energy cost for both MnOx-modified TiO2 surfaces, however the modified anatase surface is reducible at a lower energy cost. Given that the anatase surface is more easily hydroxylated, (Mu et al., 2017) which these results indicate promotes vacancy formation, one would expect more OV present on modified anatase. That OV formation is more facile for modified anatase corroborates previous experimental work on MnOx-TiO2 (Schwartzenberg et al., 2017). The most stable configurations of the heterostructures with two OV are shown in Figure 1E for Mn4O4-OH-r110 and Figure 1F for Mn4O4-OH-a101. For the Mn4O4-OH-r110 surface, the two most stable OC sites for formation of a second OV had similar formation energies. One such configuration is described here and the other is included in the Supporting Information. For the structure shown in Figure 1E, the removed OC ion was 2-fold coordinated to a cluster Mn and surface Ti ion. After vacancy formation the Mn ion binds to a bridging OS ion and remains 3-fold coordinated. In this configuration three Mn ions are reduced; the Bader charges and spin magnetizations for these sites are 11.5 electrons and 4.6 μB, respectively. Similarly, for the Ti site to which the removed OC was bound, the Bader charge and spin magnetization are 1.7 electrons and 1.0 μB. Hence, the Mn4O4-OH-r110 heterostructure with two oxygen vacancies has one Ti3+ and three Mn2+ ions.
For the modified OH-a101 surface, a 3-fold coordinated OC site, which forms a hydroxyl group bridging two Mn ions, has the lowest cost to produce a second OV. One Mn ion that was bound to the removed OC atom is 2-fold coordinated, having been originally coordinated to three OC ions and one OOH ion. The second Mn ion is 3-fold coordinated, having been 4-fold coordinated prior to vacancy formation. The H ion which was bound to the removed OC migrates to another OC ion. In this Mn4O4-OH-a101 configuration, there are four Mn2+ ions, with computed Bader charges of 11.5–11.6 electrons and spin magnetizations of 4.6 μB.
Additional structures with two OV are presented in Figure S3C for Mn4O4-OH-r110 and Figure S3D for Mn4O4-OH-a101; these are close in energy to the configurations described above, and differ in the distribution of excess charge over Mn and Ti sites. Hence, Mn and Ti sites should be present at the surface in a variety of oxidation states.
The localization of electrons at Ti and Mn sites is also accompanied by localized geometry distortions. The cation-O distances increase by ~0.1 Å after reduction, reflecting the larger ionic radii of Mn2+ and Ti3+ compared to Mn3+and Ti4+ (Shannon and Prewitt, 1969).
The projected electronic density of states (PEDOS) for the heterostructures are presented in Figure 2. Since the heterostructures are off-stoichiometric in the ground state, the PEDOS plot for Mn4O6-OH-r110 and Mn4O6-OH-a101 have been omitted from this figure and are included in the Supporting Information for completeness. The top panels of Figure 2 show the PEDOS of modified OH-r110 for (A) the ground state with one OV and (B) the reduced state with two OV. The PEDOS plots show that occupied nanocluster-derived states (Mn 3d and OC 2p) extend to 0.3 and 0.8 eV above the VBM of the rutile support for Mn4O5- and Mn4O4-OH-r110, respectively. Unoccupied Mn 3d-derived states also emerge in the titania band gap at 0.1 and 0.3 eV below the conduction band minimum (CBM) for the ground state with one OV and the reduced state with two OV. Additional states emerge in the band gap due to occupied Ti3+ states (see inset of Figure 2B), for the heterostructure with two OV.
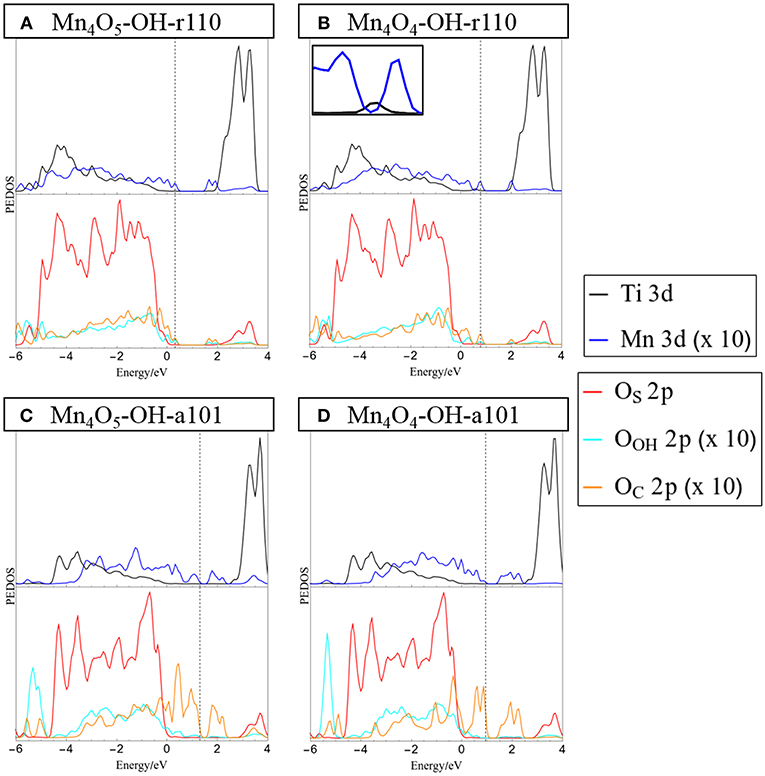
Figure 2. Projected electronic density of states (PEDOS) plots for (A) Mn4O5-, and (B) Mn4O4-OH-r110 and (C) Mn4O5-, and (D) Mn4O4-OH-a101. The computed valence band max is set to 0 eV and the Fermi energy is indicated with a dashed line. The top half of each panel displays Ti and Mn 3d derived states. The bottom halves of the panels show contributions to the DOS from oxygen 2p states of the surface (OC), surface bound hydroxyls (OOH) and nanocluster (OC). Inset in panel (B) shows the mid-gap occupied Ti 3d states in the range [0 eV, 1 eV].
The bottom panels of Figure 2 display the PEDOS of the modified OH-a101 surface for (C) the ground state, with one OV, and (D) reduced state with two OV. The PEDOS plot for the ground state, with one OV, shows that occupied Mn 3d- and OC 2p-derived states extend to 1.3 eV above the titania derived VBM, while unoccupied Mn 3d states emerge at 1 eV below the CBM, leading to a significant reduction in the computed energy gap relative to TiO2. For the reduced structure, with two OV, each of the Mn ions is reduced to Mn2+, and the highest occupied of these states is 1 eV above the VBM. The lowest energy, unoccupied state is Mn-derived and is 1 eV below the CBM. For Mn4O4-OH-a101 the energy gap is 0.6 eV, with our DFT+U set-up showing a reduction over unmodified anatase.
These features of the PEDOS for Mn4Ox-TiO2 can be attributed to formation of interfacial bonds, the presence of low-coordinated Mn and OC sites and the facile formation of OV in the supported metal oxide nanocluster. Modification pushes the VBM to higher energy and results in the emergence of empty states below the CBM; these effects, and the consequent red shift, are greater for modified anatase, consistent with previous reports (Schwartzenberg et al., 2017). These metal oxide nanocluster-modified surfaces are of interest for the oxygen evolution half reaction (OER) of the water splitting process and in this context raising the VBM from that of TiO2 and toward the water oxidation potential is a desirable effect. Lowering of the titania CBM from its favorable position straddling the water reduction potential is detrimental to the hydrogen evolution reaction (HER) activity. However, as H adsorbs too strongly at metal oxide surfaces, such heterostructures will in any case not be suitable photocathodes for water splitting.
We apply the model for the photoexcited state to the ground state systems, Mn4O5-OH-r110 and Mn4O5-OH-a101. Table 2 presents the computed vertical, singlet-triplet and electron-hole trapping energies, as discussed in the Supporting Information. As can be seen from the values listed in Table 2, the underestimation of the bandgap inherent in approximate DFT is present in our DFT+U computational set-up. Our goal in choosing +U corrections is to consistently describe the localization of electrons and holes rather than reproduce the bandgap of bulk TiO2, which is not advised. Comparison of these computed energies across different structures nonetheless yields useful qualitative information about the effect of surface modification and results for the unmodified OH-r110 and OH-a101 surfaces are included for reference. In particular, Evertical is analogous to the optical band gap, and a reduction in this value for a heterostructure relative to unmodified titania implies that modification leads to a red shift in light absorption.

Table 2. Vertical singlet-triplet energy difference (Evertical), the relaxed singlet-triplet energy difference (Eexcite) and the relaxation energy (Erelax) for Mn4O5-OH-r110 and Mn4O5-OH-a101.
When comparing Mn4O5-OH-r110 with unmodified OH-r110 we can see that the values for Evertical are similar, however Eexcite is reduced by 0.93 eV for the modified surface. Comparing Mn4O5-OH-a101 with unmodified OH-a101, decreases in Evertical and Eexcite by 0.34 and 0.57 eV, respectively, indicate that modification leads to a significant red shift in light absorption. These results corroborate the analysis of the PEDOS. Erelax is the energy gained by the system after structural relaxation in response to the triplet electronic state and is related to the stability of the trapped electron and hole. The relaxation energy is larger for Mn4O5-OH-r110 than that computed for unmodified OH-r110 (1.31 vs. 0.46 eV) and reflects the greater flexibility of the modified system in accommodating the triplet electronic state. The relaxation energies for Mn4O5-OH-a101 and unmodified OH-a101 are comparable. The mixture of Mn oxidation states and the proximity of the Mn ions to each other at the anatase surface (neighboring Mn-Mn distances are in the range 2.9–3.2 Å for Mn4O5-OH-a101 and 3.0–3.9 Å for Mn4O5-OH-r110) restricts the degree to which the nanocluster can respond structurally to the localization of photoexcited charges.
Through analysis of Bader charges and spin magnetizations we can determine the electron and hole localization sites and the results of this analysis are represented graphically in Figure 3. For Mn4O5-OH-r110, in Figures 3A,B, the electron localizes at an Mn site; the Bader charge and spin magnetization for this site are 11.5 electrons and 4.6 μB after electron localization, which are typical of Mn2+ formation. The hole localizes at an OC site which is 2-fold coordinated to the Mn2+ ion and a surface Ti. In this instance the Bader charge is 6.8 electrons and the spin magnetization is 0.8 μB, which are consistent with formation of O−. The Mn2+-O− distance increases by 0.2 Å, relative to the ground state. The Ti-O− distance decreases by 0.1 Å.
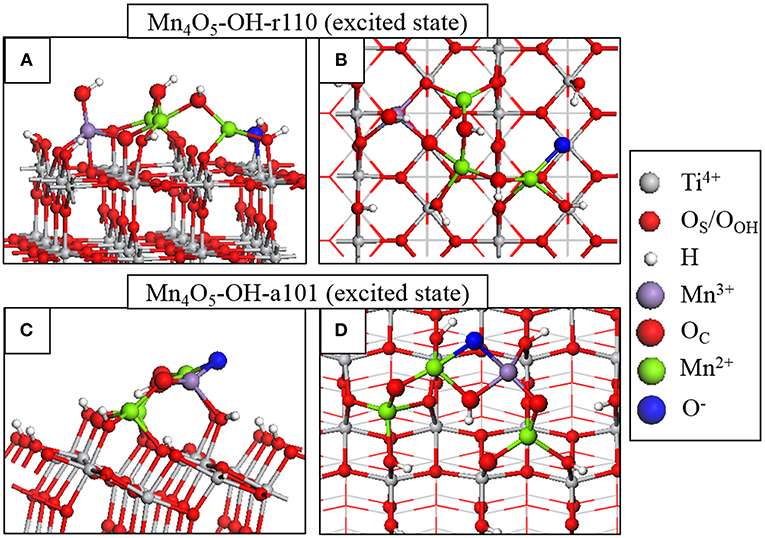
Figure 3. Atomic structure of the fully relaxed triplet electronic state imposed on Mn4O6-OH-r110 for (A) side and (B) top view and Mn4O5-OH-a101 for (C) side and (D) top view. Charge localization and changes in oxidation state are distinguished by color according to the legend on the right hand side.
For Mn4O5-OH-a101 (Figures 3C,D), the photoexcited electron localizes at an Mn site of the nanocluster, as confirmed by a computed Bader charge of 11.5 electrons and spin magnetization of 4.5 μB. The hole state localizes predominantly at an OC site which bridges Mn2+ and Mn3+ ions. After hole localization the Bader charge for the O− ion is 6.7 electrons and the spin magnetization is 0.8 μB. The Mn2+-O distances increase by 0.2–0.3 Å.
These results show that the electron localizes at an Mn site of the supported nanocluster and the hole state localizes at a neighboring OC site. Based on this model for the photoexcited state, we can conclude that modification does not necessarily promote the spatial separation of photoexcited charges. However, both electrons and holes will be available at the modified surface for transfer to adsorbed species.
For the interaction of water at the modified surfaces, only those composites with OV present were considered, as such vacancies are known to be active sites at metal oxide surfaces (Schaub et al., 2001; Wang et al., 2016; Ruiz Puigdollers et al., 2017; Zhang et al., 2017b). Water adsorption is favorable at multiple sites of both modified surfaces and the geometries of the most stable adsorption configurations are displayed in Figure 4, while the Supporting Information shows other, less stable, water adsorption structures.
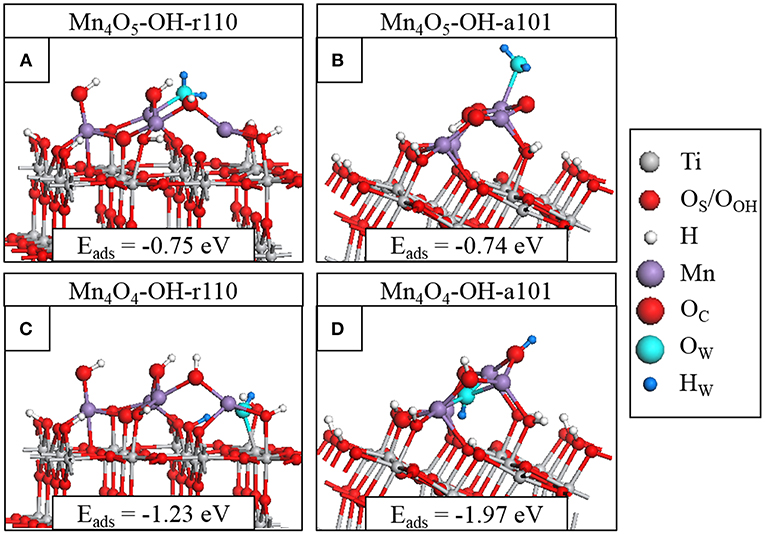
Figure 4. Relaxed atomic structures of the most stable configurations of H2O adsorbed at (A) Mn4O5-OH-r110, (B) Mn4O5-OH-a101, (C) Mn4O4-OH-r110, and (D) Mn4O4-OH-a101. Atomic species are distinguished by color according to the legend on the right hand side.
We adsorb water in molecular form at the heterostructures and relax the geometry. For Mn4O5-OH-r110, shown in Figure 4A, water is adsorbed exothermically in molecular form with a computed adsorption energy of −0.75 eV. In this instance, the water molecule binds to a 3-fold coordinated Mn2+ site with a Mn-OW distance of 2.3 Å. Figure S5A shows dissociative water adsorption at the Mn4O5-OH-r110 surface, which has an adsorption energy of −0.31 eV. Upon dissociation, an H atom migrates to a bridging OS site and the water-derived hydroxyl (OHW) is singly coordinated to an Mn site with an Mn-OW distance of 1.9 Å. The dissociation is accompanied by a transfer of charge from OW to the nanocluster modifier, indicated by a decrease of 0.4 electrons in the computed Bader charge for the OW ion. The Bader charges and spin magnetizations of cation sites are unchanged by the adsorption and dissociation.
Water adsorbs molecularly at Mn4O5-OH-a101, as shown in Figure 4B, with an adsorption energy of −0.74 eV. The H2O binds to a 4-fold coordinated Mn3+ ion with a Mn-OW distance of 2.2 Å. Since Mn4O5-OH-r110 and Mn4O5-OH-a101 are the ground states of the systems, the single OV having formed spontaneously, these composites favor non-stoichiometry so that the strength of interaction with the water molecule is not sufficient to promote spontaneous dissociation and adsorption in molecular form is favored.
The surfaces with two OV show higher reactivity to water, as indicated by the larger adsorption energies in Figures 4C,D. Water adsorbs and spontaneously dissociates at both Mn4O4-OH-r110 (Figure 4C) and Mn4O4-OH-a101 (Figure 4D). For Mn4O4-OH-r110, the water molecule adsorbs at an OV site. An H atoms migrates to a bridging OS site and the OHW group is doubly coordinated to an Mn and a surface Ti site. The Mn-OW and Ti-OW distances are 2.2 Å. Bader charge analysis reveals that 0.3 electrons are transferred from the OW to the surface. Despite this charge transfer, the Ti ion which binds to OHW and which was reduced to Ti3+ due to OV formation prior to water adsorption, remains in the Ti3+ state. This agrees with work by Henderson et al. in which no charge transfer was observed between Ti3+ and bridging hydroxyls bound at oxygen vacancy sites at the TiO2 rutile (110) surface (Henderson et al., 2003). The reduced Ti site was only oxidized after interaction of O2 with the Ti3+-OH group. After water adsorption and dissociation the distribution of cation oxidation states is unchanged so that there are three Mn2+ ions and one Ti3+. The Bader charge for the bridging OS site to which the H ion binds increases from 7.3 to 7.7 electrons which, as discussed in the methodology, is typical of hydroxyl formation.
For Mn4O4-OH-a101, the water molecule adsorbs at an OV site and after dissociation an H atom migrates to an OC ion which shows an increase in Bader charge, from 7.1 to 7.6 electrons. The OHW group binds to three Mn2+ ions; the Bader charges and spin magnetizations for cation sites are unchanged so that these ions are not involved in the charge transfer. However, for the water adsorption configuration shown in Figure S5D, an Mn2+ ion is re-oxidized to Mn3+ after dissociation of the water molecule. In this instance the adsorption energy is −1.89 eV and the OHW group is singly-coordinated to the re-oxidized Mn ion.
The properties of Mn4Ox-modified hydroxylated titania surfaces and their reduction and interaction with water depend on the phase of the TiO2 substrate. For Mn4O6 adsorbed at the hydroxylated anatase (101) surface, one interfacial bond is established between a cluster oxygen ion and the surface and Mn ions bind mostly to oxygen ions of the surface bound hydroxyls. Conversely, for Mn4O6 at hydroxylated rutile (110), the nanocluster-surface interaction is more intimate, with Mn ions binding to bridging and in-plane oxygen ions of the rutile surface.
Our results indicate that both Mn4Ox-OH-r110 and Mn4Ox-OH-a101 favor non-stoichiometry, in contrast to unhydroxylated modified TiO2 surfaces, as oxygen vacancies form spontaneously and both composites can be considered highly reducible with moderate energy costs for subsequent oxygen vacancy formation. Bader charge analysis shows that Mn ions are present in a mixture of oxidation states of at the hydroxylated surfaces. Both Mn and Ti ions are reduced in response to vacancy formation.
Modification with Mn4Ox has a significant impact on the light absorption properties. Occupied Mn 3d states extend the VBM of the composites to higher energies relative to that of the titania support and empty states emerge below the CBM. The consequent red shift in the light absorption edge is confirmed by our model for the photoexcited state. In particular, the vertical energy, analogous to the optical energy gap, decreases significantly for Mn4O5-OH-a101 relative to that computed for the unmodified, hydroxylated anatase (101) surface. Analysis of this model shows that electrons and holes localize at Mn and neighboring OC sites, respectively, so that modification may not promote separation of photoexcited charges, but the trapping energies of the electron and hole are quite high, suggesting high stability.
With regard to water adsorption and activation, the formation of oxygen vacancies has an impact on the strength of interaction and the most favorable adsorption mode of H2O at the modified surfaces. For Mn4O5-OH-a101, with a spontaneously formed OV, water adsorbs only in molecular form. With formation of reducing oxygen vacancies, water adsorption becomes more exothermic and leads to spontaneous dissociation to surface bound hydroxyls, similar to observations made for water interacting at reduced TiO2 (Schaub et al., 2001; Henderson et al., 2003) and CeO2 (Mullins et al., 2012) surfaces.
MN devised the research. SR performed the DFT modeling. Both authors analyzed the results and prepared, reviewed and approved the text of the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge support from Science Foundation Ireland through the US-Ireland R and D Parntershop program, Grant number SFI/US/14/E2915 and the ERA.Net for Materials Research and Innovation (M-ERA.Net 2), Horizon 2020 grant agreement number 685451, SFI Grant Number SFI/16/M-ERA/3418 (RATOCAT). We acknowledge access to SFI funded computing resources at Tyndall Institute and the SFI/HEA funded Irish Centre for High End Computing. We are grateful for support from the COST Action CM1104 Reducible Metal Oxides, Structure and Function.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00067/full#supplementary-material
Anisimov, V. I., Zaanen, J., and Andersen, O. K. (1991). Band theory and mott insulators: hubbard U instead of stoner I. Phys. Rev. B. 44, 943–954.
Bhachu, D. S., Sathasivam, S., Carmalt, C. J., and Parkin, I. P. (2014). PbO-modified TiO2 thin films: a route to visible light photocatalysts. Langmuir 30, 624–630. doi: 10.1021/la4038777
Bhatia, S., and Verma, N. (2017). Photocatalytic activity of ZnO nanoparticles with optimization of defects. Mater. Res. Bull. 95, 468–476. doi: 10.1016/j.materresbull.2017.08.019
Boppana, V., Jiao, B. R., Newby, F., Laverock, D., Smith, J., Lobo, K. E., et al. (2013). Analysis of visible-light-active Sn(ii)–TiO2 photocatalysts. Phys. Chem. Chem. Phys. 15, 6185–6189. doi: 10.1039/c3cp44635b
Boppana, V., and Lobo, B. R. (2011). SnOx–ZnGa2O4 photocatalysts with enhanced visible light activity. ACS Catal. 1, 923–928. doi: 10.1021/cs200137h
Chae, S. Y., Lee, C. S., Jung, H., Joo, O. S., Min, B. K., et al. (2017). Insight into charge separation in WO3/BiVO4 heterojunction for solar water splitting. ACS Appl. Mater. Interfaces. 9, 19780–19790. doi: 10.1021/acsami.7b02486
Chand, R., Obuchi, E., Katoh, K., Luitel, H. N., and Nakano, K. (2011). Enhanced photocatalytic activity of TiO2/SiO2 by the influence of Cu-doping under reducing calcination atmosphere. Catal. Commun. 13, 49–53. doi: 10.1016/j.catcom.2011.04.024
Cui, Y., Du, H., and Wen, L. (2008). Doped-TiO2 photocatalysts and synthesis methods to prepare TiO2 films. J. Mater. Sci. Technol. 24, 675–689.
Czoska, A. M., Livraghi, S., Chiesa, M., Giamello, E., Agnoli, S., Granozzi, G., et al. (2008). The nature of defects in fluorine-doped TiO2. J. Phys. Chem. C. 112, 8951–8956. doi: 10.1021/jp8004184
Di Valentin, C., Finazzi, E., Pacchioni, G., Selloni, A., Livraghi, S., and Paganini, M. C. (2007). N-doped TiO2: theory and experiment. Chem. Phys. 339, 44–56. doi: 10.1016/j.chemphys.2007.07.020
Di Valentin, C., and Selloni, A. (2011). Bulk and surface polarons in photoexcited anatase TiO2. J. Phys. Chem. Lett. 2, 2223–2228. doi: 10.1021/jz2009874
Dimitrijevic, N. M., Vijayan, B. K., Poluektov, O. G., Rajh, T., Gray, K. A., and He, H. (2011). Role of water and carbonates in photocatalytic transformation of CO2 to CH4 on Titania. J. Am. Chem. Soc. 133, 3964–3971. doi: 10.1021/ja108791u
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J., and Sutton, A. P. (1998). Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B. 57, 1505–1509. doi: 10.1103/PhysRevB.57.1505
Etacheri, V., Di Valentin, C., Schneider, J., Bahnemann, D., and Pillai, S. C. (2015). Visible-light activation of TiO2 photocatalysts: advances in theory and experiments. J. Photochem. Photobiol. C: Photochem. Rev. 25, 1–29. doi: 10.1016/j.jphotochemrev.2015.08.003
Franchini, C., Podloucky, R., Paier, J., Marsman, M., and Kresse, G. (2007). Ground-state properties of multivalent manganese oxides: density functional and hybrid density functional calculations. Phys. Rev. B. 75:195128. doi: 10.1103/PhysRevB.75.195128
Fronzi, M., Daly, W., and Nolan, M. (2016b). Reactivity of metal oxide nanocluster modified rutile and anatase TiO2: oxygen vacancy formation and CO2 interaction. App. Catal. A. 521, 240–249. doi: 10.1016/j.apcata.2015.11.038
Fronzi, M., Iwaszuk, A., Lucid, A., and Nolan, M. (2016a). Metal oxide nanocluster-modified TiO2 as solar activated photocatalyst materials. J. Phys.: Condens. Matter. 28, 074006. doi: 10.1088/0953-8984/28/7/074006
Fronzi, M., and Nolan, M. (2017). Surface modification of perfect and hydroxylated TiO2 rutile (110) and anatase (101) with chromium oxide nanoclusters. ACS Omega 2, 6795–6808. doi: 10.1021/acsomega.7b01118
Fujishima, A., and Honda, K. (1972). Electrochemical photolysis of water at a semiconductor electrode. Nature 238, 37–38. doi: 10.1038/238037a0
Fujishima, A., Zhang, X., and Tryk, D., A (2008). TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 63, 515–582. doi: 10.1016/j.surfrep.2008.10.001
Furthmüller, J., Hafner, J., and Kresse, G. (1996). Dimer reconstruction and electronic surface states on clean and hydrogenated diamond (100) surfaces. Phys. Rev. B. 53, 7334–7351.
Gai, Y., Li, J., Li, S. S., Xia, J. B., and Wei, S. H. (2009). Design of narrow-gap TiO2: a passivated codoping approach for enhanced photoelectrochemical activity. Phys. Rev. Lett. 102:036402. doi: 10.1103/PhysRevLett.102.036402
Ganduglia-Pirovano, M. V., Hofmann, A., and Sauer, J. (2007). Oxygen vacancies in transition metal and rare earth oxides: current state of understanding and remaining challenges. Surf. Sci. Rep. 62, 219–270. doi: 10.1016/j.surfrep.2007.03.002
Gordon, T. R., Cargnello, M., Paik, T., Mangolini, F., Weber, R. T., Fornasiero, P., et al. B (2012). Nonaqueous synthesis of TiO2 nanocrystals using TiF4 to engineer morphology, oxygen vacancy concentration, and photocatalytic activity. J. Am. Chem. Soc. 134, 6751–6761. doi: 10.1021/ja300823a
Graciani, J., Plata, J. J., Sanz, J. F., Liu, P., and Rodriguez, J. A. (2010). A theoretical insight into the catalytic effect of a mixed-metal oxide at the nanometer level: the case of the highly active metal/CeOx/TiO2(110) catalysts. J. Chem. Phys. 132:104703. doi: 10.1063/1.3337918
Guo, M., and Du, J. (2012). First-principles study of electronic structures and optical properties of Cu, Ag, and Au-doped anatase TiO2. Physica B 407, 1003–1007. doi: 10.1016/j.physb.2011.12.128
Habisreutinger, S. N., Schmidt-Mende, L., and Stolarczyk, J. K. (2013). Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 52, 7372–7408. doi: 10.1002/anie.201207199
Haowei, P., Jingbo, L., Shu-Shen, L., and Jian-Bai, X. (2008). First-principles study of the electronic structures and magnetic properties of 3d transition metal-doped anatase TiO2. J. Phys. 20:125207. doi: 10.1088/0953-8984/20/12/125207
He, Y., Dulub, O., Cheng, H., Selloni, A., and Diebold, U. (2009). Evidence for the predominance of subsurface defects on reduced anatase TiO2(101). Phys. Rev. Lett. 102:106105. doi: 10.1103/PhysRevLett.102.106105
Henderson, M. A., Epling, W. S., Peden, C. H. F., and Perkins, C., L (2003). Insights into photoexcited electron scavenging processes on TiO2 obtained from studies of the reaction of O2 with OH groups adsorbed at electronic defects on TiO2(110). J. Phys. Chem. B. 107, 534–545. doi: 10.1021/jp0262113
Herrmann, J. M. (2012). Detrimental cationic doping of titania in photocatalysis: why chromium Cr3+-doping is a catastrophe for photocatalysis, both under UV- and visible irradiations. New J. Chem. 36, 883–890. doi: 10.1039/c2nj20914d
Ikeda, T., Nomoto, T., Eda, K., Mizutani, Y., Kato, H., Kudo, A., et al. (2008). Photoinduced dynamics of TiO2 doped with Cr and Sb. J. Phys. Chem. C. 112, 1167–1173. doi: 10.1021/jp0752264
Iwaszuk, A., and Nolan, M. (2011). Reactivity of sub 1 nm supported clusters: (TiO2)n clusters supported on rutile TiO2 (110). Phys. Chem. Chem. Phys. 13, 4963–4973. doi: 10.1039/C0CP02030C
Iwaszuk, A., and Nolan, M. (2013). Lead oxide-modified TiO2 photocatalyst: tuning light absorption and charge carrier separation by lead oxidation state. Catal. Sci. Technol. 3, 2000–2008. doi: 10.1039/c3cy00194f
Iwaszuk, A., Nolan, M., Jin, Q., Fujishima, M., and Tada, H. (2013). Origin of the visible-light response of nickel(ii) oxide cluster surface modified titanium(iv) dioxide. J. Phys. Chem. C. 117, 2709–2718. doi: 10.1021/jp306793r
Jiang, C., Moniz, S. J. A., Wang, A., Zhang, T., and Tang, J. (2017). Photoelectrochemical devices for solar water splitting - materials and challenges. Chem. Soc. Rev. 46, 4645–4660. doi: 10.1039/C6CS00306K
Jin, Q., Fujishima, M., Nolan, M., Iwaszuk, A., and Tada, H. (2012). Photocatalytic activities of tin(iv) oxide surface-modified titanium(iv) dioxide show a strong sensitivity to the TiO2 crystal form. J. Phys. Chem. C. 116, 12621–12626. doi: 10.1021/jp302493f
Jin, Q., Fujishima, M., and Tada, H. (2011). Visible-light-active iron oxide-modified anatase titanium(iv) dioxide. J. Phys. Chem. C. 115, 6478–6483. doi: 10.1021/jp201131t
Kitchaev, D. A., Peng, H., Liu, Y., Sun, J., Perdew, J. P., and Ceder, G. (2016). Energetics of MnO2 polymorphs in density functional theory. Phys. Rev. B. 93:045132. doi: 10.1103/PhysRevB.93.045132
Kresse, G., and Hafner, J. (1994). Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B. 49, 14251–14269.
Kresse, G., and Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 59, 1758–1775.
Li, W. (2015). Influence of electronic structures of doped TiO2 on their photocatalysis. Phys. Status Solidi (RRL) – Rapid Res. Lett. 9, 10–27. doi: 10.1002/pssr.201409365
Libera, J. A., Elam, J. W., Sather, N. F., Rajh, T., and Dimitrijevic, N. M. (2010). Iron(iii)-oxo centers on TiO2 for visible-light photocatalysis. Chem. Mater. 22, 409–413. doi: 10.1021/cm902825c
Lira, E., Wendt, S., Huo, P., Hansen, J. Ø., Streber, R., Porsgaard, S., et al. (2011). The importance of Bulk Ti3+ defects in the oxygen chemistry on titania surfaces. J. Am. Chem. Soc. 133, 6529–6532. doi: 10.1021/ja200884w
Long, R., and English, N. J. (2010a). First-principles calculation of synergistic (N, P)-codoping effects on the visible-light photocatalytic activity of anatase TiO2. J. Phys. Chem. C. 114, 11984–11990. doi: 10.1021/jp100802r
Long, R., and English, N. J. (2010b). Synergistic effects on band gap-narrowing in titania by codoping from first-principles calculations. Chem. Mater. 22, 1616–1623. doi: 10.1021/cm903688z
Lucid, A., Iwaszuk, A., and Nolan, M. (2014). A first principles investigation of Bi2O3-modified TiO2 for visible light activated photocatalysis: the role of TiO2 crystal form and the Bi3+ stereochemical lone pair. Mater. Sci. Semicond. Process. 25, 59–67. doi: 10.1016/j.mssp.2014.01.005
Maeda, K., and Domen, K. (2010). Photocatalytic water splitting: recent progress and future challenges. J. Phys. Chem. Lett. 1, 2655–2661. doi: 10.1021/jz1007966
Mars, P., and van Krevelen, D. W. (1954). Oxidation carried out by means of vandium oxide catalysts. Chem. Eng. Sci. 3, 41–59.
Morgan, B. J., and Watson, G. W. (2007). A DFT + U description of oxygen vacancies at the TiO2 rutile (1 1 0) surface. Surf. Sci. 601, 5034–5041.
Mu, R., Zhao, Z.-J., Dohnálek, Z., and Gong, J. (2017). Structural motifs of water on metal oxide surfaces. Chem. Soc. Rev. 46, 1785–1806. doi: 10.1039/C6CS00864J
Muhich, C. L., Ehrhart, B. D., Al-Shankiti, I., Ward, B. J., Musgrave, C. B., and Weimer, A. W. (2016). A review and perspective of efficient hydrogen generation via solar thermal water splitting. Wiley Interdis. Rev. 5, 261–287. doi: 10.1002/wene.174
Mullins, D. R., Albrecht, P. M., Chen, L., Calaza, F. C., Biegalski, M. D., Overbury, S. H., et al. (2012). Water dissociation on CeO2(100) and CeO2(111) thin films. J. Phys. Chem. C. 116, 19419–19428. doi: 10.1021/jp306444h
Na Phattalung, S., Limpijumnong, S., and Yu, J. (2017). Passivated co-doping approach to bandgap narrowing of titanium dioxide with enhanced photocatalytic activity. Appl. Catal. B. 200, 1–9. doi: 10.1016/j.apcatb.2016.06.054
Ni, M., Leung, M., Leung, K. H., and Sumathy, K. (2007). A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 11, 401–425. doi: 10.1016/j.rser.2005.01.009
Nie, X., Zhuo, S., Maeng, G., and Sohlberg, K. (2009). Doping of polymorphs for altered optical and photocatalytic properties. Int. J. Photoenergy. 2009:294042. doi: 10.1155/2009/294042
Nolan, M. (2011a). Electronic coupling in iron oxide-modified TiO2 leads to a reduced band gap and charge separation for visible light active photocatalysis. Phys. Chem. Chem. Phys. 13, 18194–18199. doi: 10.1039/c1cp21418g
Nolan, M. (2011b). Surface modification of TiO2 with metal oxide nanoclusters: a route to composite photocatalytic materials. Chem. Commun. 47, 8617–8619. doi: 10.1039/c1cc13243a
Nolan, M. (2012). First-principles prediction of new photocatalyst materials with visible-light absorption and improved charge separation: surface modification of rutile TiO2 with nanoclusters of MgO and Ga2O3. ACS Appl. Mater. Interfaces. 4, 5863–5871. doi: 10.1021/am301516c
Nolan, M. (2018). Alkaline earth metal oxide nanocluster modification of rutile TiO2 (110) promotes water activation and CO2 chemisorption. J. Mater. Chem. A. 6, 9451–9466. doi: 10.1039/C8TA01789A
Nolan, M., Elliott, S. D., Mulley, J. S., Bennett, R. A., Basham, M., and Mulheran, P. (2008). Electronic structure of point defects in controlled self-doping of the TiO2 (110) surface: combined photoemission spectroscopy and density functional theory study. Phys. Rev. B. 77:235424. doi: 10.1103/PhysRevB.77.235424
Nolan, M., Iwaszuk, A., and Gray, K., A (2014). Localization of photoexcited electrons and holes on low coordinated Ti and O sites in free and supported TiO2 nanoclusters. J. Phys. Chem. C. 118, 27890–27900. doi: 10.1021/jp508822v
Nolan, M., Iwaszuk, A., Lucid, A. K., Carey, J. J., and Fronzi, M. (2016). Design of novel visible light active photocatalyst materials: surface modified TiO2. Adv. Mater. 28, 5425–5446. doi: 10.1002/adma.201504894
Nolan, M., Iwaszuk, A., and Tada, H. (2012). Molecular metal oxide cluster-surface modified titanium dioxide photocatalysts. Aust. J. Chem. 65, 624–632. doi: 10.1071/CH11451
Ong, C. B., Ng, L. Y., and Mohammad, A., W (2018). A review of ZnO nanoparticles as solar photocatalysts: synthesis, mechanisms and applications. Renew. Sustain. Energy Rev. 81, 536–551. doi: 10.1016/j.rser.2017.08.020
Park, J. B., Graciani, J., Evans, J., Stacchiola, D., Ma, S., Liu, P., et al. (2009). High catalytic activity of Au/CeOx/TiO2(110) controlled by the nature of the mixed-metal oxide at the nanometer level. Proc. Natl. Acad. Sci. U. S. A. 106, 4975–4980. doi: 10.1073/pnas.0812604106
Pelaez, M., Nolan, N. T., Pillai, S. C., Seery, M. K., Falaras, P., Kontos, A. G., et al. (2012). A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B. 125, 331–349. doi: 10.1016/j.apcatb.2012.05.036
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868.
Rhatigan, S., and Nolan, M. (2018a). Impact of surface hydroxylation in MgO-/SnO-nanocluster modified TiO2 anatase (101) composites on visible light absorption, charge separation and reducibility. Chin. Chem. Lett. 29, 757–764. doi: 10.1016/j.cclet.2017.11.036
Rhatigan, S., and Nolan, M. (2018b). CO2 and water activation on ceria nanocluster modified TiO2 rutile (110). J. Mater. Chem. A. 6, 9139–9152. doi: 10.1039/C8TA01270A
Ruiz Puigdollers, A., Schlexer, P., Tosoni, S., and Pacchioni, G. (2017). Increasing oxide reducibility: the role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 7, 6493–6513. doi: 10.1021/acscatal.7b01913
Scanlon, D. O., Dunnill, C. W., Buckeridge, J., Shevlin, S. A., Logsdail, A. J., Woodley, S. M., et al. (2013). Band alignment of rutile and anatase TiO2. Nat. Mater. 12, 798–801. doi: 10.1038/nmat3697
Schaub, R., Thostrup, P., Lopez, N., Lægsgaard, E., Stensgaard, I., and Nørskov, J. K. (2001). Oxygen vacancies as active sites for water dissociation on rutile TiO2(110). Phys. Rev. Lett. 87, 266104. doi: 10.1103/PhysRevLett.87.266104
Scheiber, P., Fidler, M., Dulub, O., Schmid, M., Diebold, U., Hou, W., et al. (2012). (Sub)Surface Mobility of Oxygen Vacancies at the TiO2 Anatase (101) Surface. Phys. Rev. Lett. 109:136103. doi: 10.1103/PhysRevLett.109.136103
Schwartzenberg, K. C., Hamilton, J., Lucid, W. J., Weitz, A. K., Notestein, E., Gray, K. A., et al. (2017). Multifunctional photo/thermal catalysts for the reduction of carbon dioxide. Catal. Today 280, 65–73. doi: 10.1016/j.cattod.2016.06.002
Setvin, M., Aschauer, U., Hulva, J., Simschitz, T., Daniel, B., Schmid, M., et al. (2016). Following the reduction of oxygen on TiO2 anatase (101) step by step. J. Am. Chem. Soc. 138, 9565–9571. doi: 10.1021/jacs.6b04004
Shannon, R. D., and Prewitt, C. T. (1969). Effective ionic radii in oxides and fluorides. Acta Crystallogr. Sec. B. 25, 925–946. doi: 10.1107/S0567740869003220
Sotelo-Vazquez, C., Quesada-Cabrera, R., Ling, M., Scanlon, D. O., Kafizas, A., Thakur, P. K., et al. (2017). Evidence and effect of photogenerated charge transfer for enhanced photocatalysis in WO3/TiO2 heterojunction films: a computational and experimental study. Adv. Funct. Mater. 27:1605413. doi: 10.1002/adfm.201605413
Tada, H., Jin, Q., Iwaszuk, A., and Nolan, M. (2014). Molecular-scale transition metal oxide nanocluster surface-modified titanium dioxide as solar-activated environmental catalysts. J. Phys. Chem. C. 118, 12077–12086. doi: 10.1021/jp412312m
Valdés, Á., Qu, Z. W., Kroes, G. J., Rossmeisl, J., and Nørskov, J. K. (2008). Oxidation and photo-oxidation of water on TiO2 surface. J. Phys. Chem. C. 112, 9872–9879. doi: 10.1021/jp711929d
Valentin, C. D., Pacchioni, G., Onishi, H., and Kudo, A. (2009). Cr/Sb co-doped TiO2 from first principles calculations. Chem. Phys. Lett. 469, 166–171. doi: 10.1016/j.cplett.2008.12.086
Wang, F., Wei, S., Zhang, Z., Patzke, G. R., and Zhou, Y. (2016). Oxygen vacancies as active sites for H2S dissociation on the rutile TiO2(110) surface: a first-principles study. Phys. Chem. Chem. Phys. 18, 6706–6712. doi: 10.1039/C5CP06835E
Wang, J., Li, H., Meng, S., Zhang, L., Fu, X., and Chen, S. (2017). One-pot hydrothermal synthesis of highly efficient SnOx/Zn2SnO4 composite photocatalyst for the degradation of methyl orange and gaseous benzene. Appl. Catal. B. 200, 19–30. doi: 10.1016/j.apcatb.2016.06.070
Xiong, L. B., Li, L., Yang, B., and Yu, Y. (2012). Ti3+ in the surface of titanium dioxide: generation, properties and photocatalytic application. J. Nanomater. 2013:831524. doi: 10.1155/2012/831524
Xu, J.-, Li, P., Lv, L., Zhang, Y., Chen, S., Du, W, et al. (2009). Structural and magnetic properties of Fe-doped anatase TiO2 films annealed in vacuum. Chin. Phys. Lett. 26:097502. doi: 10.1088/0256-307X/26/9/097502
Yang, K., Dai, Y., Huang, B., and 0 andWhangbo, M.- H. (2009). Density functional characterization of the visible-light absorption in substitutional C-anion- and C-cation-doped TiO2. J. Phys. Chem. C. 113, 2624–2629. doi: 10.1021/jp808483a
Yu, J., Xiang, Q., and Zhou, M. (2009). Preparation, characterization and visible-light-driven photocatalytic activity of Fe-doped titania nanorods and first-principles study for electronic structures. Appl. Catal. B. 90, 595–602. doi: 10.1016/j.apcatb.2009.04.021
Zhang, H., Yu, X., McLeod, J. A., and Sun, X. (2014). First-principles study of Cu-doping and oxygen vacancy effects on TiO2 for water splitting. Chem. Phys. Lett. 612, 106–110. doi: 10.1016/j.cplett.2014.08.003
Zhang, J., Pan, C., Fang, P., Wei, J., and Xiong, R. (2010). Mo + C codoped TiO2 using thermal oxidation for enhancing photocatalytic activity. ACS Appl. Mater. Interf. 2, 1173–1176. doi: 10.1021/am100011c
Zhang, J., Salles, I., Pering, S., Cameron, P. J., Mattia, D., and Eslava, S. (2017a). Nanostructured WO3 photoanodes for efficient water splitting via anodisation in citric acid. RSC Adv. 7, 35221–35227. doi: 10.1039/C7RA05342H
Zhang, Y., Dai, R., and Hu, S. (2017b). Study of the role of oxygen vacancies as active sites in reduced graphene oxide-modified TiO2. Phys. Chem. Chem. Phys. 19, 7307–7315. doi: 10.1039/C7CP00630F
Zheng, J. W., Bhattcahrayya, A., Wu, P., Chen, Z., Highfield, J., Dong, Z., et al. (2010). The origin of visible light absorption in chalcogen element (S, Se, and Te)-doped anatase TiO2 photocatalysts. J. Phys. Chem. C. 114, 7063–7069. doi: 10.1021/jp9115035
Keywords: TiO2, water activation, reduction, oxygen vacancies, photocatalysis
Citation: Rhatigan S and Nolan M (2019) Activation of Water on MnOx-Nanocluster-Modified Rutile (110) and Anatase (101) TiO2 and the Role of Cation Reduction. Front. Chem. 7:67. doi: 10.3389/fchem.2019.00067
Received: 12 October 2018; Accepted: 24 January 2019;
Published: 12 February 2019.
Edited by:
Javier Carrasco, CIC energigune, SpainCopyright © 2019 Rhatigan and Nolan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Nolan, bWljaGFlbC5ub2xhbkB0eW5kYWxsLmll
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.