 Moritz Schübler1
Moritz Schübler1 Bassem Sadek
Bassem Sadek Holger Stark
Holger Stark
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 12 September 2017
Sec. Medicinal and Pharmaceutical Chemistry
Volume 5 - 2017 | https://doi.org/10.3389/fchem.2017.00064
Neurleptic drugs, e.g., aripiprazole, targeting the dopamine D2S and D3 receptors (D2SR and D3R) in the central nervous system are widely used in the treatment of several psychotic and neurodegenerative diseases. Therefore, a new series of benzothiazole-based ligands (3-20) was synthesized by applying the bioisosteric approach derived from the selective D3Rs ligand BP-897 (1) and its structurally related benz[d]imidazole derivative (2). Herein, introduction of the benzothiazole moiety was well tolerated by D2SR and D3R binding sites leading to antagonist affinities in the low nanomolar concentration range at both receptor subtypes. However, all novel compounds showed lower antagonist affinity to D3R when compared to that of 1. Further exploration of different substitution patterns at the benzothiazole heterocycle and the basic 4-phenylpiperazine resulted in the discovery of high dually acting D2SR and D3R ligands. Moreover, the methoxy substitution at 2-position of 4-phenylpiperazine resulted in significantly (22-fold) increased D2SR binding affinity as compared to the parent ligand 1, and improved physicochemical and drug-likeness properties of ligands 3-11. However, the latter structural modifications failed to improve the drug-able properties in ligands having un-substituted 4-phenylpiperazine analogs (12-20). Accordingly, compound 9 showed in addition to high dual affinity at the D2SR and D3R [Ki (hD2SR) = 2.8 ± 0.8 nM; Ki (hD3R) = 3.0 ± 1.6 nM], promising clogS, clogP, LE (hD2SR, hD3R), LipE (hD2SR, hD3R), and drug-likeness score values of −4.7, 4.2, (0.4, 0.4), (4.4, 4.3), and 0.7, respectively. Also, the deaminated analog 10 [Ki (hD2SR) = 3.2 ± 0.4 nM; Ki (hD3R) = 8.5 ± 2.2 nM] revealed clogS, clogP, LE (hD2SR, hD3R), LipE (hD2SR, hD3R) and drug-likeness score values of −4.7, 4.2, (0.4, 0.4), (3.9, 3.5), and 0.4, respectively. The results observed for the newly developed benzothiazole-based ligands 3-20 provide clues for the diversity in structure activity relationships (SARs) at the D2SR and D3R subtypes.
The dopaminergic systems in the central nervous system (CNS) have been comprehensively studied over the past five decades (Missale et al., 1998). Dopamine (DA) exerts its (patho)physiological effects through five distinct G-protein coupled receptors (D1−5 receptors), grouped in two classes, Gs-coupled D1-like (D1R and D5R) and Gi-coupled D2-like (D2R, D3R, and D4R), that vary in their signal transduction, binding shape and physiological effects (Missale et al., 1998). Moreover, an alternative splicing of D2R mRNA leads to generation of two isoforms: D2 short (D2S) and D2 long (D2L), which have been linked (though not completely) with presynaptic and postsynaptic populations of D2Rs, respectively (Joyce and Millan, 2005). Accordingly, it has been found that the D2SR is mainly considered as a presynaptic receptor, whereas, the D2LR as a postsynaptic receptor (Joyce and Millan, 2005), comparable to the D3R (Lader, 2008). However, it has been proposed that D3R, in addition to the classical postsynaptic location, is also localized in the presynapse, where it modifies release and synthesis of DA (Sokoloff et al., 1990; Noeske et al., 2006; Bocker et al., 2007; Boeckler and Gmeiner, 2007). Furthermore, it has been convincingly demonstrated that hD2R and hD3R are highly homologous (Stark, 1998), sharing 78% of sequence identity in the transmembrane domains (Kaufmann and Bückmann, 1941), including the binding site (Levey et al., 1993). Thus, this sequence identity has introduced difficulties in the design of ligands selectively targeting D2sR and/or D3R, since such ligands could offer more effective neuroleptic drugs with a favorable side effect profile in therapeutic management of central diseases, e.g., schizophrenia and Parkinson's disease. Accordingly, the newer atypical antipsychotic drugs, e.g., aripiprazole, with high affinity for D2Rs and D3Rs show a unique pharmacological profile at treating the positive symptoms of schizophrenia and have the potential to treat negative and cognitive symptoms, while also mitigating risk of weight gain and movement side effects (Swainston Harrison and Perry, 2004; Casey and Canal, 2017; Tuplin and Holahan, 2017).
In the past two decades, medicinal chemists have succeeded, by using ligand-based approaches, in developing selective D2SR, D3R, and dually acting D2SR/D3R ligands such as benz[d]imidazolyl ether 2 (Pilla et al., 1999; Garcia-Ladona and Cox, 2003; Boeckler and Gmeiner, 2007; Tomic et al., 2007; Moller et al., 2014; Stucchi et al., 2015). Interestingly, ligand 1 has earlier been described as a selective D3R ligand with a 66-fold higher selectivity over D2SR (Pilla et al., 1999). This former clinical candidate bears distinctive minimal pharmacophoric structure elements for many D3R ligands, which could be divided into three substructures: a lipophilic aryl moiety connected to an amide or ether (1), which is linked by a linear alkyl spacer (2) to a basic amine residue with aryl substitution (3) (Figure 1; Pilla et al., 1999; Garcia-Ladona and Cox, 2003). The basic 4-phenylpiperazine substructure (3) also described as “privileged structural surrogate” has proved to be essential for ligand interaction with selectivity for the D2SRs and D3Rs (Hackling et al., 2003; Moller et al., 2014). Accordingly, ligand efficacy and selectivity between D2SR and D3R activation were found to be strongly influenced by regiochemistry and the nature of functional groups attached to the pyrazolo[1,5-a]pyridine heterocycle moiety. (Hackling et al., 2003; Moller et al., 2014). Also, the aliphatic tetramethylene spacer (2) in ligand 1 demonstrated the optimum distance between the two aromatic systems (1) and (3) (Hayes et al., 1992; Sokoloff et al., 1992; Noeske et al., 2006; Bocker et al., 2007; Boeckler and Gmeiner, 2007). Moreover, the amide function in numerous previously developed D2SR/D3R selective ligands, e.g., 1, has been described as a weak H-bond acceptor and was directly connected to the aryl moiety (1) (Wright and Rosowsky, 2002; Hackling et al., 2003; Moller et al., 2014). Furthermore, the lipophilic residue in the aryl moiety (1) has convincingly been demonstrated to tolerate plenty of variations including biphenyl, heteroaryl or cycloalkyl substituents, resulting in many D2R/D3R ligands (Figure 1) (Murray et al., 1996; Bettinetti et al., 2002; Moller et al., 2014). Moreover, the recently described benz[d]imidazolyl ether (2) exhibited preference to the D2SRs and D3Rs, respectively (Figure 1; Tomic et al., 2007; Moller et al., 2014). Keeping this in view and in continuation of our endeavors toward providing tools with which further exploration of the role of the dopaminergic system, the development of high-affinity D2R/D3R ligands was undertaken. It was envisaged that the heterocyclic appendage of benzothiazole, as a lipophilic bioisosterical replacement of aryl moiety (1), with the basic 4-phenylpiperazine aryl substitution (3) by an ether of varying alkyl spacer length (2) is worth the attempt. Accordingly, the replacement of the amide group present in ligand 1 with an ether structure was undertaken to investigate whether the affinity of the developed ligands will be modified toward balanced D2SR/D3R. In addition, the inclusion of different substituents at 2-position of benzothiazole (1) or 4-phenylpiperazine (3) was carried out to explore further the modulating role of hydrogen bound donors and lipophilic interaction with the heterocyclic π-system present in the novel ligands 3-20. Moreover, all the title ligands 3-20 were subjected to Molinspiration Property, Osiris Property Explorer, and MolSoft toolkits to predictively assess their physicochemical properties. Furthermore, initial metric analyses were conducted to predictably quantify the lipophilicity (clogP), the water solubility (clogS), the ligand efficiency (LE), lipophilicity-dependent ligand efficiency (LELP), and lipophilic efficiency (LipE) important for verification of drug-likeness of the novel compounds (3-20) presented in the current study.
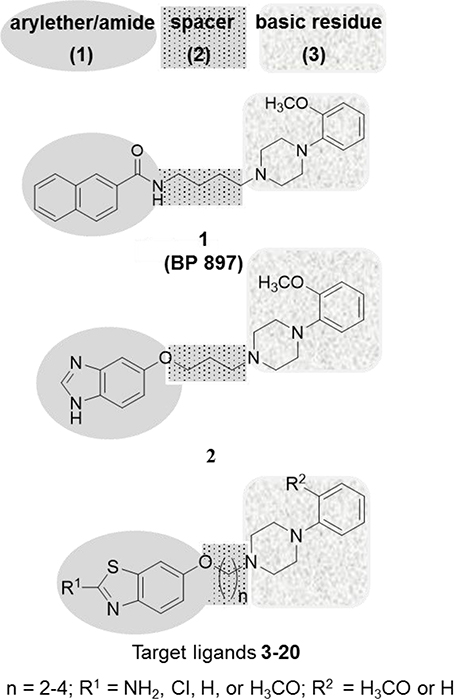
Figure 1. Features similarities between the lead structures 1, 2 and target compounds (3-20).
All starting reagents/chemicals and dry solvents of commercial quality were purchased from the commercial suppliers VWR Darmstadt/Germany, Acros, Geel/Belgium, Alfar Aesar, Ward Hill/U.S.A., Sigma-Aldrich, St. Louis/USA, Fluka, München/Germany and Carl Roth, Karlsruhe/Germany. 1H-NMR and 13C-NMR spectra were recorded on a Bruker AMX 250 (250 MHz), a Bruker AMX 300 (300 MHz) or a Bruker AMX 400 (400 MHz) spectrometer (Bruker, Germany). 1H-NMR data are reported in the following order: chemical shift (δ) in ppm downfield from tetramethylsilane as internal reference; multiplicity (br, broad; s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; m, multiplet); approximate coupling constants (J) in Hertz (Hz). ESI-MS was performed on a Fisons Instruments VG Platform II (Manchester, Great Britain) in positive polarity. Data (m/z) are listed as mass number (M+H+) and relative intensity (%). MALDI_MS was performed on Voyager DE STR (Per Septive Biosystems). High Resolution Mass Spectrometry (HRMS) was carried out on a Thermo Scientific MALDI LTQ XL Orbitrap (Waltham, USA). Thin layer chromatography was performed on silica gel 60 F254 aluminum sheets (Merck, Germany). Preparative column chromatography was performed on silica gel 63–200 μM (Merck, Germany). Melting points were determined on a Büchi B510 melting point apparatus (Büchi, Switzerland).
4-Nitrophenole (21, 3.0 g, 22 mmol) was diluted into 100 ml acetone. 1,4-dibromobutane (16.53 g, 88 mmol), anhydrous potassium carbonate (3.0 g, 22 mmol) and a catalytic amount of potassium iodide was added. The suspension was heated under reflux for 24 h. After cooling the yellow-white suspension was filtered. The yellow filtrate was evaporated to give a yellow oily raw product. The product was purified by filtration with petrolether (60–90°C) and dicloromethane over silicagel to obtain a pure yellow solid; yield 60%; C8H8BrNO3; 1H-NMR (CDCl3, 400 MHz) δ = 3.61 (t, J = 6.10 Hz, 2H, OCH2CH2Br), 4.32 (t, J = 6.11 Hz, 2H, OCH2CH2Br), 6.91 (d, J = 9.11 Hz, 2H, Ar-2-H, -6-H), 8.14 (d, J = 9.16 Hz, 2H, Ar-3-H, -5-H), ESI-MS (m/z): 246.0 (M-H+, 100), 247.4 (M-H+, 99), 249.0 (M-H+, 6).
Intermediate 22b was synthesized according to above described procedure for 22a, however, with 1,3-dibromopropne (17.77 g, 88 mmol) to yield a white solid; yield: 86%; C9H10BrNO3; 1H-NMR (CDCl3, 300 MHz): δ = 2.29 (p, J = 6.02 Hz, 2H, OCH2CH2CH2Br) 3.54 (t, J = 6.30 Hz, 2H, OCH2 CH2CH2Br), 4.14 (t, J = 5.82 Hz, 2H, OCH2CH2CH2Br), 6.89 (d, J = 9.00 Hz, 2H, Ar-2-H, -6-H), 8.12 (d, J = 9.12 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 234.9 (M-Br+NH4OH+Na+, 100), 236.0 (M-Br+NH4OH+Na+, 16).
Intermediate 22c was synthesized according to above described procedure for 1a, however, with 1,4-dibromobutane (19.00 g, 88 mmol) affording a yellow oil; yield: 90%; C10H12BrNO3; 1H-NMR (CDCl3, 400 MHz): δ = 1.90–2.05 (m, 4H, OCH2CH2CH2CH2Br) 3.43 (t, J = 6.37 Hz, 2H, OCH2CH2CH2CH2Br), 4.03 (t, J = 5.78 Hz, 2H, OCH2CH2CH2CH2Br), 6.87 (d, J = 9.15 Hz, 2H, Ar-2-H, -6-H), 8.12 (d, J = 9.13 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 297.9 (M-Na+, 100), 295.9 (M-Na+, 98), 296.7 (M-Na+, 10).
(4.2 mmol) was dissolved with 50 ml ethyl acetate. 100 mg Pd/C (10% m/m) and a few drops glacial acetic acid were added. The suspension was stirred at room temperature in a Parr apparatus with a pressure of 5 bar hydrogen for 1 h. After filtration of the catalyst the yellow solution was evaporated to dryness to give a Yellow oil (0.76 g, yield: 100%); C8H10BrNO; 1H-NMR (CDCl3, 300 MHz), δ = 3.52 (t, J = 6.34 Hz, 2H, OCH2CH2Br), 4.14 (t, J = 6.34 Hz, 2H, OCH2CH2Br), 4.38 (s, br, 2H, Ar-NH2), 6.58 (d, J = 8.86 Hz, 2H, Ar-2-H, -6-H), 6.69 (d, J = 8.85 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 215.6 (M-H+, 100), 216.6 (M-H+, 9), 217.7 (M-H+, 98).
Intermediate 23b was synthesized according to above described procedure for 23a, however, starting from 22b (4.2 mmol). Brown oil; yield: 100%; C9H12BrNO; 1H-NMR (CDCl3, 300 MHz): δ = 2.19 (p, J = 6.17 Hz, 2H, OCH2CH2CH2Br) 3.52 (t, J = 6.49 Hz, 2H, OCH2 CH2CH2Br), 3.96 (t, J = 5.82 Hz, 2H, OCH2CH2CH2Br), 4.59 (s, br, 2H, ArNH2), 6.59 (d, J = 8.79 Hz, 2H, Ar-2-H, -6-H), 6.69 (d, J = 8.84 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 229.8 (M-H+, 100), 230.7 (M-H+, 9), 231.7 (M-H+, 97).
Intermediate 23c was synthesized according to above described procedure for 23a, however, starting from 22c (4.2 mmol). Yellow oil; yield: 100%; C10H14BrNO; 1H-NMR (CDCl3, 300 MHz) δ = 1.80–2.00 (m, 4H, OCH2CH2CH2CH2Br) 3.41 (t, J = 6.62 Hz, 2H, OCH2CH2CH2CH2Br), 3.86 (t, J = 6.03 Hz, 2H, OCH2CH2CH2CH2Br), 6.59 (d, J = 9.03 Hz, 2H, Ar-2-H, -6-H), 6.67 (d, J = 8.96 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 243.7 (M-H+, 100), 245.7 (M-H+, 98).
23a (4.4 mmol) and potassium thiocyanate (1.71 g, 17.6 mmol) were solved in 25 ml glacial acetic acid. The resultant brown solution was cooled on an ice bath. Dropwise bromine (0.71 g, 4.4 mmol) dissolved in 5 ml glacial acetic acid was added, while the temperature was held constant at 4°C. Afterwards the orange suspension was stirred at room temperature overnight. The resulting solid was filtered of and the yellow filtrate was concentrated in vacuum. The yellow-brown solid was purified by column chromatography to give a yield of 68%; C9H9BrN2OS; 1H-NMR (CDCl3, 300 MHz): δ = 3.57 (t, J = 6.28 Hz, 2H, OCH2CH2Br), 4.22 (t, J = 6.28 Hz, 2H, OCH2CH2Br), 5.18 (s, br, 2H, Benzthia-2-NH2), 6.86 (d, J = 8.78 Hz, 1H, Benzthia-5-H), 7.09 (d, 4J = 2.55 Hz, 1H, Benzthia-7-H), 7.37 (d, J = 8.77 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 274.7 (M-H+, 100), 273.7 (M-H+, 12), 272.9 (M-H+, 94).
Intermediate 24b was synthesized according to above described procedure for 24a, however, starting from 23b (4.2 mmol). Brown solid; yield: 45%; C10H11BrN2OS; 1H-NMR (CDCl3, 400 MHz): δ = 2.22–2.27 (m, 2H, OCH2CH2CH2Br) 3.55 (t, J = 6.42 Hz, 2H, OCH2 CH2CH2Br), 4.05 (t, J = 5.80 Hz, 2H, OCH2CH2CH2Br), 5.05 (s, br, 2H, Benzthia-2-NH2), 6.85 (d, J = 8.75 Hz, 1H, Benzthia-5-H), 7.07 (d, 4J = 2.53 Hz, 1H, Benzthia-7-H), 7.38 (d, J = 8.78 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 288.7 (M-H+, 100), 286.7 (M-H+, 96), 289.8 (M-H+, 12).
Intermediate 24c was synthesized according to above described procedure for 24a, however, starting from 23c (4.2 mmol). Brown solid; yield: 47%; C11H13BrN2OS; 1H-NMR (d6 DMSO, 250 MHz): δ = 1.77 – 1.98 (m, 4H, OCH2CH2CH2CH2Br) 3.59 (t, J = 6.58 Hz, 2H, OCH2CH2CH2CH2Br), 3.96 (t, J = 6.13 Hz, 2H, OCH2CH2CH2CH2Br), 6.79 (d, J = 8.72 Hz, 1H, Benzthia-5-H), 7.21 (d, J = 8.67 Hz, 1H, Benzthia-4-H), 7.20 (s, br, 2H, Benzthia-2-NH2), 7.27 (d, 4J = 2.53 Hz, 1H, Benzthia-7-H); ESI-MS (m/z): 302.7 (M-H+, 100), 300.8 (M-H+, 95), 303.8 (M-H+, 13).
24a (0.6 mmol) was dissolved in acetone. 2-methoxyphenylpiperazine (0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography to give a white product which was precipitated with anhydrous oxalic acid and dried. White solid (yield: 24%; mp: 162–163°C (decomp.); C20H24N4O2S × 3 C2H2O4; 1H-NMR (d6 DMSO, 300 MHz) δ = 3.10–3.30 (m, 4H, Pip-2-H, -6-H), 3.35–3.45 (m, 4H, Pip-3-H, 5-H), 3.53 (t, J = 4.81 Hz, 2H, OCH2CH2), 3.79 (s, 3 H, OCH3), 4.34 (t, J = 4.48 Hz, 2H, OCH2CH2), 6.91 (d, J = 8.12 Hz, 2H, 2-OCH3-Ar-3-H, -6-H), 6.98–7.04 (m, 2H, 2-OCH3-Ar-4-H, -5-H), 7.26 (d, J = 8.71 Hz, 1H, Benzthia-5-H), 7.34 (s, br, 2H, Benzthia-2-NH2), 7.38–7.41 (m, 2H, Benzthia-4-H, -7-H), 9.50 (s, br,2H, NH+); 13C-NMR (d6DMSO, 75 MHz) δ = 47.16 (Pip-3-C, -5-C), 51.96 (Pip-2-C, -6-C), 54.77 (OCH2CH2), 55.36 (OCH3), 63.27 (OCH2CH2), 106.95 (Benzthia-7-C), 111.98 (Benzthia-5-C), 113.74 (2-OCH3-Ar-C-4), 117.98 (2-OCH3-Ar-C-3), 118.24 (2-OCH3-Ar-C-6), 120.83 (2-OCH3-Ar-C-5), 123.34 (Benzthia-4-C), 131.76 (Benzthia-7a-C), 139.55 (Benzthia-3a-C), 147.27 (2-OCH3-Ar-C-1), 151.87 (2-OCH3-Ar-C-2), 152.53 (Benzthia-6-C), 162.28 ((COOH)2), 165.14 (Benzthia-2-C), ESI-MS (m/z): 385.2 (M-H+, 100), 386.0 (M-H+, 22), 387.0 (M-H+, 7). CHN calc.: C 47.71, H 4.62, N 8.56; CHN found: C 47.45,H 4.82, N 8.30.
24b (0.6 mmol) was dissolved in acetone. 2-methoxyphenylpiperazine (0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography to give a white product which was precipitated with anhydrous oxalic acid and dried. White solid (yield: 82%; mp: 171°C); C21H26N4O2S; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.83–1.92 (m, 2H, OCH2CH2CH2), 2.45–2.51 (m, 6H, Pip-2-H, -6-H, OCH2 CH2CH2), 2.96 (m, 4H, Pip-3-H, -5-H), 3.76 (s, 3 H, OCH3), 3.99 (t; J = 6.33 Hz, 2H, OCH2CH2CH2), 6.81 (d, J = 8.71 Hz, 1H, Benzthia-5-H), 6.85–6.94 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.20 (s, br, 2H, Benzthia-2-NH2), 7.22 (d, J = 8.81 Hz, 1H, Benzthia-4-H), 7.29 (d, 4J = 2.57 Hz, 1H, Benzthia-7-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 26.29 (OCH2CH2CH2), 50.04 (Pip-3-C, -5-C), 53.05 (Pip-2-C, -6-C), 54.51 (OCH2CH2CH2), 55.28 (OCH3), 66.44 (OCH2CH2CH2), 106.33 (Benzthia-7-C), 111.95 (Benzthia-5-C), 113.47 (2-OCH3-Ar-C-4), 117.85 (2-OCH3-Ar-C-3), 118.03 (2-OCH3-Ar-C-6), 120.82 (2-OCH3-Ar-C-5), 122.26 (Benzthia-4-C), 131.88 (Benzthia-7a-C), 141.27 (Benzthia-3a-C), 146.83 (2-OCH3-Ar-C-1), 151.96 (2-OCH3-Ar-C-2), 153.59 (Benzthia-6-C), 164.69 (Benzthia-2-C); ESI-MS (m/z): 399.0 (M-H+, 100), 400.0 (M-H+, 26), 401.0 (M-H+, 8). CHN calc.: C 63.29, H 6.58, N 14.06; CHN found: C 63.01, H 6.56, N 13.91.
24c (0.6 mmol) was dissolved in acetone. 2-methoxyphenylpiperazine (0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography to give a white product which was precipitated with anhydrous oxalic acid and dried. White solid [yield: 56%; mp 147–150°C (decomp.)]; C22H28N4O2S × 2 C2H2O4 × 0.25 H2O; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.57–1.75 (m, 4H, OCH2CH2CH2CH2), 2.39 (t, J = 6.81 Hz, 2H, OCH2CH2CH2CH2), 2.49–2.51 (m, 4H, Pip-2-H, -6-H), 2.94 (m, 4H, Pip-3-H, -5-H), 3.76 (s, 3 H, OCH3), 3.96 (t, J = 6.32 Hz, 2H, OCH2CH2CH2CH2), 6.81 (d, J = 8.72 Hz, 1H, Benzthia-5-H), 6.85–6.94 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.19 (s, br, 2H, Benzthia-2-NH2), 7.22 (d, J = 8.75 Hz, 1H, Benzthia-4-H), 7.28 (d, 4J = 2.56 Hz, 1H, Benzthia-7-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 20.37 (OCH2CH2CH2CH2), 25.44 (OCH2CH2CH2CH2), 47.11 (Pip-3-C, -5-C), 51.27 (Pip-2-C, -6-C), 55.36 (OCH2CH2CH2CH2), 55.36 (OCH3), 67.44 (OCH2CH2CH2CH2), 106.46 (Benzthia-7-C), 111.96 (Benzthia-5-C), 113.52 (2-OCH3-Ar-4-C), 118.00 (2-OCH3-Ar-3-C), 118.24 (2-OCH3-Ar-6-C), 120.83 (2-OCH3-Ar-5-C), 123.38 (Benzthia-4-C), 131.82 (Benzthia-7a-C), 139.47 (Benzthia-3a-C), 146.84 (2-OCH3-Ar-1-C), 151.85 (2-OCH3-Ar-2-C), 153.41 (Benzthia-6-C), 162.79 ((COOH)2), 164.77 (Benzthia-2-C); ESI-MS (m/z): 413.2 (M-H+, 100), 414.1 (M-H+, 27), 415.0 (M-H+, 8). CHN calc.: C 52.69, H 5.44, N 9.45; CHN found: C 52.32, H 5.77, N 9.29.
24a (0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography to give a yellow product. Yellow solid; yield: 61%; mp 159 °C; C19H22N4OS; 1H-NMR (d6 DMSO, 300 MHz): δ = 2.61–2.65 (m, 4H, Pip-2-H, -6-H), 2.74 (t, J = 5.73 Hz, 2 H, OCH2CH2), 3.11–3.14 (m, 4H, Pip-3-H, -5-H), 4.09 (t; J = 5.77 Hz, 2H, OCH2CH2), 6.76 (t, J = 7.25 Hz, 1H, Ar-4-H), 6.83 (d, J = 8.72 Hz, 1H, Benzthia-5-H), 6.92 (d, J = 8.81 Hz, 2H, Ar-2-H, -6-H), 7.18 (d, J = 7.32 Hz, 1H, Benzthia-4-H), 7.21 (s, br, 2H, Benzthia-2-NH2), 7.20–7.24 (m, 2H, Ar-3-H, -5-H), 7.32 (d, 4J = 2.57 Hz, 1H, Benzthia-7-H); 13C-NMR (d6 DMSO, 75 MHz) δ = 48.16 (Pip-3-C, -5-C), 53.04 (Pip-2-C, -6-C), 56.67 (OCH2CH2), 66.24 (OCH2CH2), 106.42 (Benzthia-7-C), 113.50 (Benzthia-5-C), 115.29 (Ar-C-2,-C-6), 118.02 (Ar-C-4), 118.70 (Benzthia-4-C), 128.85 (Ar-C-3, -C-5), 131.87 (Benzthia-7a-C), 146.91 (Benzthia-3a-C), 151.00 (Ar-C-1), 153.41 (Benzthia-6-C), 164.73 (Benzthia-2-C); ESI-MS (m/z): 355.0 (M-H+, 100), 356.0 (M-H+, 24), 357.1 (M-H+, 7). CHN calc.: C 64.38, H 6.26, N 15.81; CHN found: C 64.33, H 6.46, N 15.59.
24b (0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography to give a white product. White solid; yield: 11%; mp 203°C; C20H24N4OS × 0.5 H2O; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.90 (p, J = 6.54 Hz, 2H, OCH2CH2CH2), 2.44–2.53 (m, 6H, Pip-2-H, -6-H, OCH2 CH2CH2), 3.10–3.13 (m, 4H, Pip-3-H, -5-H), 3.99 (t; J = 6.33 Hz, 2H, OCH2CH2CH2), 6.73–6.82 (m, 2H, Benzthia-5-H, Ar-4-H), 6.92 (d, J = 7.96 Hz, 2H, Ar-2-H, -6-H), 7.17 – 7.23 (m, 3H, Ar-3-H, -5-H, Benzthia-4-H), 7.20 (s, br, 2H, Benzthia-2-NH2), 7.29 (d, 4J = 2.57 Hz, 1H, Benzthia-7-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 26.30 (OCH2CH2CH2), 48.18 (Pip-3-C, -5-C), 52.77 (Pip-2-C, -6-C), 54.44 (OCH2CH2CH2), 66.46 (OCH2CH2CH2), 106.34 (Benzthia-7-C), 113.48 (Benzthia-5-C), 115.27 (Ar-2-C, -6-C), 118.02 (Ar-4-C), 118.86 (Benzthia-4-C), 128.85 (Ar-3-C, -5-C), 131.87 (Benzthia-7a-C), 146.86 (Benzthia-3a-C), 151.01 (Ar-1-C), 153.57 (Benzthia-6-C), 164.69 (Benzthia-2-C); ESI-MS (m/z): 369.0 (M-H+, 100), 370.1 (M-H+, 25), 371.0 (M-H+, 7). CHN calc.: C 63.63, H 6.68, N 14.84; CHN found: C 63.75, H 6.51, N 14.48.
24c (0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography to give a white product. White solid; yield 26%; mp 164°C; C21H26N4OS × 0.25 H2O; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.62–1.78 (m, 4H, OCH2CH2CH2CH2), 2.42 (t, J = 7.09 Hz, 2H, OCH2CH2CH2CH2), 2.53–2.56 (m, 4H, Pip-2-H, -6-H), 3.14–3.17 (m, 4H, Pip-3-H, -5-H), 4.02 (t, J = 6.36 Hz, 2H, OCH2CH2CH2CH2), 6.81 (t, J = 7.25 Hz, 1H, Ar-4-H), 6.86 (d, J = 8.75 Hz, 1H, Benzthia-5-H), 6.97 (d, J = 8.80 Hz, 2H, Ar-2-H, -6-H), 7.22–7.27 (m, 2H, Ar-3-H, -5-H), 7.25 (s, br, 2H, Benzthia-2-NH2), 7.27 (d, J = 8.80 Hz, 1H, Benzthia-4-H), 7.33 (d, 4J = 2.58 Hz, 1H, Benzthia-7-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 22.73 (OCH2CH2CH2CH2), 26.66 (OCH2CH2CH2CH2), 48.19 (Pip-3-C, -5-C), 52.68 (Pip-2-C, -6-C), 57.33 (OCH2CH2CH2CH2), 67.91 (OCH2CH2CH2CH2), 106.32 (Benzthia-7-C), 113.45 (Benzthia-5-C), 115.25 (Ar-2-C, -6-C), 118.02 (Ar-4-C), 118.66 (Benzthia-4-C), 128.84 (Ar-3-C, -5-C), 131.86 (Benzthia-7a-C), 146.78 (Benzthia-3a-C), 151.03 (Ar-1-C), 153.58 (Benzthia-6-C), 164.65 (Benzthia-2-C); ESI-MS (m/z): 383.2 (M-H+, 100), 384.0 (M-H+, 26), 385.1 (M-H+, 8). CHN calc.: C 65.17, H 6.90, N 14.48; CHN found: C 64.98, H 6.78, N 14.26.
White solid; yield: 10%; mp 183°C; C20H23N3O2S × 1.5 C2H2O4 × 0.5 H2O; 1H-NMR (d6 DMSO, 400 MHz): δ = 3.20 (m, 4H, Pip-2-H, -6-H), 3.30 (m, 4H, Pip-3-H, -5-H), 3.45 (m, 2H, OCH2CH2), 3.80 (s, 3 H, OCH3), 4.43 (m, 2H, OCH2CH2), 6.88–7.03 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.22 (d, J = 8.53 Hz, 1H, Benzthia-5-H), 7.81 (s, 1H, Benzthia-7-H), 8.01 (d, J = 8.80 Hz, 1H, Benzthia-4-H), 9.23 (s, 1H, Benzthia-2-H); 13C-NMR (d6DMSO, 100 MHz): δ = 47.37 (Pip-3-C, -5-C), 52.20 (Pip-2-C, -6-C), 55.02 (OCH2CH2), 55.38 (OCH3), 63.73 (OCH2CH2), 105.97 (Benzthia-7-C), 111.97 (Benzthia-5-C), 116.09 (2-OCH3-Ar-C-6), 118.20 (2-OCH3-Ar-C-3), 120.85 (2-OCH3-Ar-C-4), 123.20 (2-OCH3-Ar-C-5), 123.58 (Benzthia-4-C), 134.98 (Benzthia-7a-C), 139.89 (2-OCH3-Ar-C-1), 147.91 (Benzthia-3a-C), 151.90 (2-OCH3-Ar-C-2), 153.79 (Benzthia-2-C), 155.94 (Benzthia-6-C), 162.86 ((COOH)2); ESI-MS: 370.0 (M-H+, 100), 371.0 (M-H+, 25), 372.0 (M-H+, 8). CHN calc.: C 53.79, H 5.30, N 8.18; CHN found: C 53.92, H 5.24, N 8.16.
In a three-neck round bottom flask anhydrous copper(II)chloride (0.65 g, 4.8 mmol) was suspended in 25 ml of dry acetonitrile. t-Butylnitrite (0.62 g, 6.0 mmol) was added and stirred at room temperature for 10 min to build a black suspension. 3 (4.0 mmol) was dissolved in a mixture of 25 ml dry acetonitrile and 10 ml dry methanol and added dropwise to the black suspension in the flask. The suspension was stirred 1.5 h at room temperature and 2.5 h at 65°C. After the suspension was evaporated with the addition of silica gel to afford a dry yellow-brown solid which was purified by column chromatography to provide a white product that was precipitated with anhydrous oxalic acid and dried. White solid; yield: 7%; mp 183–185°C (decomp.); C20H22ClN3O2S × 1 C2H2O4 × 0.5 H2O; 1H-NMR (d6 DMSO, 400 MHz): δ = 3.27 (m, 4H, Pip-2-H, -6-H), 3.37 (m, 4H, Pip-3-H, -5-H), 3.53 (m, 2H, OCH2CH2), 3.85 (s, 3H, OCH3), 4.47 (m, 2H, OCH2CH2), 6.94–7.09 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.29 (d, J = 8.55 Hz, 1H, Benzthia-5-H), 7.83 (s, 1H, Benzthia-7-H), 7.96 (d, J = 8.80 Hz, 1H, Benzthia-4-H); 13C-NMR (d6 DMSO, 100 MHz): δ = 47.66 (Pip-3-C, -5-C), 52.21 (Pip-2-C, -6-C), 54.94 (OCH2CH2), 55.38 (OCH3), 63.97 (OCH2CH2), 106.23 (Benzthia-7-C), 111.99 (Benzthia-5-C), 116.49 (2-OCH3-Ar-C-6), 118.21 (2-OCH3-Ar-C-3), 120.85 (2-OCH3-Ar-C-4), 123.12 (2-OCH3-Ar-C-5), 123.21 (Benzthia-4-C), 137.05 (Benzthia-7a-C), 139.83 (2-OCH3-Ar-C-1), 145.09 (Benzthia-3a-C), 149.90 (2-OCH3-Ar-C-2), 151.90 (Benzthia-2-C), 156.13 (Benzthia-6-C), 162.86 ((COOH)2); ESI-MS (m/z): 404.0 (M-H+, 100), 405.5 (M-H+, 36), 406.0 (M-H+, 40). CHN calc.: C 52.54, H 5.01, N 8.35; CHN found: C 52.49, H 4.94, N 8.41.
White solid; yield: 35%; mp 168–170°C (decomp.); C21H25N3O2S × 1.5 C2H2O4; 1H-NMR (d6 DMSO, 300 MHz): δ = 2.17 – 2.23 (m, 2H, OCH2CH2CH2), 3.20–3.41 (m, 10H, Pip-2-H, -3-H, -5-H, -6-H, OCH2 CH2CH2), 3.79 (s, 3 H, OCH3), 4.16 (t; J = 5.85 Hz, 2H, OCH2CH2CH2), 6.90–7.03 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.16 (d, J = 8.93 Hz, 1H, Benzthia-5-H), 7.74 (d, 4J = 2.47 Hz, 1H, Benzthia-7-H), 7.99 (d, J = 8.92 Hz, 1H, Benzthia-4-H), 9.19 (s, 1H, Benzthia-2-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 23.47 (OCH2CH2CH2), 47.10 (Pip-3-C, -5-C), 51.41 (Pip-2-C, -6-C), 53.15 (OCH2CH2CH2), 55.37 (OCH3), 65.61 (OCH2CH2CH2), 105.70 (Benzthia-7-C), 112.00 (Benzthia-5-C), 115.98 (2-OCH3-Ar-C-6), 118.29 (2-OCH3-Ar-C-3), 120.84 (2-OCH3-Ar-C-4), 123.42 (2-OCH3-Ar-C-5), 123.46 (Benzthia-4-C), 134.98 (Benzthia-7a-C), 139.41 (2-OCH3-Ar-C-1), 147.71 (Benzthia-3a-C), 151.87 (2-OCH3-Ar-C-2), 153.52 (Benzthia-2-C), 156.38 (Benzthia-6-C), 162.06 ((COOH)2); ESI-MS (m/z): 384.1 (M-H+, 100), 385.1 (M-H+, 24), 386.1 (M-H+, 9). CHN calc.: C 55.59, H 5.44, N 8.10; CHN found: C 55.44, H 5.52, N 8.11.
In a three-neck round bottom flask anhydrous copper(II)chloride (0.65 g, 4.8 mmol) was suspended in 25 ml of dry acetonitrile. t-Butylnitrite (0.62 g, 6.0 mmol) was added and stirred at room temperature for 10 min to build a black suspension. 6 (4.0 mmol) was dissolved in a mixture of 25 ml dry acetonitrile and 10 ml dry methanol and added dropwise to the black suspension in the flask. The suspension was stirred 1.5 h at room temperature and 2.5 h at 65°C. Silica gel was added, and the suspension was evaporated with to provide a dry yellow-brown solid which was purified by column chromatography to provide a white product. Subsequently, the product was precipitated with anhydrous oxalic acid and dried. White solid; yield: 15%; mp 188°C; C21H24ClN3O2S × 1 C2H2O4 × 0.25 H2O; 1H-NMR (d6 DMSO, 300 MHz): δ = 2.17–2.24 (m, 2H, OCH2CH2CH2), 3.23–3.44 (m, 10H, Pip-2-H, -3-H, -5-H, -6-H, OCH2 CH2CH2), 3.79 (s, 3H, OCH3), 4.14 (t; J = 5.64 Hz, 2H, OCH2CH2CH2), 6.90–7.02 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.16 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.70 (d, 4J = 2.55 Hz, 1H, Benzthia-7-H), 7.88 (d, J = 8.95 Hz, 1H, Benzthia-4-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 23.50 (OCH2CH2CH2), 47.19 (Pip-3-C, -5-C), 51.41 (Pip-2-C, -6-C), 53.12 (OCH2CH2CH2), 55.36 (OCH3), 65.67 (OCH2CH2CH2), 105.99 (Benzthia-7-C), 112.00 (Benzthia-5-C), 116.36 (2-OCH3-Ar-C-6), 118.28 (2-OCH3-Ar-C-3), 120.84 (2-OCH3-Ar-C-4), 123.05 (2-OCH3-Ar-C-5), 123.40 (Benzthia-4-C), 137.06 (Benzthia-7a-C), 139.46 (2-OCH3-Ar-C-1), 144.86 (Benzthia-3a-C), 149.61 (2-OCH3-Ar-C-2), 151.87 (Benzthia-2-C), 156.61 (Benzthia-6-C), 162.21 ((COOH)2); ESI-MS (m/z): 418.1 (M-H+, 100), 419.4 (M-H+, 36), 420.1 (M-H+, 40). CHN calc.: C 53.90, H 5.21, N 8.20; CHN found: C 53.97, H 5.35, N 8.22.
White solid; yield: 36%; mp 159–165°C (decomp.); C22H27N3O2S × 1.5 C2H2O4; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.83 (m, 4H, OCH2CH2CH2CH2), 3.13–3.37 (m, 10H, OCH2CH2CH2CH2, Pip-2-H, -3-H, -5-H, -6-H), 3.79 (s, 3H, OCH3), 4.10 (t, J = 5.13 Hz, 2H, OCH2CH2CH2CH2), 6.87–7.02 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.15 (d, J = 8.92 Hz, 1H, Benzthia-5-H), 7.73 (d, 4J = 2.37 Hz, 1H, Benzthia-7-H), 7.97 (d, J = 8.92 Hz, 1H, Benzthia-4-H), 9.19 (s, 1H, Benzthia-2-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 19.98 (OCH2CH2CH2CH2), 26.13 (OCH2CH2CH2CH2), 46.88 (Pip-3-C, -5-C), 51.49 (Pip-2-C, -6-C), 55.35 (OCH2CH2CH2CH2), 55.35 (OCH3), 67.37 (OCH2CH2CH2CH2), 105.53 (Benzthia-7-C), 111.95 (Benzthia-5-C), 115.99 (2-OCH3-Ar-3-C), 115.99 (2-OCH3-Ar-6-C), 118.23 (2-OCH3-Ar-4-C), 120.82 (2-OCH3-Ar-5-C), 123.33 (Benzthia-4-C), 134.99 (Benzthia-7a-C), 139.47 (Benzthia-3a-C), 147.56 (2-OCH3-Ar-1-C), 151.86 (2-OCH3-Ar-2-C), 153.34 (Benzthia-2-C), 156.66 (Benzthia-6-C), 163.05 ((COOH)2); ESI-MS (m/z): 398.1 (M-H+, 100), 399.1 (M-H+, 26), 400.1 (M-H+, 8). CHN calc.: C 56.38, H 5.68, N 7.89; CHN found: C 56.12, H 5.84, N 7.88.
Yellow oil; yield: 54%; C11H12BrNOS; 1H-NMR (CDCl3, 400 MHz): δ = 1.98–2.09 (m, 4H, OCH2CH2CH2CH2Br) 3.07 (t, J = 7.02 Hz, 2H, OCH2CH2CH2CH2Br), 4.09 (t, J = 5.78 Hz, 2H, OCH2CH2CH2CH2Br), 7.11 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.38 (d, 4J = 2.44 Hz, 1H, Benzthia-7-H), 8.01 (d, J = 8.95 Hz, 1H, Benzthia-4-H), 8.83 (s, 1H, Benzthia-2-H); ESI-MS (m/z): 264.9 (M-SCN H+, 100), 265.9 (M-SCN H+, 15), 267.0 (M-SCN H+, 10).
24d (0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.09 g, 0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (yield: 17%); mp 168°C (decomp.); C21H25N3OS × 2 C2H2O4 × 0.25 H2O; 1H-NMR (d6 DMSO, 300 MHz) δ = 1.79–1.86 (m, 4H, OCH2CH2CH2CH2), 3.12–3.39 (m, 10H, OCH2CH2CH2CH2, Pip-2-H, -3-H, -5-H,-6-H), 4.10 (t, J = 5.61 Hz, 2H, OCH2CH2CH2CH2), 6.85 (t, J = 7.26 Hz, 1H, Ar-4-H), 6.99 (d, J = 7.89 Hz, 2H, Ar-2-H, -6-H), 7.15 (d, J = 8.93 Hz, 1H, Benzthia-5-H), 7.23–7.28 (m, 2H, Ar-3-H, -5-H), 7.72 (d, 4J = 2.48 Hz, 1H, Benzthia-7-H), 7.97 (d, J = 8.93 Hz, 1H, Benzthia-4-H), 9.19 (s, 1H, Benzthia-2-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 20.41 (OCH2CH2CH2CH2), 25.84 (OCH2CH2CH2CH2), 45.70 (Pip-3-C, -5-C), 50.83 (Pip-2-C, -6-C), 55.31 (OCH2CH2CH2CH2), 67.45 (OCH2CH2CH2CH2), 105.50 (Benzthia-7-C), 115.85 (Ar-2-C, -6-C), 115.98 (Benzthia-5-C), 119.84 (Ar-4-C), 123.41 (Benzthia-4-C), 129.06 (Ar-3-C, -5-C), 134.99 (Benzthia-7a-C), 147.53 (Benzthia-3a-C), 149.67 (Ar-1-C), 153.36 (Benzthia-2-C), 156.64 (Benzthia-6-C), 162.82 ((COOH)2); ESI-MS (m/z): 368.1 (M-H+, 100), 369.2 (M-H+, 25), 370.1 (M-H+, 8). CHN calc.: C 54.39, H 5.39, N 7.61; CHN found: C 54.37, H 5.22, N 7.74.
Yellow solid; yield: 72%; C8H9NO4; 1H-NMR (CDCl3, 300 MHz): δ = 3.96 (td, J = 4.60 Hz, 2H, OCH2CH2OH), 4.32 (td, J = 5.11 Hz, 2H, OCH2CH2OH), 6.93 (d, J = 9.28 Hz, 2H, Ar-2-H, -6-H), 8.16 (d, J = 9.27 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 137.6 (4-nitrophenolate−, 100), 138.6 (4-nitrophenolate−, 8), 183.5 (M−, 4).
Yellow oil; yield: 100%; C9H11H-NMR (CDCl3, 400 MHz): δ = 1.87 (p, J = 5.99 Hz, 2H, OCH2CH2CH2OH) 3.66 (t, J = 5.94 Hz, 2H, OCH2 CH2CH2OH), 4.01 (t, J = 6.10 Hz, 2H, OCH2CH2CH2OH), 6.75 (d, J = 9.22 Hz, 2H, Ar-2-H, -6-H), 7.97 (d, J = 9.21 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 197.7 (M-H+, 100), 198.7 (M-H+, 21).
22c (2.74 g, 1.0 mmol) was dissolved in 200 ml of a mixture acetone: water (7:3 v/v). After addition of the base N,N-diisopropylamine (1.1 g, 1.1 mmol) the solution was heated in a Parr apparatus to 130°C for 9 h under continuous steering. After cooling and evaporation of acetone, the brown solution was acidified with hydrochloric acid solution (2 N) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate. After evaporation of the solvent the crude product was purified by column chromatography to give a white solid (0.89 g, yield: 45%); 1H-NMR (d6 DMSO, 300 MHz) δ = 1.51–1.60 (m, 2H, OCH2CH2CH2CH2OH), 1.73–1.80 (m, 2H, OCH2CH2CH2CH2OH), 3.42–3.48 (m, 2H, OCH2CH2CH2CH2OH), 4.13 (t, J = 6.55 Hz, 2H, OCH2CH2CH2CH2OH), 4.44 (t, J = 5.14 Hz, 1H, OCH2CH2CH2CH2OH), 7.12 (d, J = 9.32 Hz, 2H, Ar-2-H, -6-H), 8.19 (d, J = 9.32 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 211.7 (M-H+, 100), 212.9 (M-H+, 23), 213.8 (M-H+, 13).
25a (4.2 mmol) was dissolved with 50 ml ethyl acetate. 100 mg Pd/C (10% m/m) and a few drops glacial acetic acid were added. The suspension was stirred at room temperature in a Parr apparatus with a pressure of 5 bar hydrogen for 1 h. After filtration of the catalyst the yellow-brown solution was evaporated to dryness to give brownish oil. Brown oil; yield: 100%; C8H11H-NMR (d6 DMSO, 300 MHz): δ = 3.63 (t, J = 5.19 Hz, 2H, OCH2CH2OH), 3.82 (t, J = 5.13 Hz, 2H, OCH2CH2OH), 6.49 (d, J = 8.97 Hz, 2H, Ar-2-H, -6-H), 6.64 (d, J = 8.92 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 153.8 (M-H+, 100), 154.7 (M-H+, 12), 175.7 (M-Na+, 87).
25b (4.2 mmol) was dissolved with 50 ml ethyl acetate. 100 mg Pd/C (10% m/m) and a few drops glacial acetic acid were added. The suspension was stirred at room temperature in a Parr apparatus with a pressure of 5 bar hydrogen for 1 h. After filtration of the catalyst the yellow-brown solution was evaporated to dryness to give brownish oil. Brown oil; yield: 100%; C9H13H-NMR (d6 DMSO, 300 MHz): δ = 1.79 (p, J = 6.34 Hz, 2H, OCH2CH2CH2OH) 3.53 (t, J = 6.30 Hz, 2H, OCH2 CH2CH2OH), 3.88 (t, J = 6.40 Hz, 2H, OCH2CH2CH2OH), 6.57 (d, J = 8.91 Hz, 2H, Ar-2-H, -6-H), 6.67 (d, J = 8.92 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 167.7 (M-H+, 100), 168.7 (M-H+, 11).
25c (0.89 g, 4.2 mmol) was dissolved with 50 ml ethyl acetate. 100 mg Pd/C (10% m/m) and a few drops glacial acetic acid were added. The suspension was stirred at room temperature in a Parr apparatus with a pressure of 5 bar hydrogen for 1 h. After filtration of the catalyst the yellow-brown solution was evaporated to dryness to give a brownish oil (0.76 g, yield: 100%). Brown oil; C10H15H-NMR (d6 DMSO, 400 MHz) δ = 1.55–1.63 (m, 2H, OCH2CH2CH2CH2OH), 1.69–1.76 (m, 2H, OCH2CH2CH2CH2OH), 3.49 (t, J = 6.48 Hz, 2H, OCH2CH2CH2CH2OH), 3.87 (t, J = 6.50 Hz, 2H, OCH2CH2CH2CH2OH), 6.55 (d, J = 8.78 Hz, 2H, Ar-2-H, -6-H), 6.69 (d, J = 8.80 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 181.8 (M-H+, 100), 182.7 (M-H+, 12).
26a (4.4 mmol) and potassium thiocyanate (1.71 g, 17.6 mmol) were solved in 25 ml glacial acetic acid. The resultant brown solution was cooled on an ice bath. Dropwise bromine (0.71 g, 4.4 mmol) dissolved in 5 ml glacial acetic acid was added, while the temperature was held constant at 4°C. Afterwards the orange suspension was stirred at room temperature overnight. The formed orange solid was filtered of and the yellow filtrate was concentrated in vacuum. The yellow-orange solid was purified by column chromatography to give a white solid (yield: 50%); C9H10N2O2S; 1H-NMR (d6 DMSO, 300 MHz): δ = 3.77 (t, J = 4.98 Hz, 2H, OCH2CH2OH), 4.02 (t, J = 5.03 Hz, 2H, OCH2CH2OH), 6.89 (d, J = 8.73 Hz, 1H, Benzthia-5-H), 7.29 (d, J = 8.71 Hz, 1H, Benzthia-4-H), 7.35 (s, br, 2H, Benzthia-2-NH2), 7.36 (s, 1H, Benzthia-7-H).
26b (4.4 mmol) and potassium thiocyanate (1.71 g, 17.6 mmol) were solved in 25 ml glacial acetic acid. The resultant brown solution was cooled on an ice bath. Dropwise bromine (0.71 g, 4.4 mmol) dissolved in 5 ml glacial acetic acid was added, while the temperature was held constant at 4°C. Afterwards the orange suspension was stirred at room temperature overnight. The resulted orange solid was filtered of and the yellow filtrate was concentrated in vacuum. The yellow-orange solid was purified by column chromatography to give a brownish oil (yield: 25%); C10H12N2O2S; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.80–1.87 (m, 2H, OCH2CH2CH2OH) 3.54 (t, J = 6.14 Hz, 2H, OCH2 CH2CH2OH), 3.99 (t, J = 6.26 Hz, 2H, OCH2CH2CH2OH), 6.83 (d, J = 8.66 Hz, 1H, Benzthia-5-H), Benzthia-7-H), 7.23 (d, J = 8.70 Hz, 1H, Benzthia-4-H), 7.30 (d, 4J = 2.05 Hz, 1H, 7.54 (s, br, 2H, Benzthia-2-NH2); ESI-MS (m/z): 224.7 (M-H+, 100), 225.8 (M-H+, 12), 226.8 (M-H+, 6).
26c (0.76 g, 4.4 mmol) and potassium thiocyanate (1.71 g, 17.6 mmol) were solved in 25 ml glacial acetic acid. The resultant brown solution was cooled on an ice bath. Dropwise bromine (0.71 g, 4.4 mmol) dissolved in 5 ml glacial acetic acid was added, while the temperature was held constant at 4°C. Afterwards the orange suspension was stirred at room temperature overnight. The resulted orange solid was filtered of and the yellow filtrate was concentrated in vacuum. The yellow-orange solid was purified by column chromatography to give a white solid (0.96 g, yield: 91%); C11H14N2O2S; 1H-NMR (d6 DMSO, 400 MHz): δ = 1.52–1.59 (m, 2H, OCH2CH2CH2CH2OH), 1.69–1.76 (m, 2H, OCH2CH2CH2CH2OH), 3.43–3.45 (m, 2H, OCH2CH2CH2CH2OH), 3.94 (t, J = 6.43 Hz, 2H, OCH2CH2CH2CH2OH), 6.80 (d, J = 8.64 Hz, 1H, Benzthia-5-H), 7.19 (s, br, 2H, Benzthia-2-NH2), 7.21 (d, J = 8.95 Hz, 1H, Benzthia-4-H), 7.27 (d, 4J = 2.07 Hz, 1H, Benzthia-7-H); ESI-MS (m/z): 238.9 (M-H+, 100), 239.9 (M-H+, 13), 240.9 (M-H+, 6).
White solid; yield: 31%; C9H9NO2S; 1H-NMR (CDCl3, 300 MHz): δ = 3.91–3.96 (m, 2H, OCH2CH2OH), 4.08–4.11 (m, 2H, OCH2CH2OH), 7.07 (d, J = 8.96 Hz, 1H, Benzthia-5-H), 7.34 (d, 4J = 2.50 Hz,1H, Benzthia-7-H), 7.95 (d, J = 8.97 Hz, 1H, Benzthia-4-H), 8.77 (s, 1H, Benzthia-2-H); ESI-MS (m/z): 195.8 (M-H+, 100), 196.8 (M-H+, 11), 197.7 (M-H+, 5).
Brown oil; yield: 40%; C10H11NO2S; 1H-NMR (CDCl3, 400 MHz): δ = 2.02 (p, J = 5.96 Hz, 2H, OCH2CH2CH2OH) 3.83 (t, J = 5.91 Hz, 2H, OCH2 CH2CH2OH), 4.13 (t, J = 6.00 Hz, 2H, OCH2CH2CH2OH), 7.04 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.32 (d, 4J = 2.43 Hz, 1H, Benzthia-7-H), 7.92 (d, J = 8.95 Hz, 1H, Benzthia-4-H), 8.75 (s, 1H, Benzthia-2-H); 13C-NMR (CDCl3): δ = 32.03 (OCH2CH2CH2), 60.10 (OCH2CH2CH2), 66.16 (OCH2CH2CH2), 104.90 (Benzthia-7-C), 116.18 (Benzthia-5-C), 124.02 (Benzthia-4-C), 135.11 (Benzthia-7a-C), 147.93 (Benzthia-3a-C), 151.55 (Benzthia-2-C), 157.27 (Benzthia-6-C); ESI-MS (m/z): 209.7 (M-H+, 100), 210.7 (M-H+, 12), 211.7 (M-H+, 5).
In a three-neck round bottom flask anhydrous copper(II)chloride (0.65 g, 4.8 mmol) was suspended in 25 ml of dry acetonitrile. t-Butylnitrite (0.62 g, 6.0 mmol) was added and stirred at room temperature for 10 min to build a black suspension. 27a (4.0 mmol) was dissolved in a mixture of 25 ml dry acetonitrile and 10 ml dry methanol and added dropwise to the black suspension in the flask. The suspension was stirred 1.5 h at room temperature and 2.5 h at 65°C. After the suspension was evaporated with the addition of silicagel to a dry yellow-brown solid, it was purified by column chromatography to yellowish oil (yield: 45%); C9H8ClNO2S; 1H-NMR (d6 DMSO, 250 MHz): δ = 3.73 (t, J = 4.76 Hz, 2H, OCH2CH2OH), 4.03 (t, J = 4.92 Hz, 2H, OCH2CH2OH), 7.14 (d, J = 8.96 Hz, 1H, Benzthia-5-H), 7.66 (d, 4J = 2.57 Hz,1H, Benzthia-7-H), 7.83 (d, J = 8.95 Hz, 1H, Benzthia-4-H); ESI-MS (m/z) 183.6 (2-chlorobenthiazole-6-O−, 100), 184.5 (2-chlorobenthiazole-6-O−, 9), 185.6 (2-chlorobenthiazole-6-O−, 37).
In a three-neck round bottom flask anhydrous copper(II)chloride (0.65 g, 4.8 mmol) was suspended in 25 ml of dry acetonitrile. t-Butylnitrite (0.62 g, 6.0 mmol) was added and stirred at room temperature for 10 min to build a black suspension. 27b (4.0 mmol) was dissolved in a mixture of 25 ml dry acetonitrile and 10 ml dry methanol and added dropwise to the black suspension in the flask. The suspension was stirred 1.5 h at room temperature and 2.5 h at 65°C. After the suspension was evaporated with the addition of silicagel to a dry yellow-brown solid, it was purified by column chromatography to yellowish oil (yield: 58%); C10H10ClNO2S; 1H-NMR (CDCl3, 400 MHz): δ = 2.02 (p, J = 5.93 Hz, 2H, OCH2CH2CH2OH) 3.82 (t, J = 5.89 Hz, 2H, OCH2 CH2CH2OH), 4.10 (t, J = 6.01 Hz, 2H, OCH2CH2CH2OH), 7.00 (d, J = 8.96 Hz, 1H, Benzthia-5-H), 7.17 (d, 4J = 2.46 Hz, 1H, Benzthia-7-H), 7.74 (d, J = 8.95 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 243.7 (M-H+, 100), 245.7 (M-H+, 46), 246.7 (M-H+, 5).
In a three-neck round bottom flask anhydrous copper(II)chloride (0.65 g, 4.8 mmol) was suspended in 25 ml of dry acetonitrile. t-Butylnitrite (0.62 g, 6.0 mmol) was added and stirred at room temperature for 10 min to build a black suspension. 27c (0.96 g, 4.0 mmol) was dissolved in a mixture of 25 ml dry acetonitrile and 10 ml dry methanol and added dropwise to the black suspension in the flask. The suspension was stirred 1.5 h at room temperature and 2.5 h at 65°C. After the suspension was evaporated with the addition of silicagel to a dry yellow-brown solid, it was purified by column chromatography to yellowish oil (0.5 g, yield: 48%); C11H12ClNO2S; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.52–1.61 (m, 2H, OCH2CH2CH2CH2OH), 1.73–1.80 (m, 2H, OCH2CH2CH2CH2OH), 3.42–3.48 (m, 2H, OCH2CH2CH2CH2OH), 4.04 (t, J = 6.40 Hz, 2H, OCH2CH2CH2CH2OH), 4.45 (t, J = 4.74 Hz, 1H, OCH2CH2CH2CH2OH), 7.12 (d, J = 8.94 Hz, 1H, Benzthia-5-H), 7.66 (d, 4J = 2.58 Hz, 1H, Benzthia-7-H), 7.83 (d, J = 8.95 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 257.8 (M-H+, 100), 258.9 (M-H+, 13), 259.9 (M-H+, 39).
Yellow oil; yield: 77%; C10H11NO4H-NMR (CDCl3, 300 MHz) δ = 3.04 (s, 3H, O-SO2-CH3), 4.24–4.27 (m, 2H, OCH2CH2O), 4.53–4.56 (m, 2H, OCH2CH2O), 7.07 (d, J = 8.96 Hz, 1H, Benzthia-5-H), 7.34 (d, 4J = 2.51 Hz,1H, Benzthia-7-H), 7.97 (d, J = 8.95 Hz, 1H, Benzthia-4-H), 8.80 (s, 1H, Benzthia-2-H); ESI-MS (m/z): 274.0 (M-H+, 100), 275.0 (M-H+, 13), 276.0 (M-H+, 11).
30a (0.5 g, 1.95 mmol) solved in 20 ml dichlormethane were added to triethylamine (0.3 g, 2.9 mmol). The solution was cooled under argon atmosphere to −10°C. Methanesulfonic chloride (0.27 g, 2.3 mmol) dissolved in 10 ml dichlormethane was added dropwise. The solution was stirred 15 min by −10°C and 45 min by room temperature, poured into ice water and twice extracted with a sodium carbonate solution. The organic layer was dried over sodium sulfate and evaporated to give a yellow oil (yield 100%); C10H10ClNO4H-NMR (CDCl3, 250 MHz): δ = 3.03 (s, 3H, O-SO2-CH3), 4.23 (m, 2H, OCH2CH2O), 4.52–4.55 (m, 2H, OCH2CH2O), 7.02 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.19 (s,1H, Benzthia-7-H), 7.78 (d, J = 8.96 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 307.8 (M-H+, 100), 308.9 (M-H+, 13), 309.8 (M-H+, 43).
Brown oil; yield: 75%; C11H13NO4H-NMR (CDCl3, 400 MHz): δ = 2.22 (p, J = 5.98 Hz, 2H, OCH2CH2CH2O), 2.94 (s, 3H, O-SO2-CH3), 4.11 (t, J = 5.87 Hz, 2H, OCH2 CH2CH2O), 4.42 (t, J = 6.09 Hz, 2H, OCH2CH2CH2O), 7.05 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.34 (d, 4J = 2.43 Hz, 1H, Benzthia-7-H), 7.95 (d, J = 8.95 Hz, 1H, Benzthia-4-H), 8.77 (s, 1H, Benzthia-2-H); ESI-MS (m/z): 287.7 (M-H+, 100), 288.9 (M-H+, 13), 289.9 (M-H+, 10).
30b (0.5 g, 1.95 mmol) solved in 20 ml dichlormethane were added to triethylamine (0.3 g, 2.9 mmol). The solution was cooled under argon atmosphere to −10°C. Methanesulfonic chloride (0.27 g, 2.3 mmol) dissolved in 10 ml dichlormethane was added dropwise. The solution was stirred 15 min by −10°C and 45 min by room temperature, poured into ice water and twice extracted with a sodium carbonate solution. The organic layer was dried over sodium sulfte and evaporated to give a yellow oil (yield 88%); C11H12ClNO4S2; 1H-NMR (CDCl3, 400 MHz): δ = 2.20 (p, J = 5.95 Hz, 2H, OCH2CH2CH2O), 2.93 (s, 3H, O-SO2-CH3), 4.08 (t, J = 5.86 Hz, 2H, OCH2 CH2CH2O), 4.41 (t, J = 6.07 Hz, 2H, OCH2CH2CH2O), 7.00 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.17 (d, 4J = 2.39 Hz, 1H, Benzthia-7-H), 7.76 (d, J = 8.95 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 321.8 (M-H+, 100), 323.8 (M-H+, 43), 324.8 (M-H+, 6).
30c (0.5 g, 1.95 mmol) solved in 20 ml dichlormethane were added to triethylamine (0.3 g, 2.9 mmol). The solution was cooled under argon atmosphere to −10°C. Methanesulfonic chloride (0.27 g, 2.3 mmol) dissolved in 10 ml dichlormethane was added dropwise. The solution was stirred 15 min by −10°C and 45 min by room temperature, poured into ice water and twice extracted with a sodium carbonate solution. The organic layer was dried over sodium sulfate and evaporated to give a yellow oil (0.54 g, yield 82%); 1H-NMR (CDCl3, 400 MHz): δ = 1.91–1.93 (m, 4H, OCH2CH2CH2CH2O), 2.95 (s, 3H, O-SO2-CH3),3.99 (t, J = 5.61 Hz, 2H, OCH2CH2CH2CH2O), 4.04 (t, J = 5.86 Hz, 2H, OCH2CH2CH2CH2O), 6.99 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.15 (d, 4J = 2.46 Hz, 1H, Benzthia-7-H), 7.75 (d, J = 8.95 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 335.8 (M-H+, 100), 336.8 (M-H+, 15), 337.7 (M-H+, 42).
(0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.09 g, 0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (0.16 g, yield: 31%); mp 215°C (decomp.); C19H21N3OS × C2H2H-NMR (d6 DMSO, 300 MHz) δ = 3.20 (m, 4H, Pip-2-H, -6-H), 3.36–3.39 (m, 6H, OCH2CH2, Pip-3-H, -5-H), 4.45 (m, 2H, OCH2CH2), 6.90 (t, J = 7.23 Hz, 1H, Ar-4-H), 7.06 (d, J = 8.04 Hz, 2H, Ar-2-H, -6-H), 7.26–7.34 (m, 3H, Benzthia-5-H, Ar-3-H, -5-H), 7.87 (d, 4J = 1.52 Hz, 1H, Benzthia-7-H), 8.08 (d, J = 8.83 Hz, 1H, Benzthia-4-H), 9.29 (s, 1H, Benzthia-2-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 45.83 (Pip-3-C, -5-C), 51.58 (Pip-2-C, -6-C), 54.73 (OCH2CH2), 63.45 (OCH2CH2), 106.01 (Benzthia-7-C), 115.79 (Ar-C-2,-C-6), 116.06 (Benzthia-5-C), 119.74 (Ar-C-4), 123.52 (Benzthia-4-C), 129.05 (Ar-C-3, -C-5), 134.95 (Benzthia-7a-C), 147.94 (Benzthia-3a-C), 149.77 (Ar-C-1), 153.82 (Benzthia-2-C), 155.81 (Benzthia-6-C), 162.14 ((COOH)2); ESI-MS (m/z): 340.1 (M-H+, 100), 341.1 (M-H+, 22), 342.1 (M-H+, 7). CHN calc.: C 57.52, H 5.52, N 9.58; CHN found: C 57.69, H 5.38, N 9.67.
31b (0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.09 g, 0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (yield: 41%); mp 168°C (decomp.); C19H20ClN3OS × 2 C2H2O4;1H-NMR (CDCl3, 400 MHz): δ = 2.68–2.70 (m, 4H, Pip-2-H, -6-H), 2.84 (t, J = 5.70 Hz, 2H, OCH2CH2), 3.15–3.17 (m, 4H, Pip-3-H, -5-H), 4.13 (t, J = 5.70 Hz, 2H, OCH2CH2), 6.79 (t, J = 7.29 Hz, 1H, Ar-4-H), 6.86 (d, J = 7.91 Hz, 2H, Ar-2-H, -6-H), 7.03 (d, J = 8.96, 1H, Benzthia-5-H), 7.18–7.20 (m, 2H, Ar-3-H, -5-H), 7.21 (d, 4J = 1.93 Hz, 1H, Benzthia-7-H), 7.75 (d, J = 8.96 Hz, 1H, Benzthia-4-H); 13C-NMR (d6 DMSO, 100 MHz): δ = 46.15 (Pip-3-C, -5-C), 51.76 (Pip-2-C, -6-C), 54.89 (OCH2CH2), 63.82(OCH2CH2), 106.24 (Benzthia-7-C), 115.74 (Ar-C-2,-C-6), 116.46 (Benzthia-5-C), 119.63 (Ar-C-4), 123.10 (Benzthia-4-C), 129.04 (Ar-C-3, -C-5), 137.03 (Benzthia-7a-C), 145.05 (Benzthia-3a-C), 149.86 (Ar-C-1), 149.92 (Benzthia-2-C), 156.13 (Benzthia-6-C), 162.18 ((COOH)2); ESI-MS (m/z): 374.1 (M-H+, 100), 376.1 (M-H+, 38), 377.1 (M-H+, 8). CHN cacl.: C 49.87, H 4.37, N 7.59; CHN found: C 49.67, H 4.38, N 7.51.
31c (0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.09 g, 0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (yield: 79%); mp 208–211°C (decomp.); C20H23N3OS × 1.5 C2H2O4 × 0.25 H2O; 1H-NMR (d6 DMSO, 300 MHz): δ = 2.17–2.22 (m, 2H, OCH2CH2CH2), 3.21–3.30 (m, 6H, Pip-2-H, -6-H, OCH2 CH2CH2), 3.40 (m, 4H, Pip-3-H, -5-H), 4.15 (t; J = 5.95 Hz, 2H, OCH2CH2CH2), 6.85 (t, J = 7.25 Hz, 1H, Ar-4-H), 7.00 (d, J = 7.99 Hz, 2H, Ar-2-H, -6-H), 7.15 (d, J = 8.93 Hz, 1H, Benzthia-5-H), 7.26 (t, J = 7.93 Hz, 2H, Ar-3-H, -5-H), 7.73 (d, 4J = 2.44 Hz, 1H, Benzthia-7-H), 7.98 (d, J = 8.93 Hz, 1H, Benzthia-4-H), 9.19 (s, 1H, Benzthia-2-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 23.66 (OCH2CH2CH2), 45.80 (Pip-3-C, -5-C), 51.00 (Pip-2-C, -6-C), 53.15 (OCH2CH2CH2), 65.68 (OCH2CH2CH2), 105.60 (Benzthia-7-C), 115.83 (Ar-2-C, -6-C), 115.98 (Benzthia-5-C), 119.81 (Ar-4-C), 123.44 (Benzthia-4-C), 129.06 (Ar-3-C, -5-C), 134.98 (Benzthia-7a-C), 147.64 (Benzthia-3a-C), 149.71 (Ar-1-C), 153.33 (Benzthia-2-C), 156.42 (Benzthia-6-C), 163.24 ((COOH)2); ESI-MS (m/z): 354.0 (M-H+, 100), 355.0 (M-H+, 24), 356.0 (M-H+, 7). CHN cacl.: C 56.03, H 5.42, N 8.52; CHN found: C 56.12, H 5.29, N 8.40.
31d (0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.09 g, 0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (0.16 g, yield: 79%); mp 159–161°C (decomp.); C20H22ClN3OS × 2 C2H2O4;1H-NMR (d6 DMSO, 300 MHz): δ = 2.17–2.24 (m, 2H, OCH2CH2CH2), 3.22–3.31 (m, 6H, Pip-2-H, -6-H, OCH2 CH2CH2), 3.40 (m, 4H, Pip-3-H, -5-H), 4.13 (t; J = 5.89 Hz, 2H, OCH2CH2CH2), 6.85 (t, J = 7.25 Hz, 1H, Ar-4-H), 7.00 (d, J = 7.99 Hz, 2H, Ar-2-H, -6-H), 7.16 (d, J = 8.96 Hz, 1H, Benzthia-5-H), 7.26 (t, J = 7.92 Hz, 2H, Ar-3-H, -5-H), 7.70 (d, 4J = 2.50 Hz, 1H, Benzthia-7-H), 7.87 (d, J = 8.96 Hz, 1H, Benzthia-4-H); 13C-NMR (d6 DMSO, 75 MHz) δ = 23.57 (OCH2CH2CH2), 45.76 (Pip-3-C, -5-C), 50.97 (Pip-2-C, -6-C), 53.07 (OCH2CH2CH2), 65.72 (OCH2CH2CH2), 105.92 (Benzthia-7-C), 115.84 (Ar-2-C, -6-C), 116.36 (Benzthia-5-C), 119.82 (Ar-4-C), 123.04 (Benzthia-4-C), 129.06 (Ar-3-C, -5-C), 137.08 (Benzthia-7a-C), 144.81 (Benzthia-3a-C), 149.60 (Ar-1-C), 149.69 (Benzthia-2-C), 156.62 (Benzthia-6-C), 162.75 ((COOH)2); ESI-MS (m/z): 388.2 (M-H+, 100), 390.1 (M-H+, 38), 391.2 (M-H+, 9). CHN calc.: C 50.74, H 4.76, N 7.16; CHN found: C 50.75, H 4.61, N 7.40.
31e (0.6 mmol) was dissolved in acetone. 2-methylphenylpiperazine (0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (0.16 g, yield: 34%); mp 126°C; C22H26ClN3O2S × 1.5 C2H2O4 × 0.7 H2O; 1H-NMR (d6 DMSO, 300 MHz): δ = 1.76 (m, 4H, OCH2CH2CH2CH2), 3.06–3.23 (m, 10H, OCH2CH2CH2CH2, Pip-2-H, -3-H, -5-H, -6-H), 3.72 (s, 3 H, OCH3), 4.02 (t, J = 5.40 Hz, 2H, OCH2CH2CH2CH2), 6.83–6.95 (m, 4H, 2-OCH3-Ar-3-H, -4-H, -5-H, -6-H), 7.10 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.63 (d, 4J = 2.46 Hz, 1H, Benzthia-7-H), 7.80 (d, J = 8.95 Hz, 1H, Benzthia-4-H);13C-NMR (d6 DMSO, 75 MHz): δ = 20.37 (OCH2CH2CH2CH2), 25.78 (OCH2CH2CH2CH2), 47.20 (Pip-3-C, -5-C), 51.31 (Pip-2-C, -6-C), 55.34 (OCH2CH2CH2CH2), 55.34 (OCH3), 67.51 (OCH2CH2CH2CH2), 105.83 (Benzthia-7-C), 111.92 (Benzthia-5-C), 116.39 (2-OCH3-Ar-6-C), 118.21 (2-OCH3-Ar-3-C), 120.81 (2-OCH3-Ar-4-C), 123.01 (2-OCH3-Ar-5-C), 123.34 (Benzthia-4-C), 137.06 (Benzthia-7a-C), 139.51 (Benzthia-3a-C), 142.00 (2-OCH3-Ar-1-C), 144.68 (2-OCH3-Ar-2-C), 151.83 (Benzthia-2-C), 156.86 (Benzthia-6-C), 163.21 ((COOH)2); ESI-MS (m/z): 432.2 (M-H+, 100), 433.6 (M-H+, 38), 434.1 (M-H+, 39). CHN calc.: C 51.80, H 5.29, N 7.25; CHN found: C 51.91, H 4.99, N 7.38.
31e (0.16 g, 0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.09 g, 0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (0.16 g, yield: 46%); mp 155–157°C (decomp.); C21H24ClN3OS × 2 C2H2O4;1H-NMR (d6 DMSO, 300 MHz): δ = 1.82 (m, 4H, OCH2CH2CH2CH2), 3.12–3.39 (m, 10H, OCH2CH2CH2CH2, Pip-2-H, -3-H, -5-H,-6-H), 4.08 (t, J = 5.55 Hz, 2H, OCH2CH2CH2CH2), 6.85 (t, J = 7.27 Hz, 1H, Ar-4-H), 6.99 (d, J = 7.93 Hz, 2H, Ar-2-H, -6-H), 7.15 (d, J = 8.97 Hz, 1H, Benzthia-5-H), 7.22 -7.28 (m, 2H, Ar-3-H, -5-H), 7.68 (d, 4J = 2.55 Hz, 1H, Benzthia-7-H), 7.86 (d, J = 8.94 Hz, 1H, Benzthia-4-H); 13C-NMR (d6 DMSO, 75 MHz): δ = 20.34 (OCH2CH2CH2CH2), 25.78 (OCH2CH2CH2CH2), 45.66 (Pip-3-C, -5-C), 50.80 (Pip-2-C, -6-C), 55.26 (OCH2CH2CH2CH2), 67.50 (OCH2CH2CH2CH2), 105.82 (Benzthia-7-C), 115.85 (Ar-2-C, -6-C), 116.38 (Benzthia-5-C), 119.85 (Ar-4-C), 123.01 (Benzthia-4-C), 129.06 (Ar-3-C, -5-C), 137.07 (Benzthia-7a-C), 144.68 (Benzthia-3a-C), 149.43 (Benzthia-2-C), 149.66 (Ar-1-C), 156.85 (Benzthia-6-C), 162.79 ((COOH)2). ESI-MS (m/z): 402.0 (M-H+, 100), 403.8 (M-H+, 30), 404.3 (M-H+, 27). CHN cacl.: C 51.59, H 4.85, N 7.22; CHN found: C 51.80, H 4.81, N 7.38.
21 (3.0 g, 22 mmol) was diluted into 100 ml acetone. 1,4-dibromobutane (19.0 g, 88 mmol), anhydrous potassium carbonate (3.0 g, 22 mmol) and a catalytic amount of potassium iodide was added. The suspension was heated under reflux for 24 h. After cooling the yellow-white suspension was filtered. The yellow filtrate was evaporated to give a yellow oily raw product. The product was purified by filtration with petrolether (60–90°C) and dicloromethane over silicagel to obtain a pure yellow oil (5.34 g, yield: 90%); 1H-NMR (CDCl3, 400 MHz): δ = 1.90–2.05 (m, 4H, OCH2CH2CH2CH2Br) 3.42 (t, J = 6.37 Hz, 2H, OCH2CH2CH2CH2Br), 4.02 (t, J = 5.78 Hz, 2H, OCH2CH2CH2CH2Br), 6.87 (d, J = 9.15 Hz, 2H, Ar-2-H, -6-H), 8.11 (d, J = 9.13 Hz, 2H, Ar-3-H, -5-H); ESI-MS m/z: 297.9 (M-Na+, 100), 295.9 (M-Na+, 98), 296.7 (M-Na+, 10).
22c (2.74 g, 1.0 mmol) was dissolved in 200 ml of a mixture acetone: water (7:3 v/v). After addition of the base N,N-diisopropylamine (1.1 g, 1.1 mmol) the solution was heated in a Parr apparatus to 130°C for 9 h under continuous steering. After cooling and evaporation of acetone, the brown solution was acidified with hydrochloric acid solution (2 N) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate. After evaporation of the solvent the crude product was purified by column chromatography to give a white solid (0.89 g, yield: 45%); 1H-NMR (d6 DMSO, 300 MHz) δ = 1.51–1.60 (m, 2H, OCH2CH2CH2CH2OH), 1.73–1.80 (m, 2H, OCH2CH2CH2CH2OH), 3.42–3.48 (m, 2H, OCH2CH2CH2CH2OH), 4.13 (t, J = 6.55 Hz, 2H, OCH2CH2CH2CH2OH), 4.44 (t, J = 5.14 Hz, 1H, OCH2CH2CH2CH2OH), 7.12 (d, J = 9.32 Hz, 2H, Ar-2-H, -6-H), 8.19 (d, J = 9.32 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 211.7 (M-H+, 100), 212.9 (M-H+, 23), 213.8 (M-H+, 13).
25c (0.89 g, 4.2 mmol) was dissolved with 50 ml ethyl acetate. One hundred milligram Pd/C (10% m/m) and a few drops glacial acetic acid were added. The suspension was stirred at room temperature in a Parr apparatus with a pressure of 5 bar hydrogen for 1 h. After filtration of the catalyst the yellow-brown solution was evaporated to dryness to give a brownish oil (0.76 g, yield: 100%)0.1H-NMR (d6 DMSO, 400 MHz) δ = 1.55–1.63 (m, 2H, OCH2CH2CH2CH2OH), 1.69–1.76 (m, 2H, OCH2CH2CH2CH2OH), 3.49 (t, J = 6.48 Hz, 2H, OCH2CH2CH2CH2OH), 3.87 (t, J = 6.50 Hz, 2H, OCH2CH2CH2CH2OH), 6.55 (d, J = 8.78 Hz, 2H, Ar-2-H, -6-H), 6.69 (d, J = 8.80 Hz, 2H, Ar-3-H, -5-H); ESI-MS (m/z): 181.8 (M-H+, 100), 182.7 (M-H+, 12).
26c (0.76 g, 4.4 mmol) and potassium thiocyanate (1.71 g, 17.6 mmol) were solved in 25 ml glacial acetic acid. The resultant brown solution was cooled on an ice bath. Dropwise bromine (0.71 g, 4.4 mmol) dissolved in 5 ml glacial acetic acid was added, while the temperature was held constant at 4°C. Afterwards the orange suspension was stirred at room temperature overnight. The formed orange solid was filtered of and the yellow filtrate was concentrated in vacuum. The yellow-orange solid was purified by column chromatography to give a white solid (0.96 g, yield: 91%); 1H-NMR (d6 DMSO, 400 MHz) δ = 1.52–1.59 (m, 2H, OCH2CH2CH2CH2OH), 1.69–1.76 (m, 2H, OCH2CH2CH2CH2OH), 3.43–3.45 (m, 2H, OCH2CH2CH2CH2OH), 3.94 (t, J = 6.43 Hz, 2H, OCH2CH2CH2CH2OH), 6.80 (d, J = 8.64 Hz, 1H, Benzthia-5-H), 7.19 (s, br, 2H, Benzthia-2-NH2), 7.21 (d, J = 8.95 Hz, 1H, Benzthia-4-H), 7.27 (d, 4J = 2.07 Hz, 1H, Benzthia-7-H; ESI-MS (m/z): 238.9 (M-H+, 100), 239.9 (M-H+, 13), 240.9 (M-H+, 6).
In a three-neck round bottom flask anhydrous copper(II)chloride (0.65 g, 4.8 mmol) was suspended in 25 ml of dry acetonitrile. t-Butylnitrite (0.62 g, 6.0 mmol) was added and stirred at room temperature for 10 min to build a black suspension. 27c (0.96 g, 4.0 mmol) was dissolved in a mixture of 25 ml dry acetonitrile and 10 ml dry methanol and added dropwise to the black suspension in the flask. The suspension was stirred 1.5 h at room temperature and 2.5 h at 65°C. After the suspension was evaporated with the addition of silicagel to a dry yellow-brown solid, it was purified by column chromatography to yellowish oil (0.5 g, yield: 48%); 1H-NMR (d6 DMSO, 300 MHz) δ = 1.52–1.61 (m, 2H, OCH2CH2CH2CH2OH), 1.73–1.80 (m, 2H, OCH2CH2CH2CH2OH), 3.42–3.48 (m, 2H, OCH2CH2CH2CH2OH), 4.04 (t, J = 6.40 Hz, 2H, OCH2CH2CH2CH2OH), 4.45 (t, J = 4.74 Hz, 1H, OCH2CH2CH2CH2OH), 7.12 (d, J = 8.94 Hz, 1H, Benzthia-5-H), 7.66 (d, 4J = 2.58 Hz, 1H, Benzthia-7-H), 7.83 (d, J = 8.95 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 257.8 (M-H+, 100), 258.9 (M-H+, 13), 259.9 (M-H+, 39).
30c (0.5 g, 1.95 mmol) solved in 20 ml dichlormethane were added to triethylamine (0.3 g, 2.9 mmol). The solution was cooled under argon atmosphere to–10°C. Methanesulfonic chloride (0.27 g, 2.3 mmol) dissolved in 10 ml dichlormethane was added dropwise. The solution was stirred 15 min by–10°C and 45 min by room temperature, poured into ice water and twice extracted with a sodium carbonate solution. The organic layer was dried over sodium sulfate and evaporated to give a yellow oil (0.54 g, yield 82%);1H-NMR (CDCl3, 400 MHz) δ = 1.91–1.93 (m, 4H, OCH2CH2CH2CH2O), 2.95 (s, 3H, O-SO2-CH3),3.99 (t, J = 5.61 Hz, 2H, OCH2CH2CH2CH2O), 4.04 (t, J = 5.86 Hz, 2H, OCH2CH2CH2CH2O), 6.99 (d, J = 8.95 Hz, 1H, Benzthia-5-H), 7.15 (d, 4J = 2.46 Hz, 1H, Benzthia-7-H), 7.75 (d, J = 8.95 Hz, 1H, Benzthia-4-H); ESI-MS (m/z): 335.8 (M-H+, 100), 336.8 (M-H+, 15), 337.7 (M-H+, 42).
31e (0.16 g, 0.6 mmol) was dissolved in acetone. N-phenylpiperazine (0.09 g, 0.6 mmol), anhydrous potassium carbonate (0.17 g, 1.2 mmol) and catalytic amounts of potassium iodide were added. The suspension was refluxed for 48 h, filtered and the yellow filtrate was evaporated to dry. The resulting yellowish raw product was purified by column chromatography. The resulting colorless oil was precipitated with anhydrous oxalic acid and dried to give a white solid (0.16 g, yield: 46%); mp 155–157°C (decomp.); 1H-NMR (d6 DMSO, 300 MHz) δ = 1.82 (m, 4H, OCH2CH2CH2CH2), 3.12–3.39 (m, 10H, OCH2CH2CH2CH2, Pip-2-H, -3-H, -5-H,-6-H), 4.08 (t, J = 5.55 Hz, 2H, OCH2CH2CH2CH2), 6.85 (t, J = 7.27 Hz, 1H, Ar-4-H), 6.99 (d, J = 7.93 Hz, 2H, Ar-2-H, -6-H), 7.15 (d, J = 8.97 Hz, 1H, Benzthia-5-H), 7.22 -7.28 (m, 2H, Ar-3-H, -5-H), 7.68 (d, 4J = 2.55 Hz, 1H, Benzthia-7-H), 7.86 (d, J = 8.94 Hz, 1H, Benzthia-4-H); 13C-NMR (d6 DMSO, 75 MHz) δ = 20.34 (OCH2CH2CH2CH2), 25.78 (OCH2CH2CH2CH2), 45.66 (Pip-3-C, -5-C), 50.80 (Pip-2-C, -6-C), 55.26 (OCH2CH2CH2CH2), 67.50 (OCH2CH2CH2CH2), 105.82 (Benzthia-7-C), 115.85 (Ar-2-C, -6-C), 116.38 (Benzthia-5-C), 119.85 (Ar-4-C), 123.01 (Benzthia-4-C), 129.06 (Ar-3-C, -5-C), 137.07 (Benzthia-7a-C), 144.68 (Benzthia-3a-C), 149.43 (Benzthia-2-C), 149.66 (Ar-1-C), 156.85 (Benzthia-6-C), 162.79 ((COOH)2); ESI-MS (m/z): 402.0 (M-H+, 100), 403.8 (M-H+, 30), 404.3 (M-H+, 27). CHN calc.: C 51.59, H 4.85, N 7.22; CHN found: C 51.80, H 4.81, N 7.38.
Competition binding data were analyzed by Graph-Pad Prism™(2000, version 3.02, San Diego, CA, USA), using nonlinear least squares fit. Affinity values (Ki) were expressed as mean ± standard deviation [nM ± S.DE.M.]. The values for Ki and pKi were calculated for compounds 3-20 are listed in Tables 1, 3.
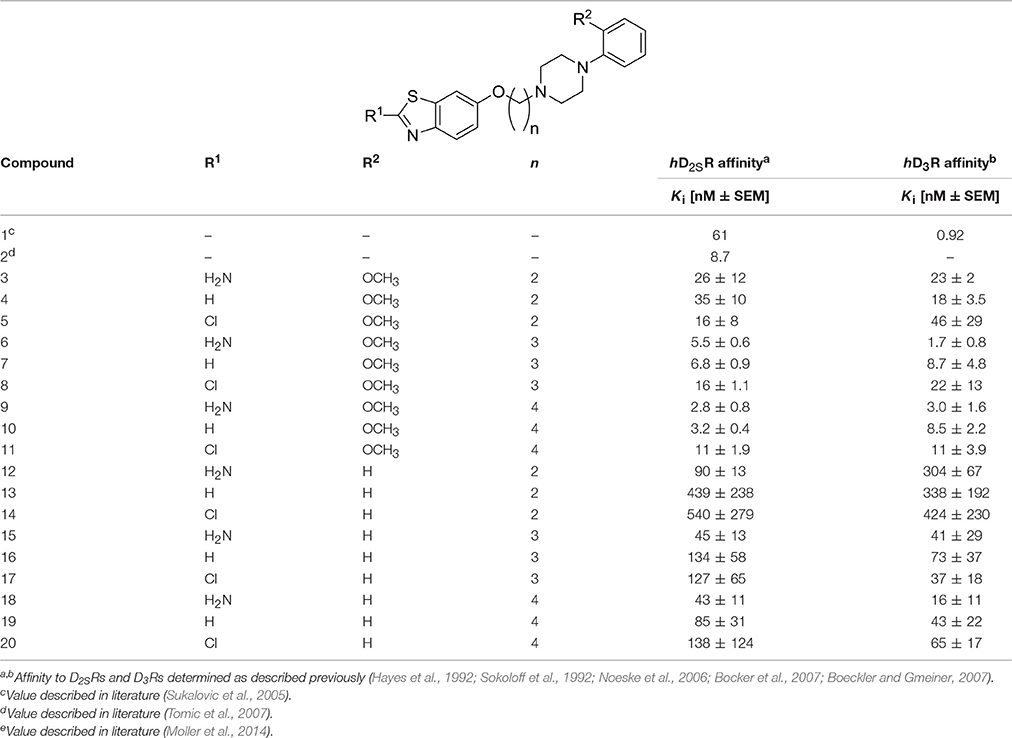
Table 1. Dopamine hD2SR and hD3R binding affinities of ligands 1, 2 and final products 3-20.
The in vitro affinity of the new compounds 3-20 for hD2SR and hD3R was determined using radioligand binding studies as described previously (Hayes et al., 1992; Sokoloff et al., 1992; Noeske et al., 2006; Bocker et al., 2007; Boeckler and Gmeiner, 2007). Membrane preparations of CHO-cells stably expressing hD2SR, the short splicing variant of that receptor subtype, and hD3R (Hopkins et al., 2004) were used for displacement studies (Sokoloff et al., 1992). In brief, [3H]spiperone (0.2 nM) served as radioligand and nonspecific binding was determined in the presence of 1 (10 μM). Stock solutions (10 mM) of test compounds were prepared with pure DMSO. They were diluted in binding puffer (120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 50 mM TRIS/HCl, pH 7.4) to give a final concentration range of 0.01–100,000 nM, and this was depending on the affinity of the test compound. The assay (containing membranes, [3H]spiperone and test compounds in a final volume of 0.2 ml) was incubated for 2 h at RT and terminated by rapid filtration through GF/B glass fiber filters (PerkinElmer Life Sciences, Rodgau, Germany) pre-treated with 0.3% polyethylenimine (Sigma–Aldrich, Taufkirchen, Germany) using an Inotech cell harvester (Inotech AG, Dottikon, Switzerland). Unbound radioligand was removed by three washing steps with 0.3 ml/well of ice-cold 50 mM Tris-HCl buffer, pH 7.4, containing 120 mM NaCl. Radioactivity was counted using a 1450 MicroBeta Trilux scintillation counter (PerkinElmer Life Sciences, Rodgau, Germany). To achieve a detailed screening, the compounds have been tested at seven concentrations in triplicates carried out in at least three independent experiments.
Lipophilicity (clogP), water solubility (clogS), molecular weight (MW), and number of rotatable bonds (NROTB) of Lipinski's rule of five were calculated using Molinspiration online property calculation toolkit, and the computational tool Osiris Property explorer (Table 2; Hopkins et al., 2004; Hou et al., 2007). Drug-likeness model score (a combined outcome of physicochemical properties, pharmacokinetics and pharmacodynamics of a compound and is represented by a numerical value was computed by MolSoft software for the 18 compounds (3-20) under study (Table 3; Drug-likeness and molecular property prediction1; Tarcsay et al., 2012). Moreover, topological polar surface area (TPSA) was calculated with software package Molinspiration Depiction Software (Table 2; Tarcsay et al., 2012).
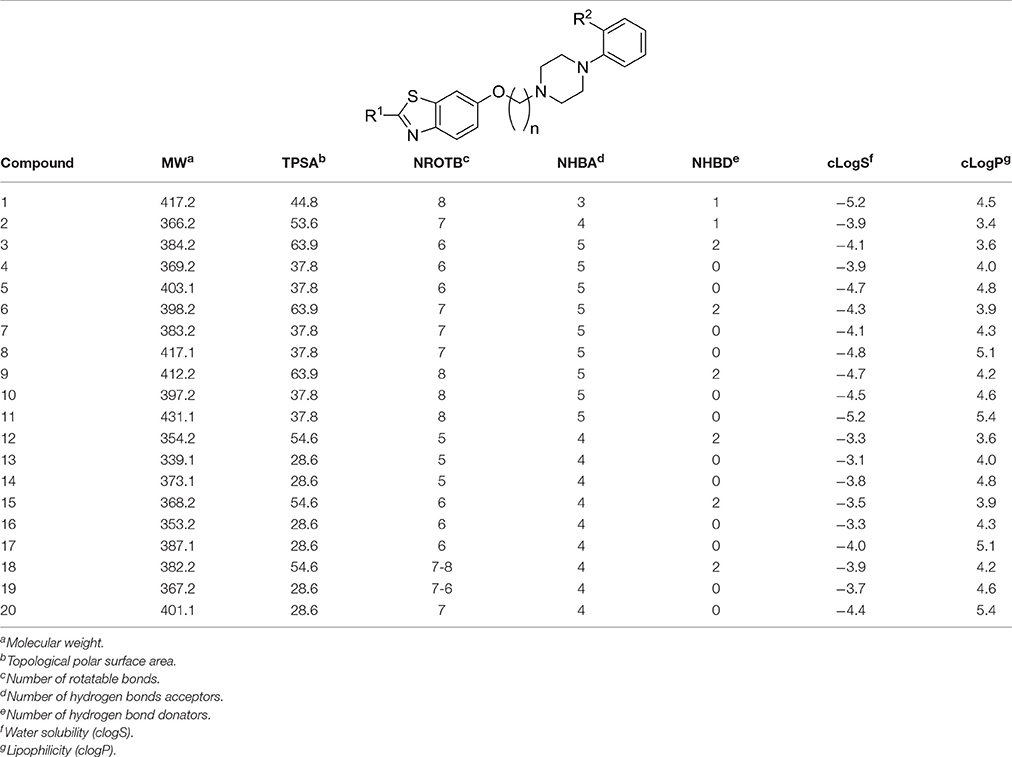
Table 2. Drug-likeness calculations and Lipinski parameters for final products 3-20.
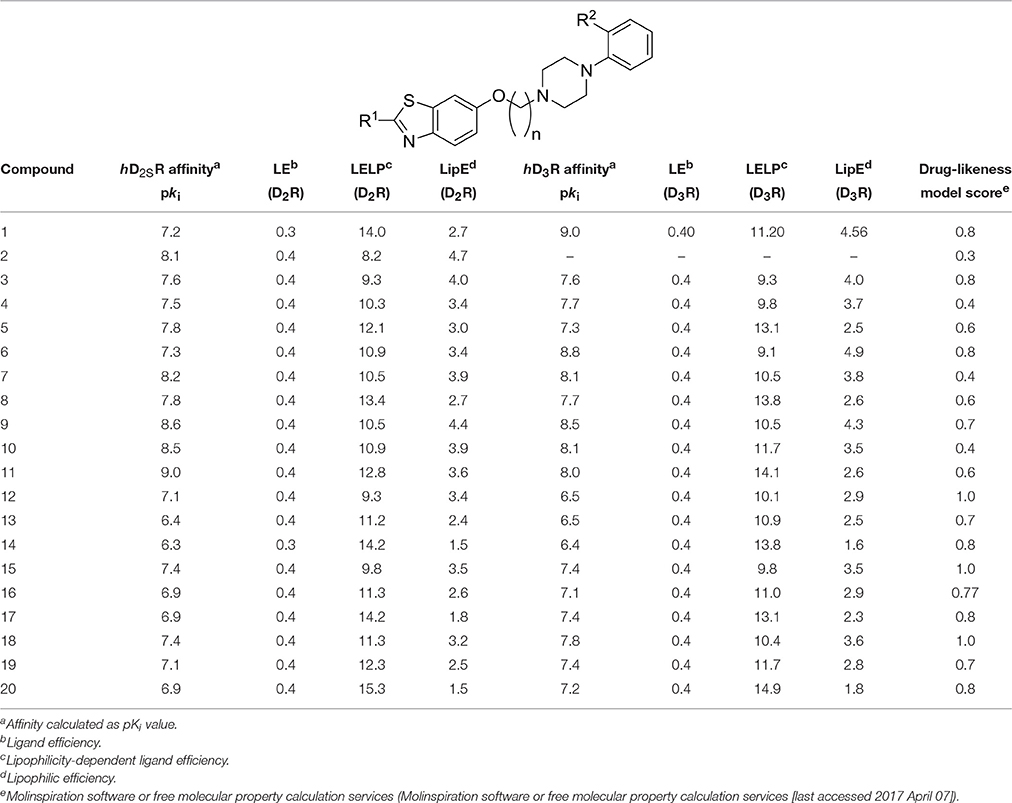
Table 3. Antagonist affinities at hD2SR and hD3R and values of LE, LELP, and LipE for final products 3-20.
The values for LE, LELP, and LipE were calculated for the compounds 3-20 based on their affinities for hD2SR and hD3R to further validate their drug-likeness and assessment as prospective new leads (Table 3). These widely used three metrics are substantial models in drug discovery and are simple for evaluating whether a ligand derives its potency from optimal fit with the target protein or simply by virtue of making many contacts (Molinspiration software or free molecular property calculation services [last accessed 2017 April 07]2; Kuntz et al., 1999; Meanwell, 2011; Tarcsay et al., 2012; Schreeb et al., 2013; Hopkins et al., 2014; Shultz, 2014). The values for LE, LELP, and LipE were calculated following equations 1, 2, and 3 (Molinspiration software or free molecular property calculation services [last accessed 2017 April 07]; Kuntz et al., 1999; Leeson and Springthorpe, 2007; Hopkins et al., 2014), respectively:
Final compounds 3-20 were prepared starting with an O-alkylation of suitable dibromoalkanes under Finkelstein conditions resulting in the formation of the key intermediates 22a-c which subsequently were reduced to their corresponding aromatic amines (23a-c), and were cyclized to their respective 2-aminobenzothiazoyl-6-ω-bromoalkyl ethers 24a-c under Kaufmann conditions and as previously described (Kaufmann and Bückmann, 1941; Sukalovic et al., 2005). The 2-aminobenz[d]thiazoyl-6-ω-bromoalkyl ethers (24a-c) were N-alkylated according to Finkelstein conditions using 2-methoxyphenylpiperazine or N-phenylpiperazine to obtain 3, 6, and 9 or 12, 15, 18, and 19, respectively. For preparation of 4, 7, and 10 the respective 2-aminobenzothiazol residue was deaminated by diazotization at 2-position using sodium nitrite and subsequent reductive cleavage of diazonium salt by means of hypophosphoric acid, whereas 5 and 7 were achieved by diazotization conducted in the presence of concentrated hydrochloric acid (Scheme 1). Finally, ligands 11, 13, 14, 16, 17, and 20 were synthesized starting from 1 which through O-alkylation under Finkelstein conditions with 2-bromoethanol, 3-chloropropan-1-ol, and 1,4-dibromobutane afforded 25a-c, respectively (Scheme 2).
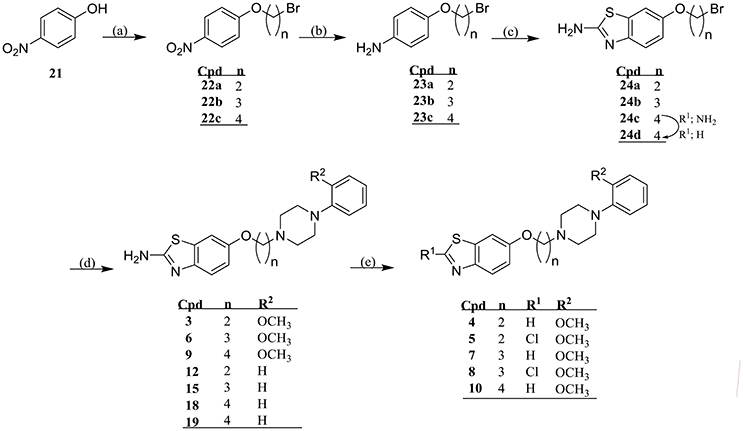
SCHEME 1. Synthesis of final products 3-10, 12, 15, 18, and 19. Reaction conditions: (a) Acetone, K2CO3, Br-(CH2)n-Br (n = 2–4), KI, 24 h reflux; (b) Methanol, Pd/C (10% m/m), 5 bar H2, 2 h, RT; (c) Acetic acid, KSCN, bromine, 4°C → RT, overnight; (d) Acetone, K2CO3, 1-(2-methoxyphenyl)piperazine (for 1-8) or N-phenylpiperazine (for 10, 13, and 16), KI, 24 h reflux; (e) HCl, NaNO2, H3PO4 (50% m/m), −30°C → RT; (f) H3PO4 conc., NaNO2, H3PO4 (50% m/m), −8°C → RT (for 17).
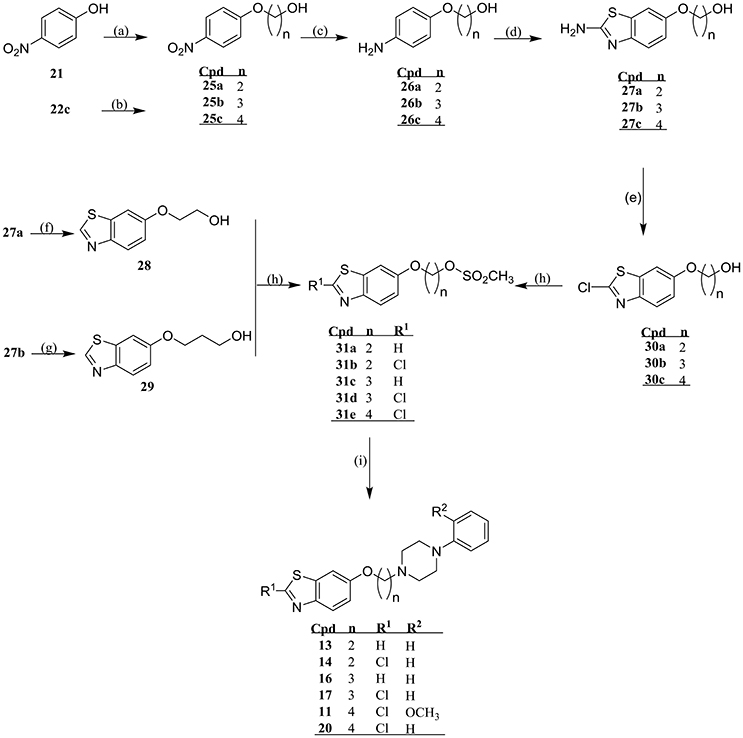
SCHEME 2. Synthesis of final products 11, 13, 14, 16, 17, and 20. Reaction conditions: (a) Acetone, K2CO3, 2-bromoethanol/3-chloropropan-1-ole, KI, 24 h reflux; (b) Acetone:water (7:3 v/v), N,N-diisopropylamine, 130°C, 9 h reflux; (c) Methanol, Pd/C (10% m/m), 5 bar H2 2 h, RT; (d) Acetic acid, KSCN, bromine, 4°C → RT, overnight; (e) Acetonitrile, Cu(II)Cl2, t-butylnitrite, 1.5 h RT, 2.5 h 65°C; (f) H3PO4 conc., NaNO2, H3PO4 (50% m/m), −8°C → RT; (h) Dichloromethane, triethylamine, methanesulfonyl chloride, −10°C → RT, 15–45 min; (i) Acetone, K2CO3, 1-(2-methoxyphenyl)piperazine (for 9)/N-phenylpiperazine (for 13, 14, 16, 17, and 20), KI, 48 h reflux.
The hD2SR and hD3R in vitro affinities observed for ligands 3-20 were in the nanomolar concentration range with Ki values of 2.3–450 [nM] and pKi values of 6.4–9.0, and Ki of 1.7–424 [nM] and pKi of 6.4–8.8 for hD2SR and hD3R, respectively. The obtained in vitro antagonist affinities for the ligands 3-20 were summarized in Table 1.
Lipophilicity (clogP), water solubility (clogS), molecular weight (MW), and number of rotatable bonds (NROTB) of Lipinski's rule of five were calculated using Molinspiration online property calculation toolkit, and the computational tool Osiris Property explorer. (Molinspiration software or free molecular property calculation services [last accessed 2017 April 07]2). The results observed for the different physicochemical properties are summarized in Table 2.
The obtained values for LE, LELP, and LipE of all novel ligands 3-20 were measured and are summarized in Table 3.
To evaluate the impact of benzothiazole appendage with the basic 4-phenylpiperazine aryl moiety by an alkyl spacer of varying length on receptor binding, human D2SR and D3R (hD2SR and hD3R) affinities of the described ligands 3-20 were determined using radioligand binding studies on membrane preparations of CHO-cells stably expressing both receptor subtypes. The hD2SR and hD3R affinities observed for ligands 3-20 were in the nanomolar concentration range with Kivalues of 2.3–450 [nM] and pKi values of 6.36–9.0, and Ki of 1.7–424 [nM] and pKi of 6.4–8.8 for hD2SR and hD3R, respectively (Tables 1–3). Notably, the results observed in the present study showed that the binding affinities of ligands bearing 2-methoxyphenylpiperazine moiety (3-11) showed significantly higher affinities to hD2SR and hD3R when compared to their corresponding ligands with an un-substituted 4-phenylpiperazine (12-20). Herein, an approximately 34-fold increase in affinity for the 2-methoxy substituted ligands [Ki (hD2SR) = 16 ± 8 [nM] for 5, Ki (hD2SR) = 540 ± 279 [nM] for 14] was observed on hD2SR, and up to 9-fold higher affinity [Ki(hD3R) = 46 ± 29 [nM] for 5 vs. Ki (hD3R) = 424 ± 230 [nM] for 15] toward the dopamine hD3R. Interestingly, these results are in agreement with previous findings that showed the strengthening effect of the 2-methoxy substitution on the binding affinity; however, without affecting selectivity profile among both receptor subtypes (Hackling et al., 2003; Boeckler and Gmeiner, 2007; Moller et al., 2014). In addition, our major findings revealed that receptor affinity observed for ligands 3-20 was roughly dependent on the length of the alkyl spacer. Accordingly, the obtained results were expressed in the order of decreasing affinity for tetramethylene ≥ trimethylene > ethylene spacer. In this regard, the ligands having trimethylene spacer showed an up to 5-times higher affinity at hD2SRs, and an up to 13-fold higher affinity at hD3Rs than their respective ethylene spacer analogs [Ki (hD2SR) = 26 ± 12 [nM] and Ki(hD3R) = 23 ± 2 [nM] for 3 vs. Ki(hD2SR) = 5.5 ± 0.6 [nM] and Ki(hD3R) = 1.7 ± 0.8 [nM] for 6]. However, ligands with tetramethylene spacer exhibited an up to 2-fold higher affinity at hD2SRs than their respective trimethylene spacer analogs, and also, with comparable affinity toward hD3Rs [Ki(hD2SR) = 6.8 ± 0.9 [nM] and Ki(hD3R) = 8.7 ± 4.8 [nM] for 7 vs. Ki (hD2SR) = 3.2 ± 0.4 [nM] and Ki(hD3R) = 8.5 ± 2.2 [nM] for 10]. Moreover, the flexible substitution pattern at the 2-position of the benzothiazole heterocycle had different effects on hD2SR and hD3R affinities. As a result, ligands of ethyl spacer series having a 2-methoxyphenylpiperazin moiety (3-5) showed no significant difference in their affinity toward hD2SRs and hD3Rs, whereas ligands of trimethylene and tetramethylene spacer (6-11) displayed an affinity toward the hD2SR in the order of H2N- = H- > Cl- with an up to 3-fold [Ki(hD2SR) = 5.5 ± 0.6 [nM] and Ki(hD3R) = 1.7 ± 0.8 [nM] for 6] vs. [Ki(hD2SR) = 16 ± 1.1 [nM] and Ki(hD3R) = 22 ± 13 [nM] for 8], and 4-fold [Ki(hD2SR) = 2.8 ± 0.8 [nM] and Ki(hD3R) = 3.0 ± 1.6 [nM] for 7] vs. [Ki(hD2SR) = 11 ± 1.9 [nM] and Ki(hD3R) = 11 ± 3.9 [nM] for 11] increase observed for the 2-amino or the deaminated ligands (6, 7, 9, and 10) when compared to their corresponding 2-chloro-analouges (8 and 11). Interestingly, the ratio values (hD2SR/hD3R) observed for the novel ligands 3-20 were found in the range of 0.3–3, indicating that when compared to the reference compound 1, which displays a 66-fold higher affinity toward hD3Rs, ligands 3-20 showed improved dual affinity toward hD2SRs and hD3Rs.
Furthermore, the results showed obviously that ligands bearing an unsubstituted (12-14) or 4-methoxy-substituted 4-phenylpiperazine moiety (3, 8, and 10) exhibited in general no significant differences in their binding affinities toward both receptor subtypes (Table 1, Figure 2). Moreover, the affinities to hD2SRs observed for ligands 12-20 were found in a high nanomolar range (43–540 [nM]) when compared with ligands 3-11 with binding affinities in the nanomolar range (2.8–26 [nM]) (Table 1). Interestingly, ligand having a tetramethylene spacer (18) showed significantly higher affinity to hD3Rs than its correspondingly 2-chloro-substituted analog 20, however, in a low nanomolar concentration range with Ki(hD3R) values of 16 ± 11 [nM] and 65 ± 17 [nM] for 18 and 20, respectively. The obtained results show that among the current series the highest affinities toward hD2SRs and hD3Rs were achieved with ligands bearing 2-methoxyphenylpiperazinpiperazine moiety connected by a tri- or tetramethylene alkyl spacer with benzothiazole heterocycle which bears an amine group or hydrogen at its 2-position. These structural characteristics were realized and are represented in ligands 6, 7, 9 and 10 with observed affinities in the low nanomolar range toward hD2SRs and hD3Rs.
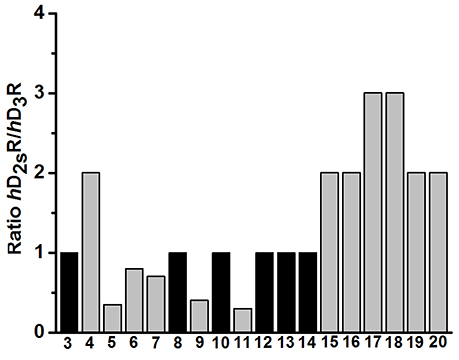
Figure 2. Dopamine hD2SR/hD3R binding affinity ratios of final products 3-20.
To assist further the drug discovery/development process and to guide the optimization from a lead compound to a successful drug candidate, various physicochemical parameters have been shown to be useful tools to aid in selecting oral drug candidates (Hopkins et al., 2004; Meanwell, 2011). Therefore, lipophilicity (clogP), water-solubility (clogS), molecular weight (MW), number of rotatable bonds (NROTB), number of hydrogen bond acceptors (NHBA), number of hydrogen bond donators (NHBD), topological polar surface area (TPSA), and drug-likeness score of Lipinski's rule of five were calculated for the current series of ligands (3-20) using Molinspiration online property calculation toolkit and Osiris Property explorer (Table 2; Meanwell, 2011; Tarcsay et al., 2012). The clogS value demonstrates the solubility of a drug candidate, which impacts its absorption and distribution properties.(Molinspiration software or free molecular property calculation services [last accessed 2017 April 07]2) Accordingly, clogS can be a useful parameter to estimate the ADME properties of a ligand (Hopkins et al., 2014) Solubility of ligands 4, 12-16, 18, and 19 were found in an acceptable range (< −4 clogS). Also, ligands 4, 12-16, 18, and 19 showed lower clogS values, indicating predictable enhanced water solubility, when compared with the reference ligand 1 (Table 2). Moreover, the models of lipophilicity-related physicochemical analyses predictively quantifying target-oriented drug-likeness have for many years been recognized as useful tools in the lead optimization process. This is not unexpected since the influence of clogP in modifying drug potency, pharmacokinetics and toxicity has been established for many years (Meanwell, 2011). Accordingly, it has been proposed that ligands with a clogP < 5 have a more promising drug-likeness profile (Schreeb et al., 2013; Shultz, 2014). Among the current series of ligands, the clogP values, except that for 8, 11, 17, and 20 were found < 5 suggesting their suitability for oral route of administration (Table 2). To promote the assessment of the drug discovery/development process from a lead ligand to a successful drug candidate, other physicochemical parameters have been publicized to be valuable tools to aid in selecting oral drug candidates (Meanwell, 2011). Consequently, it has earlier been suggested that ligands with TPSA values >60 Å2 are usually considered as poorly membrane-permeable substances with predictable reduced CNS bioavailability (Hopkins et al., 2004; Meanwell, 2011). Among the synthesized series, the observed TPSA values for compounds 3-20 were in the range of 28–63 Å2, imposing more structural optimization of the current class with the aim to generate newer derivatives with improved predictive physicochemical parameters, especially regarding TPSA (Table 2).
To further assist the drug discovery/development process, generally values of LE and LELP being >0.3 and < 7.5, respectively, are validated as promising candidates with drug-likeness (Kuntz et al., 1999). Interestingly, all ligands (3-20) showed LE >0.3 for hD2SRs and hD3Rs, whereas the correlation of observed LELP values (hD2SRs and hD3Rs) were not existing, and were found >9 (Table 3). Importantly, the LipE values which are considered to be independent on molecular size of the respective ligand were found for all ligands <5 by considering the affinity of each respective ligand for hD2SRs and hD3Rs. The LipE results observed for the current series further corroborate their drug-likeness (Table 3).
Moreover, drug-likeness model score (a combined outcome of physicochemical properties, pharmacokinetics and pharmacodynamics of a compound and is represented by a numerical value) was computed as previously described by MolSoft software for the ligands 3-20 and summarized in Table 3 (Drug-likeness and molecular property prediction) (Hopkins et al., 2004). It has been proposed that ligands having zero or negative value should not be considered as drug-like candidate. In regard to the developed series 3-20, except 11 all ligands possessed drug-likeness scores in the range of 0.4–1.00, and with ligands 12, 15, and 18 showing maximum-likeness scores of 1.0 (Table 3; Figure 2). The results of pharmacological screening and calculated metric parameters are summarized in Tables 1–3.
Several benzothiazole-based ligands targeting hD2SR and hD3R were synthesized by varying the length of alkyl spacer and the substitution at 2-position of benzothiazole and/or 4-phenylpiperazine. Most of these derivatives showed affinities to the hD2SR and hD3R in nanomolar concentration range comparable to that of the reference ligands 1 and 2. The high affinities of the new ligands, especially 9 and 10, at the hD2SR and hD3R confirm the proposed affinity-strengthening effect of benzothiazole heterocylcle toward hD2sRs and hD3Rs. A calculation of physicochemical properties may provide a good assessment to an enhanced drug-likeness of most of the ligands. Especially ligands 9 and 10 with high dual affinities at hD2SRs and hD3Rs [pKi (hD2SR) = 8.6 and pKi (hD3R) = 8.5 for 9; pKi (hD2SR) = 8.5 and pKi (hD3R) = 8.1 for 10] show reduced lipophilicity as compared to those calculated properties of 1 and 2. Combining these results, these two ligands revealed promising LE, LipE, and drug-likeness values, and showed that ligand affinity and selectivity between hD2SR and hD3R activation were strongly influenced by the appendage of benzothiazole, as a lipophilic bioisosterical replacement of aryl moiety, the length of alkyl spacer, and the nature of functional groups attached to the benzothiazole and the basic 4-phenylpiperazine moiety.
BS and HS were responsible for the study concept, design, acquisition and analysis of data. MS, TK, and LW were responsible for the generation, synthesis and pharmacological in vitro characterization the novel ligands. MS and BS drafted the manuscript. HS provided critical revision for the manuscript. All authors critically reviewed content and approved final version for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Support for this work has been kindly given to HS by the EU COST Actions BM0806, BM1007, CM1103, and CM1207 as well as the Hesse LOEWE Schwerpunkte Fh-TMP, OSF, and NEFF, the Else KrönerStiftung, TRIP, and the Deutsches Konsortium für Translationale Krebsforschung, DKTK. Support to BS was provided in form of grants received from UAE University.
1. ^Drug-likeness and molecular property prediction, a.f.h.w.m.c.m.
2. ^Molinspiration software or free molecular property calculation services [last accessed 2017 April 07], s. Available online at: URL: www.molinspiration.com/cgi-bin/properties.
Bettinetti, L., Schlotter, K., Hubner, H., and Gmeiner, P. (2002). Interactive SAR studies: rational discovery of super-potent and highly selective dopamine D3 receptor antagonists and partial agonists. J. Med. Chem. 45, 4594–4597. doi: 10.1021/jm025558r
Bocker, A., Sasse, B. C., Nietert, M., Stark, H., and Schneider, G. (2007). GPCR targeted library design: novel dopamine D3 receptor ligands. Chem. Med. Chem. 2, 1000–1005. doi: 10.1002/cmdc.200700067
Boeckler, F., and Gmeiner, P. (2007). Dopamine D3 receptor ligands: recent advances in the control of subtype selectivity and intrinsic activity. Biochim. Biophys. Acta 1768, 871–887. doi: 10.1016/j.bbamem.2006.12.001
Casey, A. B., and Canal, C. E. (2017). Classics in chemical neuroscience: aripiprazole. ACS Chem. Neurosci. 8, 1135–1146. doi: 10.1021/acschemneuro.7b00087
Garcia-Ladona, F. J., and Cox, B. F. (2003). BP 897, a selective dopamine D3 receptor ligand with therapeutic potential for the treatment of cocaine-addiction. CNS Drug Rev. 9, 141–158. doi: 10.1111/j.1527-3458.2003.tb00246.x
Hackling, A., Ghosh, R., Perachon, S., Mann, A., Holtje, H. D., Wermuth, C. G., et al. (2003). N-(omega-(4-(2-methoxyphenyl)piperazin-1-yl)alkyl)carboxamides as dopamine D2 and D3 receptor ligands. J. Med. Chem. 46, 3883–3899. doi: 10.1021/jm030836n
Hayes, G., Biden, T. J., Selbie, L. A., and Shine, J. (1992). Structural subtypes of the dopamine D2 receptor are functionally distinct: expression of the cloned D2A and D2B subtypes in a heterologous cell line. Mol. Endocrinol. 6, 920–926. doi: 10.1210/mend.6.6.1323056
Hopkins, A. L., Groom, C. R., and Alex, A. (2004). Ligand efficiency: a useful metric for lead selection. Drug Discov. Today 9, 430–431. doi: 10.1016/S1359-6446(04)03069-7
Hopkins, A. L., Keseru, G. M., Leeson, P. D., Rees, D. C., and Reynolds, C. H. (2014). The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 13, 105–121. doi: 10.1038/nrd4163
Hou, T., Wang, J., Zhang, W., and Xu, X. (2007). ADME evaluation in drug discovery. 6. Can oral bioavailability in humans be effectively predicted by simple molecular property-based rules? J. Chem. Inf. Model 47, 460–463. doi: 10.1021/ci6003515
Joyce, J. N., and Millan, M. J. (2005). Dopamine D3 receptor antagonists as therapeutic agents. Drug Discov. Today 10, 917–925. doi: 10.1016/S1359-6446(05)03491-4
Kaufmann, H. P., and Bückmann, H. J. (1941). Über die Rhodanierung des Sulfanilamids und einige Benzthiazolyl-sulfonamide. Arch. Pharm. 279:179.
Kuntz, I. D., Chen, K., Sharp, K. A., and Kollman, P. A. (1999). The maximal affinity of ligands. Proc. Natl. Acad. Sci. U.S.A. 96, 9997–10002.
Lader, M. (2008). Antiparkinsonian medication and pathological gambling. CNS Drugs 22, 407–416. doi: 10.2165/00023210-200822050-00004
Leeson, P. D., and Springthorpe, B. (2007). The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 6, 881–890. doi: 10.1038/nrd2445
Levey, A. I., Hersch, S. M., Rye, D. B., Sunahara, R. K., Niznik, H. B., Kitt, C. A., et al. (1993). Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc. Natl. Acad. Sci. U.S.A. 90, 8861–8865.
Meanwell, N. A. (2011). Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 24, 1420–1456. doi: 10.1021/tx200211v
Missale, C., Nash, S. R., Robinson, S. W., Jaber, M., and Caron, M. G. (1998). Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225.
Moller, D., Kling, R. C., Skultety, M., Leuner, K., Hubner, H., and Gmeiner, P. (2014). Functionally selective dopamine D(2), D(3) receptor partial agonists. J. Med. Chem. 57, 4861–4875. doi: 10.1021/jm5004039
Murray, P. J. H., Helden, R. M., Johnson, M. R., Robertson, G. M., Scopes, D. I. C., Stokes, M., et al. (1996). Novel 6-substituted 2-aminotetralins with potent and selective affinity for the dopamine D3 receptor. Bioorg. Med. Chem. Lett. 6, 403–408.
Noeske, T., Sasse, B. C., Stark, H., Parsons, C. G., Weil, T., and Schneider, G. (2006). Predicting compound selectivity by self-organizing maps: cross-activities of metabotropic glutamate receptor antagonists. ChemMedChem 1, 1066–1068. doi: 10.1002/cmdc.200600147
Pilla, M., Perachon, S., Sautel, F., Garrido, F., Mann, A., Wermuth, C. G., et al. (1999). Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 400, 371–375. doi: 10.1038/22560
Schreeb, A., Walter, M., Odadzic, D., Schwed, J. S., Weizel, L., and Stark, H. (2013). Piperazine modification in 2,4,6-triaminopyrimidine derivatives as histamine H4 receptor ligands. Pharmazie 68, 521–525. doi: 10.1691/ph.2013.6511
Shultz, M. D. (2014). Improving the plausibility of success with inefficient metrics. ACS Med. Chem. Lett. 5, 2–5. doi: 10.1021/ml4004638
Sokoloff, P., Andrieux, M., Besancon, R., Pilon, C., Martres, M. P., Giros, B., et al. (1992). Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: comparison with D2 receptor. Eur. J. Pharmacol. 225, 331–337.
Sokoloff, P., Giros, B., Martres, M. P., Bouthenet, M. L., and Schwartz, J. C. (1990). Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347, 146–151. doi: 10.1038/347146a0
Stucchi, M., Gmeiner, P., Huebner, H., Rainoldi, G., Sacchetti, A., Silvani, A., et al. (2015). Multicomponent synthesis and biological evaluation of a piperazine-based dopamine receptor ligand library. ACS Med. Chem. Lett. 6, 882–887. doi: 10.1021/acsmedchemlett.5b00131
Sukalovic, V., Andric, D., Roglic, G., Kostic-Rajacic, S., Schrattenholz, A., and Soskic, V. (2005). Synthesis, dopamine D2 receptor binding studies and docking analysis of 5-[3-(4-arylpiperazin-1-yl)propyl]-1H-benzimidazole, 5-[2-(4-arylpiperazin-1-yl)ethoxy]-1H-benzimidazole and their analogs. Eur. J. Med. Chem. 40, 481–493. doi: 10.1016/j.ejmech.2004.10.006
Swainston Harrison, T., and Perry, C. M. (2004). Aripiprazole: a review of its use in schizophrenia and schizoaffective disorder. Drugs 64, 1715–1736. doi: 10.2165/00003495-200464150-00010
Tarcsay, A., Nyiri, K., and Keseru, G. M. (2012). Impact of lipophilic efficiency on compound quality. J. Med. Chem. 55, 1252–1260. doi: 10.1021/jm201388p
Tomic, M., Ignjatovic, D., Tovilovic, G., Andric, D., Roglic, G., and Kostic-Rajacic, S. (2007). Two new phenylpiperazines with atypical antipsychotic potential. Bioorg. Med. Chem. Lett. 17, 5749–5753. doi: 10.1016/j.bmcl.2007.08.066
Tuplin, E. W., and Holahan, M. R. (2017). Aripiprazole, a drug that displays partial agonism and functional selectivity. Curr. Neuropharmacol. doi: 10.2174/1570159X15666170413115754. [Epub ahead of print].
Wright, J. E., and Rosowsky, A. (2002). Synthesis and enzymatic activation of N-[N(alpha)-(4-amino-4-deoxypteroyl)-N(delta)-hemiphthaloyl-L-ornithiny]-L-phenyl alanine, a candidate for antibody-directed enzyme prodrug therapy (ADEPT). Bioorg. Med. Chem. 10, 493–500. doi: 10.1016/S0968-0896(01)00298-X
Keywords: benzothiazoles, dopamine D2S/D3 receptor, in vitro activities, privileged structures, drug-likeness
Citation: Schübler M, Sadek B, Kottke T, Weizel L and Stark H (2017) Synthesis, Molecular Properties Estimations, and Dual Dopamine D2 and D3 Receptor Activities of Benzothiazole-Based Ligands. Front. Chem. 5:64. doi: 10.3389/fchem.2017.00064
Received: 28 June 2017; Accepted: 28 August 2017;
Published: 12 September 2017.
Edited by:
Alice L. Perez, University of Costa Rica, Costa RicaReviewed by:
José Carlos Menéndez, Complutense University of Madrid, SpainCopyright © 2017 Schübler, Sadek, Kottke, Weizel and Stark. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bassem Sadek, YmFzc2VtLnNhZGVrQHVhZXUuYWMuYWU=
Holger Stark, c3RhcmtAaGh1LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.