 Daniel Marx
Daniel Marx Lukas M. Wingen
Lukas M. Wingen Gregor Schnakenburg2
Gregor Schnakenburg2 Christa E. Müller
Christa E. Müller Matthias S. Scholz
Matthias S. Scholz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 18 February 2019
Sec. Organic Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00056
This article is part of the Research Topic Organic Chemistry Editor’s Pick 2021 View all 4 articles
Substituted xanthine derivatives are important bioactive molecules. Herein we report on a new, practical synthesis of 6-amino-5-carboxamidouracils, the main building blocks for the preparation of 8-substituted xanthines, by condensation of 5,6-diaminouracil derivatives and various carboxylic acids using the recently developed non-hazardous coupling reagent COMU (1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)dimethylaminomorpholinomethylene)]methanaminium hexafluorophosphate). Optimized reaction conditions led to the precipitation of pure products after only 5 to 10 min of reaction time. The method tolerates a variety of substituted 5,6-diaminouracil and carboxylic acid derivatives as starting compounds resulting in most cases in more than 80% isolated yield. Regioselectivity of the reaction yielding only the 5-carboxamido-, but not the 6-carboxamidouracil derivatives, was unambiguously confirmed by single X-ray crystallography and multidimensional NMR experiments. The described method represents a convenient, fast access to direct precursors of 8-substituted xanthines under mild conditions without the necessity of hazardous coupling or chlorinating reagents.
Xanthines are privileged structures in medicinal chemistry (Jacobson et al., 1993; Scammells et al., 1994; Kim et al., 2000; Baraldi et al., 2004; Müller and Jacobson, 2011). The methylxanthines caffeine (compound 1, Figure 1), theobromine (2) and theophylline (3) are frequently consumed and therapeutically applied natural products (Franco et al., 2013). The biological activities of 1 and 2, including central nervous system stimulatory, diuretic and antiasthmatic effects, are due to their blockade of adenosine receptors (ARs). The ARs, which belong to the family of G protein-coupled receptors (GPCRs), are (potential) drug targets for several diseases, in particular for heart and brain diseases (Baraldi et al., 2008; Müller and Jacobson, 2011; Chen et al., 2013). Recent findings point toward a great potential of A2A and A2B AR antagonists in immuno-oncology (Leone et al., 2015; Müller et al., 2018).
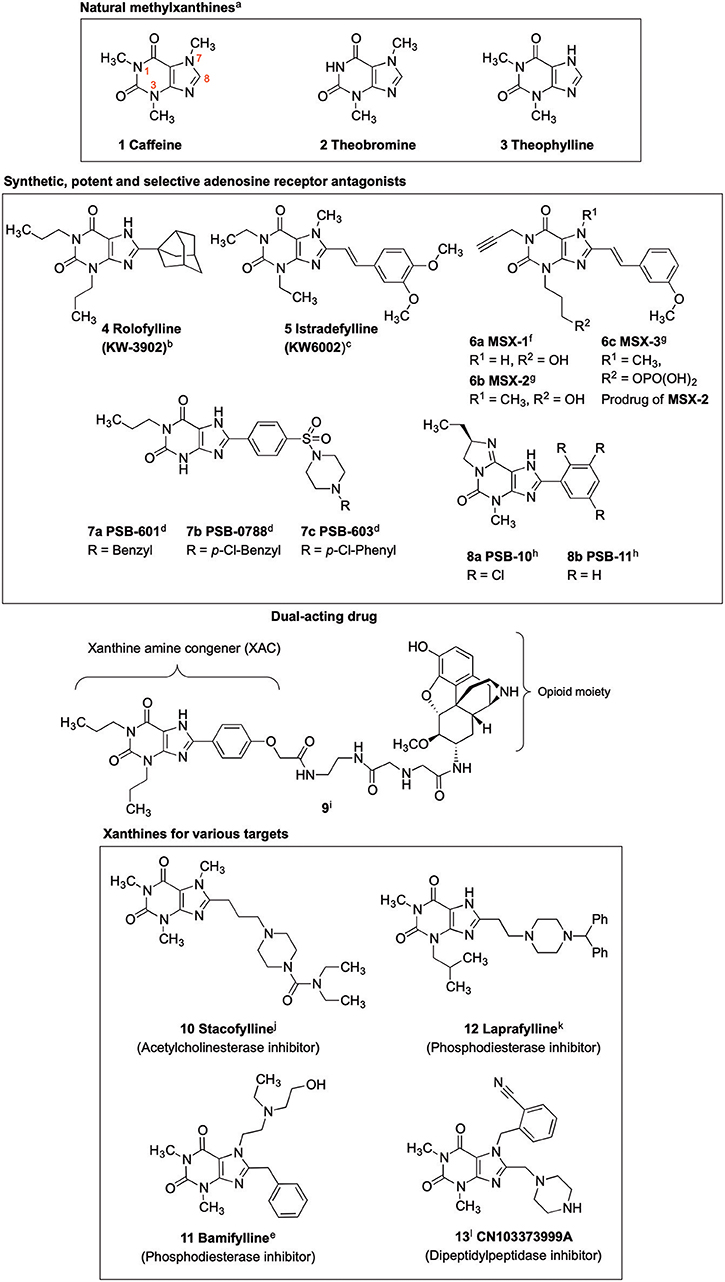
Figure 1. Xanthine-based drugs. a(Franco et al., 2013); b(Slawsky and Givertz, 2009); c(Park and Stacy, 2012); d(Borrmann et al., 2009); e(Alciato et al., 1990); f(Sauer et al., 2000); g(Hockemeyer et al., 2004); h(Ozola et al., 2003); i(Jacobson, 2009); j(Gallagher, 2004); k(Baraldi et al., 2007); l(Costante et al., 2015).
Caffeine and theophylline are weak, non-selective AR antagonists (Nieber, 2017; Oñatibia-Astibia et al., 2017). Replacing the hydrogen atom at C8 by a larger residue, in combination with suitable substituents at the xanthine nitrogen atoms, may result in highly potent and subtype-selective AR antagonists (Baraldi et al., 2007; Müller and Jacobson, 2011). Inspired by the natural methylxanthines, several drugs have been developed, which were designated by the suffix “fylline” (Figure 1) (Alciato et al., 1990; LeWitt et al., 2008). An example is rolofylline (KW-3902, 4, Figure 1), which carries a bulky noradamantanyl residue at the 8-position and acts as a selective A1 AR receptor antagonist (LeWitt et al., 2008). Istradefylline (KW-6002, 5, Figure 1), a potent, selective A2A AR antagonist was approved for the treatment of Parkinson's disease (PD) in Japan (LeWitt et al., 2008; Dungo and Deeks, 2013; Kondo and Mizuno, 2015). It features a styryl residue at the xanthine 8-position and ethyl groups at the xanthine N1 and N3 nitrogen atoms. An N1-propargyl residue in combination with a C8-styryl substitution yielded the potent and selective A2A AR antagonist MSX-2 (6b) and its prodrug MSX-3 (6c) prepared from the precursor MSX-1 (6a) (Sauer et al., 2000; Hockemeyer et al., 2004). PSB-601 (7a), PSB-0788 (7b) and PSB-603 (7c) are potent, selective A2B AR antagonists. These xanthines carry a para-sulfonamido-substituted phenyl ring at the 8-position and are potential therapeutics for the treatment of asthma, pain and cancer (Feoktistov et al., 1998; Yan et al., 2006; Singh and Yadav, 2016; Hinz et al., 2018; Müller et al., 2018). The tricyclic purine derivatives PSB-10 (8a) and PSB-11 (8b) are selective A3 AR antagonists (Müller et al., 2002; Ozola et al., 2003).
Crystal structures of the AR subtypes A1 (Cheng et al., 2017; Glukhova et al., 2017) and A2A (Doré et al., 2011; Liu et al., 2012; Sun et al., 2017) showed that large 8-substituents of xanthine derivatives point out of the receptor binding pocket toward the extracellular space. This makes C8 a privileged position for the attachment of fluorophores (Köse et al., 2018), solubilizing moieties (Daly et al., 1985), spin labels for electron paramagnetic resonance (EPR) studies (Ilaš et al., 2005) or linkers for dual-acting compounds (Jacobson, 2009). An example of a dual ligand is compound 9 (Jacobson, 2009).
Receptors other than ARs, and enzymes can also be addressed by selecting appropriate substituents at the xanthine scaffold. Stacofylline (10) inhibits the enzyme acetylcholinesterase; it contains a diethylaminocarbonylpiperazinyl residue connected via a propyl spacer to the 8-position of caffeine (Gallagher, 2004). Bamifylline (11), a phosphodiesterase inhibitor, carries a benzyl-substituent at C8 and is used as an analgesic, bronchodilatory and vasodilatory drug (Alciato et al., 1990). The phosphodiesterase inhibitor laprafylline (12) features, similar to stacofylline (10), a piperazinyl residue attached by an ethyl linker to the 8-position of 1-methyl-3-isobutylxanthine. Recently, dipeptidylpeptidase 4 (DPP-4) inhibitors have gained attention for the treatment of type 2 diabetes (Crepaldi et al., 2007; Costante et al., 2015). Xanthine-derived compounds, such as CN103373999A (13), bearing a piperazinylmethyl residue at the xanthine 8-position have been identified as potent DPP-4 inhibitors (Costante et al., 2015).
8-Substituted xanthines can be synthesized by reacting 5,6-diaminouracil derivatives with carboxylic acids or aldehydes (Scheme 1).
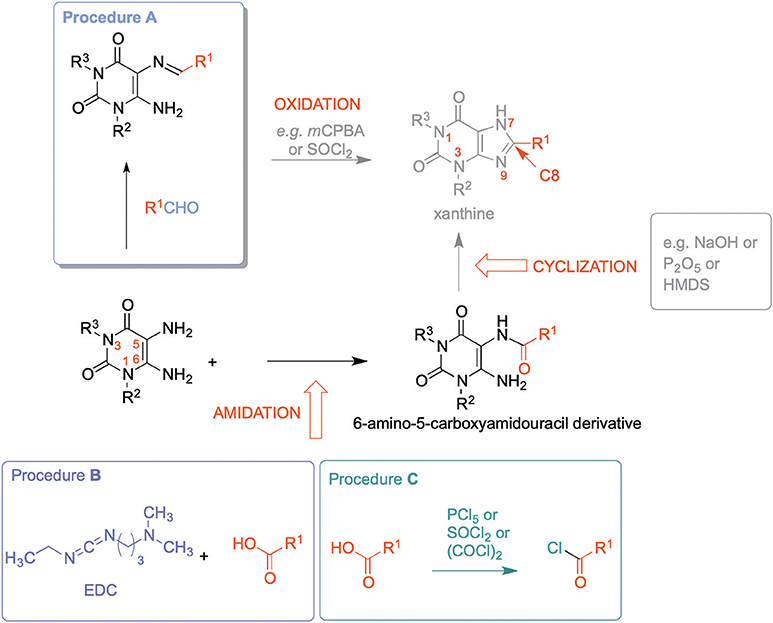
Scheme 1. Syntheses of 8-substituted xanthine derivatives.
Different routes have been employed to obtain the required xanthine precursors. Condensation of 5,6-diaminouracils with aldehydes forming the corresponding imines [5-(arylidene- or alkylidene-amino)-6-aminouracils] as precursors, followed by oxidative cyclization is a commonly used route for the synthesis of 8-substituted xanthine derivatives (Hayallah et al., 2002; El-Sabbagh et al., 2007). However, aldehydes are less stable than the corresponding carboxylic acids, and commercial availability is often limited (Procedure A, Scheme 1) (Daly et al., 1985; Hayallah et al., 2002). Alternatively, 6-amino-5-carboxamidouracils can be prepared, which are the most frequently utilized xanthine precursors, that can be cyclized using a variety of methods, e.g., by sodium hydroxide or methylate, trimethylsilyl polyphosphate (PPSE), hexamethyldisilazane (HMDS) (Hayallah et al., 2002), or phosphorus pentoxide (Müller et al., 2008), depending on their reactivity and stability.
An established method for their preparation is the coupling of 5,6-diaminouracil derivatives with carboxylic acids in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC-HCl) as a coupling reagent (Procedure B, Scheme 1) (Sauer et al., 2000; Hayallah et al., 2002; Hockemeyer et al., 2004; Basu et al., 2017). Another method requires the activation of the carboxylic acid by formation of the carboxylic acid chloride (Procedure C, Scheme 1) (Jacobson et al., 1989; Hockemeyer et al., 2004). Procedure C had been used to establish a multigram-scale synthesis of istradefylline (5). Drawbacks of this reaction are long reaction times (16 h) for the formation of the amide, only moderate yields (65%), and importantly, an additional step due to conversion of the acid into the corresponding acid chloride using hazardous chlorinating reagents. Furthermore, carboxylic acid chlorides are less stable than the corresponding carboxylic acids rendering storage and handling more demanding (Hockemeyer et al., 2004). Coupling reactions with the irritant and moisture-sensitive EDC-HCl also suffer from rather long reaction times, and typically provide moderate yields requiring tedious purification (Sauer et al., 2000; Hockemeyer et al., 2004).
All of these disadvantages motivated us to search for an alternative amide coupling procedure for the preparation of 6-amino-5-carboxamidouracil derivatives being the most stable and easily storable xanthine precursors. Our aim was to develop a fast and effective coupling method applicable to a variety of diaminouracils and carboxylic acids that would allow simple work-up and straightforward isolation of the desired product (Scheme 3).
Chemicals were purchased from Merck (Darmstadt, Germany), ABCR (Karlsruhe, Germany) or TCI (Eschborn, Germany). Analytical thin layer chromatography (TLC) was performed on TLC plates F254 (Merck) and analyzed using UV light. High resolution mass spectra (HR-MS) were recorded on a micrOTOF-Q mass spectrometer (Bruker), low resolution mass spectra (LR-MS) on an API 2000 (Applied Biosystems) mass spectrometer. 1H NMR and 13C NMR spectra were recorded in CDCl3 or (CD3)2SO on a Bruker Ascend 600 MHz NMR-spectrometer operating at 600.18 MHz (1H), and 150.93 MHz (13C). Chemical shifts (δ) are reported in ppm and are referenced to the chemical shifts of the residual solvent proton(s) present in chloroform δ [(CHCl3) = 7.26 ppm for the 1H NMR spectra and δ (CDCl3) = 77.16 ppm for the 13C NMR spectra] and in dimethylsulfoxide δ ((CH3)2SO) = 2.50 ppm for the 1H NMR spectra and δ ((CD3)2SO) = 39.52 ppm for the 13C NMR spectra. Multiplicity: s, singlet; d, doublet; q, quartet; m, multiplet. Coupling constants (J) are shown in Hertz (Hz). The infrared spectra were recorded as solid samples on an ALPHA-T (Bruker) with a Platinum ATR Module using the Opus software. The IR spectra were measured in the attenuated total reflection (ATR) mode in the region of 4,000–385 cm−1 (s, strong; m, medium; w, weak) and are reported in cm−1.
To a solution of the respective carboxylic acid (1.0 equiv.) and COMU (1.1 equiv.) dissolved in a minimum of dimethylformamide (DMF), a mixture of diaminouracil (1.1 equiv.) and N,N-diisopropylethylamine (DIPEA) (1.1 equiv.) dissolved in a minimum DMF was added dropwise. The reaction was stirred for 5–10 min at room temperature, and water was added. The resulting precipitate was filtered off, washed with water and dried under reduced pressure. Most of the reactions were performed using 300 mg of the respective diaminouracil and 4 ml of DMF. The product was precipitated using 20 ml of water and washed with small portions of water (10 ml). The reaction generally performed well from 60 mg up to 1.5 g of diaminouracil as a precursor. For the 1.5 g scale 8 ml of DMF were used for dissolution, and 40 ml of water for precipiation, and 20 ml for the subsequent washing step. All other conditions were identical, and virtually the same percentage of yield as obtained independent of the scale of the reaction.
Yield: 62% (white solid); mp 181–184°C; 1H NMR (500 MHz, DMSO-d6) δ 10.48 (s, 1H, N1-H), 8.21 (s, 1H, CONH), 7.90 (d, J = 7.5 Hz, 2H, Harom), 7.63 (d, J = 7.5 Hz, 2H, Harom), 7.42 (t, J = 7.4 Hz, 2H. Harom), 7.35 (td, J = 7.4, 1.1 Hz, 2H, Harom), 6.01 (s, 2H, NH2), 4.40 (d, J = 6.5 Hz, 2H, CH2), 4.28 (t, J = 6.4 Hz, 1H, CH), 3.40–3.32 (m, 4H, 2 × CH2), 3.06 (s, 3H, CH3), 3.05–3.01 (m, 2H, CH2), 2.48–2.37 (m, 4H, 2 × CH2). 13C NMR (126 MHz, DMSO) δ 169.5 (CON), 160.7 (C6), 154.3 (OCON), 149.9 (CO), 149.7 (CO), 143.8 (2C, Carom), 140.8 (2C, Carom), 127.6 (2C, Carom), 127.1 (2C, Carom), 124.9 (2C, Carom), 120.1 (2C, Carom), 86.7 (C5), 66.4 (CH2), 61.0 (CH2), 52.2 (2C, 2 × CH2), 46.8 (2C, 2 × CH2), 43.3 (CH), 26.4 (CH3). IR (cm−1): ṽ = 3,556 (w), 33,481 (w), 3,327 (w), 3,208 (w), 3,010 (w), 2,949 (w), 2,895 (w), 2,811 (w), 2,757 (w), 1,730 (m), 1,688 (s), 1,655 (m), 1,556 (s), 1,505 (s), 1,458 (s), 1,442 (s), 1,289 (w), 1,237 (s), 1,203 (w), 1,124 (s), 1,082 (m), 1,006 (m), 966 (m), 755 (s), 737 (s), 641 (w),621 (w), 576 (m), 499 (s), 412 (s). HRMS (ESI-QTOF) calculated for C26H28N6O5 [M+H]+: 505.2194; found: 505.2190.
Yield: 78% (white solid); mp > 320°C; 1H NMR (500 MHz, DMSO-d6) δ 10.59 (s, 1H, N3-H), 8.83 (s, 1H, CONH), 7.99 (d, J = 7.8 Hz, 2H, Harom), 7.54 (t, J = 6.8 Hz, 1H, Harom), 7.48 (q, J = 7.9, 7.3 Hz, 2H, Harom), 6.65 (s, 2H, NH2), 3.27 (s, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 166.6 (CON), 160.0 (C6), 153.7 (CO), 150.4 (CO), 134.7 (Carom), 131.2 (Carom), 128.1 (2C, Carom), 128.1(2C, Carom), 87.8 (C5), 29.2 (CH3). IR (cm−1): ṽ = 3,342 (w), 3,201 (w), 3,063 (w), 1,778 (w), 1,713 (s), 1,638 (s), 1,584 (s), 1,506 (s), 1,484 (s), 1,390 (w), 1,294 (m), 1,263 (w), 1,220 (w), 1,176 (w), 1,072 (w), 1,012 (w), 891 (w), 782 (m), 744 (w), 715 (s), 686 (w), 584 (m), 545 (s), 477 (s), 428 (w). HRMS (ESI-QTOF) calculated for C12H12N4O3 [M+H]+: 261.0982; found: 261.0981.
Yield: 87% (off-white solid); mp 109–112°C; 1H NMR (600 MHz, DMSO-d6) δ 8.73 (s, 1H, CONH), 7.95 (d, J = 8.9 Hz, 2H, Harom), 7.00 (d, J = 8.9 Hz, 2H, Harom), 6.65 (s, 2H, NH2), 3.86–3.82 (m, 2H, N1-CH2 or N3-CH2), 3.82 (s, 3H, OCH3), 3.75–3.68 (m, 2H, N1-CH2 or N3-CH2), 1.57 (dt, J = 15.1, 7.5 Hz, 2H, CH2), 1.51 (dt, J = 14.8, 7.6 Hz, 2H, CH2), 0.89 (t, J = 7.4 Hz, 3H, CH3), 0.83 (t, J = 7.5 Hz, 3H, CH3). 13C NMR (DMSO, 151 MHz) δ 166.1 (CON), 161.6 (Carom), 159.2 (C6), 151.7 (CO), 150.4 (CO), 129.8 (Carom), 126.8 (Carom), 113.1 (Carom), 87.6 (C5), 55.3 (OCH3), 43.7 (N1-CH2 or N3-CH2), 41.8 (N1-CH2 or N3-CH2), 20.8 (2C, CH2), 11.2 (CH3), 10.7 (CH3). IR (cm−1): ṽ = 3,416 (w), 3,348 (w), 3,219 (w), 2,963 (w), 2,939 (w), 2,877 (w), 2,841 (w), 1,695 (m), 1,636 (m), 1,605 (s), 1,488 (s), 1,415 (m), 1,381 (w), 1,259 (s), 1,191 (m), 1,114 (w), 1,080 (w), 1,029 (m), 901 (w), 852 (m), 762 (m), 551 (s), 513 (s). HRMS (ESI-QTOF) calculated for C18H24N4O4 [M+H]+: 361.1870; found: 361.1885.
Product was purified by column chromatography (CH2Cl2/MeOH, 95:5). Yield: 78% (yellowish solid); mp 203–206°C; 1H NMR (500 MHz, DMSO-d6) δ 10.49 (s, 1H, N1-H), 9.21 (s, 1H, CONH), 8.30–8.26 (m, 2H, Harom), 8.21–8.17 (m, 2H, Harom), 8.06–8.02 (m, 2H, Harom), 7.42–7.36 (m, 2H, Harom), 6.22 (s, 2H, NH2), 3.75 (q, J = 7.0 Hz, 2H, N3-CH2), 1.06 (t, J = 7.0 Hz, 3H, CH3).13C NMR (DMSO, 126 MHz) δ 164.8 (CON), 160.3 (C6), 153.0 (Carom), 150.5 (CO), 149.7 (CO), 146.2 (Carom), 140.6 (Carom), 135.6 (Carom), 129.4 (2C, Carom), 128.2 (2C, Carom), 125.9 (2C, Carom), 123.3 (2C, Carom), 86.4 (C5), 34.4 (N3-CH2), 13.3 (CH3). IR (cm−1): ṽ = 3,304 (w), 3,185 (w), 3,078 (w), 2,971 (w), 2,917 (w), 2,851 (w), 1,734 (m), 1,627 (m), 1,507 (s), 1,480 (s), 1,374 (s), 1,349 (s), 1,314 (m), 1,293 (m), 1,203 (s), 1,153 (s), 1,091 (m), 1,012 (w), 866 (s), 757 (s), 733 (w), 692 (m), 630 (w), 606 (s), 564 (s), 500 (s), 445 (m). HRMS (ESI-QTOF) calculated for C19H17N5O8S[M+H]+: 476.0871; found: 476.0860.
Most of the compound precipitated overnight. To increase the yield, the filtrate was extracted with diethyl ether, dried over MgSO4, and after filtration the solvent was removed in vacuo. Yield: 99% (slightly brown solid); mp 153–157°C; 1H NMR (600 MHz, DMSO-d6) δ 7.74 (s, 1H, CONH), 6.33 (s, 2H, NH2), 3.88–3.78 (m, 2H, NCH2), 3.74–3.62 (m, 2H, NCH2), 2.74–2.69 (m, 1H, CH), 2.24 (s, 2H, Hadamantyl), 2.05 (d, J = 9.8 Hz, 2H, Hadamantyl), 1.83–1.76 (m, 4H, Hadamantyl), 1.55 (p, J = 7.2 Hz, 6H, Hadamantyl and CH2CH3), 1.49 (q, J = 7.4 Hz, 2H, CH2CH3), 0.88 (t, J = 7.4 Hz, 3H, CH3), 0.82 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (DMSO, 151 MHz) δ 177.1 (CON), 158.9 (C6), 151.4 (CO), 150.3 (CO), 88.2 (C5), 54.7 (Cadamantyl), 46.8 (NCH2), 43.6 (NCH2), 43.2 (Cadamantyl), 42.3 (Cadamantyl), 41.8 (Cadamantyl), 37.0 (Cadamantyl), 34.5 (Cadamantyl), 20.8 (Cadamantyl), 11.2 (Cadamantyl), 10.7 (Cadamantyl). IR (cm−1): ṽ = 3,425 (w), 3,331 (w), 2,925 (w), 2,871 (w), 1,694 (s), 1,627 (m), 1,556 (s), 1,492 (s), 1,374 (w), 1,338 (w), 1,272 (m), 1,226 (m), 1,204 (m), 1,111 (w), 1,085 (w), 899 (w), 843 (w), 763 (w), 716 (w), 549 (m), 475 (w), 429 (w). HRMS (ESI-QTOF) calculated for C20H30N4O3 [M+H]+: 375.2391; found: 375.2389.
Yield: 70% (white solid); mp > 320°C; 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H, N1-H), 8.21 (s, 1H, CONH), 5.77 (s, 2H, NH2), 3.04 (s, 3H, NCH3), 2.74 (p, J = 8.0 Hz, 1H, CH), 1.84–1.75 (m, 2H, Hcyclopentyl), 1.74–1.66 (m, 2H, Hcyclopentyl), 1.61 (qt, J = 10.3, 4.3 Hz, 2H, Hcyclopentyl), 1.50 (dtt, J = 9.2, 5.6, 2.9 Hz, 2H, Hcyclopentyl). 13C NMR (DMSO, 126 MHz) δ 176.0 (CON), 160.9 (C6), 150.1 (CO), 150.0 (CO), 87.7 (C5), 44.1 (Ccyclopentyl), 30.1 (2C, Ccyclopentyl), 26.6 (CH3), 25.9 (2C, Ccyclopentyl). IR (cm−1): ṽ = 3,328 (w), 3,173 (w), 2,967 (w), 2,951 (w), 2,872 (w), 1,720 (s), 1,651 (s), 1,633 (s), 1,552 (s), 1,497 (s), 1,456 (s), 1,380 (w), 1,302 (w), 1,211 (m), 1,170 (w), 1,120 (w), 1,024 (w), 996 (w), 961 (w), 945 (w), 755 (s), 711 (m), 662 (m), 592 (s), 549 (m), 512 (s), 471 (m), 417 (s). HRMS (ESI-QTOF) calculated for C11H16N4O3 [M+H]+: 253.1295; found: 253.1294.
Yield: 70% (white solid); mp 108–112°C; 1H NMR (500 MHz, DMSO-d6) δ 8.49 (s, 1H, CONH), 7.39 (d, J = 15.8 Hz, 1H, CH), 7.18 (d, J = 1.9 Hz, 1H, Harom), 7.15 (dd, J = 8.3, 1.9 Hz, 1H, Harom), 7.01 (d, J = 8.4 Hz, 1H, Harom), 6.71 (d, J = 15.8 Hz, 1H, CH), 6.62 (s, 2H, NH2), 3.93 (q, J = 6.9 Hz, 2H, N1-CH2 or N3-CH2), 3.80 (d, J = 6.3 Hz, 8H, N1-CH2 or N3-CH2 and 2 × OCH3), 1.14 (t, J = 7.0 Hz, 3H, CH3), 1.07 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 165.6 (CON), 158.8 (C6), 151.0 (Carom), 150.1 (CO), 149.8 (CO), 148.9 (Carom), 138.8 (CH), 127.8 (Carom), 121.1 (Carom), 120.4 (Carom), 111.9 (Carom), 110.2 (CH), 87.8 (C5), 55.5 (OCH3), 55.4 (OCH3), 37.6 (N1-CH2), 35.4 (N3-CH2), 13.2 (2C, CH3). IR (cm−1): ṽ = 3,370 (w), 3,197 (w), 2,987 (w), 2,939 (w), 2,840 (w), 1,705 (s), 1,661 (m), 1,644 (m), 1,581 (s), 1,509 (s), 1,464 (s), 1,419 (m), 1,374 (w), 1,325 (w), 1,267 (s), 1,238 (s), 1,185 (s), 1,161 (s), 1,139 (s), 1,024 (m), 974 (m), 848 (w), 794 (m), 760 (m), 671 (m), 554 (s), 529 (s), 448 (s). HRMS (ESI-QTOF) calculated for C19H24N4O5 [M+H]+: 389.1819; found: 389.1812.
Yield: 83% (white solid); mp 295–298°C; 1H NMR (500 MHz, DMSO-d6) δ 10.59 (s, 1H, N1-H), 8.67 (s, 1H, CONH), 7.44 (d, J = 15.8 Hz, 1H, CH), 7.35 (t, J = 7.9 Hz, 1H, Harom), 7.17 (d, J = 7.7 Hz, 1H, Harom), 7.15–7.12 (m, 1H, Harom), 6.98–6.95 (m, 1H, Harom), 6.82 (d, J = 15.8 Hz, 1H, CH), 6.13 (s, 2H, NH2), 4.44 (d, J = 2.4 Hz, 2H, N3-CH2), 3.80 (s, 3H, OCH3), 3.03 (t, J = 2.4 Hz, 1H, Hpropargyl). 13C NMR (DMSO, 126 MHz) δ 165.0 (CON), 159.6 (C6), 150.3 (CO), 149.1 (CO), 138.8 (Carom or CH), 136.4 (Carom or CH), 130.0 (Carom), 122.7 (Carom), 119.7 (Carom), 115.2 (Carom), 112.7 (CH), 86.9 (C5), 79.9 (Cpropargyl), 72.4 (Cpropargyl), 55.1 (OCH3), 28.9 (N3-CH2). IR (cm−1): ṽ = 3,393 (w), 3,290 (w), 3,252 (w), 3,120 (w), 1,727 (s), 1,707 (m), 1,650 (s), 1,625 (m), 1,598 (s), 1,550 (s), 1,508 (s), 1,492 (s), 1,447 (s), 1,410 (w), 1,388 (w), 1,340 (m), 1,313 (m), 1,295 (m), 1,250 (s), 1,187 (m), 1,159 (m), 1,038 (w), 1,016 (w), 976 (s), 944 (w), 930 (w), 903 (w), 885 (w), 836 (w), 778 (m), 759 (s), 698 (s), 643 (s), 564 (s), 456 (s). HRMS (ESI-QTOF) calculated for C17H16N4O4 [M+H]+: 341.1244; found: 341.1241.
Yield: 85% (white solid); mp 258–261°C; 1H NMR (500 MHz, DMSO-d6) δ 8.58 (s, 1H, CONH), 7.38–7.33 (m, 2H, Harom), 7.32–7.27 (m, 2H, Harom), 7.21 (tt, J = 6.4, 1.1 Hz, 1H, Harom), 6.54 (s, 2H, NH2), 3.59 (s, 2H, CH2), 3.31 (s, 3H, CH3), 3.11 (s, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 170.7 (CON), 159.3 (C6), 152.0 (CO), 150.5 (CO), 136.5 (Carom), 129.2 (Carom), 128.0 (Carom), 126.1 (Carom), 87.5 (C5), 42.0 (CH2), 30.0 (CH3), 27.5 (CH3). IR (cm−1): ṽ = 3,322 (w), 3,190 (w), 1,699 (m), 1,667 (s), 1,643 (m), 1,583 (s), 1,496 (s), 1,421 (w), 1,381 (w), 1,344 (w), 1,322 (w), 1,225 (m), 1,164 (w), 1,153 (m), 1,057 (w), 1,028 (w), 979 (w), 954 (w), 935 (w), 903 (w), 837 (w), 756 (m), 728 (s), 693 (m), 557 (s), 535 (m), 487 (s), 438 (m). HRMS (ESI-QTOF) calculated for C14H16N4O3 [M+H]+: 289.1295; found: 289.1296.
Yield: 85% (off-white solid); mp. 121–124°C; 1H NMR (600 MHz, chloroform-d1) δ 8.19 (s, 1H, CONH), 7.93 (d, J = 7.4 Hz, 2H, Harom), 7.52 (t, J = 7.1 Hz, 1H, Harom), 7.43 (t, J = 7.5 Hz, 2H, Harom), 5.71 (s, 2H, NH2), 3.83 (dt, J = 14.3, 7.8 Hz, 4H, 2 × NCH2), 1.72 (q, J = 7.4 Hz, 2H, CH2), 1.61 (q, J = 7.4 Hz, 2H, CH2), 0.99 (t, J = 7.3 Hz, 3H, CH3), 0.91 (t, J = 7.4 Hz, 3H, CH3). 13C NMR (CDCl3, 151 MHz) δ 166.9 (CON), 160.2 (C6), 150.2 (CO), 148.0 (CO), 133.4 (Carom), 132.3 (Carom), 128.8 (Carom), 127.6 (Carom), 92.3 (C5), 44.9 (NCH2), 43.6 (NCH2), 21.6 (CH2), 21.3 (CH2), 11.4 (CH3), 11.3 (CH3). IR (cm−1): ṽ = 3,364 (w), 3,216 (w), 2,963 (w), 2,931 (w), 2,874 (w), 1,696 (m), 1,664 (m), 1,578 (s), 1,508 (s), 1,463 (s), 1,414 (m), 1,278 (m), 1,160 (w), 1,073 (w), 1,000 (w), 900 (w), 842 (w), 764 (m), 689 (m), 543 (s), 456 (m). HRMS (ESI-QTOF) calculated for C17H22N4O3 [M+H]+: 331.1765; found: 331.1767.
Yield: 80% (off-white solid); mp > 320°C; 1H NMR (500 MHz, DMSO-d6) δ 10.43 (s, 1H, N1-H), 8.68 (s, 1H, CONH), 7.58 (d, J = 7.4 Hz, 2H, Harom), 7.50–7.37 (m, 4H, Harom + Hvinyl), 6.83 (d, J = 15.9 Hz, 1H, Hvinyl), 5.99 (s, 2H, NH2), 3.74 (q, J = 6.5 Hz, 2H, CH2), 1.06 (t, J = 6.7 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 164.9 (CON), 160.3 (C6), 149.7 (CO), 149.5 (CO), 138.8 (Cvinyl or Carom), 135.0 (Cvinyl or Carom), 129.4 (Cvinyl or Carom), 129.0 (2C, Carom), 127.4 (2C, Carom), 122.4 (Cvinyl or Carom), 87.4 (C5), 34.4 (N3-CH2), 13.2 (CH3). IR (cm−1): ṽ = 3,315 (w), 3,166 (w), 3,065 (w), 3,026 (w), 2,976 (w), 2,940 (w), 2,913 (w), 1,723 (s), 1,646 (s), 1,617 (s), 1,557 (s), 1,490 (s), 1,427 (m), 1,381 (w), 133 (m), 1,291 (w), 1,192 (m), 1,161 (w), 1,047 (w), 999 (m), 741 (s), 713 (m), 586 (s), 543 (s), 505 (s), 487 (s), 450 (w), 433 (w). HRMS (ESI-QTOF) calculated for C15H16N4O3 [M+H]+: 301.1295; found: 301.1294.
Yield: 90% (white solid); mp > 320°C; 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H, N1-H), 8.39 (s, 1H, CONH), 7.28 (t, J = 7.4 Hz, 2H, Harom), 7.24 (d, J = 6.9 Hz, 2H, Harom), 7.18 (t, J = 7.1 Hz, 1H, Harom), 5.82 (s, 2H, NH2), 3.73 (q, J = 6.9 Hz, 2H, N3-CH2), 2.91–2.80 (m, 2H, CH2), 2.53 (dd, J = 9.2, 7.0 Hz, 2H, CH2), 1.04 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 171.7 (CON), 160.4 (C6), 149.9 (CO), 149.6 (CO), 141.5 (Carom), 128.3 (2C, Carom), 128.1 (2C, Carom), 125.8 (Carom), 87.2 (C5), 36.8 (CH2), 34.3 (N3-CH2), 30.9 (CH2), 13.2 (CH3). IR (cm−1): ṽ = 3,341 (w), 3,290 (w), 3,180 (w), 3,066 (w), 3,029 (w), 2,913 (w), 1,725 (m), 1,637 (s), 1,552 (s), 1,486 (s), 1,382 (m), 1,333 (m), 1,301 (m), 1,192 (w), 1,157 (m), 1,124 (w), 1,044 (w), 970 (w), 921 (w), 799 (w), 78 (w), 760 (s), 730 (m), 695 (m), 662 (m), 571 (s), 501 (s), 481 (s). HRMS (ESI-QTOF) calculated for C15H18N4O3 [M+H]+: 303.1452; found: 303.1454.
Yield: 89% (white solid); mp 302–305°C; 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1H, N1-H), 8.68 (s, 1H, CONH), 7.29 (t, J = 7.5 Hz, 2H, Harom), 7.19 (d, J = 7.7 Hz, 1H, Harom), 7.14 (d, J = 7.9 Hz, 2H, Harom), 5.90 (s, 2H, NH2), 3.72 (q, J = 6.9 Hz, 2H, N3-CH2), 2.28 (dt, J = 9.5, 6.0 Hz, 1H, CH), 2.09 (dt, J = 8.8, 4.7 Hz, 1H, CH), 1.37 (dt, J = 9.0, 4.5 Hz, 1H, CH), 1.26–1.20 (m, 1H, CH), 1.04(t, J = 6.9 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 171.5 (CON), 160.4 (C6), 149.9 (CO), 149.5 (CO), 141.2 (Carom), 128.2 (2C, Carom), 125.9 (3C, Carom), 87.4 (C5), 34.4 (N3-CH2), 25.6 (Ccyclopropyle), 24.3 (Ccyclopropyle), 16.1 (Ccyclopropyle), 13.2 (CH3). IR (cm−1): ṽ = 3,355 (w), 3,312 (w), 3,186 (w), 3,082 (w), 3,032 (w), 3,011 (w), 2,978 (w), 2,941 (w), 1,726 (s), 1,650 (s), 1,628 (s), 1,555 (s), 1,497 (s), 1,454 (s), 1,427 (m), 1,382 (w), 1,334 (m), 1,300 (m), 1,199 (m), 1,160 (w), 1,080 (w), 1,026 (w), 957 (w), 760 (s), 693 (m), 662 (m), 592 (m), 543 (m), 518 (s), 499 (m). HRMS (ESI-QTOF) calculated for C16H18N4O3 [M+H]+: 315.1452; found: 315.1460.
Yield: 88% (off-white solid); mp 289–293°C; 1H NMR (500 MHz, DMSO-d6) δ 10.45 (s, 1H, N1-H), 8.53 (s, 1H, CONH), 7.31 (td, J = 7.4, 2.0 Hz, 2H, Harom), 7.05–6.99 (m, 2H, Harom), 6.97 (t, J = 7.3 Hz, 1H, Harom), 6.07 (s, 2H, NH2), 4.57 (s, 2H, COCH2), 3.73 (q, J = 7.0 Hz, 2H, N3-CH2), 1.05 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 167.9 (CON), 160.3 (C6), 157.9 (Carom), 150.2 (CO), 149.6 (CO), 129.4 (2C, Carom), 121.0 (Carom), 114.7 (2C, Carom), 85.9 (C5), 66.9 (COCH2), 34.4 (N3-CH2), 13.2 (CH3). IR (cm−1): ṽ = 3,364 (w), 3,321 (w), 3,273 (w), 3,170 (w), 1,716 (m), 1,689 (m), 1,643 (m), 1,574 (s), 1,487 (s), 1,458 (m), 1,379 (w), 1,339 (w), 1,279 (w), 1,249 (w), 1,221 (s), 1,167 (w), 1,111 (w), 1,084 (w), 1,065 (w), 924 (w), 830 (w), 791 (w), 753 (s), 6,966 (w), 635 (m), 578 (w), 534 (s), 508 (m), 440 (w). HRMS (ESI-QTOF) calculated for C14H16N4O4 [M+H]+: 305.1244; found: 305.1253.
Yield: quantitative (white solid); mp 265–267°C; 1H NMR (500 MHz, DMSO-d6) δ 10.37 (s, 1H, N1-H), 8.42 (s, 1H, CONH), 7.28 (t, J = 7.4 Hz, 2H, Harom), 7.25–7.21 (m, 2H, Harom), 7.18 (t, J = 7.2 Hz, 1H, Harom), 5.51 (s, 2H, N3–NH2), 3.72 (q, J = 6.9 Hz, 2H, CH2), 2.98 (dd, J = 13.4, 6.2 Hz, 1H, CH), 2.78–2.68 (m, 1H, CH), 2.57–2.51 (m, 1H, CH), 1.04 (t, J = 7.0 Hz, 3H, CH3), 1.00 (d, J = 6.8 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 175.4 (CON), 160.3 (C6), 149.6 (CO), 149.5 (CO), 140.1 (Carom), 128.9 (2C, Carom), 128.1 (2C, Carom), 125.9 (Carom), 87.5 (C5), 41.0 (CCH3), 34.4 (N3-CH2), 16.8 (CH3), 13.2 (CH3). IR (cm−1): ṽ = 3,354 (w), 3,318 (w), 3,178 (w), 3,082 (w), 3,022 (w), 3,002 (w), 2,975 (w), 2,938 (w), 2,875 (w), 1,723 (s), 1,632 (s), 1,552 (s), 1,492 (s), 1,457 (s), 1,426 (s), 1,378 (m), 1,331 (w), 1,299 (m), 1,226 (w), 1,181 (w), 1,160 (w), 1,116 (w), 1,044 (w), 948 (w), 759 (s), 745 (m), 698 (s), 659 (m), 543 (s), 505 (s). HRMS (ESI-QTOF) calculated for C16H20N4O3 [M+H]+: 317.1608; found: 317.1617.
Yield: 87% (off-white solid); mp > 320°C; 1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H, N1-H), 8.86 (s, 1H, CONH), 7.99–7.91 (m, 2H, Harom), 7.56–7.51 (m, 1H, Harom), 7.47 (t, J = 7.5 Hz, 2H, Harom), 6.06 (s, 2H, NH2), 3.75 (q, J = 7.0 Hz, 2H, N3-CH2), 1.06 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (126 MHz, DMSO) δ 166.4 (CON), 160.5 (C6), 150.4 (CO), 149.7 (CO), 134.5 (Carom), 131.1 (Carom), 128.0 (Carom), 127.8 (Carom), 87.1 (C5), 34.4 (N3-CH2), 13.3 (CH3). IR (cm−1): ṽ = 3,302 (w), 3,166 (w), 3,061 (w), 2,976 (w), 1,718 (m), 1,627 (m), 1,552 (s), 1,504 (s), 1,481 (s), 1,456 (s), 1,426 (s), 1,381 (m), 1,334 (w), 1,299 (m), 1,165 (w), 1,047 (w), 926 (w), 883 (w), 797 (m), 760 (m), 692 (m), 657 (m), 544 (s), 503 (m), 473 (m), 445 (w). HRMS (ESI-QTOF) calculated for C13H14N4O3 [M+H]+: 275.1139; found: 275.1142.
Yield: 80% (white solid); mp > 320°C; 1H NMR (500 MHz, DMSO-d6) δ 10.39 (s, 1H, N1-H), 8.58 (s, 1H, CONH), 7.35–7.31 (m, 2H, Harom), 7.28 (m, 2H, Harom), 7.23–7.19 (m, 1H, Harom), 5.90 (s, 2H, NH2), 3.71 (q, J = 7.0 Hz, 2H, N3-CH2), 3.56 (s, 2H, CH2), 1.03 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 170.6 (CON), 160.5 (C6), 150.1 (CO), 149.7 (CO), 136.6 (Carom), 129.4 (Carom), 128.2 (Carom), 126.3 (Carom), 87.5 (C5), 42.1 (COCH2), 34.5 (N3-CH2), 13.4 (CH3). IR (cm−1): ṽ = 3,349 (w), 3,297 (w), 3,184 (w), 3,065 (w), 2,980 (w), 2,909 (w), 2,885 (w), 1,729 (m), 1,638 (s), 1,547 (s), 1,483 (s), 1,421 (s), 1,331 (m), 1,294 (m), 1,216 (w), 1,180 (m), 1,155 (m), 1,031 (w), 963 (w), 926 (w), 793 (w), 758 (s), 694 (s), 661 (m), 599 (s), 488 (s). HRMS (ESI-QTOF) calculated for C14H16N4O3 [M+H]+: 289.1295; found: 289.1304.
Yield: 81% (white solid); mp 278–281°C; 1H NMR (500 MHz, DMSO-d6) δ 10.34 (s, 1H, N1-H), 8.24 (s, 1H, CONH), 5.82 (s, 2H, NH2), 3.70 (q, J = 7.0 Hz, 2H, N3-CH2), 2.24–2.12 (m, 2H, COCH2), 1.55–1.45 (m, 3H, CH2 and CH), 1.34–1.23 (m, 2H, CH2), 1.19–1.11 (m, 2H, CH2), 1.03 (t, J = 7.0 Hz, 3H, CH3), 0.85 (d, J = 6.6 Hz, 6H, 2 × CH3). 13C NMR (DMSO, 126 MHz) δ 172.7 (CON), 160.6 (C6), 150.0 (CO), 149.7 (CO), 87.6 (C5), 38.5 (CH2), 35.4 (CH2), 34.5 (N3-CH2), 27.4 (CH2), 26.7 (CH), 25.4 (CH2), 22.7 (2 × CH3), 13.4 (CH3). IR (cm−1): ṽ = 3,341 (w), 3,302 (w), 3,186 (w), 3,075 (w), 2,957 (w), 2,915 (w), 2,875 (w), 2,851 (w), 1,728 (m), 1,637 (s), 1,551 (s), 1,488 (s), 1,424 (s), 1,379 (m), 1,333 (m), 1,294 (m), 1,200 (w), 1,159 (m), 1,111 (w), 1,048 (w), 967 (w), 925 (w), 760 (s), 729 (w), 664 (m), 580 (s), 500 (s), 444 (m). HRMS (ESI-QTOF) calculated for C14H24N4O3 [M+H]+: 297.1921; found: 297.1924.
Filtrate was extracted with Et2O, dried over MgSO4 and the solvent removed in vacuo. Yield: 75% (off-white solid); mp > 320°C; 1H NMR (500 MHz, DMSO-d6) δ 10.33 (s, 1H, N1-H), 7.77 (s, 1H, CONH), 5.64 (s, 2H, NH2), 3.71 (q, J = 7.0 Hz, 2H, N3-CH2), 1.97 (s, 3H, Hnoradamantane), 1.87 (d, J = 2.7 Hz, 6H, Hnoradamantane), 1.69–1.63 (m, 6H, Hnoradamantane), 1.04 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (DMSO, 126 MHz) δ 177.3 (CON), 160.2 (C6), 149.7 (CO), 149.6 (CO), 87.6 (C5), 38.6 (CH2noradamantane), 36.2 (CH2noradamantane), 34.3 (N3–CH2), 27.7 (4C, CHnoradamantane), 13.2 (CH3). IR (cm−1): ṽ = 3,478 (w), 3,428 (w), 3,289 (w), 3,165 (w), 3,067 (w), 2,984 (w), 2,909 (w), 2,853 (w), 1,718 (m), 1,622 (s), 1,545 (s), 1,507 (s), 1,486 (s), 1,446 (s), 1,372 (w), 1,330 (w), 1,291 (m), 1,244 (w), 1,184 (w), 1,161 (w), 1,110 (w), 1,042 (w), 989 (w), 927 (w), 760 (s), 701 (w), 653 (m), 542 (s), 499 (m). HRMS (ESI-QTOF) calculated for C17H24N4O3 [M+H]+: 333.1921; found: 333.1922.
Disadvantages of irritant and hazardous coupling procedures, long reaction times and moderate yields encouraged us to search for a new method to yield the desired 6-amino-5-carboxamidouracil derivatives. After initial experiments with various procedures, the coupling reagent COMU showed the most promising results. COMU, which was developed in 2009, does not contain a potentially explosive benzotriazole moiety, and is therefore safer than classical coupling reagents such as, for example, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate (HATU). COMU shows high solubility, is stable in typically used solvents, can be easily removed due to the water-solubility of its products, and may be used for a broad range of carboxylic acids and amines yielding the corresponding amides ((El-Faham et al., 2009; El-Faham and Albericio, 2010, 2011); Hjørringgaard et al., 2012).
The synthetic procedure which led to differently substituted 6-amino-5-carboxamidouracils is shown in Scheme 2. Diaminouracil derivatives and carboxylic acids were used as starting materials and subjected to amide coupling using COMU. N1-mono- and N1,N3-disubstituted 5,6-diaminouracil derivatives (14–20, Figure 2) were individually prepared (for details see Supporting Material Data Sheet 1) according to previously described procedures and (Maxwell and Salivar, 1952; Müller et al., 1993; Hockemeyer et al., 2004), while the employed carboxylic acid derivatives were in most cases commercially available.
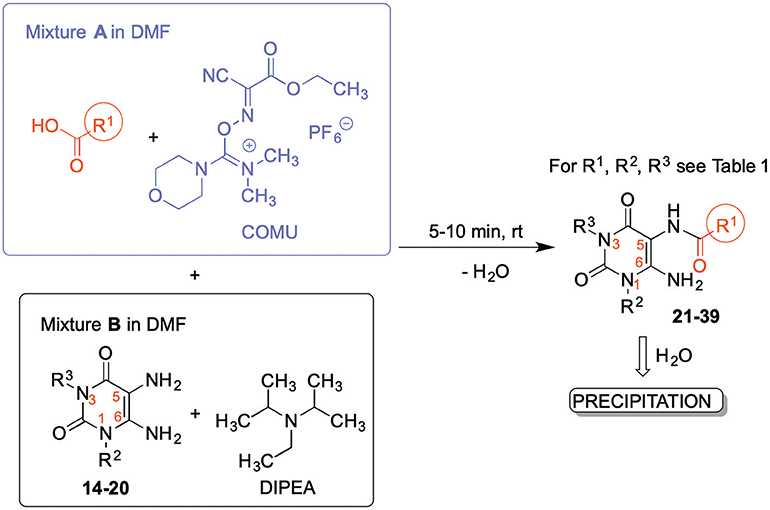
Scheme 2. Formation of 6-amino-5-carboxamidouracils using COMU as a coupling reagent.
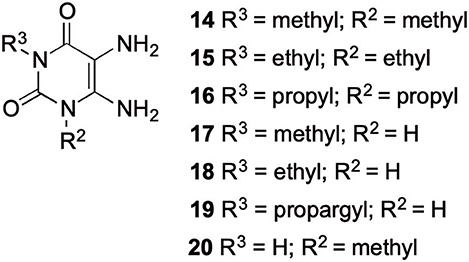
Figure 2. N1-Mono- and N1,N3-disubstituted 5,6-diaminouracil derivatives synthesized as starting materials for amide formation (for details see Supporting Material Data Sheet 1).
Amide formation with the coupling reagent required the adjustment of different parameters, including solvent, reaction time, temperature and base. With DMF, DIPEA and COMU the optimal conditions were found (Scheme 2). The reaction may also be performed in other solvents, such as CH2Cl2, ethyl acetate or tetrahydrofurane (MacMillan et al., 2013), however, DMF is preferred resulting in short reaction times, and, importantly, the product can easily be precipitated in high purity by the addition of water. This renders a tedious isolation and purification procedure dispensable.
Scheme 3 depicts the proposed reaction mechanism, which is based on the mechanism proposed for the synthesis of esters using COMU (Twibanire and Grindley, 2011). The first step is the nucleophilic attack of the carboxylic acid (A) at the uronium moiety of COMU (B) resulting in intermediate C. Decomposition of C, followed by addition of the resulting anion E to the carbonyl group of D and subsequent elimination of the urea derivative F leads to the activated carboxylic acid G. Finally, the corresponding amide derivative is formed by nucleophilic attack of an amine and elimination of the water-soluble side product H.
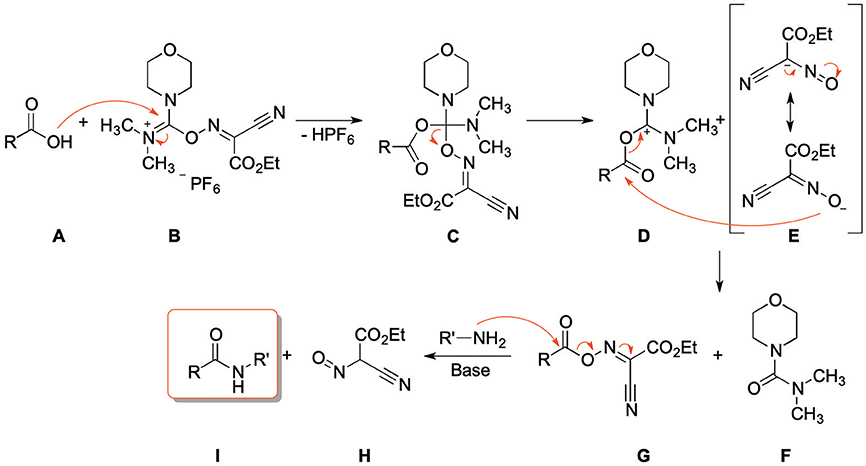
Scheme 3. Proposed reaction mechanism of amide coupling with COMU.
According to the proposed reaction mechanism, the carboxylic acid was converted to its active ester after dissolving it (1.0 equiv) together with COMU (1.1 equiv) in a minimum of DMF (mixture A, Scheme 2). Then, a solution of the 5,6-diaminouracil derivative (1.2 equiv) and diisopropylethylamine (DIPEA, 1.1 equiv) as a base dissolved in a minimum of DMF (mixture B) was added, followed by 5–10 min of stirring at room temperature (Scheme 2). Upon addition of cold water, the product precipitated. It was filtered off, washed with cold water, and dried under reduced pressure yielding the target compounds 21–39 (Table 1) in high purity and with yields ranging from 62 to 99%. Due to our interest in AR antagonists, we prepared various precursors for 8-substituted xanthines, which we could obtain in high yields and isolate by simple precipitation as shown for various examples (22–29). The 1,3-dipropyl derivatives 23 and 30 were formed in 87 and 85% yield, with 98 and 99% purity, respectively. Compound 23 is a precursor of the dual-acting A1 AR-opioid receptor ligands, such as 9. Compound 22 was obtained in 78% yield and provides access to the A3 AR antagonists PSB-11 (8b). Compound 24, the key compound for the synthesis of highly potent and selective A2B AR antagonists, was successfully condensed and precipitated. The carboxylic acid for the synthesis of 24 was not commercially available and was therefore prepared according to a literature procedure (Borrmann et al., 2009). To gain a purity of over 95% for 24, an additional chromatographic purification procedure was required. Compound 25, the precursor of the A1 AR antagonist rolofylline (4), which contains an 8-noradamantanyl substituent, and propyl residues on N1 and N3, precipitated in high purity (99%); fractional precipitation after cooling to 0°C was required to give a final yield of 79%. The less bulky and less hydrophobic cyclopentanecarboxylic acid was reacted with 5,6-diamino-3-methyluracil to obtain amide 26 as a precursor for 8-cyclopentyltheophylline (CPX), and was isolated in 69% yield with 99% purity. The additional substituent on N1 can be easily introduced subsequently by alkylation according to literature procedures (Hockemeyer et al., 2004). The precursor 29 of the A1 AR antagonist bamifylline (11), with methyl groups at both uracil nitrogen atoms, precipitated immediately in 85% yield and 99% purity. Compound 27, the precursor of the A2A AR antagonist and anti-Parkinson drug istradefylline (5), precipitated in 70% yield with 97% purity. Amide formation with 3-methoxycinnamic acid, carrying the styrene moiety, which is required for the preparation of the potent and selective A2A AR antagonists of the MSX series (6a-c), gave the 6-amino-5-carboxamidouracil precursor 28 in 83% isolated yield after precipiation.
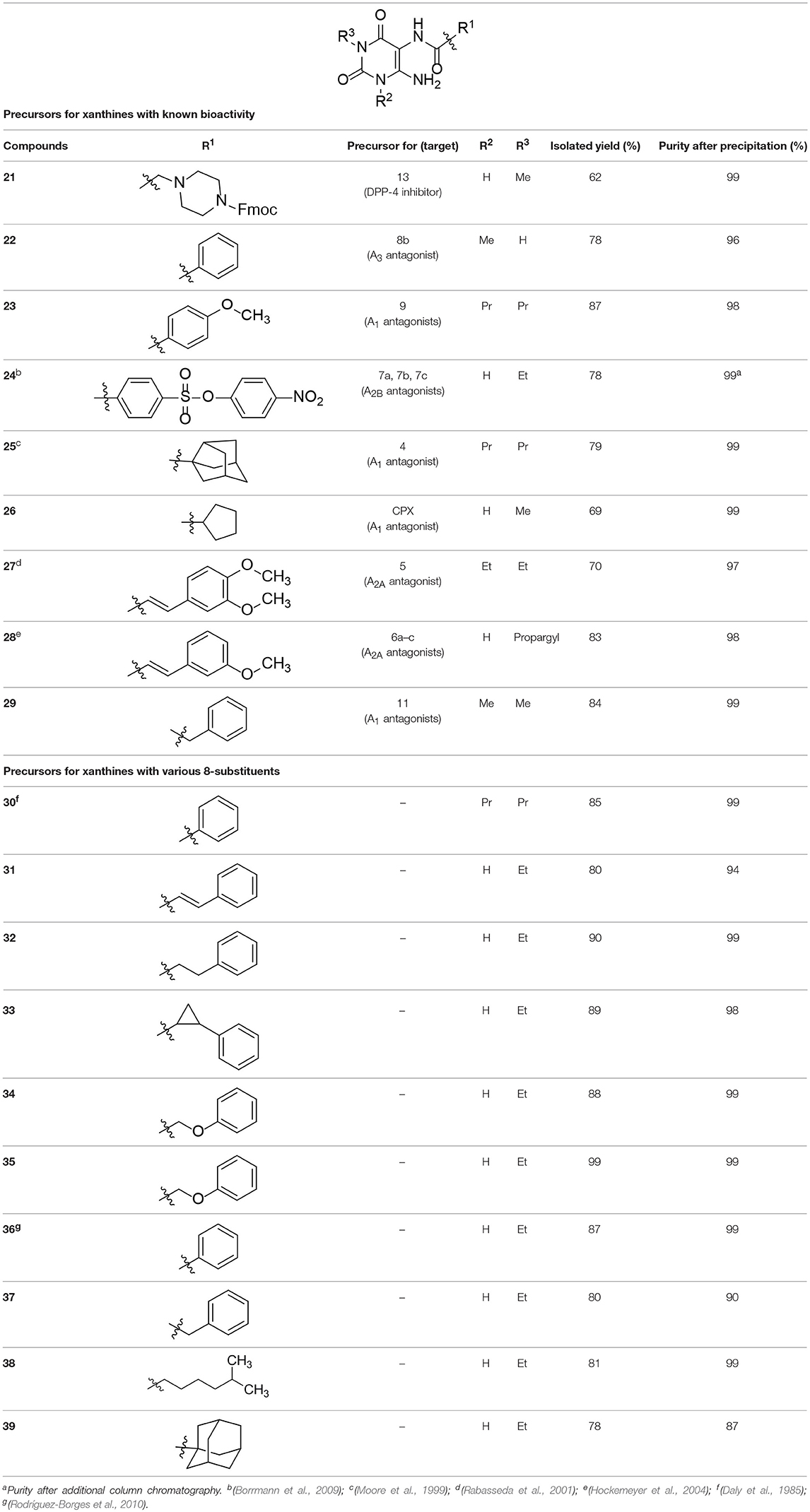
Table 1. Formation of 6-amino-5-carboxamidouracil derivatives.
To investigate the impact of different carboxylic acid derivatives regarding precipitation of the product, we used 3-ethyldiaminouracil and various carboxylic acids as a test system for the formation of differently substituted 6-amino-5-carboxamidouracils (Table 1). Compound 32, with a phenylpropionyl residue, was isolated in 90% yield. The analogous compound 33 containing a rigidified cyclopropyl ring gave a similar yield of 89%, as did the ether analog 34. The presence of an α-methyl group in compound 35 resulted in quantitative product formation and precipitation. The 6-amino-5-carboxamidouracil 38 bearing an alkyl residue was isolated in 81% yield with 99% purity.
Comparing all reactions, we observed the following trends: 1,3-disubstituted uracils could be formed best in case of a bulky, hydrophobic carboxylic acid derivative, which favors precipitation from the DMF/H2O solution. Reactions of N1-unsubstituted diaminouracils generally gave higher product yields, and the products were easily precipitated. The melting points of those products were high indicating the formation of intermolecular hydrogen bonds in the solid state, which was confirmed by the crystal structure of 32 (see below).
Since 5,6-diaminouracil carries two amino groups, the question arises, which one forms the amide bond (Yang et al., 2015). Due to literature reports, the 5-amino group is proposed to react (Sauer et al., 2000; Hayallah et al., 2002; Hockemeyer et al., 2004). We checked this assumption by NMR and small single molecule X-ray crystallography, comparing the NMR signals of 6-aminouracil, 6-amino-5-nitrosouracil, 5,6-diaminouracil, and 5-amino-6-carboxamidouracil. We additionally applied 2-dimensional NMR spectroscopy, namely heteronuclear multiple bond correlation (HMBC) and nuclear Overhauser enhancement spectroscopy (NOESY), for determining the structure of amide 25.
In literature, the product of the first reaction step has been described as a 5-nitroso derivative. Based on our NMR experiments, the 5-(hydroxyimino)-6-imino derivative is the tautomer that is present in chloroform employed as a solvent (Scheme 4). The chemical shift of the 5-amino group in compound 16 indicates a magnetic shielding of the hydrogen atoms giving the nitrogen atom a more nucleophilic character, which is in accordance with our regioselectivity studies.
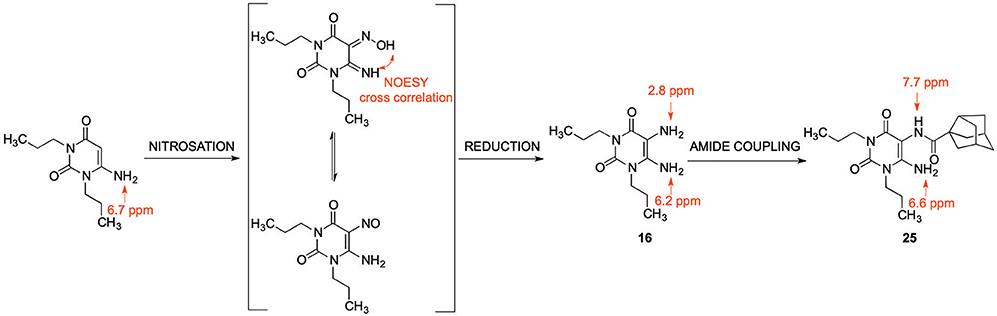
Scheme 4. NMR signals of 6-aminouracil derivatives with various substituents in the 5-position, and NOESY cross correlation for structure/tautomer analysis determined in chloroform-d1.
Finally, we tried to obtain a crystal structure of 25. Different crystallization experiments were performed but the crystallization of 25 has not been successful. Fortunately, compound 32, crystallized from DMSO solution at room temperature, yielding a crystal of the size 0.4 × 0.2 × 0.08 mm. Measurement and analysis of the resulting crystal structure using a Bruker X8-KappaApexII instrument showed a monocline crystal system within the space group P21. In accordance with the NMR experiment of 25 the crystal structure of 32 confirmed a regioselective amide coupling of the carboxylic acid with the 5,6-diaminouracil derivative in position 5. The crystal is mainly formed by intermolecular hydrogen bonds. π-Stacking or interaction with the solvent could not be observed. The most important intermolecular hydrogen bonds are summarized in Figure 3. All NH groups showed a donor functionalization and all oxygen atoms showed acceptor properties to surrounding molecules. Figure 3 visualizes these intermolecular interactions. The surrounding molecules are shaded while the intermolecular interactions are shown in turquoise. All bond lengths were in the expected range.
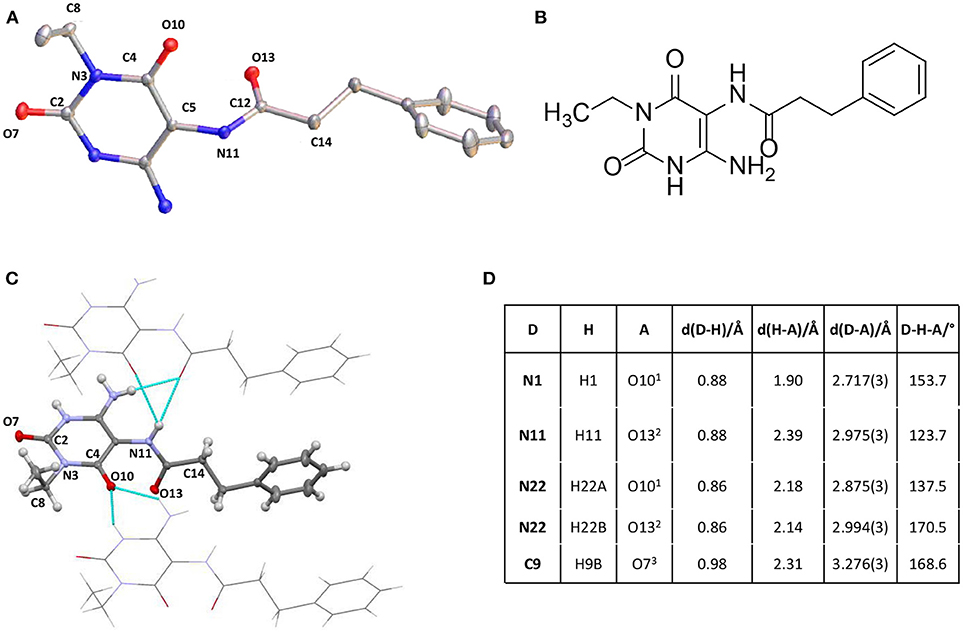
Figure 3. (A) Crystal structure of 6-amino-5-carboxamidouracil 32. (B) Structure of 32. (C) Intermolecular interactions building the crystal structure of 32. (D) Most important intermolecular hydrogen bonds of 32. Supporting Material Data Sheet 2.
In summary, we report on a new regioselective amide formation of 5,6-diaminouracil derivatives with carboxylic acids using the coupling reagent COMU which leads to the preparation of important precursors for xanthine derivatives. The reaction is completed after only 5–10 min of stirring at room temperature in DMF, followed by straightforward isolation of the formed amides by precipitation through the addition of water. After filtration, the 6-amino-5-carboxamidouracils were obtained in high isolated yields and showed in most cases purities of 90% or higher requiring no further chromatographic purification. The new procedure is advantageous with regard to reaction time and yields, and it avoids hazardous coupling or chlorinating reagents. In addition to several new derivatives, we synthesized the 6-amino-5-carboxamidouracil precursors of important, biologically active and literature-known xanthines utilizing the new method. The regioselectivity of the amide formation with the 5- rather than the 6-amino group of the uracil derivatives was proven by 2D-NMR spectroscopy and X-ray crystallography. The new regioselective amide coupling procedure allows the preparation of a variety of xanthine precursors. Moreover, the procedure will be well-suitable for automated and parallel synthesis.
DM performed most of the experiments. CM supervised the experiments. LW and MS had the idea to use COMU for the synthesis of xanthine precursors, and performed initial experiments elaborating reaction and workup conditions. GS determined the X-ray crystal structure. MS supervised experiments performed by LW. All authors contributed to writing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Jörg Hockemeyer for helpful discussions, and Marion Schneider, Annette Reiner, and Sabine Terhart-Krabbe for recording NMR and MS data. MS and LW were supported by the German Research Foundation DFG (SCHO 1593/1-1).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00056/full#supplementary-material
For details of synthetic procedures, crystallographic parameters, and NMR and IR spectra see Supporting Material Data Sheets 1 and 2.
Alciato, P., Cantone, P. A., Fico, D., Gagliardini, R., and Petrella, V. (1990). Bamifylline in the therapy of asthmatic syndromes. Efficacy and side effects vs delayed-action theophylline anhydride. Minerva Med. 81, 93–97.
Baraldi, P. G., Fruttarolo, F., Tabrizi, M. A., Romagnoli, R., and Preti, D. (2007). Novel 8-heterocyclyl xanthine derivatives in drug development–an update. Expert Opin. Drug. Discov. 2, 1161–1183. doi: 10.1517/17460441.2.9.1161
Baraldi, P. G., Tabrizi, M. A., Gessi, S., and Borea, P. A. (2008). Adenosine receptor antagonists: translating medicinal chemistry and pharmacology into clinical utility. Chem. Rev. 108, 238–263. doi: 10.1021/cr0682195
Baraldi, P. G., Tabrizi, M. A., Preti, D., Bovero, A., Romagnoli, R., Fruttarolo, F., et al. (2004). Design, synthesis, and biological evaluation of new 8-heterocyclic xanthine derivatives as highly potent and selective human A2B adenosine receptor antagonists. J. Med. Chem. 47, 1434–1447. doi: 10.1021/jm0309654
Basu, S., Barawkar, D. A., Ramdas, V., Patel, M., Waman, Y., Panmand, A., et al. (2017). Design and synthesis of novel xanthine derivatives as potent and selective A2B adenosine receptor antagonists for the treatment of chronic inflammatory airway diseases. Eur. J. Med. Chem. 134, 218–229. doi: 10.1016/j.ejmech.2017.04.014
Borrmann, T., Hinz, S., Bertarelli, D. C., Li, W., Florin, N. C., Scheiff, A. B., et al. (2009). 1-Alkyl-8-(piperazine-1-sulfonyl)phenylxanthines: development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J. Med. Chem. 52, 3994–4006. doi: 10.1021/jm900413e
Chen, J. F., Eltzschig, H. K., and Fredholm, B. B. (2013). Adenosine receptors as drug targets–what are the challenges? Nat. Rev. Drug. Disc. 12, 265–286. doi: 10.1038/nrd3955
Cheng, R. K. Y., Segala, E., Robertson, N., Deflorian, F., Doré, A. S., Errey, J. C., et al. (2017). Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 25, 1275–1285.e1274. doi: 10.1016/j.str.2017.06.012
Costante, R., Stefanucci, A., Carradori, S., Novellino, E., and Mollica, A. (2015). DPP-4 inhibitors: a patent review (2012–2014). Expert Opin. Ther. Pat. 25, 209–236. doi: 10.1517/13543776.2014.991309
Crepaldi, G., Carruba, M., Comaschi, M., Del Prato, S., Frajese, G., and Paolisso, G. (2007). Dipeptidyl peptidase 4 (DPP-4) inhibitors and their role in type 2 diabetes management. J. Endocrinol. Invest. 30, 610–614. doi: 10.1007/bf03346357
Daly, J. W., Padgett, W., Shamim, M. T., Butts-Lamb, P., and Waters, J. (1985). 1,3-Dialkyl-8-(p-sulfophenyl)xanthines: potent water-soluble antagonists for A1- and A2-adenosine receptors. J. Med. Chem. 28, 487–492. doi: 10.1021/jm00382a018
Doré, A. S., Robertson, N., Errey, J. C., Ng, I., Hollenstein, K., Tehan, B., et al. (2011). Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19, 1283–1293. doi: 10.1016/j.str.2011.06.014
Dungo, R., and Deeks, E. D. (2013). Istradefylline: first global approval. Drugs 73, 875–882. doi: 10.1007/s40265-013-0066-7
El-Faham, A., and Albericio, F. (2010). COMU: a third generation of uronium-type coupling reagents. J. Pept. Sci. 16, 6–9. doi: 10.1002/psc.1204
El-Faham, A., and Albericio, F. (2011). Peptide coupling reagents, more than a letter soup. Chem. Rev. 111, 6557–6602. doi: 10.1021/cr100048w
El-Faham, A., Subirós Funosas, R., Prohens, R., and Albericio, F. (2009). COMU: a safer and more effective replacement for benzotriazole-based uronium coupling reagents. Chem. Eur. J. 15, 9404–9416. doi: 10.1002/chem.200900615
El-Sabbagh, O. I., El-Sadek, M. E., El-Kalyoubi, S., and Ismail, I. (2007). Synthesis, DNA binding and antiviral activity of new uracil, xanthine, and pteridine derivatives. Arch. Pharm. 340, 26–31. doi: 10.1002/ardp.200600149
Feoktistov, I., Biaggioni, I., Polosa, R., and Holgate, S. T. (1998). Adenosine A2B receptors: a novel therapeutic target in asthma? Trends. Pharmacol. Sci. 19, 148–153. doi: 10.1016/S0165-6147(98)01179-1
Franco, R., Oñatibia-Astibia, A., and Martínez-Pinilla, E. (2013). Health benefits of methylxanthines in cacao and chocolate. Nutrients 5, 4159–4173. doi: 10.3390/nu5104159
Gallagher, M. (2004). Method for Improving Cognitive Function by Co-administration of a GABAB Receptor Antagonist and an Acetylcholinesterase Inhibitor (Baltimore: Michela Gallagher), US20050267077A1.
Glukhova, A., Thal, D. M., Nguyen, A. T., Vecchio, E. A., Jörg, M., Scammells, P. J., et al. (2017). Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 168, 867–877. doi: 10.1016/j.cell.2017.01.042
Hayallah, A. M., Sandoval-Ramírez, J., Reith, U., Schobert, U., Preiss, B., Schumacher, B., et al. (2002). 1,8-Disubstituted xanthine derivatives: synthesis of potent A2B-selective adenosine receptor antagonists. J. Med. Chem. 45, 1500–1510. doi: 10.1021/jm011049y
Hinz, S., Alnouri, W. M., Pleiss, U., and Müller, C. E. (2018). Tritium-labeled agonists as tools for studying adenosine A2B receptors. Purinergic Signal. 14, 223–233. doi: 10.1007/s11302-018-9608-5
Hjørringgaard, C. U., Brust, A., and Alewood, P. F. (2012). Evaluation of COMU as a coupling reagent for in situ neutralization Boc solid phase peptide synthesis. J. Pept. Sci. 18, 199–207. doi: 10.1002/psc.1438
Hockemeyer, J., Burbiel, J. C., and Müller, C. E. (2004). Multigram-scale syntheses, stability, and photoreactions of A2A adenosine receptor antagonists with 8-styrylxanthine structure: potential drugs for Parkinson's disease. J. Org. Chem. 69, 3308–3318. doi: 10.1021/jo0358574
Ilaš, J., Pečar, S., Hockemeyer, J., Euler, H., Kirfel, A., and Müller, C. E. (2005). Development of spin-labeled probes for adenosine receptors. J. Med. Chem. 48, 2108–2114. doi: 10.1021/jm049513x
Jacobson, K. A. (2009). Functionalized congener approach to the design of ligands for G protein-coupled receptors (GPCRs). Bioconjug. Chem. 20, 1816–1835. doi: 10.1021/bc9000596
Jacobson, K. A., Kiriasis, L., Barone, S., Bradbury, B. J., Kammula, U., Campagne, J. M., et al. (1989). Sulfur-containing 1,3-dialkylxanthine derivatives as selective antagonists at A1-adenosine receptors. J. Med. Chem. 32, 1873–1879. doi: 10.1021/jm00128a031
Jacobson, K. A., Shi, D., Gallo-Rodriguez, C., Manning, M., Müller, C., Daly, J. W., et al. (1993). Effect of trifluoromethyl and other substituents on activity of xanthines at adenosine receptors. J. Med. Chem. 36, 2639–2644. doi: 10.1021/jm00070a007
Kim, Y. C., Ji, X., Melman, N., Linden, J., and Jacobson, K. A. (2000). Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. J. Med. Chem. 43, 1165–1172. doi: 10.1021/jm990421v
Kondo, T., and Mizuno, Y. (2015). A long-term study of istradefylline safety and efficacy in patients with Parkinson's disease. Clin. Neuropharmacol. 38, 41–46. doi: 10.1097/wnf.0000000000000073
Köse, M., Gollos, S., Karcz, T., Fiene, A., Heisig, F., Behrenswerth, A., et al. (2018). Fluorescent-labeled selective adenosine A2B receptor antagonist enables competition binding assay by flow cytometry. J. Med. Chem. 61, 4301–4316. doi: 10.1021/acs.jmedchem.7b01627
Leone, R. D., Lo, Y.-C., and Powell, J. D. (2015). A2AR antagonists: next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 13, 265–272. doi: 10.1016/j.csbj.2015.03.008
LeWitt, P. A., Guttman, M., Tetrud, J. W., Tuite, P. J., Mori, A., Chaikin, P., et al. (2008). Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson's disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 63, 295–302. doi: 10.1002/ana.21315
Liu, W., Chun, E., Thompson, A. A., Chubukov, P., Xu, F., Katritch, V., et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337:232. doi: 10.1126/science.1219218
MacMillan, D. S., Murray, J., Sneddon, H. F., Jamieson, C., and Watson, A. B. J. (2013). Evaluation of alternative solvents in common amide coupling reactions: replacement of dichloromethane and N,N-dimethylformamide. Green Chem. 15, 596–600. doi: 10.1039/c2gc36900a
Maxwell, C. E., and Salivar, C. J. (1952). Method of Preparing 4-Aminouracils. Brooklyn, NY: Pfizer & Co., Inc.
Moore, A. G., Schow, S. R., Lum, R. T., Nelson, M. G., and Melville, C. R. (1999). Convenient one-pot synthesis of 8-substituted xanthines from 6-amino-5-nitrosouracils. Synthesis 7, 1123–1126. doi: 10.1055/s-1999-3513
Müller, C., and Jacobson, K. A. (2011). Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 200, 151–199. doi: 10.1007/978-3-642-13443-2_6
Müller, C. E., Baqi, Y., Hinz, S., and Namasivayam, V. (2018). “Medicinal chemistry of A2B adenosine receptors,” in The Adenosine Receptors, eds. P. A. Borea, K. Varani, S. Gessi, S. Merighi, and F. Vincenzi (Cham: Springer International Publishing), 137–168.
Müller, C. E., Diekmann, M., Thorand, M., and Ozola, V. (2002). [3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H]PSB-11), a novel high-affinity antagonist radioligand for human A3 adenosine receptors. Bioorg. Med. Chem. Lett. 12, 501–503. doi: 10.1016/S0960-894X(01)00785-5
Müller, C. E., Hockemeyer, J., Tzvetkov, N., and Burbiel, J. (2008). New 8-Ethinylxanthine Compounds Useful for Treating e.g. Parkinson's Disease, Catalepsy, Dystonia, Dyskinetic Syndrome, Restless Legs Syndrome, Migraine, Pain, Dementia, Neurodegenerative Disorders, Alcohol Withdrawal, and Stroke (Monheim), WO002008077557A1.
Müller, C. E., Shi, D., Manning Jr., M., and Daly, J. W. (1993). Synthesis of paraxanthine analogs (1, 7-disubstituted xanthines) and other xanthines unsubstituted at the 3-position: structure-activity relationships at adenosine receptors. J. Med. Chem. 36, 3341–3349. doi: 10.1021/jm00074a015
Nieber, K. (2017). The impact of coffee on health. Planta Med. 83, 1256–1263. doi: 10.1055/s-0043-115007
Oñatibia-Astibia, A., Franco, R., and Martínez-Pinilla, E. (2017). Health benefits of methylxanthines in neurodegenerative diseases. Mol. Nutr. Food Res. 61, 1–14. doi: 10.1002/mnfr.201600670
Ozola, V., Thorand, M., Diekmann, M., Qurishi, R., Schumacher, B., Jacobson, K. A., et al. (2003). 2-Phenylimidazo[2,1-i]purin-5-ones: Structure-activity relationships and characterization of potent and selective inverse agonists at human A3 adenosine receptors. Bioorg. Med. Chem. 11, 347–356. doi: 10.1016/s0968-0896(02)00456-x
Park, A., and Stacy, M. (2012). Istradefylline for the treatment of Parkinson's disease. Expert Opin. Pharmacother. 13, 111–114. doi: 10.1517/14656566.2012.643869
Rabasseda, X., Sorbera, L., Martin, L., Leeson, P., and Castañer, J. (2001). Monographs-KW-6002. Antiparkinsonian, antidepressant, adenosine A2 antagonist. Drug Fut. 26, 20–24.
Rodríguez-Borges, J. E., García-Mera, X., Balo, M. C., Brea, J., Caamaño, O., Fernández, F., et al. (2010). Synthesis and pharmacological evaluation of novel 1,3,8- and 1,3,7,8-substituted xanthines as adenosine receptor antagonists. Bioorg. Med. Chem. 18, 2001–2009. doi: 10.1016/j.bmc.2010.01.028
Sauer, R., Maurinsh, J., Reith, U., Fülle, F., Klotz, K. N., and Müller, C. E. (2000). Water-soluble phosphate prodrugs of 1-propargyl-8-styrylxanthine derivatives, A2A-selective adenosine receptor antagonists. J. Med. Chem. 43, 440–448. doi: 10.1021/jm9911480
Scammells, P. J., Baker, S. P., Belardinelli, L., and Olsson, R. A. (1994). Substituted 1,3-dipropylxanthines as irreversible antagonists of A1 adenosine receptors. J. Med. Chem. 37, 2704–2712. doi: 10.1021/jm00043a010
Singh, S., and Yadav, R. (2016). Bronchial asthma: a global health problem. Int. J. Pharm. Bio. Sci. 7, 163–173. doi: 10.22376/ijpbs.2016.7.4.p163-173
Slawsky, M. T., and Givertz, M. M. (2009). Rolofylline: a selective adenosine 1 receptor antagonist for the treatment of heart failure. Expert Opin. Pharmacother. 10, 311–322. doi: 10.1517/14656560802682213
Sun, B., Bachhawat, P., Chu, M. L., Wood, M., Ceska, T., Sands, Z. A., et al. (2017). Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. U.S.A. 114, 2066–2071. doi: 10.1073/pnas.1621423114
Twibanire, J. A. K., and Grindley, T. B. (2011). Efficient and controllably selective preparation of esters using uronium-based coupling agents. Org. Lett. 13, 2988–2991. doi: 10.1021/ol201005s
Yan, L., Bertarelli, D. C. G., Hayallah, A. M., Meyer, H., Klotz, K. N., and Müller, C. E. (2006). A new synthesis of sulfonamides by aminolysis of p-nitrophenylsulfonates yielding potent and selective adenosine A2B receptor antagonists. J. Med. Chem. 49, 4384–4391. doi: 10.1021/jm060277v
Keywords: amide, COMU, purine, uracil, xanthine, X-ray crystal structure
Citation: Marx D, Wingen LM, Schnakenburg G, Müller CE and Scholz MS (2019) Fast, Efficient, and Versatile Synthesis of 6-amino-5-carboxamidouracils as Precursors for 8-Substituted Xanthines. Front. Chem. 7:56. doi: 10.3389/fchem.2019.00056
Received: 15 November 2018; Accepted: 21 January 2019;
Published: 18 February 2019.
Edited by:
Naohiko Yoshikai, Nanyang Technological University, SingaporeReviewed by:
Mariafrancesca Fochi, University of Bologna, ItalyCopyright © 2019 Marx, Wingen, Schnakenburg, Müller and Scholz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christa E. Müller, Y2hyaXN0YS5tdWVsbGVyQHVuaS1ib25uLmRl
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.