 Diamantoula Maniaki
Diamantoula Maniaki Evangelos Pilichos
Evangelos Pilichos Spyros P. Perlepes
Spyros P. Perlepes- Department of Chemistry, University of Patras, Patras, Greece
The area of 3d-metal coordination clusters that behave as Single-Molecule Magnets (SMMs) is now quite mature within the interdisciplinary field of Molecular Magnetism. This area has created a renaissance in Inorganic Chemistry. From the synthetic Inorganic Chemistry viewpoint, the early years of “try and see” exercises (1993–2000) have been followed by the development of strategies and strict approaches. Our review will first summarize the early synthetic efforts and routes for the preparation of polynuclear 3d-metal SMMs, and it will be then concentrated on the description of the existing strategies. The former involve the combination of appropriate 3d-metal-containing starting materials (simple salts with inorganic anions, metal cardoxylates, and pre-formed carboxylate clusters, metal phosphonates) and one or two primary organic ligands; the importance of the end-on azido group as a ferromagnetic coupler in 3d-metal SMM chemistry will be discussed. The utility of comproportionation reactions and the reductive aggregation route for the construction of manganese SMMs will also be described. Most of the existing strategies for the synthesis of SMMs concern manganese. These involve substitution of carboxylate ligands in pre-formed SMMs by other carboxylate or non-carboxylate groups, reduction procedures for the {} SMMs, spin “tweaking,” “switching on” SMM properties upon conversion of low-spin clusters into high-spin ones, ground-state spin switching and enhancing SMM properties via targeted structural distortions, the use of radical bridging ligands and supramolecular approaches. A very useful strategy is also the “switching on” of SMM behavior through replacement of bridging hydroxide groups by end-on azido or isocyanato ligands in clusters. Selected examples will be mentioned and critically discussed. Particular emphasis will be given on the criteria for the choice of ligands.
Introduction
Today there is a multi-billion euro industry concerning magnets due to their applications in many useful and modern technologies, such as televisions, credit/debit/ATM cards, motors, computer hard drives, switches and medical equipment, among others. Magnetism has its roots in Ancient Greece; in the province of Magnesia at central Greece, the first discovery of the attraction between lodestone and iron was made. This, however, is a matter of debate. The first observations were made near the city of “Magnesia” (close to the Sipylum mountain) in the old, Asian Minor Greek province of Lydia. This old city was near the modern city Manisa, close to Smirna (Izmir). Sophocles and other Ancient Greek writers reported on the “Lydian stone” in reference to the lodestone (Guimaraes, 2005). There is also a strong piece of evidence that Chinese sailors used the compass. The principles of Magnetism were established through the excellent work of Ampeére, Oersted, Faraday, Maxwell and Pierre Curie, just to name few legendary scientists who contributed in this field of Physics. The discovery of the electron, the seminal ideas of Lewis who wrote simple electronic structures for molecules and ions, and the development of quantum mechanics provided scientists with the appropriate tools for the in-depth understanding of the magnetic properties of the matter. Until ~1960, the popular magnets consisted mainly of metallic alloys and ceramics, e.g., Alnico and ferrites.
Chemists entered the field in the late 50s developing the field of Magnetochemistry. They used experimentally determined magnetic moments and their variation with temperature to draw chemical conclusions and make structural proposals in molecular compounds. Famous examples include: (i) the distinction between the two stereochemistries (tetrahedral, square planar) in 4-coordinate Ni(II) complexes based on the value of the effective magnetic moment, and (ii) the determination by Bleaney and Bowers of the singlet-triplet gap in copper(II) acetate hydrate, before the crystallographic determination of its dinuclear [Cu2(O2CMe)4(H2O)2] structure.
Molecular Magnetism (Kahn, 1993) came into the scientific scene in the early 80s. It is an interdisciplinary field in which chemists, physicists, material scientists, and chemical engineers, both experimentalists and theoreticians, actively participate. A NATO ASI in Italy in 1983 is widely considered as the beginning of the Molecular Magnetism era. The chemists presented magnetic properties of dinuclear, polynuclear, and polymeric molecular species, whereas the contribution from physicists was based on infinite atom-based arrays with a special focus on 1D systems which were popular at that time (mainly due to the availability of spin Hamiltonians which could be solved). The ASI, which gave the birth of a common scientific language between the two communities, convinced (a) chemists to dare to synthesize complex molecular compounds, and (b) physicists that it was possible to discover new magnetic phenomena working with molecule-based materials (Benelli and Gatteschi, 2015). The development of Molecular Magnetism has been tremendous since then. Milestones include-among others-the synthesis of molecular ferrimagnets, examples of molecular ferromagnets, the discovery that pure organic matter can order ferromagnetically, the attainment of room temperature molecular magnets, the development of spin-crossover systems, the discovery that the magnetic state of several molecular magnets can be photoinduced in a reversible manner, the isolation of systems exhibiting a coexistence of magnetic and other (e.g., conductivity) properties, the development of “magnetic nanowires” (Single-Chain Magnets) and the revolution with Single-Molecule Magnets (Coronado and Gatteschi, 2006; Coronado and Dunbar, 2009; Dunbar, 2012; Palacio, 2014; Benelli and Gatteschi, 2015). Some synthetic aspects of 3d-metal coordination clusters that behave as SMMs are presented in this Review.
An important milestone in the field of Molecular Magnetism was the discovery of the magnetic properties of complex [O12(O2CMe)16(H2O)4] (1) (Caneschi et al., 1991; Sessoli et al., 1993a), which indicated that this molecule can be considered as a small piece of manganese oxide whose further growth is inhibited by the presence of the acetate groups. The molecule possesses 12 spin centers; the 8 MnIII atoms have their spins up and the 4 MnIV ones down and this coupling scheme result in a S = 10 spin ground state (8 × 2 – 4 × 3/2 = 10). The impressive discovery was that, at very low temperatures, the magnetization relaxes slowly and the complex displays hysteresis (of molecular origin) like a tiny magnet. Most remarkably, a negative value for the axial zero-field splitting, D, results in the loss of the degeneracy of the corresponding MS sublevels, and so the MS = +10 and MS = −10 levels become lowest in energy. Since the selection rule is ΔMS = ±1, an energy barrier of U = S2 |D| results for “spin up” to “spin down” conversion in the ground state. In the case of 1, the S and D values are 10 and −0.5 cm−1, respectively; these values give a U ≈ 50 cm−1 and this results in magnetic blocking below 4 K, as proved by magnetic hysteresis loops. The behavior of 1 gave the idea that information could in the future be stored as the magnetization direction in discrete molecules. Assuming that a diameter for each bit is ~1 nm, a dense array of molecules might achieve a very high surface density (~200,000 gigabits/in2), and this is an advantage of several orders of magnitude over the existing magnetic alloy film technologies, a very important consequence for computer hard drives (Beltran and Long, 2005). The ability of a single molecule to behave as a tiny magnet represents another approach, the so-called “bottom-up” approach, to nanoscale magnetic materials, in which the need for smaller and smaller magnetic particles increases continuously (the so-called “top-down” approach). The “bottom-up” route provides this scientific area with many advantages of molecular chemistry, for example good solubility (and not colloid formation observed in magnetic nanoparticles), monodispersity, crystallinity, and avoidance of close contacts between the magnetic cores due to the intervention by an organic shell from the ligands (Bagai and Christou, 2009).
Polynuclear complexes (or coordination clusters) with properties similar to those of 1 are called Single-Molecule Magnets (SMMs); however, some scientists prefer the term Molecular Nanomagnets. In brief, SMMs are individual polynuclear molecules that function as magnets (i.e., they display magnetization hysteresis) below a characteristic blocking temperature, TB (which depends on the sweep rate of the magnetic field). TB is the highest temperature at which an SMM displays hysteresis in plots of magnetization (M) vs. magnetic field (H). However, since the TB value depends strongly on the sweep rate of H, comparisons of blocking temperatures in different SMMs should be done with caution (Woodruff et al., 2013). In the now classical book of Gatteschi, Sessoli, and Villain (Gatteschi et al., 2006), the authors define TB as the temperature at which the time for the magnetization relaxation is 100 s. This property arises from a simultaneous presence of a high-spin ground state, S, and a large and negative Ising (or easy-axis) type of magnetoanisotropy, as measured by the parameter D. This combination gives a significant energy barrier to magnetization reversal (U), the maximum value of which is given by S2 |D| (see above) and (S2-1/4) |D| for integer and non-integer total ground state spins, respectively. However, quantum tunneling of the magnetization vector (QTM) (Friedman et al., 1996; Thomas et al., 1996) through the barrier via MS levels of higher energy leads to a smaller effective energy barrier (Ueff) than the calculated U. In addition to QTM, some SMMs display other interesting quantum properties such as quantum phase interference (Wernsdorfer and Sessoli, 1999; Lecren et al., 2005; Wernsdorfer et al., 2005), exchange-biased QTM (Wernsdorfer et al., 2002; Pinkowicz et al., 2015) and others with potential technological interest (Leuenberger and Loss, 2001; Affronte et al., 2007; Bogani and Wernsdorfer, 2008; Affronte, 2009; Urdampilleta et al., 2011). For all these reasons, SMMs are candidates for a number of specialized applications (Bagai and Christou, 2009). Excellent books (Gatteschi et al., 2006; Benelli and Gatteschi, 2015) and reviews (Gatteschi and Sessoli, 2003; Beltran and Long, 2005; Aromi and Brechin, 2006; Bagai and Christou, 2009; Wang et al., 2011; Milios and Winpenny, 2015) for the properties of transition-metal SMMs are available.
In the last 10 years or so, there has been strong evidences that the most important parameter when designing SMMs is the single-ion anisotropy which should be large. Thus the research focus has been shifted to lanthanides (Ln) and actinides (An) which, in general, have large anisotropies. In Ln(III) SMMs, the required electronic ground-state bistability is due to the [2J + 1] MJ microstates within the ground term 2S+1Lj which is spin-orbit-coupled (Woodruff et al., 2013). The advantages of working with An-based SMMs are their large magnetic anisotropy (like the LnIII ions) and the possibility for covalency (like the transition metal ions), advantages which, however, add some complexity (Meinhaus and Long, 2015). Very interesting reviews have been published on the SMM properties of dinuclear and polynuclear Ln(III) (Sessoli and Powell, 2009; Guo et al., 2011; Rinehart and Long, 2011; Habib and Murugesu, 2013; Woodruff et al., 2013; Zhang et al., 2013a,b, 2015; Liddle and van Slageren, 2015; Pedersen et al., 2015; Pointillart et al., 2017; Liu et al., 2018), An (Meinhaus and Long, 2015), 3d/4f (Polyzou et al., 2013; Feltham and Brooker, 2014; Sharples and Collison, 2014; Liu et al., 2015; Rosado Piquer and Sañudo, 2015), and organometallic Ln(III) and An (Layfield, 2014) SMMs.
Before going to the main “menu” of this Review, we would like to say few words about the so-called Single-Ion Magnets (SIMs), which is currently a “hot” research area. These are mainly mononuclear d- or f-metal complexes whose magnetization relaxes slowly not because of cooperative exchange phenomena (as in dinuclear and polynuclear SMMs), but due to the intrinsic properties of the metal ion under an appropriate ligand field. The origin of the barrier here is the magnetic anisotropy. Excellent reviews on transition- (Craig and Murrie, 2015; Bar et al., 2016; Frost et al., 2016), 4f- (Woodruff et al., 2013; Gupta and Murugavel, 2018; Liu et al., 2018) and 5f-metal (Meinhaus and Long, 2015) SIMs are available in the literature. The very recent report of the SIM [()2Dy][B(C6F5)4] (2) (Goodwin et al., 2017a,b; Guo et al., 2017) with a record TB value of ~60 K (Ueff = 1277 cm−1 in zero field) is a revolutionary result, and it predicts a brilliant future in this area; is the 1,2,4-tri(tert-butyl)cyclopentadienide, i.e., {C5H2tBu3-1,2,4} with tBu = C(Me)3. It should be emphasized at this point that the SMM properties of many dinuclear and polynuclear Ln(III) complexes are in fact due to single-ion properties and such molecules can be regarded as a collection of SIMs. This is a consequence of the weak or very weak LnIII…LnIII exchange interactions due to the well-known internal character of the 4f orbitals which are severely shielded from the environment; however, most LnIII ions easily produce large anisotropy due to unquenched orbital moment.
The above mentioned developments in the areas of SMMs and SIMs created an explosive growth in synthetic Inorganic (both coordination and organometallic) Chemistry in the first quarter-century of this new “chapter” in the “book” of Molecular Magnetism. New reaction schemes and new types of metal complexes have been discovered. This Review covers the most important (a subjective opinion!) synthetic routes and strategies that have led to SMMs that belong to the family of 3d-metal coordination clusters. This means that synthetic aspects related to SIMs and other types of SMMs (4d-, 5d-, 4f-, 5f-, and d/f-metal-based) are not covered. The content of the Review is purely chemical and it is assumed that the readers have a basic knowledge of structural inorganic chemistry, as well as of the properties of SMMs and the methods of their study. Structural and magnetic descriptions will be confined to the necessary minimum. To avoid long synthetic descriptions, balanced chemical equations (written using molecular-and not ionic-formulae) will be used. In the text, we shall try to explain the synthetic rationale and philosophy behind the reactions with particular emphasis on the choice of reactants (ligands and metal-containing starting materials). Synthetic schemes, routes and strategies for the preparation of 3d-metal SMMs have appeared in several books, reviews, and papers along with detailed structural and magnetic descriptions; this is, however, the first review which is concentrated exclusively on the synthetic and reactivity chemistry of 3d-metal SMMs. Since there are thousands of papers describing the syntheses and properties of polynuclear 3d-metals SMMs, it is inevitable that we cannot cover all the existing literature. Thus, we express our apologies to the scientists whose excellent research will not be cited here.
The popular Harris notation (Coxall et al., 2000) will be used to describe the ligands' coordination modes. The Harris notation describes the ligation mode as X.Y1Y2Y3…Yn, where X is the total number of metal centers bound by the ligand, and each of Y refers to the number of metal ions attached to the different donor atoms. The order of the Y groups follows the well-known Cahn-Ingold-Prelog priority rules; therefore, for the most ligands reported in this work O comes before N. From time to time the traditional η/μ notation will be also used.
Synthesis of 3d-Metal SMMs: General Considerations
The 3d-metal systems that have been used in the preparation of SMMs are V(III), V(IV), Mn(II/III/IV), Mn(III), Mn(II/III), Mn(III/IV), Fe(II), Fe(III), Co(II), Co(II/III), Ni(II), and Cu(II). The most effective and well-studied SMMs are those containing high-spin Mn(III). The presence of high-spin octahedral MnIII atoms in molecules is valuable for making SMMs; the reason is that this 3d4, Jahn-Teller elongated ion has a large easy-axis anisotropy (negative D) which is desirable. Thus, many of the routes and strategies below involve examples from Mn chemistry.
All 3d-metal SMMs are dinuclear or polynuclear complexes. We are doing a parenthesis here to mention that the exchange coupling, J, is an important parameter to consider in the preparation of SMMs. This should be strong, because otherwise the spin ground state is not energetically isolated. Single-molecule magnetism is a ground-state phenomenon, and thus in order SMM properties to be observed at a certain temperature, the ground state must be ideally occupied at this temperature. The “translation” of this is that the bridging ligand or ligands should propagate strong ferromagnetic exchange interactions between the metal centers resulting in high-spin ground states (large S values). However, large S values can also arise from competing antiferromagnetic interactions in some topologies that prevent (“frustrate”) exactly antiparallel spin alignments. Organic ligands discussed in this Review are shown in Figure 1. Many of these ligands can provide monoatomic bridges which often promote ferromagnetic exchange interactions.
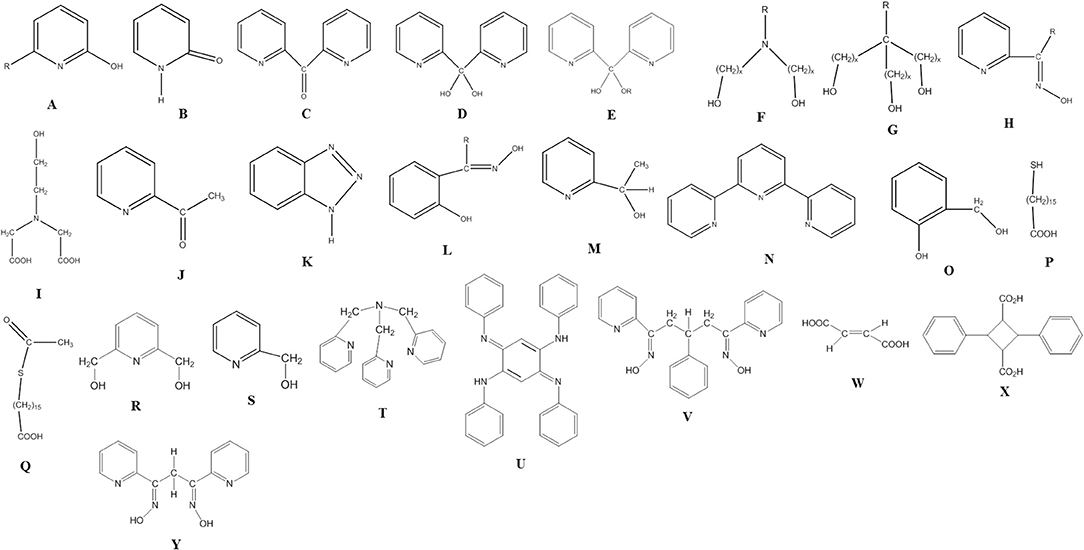
Figure 1. The structural formulae of some of the organic ligands discussed in this review (R = various).
A great development in synthetic chemistry during the twentieth century has been the ability of organic chemists to make large, complicated molecules in a systematic, step by step controlled manner, based on the known behavior of carbon and well-established reaction patterns. Sometimes, three or more different parts of a molecule are synthesized separately before the whole species is assembled in two or three final steps. Unfortunately, this is not possible for the transition-metal synthetic chemists, whose efforts are complicated by the non-predictable nature of the metal (e.g., variable oxidation state and coordination number), and the influence of reaction stoichiometry and solvent on product identity. For this reason, many synthetic schemes for SMMs are based on one- or two-step reactions. The lack of control in transition-metal chemistry has led to the adoption of the neologism “self-assembly” (Winpenny, 1999). The situation is becoming worse for the inorganic chemist who must crystallize her/his products (and SMM candidates) and determine their structures by single-crystal X-ray crystallography; this is often a difficult task. On the contrary, organic chemists can use all the “arsenal” of NMR and mass spectroscopic methods (which have undergone an explosive growth in lase decades) to firmly establish the structures, with no need to crystallize the compounds.
The Years of Innocence: “Try and See” Exercises
The discovery of the exciting magnetic properties of 1 ignited many efforts to synthesize dinuclear and polynuclear (coordination clusters) 3d-metal complexes with SMM properties. Most research groups, including our group, used simple 3d-metal “salts” with coordinating (e.g., Cl−, Br−, , SCN−, ,…) or non-coordinating (e.g., , , ,…) inorganic anions and flexible polydentate organic ligands. Such reaction schemes permit a “conversation” between the preferred coordination geometry of the metal ion and the coordination mode of the ligand. If exclusive formation of 5- or 6-membered chelating rings is avoided, the coordinative flexibility of the polydentate ligand is large. This allows the isolation of coordination clusters with a variety of unpredictable structures (and hopefully SMM properties), almost always incorporating further auxiliary bridging groups formed in situ, e.g., OH−, RO−, O2−, depending on the solvent and the “pH” of the reaction medium. Organic ligands possessing one, two, three or more ionizable hydrogens exhibit better bridging capabilities, if fully or in part deprotonated (with the help of an external base). The fate of the inorganic anion, present in the 3d-metal starting material, depends on the reaction conditions. It can participate in the cluster (either as a ligand or a counterion) or it can be absent. If there is evidence (or suspicion) for the presence of anionic cluster species in solution, bulky cations, e.g., Bun4N+, should be used to precipitate the product.
In a number of reaction schemes, the 3d-metal starting material contains a strongly basic anionic group, e.g., a carboxylate group (, ,…) or a β-diketonate ion (acac−, bzac−, dbm−,…), where Me = methyl, Ph = phenyl, acacH = acetylacetone, bzacH = benzoylacetone, and dbmH = dibenzoylmethane. If the primary organic ligand LHx (x = 1,2,3,…) is not in excess, then it can be deprotonated by the auxiliary organic anion while an amount of the latter remains in solution. This organic “blend,” which contains the deprotonated primary organic ligand and the anionic auxiliary organic group, leads often to dinuclear or polynuclear 3d-metal complexes which often exhibit interesting magnetic properties. This route is particularly useful when external bases for ligand deprotonation perplex the reaction, e.g., by precipitating amorphous hydroxido- or/and oxido-containing materials.
It would be naïve to believe that the above described “try and see” approach leads directly to SMMs. Considerable forethought and hindsight in the metal ions, their inorganic or auxiliary organic anions, primary organic ligands and reaction conditions (reaction ratio, solvent, “pH,” nature of external base, temperature, pressure, crystallization technique, etc.) are necessary to “switch on” or improve the SMM properties in the products.
The idea of preparing coordination clusters without strictly designing the product has been termed “serendipitous assembly” (Winpenny, 2002) and it is a productive means of preparing SMMs. The seminal review-type article by Winpenny has made inorganic chemists guiltless of the “crime,” the crime being their ability to isolate complexes with exciting molecular structures and impressive magnetic properties within few days!
Before proceeding to representative examples of the “try and see” route, we would like to mention that among the large number of primary organic ligands used in this chemistry are 2-hydroxypyridines (A)/2-pyridones (B) (Brechin et al., 1999; Winpenny, 2002, 2004), di-2-pyridyl ketone (C) and its related gem-diol (D) and hemiketal (E) ligands (Papaefstathiou and Perlepes, 2002; Stamatatos et al., 2009), amino diol-type ligands (F) (Tasiopoulos and Perlepes, 2008), tripodal alcohols (G) (Brechin, 2005), 2-pyridyl oximes (H) and related ligands (Milios et al., 2006), mixed hydroxyl-carboxylic acid ligands (e.g., I) (Powell et al., 1995), shown in Figure 1, and myriads of polydentate Schiff-base ligands (Hernández-Molina and Mederos, 2004).
A final point of general discussion is the importance of the presence of terminal ligands in the reaction media; these are necessary for the termination of a possible polymerization process which would lead to 1D, 2D, or 3D coordination polymers. In the chemistry of 3d-metal coordination clusters, this role is often fulfilled by the solvent molecules, the inorganic anions of the 3d-metal starting materials, the auxiliary organic ligands and the “chelating” part of the bridging organic ligands, or by the purposefully addition of a chelating ligand in the reaction systems.
Simple 3d-Metal Starting Materials With Inorganic Anions
Commonly used 3d-metal “salts” with inorganic anions include halides, nitrates, sulfates, perchlorates, etc. Ni(II) is promizing in the synthesis of SMMs due to its significant single-ion anisotropy. For example, the reaction between Ni(NO3)2 ·6H2O, 1,1,1-tris(hydroxymethyl)ethane (H3thme; G with × = 1 and R = Me in Figure 1) and NaOMe in MeOH gives complex [Ni4(H2thme)4(MeCN)4](NO3)4 (3), Equation (1) (Moragues-Cánovas et al., 2004). The initial reaction solution was evaporated to give a solid residue that was extracted with MeCN to give blue crystals of the product. The cation has a cubane structure. Each NiII atom is surrounded by two neutral oxygen atoms of H2thme− that act as terminal donors and three alkoxido-like deprotonated oxygen atoms from three ligands; thus, each monoanion behaves as a 3.311 ligand (AA in Figure 2); one MeCN molecule completes six coordination at each metal center. Intramolecular ferromagnetic NiII…NiII exchange interactions give an S = 4 ground state which is 40 cm−1 lower in energy from the first S = 3 excited state. Single-crystal magnetization vs. field (H) measurements reveal an SMM behavior below 0.5 K with fast magnetization relaxation due to QTM. The width of the hysteresis loop depends on the “history” of the sample, i.e., the starting magnetic state before varying the magnetic field.
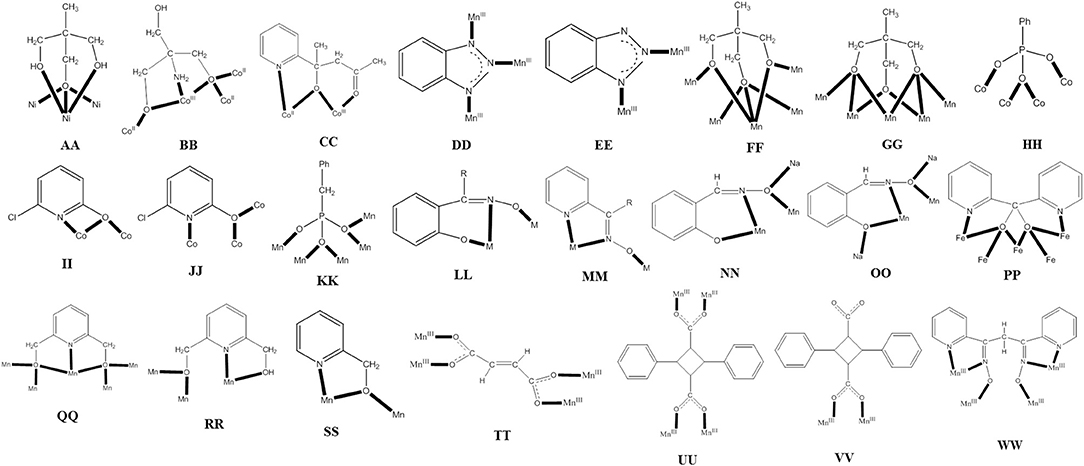
Figure 2. The coordination modes of some ligands discussed in this review; see the text for further details.
Another member of the ligand family G (see Figure 1), but in Co chemistry this time, gives a structurally and magnetically interesting mixed-valence coordination cluster. The D parameter for the CoII single ion in a tetragonally distorted octahedral environment is positive, because the M = ±1/2 ground state is lower in energy than the M = ±3/2 state, these two states being the Kramers' doublets that derive from the 4A2g ground state in ideal D4h symmetry due to spin-orbit coupling However, the orientation of the axial zero-field tensors on the CoII atoms, that are part of a cluster, may be favorable with respect to the molecular easy axis giving an overall negative D-value (Ising type anisotropy) and leading to SMM behaviors (Kostakis et al., 2012). The 1:1 reaction between Co(NO3)2 · 6H2O and 2-amino-2(hydroxymethyl)propane-1,3-diol (H3ahmpd; G with R = NH2 in Figure 1) in H2O, followed by increase of the pH to 7.5 by addition of (Bun4N)OH· 5H2O gives a solution; removal of water under vacuum and dissolution of the residue in MeOH leads to cluster [(Hahmpd)6(NO3)3(H2O)3](NO3)2 (4), Equation (2) (Ferguson et al., 2007). The yield is very low (~5%) but the reaction is reproducible. The cation is disk-like with the octahedral metal ions approximately coplanar and bridged exclusively by the alkoxido groups of the doubly deprotonated ligands, which adopt the 4.3201 coordination mode (BB in Figure 2). One alkoxido oxygen atom of each ligand is μ3, bridging the central CoII to one outer CoII and one CoIII, with the second alkoxido oxygen bridging one outer CoII to a CoIII center; the amine group is bound to CoIII. The coordination at each outer CoII is completed by a H2O molecule and a monodentate nitrato group. The complex is an exchange-biased SMM. For a well-isolated SMM, the first step in the hysteresis loop due to QTM should occur at H = 0 T. For 4, due to weak intermolecular interactions, this step is shifted to ±30 mT, suggesting a small antiferromagnetic exchange bias between cations of about 30 mT.
Several Co7 coordination clusters have a wheel-shaped (or disk-like) metal topology similar to that found in 4 (Kitos et al., 2011). The metal oxidation levels in these compounds are , CoIII, , and ; few of them are SMMs. In another example (Kitos et al., 2011), the reaction of Co(ClO4)2 · 6H2O and 2-acetylpyridine [(py)(me)CO; J in Figure 1] in the presence of (Bun4N)OMe (~1:1:1) in Me2CO at room temperature under aerobic conditions gave a brown solution, from which the green compound [CoIII(OH)6(L)6](ClO4)3 (5) was isolated in a moderate yield, Equation (3). The ligand L− which participates in the complex is the anion of 2-(pyridine-2-yl)pentane-2-ol-4-one, (py)(me)C(CH2COCH3)(O)−, formed in solution (in situ) through a metal ion-assisted, crossed-aldol reaction in Me2CO under strongly basic conditions (see Figure 3). The disk-like cation of 5 constists of a central octahedral CoIII atom linked to six peripheral CoII atoms (also octahedral) by six 3.3 hydroxido groups; the six CoII centers on the rim are held together by six 2.211 L− ligands (CC in Figure 2) and the oxygen atoms of the hydroxido groups. Each heptanuclear cation is a weak SMM but there are also intermolecular interactions (proven by crystallography) leading to a 3D architecture; this gives magnetization hysteresis at 5 K.
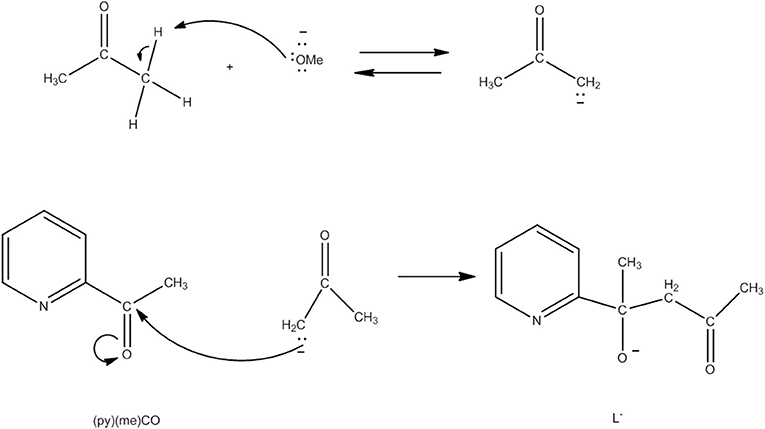
Figure 3. Simplified view of the mechanism that gives the ligand L− which participates in complex 5 (Schematic drawing inspired by Kitos et al., 2011).
When 3d-metal fluorides are used as starting materials in SMM chemistry, their insolubility in common organic solvents is a problem. In order to overcome this disadvantage, “melt” reactions are often used. For example, Collison, Brechin, and co-workers (Jones et al., 2004) mixed together and heated MnF3 and benzotriazole (Hbta; K in Figure 1), under nitrogen, to the melting point of the organic ligand (100°C). As the ligand melts, the MnF3 dissolved and the reaction takes place. After drying of the resulting black “melt” under vacuum and extraction of the solid mixture into MeOH at 50°C, black crystals of [O17(OH)8(OMe)4(bta)22(MeOH)14(H2O)2] (6) are obtained in a low yield (~10%), Equation (4). The molecule of 6 consists of an array of MnIII tetrahedra linked by 3.3-O2− ions, 3.3- and 3.2-OH− ions, 2.2-MeO− ions, and 3.111 and 2.110 bta− ligands (DD and EE in Figure 2). Single-crystal magnetic studies at low temperatures performed on 6 (S = 4, D = −0.90 cm−1) revealed the time- and temperature-dependent magnetization hysteresis loops indicative of SMM behavior. The SMM parameters are Ueff = 10.4 cm−1 and τ0 = 3 × 10−9 s. Below ~0.2 K, there is strong evidence for the presence of QTM in the ground state because the relaxation rates are independent of the temperature.
Simple 3d-Metal Carboxylates and Carboxylate Triangles and Butterflies as Starting Materials
Simple 3d-metal carboxylates are very popular in the synthesis of coordination clusters and SMMs, especially when the primary organic ligand possesses ionizable hydrogens. If the carboxylate to primary organic ligand reaction ratio is large enough, carboxylate groups are involved in the products either as terminal or (more often) as bridging ligands. We give two examples of Mn SMMs prepared using this route.
Complex [O4(OMe)4(O2CMe)3(mesao)6(H2O)2] (7) can be prepared successfully (and in a serendipitous way) by adding a variety of highly charged anions or cations to a reaction solution comprising Mn(O2CMe)2·4H2O, mesaoH2 and Et3N in MeOH, Equation (5); mesao2− is the dianion of the R = Me derivative of salicylaldoxime (L in Figure 1). Equivalent reactions without the presence of highly charged anions or cations affords hexanuclear Mn(III) complexes. It has been suggested (Inglis et al., 2011) that these ions simply enforce changes in the self-assembly process (thus avoiding the clusters) by their mere presence in the reaction mixtures. The metal topology can be described as a partial supertetrahedron in which the upper vertex is absent. The complex is an SMM with a Ueff value of ~21 cm−1 and-at the time of publication- represented the first chiral SMM obtained from achiral starting materials. Fitting of the dc magnetization data give the parameters S = 6, g = 1.98, and D = −0.60 cm−1.
The use of α-methyl-2-pyridine-methanol (mpmH; M in Figure 1) in Mn carboxylate chemistry has provided access to the first {Mn31} coordination cluster (Abasi et al., 2017). The 2:1:2 Mn(O2CPh)2·2H2O/rac-mpmH/Et3N reaction mixture in MeOH gives a dark red solution, from which complex [MnIVO24(OH)2(OMe)24(O2CPh)16(rac-mpm)2] (8) can be crystallized in 30% yield, Equation (6). The core can be described as a consecutive array of edge-sharing {Mn4(μ4-O)} tetrahedra and {Mn3(μ3-O)} triangles that are connected to each other via bridging oxido and methoxido groups. Complex 8 is not only structurally novel due to its nano-size dimensions, but also behaves as SMM with one of the largest Ueff values (~40 cm−1) yet reported for a 3d-metal SMM; moreover, it presents magnetization loops below 5 K, one of the highest TB temperatures at which molecular hysteresis has been observed for 3d-metal-based SMMs. Ac magnetic susceptibility measurements indicate an S = 23/2 ground state for this giant cluster.
A common synthetic route for preparing Mn coordination clusters and SMMs is the reaction of a pre-formed carboxylate cluster with a polydentate organic ligands (either bridging or chelating). Two frequently used Mn(II) carboxylate sources are the family of triangular oxido-centered complexes of the general formulation [Mn3O(O2CR)6L3]0,+ (Vincent et al., 1987), where L is a neutral monodentate ligand or solvent and R = Me, Ph,…, and the butterfly anionic complex (Bun4N)[O2(O2CPh)9(H2O)] (Wang et al., 1991). Two examples (from hundreds in the literature) are illustrated below.
The reaction of [O(O2CPh)6(py)3] (py = pyridine) and 1,1,1-tris(hydroxymethyl)ethane (H3thme; G with x = 1 and R = Me in Figure 1) in MeCN gives complex [O4(OH)2(O2CPh)12(thme)4(py)2] (9) in 20% yield, Equation (7). The molecule has a rod- or ladder-like structure and its core can be described as consisting of 10 edge-sharing {Mn3(μ3-O)} triangles or five edge-sharing {Mn4(μ3-O)2} units. The four triply deprotonated ligands sit above and below the {O4(OH)2} plane and are two types: two use two of their arms in a μ2-fashion with the third arm acting as a μ3-bridge adopting the 5.322 coordination mode (FF in Figure 2); the other two thme3− ligands have two μ3-arms and one μ2-arm adopting the 5.332 coordination mode (GG in Figure 2). The complex has an S = 7 ground state with g = 1.98 and D = −0.09 cm−1. There are low-lying excited states with S values greater than the S = 7 ground state. The appearance of single-crystal magnetization hysteresis loops and relaxation measurements shows this complex to be an SMM (Rajaraman et al., 2004).
The (Bun4N)[O2(O2CPh)9(H2O)]/terpy (2.5:1) reaction mixture in MeCN/PhCH2OH gives a brown solution, from which complex [MnIIMnIVO5(OCH2Ph)2(O2CPh)9(terpy)] (10) is subsequently isolated in ~50% yield, Equation (8); terpy is the rigid tridentate chelating ligand 2,2′:6′, 2″-terpypyridine (N in Figure 1) (Mishra et al., 2006). A Mn: terpy reaction ratio of ~6:1 or higher facilitates the formation of a polynuclear complex. The mixed MeCN/PhCH2OH solvent mixture is necessary to ensure solubility of the reagents and to provide the alkoxido ligands. The metal centers are held together by nine 2.11 benzoato ligands, one 5.5-O2− ion, four 3.3-O2− ions and two 2.2-PhCH2O− groups. The single terpy molecule is attached to the MnII center. The core topology (see Figure 4) is unique, comprising a {MnIIO2} butterfly and a {MnIVO4} cubane sharing a MnIII ion and the 5.5 (μ5) oxido group. The complex possesses an S = 6 ground state spin (D = −0.18 cm−1, g = 1.86). Single-crystal magnetization vs. dc field scans show only very weak hysteresis at 0.1 K.
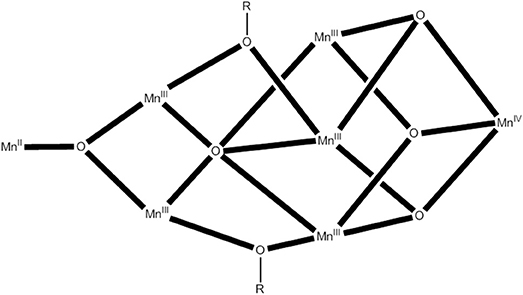
Figure 4. The novel manganese-oxygen core of complex 10; coordination bonds are drawn in bold. R– = PhCH2- (Schematic drawing inspired by Mishra et al., 2006).
Phosphonates, a Useful Alternative to Carboxylates
Phosphonates () are ideal ligands for stabilizing 3d-metal coordination clusters, some of which are SMMs. They possess three O donor atoms and can, in principle link to a maximum of nine metal centers. However, polymeric 3d-metal phosphonates are very insoluble and the isolation of phosphonato complexes is problematic. Three routes have been used to overcome the insolubility problem and stabilize discrete molecular species (Langley et al., 2008). First, solvothermal techniques can improve the solubilities. Second, a “co-ligand” can be used to maintain some solubility. And third, pre-formed carboxylato clusters can be linked in various ways by displacing some carboxylate groups by phosphonato ligands. For preparing 3d-metal phosphonate SMMs, there is one disadvantage and one advantage. The disadvantage is that phosphonato ligands propagate weak magnetic exchange interactions between metal centers, but the presence of bridging co-ligands (including monoatomic ones) can improve the situation. The advantage is that most metal phosphonates form 2D lattices; if polymerization can be stopped, e.g., by using other ligands, this would be ideal for creating structural anisotropy which, in turn, can lead to the desirable magnetic anisotropy.
Compound (Et3NH)[(NO3)3(O3PPh)2(chp)10(Hchp)2] (11), where Hchp is 6-chloro-2-hydroxypyridine (A with R = Cl in Figure 1) and is the dianion of phenylphosphonic acid, is prepared by the reaction of Co(NO3)2·6H2O, Hchp and PhPO3H2 in MeCN in the presence of Et3N, Equation (9); the yield is ~50%. To retain the reagents in solution, the phenylphosphonic acid: Co(II) ratio should be low and Hchp has to be added before PhPO3H2. The base is necessary to deprotonate the two ligands. The two P atoms and the four CoII ions lie on the vertices of a trigonal prism. The remaining four metal ions lie above and below the triangular faces of the prism. The phosphonato ligands both adopt the 4.211 mode (HH in Figure 2), while eight of the chp− ligands bind with the 2.21 mode (II in Figure 2) and two with the 3.21 mode (JJ in Figure 2). The two neutral Hchp molecules are terminally coordinated exhibiting the 1.10 mode. The structure also contains two chelating nitrato groups (1.110) and a bridging 2.110 nitrato group (Langley et al., 2008). Study of the dynamic behavior of the magnetization shows that 11 displays slow magnetization relaxation. It was found that different samples gave different Ueff values and a range of pre-exponential factors, τ0. For example, one sample gave Ueff = 58.4(±2) cm−1 and τ0 = 1.8(±3) × 10−12 s, and another gave Ueff = 55.6(±2) cm−1 and τ0 = 2.1 (±3) × 10−11s.
The reaction between the pre-formed oxido-centered triangle [O(O2CPh)6(py)2(H2O)] and benzylphosphonic acid (PhCH2PO3H2) in MeOH/MeCN, in the presence of LiOMe, in an 1:1:1 ratio, gives complex [O4(OMe)4(O2CPh)15(O3PCH2Ph)4(py)2] (12) in 60% yield, Equation (10). The core of the complex consists of a central trigonal prism. Two of the square faces of the prism are each bridged to one edge of a peripheral triangular unit by two 5.221 phosphonato ligands (KK in Figure 2) and two 2.2 methoxido groups. The LiI atom sits in the cavity of the central trigonal prism and forms six bonds to oxygen atoms. The remaining edges of the peripheral triangular units are unchanged from those in the starting material, i.e., are bridged by 2.11 groups. The oxido atom in the triangular Mn units are μ3 (3.3), again as found in the starting Mn triangle. Interestingly, the crystal lattice contains two different {} molecules which have the same connectivity, but which are Jahn-Teller isomers of one another. The complex has a spin ground state of S = 6, but the small anisotropy of the system makes the compound a poor SMM with a very low Ueff (Shanmugam et al., 2006).
“Dancing” With Azides
The azide () ion is a very popular inorganic ligand in transition-metal cluster chemistry (Escuer and Aromi, 2006; Stamatatos and Christou, 2009; Escuer et al., 2014). In addition to its terminal coordination mode (1.10), it often participates as a bridging ligand in transition-metal chemistry. No less than 8 bridging modes have to-date been crystallographically characterized for the azido ligand. These are end-to-end (EE) (syn, syn-2.11, and syn, anti-2.11), end-on (EO) (2.20, 3.30, 4.40) and mixed EE/EO (3.21, 4.22, 6.33) modes. The bridging azido group is an effective magnetic coupler and it is thus not surprising that it is an ingredient in many 3d-metal SMMs. EE azido groups generally promote antiferromagnetic exchange interactions, whereas the EO ligation mode is responsible for ferromagnetic exchange interactions; however, there are exceptions to this general rule. There have been reported many reactions schemes that lead to azido-bridged 3d-metal coordination clusters and SMMs. These include, among others, systems intended to provide azides as the only bridging ligands in the cluster, primary organic ligand/azide “blends,” and reaction mixtures containing two different organic ligands and ions. In the following, we give a representative example of SMMs from each reaction system.
The 1:1:4 reaction between Co(ClO4)2·6H2O, Et3N, and Me3SiN3 in MeCN gives a pink solution which, upon layering with Et2O, leads to the heptanuclear cluster [(N3)12(MeCN)12](ClO4)2 (13), Equation (11), in ~75% yield. Replacement of Co(ClO4)2·6H2O with Ni(ClO4)2·6H2O in the reaction gives the isomorphous compound [(N3)12(MeCN)12](ClO4)2 (14) (Alexandropoulos et al., 2014). The presence of Et3N is necessary for the isolation of the cluster, but its role appears unclear. The heptanuclear cations of 13 and 14 consist of an hexagon of MII atoms (M = Co, Ni) surrounding a seventh metal ion in the center of the hexagon. The disk-like core is completed by 12 N atoms of six 3.30 and six 2.20 EO groups. The 3.30 azido groups bridge the hexagon with the central metal ion, while the 2.20 ones bridge the pairs of the hexagon. Two MeCN molecules are coordinated to each external metal center (see Figure 5). The cations have a layered structure, with layers of azido N atoms above and below the plane. There are strong intracluster ferromagnetic CoII…CoII interactions in 13, which leads to an effective spin ground state of S = 7/2; the complex is SMM under an external dc field of 0.1 T (Ueff = ~20 cm−1, τ0 = 8.0 × 10−8 s). The cluster cation has a very well-isolated ground-state spin value as indicated by the similar EPR spectra at 4 and 23 K.
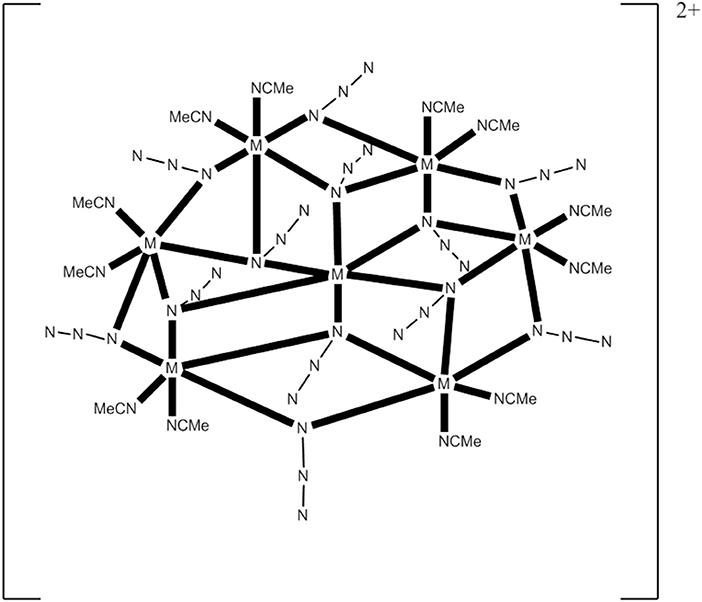
Figure 5. Schematic molecular structure of the heptanuclear cations of complexes 13 and 14; coordination bonds are drawn in bold (Schematic drawing inspired by Alexandropoulos et al., 2014).
The ligand “blend” sao2−/ where sao2− is the dianion of salicylaldoxime (saoH2; L with R = H in Figure 1), in Mn chemistry has provided access to a structurally interesting SMM (Yang et al., 2007). The reaction of MnCl2·6H2O, NaN3, saoH2, and (Et4N)OH in MeOH gives a dark green solution from which (Et4N)3[O(N3)6Cl2(sao)3] (15) can be crystallized in low yield (~15%), Equation (12). The five metal ions define a trigonal bipyramid in which the two tetrahedral MnII atoms occupy the axial positions and the distorted octahedral MnIII atoms are in the equatorial plane linked by a 3.3-O2− group. Each MnII atom is connected to the equatorial MnIII centers with three 2.20 azido groups, while a terminal chlorido group completes four-coordination at each MnII. The MnIII atoms are bridged by three 2.111 sao2− ligands (LL with M = MnIII and R = H in Figure 2). The anion has a ground-state spin of S = 11, with a first excited state S = 10 being 3.2 cm−1 higher in energy. Fit of the magnetization data gave a D value of−0.22 cm−1. The cluster is SMM with a Ueff value of 28 cm−1.
A modern trend in 3d-metal azide cluster chemistry is the employment of two organic ligands (a primary and an ancillary) and ions in the reaction systems. The loss of a degree of synthetic control seems to be, at first glance, disadvantageous; however, this loss is more than compensated for by the large diversity of structural types expected. We present an example in which the ancillary organic ligand is a carboxylate ion. The reaction of Ni(ClO4)26H2O, phenyl 2-pyridyl ketoxime (ppkoH; H with R = Ph in Figure 1), NaN3 and NaO2CH in a ~1:1:1:1.5 ratio in MeOH gives a red-brown solution, from which can be subsequently isolated complex [Ni5(N3)4(O2CH)2(ppko)4(MeOH)4] (16) in ~50% yield, Equation (13). The structurally novel {Ni5(2.20-N3)2(3.30-N3)2}6+ core (see Figure 6A) contains the five metal ions in a bowtie topology (two Ni3 triangles with a common vertex). Each diatomic oximate group of a 2.111 ppko− (MM with R = Ph and M = Ni in Figure 2) bridges the central NiII atom and a peripheral one (see Figure 6B), while a 2.11 formato ligand spans the base of each isosceles triangle. Magnetic data indicate an overall ferromagnetic behavior (S = 5 ground state). Ac magnetic susceptibility studies reveal non-zero, frequency-dependent out-of-phase () signals at temperatures below ~3.5 K. However, single-crystal magnetization vs. dc field scans at variable temperatures and variable sweep rates down to 0.04 K show no significant hysteresis loops, except minor ones at 0.04 K that are assigned to weak intermolecular interactions (Papatriantafyllopoulou et al., 2010).
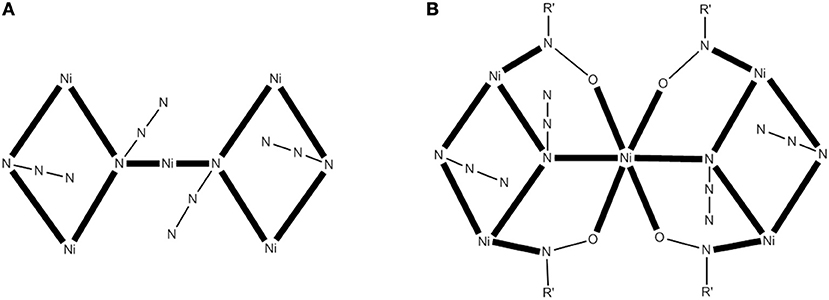
Figure 6. The {Ni5(2.20-N3)2 (3.30-N3)2}6+ core of the complex 16 (A) and a more detailed representation of the core (B) emphasizing its {Ni5(2.20-N3)2 (3.30-N3)2 (2.11-0NR')4}2+ description (R′NO− = ppko−); coordination bonds are drawn in bold (Schematic drawing inspired by Papatriantafyllopoulou et al., 2010).
Comproportionation Reactions in Manganese SMM Chemistry
A commonly used synthetic route for the preparation of high-oxidation state Mn clusters and SMMs is the comproportionation reaction of a MnII source and MnVII (i.e., MnVII) in the presence of simple carboxylate ions and/or chelating/bridging ligands. Such reactions are very useful because they normally give products containing some Mn(IV), thus preventing complication from low-lying excited states which is a common situation with MnII…MnIII and MnIII…MnIII couplings. This was the original method for the synthesis of the {} complex 1 which, in a sense, initiated the area of SMMs (Lis, 1980). The reaction of MnII(O2CMe)·4H2O and KMnO4 in 60% v/v MeCO2H/H2O, in a ratio appropriate for the final +3.33 Mn oxidation level and under warm heating, provides access to high yields of crystalline 1·2H2O·4MeCO2H, Equation (14), in the form of reddish-black crystals. The crystals were of X-ray quality and the structure of the cluster was solved by single-crystal, X-ray crystallography.
The prototype SMM 1 can also be isolated in high yields by the oxidation of [O(O2CMe)6(py)3](ClO4) (Vincent et al., 1987) by KMnO4 in 60% v/v MeCO2/H2O in a ratio designed to 1 (Sessoli et al., 1993b), Equation (15).
An ideal high-oxidation Mn(VII) reagent for such reactions in organic solvents has been documented to be (Bun4N)(MnO4) (Vincent et al., 1986); this oxidizing reagent is soluble in many organic solvents. It is easily prepared by mixing aqueous solutions of KMnO4 and (Bun4N)Br in slight excess; it is pure enough for use without recrystallization and storage at −5°C increases its stability (storage at room temperature leads to slow decomposition in a period of ~1 month give a brown sticky material). The benzoato analog of 1, i.e., [O12(O2CPh)16(H2O)] (17), also a good SMM, can be prepared in low yield (~10%) by comproportionation of Mn(O2CMe)4H2O with (Bun4N)(MnO4) in the presence of PhCO2H in py/MeCN (Sessoli et al., 1993b). Although the MnII: ratio was identical to that in Equation (14), the major product of this reaction is [O(O2CPh)6(py)2(H2O)], i.e., in this case only a small amount of higher (>III) average oxidation product is obtained.
An interesting comproportionation reaction to manganese (III/IV) pivalato clusters gave a half-integer spin SMM (Mukherjee et al., 2013). The 1:1:1:1 comproportionation reaction of MnII(O2CBut)2·2H2O, MnII(ClO4)2·6H2O, (Bun4N)Cl and (Bun4N)(MnO4) in the presence of a large excess of ButCO2H (pivalic acid; its excess is necessary to prevent precipitation of amorphous Mn oxides) in hot MeCN gives a dark brown solution that leads to the isolation of [MnIVO6(OH)Cl3(O2CBut)9(ButCO2H)0.5(MeCN)0.5] (18) in ~25% yield. The average metal oxidation state in the reaction is +4.5, Equation (16), but in the product it is less (~+3.1). The low yield of 18 is an evidence that there are probably other higher oxidation-state products in the mother liquor which are not precipitated. Use of MnCl2 or Mn(NO3)2, instead of the manganese(II) perchlorate salt, also give 18, but in lower yields. It was seemed possible that the low Mn(IV) content in 18 is due to the fact that oxidizes not only Mn(II) but also the solvent. The use of an even lower Mn(II):Mn(VII) reaction ratio of 2:3 to increase the average oxidation state to +5, Equation (17), gives a mixture of 18 and [O9(O2CBut)12] (19); the latter can be prepared pure from a simplified comproportionation reaction between Mn(NO3)2 and (Bun4N)(MnO4) (2:3) in the presence of pivalic acid in hot MeCN in ~35% yield. The average metal oxidation state in 19 is now +3.75.
Complex 18 possesses a {MnIV(3.3-O)4(4.4-O)2(3.3-OH)(4.4-Cl)(2.2-Cl)}10+ core containing two body-fused Mn4 butterfly subunits attached to the remaining Mn ions through bridging oxido, hydroxide, and chlorido groups. Cluster 19 possesses a {(3.3-O)6(2.2-O)3}12+ core consisting of two incomplete {O4} cubanes linked to two MnIII atoms. Solid-state dc and ac magnetic susceptibility data have indicated an S = 15/2 ground state for 18; the D value is −0.22(2) cm−1. The ac susceptibility data revealed non-zero, frequency-dependent χ”M signals for this complex below 3 K, providing a strong evidence for SMM behavior; this behavior was confirmed by the appearance of hysteresis loops in single-crystal magnetization vs. dc field studies. The loops display two well-resolved QTM steps. The complex is a half-integer cluster and it is not expected to display QTM in the absence of transverse fields; the latter are provided by the transverse components of dipolar and exchange fields from neighboring molecules, and hyperfine fields from 55Mn (I = 15/2) nuclei. The relaxation rate at zero applied field is very fast at several temperatures, and this prevented the determination of Ueff. Complex 19 does not exhibit χ“M signals above 1.8 K.
The Reductive Aggregation Route and Its Modifications in Manganese SMM Chemistry
The reductive aggregation route in Mn chemistry represents a modification of the comproportionation procedure. The Mn(II) source is omitted and MeOH is used not only as a solvent, but also both as the reducing agent for Mn(IV) and as a potential source of bridging MeO− groups, in the presence of an excess of carboxylic acid that prevents precipitates of Mn hydroxides and/or oxides, and provides carboxylato ligands.
The reaction of phenylacetic acid (PhCH2CO2H) with (Bun4N)(MnO4) in MeOH leads to a dark brown solution and isolation of [O16(OMe)6(O2CCH2Ph)16(MeOH)6] (20) (King et al., 2004), eqn. (18). In Equation (18), it is assumed that MeOH is oxidized to formaldehyde. The yield is low (~10%), but the reaction is perfectly reproducible and gives crystalline material. Such reactions are certainly complicated with several cluster species in solution; the final outcome depends on several factors, such as relative solubilities, kinetics of crystallization and lattice energies. An important issue of this reaction (and several reactions of this type) is the necessity of using small volumes of MeOH; dilute solutions lead to Mn hydroxides and/or oxides. In the reaction that leads to 20, use of EtOH instead of MeOH leads also to hydroxides/oxides, whereas 2-propanol leads to pale yellow solutions indicative of full reduction of Mn(VII) to Mn(II). The metal centers are linked through 14 3.3-O2−, 2.2-O2−, 4 2.2-MeO−, and 2 2.11-O2CCH2Ph ligands, the remaining 14 2.11-O2CCH2Ph−, 2 2.2-MeO−, and 6 1.1-MeOH ligands providing peripheral ligation. A useful description of the complex is as a very small piece of a Mn oxide mineral held within a nonplanar ring of MnIII atoms. There are dominant antiferromagnetic exchange interactions in the molecule. Low-lying states make a valid assignment for the total ground-state spin of 20 difficult; it is most probably S = 2. Hysteresis behavior is clearly visible below 1 K, indicating SMM properties.
An analogous reaction, but using BunCO2H this time, leads to the low-yield preparation of [O10(OMe)4(O2CBut)16(MeOH)2] (21), Equation (19). The 12 Mn centers are held together by 10 3.3-O2− and 4 2.2-MeO− groups. The molecule possesses a central face-fused defective dicubane core of four MnIV atoms held within a non-planar ring of eight MnIII atoms to give an overall chair conformation. The complex has an S = 9 ground state and is SMM (King et al., 2005). No sign of any steps was observed in the hysteresis loops as a result of broadening effects due to extensive disorder of most But groups. The reductive aggregation route has also led to anionic {}2− SMMs (Tasiopoulos et al., 2005).
A modification of the reductive aggregation route involves employment of a second (in addition to MnVII) high oxidation-state Mn cluster. Thus, the [O12(O2CMe)16(H2O)4] (1)/(Bun4N)(MnVIIO4)/MeCO2H reaction mixtures in MeOH and EtOH lead to the giant torus-like SMMs [O72(OH)6(OMe)24(O2CMe)78(H2O)42(MeOH)12] (22) (Tasiopoulos et al., 2004) and [O60(OEt)20(O2CMe)70(H2O)22(EtOH)16] (23) (Vinslava et al., 2016), respectively, Equations (20) and (21). In these chemical equations, it is assumed that MeOH and EtOH are oxidized to formaldehyde and acetaldehyde, respectively. The Ueff value of the {} cluster is ~16 cm−1.
Procedures Involving Two Different Primary Organic Ligands
The combined use of two primary organic ligands with different functionalities in the reaction systems is another route for the construction of clusters and SMMs. This method has developed mainly by the groups of Tasiopoulos and Brechin. In the most of the reported examples, one of the primary organic ligands does not appear in the final product, but its role in solution appears essential for the reaction. This route has led, among others, to a giant {} double-decker wheel with an S = 11 or 12 ground state and SMM behavior (Ueff = ~31 cm−1, τ0 = 3.5 × 10−12 s); the small τ0 value has been attributed to low-lying excited states and weak intermolecular interactions (Manoli et al., 2011). We give below a representative, simpler example to illustrate the method; the product however, lacks SMM behavior, even to temperatures as low as 30 mK.
The 1:1:1:1 reaction of MnBr2·4H2O, 2-(hydroxymethyl)phenol (hpH2; O in Figure 1), saoH2 (L with R = H in Figure 1), and NaOCN in MeCN/DMF (4:1 v/v) leads to [O6Br12(sao)18(H2O)18(DMF)6] (24) in typical yields of ~50% (Manoli et al., 2016), Equation (22). Reactions performed without the presence of the diol do not give this cluster, emphasizing its important role in the formation of 24. The structure of 24 consists of a {} wheel-like cluster comprising oximate-based {O}7+ triangular units linked through six Na+ ions. The {NaI} repeating unit possesses an oxo-centered triangular arrangement of MnIII atoms and a Na+ ion attached to it via oxygen atoms (see Figure 7) of oximate groups which occupy the edges of the triangle. Two 3.211 (NN in Figure 2) and one 4.221 (OO in Figure 2) sao2− ligands, as well as one 2.2 H2O molecule connect each Mn(III) triangle with two neighboring Na+ ions, which in turn are linked to the next triangular subunits forming the wheel.
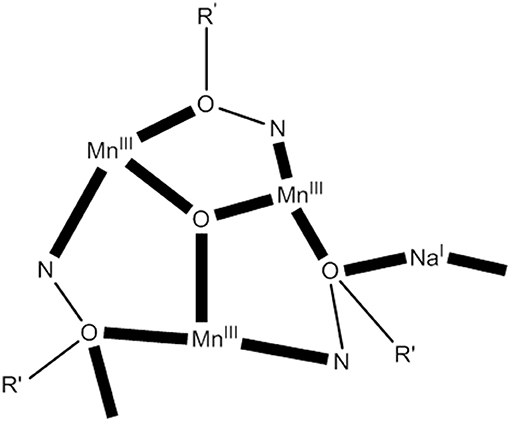
Figure 7. A simplified view of the { NaI} repeating unit that forms the core of complex 24 (R′ NO = saph2−); coordination bonds are drawn in bold (Schematic drawing inspired by Manoli et al., 2016).
The Years of Design: Strict and Less Strict Synthetic Strategies
All the routes described in section The Years of Innocence: “Try and See” Exercises depend on serendipity rather than on strict design; this means that the final product and its properties could not be predicted. However, those routes (and others not mentioned) were the basis of the great developments in the area of SMMs.
After ~2,000, scientists were more interested in designing the chemical and structural identity of the coordination clusters resulting from their efforts, and in predicting and controlling the magnetic properties of the products. Design principles and examples, which have been developed mainly in Mn SMM chemistry, will be presented in this chapter. The strategies are based on the modification of structural types already known that may or may not already be SMMs in their own right. From the structural viewpoint, two are the important parameters to be considered for such modifications: (a) the nuclearity of the coordination cluster, and (b) the core. Each of these parameters, or both, may change or may not change in the product with respect to the starting material (which is always a pre-formed cluster). From the magnetic viewpoint, the starting material may or may not be an SMM. In the former case, the primary goal of the synthetic strategy is to improve the SMM properties. In the latter, the main goal is to “switch on” SMM properties.
The choice of the below mentioned strategies is subjective and again we owe our apologies to many synthetic chemists whose elegant work will not be illustrated here.
Substitution of Carboxylato Ligands by Other Carboxylato Groups
Several groups (mainly Christou's group) have developed procedures to replace ligands in Mn SMMs with a variety of other R′ groups. The replacement can be complete or partial (vide infra). The most studied SMM for such reactivity studies is 1. As described in section Comproportionation Reactions in Manganese SMM Chemistry, complex 17 can be prepared using a comproportionation reaction, but in low yield (~10%). Treatment of 1 with 32 equivs of PhCO2H (100% excess) in CH2Cl2 leads to the exchange of most ligands. For full replacement another ligand substitution cycle can be performed; the partially exchanged solid from the first cycle can be treated with other 32 equivs of PhCO2H giving the desired product in an overall >50% yield (Sessoli et al., 1993b), Equation (23).
In general, such reactions are equilibria, Equation (24). The substitution reaction is facilitated by the higher acidity of the incoming carboxylic acid, R′CO2H, and its presence in excess. In the case of R = H (i.e., 1), MeCO2H can be removed as its toluene azeotrope shifting the equilibrium represented by Equation (23) to the right. The distillation of the azeotrope of MeCO2H and toluene is a very valuable method for introducing R′ groups whose conjugate acid has a pKa value higher than that of MeCO2H or comparable to this (Bagai and Christou, 2009). The nuclearity and the core in the product are the same with those existing in the starting material. Such carboxylate substitution reactions often lead to better solubility in organic solvents and altered redox properties of the products compared to 1.
This type of carboxylate substitution is of important utility in several technological aspects of the chemistry of 1 and its derivatives (Domingo et al., 2012). We are doing a parenthesis here to mention that applications of SMMs require the development of strategies to move from bulk crystals to molecules that can be grafted on surfaces, sensors or other systems able to behave as a device; the challenge here is that the SMMs should retain their properties in the device. There are three routes for the nanostructuration of SMMs on surfaces from solution: (a) SMM direct deposition on a bare surface in order to immobilize the SMM through weak non-covalent interactions, (b) pre-functionalization of SMMs with groups that are able to interact chemically with the bare surface, and (c) pre-functionalization of the surface with appropriate groups that are able to interact with the SMM. An early example of pre-functionalization of SMMs for deposition onto an Au(111) surface involves replacement of the groups of 1 by carboxylato ligands containing thiol groups (Cornia et al., 2003). The 16-sulfanylhexadecanoate (P in Figure 1) derivative of 1 was designed, in which the thiol groups were acetyl-protected (Q in Figure 1) to avoid undesirable reactivity in the presence of the MnIII and MnIV centers. The Ueff value of the product is ~38 cm−1, close to that of 1. The deposition was achieved by incubating the Au substrate in an SMM solution in a basic environment to ensure the deprotection of the thiol groups. The SMMs covered completely the surface with disordered layers, which could be disrupted by a continuous scanning of the area allowing the first imaging of discrete dodecanuclear Mn molecules by Scanning Tunneling Microscopy.
The carboxylate substitution can be partial and site-selective. In the case of the {} SMMs, the products have the formula [O12(O2CR)8(O2CR')8(H2O)4], i.e., x = 8 in Equation (24). The site-selective (or specific) substitution results from the different reactivity to electrophiles of some carboxylate ligands in 1, 17 and related SMMs. Of the 16 carboxylates, 4 (type I) have both their O atoms on Jahn-Teller elongation axes of MnIII atoms, 4 (type I′) have one, and 8 (type I″) have none being equatorial. Since the MnIII-O bonds on the Jahn-Teller axis are weaker than those on the axes of the equatorial plane, the relative susceptibility of the carboxylate groups to electrophilic attack would be expected to be I>I′> I″ (Bagai and Christou, 2009) and this has been nicely demonstrated in several cases. Another relevant synthetic hypothesis is that the more basic carboxylate groups would thermodynamically prefer to bind at the non-Jahn-Teller equatorial sites (type I″) where they can form the strongest MnIII-O bonds.
Examples confirming these synthetic hypotheses are the reactions illustrated by Equations (25–28). Addition of 8 equivs of CHCl2CO2H (pKa = 1.48) to [O12(O2CCH2tBu)16(H2O)4] (25) or [O12(O2CEt)16(H2O)3] (26) in CH2Cl2 gives [O12(O2CCHCl2)8(O2CCH2tBu)8(H2O)3] (27) or [O12(O2CCHCl2)8(O2CEt)8(H2O)3] (28), respectively; the pKa values of ButCH2COOH and EtCO2H are 5.24 and 4.86, respectively, and the order of basicities is thus ButCH2 > >> CHCl2 (Soler et al., 2001). In the structures of both 27 and 28, the CHCl2 groups are in axial sites and the pre-existed groups (R = ButCH2, Et) are equatorial. Thus, the CHCl2 ligands are either bridging MnIII/MnIII pairs with both their O atoms lying on the Jahn-Teller axes, or they are bridging MnIII/MnIV pairs with only one O atom on a MnIII Jahn-Teller elongation axis. The 8 equatorial, more basic ligands bridge MnIII/MnIII pairs and there are no O atoms on the Jahn-Teller axes. Complexes 27 and 28 can also be prepared by the 1:1 reaction of the corresponding homo-carboxylato complexes 25, 26, and [O12(O2CCHCl2)16(H2O)4] (29), the latter prepared by the reaction of 1 with a large excess of CHCl2CO2H in CH2Cl2 and removal of MeCO2H as its toluene azeotrope, Equation (29). This method involves ligand redistribution and the isolated products are the 8:8 complexes. Complexes 27 and 28 are SMMs with Ueff values of ~50 cm−1.
Substitution of Carboxylato Ligands by Non-Carboxylate Groups
Work from Christou's group has led to the development of procedures for partial (and sometimes site-selective) non-carboxylato substitution in {} SMMs. Site-selective carboxylato abstraction has been become possible from [O12(O2CR)16(H2O)4] (R = Ph, 17; R = ButCH2, 25) by treatment with 4 equivs of HNO3 in MeCN, Equation (30). The products are [O12(O2CR)12(NO3)4(H2O)4] (R = Ph, 30; R = ButCH2, 31). The four nitrato groups are coordinated in the same 2.11 mode as the carboxylato ligands at the type I (section Substitution of Carboxylato Ligands by Other Carboxylato Groups) MnIII axial Jahn-Teller positions (Artus et al., 2001). The reaction represented by Equation (30) can be reversed; reactions of 30 and 31 with 4 equivs of NaO2CR in CH2Cl2/MeOH give complexes 17 and 25, respectively, Equation (31). This makes complexes such as 30 and 31 candidates for reactivity centered at the nitrato positions, taking advantage of the good leaving properties of bound ions. Complex 31 (S = 10) is an SMM with a Ueff value of 50.0 cm−1. The complex also shows hysteresis in magnetization vs. dc field scans; the hysteresis loops show steps at regular intervals of magnetic field, the diagnostic evidence of QTM.
In a similar way, the reaction of 1 with 8 equivs of benzenesulfonic acid (PhSO3H) in MeCN, followed by several cycles of the removal of generated MeCO2H as the toluene azeotrope to ensure complete reaction, gives complex [O12(O2CMe)8(O3SPh)8(H2O)4] (32) in a >95% yield, Equation (32) (Chakov et al., 2003). The Ph ligands are at the eight axial (I and I′, see in the “Substitution of Carboxylato Ligands by Other Carboxylato Groups” section above) MnIII Jahn-Teller positions, again as expected on the basis of relative basicities (pKa of MeCO2H = 4.76, pKa of PhSO3H = 2.55). In other words, the more basic (stronger donors) ligands prefer the equatorial sites where shorter, stronger Mn-O bonds can be formed stabilizing better the molecule. The less basic Ph ligands are located at axial positions where they bridge either MnIII/MnIII or MnIII/MnIV pairs and therefore have one or both of their O atoms on the Jahn-Teller elongation axes of the MnIII atoms. The mixed-ligand cluster 32 retains both the high ground-state spin (S = 10) and SMM behavior (Ueff = ~47 cm−1) of the parent SMM 1 and its carboxylato derivatives. Hysteresis loops were observed below 4.0 K; their coercivities increase with decreasing temperature, a typical behavior of SMMs. The synthetic importance of reactions, such as that represented by Equation (32), is that regioselective chemistry at the axial positions with anionic ligands can become a reality.
In the non-carboxylato substitution of {} SMMs i.e., 1 and its derivatives, sometimes steric effects overcome basicity effects. A typical example is the reaction of 1 with 8 equivs of diphenylphosphinic acid (Ph2PO2H) in MeCN that gives the 8:8 cluster [O12(O2CMe)8(O2PPh2)8(H2O)4] (33) in 60% yield. In 33, the 4 ligands are located at the four axial MnIII/MnIII and 4 of the eight equatorial MnIII/MnIII carboxylate sites have been replaced by Ph2 groups, while the remaining equatorial sites and the four axial MnIII/MnIV sites remain occupied by ligands (Boskovic et al., 2001). In other words, the large steric bulk of the Ph2 ligands has, as a consequence, their equal distribution between axial and equatorial sites. The SMM properties are retained in 33 (Ueff = ~42 cm−1). Magnetization hysteresis loops were observed for oriented crystals of 33·12CH2Cl2.
Reduction Pathways for the {} SMMs
Detailed electrochemical studies on many members of the {} family of SMMs (1, 17, 25, 26,…) have revealed several oxidation and reduction processes, with some of the latter being reversible. The oxidation processes are at rather high potentials, but the reduction processes are accessible by chemical means. Electron-withdrawing R groups in the carboxylato ligands (e.g., R = CHCl2 or C6F5) favor the reduction processes, because they reduce the electron density in the core and make reductions easier. An ideal reducing agent is the iodide (I−) because: (i) Its reducing strength is sufficient, but mild; (ii) there are many organic salts C+I−, the majority of which have good solubility in a variety of organic solvents; and (iii) the only by-product of the redox processes is elemental I2 which can be easily removed.
Reduction of complex 26 with 1 equiv of Ph4PI in CH2Cl2 leads to complex (Ph4P)[MnIIO12(O2CEt)16(H2O)4] (34) in ~70% yield, Equation (33). The structure reveals that the extra electron is localized on an outer (originally MnIII) ion rather than an inner (cubane) MnIV center (see Figure 8), and the product is a trapped-valence {MnII}− ion (Eppley et al., 1995). Cluster 34 has an S = 19/2 ground state being SMM. The complex exhibits well-developed hysteresis loops. The benzoate analog of 34, i.e., cluster (Ph4P)[Mn12O12(O2CPh)16(H2O)4] (35) can be prepared in a similar way (Aubin et al., 1999). For the eight “external” Mn ions (formerly MnIII), it was not possible to decide whether a trapped-valence {MnII} or an electronically delocalized description is the most scientifically correct. Complexes such as 34 and 35 (both with S = 19/2) are ideal candidates for the study of QTM in half-integer spin systems; QTM is not allowed in such compounds in the absence of an applied magnetic field.
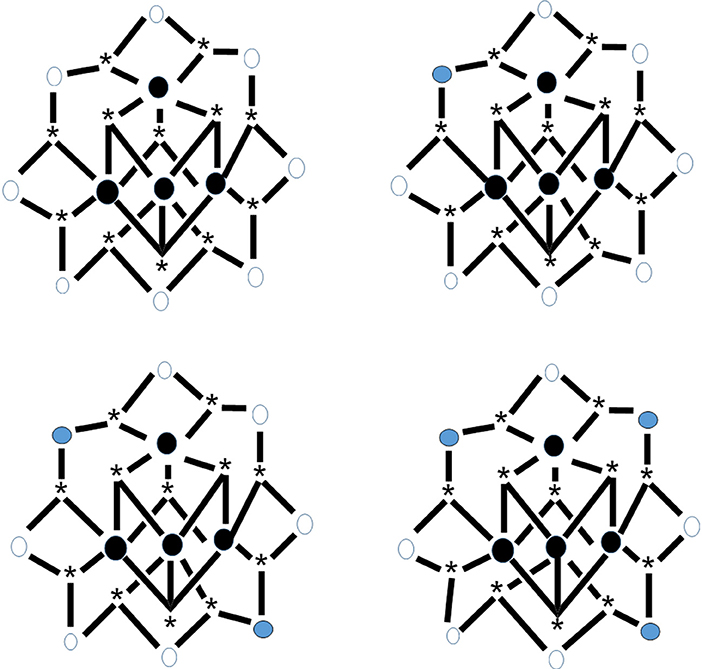
Figure 8. The crystallographically established {Mn12O12} cores of the {Mn12}, {Mn12}−, and {Mn12}2− SMMs, and proposed core for the {Mn12}3− SMMs. The symbol  represents MnIV, the symbol
represents MnIV, the symbol  represents MnIII, the symbol
represents MnIII, the symbol  represents MnII, and the asterisk illustrates the triply bridging O2− group (Schematic drawing inspired by Bagai and Christou, 2009). Coordination bonds are drawn in bold.
represents MnII, and the asterisk illustrates the triply bridging O2− group (Schematic drawing inspired by Bagai and Christou, 2009). Coordination bonds are drawn in bold.
The carboxylato substitution reactions described in section Substitution of Carboxylato Ligands by Other Carboxylato Groups have permitted the incorporation onto the {Mn12} unit of carboxylate ligands possessing strongly electron-withdrawing groups, and these can raise the reduction potentials so that the second reduction is feasible with I−. For example, the reaction of 29 with two equivs of Ph4PI in MeCN provides access to complex (Ph4P)2[O12(O2CCHCl2)16(H2O)4] (36) in very good yield (Soler et al., 2003), Equation (34). Crystallization of the bulk material from CH2Cl2/hexanes yields a mixture of two crystals forms, 36·4CH2Cl2·H2O (36a) and 36·6CH2Cl2 (36b), both of which have been structurally characterized by single-crystal, X-ray crystallography. The molecular structures are almost identical, with the two extra electrons localized on former MnIII centers (see Figure 8) generating a trapped-valence {}2− ion. Bulk dc magnetization data for dried 36 in the 1.8–4.0 K and 1–7 T ranges were fit to give S = 10, D = −0.275 cm−1 and g = 2.00. The Ueff values for 36a and 36b are ~13 and ~21 cm−1, respectively. Magnetization vs. dc field scans on single crystals of the two samples gave hysteresis loops containing steps due to QTM. The step separations yielded |D|/g values of 0.087 and 0.14 cm−1 for 36a and 36b, respectively, indicating that the differences in Ueff are caused by changes to D.
The electrochemical properties of 29 exhibit a third reversible reduction accessible to I−. Thus, the {Mn12} family of SMMs has been extended to a fourth isolated member. The reaction of 29 with 3 equivs of Prn4NI or Me4NI in MeCN for a long time (~2 days) give complexes (Prn4N)3[O12(O2CCHCl2)16(H2O)4] (37) and (Me4N)3[O12(O2CCHCl2)16(H2O)4] (38), respectively (Bagai and Christou, 2007), Equation (35) (C = Prn4N, Me4N); the yields are high (~80%). The complexes are not stable in solution and all the attempts to grow single-crystals of the salts were unsuccessful. Dc magnetization data on dried microcrystalline samples were fit by matrix diagonalization methods to give S = 17/2, D = −0.25 cm−1 and g = 1.91 for 37 and S = 17/2, D = −0.23 cm−1 and g = 1.90 for 38. The two complexes exhibit frequency-dependent out-of-phase magnetic susceptibility signals at ≤2.5 K, indicating them to be SMMs, albeit at lower temperatures compared with the {Mn12} SMMs (6–8 K range), {Mn12}− SMMs (4–6 K range), and {Mn12}2− SMMs (2–4 K range); the shifts to lower temperatures are indicative of the decreasing S and D-values upon successive reduction and therefore the decreasing Ueff value. The decreased |D| values in the order {Mn12} >{Mn12}− >{Mn12}2− >{Mn12}3− indicate that the third added electron is localized on a formerly MnIII ion, since the Jahn-Teller-distorted MnIII ions are the main source of the molecular anisotropy (see Figure 8).
The preferential reduction of MnIII rather than MnIV can be interpreted in terms of the different environments which these metals have within the {Mn12} unit. Thus: (i) The MnIV centers are each bound to five hard O2− groups which favor higher oxidation states and preclude reduction to MnIII; and (ii) the reduction of a MnIV would generate a MnIII center whose Jahn-Teller distortion axis would have to include at least one MnIII-O2− bond (an inherently strong bond) and would introduce a severe strain into the rigid {O4} subcore.
“Switching On” SMM Behavior Through Substitution of Bridging Hydroxido Groups by End-on Azido or Isocyanato Ligands in Pre-formed Clusters
One of the prerequisities of a 3d-metal cluster to behave as SMM is a high or a relatively high total spin in the ground state. The high-spin ground state can result from either ferromagnetic (or ferrimagnetic) metal···metal exchange interactions and/or topologically frustrated antiferromagnetic exchange interactions. In general, however, it is difficult to predict which topology will lead to high-spin ground states, and more difficult to design and achieve the syntheses of such compounds. In many coordination clusters, the exchange interactions between the paramagnetic metal ions is propagated by hydroxido (OH−), oxido (O2−), alkoxido or alkoxido-type (RO−), or carboxylato () ligands, or a combination of two or more such ligands. These ligands most often propagate antiferromagnetic exchange interactions. Our group (Papaefstathiou et al., 2001a,b; Boudalis et al., 2004, 2008), in collaboration with Escuer's group, have developed a general strategy for the synthesis of high-spin clusters, which often “switches on” SMM behavior. The strategy is based on the substitution of bridging hydroxido or/and alkoxido groups in pre-formed coordination clusters (generally low-spin due to antiferromagnetic interactions) by EO azido groups (2.20, 3.30, 4.40) or N-bonded cyanato (isocyanato) bridging groups (2.02, 3.03, 4.04). The nuclearity does not change, but the core does. The entering bridging groups introduce ferromagnetic components in the superexchange scheme of the cluster, always increasing the ground-state spin and sometimes “switching on” SMM properties. The method works better for divalent 3d metals. The strategy is illustrated with one example (Boudalis et al., 2004, 2008).
Cluster [(OH)2(O2CMe)8{(py)2CO2}4] (39), where (py)2CO is di-2-pyridyl ketone (C in Figure 1) and (py)2 is the dianion of the gem-diol form of (py)2CO (D in Figure 1) is prepared under an inert atmosphere by the reaction outlined in eqn. (36) in ~30% yield. The nine FeII atoms adopt the topology of two square pyramids sharing a common apex and are held together by four 5.3311 (py)2 ligands (PP in Figure 2). Each FeII···FeII edge of the bases of the pyramids is further bridged by one syn, syn-2.11 group. The four groups create a concave cavity in the base of each pyramid, into which a rare 4.4 OH− group is trapped capping the square base. The magnetic study of 39 reveals an overall antiferromagnetic behavior with an S = 2 ground state.
Anaerobic reactions of 39 with a slight excess of NaN3 or KOCN in refluxing MeCN provides access to clusters [(N3)2(O2CMe)8{(py)2CO2}4] (40) and [(NCO)2(O2CMe)8{(py)2CO2}4] (41), respectively, in moderate yields, Equation (37). Complexes 40 and 41 have striking structurally similarity to 39, the main difference being the replacement of the 4.4 OH− groups in the latter by the 4.40 and 4.04 OCN− groups in the products (see Figure 9).1H paramagnetic NMR spectroscopic studies in CD3CN reveal that the three enneanuclear Fe(II) complexes are stable in solution. Dc magnetic susceptibility studies show that the ground-state spin values are higher in 40 and 41 than in 39; variable-field experiments have shown that the ground state is not well isolated from the low-lying excited states, and it cannot thus be determined with accuracy. X-band EPR spectroscopy at 4.2 K reveals characteristic integer-spin signals for 39, but not for the azido- and isocyanato-bridged clusters. Complexes 40 and 41 are SMMs with Ueff values of 28.5 and 30.5 cm−1, respectively. The slow magnetic relaxation has also been observed by 57Fe Mössbauer spectroscopy.
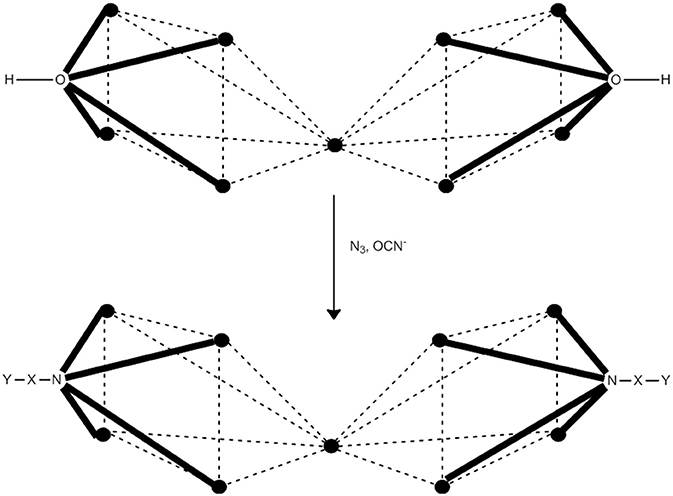
Figure 9. Inorganic core change upon conversion of cluster 39 to the azido- and isocyanato-bridged clusters 40 and 41. The symbol represents FeII. The dashed lines do not represent chemical bonds, but they help the understanding of the metal topology. Coordination bonds are drawn in bold. X, Y = N for 40; X = C and Y = 0 for 41.
“Tweaking” the Spin
The strategy reported in section “Switching on” SMM Behavior Through Substitution of Bridging Hydroxido Groups by End-on Azido or Isocyanato Ligands in Pre-formed Clusters involves conversion of a low-spin 3d-metal cluster into a high-spin one with the simultaneous “switching on” of SMM properties. This approach changes the core. An alternative approach is to start with a pre-formed, high-spin molecule (and perhaps an SMM) and then perturb it without significant core change by targeted modification of the peripheral ligands, in order to modify some exchange parameters, and slightly increase the ground-state spin and “switch on” or enhance the SMM properties. This strategy has been termed “spin tweaking” (Stamatatos et al., 2007a,b). We give an example of this approach from Mn chemistry.
The starting material is [MnIV(OH)2O18(N3)12(pdm)6(pdmH)6](N3)2 (42) (Murugesu et al., 2004, 2008), where pdm2− and pdmH− are the di- and the monoanion, respectively, of pyridine-2,6-dimethanol (pdmH2; R in Figure 1). The core is held together by 12 4.4 O2−, 6 3.3 O2− and two 3.3 OH− groups, as well as 6 2.20 ions and the deprotonated alkoxido arms of the 5.331 pdm2− (QQ in Figure 2) and 3.211 pdmH− ligands (RR in Figure 2). The complex has a total spin of S = 51/2 in the ground state, being an SMM (Ueff = ~8 cm−1). The target was to replace the peripheral 6 2.20 and 6 terminal (1.10) azido ligands by 6 2.21 hmp− ligands (SS in Figure 2), where hmp− is the monoanion of 2-(hydroxymethyl)pyridine (hmpH; S in Figure 1). The synthetic idea was that the bridging deprotonated oxygen atom of hmp− (known to promote ferromagnetic exchange between Mn ions) can be considered as the equivalent of the bridging N atom of the 2.20 ligands, while the 2-pyridyl nitrogen atom of hmp− is the equivalent of the terminal N atom of the 1.10 ligands. The reaction of 42, Na(hmp) and NaClO4 in 1:6:6 molar ratio in MeCN/MeOH gives a dark brown solution, from which is subsequently crystallized [MnIV(OH)(OMe)O18(hmp)6(pdm)6(pdmH)6](N3)2(ClO4)6 (43) in ~65% yield, Equation (38). The crystal structure shows the core of 43 to be isostructural with that of 42. The main differences are that the 12 coordinated azido groups of 42 are replaced by 6 2.21 hmp− ligands in 43 (see Figure 10) and also a 2.2 OH− group of the former is substituted by a 2.2 MeO− group in the latter. The result is that all bridging atoms in the product are oxygen atoms and there are thus changes in many exchange interactions. The ground-state spin increases from S = 51/2 in 42 to S = 61/2 in 43. Despite the intended increase of S, 43 is not a better SMM compared to 42 (Stamatatos et al., 2007a,b). The conversion of 42 to 43 is the first time in which removal of EO azido groups leads to an increase in the ground-state S-value in a 3d-metal coordination cluster; usually their addition is a method of increasing the S value (section “Switching on” SMM Behavior through Substitution of Bridging Hydroxido Groups by End-on Azido or Isocyanato Ligands in Pre-formed Clusters).
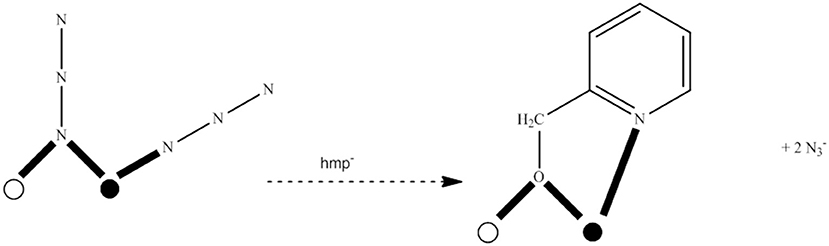
Figure 10. Schematic drawing of the synthetic approach that leads to the 42→43 conversion. The symbols  and represent MnII and MnIII, respectively. Coordination bonds are drawn in bold.
and represent MnII and MnIII, respectively. Coordination bonds are drawn in bold.
“Switching On” SMM Properties Upon Conversion of Low-Spin Complexes Into High-Spin Ones Without Changing the Core
The strategy described in section “Switching on” SMM Behavior through Substitution of Bridging Hydroxido Groups by End-on Azido or Isocyanato Ligands in Pre-formed Clusters involves structural change of the core, while in the approach detailed in section “Tweaking” the Spin the core remains the same but the starting materials are already high-spin complexes. A distinctly different approach within the general strategy of modifying known non-SMM complexes is the conversion of certain low-spin complexes into high-spin molecules without change of the core; this conversion results often in the appearance of SMM properties. Our group, in collaboration with Christou's group have contributed into this approach. As mentioned in section Simple 3d-Metal Carboxylates and Carboxylate Triangles and Butterflies as Starting Materials, triangular oxido-centered complexes of the general formulation [Mn3O(O2CR)6L3]0, + are useful starting materials in Mn carboxylate chemistry. Antiferromagnetic exchange interactions within the core lead to small S values, and the complexes are therefore non-SMMs. It was believed that this common triangular topology could never lead to SMMs. However, it has now been well established that relatively small, ligand-induced structural distortions of the core can alter the sign of the exchange interactions and “switch on” SMM properties.
The reactions between the oxido-centered, triangular complexes [O(O2CR)6(py)3](ClO4) [R = Me (44a), Et (44b), and Ph (44c)] and 3 equivs of methyl 2-pyridyl ketoxime (mpkoH; H with R = Me in Figure 1) in MeCN/MeOH give complexes [O(O2CR)3(mpko)3](ClO4) [R = Me (45a), Et (45b), and Ph (45c)] in almost quantitative yields (Stamatatos et al., 2005, 2007c), Equation (39). The 1:3 ratio was chosen to allow for the addition of one mpko− ligand onto each edge of the triangle. The process is a simple substitution reaction involving replacement of 3 groups and 3 py ligands by three mpko− ones. As in 44a–44c, the cations of 45a–45c contain a {(μ3-O)}7+ triangular core, but with each edge now bridged by a 2.11 group and a diatomic oximato group of one mpko− ligand (see Figure 11). The 3 groups lie on one side of the {} plane and the three oximato groups on the other. The 2.111 coordination mode of the mpko− ligands (MM with R = Me and M = MnIII in Figure 2) results in a buckling of the formerly planar {(μ3-O)}7+ core; the consequences are a relative twisting of the three metal octahedra and the displacement of the central O2− group ~0.3 Å above the {} plane on the same side as the groups. The MnIII···MnIII exchange interactions are ferromagnetic in 45a–45c resulting in an S = 6 ground state. The complexes are SMMs with Ueff values of ~8 cm−1. Magnetization vs. dc field experiments on single crystals of 45a·3CH2Cl2 display hysteresis loops that exhibit steps due to QTM. The loops are temperature-independent below 0.3 K, suggesting only QTM in the ground state between the lowest-lying MS = ±6 levels. High-frequency EPR spectra of single crystals of 45a·3CH2Cl2 provide strong evidence for a non-negligible transverse anisotropy of E ≥ 0.015 cm−1.
Figure 11. Schematic drawing of the synthetic strategy that leads to the 44→45 conversion and the ”switching on” of SMM properties. The symbol represents MnIII. The curved solid and dashed lines represent triatomic carboxylate and diatomic oximato bridging groups, respectively. Coordination MnIII-O2− bonds are drawn in bold.
DFT calculations reveal that the unusual ferromagnetic exchange interactions in complexes 45 may originate from a combination of the “non-planarity” (with respect to the {} plane) of the bridging oximato groups and the non-parallel alignment of the Jahn-Teller axes; however, several factors are at play (Cano et al., 2008; Atanasov et al., 2011) including the {} out-of-phase shift of the triply bridging oxido group.
Ground-State Spin Switching and Enhancing SMM Properties Via Targeted Structural Distortion
In 2004, our group with the collaboration of Escuer's group reported cluster [O2(O2CMe)2(sao)6(EtOH)4] (46), where sao2− is the dianion of salicylaldoxime (saoH2; L with R = H in Figure 1). The complex can be synthesized by the 1:1 reaction between Mn(O2CMe)2·4H2O and saoH2 in EtOH at room temperature in moderate yields (~50%), Equation (40). Another route to good yields of pure 46 involves a comproportionation reaction between (Bun4N)(MnO4) and Mn(O2CMe)2·4H2O in the presence of saoH2 in EtOH (Milios et al., 2004).
The molecular structure of the complex, which is common to all members of this family (vide infra) consists of two off-set, stacked triangular {(μ3-O)} subunits held together via six sao2− ligands, which span each MnIII···MnIII edge of the two triangles, forming MnIII-N-O-MnIII bridges, as well as dimerizing the two {(μ3-O)} subunits via their phenolato and oximato O atoms (see Figure 12). The two groups are 2.11 (this also occurs in the firstly reported members of the family with formato, benzoate etc. groups) and sit “above” the plane of the symmetry equivalent triangles. Four of the sao2− ligands show the common 2.111 coordination mode (LL with R = H and M = MnIII in Figure 2), while the other two adopt the rather rare 3.211 mode (this is similar to the coordination mode NN observed in cluster 24, section 3.8, with a MnIII atom replacing NaI). The core is {(3.3-O)2(3.21-ONR″)2(2.11-ONR″)4}8+, where R″NO− = sao2−. The four “central” MnIII ions are 6-coordinate with distorted octahedral geometries, while the outermost MnIII ions exhibit square pyramidal geometries with an axial weak bonding interaction of ~3.5 Å to a phenolato O atom. Magnetic studies reveal that the compound has an S = 4 ground state as a result of ferromagnetic exchange between the two antiferromagnetically coupled {} triangles; it is also an SMM with Ueff = ~20 cm−1. While the S and Ueff values are not spectacular, the D-value (−1.2 cm−1) certainly is.
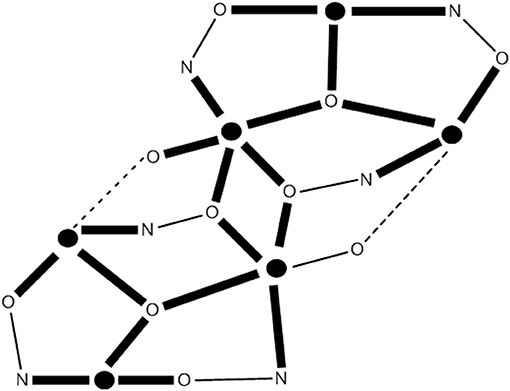
Figure 12. The magnetic core common to the {} family of SMMs. The symbol represents MnIII. The dashed lines represents the phenolato-o to square-based pyramidal MnIII interaction (Schematic drawing inspired by Milios et al., 2008).
In an elegant series of studies (Milios et al., 2007a,b,c,d, 2008), Brechin's group showed that by employing derivatised versions of the salicylaldoximate ligand (Rsao2−; R = Me, Et) and bulky carboxylates [R′; R = Ph, CMe3, CPh3, Ph2OPh, Ph4OPh, PhBr, Ph(Me)2, 3-thiophene…], it was possible to significantly distort the core of the molecule synthesizing family members of general formula [O2(O2CR′)2(Rsao)6(solvents)4−6] in which the planarity of the {MnIII-N-O-MnIII} moieties is significantly disrupted and the carboxylate groups become monodentate. There is a dramatic effect on the magnetic properties; the ground-state spin increases from S = 4 to S = 12 and the SMM behavior improves. Studies on ~30 family members suggest that the switch from antiferromagnetic to ferromagnetic pairwise exchange occurs when the MnIII-N-O-MnIII torsion angle becomes larger than ~31°. The more puckered the ring (-Mn-N-O-)3, the more ferromagnetic the exchange and the larger the barrier to magnetization reversal (Ueff) becomes. These studies have allowed for a rare semi-quantitive magnetostructural correlation in SMMs which enables explanation and prediction of the magnetic properties of family members. Examples include clusters [O2(O2CPh)2(etsao)6(EtOH)4(H2O)2] (47; S = 12, D = −0.43 cm−1, Ueff = 36.9 cm−1), [O2{O2CPh(Me)2}2(etsao)6(EtOH)6] (48; S = 12, D = −0.43 cm−1, Ueff = 60.1 cm−1), [O2(O2CCMe3}2(etsao)6(EtOH)5] (49; S = 7, D = −0.75 cm−1, Ueff = 20.9 cm−1), [O2(O2CPh4OPh)2(etsao)6(EtOH)4(H2O)2] (50; S = 9, D = −0.37 cm−1, Ueff = 39.6 cm−1) and [O2(O2CPhBr)2(mesao)6(EtOH)6] (51; S = 11, D = −0.50 cm−1, Ueff = 34.9 cm−1); mesao2− and etsao2− are the dianions of 2-hydroxyphenylethanone oxime and 2-hydroxyphenylpropanone oxime, respectively (L with R = Me, Et in Figure 1). Complex 48 holds the record Ueff (60.1 cm−1) and TB (~4.5 K) values for a 3d-metal cluster with SMM properties.
It should be mentioned at this point that the strategy detailed in this section is different from the approaches described in sections “Switching on” SMM Behavior through Substitution of Bridging Hydroxido Groups by End-On Azido or Isocyanato Ligands in Pre-formed Clusters, “Tweaking” the Spin, and “Switching on” SMM properties upon Conversion of Low-Spin Complexes Into High-Spin Ones Without Changing the Core. The present strategy is strictly speaking not a switching on of the SMM property (sections “Switching on” SMM Behavior Through Substitution of Bridging Hydroxido Groups by End-on Azido or Isocyanato Ligands in Pre-formed Clusters and “Switching on” SMM properties upon Conversion of Low-Spin Complexes Into High-Spin Ones Without Changing the Core), since the S = 4 {} cluster is also an SMM. Also, the increase of the total ground-state spin here is large (~200%) and not small (~20%) as in the “spin tweaking” strategy (section “Tweaking” the Spin); in both strategies the starting materials are SMMs.
Synthesis of Radical-Bridged 3d-Metal SMMs
As it has been clearly stated in sections Introduction and Synthesis of 3d-Metal SMMs: General Considerations, the continuing research efforts to prepare 3d-metal SMMs with higher Ueff values take into consideration the ground-state spin, S, and the axial zero-field splitting parameter, D; both these key parameters are correlated to the energy barrier height. A third important parameter, which is often overlooked, is the exchange coupling constant, J. The value of this physical parameter determines the separation between the spin of the ground state and the spins of the excited states. The J-value must be large (strong coupling) in an SMM to maintain slow magnetization relaxation at elevated temperatures and also to decrease fast magnetization relaxation through pathways involving excited spin states. An excellent synthetic strategy to achieve strong magnetic exchange is the incorporation of radical ligands into coordination complexes. The direct exchange between paramagnetic 3d-metal ions and paramagnetic radical ligands can result in very strong magnetic coupling, stronger than the more common superexchange interactions between the metal centers (Demir et al., 2015). In the coordination clusters of 3d-metal ions, the radical ligand is often bridging. Transition metal-based polynuclear SMMs possessing a radical ligand are extremely rare, most of them being dinuclear. We give an example of a dinuclear complex below.
Benzoquinonoid groups (A in Figure 13) are ideal ligands for the synthesis of radical-bridged SMMs with strong magnetic coupling. They can easily undergo redox reactions providing paramagnetic and diamagnetic electron-transfer isomeric species. The reaction of Fe(CF3SO3)2 with tris(2-pyridylmethyl)amine (tpya; T in Figure 1) in anhydrous MeCN, followed by treatment with a mixture of Li{N(SiMe3)2} and NPhLH2 (U in Figure 1) gives a dark brown solution, from which dark yellow crystals of the dinuclear complex [(NPhL2−)(tpya)2](CF3SO3)2 (52) are isolated, Equation (41). Subsequent reaction of this complex with a strong reducing agent, e.g., [CoII(C5Me5)2] in MeCN/MeOH at −35°C leads to the dark blue complex [(NPhL3−•)(tpya)2](CF3SO3) (53), Equation (42). Compound 53 is the one-electron reduced analog of 52 (Jeon et al., 2013).
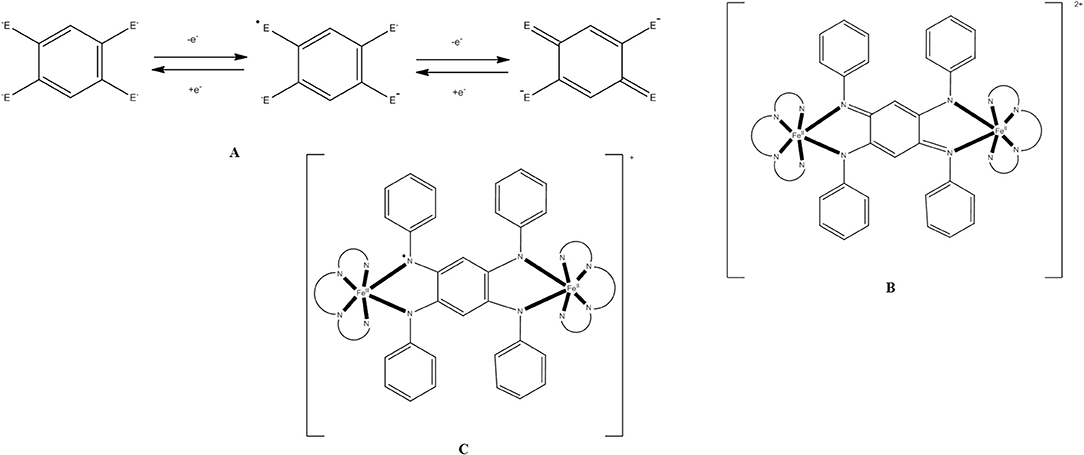
Figure 13. (A) Redox transformations of anionic benzoquinonoid ligands (E = O and NR; R is the Ph group in the present example). (B) Schematic representation of the molecular structure of the dication that is present in 52. (C) Schematic representation of the molecular structure of the monocation that is present in 53. N∧N∧N∧N∧ represents the tetradentate chelating ligand tpya. Coordination bonds are drawn in bold (Schematic drawing inspired by Jeon et al., 2013; Demir et al., 2015).
The molecular structures of both unreduced (52) and reduced (53) complexes show two {(tpya)FeII}2+ subunits connected by a deprotonated azophenine bridging ligand, with two N atoms occupying cis sites of each octahedral FeII ion; the four N atoms of the tetradentate chelating tpya ligand complete the coordination sphere at each metal center (see B for 52 and C for 53 in Figure 13). While the gross structural features of the cations of 52 and 53 are similar, bond lengths in the backbone of the bridging ligand show key differences which permit the assignment of a trianionic bridging ligand with an unpaired electron (NPhL3−•) in the case of 53. This assignment is supported by 57Fe-Mössbauer studies; the identical isomer shifts, δ, in the two complexes suggest that the reduction is ligand-centered, while the larger quadrupole splitting, ΔEQ, in 53 is due to the change of the ligand field at FeII associated with ligand reduction and the larger distortion from the ideal octahedral environment at the metal ion in 53 compared to 52. Variable-temperature dc magnetic susceptibility studies for solid 52 show a weak antiferromagnetic exchange interaction between the high-spin FeII ions through the diamagnetic NPhL2− ligand leading to an S = 0 ground state; the J value is −2.90(2) cm−1 based on the spin Hamiltonian Ĥ = −2J(ŜFe1·ŜFe2). Data for 53 reveal an exceptionally strong antiferromagnetic exchange between FeII centers and the NPhL3−• radical ligand (J ≤ −900 cm−1 based on the same Hamiltonian). SMM behavior was confirmed for 53 through ac susceptometry with a Ueff value of 50 cm−1. The |J| ≥ 900 cm−1 value is the strongest magnetic exchange ever observed in an SMM.
Supramolecular Approaches in Manganese SMM Chemistry
For several technological applications of SMMs, coupling of two or more SMMs to each other or to components of a device is very important. The coupling must be very weak in order to preserve the intrinsic single-molecule properties of each SMM. This coupling can be, in principle, achieved by H bonds and many H-bonded dimers, 1D, 2D, and 3D networks have been reported in the literature. However, reliance on H bonds to obtain the desired inter-SMM associations is a synthetic weakness, because it is difficult to control the oligomerization and to achieve retention of the supramolecular structure in solution. The answer to the problem is the linking of SMMs via coordination bonds. This approach gives often coordination polymers (1D, 2D, 3D) in which the coupling between the SMMs is strong leading to loss of the SMM property and appearance of single-chain magnetism (for 1D compounds) or ordering (for the 2D and 3D compounds). A synthetic challenge is thus the connection of “good” SMMs into discrete oligomers and the achievement of very weak exchange coupling between them through the bridging ligands, so that the properties of the oligomer can be considered as a small perturbation of the properties of the constituent SMM subunits. The desired oligomers can be prepared by direct mixing simple starting materials, but such a route offers limited synthetic control and there is no guarantee that the magnetic properties will be the targeted ones. A good strategy is the use of building-block approaches to link SMMs together employing carefully chosen linker groups that will provide weak inter-SMM interactions and ensure that discrete oligomeric species (and not polymers) are obtained. The groups of Papaefstathiou, Brechin, Christou, and Escuer, among others, have successfully contributed into this general synthetic goal (Stoumpos et al., 2009; Inglis et al., 2010; Cordero et al., 2011; Nguyen et al., 2011, 2015, 2016; Mowson et al., 2013). We herein present examples of the supramolecular approaches which have been almost exclusively developed in Mn SMM chemistry.
Complexes 45a, 45b, and 45c (section “Switching on” SMM properties upon Conversion of Low-Spin Complexes Into High-Spin Ones Without Changing the Core) are good SMM building blocks for the application of the supramolecular approach. They have an S = 6 ground state and are SMMs. Their triangular cations have C3 symmetry with the carboxylate and mpko− ligands on opposite sites of the plane defined by the three metal ions. This tripodal arrangement of the three oximato groups suggested (Nguyen et al., 2011, 2016) that their replacement with dioximate groups to link multiple {} units together might give oligomeric complexes rather than polymeric species. The linker of choice has been 3-phenyl-1,5-bis(pyridin-2-yl)pentane-1,5-dione (pdpdH2; V in Figure 1). This ligand consists of two mpkoH (H with R = Me in Figure 1) groups that are connected by a benzyl unit. The 1:2 reaction of 44a and pdpdH2 in CH2Cl2 gives complex [O4(O2CMe)12(pdpd)6](ClO4)4 (54) in 35% yield, Equation (43). The excess of pdpdH2 (the stoichiometric ratio is 2:3) gives the maximum possible yield.
The cation of 54 contains 4 {(μ3-O)}7+ subunits connected by 6 pdpd2− ligands to give a rectangular {}4 supramolecular species with each triangular unit at a corner. One of the two 2.11- groups bridging each edge of 44a has been replaced by a bridging oximato group from a pdpd2− ligand. The short and long sides of the rectangle are bridged by two and one pdpd2− ligands, respectively. In addition, the 2-pyridyl N atoms of pdpd2− have replaced the terminal py molecules of 44a. The local structure of each {} unit of 54 is similar to that of the mpko− - containing complex 45a. As in 45a, the 3 bridging oximato groups are on the same side of the {} plane, and this leads to a tetramer of triangles rather than a coordination polymer. Solid-state dc magnetic susceptibility studies show that the {} subunits of 54 possess an S = 6 ground state. Ac susceptibility studies indicate the dodecanuclear cluster to be a tetramer of {} SMMs. Magnetization vs. dc field studies on a single crystal of 54·xCH2Cl2 give hysteresis loops below 1 K that exhibit exchange-biased QTM with a bias field of 0.19 T. Simulation of the loops reveals that each {} subunit is exchange-coupled to the two neighboring subunits connected to it by the pdpd2− bridging ligands, with an antiferromagnetic inter-{} exchange interaction of J/KB = −0.008 cm−1. Thus, the synthesis of 54 confirms the feasibility of connecting multiple Mnx SMMs to isolate a discrete supramolecular “cluster of SMMs” with only weak coupling between them.
Supramolecular aggregates of the {} SMMs 45 can also be prepared through the carboxylato sites of the staring materials, thus complementing the reports of aggregation at the oximato positions using dioximate linkers (Mowson et al., 2013). The latter strategy is not by substitution on pre-formed 45, but the former is. The 2:3 reaction of 45a and fumaric acid (fumH2; W in Figure 1) in MeCN gives complex [O4(fum)6(mpko)12](ClO4)4 (55) in ~35% yield, Equation (44). The reaction of 45a and bulky α-truxillic acid (atxH2; X in Figure 1) in a 2:3 ratio in MeCN/EtOH leads to cluster [O2(atx)4(mpko)6] (56) in 30% yield, Equation (45).
The cation of 55 consists of a {}4 tetrahedron with a 4.1111 fum2− (TT in Figure 2) bridging each edge to give an adamantane-like motif, which is a common unit in many supramolecular systems. Each carboxylate group of fum2− ligand is 2.11 and thus bridges within a {} subunit like the group that it replaces. Cluster 55 can be described as a tetramer of 45a held together by fum2− linkers. Complex 56 is a {}2 dimer (see Figure 14 for its core); for steric reasons (two bulky Ph substituents per ligand) it is bridged by only two 4.1111 atx2− groups (UU in Figure 2), with two additional atx2− groups coordinated only through one carboxylate at each {} subunit (VV in Figure 2). The mpko− ligands in 55 and 56 bind in the familiar 2.111 mode (MM with R = Me and M = MnIII in Figure 2). Detailed magnetic studies indicate that the S = 6 {} subunits within the “tetramer” (55) and “dimer” (56) aggregates are only weakly interacting and retain the intrinsic SMM properties of the “monomeric” {} complex (45a). Ac magnetic susceptibility studies of 55 and 56 show the appearance of out-of-phase signals below 3.0 K, indicating the slow relaxation of SMMs.
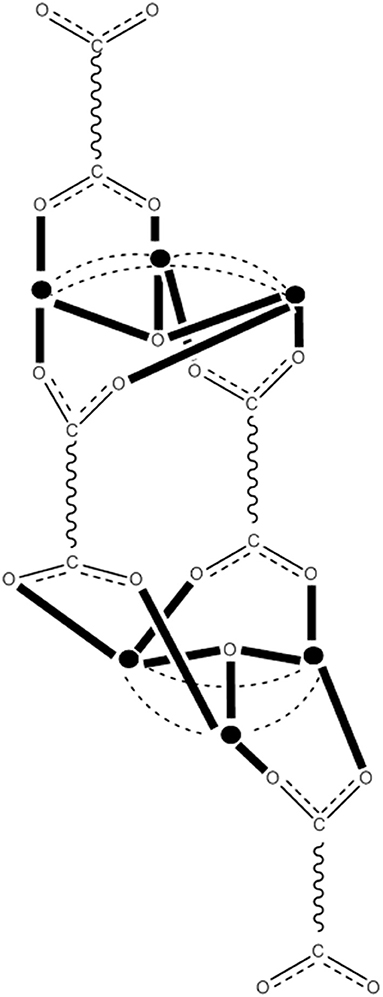
Figure 14. An extended view of the core of cluster 56. The symbol represents MnIII. The dashed curve represents the diatomic oximato groups of mpko−. The line  represents the aromatic and aliphatic backbone of the atx2− ligands. Coordination bonds are drawn in bold (Schematic drawing inspired by Mowson et al., 2013).
represents the aromatic and aliphatic backbone of the atx2− ligands. Coordination bonds are drawn in bold (Schematic drawing inspired by Mowson et al., 2013).
Another attractive ligand for the supramolecular synthesis of Mn SMM is 1,3-di(pyridin-2-yl)propane-1,3-dione dioxime (dpdH2; Y in Figure 1). This ligand represents a fusion of two mpkoH (H with R = Me in Figure 1) groups at the methyl group. The single sp3 C atom at the center of the molecule reduces the conformational flexibility with respect to pdpdH2 (V in Figure 1), and the combination of a ~109° ditopic dioximate with a tritopic {} unit should yield a {}2 “dimer” with three linker groups and parallel {} planes (to facilitate study of quantum properties). Another parameter for the selection of dpdH2 is the fact that binding by tridentate 2-pyridyloximate groups of three doubly deprotonated linkers would provide robustness and rigidity in the “dimer,” resulting in retention of the structure in solution. Moreover, the single sp3 carbon atom was expected to propagate a very weak (but non-zero) superexchange interaction between the two {} SMMs and therefore a small perturbation on a two-SMM system (Nguyen et al., 2015). The reaction of 44a with dpdH2 and I2 in a 2:3:6 molar ratio in EtOH/CH2Cl2 gives [O2(O2CMe)6(dpd)3](I3)2 (57) in 22% yield, Equation (46). I2 is added to stop reduction of some of the MnIII to MnII, as sometimes observed in such reactions.
The structure of the hexanuclear dication consists of two {(μ3-O)}7+ subunits linked by three dpd2− ligands to give a {} “dimer,” with the two {} planes being parallel. Each trinuclear subunit is structurally similar to that of “monomer” 45a. The dianionic ligand adopts the 4.111111 coordination mode (WW in Figure 2), each 2-pyridyloximate subgroup bridging two MnIII ions belong to a {} subunit. Solid-state dc and ac magnetic susceptibility studies indicate that each {} subunit of the “dimer” is a separate SMM with an S = 6 ground state and that the two SMM subunits interact very weakly in a ferromagnetic fashion. The ferromagnetic nature of the coupling arises from a spin polarization mechanism through the central sp3 carbon atom, caused by spin density delocalized into the dpd2− ligand π systems from the MnIII dπ magnetic orbitals. High-frequency EPR spectra on a single crystal of 57 exhibit signal splittings that indicate quantum superposition of two SMMs. Studies on toluene/MeCN (1:1 v/v) solutions display the same spectra, indicating that the “dimer” retains its structure and the weak inter-{} coupling in solution. This is of particular importance in the currently intense efforts for the deposition of SMMs on surfaces and other substrates.
Brief Summary and Outlook
We hope that this review has provided the readers with a “flavor” of the synthetic chemistry and reactivity studies of 3d-metal coordination clusters with SMM properties. Empirical routes (section The Years of Innocence: “Try and See” Exercises) and strategies (section The Years of Design: Strict and Less Strict Synthetic Strategies) have been discussed and critically analyzed through selected examples; often the distinction between an empirical route and a strict strategy is blurred. Particular emphasis has been given in the synthetic rationale behind the reactions and in the criteria for the selection of the metal ions and the ligands. Most strategies refer to Mn chemistry, because the relatively large number of unpaired electrons (four) and the large easy-axis anisotropy (negative D) for the Jahn-Teller elongated high-spin MnIII make this ion an ideal candidate for the construction of SMMs.
The extremely low temperatures at which the polynuclear 3d-metal complexes operate as SMMs (<4.5 K) make these molecules not suitable for technological applications. Their future utility is in low-temperature and specialized applications that take full advantage of their unique molecular characteristics (size, monodispersity, crystallinity, good solubility, feasibility of altering their peripheral ligation, and well-defined quantum properties). However, the inability of polynuclear 3d-metal SMMs for every day applications does not underestimate their importance. These compounds are responsible for great revolutions in synthetic (for their preparation and reactivity) and physical (for their characterization and study) Inorganic Chemistry, a field that has undergone a second renaissance (the first was in the 50's and early 60's) after the second world war.
Most research efforts in the chemistry of molecular compounds exhibiting slow magnetization relaxation (at zero field) are now focused on 4f- and 5f-metal SIMs for reasons outlined in section Introduction. However, we do believe that there is still enough room for the improvement of the existing strategies and the development of new ones in the chemistry of 3d-metal clusters with SMM properties. We expect that the synthesis and the study of radical-bridged 3d-metal SMMs (section Synthesis of Radical-Bridged 3d-Metal SMMs) will be intensified in the future because of the potential of huge J-values. We also predict new developments in the supramolecular chemistry of 3d-metal SMMs (section Supramolecular Approaches in Manganese SMM Chemistry), since this is related to the linking of SMMs on surface and other substrates. A final point of synthetic interest is that the larger radial extension of the 4d and 5d orbitals (compared to the 3d ones) allows for stronger exchange coupling through diamagnetic bridging ligands. Since the strong exchange is a key factor in SMM research, we anticipate an increased synthetic activity in the area of 4d- and 5d-metal cluster SMMs (not covered in this review).
We conclude by pointing out that it is hoped that this review will be proven useful for inorganic chemists who are already active in SMM research or just enter into this area, and we shall be happy if the readers enjoy the review as we enjoyed writing it.
Author Contributions
DM and EP jointly studied the literature, proposed some of the cited examples, prepared the figures, and typed the manuscript. SP developed the concept of the review and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SP is grateful to the COST Action: CA15128-Molecular Spintronics (MOLSPIN) for encouraging his group activities in Patras.
References
Abasi, P., Quinn, K., Alexandropoulos, D. I., Damjanovich, M., Wernsdorfer, W., Escuer, A., et al. (2017). Transition metal single-molecule magnets: a {Mn31} nano-sized cluster with a large energy barrier of ~60 K and magnetic hysteresis at ~5 K. J. Am. Chem. Soc. 139, 15644–15647. doi: 10.1021/jacs.7b10130
Affronte, M. (2009). Molecular nanomagnets for information technologies. J. Mater. Chem. 19, 1731–1737. doi: 10.1039/B809251F
Affronte, M., Troiani, F., Ghirri, A., Candini, A., Evangelisti, M., Corradini, V., et al. (2007). Single molecule magnets for quantum computation. J. Phys. D 40, 2999–3004. doi: 10.1088/0022-3727/40/10/S01
Alexandropoulos, D. I., Cunha-Silva, L., Escuer, A., and Stamatatos, T. C. (2014). New classes of ferromagnetic materials with exclusively end-on azido bridges: from single-molecule magnets to 2D molecule-based magnets. Chem. Eur. J. 20, 13860–13864. doi: 10.1002/chem.201403815
Aromi, G., and Brechin, E. K. (2006). Synthesis of 3d metallic single-molecule magnets. Struct. Bond. 122, 1–67. doi: 10.1007/430_022
Artus, P., Boskovic, C., Yoo, J., Streib, W. E., Brunel, L.-C., Hendrickson, D. N., et al. (2001). Single-molecule magnets: site-specific ligand abstraction from [Mn12O12(O2CR)16(H2O)4] and the preparation and properties of [Mn12O12(NO3)4(O2CCH2 tBu)12(H2O)4]. Inorg. Chem. 40, 4199–4210. doi: 10.1021/ic010252g
Atanasov, M., Delley, B., Neese, F., Tregenna-Piggott, P. L., and Sigrist, M. (2011). Theoretical insights into the magnetostructural correlations in Mn3-based single-molecule magnets. Inorg. Chem. 50, 2112–2124. doi: 10.1021/ic1023482
Aubin, S. M. J., Sun, Z., Pardi, L., Krzystek, J., Folting, K., Brunel, L. C., et al. (1999). Reduced anionic Mn12 molecules with half-integer ground states as single-molecule magnets. Inorg. Chem. 38, 5329–5340. doi: 10.1021/ic990613g
Bagai, R., and Christou, G. (2007). A fourth isolated oxidation level of the [Mn12O12(O2CR)16(H2O)4] family of single-molecule magnets. Inorg. Chem. 46, 10810–10818. doi: 10.1021/ic7015758
Bagai, R., and Christou, G. (2009). The drosophila of single-molecule magnetism: [Mn12O12(O2CR)16(H2O)4]. Chem. Soc. Rev. 38, 1011–1026. doi: 10.1039/b811963e
Bar, A. K., Pichon, C., and Sutter, J.-P. (2016). Magnetic anisotropy in two- to eight-coordinated transition-metal complexes: recent developments in molecular magnetism. Coord. Chem. Rev. 308, 346–380. doi: 10.1016/j.ccr.2015.06.013
Beltran, L. M., and Long, J. R. (2005). Directed assembly of metal-cyanide cluster magnets. Acc. Chem. Res. 38, 325–334. doi: 10.1021/ar040158e
Benelli, C., and Gatteschi, D. (2015). Introduction to Molecular Magnetism-From Transition Metals to Lanthanides. Weinheim: Wiley-VCH.
Bogani, L., and Wernsdorfer, W. (2008). Molecular spintronics using single-molecule magnets. Nature Mater. 7, 176–186. doi: 10.1038/nmat2133
Boskovic, C., Pink, M., Huffman, J. C., Hendrickson, D. N., and Christou, G. (2001). Single-molecule magnets: ligand-induced core distortion and multiple Jahn-Teller isomerism in [Mn12O12(O2CMe)8(O2PPh2)8(H2O)4]. J. Am. Chem. Soc. 123, 9914–9915. doi: 10.1021/ja016341
Boudalis, A. K., Donnadieu, B., Nastopoulos, V., Clemente-Juan, J. M., Mari, A., Sanakis, Y., et al. (2004). A nonanuclear iron(II) single-molecule magnet. Angew. Chem. 116, 2316–2320. doi: 10.1002/ange.200353147
Boudalis, A. K., Sanakis, Y., Clemente-Juan, J. M., Donnadieu, B., Nastopoulos, V., Mari, A., et al. (2008). A family of enneanuclear iron(II) single-molecule magnets. Chem. Eur. J. 14, 2514–2526. doi: 10.1002/chem.200701487
Brechin, E. K. (2005). Using tripodal alcohols to build high-spin molecules and single-molecule magnets. Chem. Commun. 7, 5141–5153. doi: 10.1039/b510102f
Brechin, E. K., Graham, A., Milne, P. E. Y., Murrie, M., Parsons, S., and Winpenny, R. E. P. (1999). New high-spin clusters featuring transition metals. Philos. Trans. R. Soc. Lond. A 357, 3119–3137. doi: 10.1098/rsta.1999.0484
Caneschi, A., Gatteschi, D., Sessoli, R., Barra, A. L., Brunel, L. C., and Guillot, M. (1991). Alternating current susceptibility, high field magnetization, and millimeter band EPR evidence for a ground S = 10 state in [Mn12O12(CH3COO)16(H2O)4]2CH3COOH4H2O. J. Am. Chem. Soc. 113, 5873–5874. doi: 10.1021/ja00015a057
Cano, J., Cauchy, J., Ruiz, E., Milios, C. J., Stoumpos, C. C., Stamatatos, T. C., et al. (2008). On the origin of ferromagnetism in oximato-based [Mn3O]7+ triangles. Dalton Trans. 14, 234–240. doi: 10.1039/B710055H
Chakov, N. E., Wernsdorfer, W., Abboud, K. A., Hendrickson, D. N., and Christou, G. (2003). Single-molecule magnets. A Mn12 complex with mixed carboxylate-sulfonate ligation: [Mn12O12(O2CMe)8(O3SPh)8(H2O)4]. Dalton Trans. 2243–2248. doi: 10.1039/B301785K
Cordero, B., Roubeau, O., Teat, S. J., and Escuer, A. (2011). Building of a novel Mn12 single molecule magnet by assembly of anisotropic {Mn3(μ3-O)(salox)3} triangles. Dalton Trans. 40, 7127–7129. doi: 10.1039/c1dt10782h
Cornia, A., Fabretti, A. C., Pacchioni, M., Zobbi, L., Bonacchi, D., Caneschi, A., et al. (2003). Direct observation of single-molecule magnets organized on gold surfaces. Angew. Chem. Int. Ed. 42, 1645–1648. doi: 10.1002/anie.200350981
Coronado, E., and Dunbar, K. R. (2009). Preface for the forum on molecular magnetism: the role of inorganic chemistry. Inorg. Chem. 48, 3293–3295. doi: 10.1021/ic900218f
Coronado, E., and Gatteschi, D. (2006). Trends and challenges in molecule-based magnetic materials. J. Mater. Chem. 16, 2513–2515. doi: 10.1039/b608099p
Coxall, R. A., Harris, S. G., Henderson, D. K., Parsons, S., Tasker, P. A., and Winpenny, R. E. P. (2000). Inter-ligand reactions: in situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2349–2356. doi: 10.1039/b001404o
Craig, G. A., and Murrie, M. (2015). 3d single-ion magnets. Chem. Soc. Rev. 44, 2135–2147. doi: 10.1039/C4CS00439F
Demir, S., Jeon, I.-R., Long, J. R., and Harris, T. D. (2015). Radical ligand-containing single-molecule magnets. Coord. Chem. Rev. 289–290, 149–176. doi: 10.1016/j.ccr.2014.10.012
Domingo, N., Bellido, E., and Ruiz-Molina, D. (2012). Advances on structuring, intergration and magnetic characterization of molecular nanomagnets on surfaces and devices. Chem. Soc. Rev. 41, 258–302. doi: 10.1039/C1CS15096K
Dunbar, K. R. (2012). Editorial for the virtual issue on quantum molecular magnets. Inorg. Chem. 51, 12055–12058. doi: 10.1021/ic302312m
Eppley, H. J., Tsai, H.-L., De Vries, N., Folting, K., Christou, G., and Hendrickson, D. N. (1995). High-spin molecules: Unusual magnetic susceptibility relaxation effects in [Mn12O12(O2CEt)16(H2O)3] (S = 9) and the one-electron reduction product (PPh4) [Mn12O12(O2CEt)16(H2O)4] (S = 19/2). J. Am. Chem. Soc. 117, 301–317. doi: 10.1021/ja00106a033
Escuer, A., and Aromi, G. (2006). Azide as a bridging ligand and magnetic coupler in transition metal clusters. Eur. J. Inorg. Chem. 2006, 4721–4736. doi: 10.1002/ejic.200600552
Escuer, A., Esteban, J., Perlepes, S. P., and Stamatatos, T. C. (2014). The bridging azido ligand as a central “player” in high-nuclearity 3d-metal cluster chemistry. Coord. Chem. Rev. 275, 87–129. doi: 10.1016/j.ccr.2014.04.001
Feltham, H. L. C., and Brooker, S. (2014). Review of purely 4f and mixed-metal nd-4f single-molecule magnets containing only one lanthanide ion. Coord. Chem. Rev. 276, 1–33. doi: 10.1016/j.ccr.2014.05.011
Ferguson, A., Parkin, A., Sanchez-Benitez, J., Kamenev, K., Wernsdorfer, W., and Murrie, M. (2007). A mixe-valence Co7 single-molecule magnet with C3 symmetry. Chem. Commun. 2007, 3473–3475. doi: 10.1039/b706238a
Friedman, J. R., Sarachik, M. P., Tejada, J., and Ziolo, R. (1996). Macroscopic measurement of resonant magnetization tunneling in high-spin molecules. Phys. Rev. Lett. 76, 3830–3833. doi: 10.1103/PhysRevLett.76.3830
Frost, J. M., Harriman, K. L. M., and Murugesu, M. (2016). The rise of 3-d single-ion magnets in molecular magnetism: towards materials from molecules? Chem. Sci. 7, 2470–2491. doi: 10.1039/c5sc03224e
Gatteschi, D., and Sessoli, R. (2003). Quantum tunneling of magnetization and related phenomena in molecular materials. Angew. Chem. Int. Ed. 42, 268–297. doi: 10.1002/anie.200390099
Gatteschi, D., Sessoli, R., and Villain, J. (2006). Molecular Nanomagnets. Oxford: Oxford Univercity Press.
Goodwin, C. A. P., Ortu, F., Reta, D., Chilton, N. F., and Mills, D. P. (2017a). Molecular magnetism hysteresis at 60 Kelvin in dysprocenium. Nature 548, 439–442. doi: 10.1038/nature23447
Goodwin, C. A. P., Reta, D., Ortu, F., Chilton, N. F., and Mills, D. P. (2017b). Synthesis and electronic structures of heavy lanthanide metallocenium cations. J. Am. Chem. Soc. 139, 18714–18724. doi: 10.1021/jacs.7b11535
Guo, F. S., Day, B. M., Chen, Y. C., Tong, M. L., Mansikkamäki, A., and Layfield, R. A. (2017). A dysprosium metallocene single-molecule magnet functioning at the axial limit. Angew. Chem. Int. Ed. 56, 11445–11449. doi: 10.1002/anie.201705426
Guo, Y. N., Xu, G. F., Guo, Y., and Tang, J. (2011). Relaxation dynamics of dysprosium(III) single-molecule magnets. Dalton Trans. 40, 9953–9963. doi: 10.1039/c1dt10474h
Gupta, S. K., and Murugavel, R. (2018). Enriching lanthanide single-ion magnetism through symmetry and axiality. Chem. Comm. 54, 3685–3696. doi: 10.1039/C7CC09956H
Habib, F., and Murugesu, M. (2013). Lessons learned from dinuclear lanthanide nano-magnets. Chem. Soc. Rev. 42, 3278–3288. doi: 10.1039/c2cs35361j
Hernández-Molina, R., and Mederos, A. (2004). “Acyclic and macrocyclic Schiff base ligands”, in Comprehensive Coordination Chemistry II, Vol. 1, eds J. A. McCleverty and T. J. Meyer (Elsevier: Amsterdam), 411–446.
Inglis, R., Katsenis, A. D., Collins, A., White, F., Milios, C. J., Papaefstathiou, G. S., et al. (2010). Assembling molecular triangles into discrete and infinite architectures. Cryst. Growth Des. 12, 2064–2072. doi: 10.1039/C002761H
Inglis, R., White, F., Piligkos, S., Wernsdorfer, W., Brechin, E. K., and Papaefstathiou, G. S. (2011). Chiral single-molecule magnets: a partial Mn(III) supertetrahedron from achiral components. Chem. Commun. 47, 3090–3092. doi: 10.1039/c0cc05750a
Jeon, I. R., Park, J. G., Xiao, D. J., and Harris, T. D. (2013). An azophenine radical-bridged Fe2 single-molecule magnet with record magnetic exchange coupling. J. Am. Chem. Soc. 135, 16845–16848. doi: 10.1021/ja409927v
Jones, L. F., Rajaraman, G., Brockman, J., Murugesu, M., Sañudo, E. C., Raftery, J., et al. (2004). New routes to polymetallic clusters: fluoride-based tri-, deca- and hexaicosametallic MnIII clusters and their magnetic properties. Chem. Eur. J. 10, 5180–5194. doi: 10.1002/chem.200400301
King, P., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2004). A family of Mn16 single-molecule magnets from a reductive aggregation route. Inorg. Chem. 43, 7315–7323. doi: 10.1021/ic048906j
King, P., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2005). Single-molecule magnets: a reductive aggregation route to new types of Mn12 complexes. Inorg. Chem. 44, 8659–8669. doi: 10.1021/ic051150j
Kitos, A. A., Efthymiou, C. G., Papatriantafyllopoulou, C., Nastopoulos, V., Tasiopoulos, A. J., Manos, M. J., et al. (2011). The search for cobalt single-molecule magnets. A disk-like CoIII with a ligand derived from a novel transformation of 2-acetylpyridine. Polyhedron 30, 2987–2996. doi: 10.1016/j.poly.2011.02.013
Kostakis, G. E., Perlepes, S. P., Blatov, V. A., Proserpio, D. M., and Powell, A. K. (2012). High-nuclearity cobalt coordination clusters: synthetic, topological and magnetic aspects. Coord. Chem. Rev. 256, 1246–1278. doi: 10.1016/j.ccr.2012.02.002
Langley, S., Heliwell, M., Sessoli, R., Teat, S. J., and Winpenny, R. E. (2008). Synthesis and structural and magnetic characterization of cobalt(II) phosphonate cage compounds. Inorg. Chem. 47, 497–507. doi: 10.1021/ic700984r
Layfield, R. A. (2014). Organometallic single-molecule magnets. Organometallics 33, 1084–1099. doi: 10.1021/om401107f
Lecren, L., Wernsdorfer, W., Li, Y. G., Roubeau, O., Miyasaka, H., and Clerac, R. (2005). Quantum tunneling and quantum phase interference in a [] single-molecule magnet. J. Am. Chem. Soc. 127, 11311–11317. doi: 10.1021/ja050994z
Leuenberger, M. N., and Loss, D. (2001). Quantum computing in molecular magnets. Nature 410, 789–793. doi: 10.1038/35071024
Liddle, S. T., and van Slageren, J. (2015). Improving f-element single molecule magnets. Chem. Soc. Rev. 44, 6655–6669. doi: 10.1039/C5CS00222B
Lis, T. (1980). Preparation, structure, and magnetic properties of a dodecanuclear mixed-valence manganese carboxylate. Acta Crystalogr. Sect. B Struct. Sci. 36, 2042–2046.
Liu, J. L., Chen, Y. C., and Tong, M.-L. (2018). Symmetry strategies for high performance lanthanide-based single-molecule magnets. Chem. Soc. Rev. 47, 2431–2453. doi: 10.1039/C7CS00266A
Liu, K., Shi, W., and Cheng, P. (2015). Toward heterometallic single-molecule magnets: synthetic strategy, structures and properties of 3d-4f discrete complexes. Coord. Chem. Rev. 289–290, 74–112. doi: 10.1016/j.ccr.2014.10.004
Manoli, M., Inglis, R., Manos, M. J., Nastopoulos, V., Wernsdorfer, W., Brechin, E. K., et al. (2011). A [Mn32] double-decker wheel. Angew. Chem. Int. Ed. 50, 4441–4444. doi: 10.1002/anie.201100976
Manoli, M., Inglis, R., Piligkos, S., Yanhua, L., Wernsdorfer, W., Brechin, E. K., et al. (2016). A hexameric [Na6] wheel based on [O]7+ sub-units. Chem. Commun. 52, 12829–12832. doi: 10.1039/C6CC06644E
Meinhaus, K. R., and Long, J. R. (2015). Actinide-based single-molecule magnets. Dalton Trans. 44, 2517–2528. doi: 10.1039/C4DT02391A
Milios, C. J., Inglis, R., Vinslava, A., Bagai, R., Wernsdorfer, W., Parsons, S., et al. (2007a). Toward a magnetostructural correlation for a family of Mn6 SMMs. J. Am. Chem. Soc. 129, 12505–12511. doi: 10.1021/ja0736616
Milios, C. J., Piligkos, S., and Brechin, E. K. (2008). Ground state spin-switching via targeted structural distortion: twisted single-molecule magnets from derivatised salicylaldoximes. Dalton Trans. 2008, 1809–1817. doi: 10.1039/B716355J
Milios, C. J., Raptopoulou, C. P., Terzis, A., Lloret, F., Vicente, R., Perlepes, S. P., et al. (2004). Hexanuclear manganese(III) single-molecule magnets. Angew. Chem. Int. Ed. 43, 210–212. doi: 10.1002/anie.200351079
Milios, C. J., Stamatatos, T. C., and Perlepes, S. P. (2006). The coordination chemistry of pyridyl oximes. Polyhedron 22, 134–194. doi: 10.1016/j.poly.2005.07.022
Milios, C. J., Vinslava, A., Wernsdorfer, W., Moggach, S., Parsons, S., Perlepes, S. P., et al. (2007b). A record anisotropy barrier for a single-molecule magnet. J. Am. Chem. Soc. 129, 2754–2755. doi: 10.1021/ja068961m
Milios, C. J., Vinslava, A., Wernsdorfer, W., Prescimone, A., Wood, P. A., Parsons, S., et al. (2007c). Spin switching via targeted structural distortion. J. Am. Chem. Soc. 129, 6547–6561. doi: 10.1021/ja070411g
Milios, C. J., Vinslava, A., Wood, P. A., Parsons, S., Wernsdorfer, W., Christou, G., et al. (2007d). A single-molecule magnet with a “twist”. J. Am. Chem. Soc. 129, 8–9. doi: 10.1021/ja0666755
Milios, C. J., and Winpenny, R. E. P. (2015). Cluster-based single-molecule magnets. Struct. Bond. 164, 1–109. doi: 10.1007/430_2014_149
Mishra, A., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2006). [Mn7O5(OR)2(O2CPh)9(terpy)] (R = Me, CH2Ph) complexes with a fused cubane/butterfly core and an S = 6 ground-state spin. Inorg. Chem. 45, 10197–10206. doi: 10.1021/ic061334d
Moragues-Cánovas, M., Helliwell, M., Ricard, L., Riviére, E., Wernsorfer, W., Brechin, E., et al. (2004). An Ni4 single-molecule magnet: Synthesis, structure and low-temperature magnetic behavior. Eur. J. Inorg. Chem. 2004, 2219–2222. doi: 10.1002/ejic.200400129
Mowson, A. M., Nguyen, T. N., Abboud, K. A., and Christou, G. (2013). Dimeric and tetrameric supramolecular aggregates of single-molecule magnets via carboxylate substitution. Inorg. Chem. 52, 12320–12322. doi: 10.1021/ic402155h
Mukherjee, S., Abboud, K. A., Wernsdorfer, W., and Christou, G. (2013). Comproportionation reactions to manganese(III/IV) pivalate clusters: a new half-integer spin single-molecule magnet. Inorg. Chem. 52, 873–884. doi: 10.1021/ic302021a
Murugesu, M., Habrych, M., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2004). Single-molecule magnets: a Mn25 complex with a record S = 51/2 spin for a molecular species. J. Am. Chem. Soc. 126, 4766–4767. doi: 10.1021/ja031682
Murugesu, M., Takahasi, S., Wilson, A., Abboud, K. A., Wernsdorfer, W., Hill, S., et al. (2008). Large Mn25 single-molecule magnet with spin S = 51/2: magnetic and high-frequency electron paramagnetic resonance spectroscopic characterization of a giant spin state. Inorg. Chem. 47, 9459–9470. doi: 10.1021/ic801142p
Nguyen, T. N., Shiddiq, M., Ghosh, T., Abboud, K. A., Hill, S., and Christou, G. (2015). Covalently linked dimer of Mn3 single-molecule magnets and retention of its structure and quantum properties in solution. J. Am. Chem. Soc. 137, 7160–7168. doi: 10.1021/jacs.5b02677
Nguyen, T. N., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2011). A supramolecular aggregate of four exchange-biased single-molecule magnets. J. Am. Chem. Soc. 133, 20688–20691. doi: 10.1021/ja2087344
Nguyen, T. N., Wernsdorfer, W., Shiddiq, M., Abboud, K. A., Hill, S., and Christou, G. (2016). Supramolecular aggregates of single-molecule magnets: exchange-biased quantum tunneling of magnetization in a rectangular [Mn3]4 tetramer. Chem. Sci. 7, 1156–1173. doi: 10.1039/C5SC02599K
Palacio, F. (2014). In the dawn of magnets made from molecules. Chem. Commun. 50, 11437–11439. doi: 10.1039/C3CC49087D
Papaefstathiou, G. S., Escuer, A., Vicente, R., Font-Barbia, M., Solans, X., and Perlepes, S. P. (2001a). Reactivity in polynuclear transition metal chemistry as a means to obtain high-spin molecules: substitution of μ4-OH− by η1, μ4- increases nine times the ground state S value of a nonanuclear nickel(II) cage. Chem. Commun. 2414–2415. doi: 10.1039/b106472j
Papaefstathiou, G. S., and Perlepes, S. P. (2002). Families of polynuclear manganese, cobalt, nickel and copper complexes stabilized by various forms of di-2-pyridyl ketone. Comments Inorg. Chem. 23, 249–274. doi: 10.1080/02603590213135
Papaefstathiou, G. S., Perlepes, S. P., Escuer, A., Vicente, R., Font-Barbia, M., and Solans, X. (2001b). Unique single-atom binding of pseudohalogeno ligands to four metal ions induced by their trapping into high-nuclearity cages. Angew. Chem. Int. Ed. 40, 884–886. doi: 10.1002/1521-3773(20010302)40:5<884::AID-ANIE884>3.0.CO;2-C
Papatriantafyllopoulou, C., Stamatatos, T. C., Wernsdorfer, W., Teat, S. J., Tasiopoulos, A. J., Escuer, A., et al. (2010). Combining azide, carboxylate, and 2-pyridyloximate ligands in transition-metal chemistry: ferromagnetic clusters with a bowtie skeleton. Inorg. Chem. 49, 10486–10496. doi: 10.1021/ic1014829
Pedersen, K. S., Woodruff, D. N., Bendix, J., and Clérac, R. (2015). “Experimental aspects of lanthanide single-molecule magnet physics,” in Lanthanides and Actinides in Molecular Magnetism, 1st edn, eds R. A. Layfield and M. Murugesu (Weinheim: Wiley VCH), 125–152.
Pinkowicz, D., Southerland, H. I., Avendaño, C., Prosvirin, A., Sanders, C., Wernsdorfer, W., et al. (2015). Cyanide single-molecule magnets exhibiting solvent dependent reversible “on” and “off” exchange bias behavior. J. Am. Chem. Soc. 137, 14406–14422. doi: 10.1021/jacs.5b09378
Pointillart, F., Cador, O., Le Guennic, B., and Quahab, L. (2017). Uncommon lanthanide ions in purely 4f single molecule magnets. Coord. Chem. Rev. 346, 150–175. doi: 10.1016/j.ccr.2016.12.017
Polyzou, C. D., Efthymiou, C. G., Escuer, A., Cunha-Silva, L., Papatriantafyllopoulou, C., and Perlepes, S. P. (2013). In search of 3d/4f-metal single-molecule magnets: nickel(II)/lanthanide(III) coordination clusters. Pure Appl. Chem. 85, 315–327. doi: 10.1351/PAC-CON-12-09-08
Powell, A. K., Heath, S. L., Gatteschi, D., Pardi, L., Sessoli, R., Spina, G., et al. (1995). Synthesis, structures, and magnetic properties of Fe2, Fe17 and Fe19 oxo-bridged iron clusters: the stabilization of high ground state spins by cluster aggregates. J. Am. Chem. Soc. 117, 2491–2502. doi: 10.1021/ja00114a012
Rajaraman, G., Murugesu, M., Sañudo, E. C., Soler, M., Wernsdorfer, W., Heliwell, M., et al. (2004). A family of manganese rods: syntheses, structures, and magnetic properties. J. Am. Chem. Soc. 126, 15445–15457. doi: 10.1021/ja0471929
Rinehart, J. D., and Long, J. R. (2011). Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2, 2078–2085. doi: 10.1039/c1sc00513h
Rosado Piquer, L., and Sañudo, E. C. (2015). Heterometallic 3d-4f single-molecule magnets. Dalton Trans. 44, 8771–8780. doi: 10.1039/C5DT00549C
Sessoli, R., Gatteschi, D., Caneshi, A., and Novak, M. A. (1993a). Magnetic bistability in a metal-ion cluster. Nature 365, 141–143. doi: 10.1038/365141a0
Sessoli, R., and Powell, A. K. (2009). Strategies towards single molecule magnets based on lanthanide ions. Coord. Chem. Rev. 253, 2328–2341. doi: 10.1016/j.ccr.2008.12.014
Sessoli, R., Tsai, H.-L., Schake, A. R., Wang, S., Vincent, J. B., Folting, K., et al. (1993b). High-spin molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 115, 1804–1816. doi: 10.1021/ja00058a027
Shanmugam, M., Shanmugam, M., Chastanet, G., Sessoli, R., Mallah, T., Wernsdorfer, W., et al. (2006). Minor changes in phosphonate ligands leads to new hexa- and dodeca-nuclear Mn clusters. J. Mater. Chem. 16, 2576–2578. doi: 10.1039/b606571f
Sharples, J. W., and Collison, D. (2014). The coordination chemistry and magnetism of some 3d-4f and 4f amino-polyalcohol compounds. Coord. Chem. Rev. 260, 1–20. doi: 10.1016/j.ccr.2013.09.011
Soler, M., Artus, P., Folting, K., Huffman, J. C., Hendrickson, D. N., and Christou, G. (2001). Single-molecule magnets: preparation and properties of mixed-carboxylate complexes [Mn12O12(O2CR)8(O2CR')8(H2O)4]. Inorg. Chem. 40, 4902–4912. doi: 10.1021/ic0104048
Soler, M., Wernsdorfer, W., Abboud, K. A., Huffman, J. C., Davidson, E. R., Hendrickson, D. N., et al. (2003). Single-molecule magnets: two-electron reduced version of a Mn12 complex and environmental influences on the magnetization relaxation of [PPh4]2 [Mn12O12(O2CCHCl2)16(H2O)4]. J. Am. Chem.Soc. 125, 3576–3588. doi: 10.1021/ja021066s
Stamatatos, T. C., Abboud, K. A., Wernsdorfer, W., and Christou, G. (2007a). “Spin tweaking” of a high-spin molecule. An Mn25 single-molecule magnet with an S = 61/2 ground state. Angew. Chem. Int. Ed. 46, 884–888. doi: 10.1002/anie.200603254
Stamatatos, T. C., Abboud, K. A., Wernsdorfer, W., and Christou, G. (2007b). A new Mn25 single-molecule magnet with an S = 61/2 ground state arising from ligand-induced ‘spin-teaking' in a high-spin molecule. Polyhedron 26, 2095–2100. doi: 10.1016/j.poly.2006.10.011
Stamatatos, T. C., and Christou, G. (2009). Azide groups in higher oxidation state manganese cluster chemistry: from structural aesthetics to single-molecule magnets. Inorg. Chem. 48, 3308–3322. doi: 10.1021/ic801217j
Stamatatos, T. C., Efthymiou, C. G., Stoumpos, C. C., and Perlepes, S. P. (2009). Adventures in the coordination chemistry of di-2-pyridyl ketone and related ligands: from high-spin molecules and single-molecule magnets to coordination polymers, and from structural aesthetics to an exciting new reactivity chemistry of coordinated ligands. Eur. J. Inorg. Chem. 2009, 3361–3391. doi: 10.1002/ejic.200900223
Stamatatos, T. C., Foguet-Albiol, D., Lee, S. C., Stoumpos, C. C., Raptopoulou, C. P., Terzis, A., et al. (2007c). “Switching on” the properties of single-molecule magnetism in triangular manganese(III) complexes. J. Am. Chem. Soc. 129, 9484–9499. doi: 10.1021/ja072194p
Stamatatos, T. C., Foguet-Albiol, D., Stoumpos, C. C., Raptopoulou, C. P., Terzis, A., Wernsdorfer, W., et al. (2005). Initial example of a triangular single-molecule magnet from ligand-induced structural distortion of a [O]7+ complex. J. Am. Chem. Soc. 127, 15380–15381. doi: 10.1021/ja0558138
Stoumpos, C. C., Inglis, R., Karotsis, G., Jones, L. F., Collins, A., Parsons, S., et al. (2009). Supramolecular entanglement from interlocked molecular nanomagnets. Cryst. Growth Des. 9, 24–27. doi: 10.1021/cg800947z
Tasiopoulos, A. J., and Perlepes, S. P. (2008). Diol-type ligand as central ‘players' in the chemistry of high-spin molecules and single-molecule magnets. Dalton Trans. 7, 5537–5555. doi: 10.1039/b805014g
Tasiopoulos, A. J., Vinslava, A., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2004). Giant single-molecules magnets: a [Mn84] torus and its supramolecular nanotubes. Angew. Chem. Int. Ed. 43, 2117–2121. doi: 10.1002/anie.200353352
Tasiopoulos, A. J., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2005). [Mn12O12(OMe)2(O2CPh)16(H2O)2]2− single-molecule magnets and other manganese compounds with a reductive aggregation procedure. Inorg. Chem. 44, 6324–6338. doi: 10.1021/ic050868w
Thomas, L., Lionti, F., Ballou, R., Gatteschi, D., Sessoli, R., and Barbara, B. (1996). Macroscopic quantum tunneling of magnetization in a single crystal of nanomagnets. Nature 383, 145–147. doi: 10.1038/383145a0
Urdampilleta, M., Klyatskaya, S., Cleuziou, J.-P., Ruben, M., and Wernsdorfer, W. (2011). Supramolecular spin values. Nature Mater. 10, 502–506. doi: 10.1038/nmat3050
Vincent, J. B., Chang, H.-R., Folting, K., Huffman, J. C., Christou, G., and Hendrickson, D. N. (1987). Preparation and physical properties of the trinuclear oxo-centered manganese complexes of the general formulation [Mn3O(O2CR)6L3]0,+ (R = Me or Ph; L = a neutral donor group) and the crystal structures of [Mn3O(O2CMe)6(pyr)3](pyr) and [Mn3O(O2CPh)6(pyr)2(H2O)]·0.5MeCN. J. Am. Chem. Soc. 109, 5703–5711. doi: 10.1021/ja00253a023
Vincent, J. B., Folting, K., Huffman, J. C., and Christou, G. (1986). Use of tetra-n-butylammonium permanganate for inorganic syntheses in nonaqueous solvents. Preparation and structure of a manganese(III) dimer containing bridging phenoxo oxygen atoms. Inorg. Chem. 25, 996–999. doi: 10.1021/ic00227a022
Vinslava, A., Tasiopoulos, A. J., Wernsdorfer, W., Abboud, K. A., and Christou, G. (2016). Molecules at the quantum-classical nanoparticle interface: giant Mn70 single-molecule magnets of ~4 nm diameter. Inorg. Chem. 55, 3419–3430. doi: 10.1021/acs.inorgchem.5b02790
Wang, S., Huffman, J. C., Folting, K., Streib, W. E., Lobkovsky, E. B., and Christou, G. (1991). (NBu4)[Mn4O2(H2O)(O2CPh)9], a butterfly complex with bound H2O, and its use to prepare octanuclear and undecanuclear metal complexes. Angew. Chem. Int. Ed. Engl. 30, 1672–1674. doi: 10.1002/anie.199116721
Wang, X.-Y., Avendaño, C., and Dunbar, K. R. (2011). Molecular magnetic materials based on 4d and 5d transition metals. Chem. Soc. Rev. 40, 3213–3238. doi: 10.1039/c0cs00188k
Wernsdorfer, W., Aliaga-Alcalde, N., Hendrickson, D. N., and Christou, G. (2002). Exchange-biased quantum tunneling in a supramolecular dimer of single-molecule magnets. Nature 416, 406–409. doi: 10.1038/416406a
Wernsdorfer, W., Chakov, N. E., and Christou, G. (2005). Quantum phase interference and spin-parity in Mn12 single-molecule magnets. Phys. Rev. Lett. 95:037203. doi: 10.1103/PhysRevLett.95.037203
Wernsdorfer, W., and Sessoli, R. (1999). Quantum phase interference and parity effects in magnetic molecular clusters. Science 284, 133–135. doi: 10.1126/science.284.5411.133
Winpenny, R. E. P. (1999). “Design and serendipity in the synthesis of polymetallic complexes of the 3d-metals,” in Transition Metals in Supramolecular Chemistry-Perspectives in Supramolecular Chemistry, Vol. 5, ed J.-P. Sauvage (Chichester: Wiley), 193–223
Winpenny, R. E. P. (2002). Serendipitous assembly of polynuclear cage compounds. J. Chem. Soc. Dalton Trans. 2, 1–10. doi: 10.1039/b107118c
Winpenny, R. E. P. (2004). “High nuclearity clusters: clusters and aggregates with paramagnetic centers: oxygen and nitrogen bridged systems,” in Comprehensive Coordination Chemistry II, Vol. 7, eds J. A. McCleverty and T. J. Meyer (Amsterdam: Elsevier), 125–175
Woodruff, D. N., Winpenny, R. E. P., and Layfield, R. A. (2013). Lanthanide single-molecule magnets. Chem. Rev. 113, 5110–5148. doi: 10.1021/cr400018q
Yang, C. I., Wernsdorfer, W., Lee, G. H., and Tsai, H. L. (2007). A pentanuclear manganese single-molecule magnet with a large anisotropy. J. Am. Chem. Soc. 129, 456–457. doi: 10.1021/ja066533a
Zhang, P., Guo, Y. N., and Tang, J. (2013a). Recent advances in dysprosium-based single molecule magnets: structural overview and synthetic strategies. Coord. Chem. Rev. 257, 1728–1763. doi: 10.1016/j.ccr.2013.01.012
Zhang, P., Zhang, L., and Tang, J. (2013b). Hydrazone-based dysprosium single molecule magnets. Curr. Inorg. Chem. 3, 101–111. doi: 10.2174/1877944111303020005
Keywords: 3d-metal single-molecule magnets, synthetic routes, synthetic strategies, coordination clusters, magnetic properties, reactivity studies, manganese single-molecule magnets, ligand design
Citation: Maniaki D, Pilichos E and Perlepes SP (2018) Coordination Clusters of 3d-Metals That Behave as Single-Molecule Magnets (SMMs): Synthetic Routes and Strategies. Front. Chem. 6:461. doi: 10.3389/fchem.2018.00461
Received: 03 July 2018; Accepted: 14 September 2018;
Published: 09 October 2018.
Edited by:
Matilde Fondo, Universidade de Santiago de Compostela, SpainReviewed by:
Ramon Vicente, University of Barcelona, SpainGiannis Papaefstathiou, National and Kapodistrian University of Athens, Greece
Copyright © 2018 Maniaki, Pilichos and Perlepes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Spyros P. Perlepes, cGVybGVwZXNAcGF0cmVhcy51cGF0cmFzLmdy