 Xiaoqian Zhang
Xiaoqian Zhang Xiao-Xi Li
Xiao-Xi Li Yufang Liu1
Yufang Liu1 Yong Wang
Yong Wang- 1College of Physics and Materials Science, Henan Normal University, Xinxiang, China
- 2State Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of LICP, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, China
N-benzyl-N-cyclopropylamine (BCA) has been attracting great interests for decades for its partial suicide inactivation role to cytochrome P450 (P450) via a ring-opening mechanism besides acting as a role of normal substrates. Understanding the mechanism of such partial inactivation is vital to the clinical drug design. Thus, density functional theoretical (DFT) calculations were carried out on such P450-catalyzed reactions, not only on the metabolic pathway, but on the ring-opening inactivation one. Our theoretical results demonstrated that, in the metabolic pathway, besides the normal carbinolamine, an unexpected enamine was formed via the dual hydrogen abstraction (DHA) process, in which the competition between rotation of the H-abstracted substrate radical and the rotation of hydroxyl group of the protonated Cpd II moiety plays a significant role in product branch; In the inactivation pathway, the well-noted single electron transfer (SET) mechanism-involved process was invalidated for its high energy barrier, a proton-coupled electron transfer [PCET(ET)] mechanism plays a role. Our results are consistent with other related theoretical works on heteroatom-hydrogen (X-H, X = O, N) activation and revealed new features. The revealed mechanisms will play a positive role in relative drug design.
Introduction
Suicide inhibitors, or called mechanism-based (kcat) inactivators, are attractive of great interests in enzymology and drug industry, because the action of such inhibitors is intimately related to the enzymatic mechanism, well knowledge of the latter often provides an excellent starting point for the rational design of highly specific and effective drugs in clinical use (e.g., penicillin) (Meunier et al., 2004; Ortiz de Montellano and De Voss, 2005). Besides, discovery of such inhibitors for an enzyme, e.g., cytochrome P450, whose mechanisms are not well-characterized should provide an equally specific and effective probe. Metabolism of these alternative substrates could result in the generation of reactive species that inactivates the enzyme through either covalent modification or tight binding (Orr et al., 2012). Cyclopropylamines are one kind of such prototypical inhibitors for cytochrome P450 enzymes (P450s), which are widely found in biologically active natural products, synthetic drugs and also widely used as mechanistic probes to reveal elusive reaction mechanisms, such as that involved in P450-catalyzed amine oxidations (Bhakta and Wimalasena, 2002; Ortiz de Montellano and De Voss, 2002; Totah and Hanzlik, 2002; Bhakta et al., 2005).
N-benzyl-N-cyclopropylamine 1 (Figure 1, hereafter as BCA in brief) functioning as a suicide inhibitor of P450 was first reported by Hanzlik et al. (1979), and toward understand the special inactivation role, they proposed a Cα-hydrogen atom transfer (Cα-HAT, I in Figure 1) mechanism in which a Schiff base intermediate 3 was deemed to account for the inactivation. However, Hanzlik (Hanzlik and Tullman, 1982) and Guegerich (Macdonald et al., 1982) further found that the 1'-methyl-substituted analog of 1, N-benzyl-N-(1′-methylcyclopropyl)amine lacking hydrogen atom at the Cα position of cyclopropyl group, was also capable of inactivating P450 almost as effectively as 1. Thus, the Cα-HAT mechanism was ruled out. Instead, a single electron transfer (SET) mechanism (Macdonald et al., 1982) (II in Figure 1) was postulated that the inactivation involved an initial heteroatom oxidation to aminium cation radical 4, which could undergo rapid ring-opening process to form highly reactive carbon-centered radical species 5 and subsequently covalently bound to the amino residue within the enzyme active site (Macdonald et al., 1982).
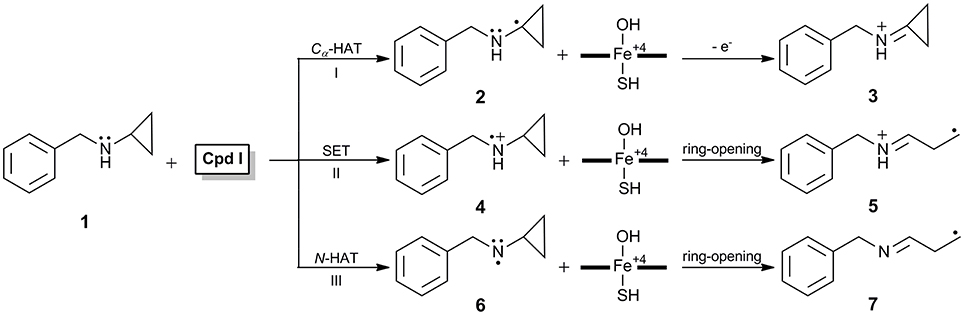
Figure 1. Proposed mechanisms involved in BCA-induced inactivation of cytochrome P450.
The SET mechanism can also explain some other observations for amine oxidations, such as the inhibition role of 4-alkyl-1,4-dihydropyridine derivatives which could extrude an alkyl radical during the P450-catalyzed oxidation (Augusto et al., 1982), the small deuterated kinetic isotope effect (KIE) (Miwa et al., 1983), the correlation of free energy relationship to one-electron oxidation potential (Guengerich et al., 1984), the large negative Hammett ρ in N-demethylations of para-substituted N,N-dimethylanilines by both P450 (Burka et al., 1985) and its nonheme biomimics (Nehru et al., 2007), thusly was widely accepted as a general mechanism involved in amine oxidation (Guengerich et al., 1996). However, the validity of such evidence for the SET mechanism was subsequently challenged. Dinnocenzo et al. found that the KIE profiles for a series of dimethylaniline (DMA) oxidations catalyzed by P450 correlated linearly with those measured in the reactions of the same DMAs with tert-butoxyl radical which actually involved a typical hydrogen atom transfer (HAT) mechanism (Dinnocenzo et al., 1993; Karki and Dinnocenzo, 1995; Karki et al., 1995; Manchester et al., 1997). Shaffer et al. reported that the oxidation of N-methyl-N-cyclopropylaniline by horseradish peroxidase (HRP), a conventional SET oxidant, led to yields of ring-opened intermediates and subsequently inactivated the enzyme, conversely, as an oxidant whose reduction potential was higher, the P450-catalyzed oxidation of N-methyl-N-cyclopropylaniline produced ring-intact metabolites exclusively and no inactivation was observed (Shaffer et al., 2001a,b). In addition, the use of clock substrate and other experimental criteria seemed to rule out the involvement of the amine cation radical intermediate (Shaik et al., 2010). To determine the complete fate of 1 in vivo by P450 as well as to reconcile the discrepant behaviors in HRP- and P450-catalyzed reactions, Cerny and Hanzlik (2005, 2006) performed a series of experiments, and eventually proposed an N-HAT mechanism (III in Figure 1) through the product study. This novel N-HAT mechanism was proposed upon a ring-opening process of the cyclopropyl group, i.e., hydrogen abstraction from the N-H bond of the secondary cyclopropylamine 1 gave a neutral aminyl radical species 6 which could undergo rapid ring-opening process to form 7, such reactive C-centered radical species accounted for the enzyme inactivation by means of covalently binding to the amino residue in the active site. Moreover, such N-HAT mechanism was subsequently validated by Hirao et al. in their investigation on P450 inactivation by 1,1-dimethylhydrazine (Hirao et al., 2013a). However, previous investigations have revealed that there might be an alternative proton-coupled electron transfer (PCET) mechanism for the polar X-H (X = O, N) bond activation (Mayer et al., 2002; Usharani et al., 2013; Hirao and Chuanprasit, 2015; Li et al., 2015). Thus, an obvious question to be answered is: Ring-opening of BCA is initiated by a direct HAT mechanism or a PCET one?
Furthermore, according to Hanzlik, the incubation of 1 with microsomes indeed caused a time-, concentration- and cofactor-dependent loss of cytochrome P450 activity, which was, however, not vanished completely but instead remained 25–30% of it (Cerny and Hanzlik, 2005). In an effort to reveal the genuine mechanism of P450 inactivation by N-benzyl-N-cyclopropylamine that involved in the physiological process, and thereby making contributions to pharmacy, we performed a series of density functional theory (DFT) calculations. Two distinct pathways, inactivation and metabolic ones, were examined in which BCA reacted with P450 efficiently. Moreover, we also demonstrated that in the inactivation pathway, the N-H bond activation/ring-opening process was a prior reaction route for the low energy barrier of its rate-limiting step, 0.6/0.4 kcal/mol for the high quartet spin-state (HS)/low doublet spin-state (LS). Essentially, the validity of the PCET mechanism that involved in the H-abstraction at the N-H bond of BCA by P450 was confirmed.
Theoretical Methods
Due to the key role of the computational modeling on the investigation of enzyme reaction (Li et al., 2012; de Visser et al., 2014), we employed an iron-oxo open-shell porphyrin with an axial thiolate ligand to model CpdI [Fe4+O2−(C20N4H12)−(SH)−] (Shaik et al., 2005; Wang et al., 2006, 2007a, 2010), and used the N-benzyl-N-cyclopropylamine as the substrate. All DFT calculations were performed with the Gaussian 03 suite of quantum chemical packages (Frisch et al., 2004). The spin-unrestricted B3LYP (Lee et al., 1988; Becke, 1992a,b, 1993) functional was employed with two basis sets: (a) The LACVP(Fe)/6-31G*(H, C, N, O, S) (denoted as LACVP*, henceforth B1) for geometry optimizations without symmetry constraint; (b) The LACV3P(Fe)/6-311++G**(H, C, N, O, S) (denoted as LACV3P++**, henceforth B2) for single point energy (SPE) calculations (Hay and Wadt, 1985). Transition states were ascertained by vibrational frequency analysis to possess a single mode along the reaction path with only one imaginary frequency. Bulk polarity effects of the active site in the protein environment were evaluated with the polarizable continuum model (PCM) using a nonpolar solvent, chlorobenzene (ε = 5.697).
The 4(2)IMSET species (whose occupation diagram and Mulliken spin density are shown in Supplementary Figure 2 and Supplementary Table 3) involved in the SET process were obtained by shifting one electron from the highest occupied orbital of N-benzyl-N-cyclopropylamine (σCH) to the lowest vacant orbital of porphyrin moiety (a2u) of CpdI on the reactant complex (4(2)RC) (Li et al., 2006). Any attempt to optimize the structure of 4(2)IMSET species will result in their collapse to the ground state.
The kinetic isotope effect for the hydrogen abstraction process was determined using Gaussian frequency data based on the semi-classical Eyring equation (Melander and Saunders, 1987), where the KIE is given as.
k denotes the reaction rate constant, G is the Gibbs free energy of abstraction, R is the gas constant, T is the absolute temperature.
All data were collected in the Supplementary Material document, while we discussed the highest-level results, UB3LYP/B2//B1, in the text. Thus, the SPE B2//B1 value with the zero point energy (ZPE) correction referred to E1, whereas E2 included the bulk polarity effects and ZPE corrections.
Results and Discussion
The Inactivation Pathway
In retrospect, Hirao et al. recently investigated the mechanism-based inactivation of cytochrome P450 by 1,1-dimethylhydrazine (UMDH) (Hirao et al., 2013a), they examined four possible reaction pathways including H-abstractions from the primary amine moiety and the methyl moiety, as well as the direct oxidations on two nitrogen atoms. Owing to the lower energy barriers, they concluded that the H-abstraction from the N-H bond of the primary amine moiety was the most favorable pathway, on which our investigation is based.
As for the inactivation of P450 by BCA (Figure 2) which involves the PCET mechanism, the substrate is initially bound to CpdI through the N-H…O hydrogen-bonding interaction, the subsequent H-abstraction from the N-H bond results in an amino radical intermediate (IM species). Owing to the newly-formed hydrogen-bonding between O-H and N, dichotomous behaviors are encountered at the following step. The amino radical IM species could undergo either a traditional O-rebound process to yield hydroxylamine which has been deemed to be the precursor to oxime (Cerny and Hanzlik, 2005; Hirao et al., 2013b), or a rapid ring-opening of the cyclopropyl group forming a C-centered radical species, which is essential for the enzyme inactivation and could be converted to 3HP via a further oxidation.
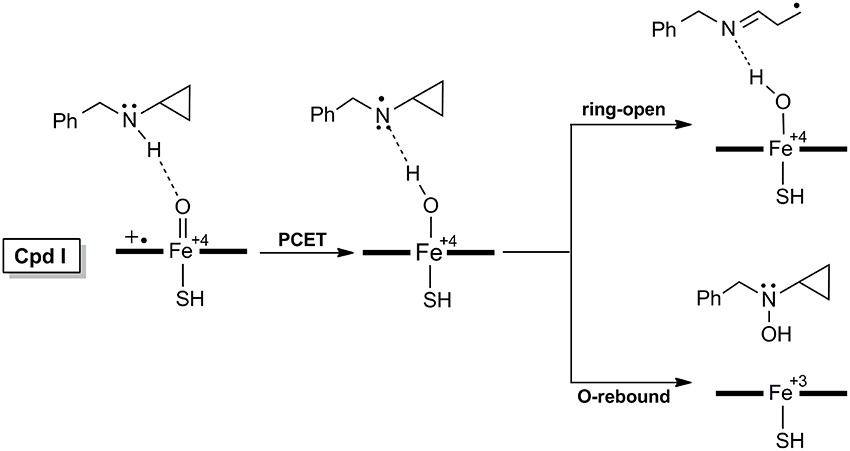
Figure 2. Proposed mechanisms of H-abstraction and the following ring-opening/O-rebound processes of BCA by P450.
On the exploration of the inactivation mechanism of P450 by BCA, two controversial mechanisms (II and III in Figure 1) have been postulated by Guengerich (Macdonald et al., 1982) and Hanzlik (Cerny and Hanzlik, 2006), respectively. Figure 3 shows the energy profiles for the SET and PCET processes, accompanied by the geometries of the involved key intermediates. First of all, we evaluate the energy barriers of the SET process for both quartet and doublet spin states through DFT calculations. It is obvious that the SET energy barrier (IMSET, 31.0/38.2 kcal/mol for the HS/LS) is much higher compared with the direct hydrogen abstraction from N-H bond via a PCET mechanism (0.6/0.4 kcal/mol for the HS/LS). Such sharp energetic comparison between SET and PCET processes definitely rules out the former and favors the latter. On RC, two spin states are nearly degenerated with the LS lying under the HS by -0.3/-0.1 kcal/mol at the E1/E2 level. Intriguingly, the following essential H-abstraction from the N-H bond of the secondary amine moiety proceeds through an extremely low energy barrier (0.6/0.4 kcal/mol for the HS/LS at the E1 level, indicating an involvement of two-state reactivity mechanism (Shaik et al., 1998; Schröder et al., 2000; de Visser and Tan, 2008), which is similar to our previous finding about the oxidation of 4-alkylated DHPs (Li et al., 2015) as well as that reported by Hirao et al. (2013a) but different from that obtained by Rydberg and Olsen (2011). Hirao et al. attributed the tiny energy barrier to weak bond disassociation energy (BDE) of N-H bond rather than the involvement of PCET mechanism, which has been proved in our previous work. Whereas, in Rydberg's work which studies the P450-catalyzed hydroxylation of propan-2-amine, the energy barrier of such H-abstraction was much higher, 12.6/11.6 kcal for the HS/LS in the gas phase, such significant energetic discrepancy is attributed to that the N-H bond BDE decreases when going from primary to secondary amines (Lalevée et al., 2002; Luo, 2002). The inclusion of the bulk polar effect even changes the H-abstraction into a barrierless process (−2.9/−2.7 kcal/mol for the HS/LS). The IM species which comprises the N-centered radical substrate and the ferryl-hydroxyl closed-shell porphyrin (protonated CpdII) is generated after the H-abstraction at the N-H bond. Unlike the IMCH species in most P450-catalyzed C-H bond hydroxylation which always lies upon the RCCH species (Ogliaro et al., 2000; de Visser et al., 2002, 2004; Kumar et al., 2003; Li et al., 2006; Olsen et al., 2006; Wang et al., 2006, 2007a,b, 2010), the IM species in N-H bond activation lies under the RC species by −7.7/−7.9 kcal/mol for the HS/LS at the E1 level. That is because compared with the IMCH in which the carbon radical interacts with the protonated CpdII, the OH…N hydrogen bonding in IM is quite significant for stabilizing the amino radical, and consequently lowering the systematic energy (Korzekwa et al., 1990; Jones et al., 2002; Olsen et al., 2006). As mentioned above, the following step is dichotomous. Through the novel ring-opening route, the N-centered radical rapidly rearranges to a ring-opened C-centered radical that could covalently bind to the amino residue within the active site, and therefore inactivating the enzyme. The TSring species is more stable than the RC species in an energy of −3.6/−3.6 kcal/mol at the E1 level and −2.0/−1.8 kcal/mol at the E2 level for the HS/LS, while lies 4.1/4.3 kcal/mol higher relative to the IM for the HS/LS at the E1 level. Thus, the ring-opening process should be the rate-limiting step on such hydrogen abstraction/ring-opening route. Additionally, the relative “high” energy of the ring-opened product PCring (−13.4/−13.5 kcal/mol for the HS/LS at the E1 level) implies that it may be reactive and could be further oxidized to 3HP. Alternatively on the conventional O-rebound route, it would form hydroxylamine in a same manner as the well-known C-H bond hydroxylation by CpdI (Ogliaro et al., 2000; de Visser et al., 2002; Kumar et al., 2003; Wang et al., 2006). During the O-rebound process, the spin state order exchanges with an energy barrier of 2.4/3.6 kcal/mol for the HS/LS at the E1 level. Such energy barrier is higher than that of the PCET process, thus making the O-rebound process the rate-limiting step on this hydrogen abstraction/O-rebound route. While in P450-catalyzed C-H bond hydroxylations (Ogliaro et al., 2000; de Visser et al., 2002; Kumar et al., 2003; Wang et al., 2006), the O-rebound process proceeds via a quite low energy barrier, even it often becomes barrierless on the doublet spin state. Rydberg et al. also reported the similar higher rebound energy barrier in their recent investigation regarding the P450-catalyzed hydroxylation of primary amines, such distinct energetic discrepancy between C-H bond and N-H bond hydroxylations was ascribed to different radical orientations and the existence of strong O-H…N hydrogen-bonding interaction in the intermediate species (Rydberg and Olsen, 2011). On the whole, the energy barrier of O-rebound process is significantly higher than that of ring-opening process by 6–7 kcal/mol, indicating that the hydrogen abstraction/ring-opening route is prior to the hydrogen abstraction/O-rebound one. Such conclusion also coincides with the observation of the major ring-opened product, i.e., 3HP, in Hanzlik's experiments (Cerny and Hanzlik, 2006).
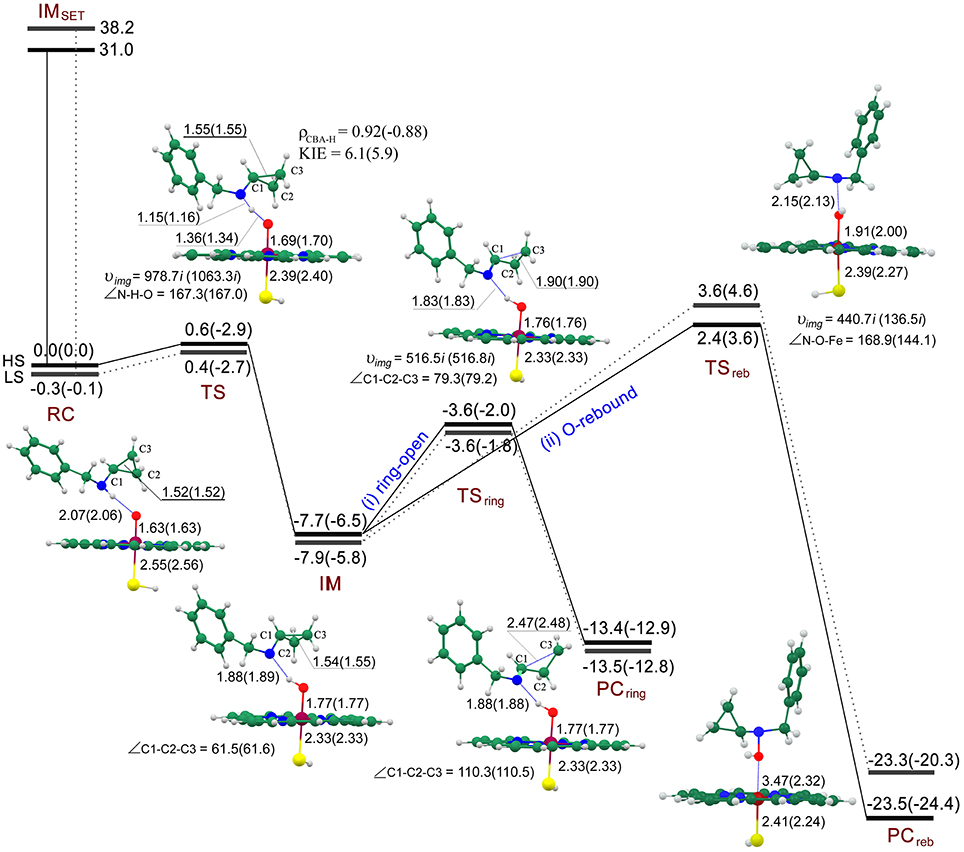
Figure 3. Energy profiles (UB3LYP/B2//B1) for SET process and the competitive ring-opening/O-rebound step after H-abstraction process at the E1(E2) level and the geometries (UB3LYP/B1) of the key intermediates for the HS(LS) involved. The relative energy is in kcal/mol, bond length is in Å, bond angle is in degree and the imaginary frequency is in cm−1 unit.
Geometrically on RC (see Figure 3), the H-O distance along the reaction coordinate is 2.07/2.06 Å for the HS/LS, thus an N-H…O hydrogen-bonding interaction exists which might be essential for the following step. Such hydrogen-bonding interaction not only orientates the substrate, therefore facilitating the H-abstraction from the N-H bond, but might weaken the N-H bond in some degree, due to the larger electronegativity of oxygen than nitrogen. On TS, the N-H distance (1.15/1.16 Å for the HS/LS) is much shorter than H-O distance (1.36/1.34 Å for the HS/LS), such transition states are asymmetric and exhibit an significant “earlier” characteristic. Similarly in Hirao's work, the transition states of the H-abstraction from the N-H bond were even much earlier, due to its shorter N-H distance (1.04/1.06 Å for the HS/LS) and longer H-O distance (1.76/1.71 Å for the HS/LS).(Hirao et al., 2013a) On the contrary, the H-O distance in P450-catalyzed H-abstraction from the C-H bond was shorter (Ogliaro et al., 2000; de Visser et al., 2002; Kumar et al., 2003; Wang et al., 2006), and thus corresponding to a “later” transition state compared with ours. As expected, the “later” transition state in C-H bond H-abstraction with a higher energy barrier follows the correlation between the transition state and the energy barrier (Shaik et al., 2008; Kumar et al., 2013). Additionally, along the reaction coordinate, the angle of the N-H-O moiety is 167.3°/167.0°For the HS/LS, which is not as linear as the C-H-O moiety in C-H bond H-abstraction. While on IM, the newly-formed O-H…N hydrogen-bonding interaction with a H-N distance of 1.88/1.89 Å for the HS/LS, becomes stronger than that in RC. Such strong hydrogen-bonding interaction, as Rydberg concluded, results in the disadvantage of the following O-rebound process (Rydberg and Olsen, 2011). Besides, the weak nucleophilicity of nitrogen might somehow account for such interference as well. On TSreb, the Fe-O distance is 1.91/2.00 Å for the HS/LS, whereas the O-N distance is slightly longer (2.15/2.13 Å for the HS/LS). Along the rebound reaction coordinate, the angles of the Fe-O-N moiety is quite different for the quartet (168.9°) and doublet (144.1°) states, the higher energy barrier for the LS of this O-rebound process may be a consequence of such bent TS geometry. Such distinct linearities of the Fe-O-N moiety for HS and LS during the O-rebound process were also obtained by Rydberg and Olsen (2011). Whereas, in C-H bond hydroxylations (Ogliaro et al., 2000; de Visser et al., 2002; Kumar et al., 2003; Wang et al., 2006), the transition states of O-rebound process are not as symmetric as those in N-H bond hydroxylations, the Fe-O distance (~1.8 Å) is clearly shorter than the O-C distance (~2.4 Å), and as for the linearity, the rebound transition states in N-H bond hydroxylation are more bent, the angle of Fe-O-C moiety is approximate 158°For the HS. On hydrogen abstraction/ring-opening route, the C1-C3 (~1.55 Å) distance does not increase until the IM species is generated which involves an amino radical. On TSring, the C1-C3 distance increases to 1.90Å for both spin states, however the O-H…N hydrogen-bonding distance remains almost permanent, thus indicating the radical substrate is always anchored during the ring-opening process.
Inspection of the spin density (Supplementary Table 2) on TSs reveals a confusing result that the distribution on “substrate” is 0.92/-0.88 for the HS/LS, which is suspected exhibiting a PCET character, even if the large KIE values (6.1/5.9 for the HS/LS) supports the HAT mechanism. In retrospect, Hirao et al. obtained the similar tiny energy barrier and distribution on TS1A in the favorable N-HAT pathway, but they ruled out the involvement of PCET mechanism eventually (Hirao et al., 2013a). Thus, to reveal the genuine mechanism involved in the present H-abstraction from N-H bond, the spin-natural orbitals (SNOs), which are useful for distinguishing between the hydrogen atom transfer (HAT) transition state and the proton-coupled electron transfer transition state, have been analyzed for 2,4TS species (Figure 4). It is obvious that two π-type lobes on both of the substrate nitrogen atom and the oxidant oxygen atom are perpendicular to the N-H-O axis, which is a typical feature of the PCET transition state. This is similar to the findings of previous studies (Mayer et al., 2002; Usharani et al., 2013; Hirao and Chuanprasit, 2015).
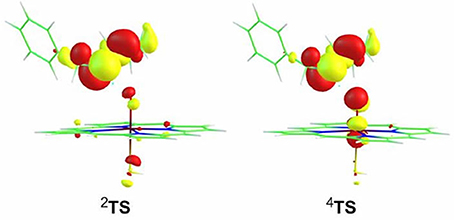
Figure 4. Single occupied spin-natural orbitals for transition state species of the H-abstraction process.
Furthermore, Figure 5 depicts the spin density on ferryl-oxo (green line), “substrate” (red line) and “porpine+thiolate” (blue line) moieties along the reaction coordinate in a stepwise manner. As the hydrogen approaching the oxygen by every 0.05 Å, the spin density on “substrate” is decreasing from 0 to −1 gradually, contrarily, that on “porphyrin+thiolate” is increasing from −1 to 0. Besides, the spin density on ferryl-oxo remains about 2 all along. Inspection of Figure 5, it can be seen that the electron transfer occurs at a H-O distance of 1.4 Å with an obvious change of spin densities on both “substrate” and “porphyrin+thiolate” moieties. From Figure 3, we see that the H-O distance of the transition state is 1.34 Å. This indicates that the electron transfer and the proton transfer are concerted but asynchronous. In other words, the present N-H bond activation should be achieved via a PCET(ET) mechanism as the findings of Usharani et al. (2013).
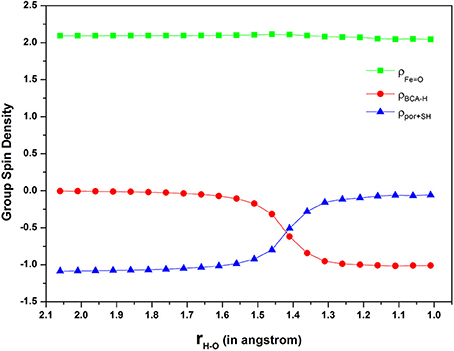
Figure 5. Diagram of group spin density during the H-abstraction from nitrogen.
As mentioned by Cerny and Hanzlik (2005), cytochrome P450 activity was not vanished completely but instead remained 25–30% during the incubation with BCA. Consequently, they attributed this partial inactivation to that BCA reacted with P450 in two reaction pathways (Supplementary Figure 1): a conventional metabolic one on the methylene (-CH2-) and cyclopropyl groups, respectively, and a novel inactivation one on the heteroatom, on which our theoretical investigations are based.
The Metabolic Pathway
Figure 6 depicts the mechanisms involved in the reactions on the methylene (route A) and cyclopropyl (route B) groups of BCA. Resembling each other, an initial hydrogen atom transfer (HAT) occurs to form the radical and a ferryl-hydroxyl closed-shell porphyrin complex which is unstable. The subtle geometric feature of the radical moiety is responsible for the dichotomous behaviors of the reactions in the second step, which are determined by the action of the radical or the newly-generated hydroxyl group as shown in Figure 7. Specifically, when the hydroxyl rotates independently, it will generate carbinolamine via a direct O-rebound process, while the rotation of the hydroxyl and the radical is coupled, an unexpected enamine product will be produced via another HAT process from the adjacent heteroatom (the overall reaction is a dual hydrogen abstraction process, DHA). The competitive DHA mechanism is also a conventional mechanism involved in the P450-catalyzed cholesterol side-chain cleavage reaction and other oxidations (Vanlier and Rousseau, 1976; Sono et al., 1996; Kumar et al., 2004; Oh et al., 2005; Wang et al., 2007b; Ji et al., 2015).
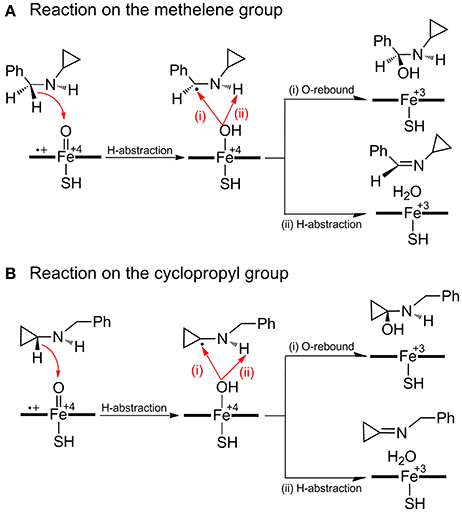
Figure 6. Proposed mechanisms of hydroxylation and dual hydrogen abstraction (DHA) on both methylene (route A) and cyclopropyl (route B) groups of BCA by P450.
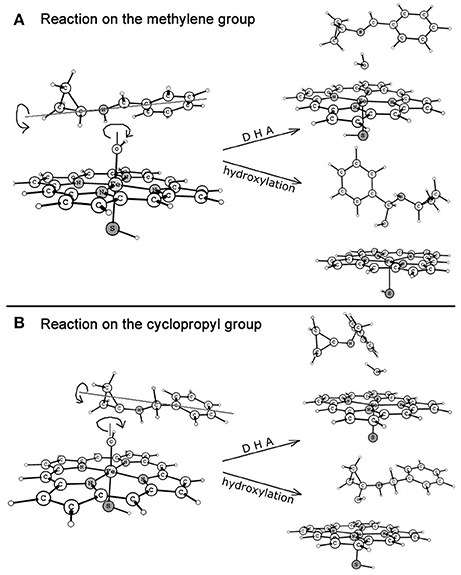
Figure 7. Dichotomous reaction routes after HAT process on the methylene (route A) and cyclopropyl (route B) groups.
Figure 8 shows the energy profiles for both reactions on routes A and B accompanied by the geometries of the key intermediates involved. On RC, the high-spin quartet state (HS) and the low-spin doublet state (LS) are nascent from the degenerate states of CpdI, with the LS lying 0.1/0.2 kcal/mol lower under the HS on route a/b in the gas phase. The energy differences are eliminated by the inclusion of bulk polar effect.
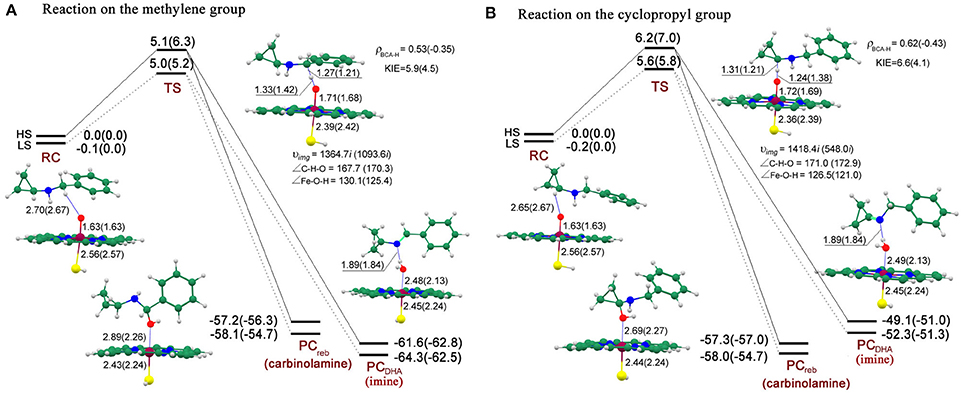
Figure 8. Energy profiles (UB3LYP/B2//B1) for both reactions on the methylene (route A) and cyclopropyl (route B) groups at the E1(E2) level and the geometries (UB3LYP/B1) of the key intermediates for the HS(LS) involved. The relative energy is in kcal/mol, bond length is in Å, bond angle is in degree and the imaginary frequency is in cm−1 unit.
On route A, the energy barrier of the rate-limiting step, which refers to HAT process, is 5.1/5.0 kcal/mol for the HS/LS in the gas phase, which increases to 6.3/5.2 kcal/mol when bulk polar effect is included. As discussed above, the following step is competitive and depends on the rotation of the hydroxyl and the radical. Once after the barrierless O-rebound process, the carbinolamine (−57.2/−58.1 kcal/mol for the HS/LS) is produced, which might subsequently decompose to benzaldehyde and cyclopropylamine, probably in a non-enzymatic environment assisted by water molecule. By contrast, the DHA process, in which the second H-abstraction from nitrogen atom is also barrierless, is a more exothermic process due to the lower energy (−61.6/−64.3 kcal/mol for the HS/LS) of enamine product complex (PC). Along the C-H-O reaction coordinate, the C-H distance is 1.27/1.21 Å for the HS/LS which is shorter than 1.33/1.42 Å of the H-O distance, and consequently making 2TS an earlier transition state compared to 4TS. Inspection of the spin density (Supplementary Table 6) reveals that, for the HS/LS, the spin density populated on “substrate” (BCA-H moiety) is 0.53/−0.35, while “porphyrin+thiolate” moiety holds 0.45/−0.48. Thus, the TS species are deemed to exhibit an “HAT-type” character. In addition, the semi-classical Eyring KIE value (see Computational methods above) for each state is 5.9/4.5, which is relatively large and supports the HAT mechanism.
Whereas on route B, there are still a few discrepancies compared to those on the similar route A. The energy barrier of the rate-limiting step (6.2/5.7 kcal/mol in the gas phase and 7.0/5.8 kcal/mol in solvent for the HS/LS) is about 1 kcal/mol higher than that on route A. However, given that the tiny difference of the energy barriers between these two reactions, they probably occur competitively. Alternatively on route B, the carbinolamine product is generated via a more exothermic HAT/O-rebound process compared to the competitive DHA process. The carbinolamine from this route would undergo following decomposition to yield cyclopropanone and benzylamine identified in Hanzlik's experiments (Cerny and Hanzlik, 2005, 2006). Geometrically, the 4TS species is the only one that possesses a “later” transition state geometry in both reactions, in which C-H distance (1.31 Å) is longer than H-O distance (1.24 Å).
Conclusion
For decades, the mechanism-based inactivation role of N-benzyl-N-cyclopropylamine to cytochrome P450 has been attracting great interests. Theoretical investigation on the way they function is contributory to clinical drug design. Thereby we performed DFT calculation on P450-catalyzed BCA reaction, in which it proceeded on dichotomous metabolic and inactivation pathways. In metabolic pathway, besides the carbinolamine, which is the precursor to the product identified experimentally, an unexpected enamine product was formed via the competitive DHA route switched by the coupled rotation of the radical and hydroxyl group. In inactivation pathway, the SET process was invalidated for its high energy barrier. Whereas, an amino radical was formed after the initial H-abstraction from nitrogen atom, the energy barrier involved was 0.6/0.4 kcal/mol for two spin states, and then dichotomous behaviors were encountered again. Owing to the steric hindrance caused by the hydrogen-bonding between O-H and N on IM, the reaction would primarily proceed through the rapid ring-opening rather than the O-rebound process to generate a C-centered radical species. Such carbon radical species may not only subsequently convert to 3HP, which was identified as the major product experimentally, but essentially account for the inactivation by covalent binding to amino residue. Intriguingly, in addition to the extremely low energy barrier of the H-abstraction in inactivation pathway, the spin density distribution on “substrate” moiety is approaching ±1, exhibiting several PCET characters. Furthermore, the SNOs for the transition state in such H-abstraction together with the analysis of spin densities on “substrate” and “porpine+thiolate” moieties along the reaction coordinate definitely demonstrate a PCET(ET) mechanism.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by National Natural science Foundation of China (Grant no. 21003116, 21173211 and 11274096), Innovation Scientists and Technicians Troop Construction Projects of Henan Province (Grant no. 124200510013) and Innovative Research Team in Science and Technology in University of Henan Province (Grant no. 13IRTSTHN016).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fchem.2017.00003/full#supplementary-material
References
Augusto, O., Beilan, H. S., and Demontellano, P. R. (1982). The catalytic mechanism of cytochrome-P-450. Spin-trapping evidence for one-electron substrate oxidation. J. Biol. Chem. 257, 1288–1295.
Becke, A. D. (1992a). Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 96, 2155–2160. doi: 10.1063/1.462066
Becke, A. D. (1992b). Density-functional thermochemistry. II. The effect of the Perdew-Wang generalized-gradient correlation correction. J. Chem. Phys. 97, 9173–9177. doi: 10.1063/1.463343
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Bhakta, M. N., Hollenberg, P. F., and Wimalasena, K. (2005). P450/NADPH/O2- and P450/PhIO-catalyzed N-dealkylations are mechanistically distinct. J. Am. Chem. Soc. 127, 1376–1377. doi: 10.1021/ja0436143
Bhakta, M. N., and Wimalasena, K. (2002). Microsomal P450-catalyzed N-dealkylation of N,N-dialkylanilines: evidence for a Cα-H abstraction mechanism. J. Am. Chem. Soc. 124, 1844–1845. doi: 10.1021/ja011041d
Burka, L. T., Guengerich, F. P., Willard, R. J., and Macdonald, T. L. (1985). Mechanism of cytochrome P-450 catalysis. Mechanism of N-dealkylation and amine oxide deoxygenation. J. Am. Chem. Soc. 107, 2549–2551. doi: 10.1021/ja00294a057
Cerny, M. A., and Hanzlik, R. P. (2005). Cyclopropylamine inactivation of cytochromes P450: role of metabolic intermediate complexes. Arch. Biochem. Biophy. 436, 265–275. doi: 10.1016/j.abb.2005.02.020
Cerny, M. A., and Hanzlik, R. P. (2006). Cytochrome P450-catalyzed oxidation of N-benzyl-N-cyclopropylamine generates both cyclopropanone hydrate and 3-hydroxypropionaldehyde via hydrogen abstraction, not single electron transfer. J. Am. Chem. Soc. 128, 3346–3354. doi: 10.1021/ja054938+
de Visser, S. P., Kumar, D., Cohen, S., Shacham, R., and Shaik, S. (2004). A predictive pattern of computed barriers for C-H hydroxylation by compound I of cytochrome P450. J. Am. Chem. Soc. 126, 8362–8363. doi: 10.1021/ja04858h
de Visser, S. P., Ogliaro, F., Sharma, P. K., and Shaik, S. (2002). What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 124, 11809–11826. doi: 10.1021/ja026872d
de Visser, S. P., Quesne, M. G., Martin, B., Comba, P., and Ryde, U. (2014). Computational modelling of oxygenation processes in enzymes and biomimetic model complexes. Chem. Commun. 50, 262–282. doi: 10.1039/c3cc47148a
de Visser, S. P., and Tan, L. S. (2008). Is the bound substrate in nitric oxide synthase protonated or netural and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 130, 12961–12974. doi: 10.1021/ja8010995
Dinnocenzo, J. P., Karki, S. B., and Jones, J. P. (1993). On isotope effects for the cytochrome P-450 oxidation of substituted N,N-dimethylanilines. J. Am. Chem. Soc. 115, 7111–7116. doi: 10.1021/ja00069a007
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2004). Gaussian 03, Revision E.01. Wallingford, CT: Gaussian, Inc.
Guengerich, F. P., Willard, R. J., Shea, J. P., Richards, L. E., and Macdonald, T. L. (1984). Mechanism-based inactivation of cytochrome P-450 by heteroatom-substituted cyclopropanes and formation of ring-opened products. J. Am. Chem. Soc. 106, 6446–6447. doi: 10.1021/ja00333a071
Guengerich, F. P., Yun, C. H., and Macdonald, T. L. (1996). Evidence for a 1-electron oxidation mechanism in N-dealkylation of N,N-dialkylanilines by cytochrome P450 2B1. Kinetic hydrogen isotope effects, linear free energy relationships, comparisons with horseradish peroxidase, and studies with oxygen surrogates. J. Biol. Chem. 271, 27321–27329.
Hanzlik, R. P., Kishore, V., and Tullman, R. (1979). Cyclopropylamines as suicide substrates for cytochromes P-450. J. Med. Chem. 22, 759–761. doi: 10.1021/jm00193a002
Hanzlik, R. P., and Tullman, R. H. (1982). Suicidal inactivation of cytochrome P-450 by cyclopropylamines. Evidence for cation-radical intermediates. J. Am. Chem. Soc. 104, 2048–2050. doi: 10.1021/ja00371a055
Hay, P. J., and Wadt, W. R. (1985). Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310. doi: 10.1063/1.448975
Hirao, H., and Chuanprasit, P. (2015). An attempt to evaluate the effect of proton-coupled electron transfer on the H-abstraction step of the reaction between 1,1-dimethylhydrazine and cytochrome P450 compound I. Chem. Phys. Lett. 621, 188–192. doi: 10.1016/j.cplett.2014.12.027
Hirao, H., Chuanprasit, P., Cheong, Y. Y., and Wang, X. (2013a). How is a metabolic intermediate formed in the mechanism-based inactivation of cytochrome P450 by using 1,1-dimethylhydrazine: hydrogen abstraction or nitrogen oxidation? Chem. Eur. J. 19, 7361–7369. doi: 10.1002/chem.201300689
Hirao, H., Thellamurege, N. M., Chuanprasit, P., and Xu, K. (2013b). Importance of H-abstraction in the final step of nitrosoalkane formation in the mechanism-based inactivation of cytochrome P450 by amine-containing drugs. Int. J. Mol. Sci. 14, 24692–24705. doi: 10.3390/ijms141224692
Ji, L., Faponle, A. S., Quesne, M. G., Sainna, M. A., Zhang, J., Franke, A., et al. (2015). Drug metabolism by cytochrome P450 enzymes: what distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 21, 9083–9092. doi: 10.1002/chem.201500329
Jones, J. P., Mysinger, M., and Korzekwa, K. R. (2002). Computational models for cytochrome P450: a predictive electronic model for aromatic oxidation and hydrogen atom abstraction. Drug Metab. Dispos. 30, 7–12. doi: 10.1124/dmd.30.1.7
Karki, S. B., and Dinnocenzo, J. P. (1995). On the mechanism of amine oxidations by P450. Xenobiotica 25, 711–724. doi: 10.3109/00498259509061887
Karki, S. B., Dinnocenzo, J. P., Jones, J. P., and Korzekwa, K. R. (1995). Mechanism of oxidative amine dealkylation of substituted N,N-dimethylanilines by cytochrome P-450: application of isotope effect profiles. J. Am. Chem. Soc. 117, 3657–3657. doi: 10.1021/ja00118a001
Korzekwa, K. R., Jones, J. P., and Gillette, J. R. (1990). Theoretical studies on cytochrome P-450 mediated hydroxylation: a predictive model for hydrogen-atom abstractions. J. Am. Chem. Soc. 112, 7042–7046. doi: 10.1021/ja00175a040
Kumar, D., de Visser, S. P., and Shaik, S. (2003). How does product isotope effect prove the operation of a two-state “rebound” mechanism in C-H hydroxylation by cytochrome P450? J. Am. Chem. Soc. 125, 13024–13025. doi: 10.1021/ja036906x
Kumar, D., de Visser, S. P., and Shaik, S. (2004). Oxygen economy of cytochrome P450: what is the origin of the mixed functionality as a dehydrogenase-oxidase enzyme compared with its normal function? J. Am. Chem. Soc. 126, 5072–5073. doi: 10.1021/ja0318737
Kumar, D., Latifi, R., Kumar, S., Rybak-Akimova, E. V., Sainna, M. A., and de Visser, S. P. (2013). Rationalization of the barrier height for p-Z-styrene epoxidation by iron(IV)-oxo porphyrin cation radicals with variable axial ligands. Inorg. Chem. 52, 7968–7979. doi: 10.1021/ic4005104
Lalevée, J., Allonas, X., and Fouassier, J. P. (2002). N-H and α(C-H) bond dissociation enthalpies of aliphatic amines. J. Am. Chem. Soc. 124, 9613–9621. doi: 10.1021/ja0204168
Lee, C. T., Yang, W. T., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B Condens. Matter 37, 785–789. doi: 10.1103/PhysRevB.37.785
Li, C. S., Wu, W., Kumar, D., and Shaik, S. (2006). Kinetic isotope effect is a sensitive probe of spin state reactivity in C-H hydroxylation of N,N-dimethylaniline by cytochrome P450. J. Am. Chem. Soc. 128, 394–395. doi: 10.1021/ja055987p
Li, D. M., Wang, Y., and Han, K. L. (2012). Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coor. Chem. Rev. 256, 1137–1150. doi: 10.1016/j.ccr.2012.01.016
Li, X. -X., Zhang, X., Zheng, Q. -C., and Wang, Y. (2015). Bio-activation of 4-alkyl analogs of 1,4-dihydropyridine mediated by cytochrome P450 enzymes. J. Biol. Inorg. Chem. 20, 665–673. doi: 10.1007/s00775-015-1252-8
Luo, Y. -R. (2002). Handbook of Bond Dissociation Energies in Organic Compounds. Boca Raton, FL: CRC press.
Macdonald, T. L., Zirvi, K., Burka, L. T., Peyman, P., and Guengerich, F. P. (1982). Mechanism of cytochrome P-450 inhibition by cyclopropylamines. J. Am. Chem. Soc. 104, 2050–2052. doi: 10.1021/ja00371a056
Manchester, J. I., Dinnocenzo, J. P., Higgins, L. A., and Jones, J. P. (1997). A new mechanistic probe for cytochrome P450: an application of isotope effect profiles. J. Am. Chem. Soc. 119, 5069–5070. doi: 10.1021/ja9705250
Mayer, J. M., Hrovat, D. A., Thomas, J. L., and Borden, W. T. (2002). Proton-coupled electron transfer versus hydrogen atom transfer in benzyl/toluene, methoxyl/methanol, and phenoxyl/phenol self-exchange reactions. J. Am. Chem. Soc. 124, 11142–11147. doi: 10.1021/ja012732c
Melander, L., and Saunders, W. H. (1987). Reaction Rates of Isotopic Molecules. Malabar, FL: RE Krieger Pub. Co.
Meunier, B., de Visser, S. P., and Shaik, S. (2004). Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 104, 3947–3980. doi: 10.1021/cr020443g
Miwa, G. T., Walsh, J. S., Kedderis, G. L., and Hollenberg, P. F. (1983). The use of intramolecular isotope effects to distinguish between deprotonation and hydrogen-atom abstraction mechanisms in cytochrome P-450-catalyzed and peroxidase-catalyzed N-demethylation reactions. J. Biol. Chem. 258, 4445–4449.
Nehru, K., Seo, M. S., Kim, J., and Nam, W. (2007). Oxidative N-dealkylation reactions by oxoiron(IV) complexes of nonheme and heme ligands. Inorg. Chem. 46, 293–298. doi: 10.1021/ic0614014
Ogliaro, F., Harris, N., Cohen, S., Filatov, M., de Visser, S. P., and Shaik, S. (2000). A model “rebound” mechanism of hydroxylation by cytochrome P450: stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 122, 8977–8989. doi: 10.1021/ja991878x
Oh, N. Y., Suh, Y., Park, M. J., Seo, M. S., Kim, J., and Nam, W. (2005). Mechanistic insight into alcohol oxidation by high-valent iron-oxo complexes of heme and nonheme ligands. Angew. Chem. Int. Ed Engl. 44, 4235–4239. doi: 10.1002/anie.200500623
Olsen, L., Rydberg, P., Rod, T. H., and Ryde, U. (2006). Prediction of activation energies for hydrogen abstraction by cytochrome P450. J. Med. Chem. 49, 6489–6499. doi: 10.1021/jm060551l
Orr, S. T. M., Ripp, S. L., Ballard, T. E., Henderson, J. L., Scott, D. O., Obach, R. S., et al. (2012). Mechanism-based inactivation (MBI) of cytochrome P450 enzymes: structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J. Med. Chem. 55, 4896–4933. doi: 10.1021/jm300065h.
Ortiz de Montellano, P. R., and De Voss, J. (2005). Cytochrome P450: Structure, Mechanism and Biochemistry. New York, NY: Klumer Academic/Plenum Publisher.
Ortiz de Montellano, P. R., and De Voss, J. J. (2002). Oxidizing species in the mechanism of cytochrome P450. Nat. Prod. Rep. 19, 477–493. doi: 10.1039/b101297p
Rydberg, P., and Olsen, L. (2011). Do two different reaction mechanisms contribute to the hydroxylation of primary amines by cytochrome P450? J. Chem. Theory Comput. 7, 3399–3404. doi: 10.1021/ct200422p
Schröder, D., Shaik, S., and Schwarz, H. (2000). Two-state reactivity as a new concept in organometallic chemistry. Acc. Chem. Res. 33, 139–145. doi: 10.1021/ar990028j
Shaffer, C. L., Morton, M. D., and Hanzlik, R. P. (2001a). Enzymatic N-dealkylation of an N-cyclopropylamine: an unusual fate for the cyclopropyl group. J. Am. Chem. Soc. 123, 349–350. doi: 10.1021/ja003048l
Shaffer, C. L., Morton, M. D., and Hanzlik, R. P. (2001b). N-dealkylation of an N-cyclopropylamine by horseradish peroxidase. Fate of the cyclopropyl group. J. Am. Chem. Soc. 123, 8502–8508. doi: 10.1021/ja0111479
Shaik, S., Cohen, S., Wang, Y., Chen, H., Kumar, D., and Thiel, W. (2010). P450 Enzymes: their structure, reactivity, and selectivity-modeled by QM/MM calculations. Chem. Rev. 110, 949–1017. doi: 10.1021/cr900121s
Shaik, S., Filatov, M., Schröder, D., and Schwarz, H. (1998). Electronic structure makes a difference: cytochrome P-450 mediated hydroxylations of hydrocarbons as a two-state reactivity paradigm. Chem. Eur. J. 4, 193–199. doi: 10.1002/(sici)1521-3765(19980210)4:2<193::aid-chem193>3.0.co;2-q
Shaik, S., Kumar, D., and de Visser, S. P. (2008). Valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 130, 10128–10140. doi: 10.1021/ja8019615
Shaik, S., Kumar, D., de Visser, S. P., Altun, A., and Thiel, W. (2005). Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 105, 2279–2328. doi: 10.1021/cr030722j
Sono, M., Roach, M. P., Coulter, E. D., and Dawson, J. H. (1996). Heme-containing oxygenases. Chem. Rev. 96, 2841–2887. doi: 10.1021/cr9500500
Totah, R. A., and Hanzlik, R. P. (2002). Non-oxidative decarboxylation of glycine derivatives by a peroxidase. J. Am. Chem. Soc. 124, 10000–10001. doi: 10.1021/ja020557u
Usharani, D., Lacy, D. C., Borovik, A. S., and Shaik, S. (2013). Dichotomous hydrogen atom transfer vs proton-coupled electron transfer during activation of X-H bonds (X = C, N, O) by nonheme iron-oxo complexes of variable basicity. J. Am. Chem. Soc. 135, 17090–17104. doi: 10.1021/ja408073m
Vanlier, J. E., and Rousseau, J. (1976). Mechanism of cholesterol side-chain cleavage: enzymic rearrangement of 20β-hydroperoxy-20-isocholesterol to 20β,21-dihydroxy-20-isocholesterol. FEBS Lett. 70, 23–27. doi: 10.1016/0014-5793(76)80718-1
Wang, Y., Kumar, D., Yang, C. L., Han, K. L., and Shaik, S. (2007a). Theoretical study of N-demethylation of substituted N,N-dimethylanilines by cytochrome P450: the mechanistic significance of kinetic isotope effect profiles. J. Phys. Chem. B 111, 7700–7710. doi: 10.1021/jp072347v
Wang, Y., Li, D. M., Han, K. L., and Shaik, S. (2010). An acyl group makes a difference in the reactivity patterns of cytochrome P450 catalyzed N-demethylation of substituted N,N-dimethylbenzamides-High spin selective reactions. J. Phys. Chem. B 114, 2964–2970. doi: 10.1021/jp9097974
Wang, Y., Wang, H. M., Wang, Y. H., Yang, C. L., Yang, L., and Han, K. L. (2006). Theoretical study of the mechanism of acetaldehyde hydroxylation by compound I of CYP2E1. J. Phys. Chem. B 110, 6154–6159. doi: 10.1021/jp060033m
Keywords: cytochrome P450, hydrogen atom transfer, proton-coupled electron transfer, suicide inhibition, mechanism-based inactivation
Citation: Zhang XQ, Li X-X, Liu YF and Wang Y (2017) Suicide Inhibition of Cytochrome P450 Enzymes by Cyclopropylamines via a Ring-Opening Mechanism: Proton-Coupled Electron Transfer Makes a Difference. Front. Chem. 5:3. doi: 10.3389/fchem.2017.00003
Received: 12 July 2016; Accepted: 10 January 2017;
Published: 31 January 2017.
Edited by:
Antonio Monari, Université de Lorraine, FranceReviewed by:
Sam P. De Visser, University of Manchester, UKYohann Moreau, Université Grenoble Alpes, France
Copyright © 2017 Zhang, Li, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Xi Li, bGl4aWFveGlAbGljcC5jYXMuY24=
Yong Wang, d2FuZ3lvbmdAbGljcC5jYXMuY24=