 Amy Timmins
Amy Timmins Sam P. de Visser*
Sam P. de Visser*- School of Chemical Engineering and Analytical Science, Manchester Institute of Biotechnology, The University of Manchester, Manchester, United Kingdom
Prolyl-4-hydroxylase is a vital enzyme for human physiology involved in the biosynthesis of 4-hydroxyproline, an essential component for collagen formation. The enzyme performs a unique stereo- and regioselective hydroxylation at the C4 position of proline despite the fact that the C5 hydrogen atoms should be thermodynamically easier to abstract. To gain insight into the mechanism and find the origin of this regioselectivity, we have done a quantum mechanics/molecular mechanics (QM/MM) study on wildtype and mutant structures. In a previous study (Timmins et al., 2017) we identified several active site residues critical for substrate binding and positioning. In particular, the Glu127 and Arg161 were shown to form multiple hydrogen bonding and ion-dipole interactions with substrate and could thereby affect the regio- and stereoselectivity of the reaction. In this work, we decided to test that hypothesis and report a QM/MM and molecular dynamics (MD) study on prolyl-4-hydroxylase and several active site mutants where Glu127 or Arg161 are mutated for Asp, Gln, or Lys. Thus, the R161D and R161Q mutants give very high barriers for hydrogen atom abstraction from any proline C–H bond and therefore will be inactive. The R161K mutant, by contrast, sees the regio- and stereoselectivity of the reaction change but still is expected to hydroxylate proline at room temperature. By contrast, the Glu127 mutants E127D and E127Q show possible changes in regioselectivity with the former being more probable to react compared to the latter.
Introduction
Metalloenzymes play vital roles in nature and are involved in biosynthesis as well as biodegradation of compounds (Solomon et al., 2000; Costas et al., 2004; Abu-Omar et al., 2005; Kryatov et al., 2005; Bruijnincx et al., 2008; Kadish et al., 2010). Due to its large natural abundance often metalloenzymes contain one or more iron centers; however, in this work we will restrict ourselves to mononuclear iron enzymes only and particularly those that utilize molecular oxygen. In general, iron containing dioxygenases and monoxygenases use one molecule of molecular oxygen in their catalytic cycle and either transfer both oxygen atoms to substrate(s) or a single one with a water molecule as by-product (Sub = substrate), Equations 1, 2.
Thus, heme monoxygenases, like the cytochromes P450, react as monoxygenases and proceed through a catalytic cycle starting from an iron(III)-heme resting state with a protein cysteinate and a water molecule in the fifth and sixth iron ligand positions, respectively (Meunier et al., 2004; Ortiz de Montellano, 2004, 2010; Denisov et al., 2005; Kadish et al., 2010). The water molecule is released after substrate binding, which triggers a spin state change from low-spin to high-spin and enables molecular oxygen binding to the iron center. The iron-superoxo is subsequently reduced and protonated to form an iron(III)-hydroperoxo(heme) complex, also called Compound 0 (Meunier et al., 2004; Denisov et al., 2005; Shaik et al., 2005; Ortiz de Montellano, 2010). A final protonation step gives water and an iron(IV)-oxo(heme cation radical) species called Compound I (CpdI) (de Visser et al., 2003; Rittle and Green, 2010; de Visser and Kumar, 2011). Now, CpdI is the active species of P450 enzymes and reacts with substrates through oxygen atom transfer and hence converts aliphatic groups to alcohols (Ogliaro et al., 2000b; de Visser et al., 2004; Ji et al., 2015), C = C double bonds to epoxides (de Visser et al., 2001, 2002b; Sainna et al., 2015), sulfides to sulfoxides (Sharma et al., 2003; Kumar et al., 2005b, 2011a), and arenes to phenols (de Visser and Shaik, 2003; de Visser, 2006c; Cantú Reinhard et al., 2016b). The mechanism of these reactions has been established with computational modeling including density functional theory (de Visser, 2012; Blomberg et al., 2014). In recent years full calculations on enzymatic structures were done and identified the effect of the protein, substrate orientation and hydrogen bonding interactions on the kinetics and thermodynamics of the reaction and the product distributions of P450 catalyzed reaction mechanisms. Thus, it was shown with small model complexes that hydrogen bond donations toward the axial thiolate ligand affected the electron affinity of this cysteinate residue, which led to a push-effect of electrons to the heme that influenced its redox potential and hence catalytic potential (Ogliaro et al., 2000a; de Visser et al., 2002a; Schöneboom et al., 2002). In the heme enzyme cytochrome c peroxidase, a model that included a cation binding site reproduced the experimentally characterized electronic configuration, and highlighted the importance of long-range electrostatic effects in enzyme models (de Visser, 2005). Because of these long-range effects, more and more computational studies are done using full enzymatic systems using the Quantum Mechanics/Molecular Mechanics (QM/MM) technique.
Figure 1 displays the active site structures of (Figure 1A) cytochrome P450 and (Figure 1B) taurine/α-ketoglutarate dioxygenase (TauD) as a structural comparison (O'Brien et al., 2003; Guo and Sevrioukova, 2017). Thus, the P450s are heme enzymes, where the heme is linked to the protein backbone through an interaction of the metal with a cysteinate residue (the axial ligand). On the distal site of the heme the substrate binds, which is the drug molecule metformin in the 5G5J protein databank (pdb) file. The distal site of the heme has several hydrogen bonding and polar residues, such as Ser119 and Arg212, the former has been proposed to be involved in the proton relay mechanisms during the catalytic cycle (Kumar et al., 2005a), whereas the latter holds the substrate through a salt bridge into position.
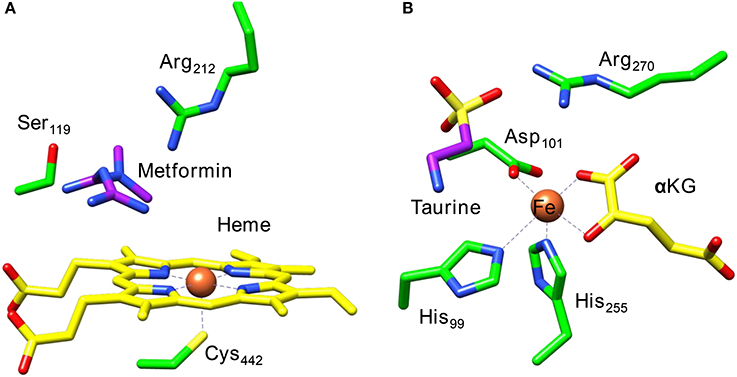
Figure 1. Extracts from the crystal structure coordinates of cytochrome P450 (A) and taurine/α-ketoglutarate dioxygenase (B) as taken from the 5G5J and 1OS7 pdb files.
A second iron enzyme class that utilizes molecular oxygen is the nonheme iron dioxygenases (Bugg, 2001; Ryle and Hausinger, 2002; Solomon et al., 2013). These dioxygenases are found in all forms of life and are involved in the biosynthesis of antibiotics (Choroba et al., 2000; Higgins et al., 2005; Siitonen et al., 2016), DNA and RNA repair enzymes (O'Brien, 2006; Yi et al., 2009), as well as the metabolism of toxic natural compounds such as cysteine (Stipanuk, 2004; Straganz and Nidetzky, 2006; de Visser, 2009; Buongiorno and Straganz, 2013). These enzymes are structurally very different from the heme monoxygenases as they link the iron atom to the protein with only amino acid side chains such as His, Asp, or Glu residues. Usually, the nonheme iron dioxygenases contain a facial triad of amino acid ligands with two histidine and one carboxylate group, i.e., 2-His/1-Asp, Glu (Que, 2000; Kovaleva and Lipscomb, 2008). As an example of a dioxygenase with these ligand features we show in Figure 1B the active site of taurine/α-ketoglutarate dioxygenase (TauD). TauD is a dioxygenase involved in the metabolism of cysteine, whereby it converts taurine to hydroxy-taurine. The 1OS7 pdb file (O'Brien et al., 2003) is a substrate and α-ketoglutarate (α-KG) bound structure of TauD with the iron bound to the protein through the side chains of residues His99, Asp101 and His255. Substrate taurine is located nearby the metal and is held in position through a salt bridge with residue Arg270. Co-substrate α-KG is bound to the metal as a bidentate ligand through the keto and acid groups.
The catalytic cycle of TauD has been established through a combination of experimental and computational studies (Borowski et al., 2004; Bollinger et al., 2005; de Visser, 2006a,b, 2007; Godfrey et al., 2008). Figure 2 schematically depicts the catalytic cycle of TauD specifically and starts from the resting state structure where iron is bound to the 2-His/1-Asp ligand system and the other ligand positions of the metal are occupied by three water molecules (structure A). When α-KG enters the pocket two water molecules are displaced and replaced by the keto and acid groups of α-KG (structure B). In the next step, substrate taurine binds, which displaces the last water molecule from iron (structure C) and is replaced by molecular oxygen that binds as an iron(III)-superoxo (structure D). Subsequently, the superoxo group attacks the α-keto position of α-KG to form a bicyclic ring-structure (structure E). In the next step, the dioxygen bond breaks to form a peracid succinate with the release of CO2. Finally, the peracid bond breaks and splits into an iron(IV)-oxo species and succinate (structure F). Iron(IV)-oxo is known to be a powerful oxidant that abstracts a hydrogen atom from taurine to give an iron(III)-hydroxo group (structure G) and the hydroxyl radical is then rebound to form hydroxy-taurine as product (structure H). Products hydroxy-taurine and succinate are released from the iron center and their positions are replaced by water molecules to bring the catalytic cycle back into the resting state.
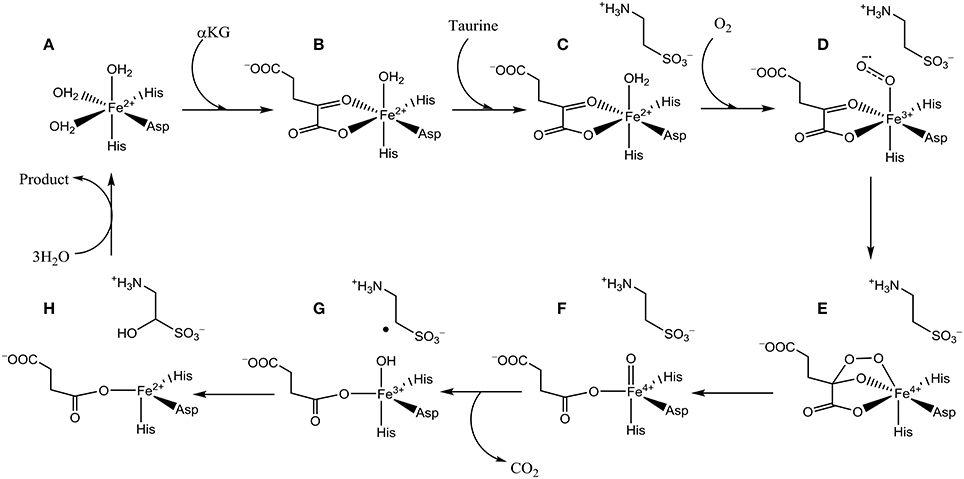
Figure 2. Catalytic cycle of nonheme iron dioxygenases. Structures (A–H) are defined in the text.
Another nonheme iron dioxygenase with a catalytic cycle similar to TauD is prolyl-4-hydroxylase (P4H), which regio- and stereospecifically hydroxylates a proline residue in a protein to R-4-hydroxyproline, Scheme 1. Product R-4-hydroxyproline is a common amino acid in animals and plants and has functions in collagen, where it enables crosslinking between individual strands. In addition, it is relevant to the synthesis of the hypoxia induced factor in animals (McDonough et al., 2006).
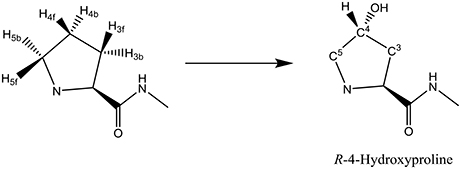
Scheme 1. Conversion of a proline residue in a protein to R-4-hydroxyproline by P4H.
A range of biochemical and spectroscopic studies on P4H established key details of the catalytic cycle. Thus, reactions of P4H with (de Visser et al., 2001) 18O2 provided evidence of the transfer of one atom of molecular oxygen to proline (Myllyharju and Kivirikko, 1997). Low-temperature Mössbauer, electron paramagnetic resonance (EPR) and UV-Vis absorption spectroscopic studies characterized several intermediates in the catalytic cycle, including the iron(IV)-oxo species (Hoffart et al., 2006). It was shown that the iron(IV)-oxo species has a quintet spin ground state and reacts with the substrate through a rate-determining hydrogen atom abstraction. In particular, rate constants for the reaction with taurine and taurine-d2 gave a large kinetic isotope effect. To confirm the reaction mechanism computational studies on the catalytic cycle of P4H were performed: One study using an active site model complex (Karamzadeh et al., 2010) and another using the full enzyme structure with QM/MM (Timmins et al., 2017). These studies established the technical details of the catalytic cycle and confirmed the mechanism shown above in Figure 2. Furthermore, key functions of several amino acids were identified related to substrate positioning and product release as will be described in more detail later.
The key step in the catalytic cycle of P4H is the hydrogen atom abstraction of substrate by the iron(IV)-oxo intermediate (Hoffart et al., 2006), which was shown to be rate-determining. In principle, substrate proline has six aliphatic C–H bonds at positions C3, C4, and C5 that could lead to six different product isomers, Scheme 2. We label the two hydrogen atoms on C3, C4, and C5 as front (f) or back (b). Also shown in Scheme 2 are bond dissociation free energies (BDFE) of each of these C–H bonds as calculated at UB3LYP/6-311+G* as the difference in free energy of proline and the sum of a hydrogen atom and [proline – H•]. As can be seen, the C–H bond strength at the C3 and C4 positions in proline are comparable, while the one at the C5 position is much weaker in energy. Therefore, in the gas-phase proline hydroxylation should happen at the C5 position as it is the weakest bond to break rather than at the thermodynamically unfavorable C4 position. How P4H prevents hydroxylation of the weaker C5 position in favor of hydroxylation at the C4 position is the topic of this paper.
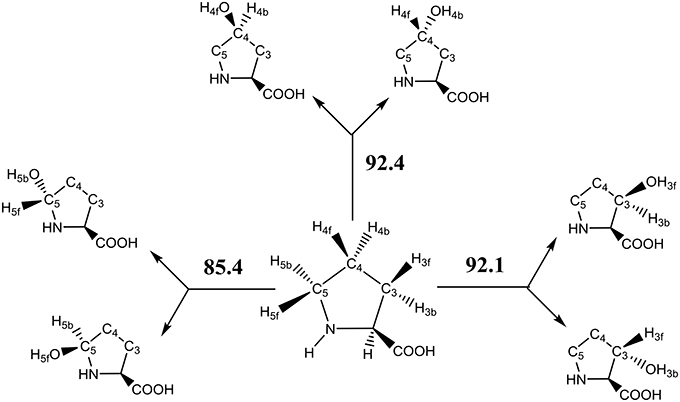
Scheme 2. Aliphatic C–H bonds of proline and definition of atom labels and their bond dissociation free energies (BDFE in kcal mol−1).
In addition to studies on wildtype (WT) P4H, we looked at the structure and catalytic properties of two active site mutants where the Glu127 and Arg161 residues were mutated to alternative groups. A previous study (Timmins et al., 2017) identified these two amino acids as key for substrate positioning in the substrate binding pocket and hence mutating them to a different amino acid should have a considerable effect.
Methods
The calculations presented in this work follow previously described and benchmarked methods using QM/MM (Porro et al., 2009; Kumar et al., 2011b; Quesne et al., 2014, 2016a; Faponle et al., 2017; Li et al., 2017). Specifically, our previous work on the mechanism of the possible reaction channels of cytochrome P450 decarboxylase leading to decarboxylation of fatty acids or hydroxylation of fatty acids, predicted the correct regioselectivity of the reaction as compared to experiment and reproduced experimentally determined kinetic isotope effects (Faponle et al., 2016). Furthermore, QM/MM studies on 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase focused on the rate-determining step of the co-factor independent reaction of substrate with molecular oxygen and predicted a rate constant in good agreement with experiment and explained how this enzyme functions without a metal cofactor present (Hernández-Ortega et al., 2014, 2015). Very recently, we used QM/MM modeling to predict spectroscopic fingerprints of short-lived catalytic cycle intermediates and used this on cysteine dioxygenase enzymes. Calculated UV-Vis absorption spectra and Mössbauer and EPR parameters enabled the experimental characterization of a short-lived oxygen-bound intermediate (Fellner et al., 2016; Tchesnokov et al., 2016).
Model Set-Up
Our QM/MM starting point structures were set-up using previously described methods and procedures (Quesne et al., 2016a; Timmins et al., 2017), which we will summarize briefly here. The crystal structure coordinates from the 3GZE pdb file was used as a starting point for all models (Koski et al., 2009). The 3GZE pdb file represents a resting state P4H structure with Zn2+, pyridine-dicarboxylate co-substrate mimic and the (Ser-Pro)5 peptide chain bound. The active site zinc(II)-water(pyridine-dicarboxylate) was replaced with iron(IV)-oxo(succinate) manually with an Fe–O distance of 1.63 Å: a typical distance found for analogous nonheme iron(IV)-oxo complexes in enzymes and model complexes (de Visser, 2006a,b,d, 2007; Godfrey et al., 2008; Quesne et al., 2016b; Cantú Reinhard and de Visser, 2017). The short peptide chain (Ser-Pro)5 we retained in the model as it has its proline residue tightly packed nearby the iron(IV)-oxo group.
Subsequently, hydrogen atoms were added to the protein structure using the pdbtopqr program package assuming a pH = 7 (Dolinsky et al., 2007). Thus, all acid residues, i.e., Glu and Asp, were deprotonated whereas the basic residues, i.e., Arg and Lys, were protonated. The protonation state of each individual histidine residue was decided upon visual inspection of its local environment (donating/accepting hydrogen bonds) and we chose to assign all as singly protonated. Thereafter, the protein structure was solvated in a sphere with radius of 40 Å and energy minimized with the Charmm forcefield (Brooks et al., 1983). The solvation procedure was repeated a number of times until a situation was reached (Figure 3), whereby <20 water molecules were added to the chemical system. The saturated structure was then minimized without geometric constraints and heated to a temperature of 298 K. Finally, a full molecular dynamics (MD) simulation was run for 10 ns. The full set-up procedures were repeated for the mutant structures, whereby one amino acid was manually replaced. As follows from the MD simulations shown in Figure 3B, all converge well within 10ns. For each of the structures, we started QM/MM calculations using the snapshots taken after 5 ns (Sn5ns).
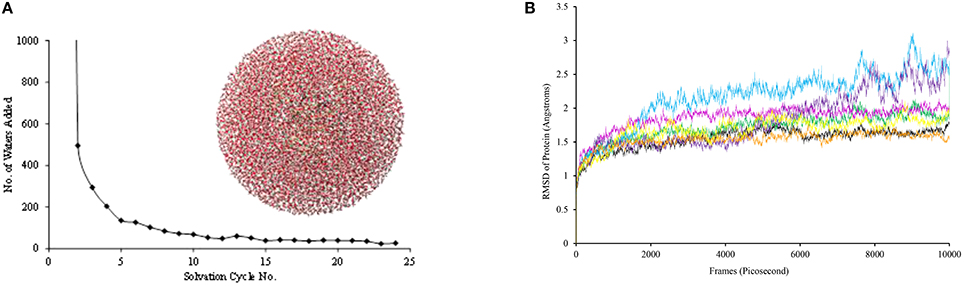
Figure 3. (A) Iterative solvation procedure for WT P4H. (B) MD simulations for WT and mutants of P4H. Colors represent WT (black), E127D (green), E127Q (pink), R161C (blue), R161D (purple), R161K (orange), and R161Q (yellow).
QM/MM Procedures
Density functional theory (DFT) methods were used to describe the QM region of the QM/MM calculation. In particular, we used the unrestricted hybrid density functional method B3LYP (Lee et al., 1988; Becke, 1993) in all cases as recent benchmark studies from our group showed this procedure to give rate constants in very good agreement with experiment (Cantú Reinhard et al., 2016a). In particular, QM methods with dispersion included were shown to underestimate free energies of activation considerably (Cantú Reinhard et al., 2016a). Furthermore, B3LYP was shown previously to predict regioselectivities and bifurcation pathways well as compared to experiment (Kumar et al., 2004; Barman et al., 2016; Brazzolotto et al., 2017). Also, DFT calculated free energies of activation were shown to match experimentally determined ones of biomimetic model complexes containing iron and manganese very well and reproduced Hammett trends (Vardhaman et al., 2011, 2013; Kumar et al., 2014; Yang et al., 2016). Here, DFT calculations were run in Turbomole (Ahlrichs et al., 1989), and the MM ones in DL-Poly with the Charmm forcefield (Smith and Forester, 1996). The ChemShell software package(Sherwood et al., 2003) interfaced Turbomole and DL-Poly and was used to obtain QM/MM energies and derivatives. The link-atom approach was used to describe atoms on the border between the QM and MM regions and essentially replaced a covalent bond with a C–H bond (Bakowies and Thiel, 1996). All calculations use electronic embedding of the charges of the MM region included into the QM Hamiltonian.
Geometry optimizations and reaction coordinate scans were done with an SV(P) basis set on all atoms: basis set BSI (Schafer et al., 1992). Reaction coordinate scans were run with one degree of freedom fixed and explored the potential energy surface between reactants, intermediates and products. The maxima of these scans were used as starting points for transition state searches. The energies of the stationary points were improved by running a single point calculation with an all-electron Wachters-type basis set on iron and def2-TZVP on the rest of the atoms: basis set BSII (Wachters, 1970).
QM Region
As the active site region of the protein contains many hydrogen bonding and π-stacking interactions we considered two QM regions: a minimal QM region A and an expanded QM region AB, see Figure 4. Thus, the minimal QM region A contains the iron(IV)-oxo group and its direct ligands (His143, His227, Asp145, and succinate) as well as the proline ring of the peptide substrate. The larger QM region AB was expanded with the indole ring of Trp243, the phenol group of Tyr140 and three water molecules.
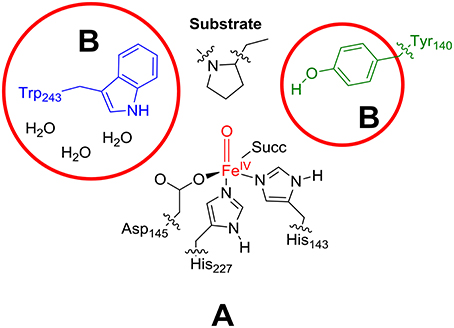
Figure 4. Description of QM region A and AB.
Results and Discussion
P4H WT Structure
The iron(IV)-oxo species (structure G in the catalytic cycle of Figure 2) was fully optimized with QM/MM methods using B3LYP/BSI and QM region A and AB, see Figure 5. In agreement with experimental EPR and Mössbauer spectroscopic studies (Hoffart et al., 2006) the quintet spin state is the ground state, while we located the triplet and singlet spin states higher in energy by 16.0 and 33.1 kcal mol−1, respectively. Geometrically, the iron(IV)-oxo species is bound to the protein with two Fe–NHis interactions of 2.06 and 2.08 Å, which is typical for metal-histidine interactions in proteins (de Visser et al., 2009). The carboxylate group of Asp145 binds as a monodentate ligand at a distance of 2.01 Å, whereas the succinate carboxylate group binds as a bidentate ligand with distances of 2.20 and 2.30 Å. Again, these distances match previous calculations on similar complexes nicely (Pratter et al., 2013). Substrate proline is not bound directly to the iron center but its transferring C–H4b hydrogen atom is found at a distance of 2.86 Å from the oxo group and hence is positioned in the ideal orientation for oxidation. The triplet and singlet spin states give analogous ligand distances but are distinguished by their differences in iron(IV)-oxo bond length due to differences in molecular orbital occupation.
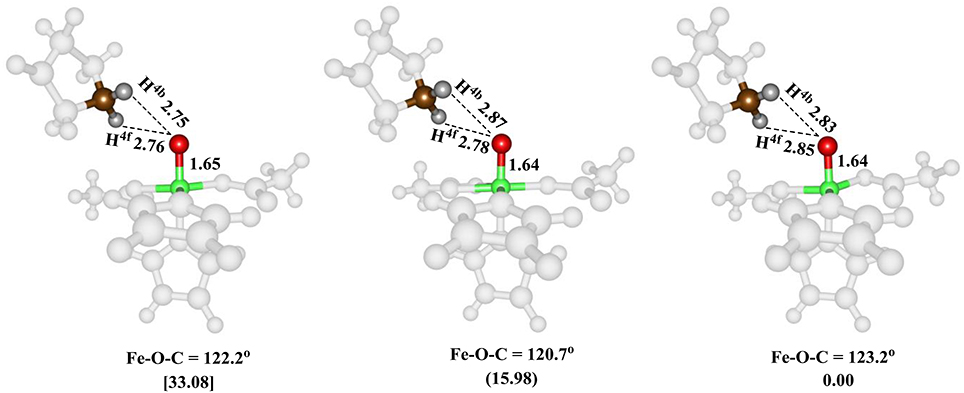
Figure 5. QM/MM optimized geometries of the low-lying spin states of the iron(IV)-oxo species of WT P4H for snapshot Sn5ns and QM region A. From left-to-right, the data reported are for the singlet, triplet and quintet spin states. Bond lengths are in angstroms and relative energies in kcal mol−1.
High-lying occupied and low-lying virtual orbitals of the iron(IV)-oxo species are shown in Figure 6, which gives the molecular z-axis along the Fe–O bond. The lowest lying orbital is the π*xy orbital that represents the interactions of the metal 3dxy orbital with the equatorial ligands, namely His143, Asp145 and succinate (Succ). A bit higher in energy are the two orthogonal π* orbitals along the Fe–O bond that correspond to the mixing of the 3dxz on iron with 2px on oxygen, i.e., π*xz, and the 3dyz on iron with the 2py on oxygen, i.e., π*yz. Higher in energy still are two σ-type orbital interactions. The first one along the z-axis for the mixing of the metal 3dz2 with a 2pz on oxygen: . The second one is located in the xy-plane and results from the interaction of the 3dx2−y2 orbital on iron with orbitals on the ligands: σ*x2−y2. The quintet spin state has orbital occupation , while the triplet spin state has configuration . Generally, enzymatic iron(IV)-oxo species tend to have a quintet spin ground state (Latifi et al., 2009), while most synthetic biomimetic models have a triplet spin ground state (de Visser et al., 2012). Computational studies showed that this is the result of differences in coordination system, where biomimetic models are often in octahedral coordination, whereas enzymatic structures have the iron(IV)-oxo in pentacoordination (Latifi et al., 2013).
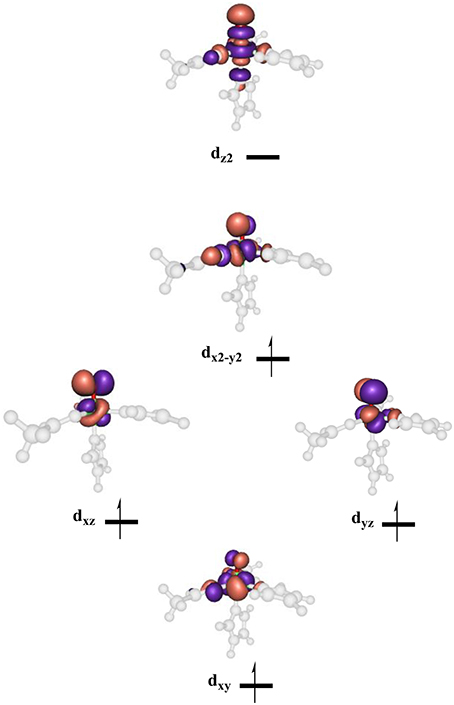
Figure 6. Molecular valence orbitals of the iron(IV)-oxo species of P4H.
P4H Hydroxylation Mechanisms
Subsequently, we investigated proline hydroxylation at the C3, C4 and C5 position of P4H and considered both hydrogen atoms at each of these positions. All reactions were found to be stepwise with an initial hydrogen atom abstraction via transition state TSHA to form a radical intermediate IH. A radical rebound step via TSreb then produced the alcohol product complexes P. In all cases the rebound step was small and the hydrogen atom abstraction was rate-determining. The QM/MM calculated energy landscapes are given in Figure 7 for hydrogen atom abstraction from C4f, C4b, C5f, C5b, C3f, and C3b. The lowest energy barrier height is the one for C4b and after rebound will give the R-4-hydroxyproline product complex, which is the experimentally determined stereo- and regioselective product found. Although the difference in relative energies is small, the calculations predict the correct regio- and stereoselectivity. Slightly higher in energy (1 kcal mol−1 using basis set BSII and 2.9 kcal mol−1 using basis set BSI) we find the pathway for hydrogen atom abstraction from C5b, the thermodynamically more favorable pathway. Indeed the radical intermediate for C5b is much lower in energy than the one for C4b in agreement with the thermodynamic predictions. About 5.5 kcal mol−1 higher in energy than 5 TSHA,C4b is the barrier 5 TSHA,C4f, which implies that there are strong energetic differences between hydrogen atom abstraction of the two hydrogen atoms on carbon center C4. The three barriers for hydrogen atom abstraction from C5f, C3f, and C3b are all well higher in energy than 5 TSHA,C4b by at least 15 kcal mol−1 and hence will play little role of importance. As the singlet and triplet spin states were already considerably higher in energy at the reactant stage, they remain well higher in the hydrogen atom abstraction transition states as well. The triplet and singlet spin barriers for abstracting the C4b hydrogen atoms are 34.0 and 40.2 kcal mol−1 at UB3LYP/BSI in QM/MM. As such the reactivity takes place on a single spin state only, the quintet spin state, and other spin states play no role in the rate-determining pathway.
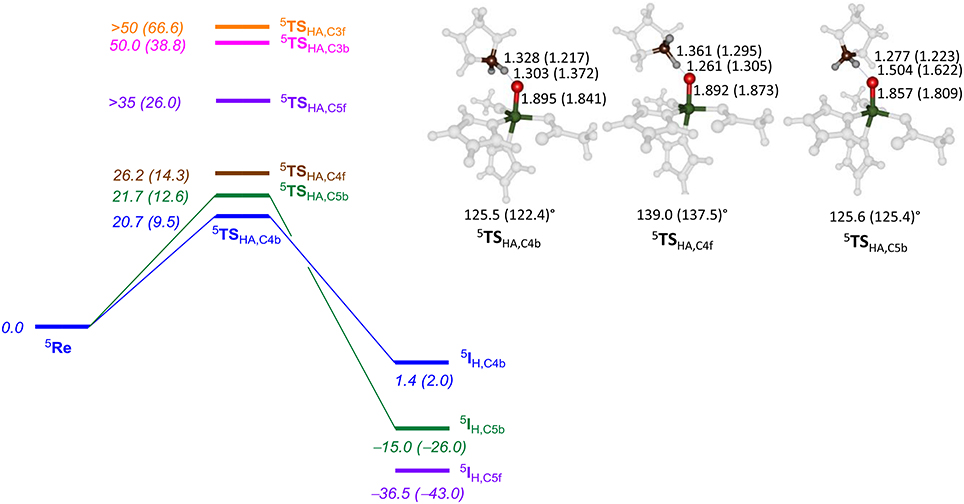
Figure 7. QM/MM calculated hydrogen atom abstraction barriers for P4H WT with energies in kcal mol−1 as obtained with QM region AB (A). Optimized geometries of the lowest energy hydrogen atom abstraction transition states are given with bond lengths in angstroms and angles in degrees.
The optimized geometries of 5 TSHA,C4b, 5 TSHA,C4f, and 5 TSHA,C5b (right-hand-side of Figure 7) give insight into their energetic ordering and relative energies. Thus, in 5 TSHA,C4b the transferring hydrogen atom is almost midway in between donor and acceptor atom and the Fe–O–C4b angle is about 125°. In 5 TSHA,C4f, by contrast, the substrate is oriented along a much larger angle of 139.0°. The 5 TSHA,C5b structure, on the other hand, has the transferring hydrogen atom at a relatively large distance from the accepting oxygen atom and hence is destabilized considerably.
P4H Mutations of Arg161
To find out the effect of substrate positioning and catalytic turnover of active site mutations, we investigated several P4H models, where amino acids were replaced. Thus, in our previous studies (Timmins et al., 2017) we implicated an important role of Arg161 and Glu127 through hydrogen bonding interactions. In this section we will look into the structure and catalytic activity of P4H mutants with Arg161 replaced by either Asp, Gln, or Lys. These changes could be dramatic as the Arg161Asp (R161D) mutation will replace a positively charged residue with a negatively one. Similarly, the Arg161Gln (R161Q) mutation changes a cationic residue into a neutral one.
Figure 8 displays an overlay of the structures of the iron(IV)-oxo species for WT and R161K mutation after a full QM/MM geometry optimization, where the positively charged Arg residue is replaced by the positively charged Lys amino acid. As can be seen the mutation displaces the salt bridge between Arg161 and Glu127, which bends outward. The space provided by the Glu127 migration is filled up with extra water molecules. However, the removal of the strong hydrogen bonding interactions of the Arg161-Glu127 couple toward the substrate has a major effect on the stability of the substrate and its positioning. Thus, substrate is lesser tight bound in the R161K mutant than in WT and hence its regio- and stereoselective substrate activation may be affected.
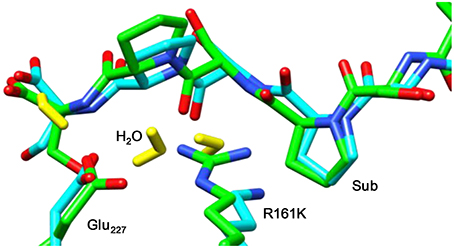
Figure 8. Overlay of the iron(IV)-oxo structures of WT P4H and R161K mutants as optimized with QM/MM. Bond lengths are in angstroms.
Subsequently, we studied the hydrogen atom abstraction mechanisms of the R161K mutant from the C3, C4, and C5 positions of proline for the back and front protons. Figure 9 displays relative energies and optimized geometries of selected hydrogen atom abstraction transition states for the R161K mutant. The hydrogen atom abstraction barrier from the C4b position (5 TSR161K,C4b) has an energy of 14.0 kcal mol−1 (UB3LYP/BSI), which is almost identical to the one observed for WT of 14.3 kcal mol−1. Indeed, the optimized geometries are very similar: C–H and O–H distances are found of 1.24 and 1.40 Å for R161K, whereas they are 1.22 and 1.37 Å, respectively, for WT (Figure 7 above). However, a much lower transition state is found for activation of the C4f position of only 2.7 kcal mol−1. Therefore, the R161K mutation will not affect the catalytic performance of the enzyme: It should react faster than WT, but will give a reversal of stereochemistry and predominantly produce the S-4-hydroxyproline product instead.
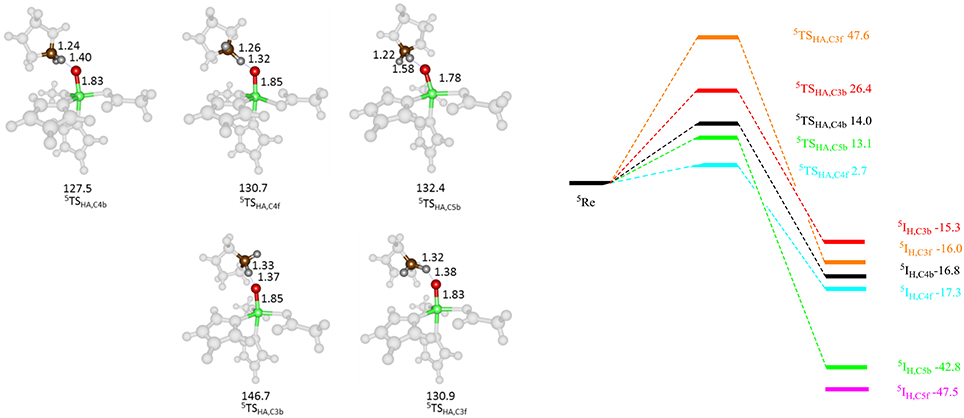
Figure 9. QM/MM calculated hydrogen atom abstraction barriers for P4H R161K with energies in kcal mol−1 as obtained with QM region A. Optimized geometries of the lowest energy hydrogen atom abstraction transition states are given with bond lengths in angstroms and angles in degrees.
Comparison of 5 TSC4b transition state structures in R161K to WT shows that the positions of the active site components change little, however, the Fe-O-C4 angle and NHis-Fe-O-C4 dihedral angle are different, namely 137.5° and −73.4° for WT and 130.7° and −67.5° for R161K, respectively. Therefore, this mutation allows for the orientation of the substrate relative to the iron(IV)-oxo to change in such a way that the C4f position is now accessible to it. Hydrogen atom abstraction from the C3 position is seen to be lowered as compared to WT but is significantly higher in energy than the barrier 5 TSR161K,C4f. The barrier for C5b hydrogen atom abstraction is similar to that for C4b but now slightly lower in energy.
Thereafter, we studied the R161Q and R161D mutants and Figure 10 gives structures of the iron(IV)-oxo species as compared to WT. As can be seen both mutations have a dramatic effect on substrate binding and positioning as a result of changes in the hydrogen bonding network between Glu127, R161D and surrounding residues located in the βII-βIII and β3-β4 loops. We then attempted to abstract hydrogen atoms from proline by the R161Q and R161D mutants. Table 1 gives data with calculated barrier heights for several hydrogen atoms of proline. None of these barrier heights, however, is low enough in energy to make them accessible at room temperature. Therefore, the R161Q and R161D mutants will be catalytically inactive. As a result, the Arg161 residue has a critical function in P4H enzymes in positioning the substrate in the correct orientation. This is done in conjunction with the Glu127 residue that hydrogen bonds the protein loop of the substrate and makes sure it can approach the iron(IV)-oxo species.
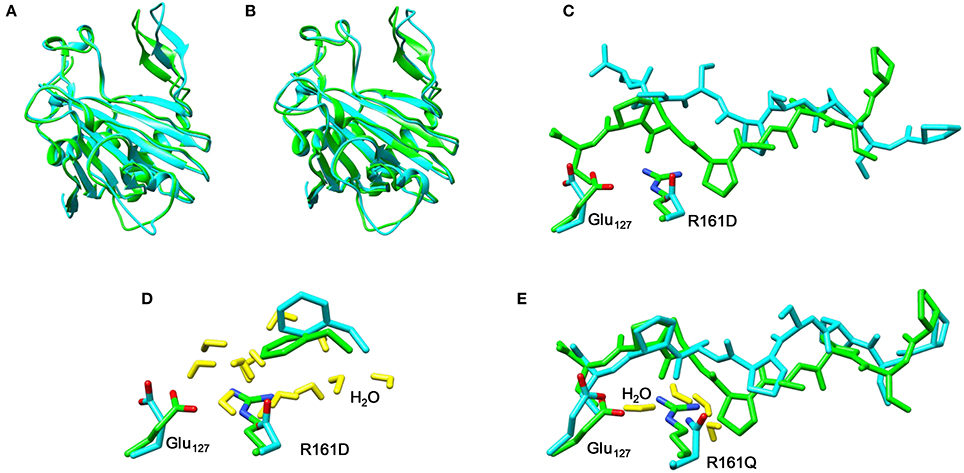
Figure 10. Overlay of the iron(IV)-oxo structures of WT P4H (in green) and Arg161 mutants (in blue) as optimized with QM/MM. (A) WT vs R161D (B) WT vs R161Q (C) WT vs R161D (D) WT vs R161D (E) WT vs R161Q.
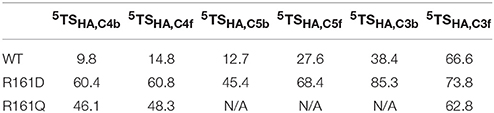
Table 1. Calculated barrier heights (kcal mol−1) for several hydrogen atom transfers from the substrate proline residue to the iron(IV)-oxo oxidant in prolyl-4-hydroxylase.
P4H Mutations of Glu127
In a final set of calculations we investigated P4H mutants where Glu127 is replaced by either Asp or Gln. Figure 11 displays the QM/MM optimized iron(IV)-oxo species of WT version E127D and E127Q mutants. In E127D the hydrogen bond between Asp127 and Arg161 is broken and as a result as Asp127 swings out, to subsequently effecting the positions of surrounding residues and the substrate. Interestingly, the hydrogen bond between Arg161 and the substrate is maintained; however, the position of this residue corresponds to the change in the substrate position. It suggests that the function of Glu127 is to anchor Arg161 in a fixed position, which is essential for proper substrate positioning. In E127Q, there are notable changes around the active site with, e.g., Tyr140 rotating out of its WT position breaking its hydrogen bond to the iron(IV)-oxo which has previously been shown to be important for correct substrate positioning and release. Previous research (Koski et al., 2009) has suggested that any disturbance of this “conformational switch” would result in the inactivation of the enzyme as shown by its mutation to alanine in experiment (Koski et al., 2009). The position of Trp243 is also altered affecting substrate positioning even more, and consequently the E127Q mutation is likely to lead to an inactive form of the enzyme. To test the hypothesis, we explored hydrogen atom abstraction from various positions of proline residue which are given in Table 2.
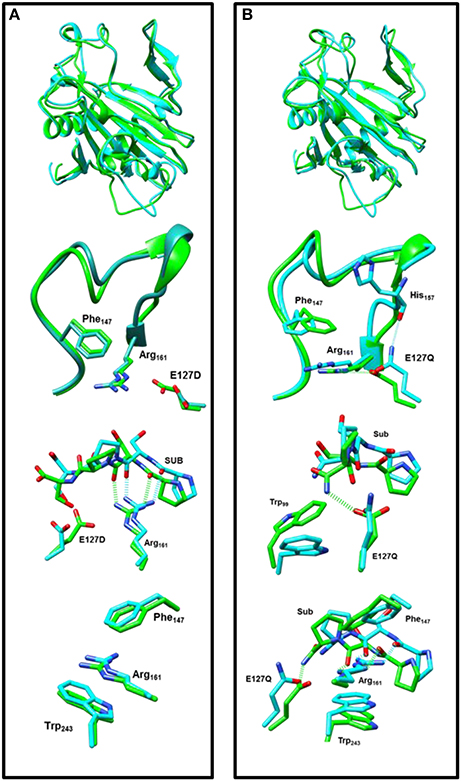
Figure 11. Overlay of the iron(IV)-oxo structures of WT P4H (in green) and Glu127 mutants (in cyan) as optimized with QM/MM. (A) WT vs. E127D and (B) WT vs E127Q.
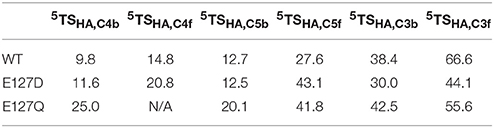
Table 2. Calculated barrier heights (kcal mol−1) for several hydrogen atom transfers from the substrate proline residue to the iron(IV)-oxo oxidant in prolyl-4-hydroxylase.
In E127D, no longer is hydrogen atom abstraction (HAT) from C4b the favored pathway, but now, HAT from C5b is within 1 kcal mol−1 and hence the two pathways are competitive. Considering the energies of the intermediate radical species in the mutant, it becomes clear that the C5b structure (−31.8 kcal mol−1) product will be the major product over the C4b (−2.9 kcal mol−1), therefore the E127D mutation leads to a change in the regioselectivity of the reaction. This is similar to what has been seen in previous DFT calculations which showed that if given the choice HAT from the C5 position will always be favored over the C4 position as the BDE at the former position is weaker compared to the latter (Karamzadeh et al., 2010). Comparison of the 5TSC5b transition state structures in WT and E127D reveal similar geometric parameters, however, the Fe-O-C5 angle and NHis-Fe-O-C5 dihedral angle are different, namely 125.4° and −77.8° for WT and 132.1° and −88.2° for E127D, respectively. As such, the orientation of the substrate relative to the iron(IV)-oxo has changed, allowing the substrate in E127D to adopt a more favorable position for HAT from the C5b position as compared to WT. In E127Q, HAT from the C5b position is favored compared to that from C4b, as shown also for E127D. However, as the transition state barrier is 20.1 kcal mol−1 for HAT from the C4b position, the mutant will result in much slower reactivity as compared to WT.
In conclusion, previous QM/MM studies on P4H have elucidated the reasoning behind its observed regioselectivity and stereoselectivity in the WT through various mutations to the protein. The results of that study highlighted the role of Glu127 and Arg161 in substrate positioning and suggested how mutating those residues could alter the regioselectivity and stereoselectivity with minimal influence on the enzymes stability and catalytic ability, an important consideration for future biotechnological applications. This study has revealed that mutations E127D and R161K are possible mutation candidates which will result in a change in the regioselelctivity and stereoselectivity. Additionally, the work highlights the importance in conserving the charge in the substrate binding residues of the enzyme around the substrate cavity to ensure interactions between the substrate and protein are maintained when an amino acid is mutated.
Conclusion
Here we describe a detailed computational study into the activity of prolyl-4-hydroxylase enzymes and several Glu127 and Arg161 mutants. In particular, a comprehensive QM/MM study is presented, whereby we investigated hydrogen atom abstraction channels of each pair of hydrogen atoms bound to C3, C4, and C5 of the proline residue of the substrate. Studies on WT predict the experimentally observed product distributions and give regio- and enantioselective R-4-hydroxyproline as a product. Analysis of the structure and electronic configurations show the regioselectivity to be guided by substrate positioning and hydrogen bonding interactions. Mutations of Glu127 and Arg161 have major effects and lead to inactivity of the protein in several cases, in particular, when an anionic residue is replaced by a cationic one. Only in the case of the E127D mutant significant activity remains although competitive C4b and C5b hydroxylation is predicted.
Author Contributions
AT and SdV designed and developed the project. AT performed the calculations. AT and SdV wrote the paper.
Funding
The work was funded through a studentship from the Biotechnology and Biological Sciences Research Council (BBSRC) under grant code BB/J014478/1.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, TM, and handling Editor declared their shared affiliation.
References
Abu-Omar, M. M., Loaiza, A., and Hontzeas, N. (2005). Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 105, 2227–2252. doi: 10.1021/cr040653o
Ahlrichs, R., Bär, M., Häser, M., Horn, H., and Kölmel, C. (1989). Electronic structure calculations on workstation computers: the program system Turbomole. Chem. Phys. Lett. 162, 165–169. doi: 10.1016/0009-2614(89)85118-8
Bakowies, D., and Thiel, W. (1996). Hybrid models for combined quantum mechanical and molecular mechanical approaches. J. Phys. Chem. 100, 10580–10594. doi: 10.1021/jp9536514
Barman, P., Upadhyay, P., Faponle, A. S., Kumar, J., Nag, S. S., Kumar, D., et al. (2016). Deformylation reaction by a nonheme manganese(III)-peroxo complex via initial hydrogen atom abstraction. Angew. Chem. Int. Ed. 55, 11091–11095. doi: 10.1002/anie.201604412
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Blomberg, M. R. A., Borowski, T., Himo, F., Liao, R.-Z., and Siegbahn, P. E. M. (2014). Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 114, 3601–3658. doi: 10.1021/cr400388t
Bollinger, J. M. Jr., Price, J. C., Hoffart, L. M., Barr, E. W., and Krebs, C. (2005). Mechanism of taurine: α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem. 4245–4254. doi: 10.1002/ejic.200500476
Borowski, T., Bassan, A., and Siegbahn, P. E. M. (2004). Mechanism of Dioxygen activation in 2-oxoglutarate-dependent enzymes: a hybrid DFT Study. Chem. Eur. J. 10, 1031–1041. doi: 10.1002/chem.200305306
Brazzolotto, D., Cantú Reinhard, F. G., Smith-Jones, J., Retegan, M., Amidani, L., Faponle, A. S., et al. (2017). A high-valent non heme μ-oxo MnIV dimer generated from a thiolate-bound MnII complex and O2. Angew. Chem. Int. Ed. 56, 8211–8215. doi: 10.1002/anie.201703215
Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S., and Karplus, M. (1983). CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217. doi: 10.1002/jcc.540040211
Bruijnincx, P. C. A., van Koten, G., and Klein Gebbink, R. J. M. (2008). Mononuclear non-heme iron enzymes with the 2-His-1-carboxylate facial triad: recent developments in enzymology and modeling studies. Chem. Soc. Rev. 37, 2716–2744. doi: 10.1039/b707179p
Bugg, T. D. H. (2001). Oxygenases: mechanisms and structural motifs for O(2) activation. Curr. Opin. Chem. Biol. 5, 550–555. doi: 10.1016/S1367-5931(00)00236-2
Buongiorno, D., and Straganz, G. D. (2013). Structure and function of atypically coordinated enzymatic mononuclear non-heme-Fe(II) centers. Coord. Chem. Rev. 257, 541–563. doi: 10.1016/j.ccr.2012.04.028
Cantú Reinhard, F. G., and de Visser, S. P. (2017). Oxygen atom transfer using an iron(IV)-oxo embedded in a tetracyclic N-heterocyclic carbene system: how does the reactivity compare to Cytochrome P450 Compound? Chem. Eur. J. 23, 2935–2944. doi: 10.1002/chem.201605505
Cantú Reinhard, F. G., Faponle, A. S., and de Visser, S. P. (2016a). Substrate sulfoxidation by an iron(IV)-oxo complex: benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 120, 9805–9814. doi: 10.1021/acs.jpca.6b09765
Cantú Reinhard, F. G., Sainna, M. A., Upadhyay, P., Balan, G. A., Kumar, D., Fornarini, S., et al. (2016b). A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: a low-pressure mass spectrometry and computational study. Chem. Eur. J. 22, 18608–18619. doi: 10.1002/chem.201604361
Choroba, O. W., Williams, D. H., and Spencer, J. B. (2000). Biosynthesis of the vancomycin group of antibiotics: involvement of an unusual dioxygenase in the pathway to (s)-4-hydroxyphenylglycine. J. Am. Chem. Soc. 122, 5389–5390. doi: 10.1021/ja000076v
Costas, M., Mehn, M. P., Jensen, M. P., and Que, L. Jr. (2004). Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986. doi: 10.1021/cr020628n
Denisov, I. G., Makris, T. M., Sligar, S. G., and Schlichting, I. (2005). Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277. doi: 10.1021/cr0307143
de Visser, S. P. (2005). What affects the quartet-doublet energy splitting in peroxidase enzymes? J. Phys. Chem. A. 109, 11050–11057. doi: 10.1021/jp053873u
de Visser, S. P. (2006a). Differences in and comparison of the catalytic properties of heme and non-heme enzymes with a central oxo-iron group. Angew. Chem. Int. Ed. 45, 1790–1793. doi: 10.1002/anie.200503841
de Visser, S. P. (2006b). Propene activation by the oxo-iron active species of taurine/α-ketoglutarate dioxygenase (TauD) enzyme. How does the catalysis compare to heme-enzymes? J. Am. Chem. Soc. 128, 9813–9824. doi: 10.1021/ja061581g
de Visser, S. P. (2006c). Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. Eur. J. 12, 8168–8177. doi: 10.1002/chem.200600376
de Visser, S. P. (2006d). What factors influence the ratio of C–H hydroxylation versus C = C epoxidation by a nonheme cytochrome P450 biomimetic? J. Am. Chem. Soc. 128, 15809–15818. doi: 10.1021/ja065365j
de Visser, S. P. (2007). Can the peroxosuccinate complex in the catalytic cycle of taurine/α-ketoglutarate dioxygenase (TauD) act as an alternative oxidant? Chem. Commun. 171–173. doi: 10.1039/B611273K
de Visser, S. P. (2009). Elucidating enzyme mechanism and intrinsic chemical properties of short-lived intermediates in the catalytic cycles of cysteine dioxygenase and taurine/α-ketoglutarate dioxygenase. Coord. Chem. Rev. 253, 754–768. doi: 10.1016/j.ccr.2008.05.001
de Visser, S. P. (2012). Predictive studies of oxygen atom transfer reactions by Compound I of cytochrome P450: aliphatic and aromatic hydroxylation, epoxidation and sulfoxidation. Adv. Inorg. Chem. 64, 1–31. doi: 10.1016/B978-0-12-396462-5.00001-5
de Visser, S. P., and Kumar, D. (2011). Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reaction in Nature. Cambridge: RSC Publishing.
de Visser, S. P., Kumar, D., Cohen, S., Shacham, R., and Shaik, S. (2004). A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 126, 8362–8363. doi: 10.1021/ja048528h
de Visser, S. P., Ogliaro, F., Harris, N., and Shaik, S. (2001). Multi-state epoxidation of ethene by cytochrome P450: a quantum chemical study. J. Am. Chem. Soc. 123, 3037–3047. doi: 10.1021/ja003544+
de Visser, S. P., Ogliaro, F., Sharma, P. K., and Shaik, S. (2002a). Hydrogen bonding modulates the selectivity of enzymatic oxidation by P450: a chameleon oxidant behavior of Compound I. Angew. Chem. Int. Ed. 41, 1947–1951.
de Visser, S. P., Ogliaro, F., Sharma, P. K., and Shaik, S. (2002b). What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 124, 11809–11826. doi: 10.1021/ja026872d
de Visser, S. P., Rohde, J.-U., Lee, Y.-M., Cho, J., and Nam, W (2012). Intrinsic properties and reactivities of mononuclear nonheme iron–oxygen complexes bearing the tetramethylcyclam ligand. Coord. Chem. Rev. 257, 381–393. doi: 10.1016/j.ccr.2012.06.002
de Visser, S. P., and Shaik, S. (2003). A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. J. Am. Chem. Soc. 125, 7413–7424. doi: 10.1021/ja034142f
de Visser, S. P., Shaik, S., Sharma, P. K., Kumar, D., and Thiel, W. (2003). Active species of horseradish peroxidase (HRP) and cytochrome P450: two electronic chameleons. J. Am. Chem. Soc. 125, 15779–15788. doi: 10.1021/ja0380906
de Visser, S. P., Tahsini, L., and Nam, W. (2009). How does the axial ligand of cytochrome P450 biomimetics influence the regioselectivity of aliphatic versus aromatic hydroxylation? Chem. Eur. J. 15, 5577–5587. doi: 10.1002/chem.200802234
Dolinsky, T. J., Czodrowski, P., Li, H., Nielsen, J. E., Jensen, J. H., Klebe, G., et al. (2007). PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucl. Acids Res. 35, 522–525. doi: 10.1093/nar/gkm276
Faponle, A. S., Quesne, M. G., and de Visser, S. P. (2016). Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: what drives the reaction to biofuel production? Chem. Eur. J. 22, 5478–5483. doi: 10.1002/chem.201600739
Faponle, A. S., Seebeck, F. P., and de Visser, S. P. (2017). Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J. Am. Chem. Soc. 139, 9259–9270. doi: 10.1021/jacs.7b04251
Fellner, M., Siakkou, E., Faponle, A. S., Tchesnokov, E. P., de Visser, S. P., Wilbanks, S. M., et al. (2016). Influence of cysteine 164 on active site structure in rat cysteine dioxygenase. J. Biol. Inorg. Chem. 21, 501–510. doi: 10.1007/s00775-016-1360-0
Godfrey, E., Porro, C. S., and de Visser, S. P. (2008). Comparative quantum mechanics / molecular mechanics (QM/MM) and density functional theory calculations on the oxo-iron species of taurine/α-ketoglutarate dioxygenase. J. Phys. Chem. A. 112, 2464–2468. doi: 10.1021/jp710999v
Guo, Z., and Sevrioukova, I. (2017). Crystal structure of human Cyp3A4 bound to metformin. doi: 10.2210/pdb5g5j/pdb
Hernández-Ortega, A., Quesne, M. G., Bui, S., Heuts, D. P. H. M., Steiner, R. A., Heyes, D. J., et al. (2014). Origin of the proton-transfer step in the cofactor-free 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase: effect of the basicity of an active site His residue. J. Biol. Chem. 289, 8620–8632. doi: 10.1074/jbc.M113.543033
Hernández-Ortega, A., Quesne, M. G., Bui, S., Heyes, D. J., Steiner, R. A., Scrutton, N. S., et al. (2015). Catalytic mechanism of cofactor-free dioxygenases and how they circumvent spin-forbidden oxygenation of their substrates. J. Am. Chem. Soc. 137, 7474–7487. doi: 10.1021/jacs.5b03836
Higgins, L. J., Yan, F., Liu, P., Liu, H.-W., and Drennan, C. L. (2005). Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. Nature 437, 838–844. doi: 10.1038/nature03924
Hoffart, L. M., Barr, E. W., Guyer, R. B., Bollinger Jr, J. M., and Krebs, C. (2006). Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 103, 14738–14743. doi: 10.1073/pnas.0604005103
Ji, L., Faponle, A. S., Quesne, M. G., Sainna, M. A., Zhang, J., Franke, A., et al. (2015). Drug metabolism by cytochrome P450 enzymes: what distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 21, 9083–9092. doi: 10.1002/chem.201500329
Kadish, K. M., Smith, K. M., and Guilard, R. (2010). Handbook of Porphyrin Science. Hackensack, NJ: World Scientific Publishing Co.
Karamzadeh, B., Kumar, D., Sastry, G. N., and de Visser, S. P. (2010). Steric factors override thermodynamic driving force in regioselectivity of proline hydroxylation by prolyl-4-hydroxylase enzymes. J. Phys. Chem. A 114, 13234–13243. doi: 10.1021/jp1089855
Koski, M. K., Hieta, R., Hirsila, M., Ronka, A., Myllyharju, J., and Wierenga, R. K. (2009). The crystal structure of an algal prolyl 4-hydroxylase complexed with a proline-rich peptide reveals a novel buried tripeptide binding motif. J. Biol. Chem. 284, 25290–25301. doi: 10.1074/jbc.M109.014050
Kovaleva, E. G., and Lipscomb, J. D. (2008). Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol. 4, 186–193. doi: 10.1038/nchembio.71
Kryatov, S. V., Rybak-Akimova, E. V., and Schindler, S. (2005). Kinetics and mechanisms of formation and reactivity of non-heme iron oxygen intermediates. Chem. Rev. 105, 2175–2226. doi: 10.1021/cr030709z
Kumar, D., de Visser, S. P., Sharma, P. K., Cohen, S., and Shaik, S. (2004). Radical clock substrates, their C–H hydroxylation mechanism by cytochrome P450 and other reactivity patterns: what does theory reveal about the clocks' behavior? J. Am. Chem. Soc. 126, 1907–1920. doi: 10.1021/ja039439s
Kumar, D., de Visser, S. P., Sharma, P. K., Hirao, H., and Shaik, S. (2005b). Sulfoxidation mechanisms catalyzed by cytochrome P450 and horseradish peroxidase models: spin selection induced by the ligand. Biochemistry 44, 8148–8158. doi: 10.1021/bi050348c
Kumar, D., Hirao, H., de Visser, S. P., Zheng, J., Wang, D., Thiel, W., et al. (2005a). New features in the catalytic cycle of cytochrome P450 during the formation of Compound I from Compound 0. J. Phys. Chem. B 109, 19946–19951. doi: 10.1021/jp054754h
Kumar, D., Sastry, G. N., and de Visser, S. P. (2011a). Effect of the axial ligand on substrate sulfoxidation mediated by iron(IV)-oxo porphyrin cation radical oxidants. Chem. Eur. J. 17, 6196–6205. doi: 10.1002/chem.201003187
Kumar, D., Thiel, W., and de Visser, S. P. (2011b). Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J. Am. Chem. Soc. 133, 3869–3882. doi: 10.1021/ja107514f
Kumar, S., Faponle, A. S., Barman, P., Vardhaman, A. K., Sastri, C. V., Kumar, D., et al. (2014). Long-range electron transfer triggers mechanistic differences between iron(IV)-oxo and iron(IV)-imido oxidants. J. Am. Chem. Soc. 136, 17102–17115. doi: 10.1021/ja508403w
Latifi, R., Bagherzadeh, M., and de Visser, S. P. (2009). Origin of the correlation of the rate constant of substrate hydroxylation by nonheme iron(IV)-oxo complexes with the bond-dissociation energy of the C–H bond of the substrate. Chem. Eur. J. 15, 6651–6662. doi: 10.1002/chem.200900211
Latifi, R., Sainna, M. A., Rybak-Akimova, E. V., and de Visser, S. P. (2013). Does hydrogen bonding-donation to manganese(IV)-oxo and iron(IV)-oxo oxidants affect the oxygen atom transfer ability? A computational study. Chem. Eur. J. 19, 4058–4068. doi: 10.1002/chem.201202811
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi: 10.1103/PhysRevB.37.785
Li, X.-X., Postils, V., Sun, W., Faponle, A. S., Solà, M., Wang, Y., et al. (2017). Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: which is the better oxidant? Chem. Eur. J. 23, 6406–6418. doi: 10.1002/chem.201700363
McDonough, M. A., Li, V., Flashman, E., Chowdhury, R., Mohr, C., Lienard, B. M., et al. (2006). Cellular oxygen sensing: crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. U.S.A. 103, 9814–9819. doi: 10.1073/pnas.0601283103
Meunier, B., de Visser, S. P., and Shaik, S. (2004). Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 104, 3947–3980. doi: 10.1021/cr020443g
Myllyharju, J., and Kivirikko, K. I. (1997). Characterization of the iron- and 2-oxoglutarate-binding sites of human prolyl 4-hydroxylase. EMBO J. 16, 1173–1180. doi: 10.1093/emboj/16.6.1173
O'Brien, J. R., Schuller, D. J., Yang, V. S., Dillard, B. D., and Lanzilotta, W. N. (2003). Substrate-induced conformational changes in escherichia coli taurine/alpha-ketoglutarate dioxygenase and insight into the oligomeric structure. Biochemistry 42, 5547–5554. doi: 10.1021/bi0341096
O'Brien, P. J. (2006). Catalytic promiscuity and the divergent evolution of DNA repair enzymes. Chem. Rev. 106, 720–752. doi: 10.1021/cr040481v
Ogliaro, F., Cohen, S., de Visser, S. P., and Shaik, S. (2000a). Medium polarization and hydrogen bonding effects on Compound I of cytochrome P450: what kind of a radical is it really? J. Am. Chem. Soc. 122, 12892–12893. doi: 10.1021/ja005619f
Ogliaro, F., Harris, N., Cohen, S., Filatov, M., de Visser, S. P., and Shaik, S. (2000b). A model “rebound” mechanism of hydroxylation by cytochrome P450: stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 122, 8977–8989. doi: 10.1021/ja991878x
Ortiz de Montellano, P. R. (2004). Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd Edn., New York, NY: Kluwer Academic/Plenum Publishers.
Ortiz de Montellano, P. R. (2010). Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 110, 932–948. doi: 10.1021/cr9002193
Porro, C. S., Sutcliffe, M. J., and de Visser, S. P. (2009). Quantum mechanics/molecular mechanics studies on the sulfoxidation of dimethyl sulfide by Compound I and Compound 0 of Cytochrome P450: which is the better oxidant? J. Phys. Chem. A 113, 11635–11642. doi: 10.1021/jp9023926
Pratter, S. M., Konstantinovics, C., DiGiuro, C. L. M., Leitner, E., Kumar, D., de Visser, S. P., et al. (2013). Inversion of enantio-selectivity of a mononuclear non-heme iron(II)-dependent hydroxylase by tuning the interplay of metal-center geometry and protein structure. Angew. Chem. Int. Ed. 52, 9677–9681. doi: 10.1002/anie.201304633
Que, L. Jr. (2000). One motif — many different reactions. Nat. Struct. Biol. 7, 182–184. doi: 10.1038/73270
Quesne, M. G., Borowski, T., and de Visser, S. P. (2016a). Quantum mechanics/molecular mechanics modelling of enzymatic processes: caveats and breakthroughs. Chem. Eur. J. 22, 2562–2581. doi: 10.1002/chem.201503802
Quesne, M. G., Latifi, R., Gonzalez-Ovalle, L. E., Kumar, D., and de Visser, S. P. (2014). Quantum mechanics/molecular mechanics study on the oxygen binding and substrate hydroxylation step in AlkB repair enzymes. Chem. Eur. J. 20, 435–446. doi: 10.1002/chem.201303282
Quesne, M. G., Senthilnathan, D., Singh, D., Kumar, D., Maldivi, P., Sorokin, A. B., et al. (2016b). Origin of the enhanced reactivity of μ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 Compound I. ACS Catal. 6, 2230–2243. doi: 10.1021/acscatal.5b02720
Rittle, J., and Green, M. T. (2010). Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 330, 933–937. doi: 10.1126/science.1193478
Ryle, M. J., and Hausinger, R. P. (2002). Non-heme iron oxygenases. Curr. Opin. Chem. Biol. 6, 193–201. doi: 10.1016/S1367-5931(02)00302-2
Sainna, M. A., Kumar, S., Kumar, D., Fornarini, S., Crestoni, M. E., and de Visser, S. P. (2015). A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 6, 1516–1529. doi: 10.1039/C4SC02717E
Schafer, A., Horn, H., and Ahlrichs, R. (1992). Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577. doi: 10.1063/1.463096
Schöneboom, J. C., Lin, H., Reuter, N., Thiel, W., Cohen, S., Ogliaro, F., et al. (2002). The elusive oxidant species of cytochrome P450 enzymes: characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 124, 8142–8151. doi: 10.1021/ja026279w
Shaik, S., Kumar, D., de Visser, S. P., Altun, A., and Thiel, W. (2005). Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 105, 2279–2328. doi: 10.1021/cr030722j
Sharma, P. K., de Visser, S. P., and Shaik, S. (2003). Can a single oxidant with two spin states masquerade as two different oxidants? A study of the sulfoxidation mechanism by cytochrome P450. J. Am. Chem. Soc. 125, 8698–8699. doi: 10.1021/ja035135u
Sherwood, P., de Vries, A. H., Guest, M. F., Schreckenbach, G., Catlow, C. R. A., French, S. A., et al. (2003). QUASI: a general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct. 632, 1–28. doi: 10.1016/S0166-1280(03)00285-9
Siitonen, V., Selvaraj, B., Niiranen, L., Lindqvist, Y., Schneider, G., and Metsä-Ketelä, M. (2016). Divergent non-heme iron enzymes in the nogalamycin biosynthetic pathway. Proc. Natl. Acad. Sci. U.S.A. 113, 5251–5256. doi: 10.1073/pnas.1525034113
Smith, W., and Forester, T. R. (1996). DL_POLY_2.0: a general-purpose parallel molecular dynamics simulation package. J. Mol. Graphics 14, 136–141. doi: 10.1016/S0263-7855(96)00043-4
Solomon, E. I., Brunold, T. C., Davis, M. I., Kemsley, J. N., Lee, S.-K., Lehnert, N., et al. (2000). Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 100, 235–349. doi: 10.1021/cr9900275
Solomon, E. I., Light, K. M., Liu, L. V., Srnec, M., and Wong, S. D. (2013). Geometric and electronic structure contributions to function in non-heme iron enzymes. Acc. Chem. Res. 46, 2725–2739. doi: 10.1021/ar400149m
Stipanuk, M. H. (2004). Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr. 24, 539–577. doi: 10.1146/annurev.nutr.24.012003.132418
Straganz, G. D., and Nidetzky, B. (2006). Variations of the 2-His-1-carboxylate Theme in Mononuclear Non-Heme FeII Oxygenases. Chembiochem 7, 1536–1548. doi: 10.1002/cbic.200600152
Tchesnokov, E. P., Faponle, A. S., Davies, C. G., Quesne, M. G., Turner, R., Fellner, M., et al. (2016). An iron-oxygen intermediate formed during the catalytic cycle of cysteine dioxygenase. Chem. Commun. 52, 8814–8817. doi: 10.1039/C6CC03904A
Timmins, A., Saint-André, M., and de Visser, S. P. (2017). Understanding how prolyl-4-hydroxylase structure steers a ferryl oxidant toward scission of a strong C-H bond. J. Am. Chem. Soc. 139, 9855–9866. doi: 10.1021/jacs.7b02839
Vardhaman, A. K., Barman, P., Kumar, S., Sastri, C. V., Kumar, D., and de Visser, S. P. (2013). Comparison of the reactivity of nonheme iron(IV)-oxo versus iron(IV)-imido complexes: which is the better oxidant? Angew. Chem. Int. Ed. 52, 12288–12292. doi: 10.1002/anie.201305370
Vardhaman, A. K., Sastri, C. V., Kumar, D., and de Visser, S. P. (2011). Nonheme ferric hydroperoxo intermediates are efficient oxidants of bromide oxidation. Chem. Commun. 47, 11044–11046. doi: 10.1039/c1cc13775a
Wachters, A. J. H. (1970). Gaussian basis set for molecular wavefunctions containing third-row atoms. J. Chem. Phys. 52, 1033–1036. doi: 10.1063/1.1673095
Yang, T., Quesne, M. G., Neu, H. M., Cantú Reinhard, F. G., Goldberg, D. P., and de Visser, S. P. (2016). Singlet versus triplet reactivity in an Mn(V)-Oxo species: testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 138, 12375–12386. doi: 10.1021/jacs.6b05027
Keywords: quantum mechanics/molecular mechanics, enzyme mechanism, enzyme catalysis, mutations, density functional theory
Citation: Timmins A and de Visser SP (2017) How Are Substrate Binding and Catalysis Affected by Mutating Glu127 and Arg161 in Prolyl-4-hydroxylase? A QM/MM and MD Study. Front. Chem. 5:94. doi: 10.3389/fchem.2017.00094
Received: 13 September 2017; Accepted: 24 October 2017;
Published: 09 November 2017.
Edited by:
Nino Russo, Università della Calabria, ItalyReviewed by:
Tiziana Marino, University of Calabria, ItalyXavier Assfeld, Université de Lorraine, France
Copyright © 2017 Timmins and de Visser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sam P. de Visser, c2FtLmRldmlzc2VyQG1hbmNoZXN0ZXIuYWMudWs=