 Xuguang Gao
Xuguang Gao Qi Ren
Qi Ren Sun Choi
Sun Choi Zhengshuang Xu
Zhengshuang Xu Tao Ye
Tao Ye
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 17 March 2015
Sec. Chemical Biology
Volume 3 - 2015 | https://doi.org/10.3389/fchem.2015.00019
This article is part of the Research Topic Natural Product Synthesis View all 7 articles
The first total synthesis of four possible isomers of a molecule possessing the configuration proposed for Banyasin A is described. The structure synthesized appears to be different from that of the natural product.
Secondary metabolites produced by cyanobacteria are key synthetic targets in the quest for new leads in the pharmaceutical industry (Singh et al., 2011). One particular species, Nostoc sp., (Rawat and Bhargava, 2011; Niedermeyer, 2014) is responsible for the production of a considerable of bioactive cyclopeptide and cyclodepsipeptides with a variety of significant associated biological activities, including antiproliferative activities (Kang et al., 2012) antitoxin against Microcystins (Jokela et al., 2010), inhibition of chymotrypsin (Ploutno and Carmeli, 2002) antimicrobial activities (Ploutno and Carmeli, 2000), selective cytotoxicity (Trimurtulu et al., 1994). Some of the metabolic signatures of cyanobacterial peptides include a high degree of N-methylated amino acids, D-amino acids, and β-amino acids. We have been interested in marine secondary metabolites, especially the cyclopeptides and cyclodepsipeptides, and view their syntheses as playing a key role in structural confirmation, structural modification, and subsequent activity control. Previous work in our group led to the assignment/revision of a number of natural products including mandelalide A (Lei et al., 2014), lagunamide A (Dai et al., 2012), nocardioazine B (Wang et al., 2012), burkholdac A (Liu et al., 2012), bisebromoamide (Gao et al., 2010), and yanucamide A (Xu et al., 2003). Thus, we were encouraged to consider the synthesis of other natural products of uncertain configuration.
In 2005, Carmeli and Pluotno reported the isolation of banyasin A (1) (Figure 1), from cyanobacterium Nostoc sp. (Pluotno and Carmeli, 2005). Structurally, banyasin A possesses a 16-membered macrocyclic peptide backbone which is composed of the unique, non-proteinogenic amino acids residues such as 3-amino-2-methyl-5E-octenoic acid and L-N8-(N-methylcarboxyaminde)arginine. Both the relative stereochemistry and absolute configuration of 3-amino-2-methyl-5E-octeneoic acid were unassigned in the isolation paper. The final structural determination had to await the total synthesis of the four possible diastereomeric structures proposed for the natural product. Banyasin A was isolated followed by a serine protease inhibition-guided protocol, however, pure banyasin A was found to be inactive toward trypsin inhibition. Banyasin A is structurally closely related to the known chitinase inhibitors argifin and argadin (Figure 1) (Arai et al., 2000; Omura et al., 2000). All these three natural products contained a highly modified arginine at the guanidine moiety. It is known that the dimethylguanylurea fragment of argifin was found to harbor all significant interactions with the chitinase and binds with unusually high efficiency. (Andersen et al., 2008). L-N8-(N-methylcarboxyaminde)arginine moiety played the critical role in binding of argifin to its cognate target (Dixon et al., 2009). The 16-membered macrolactam ring of banyasin A resemble the macrocyclic core of the known chitinase inhibitor argifin. Both banyasin A and argifin are comprised of L-N8-(N-methylcarboxyaminde)arginine residue. Modeling studies based on conformational analysis and manual docking yielded a binding model of banyasin A to a fungal chitinase (AfChiB1) active site, which has the most similar binding mode of argifin to AfChiB1 (Rao et al., 2005). As a representative compound, 1b was displayed in Figure 2. The rest binding modes of banyasin's diastereomers (1a, 1c, and 1d) in AfChiB1 active site were included in support information. Taken together, these studies implied that banyasin A may serve as a potential lead compound for the development of antifungals and pesticides. The limited supply of banyasin A from its natural source has prevented its full biological characterization. In order to obtain sufficient material for more extensive biological evaluation as well as to determine the absolute configuration of banyasin A, we undertook research to develop a total synthesis of banyasin A with flexibility to enable future SAR development. Herein, we describe our efforts toward the total synthesis of banyasin A.
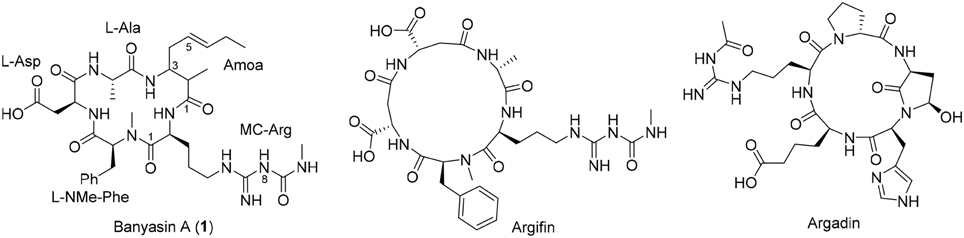
Figure 1. The Structures of banyasin A (1), argifin and argadin.
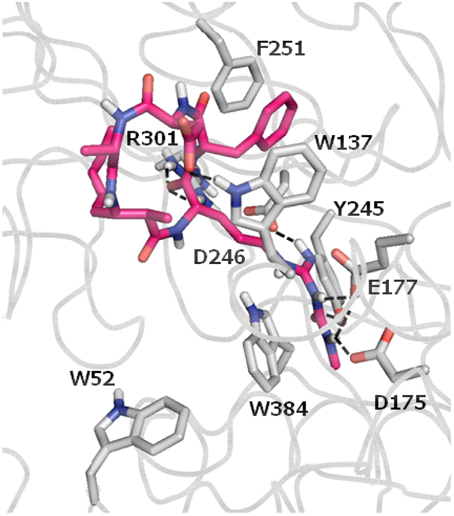
Figure 2. Binding mode of banyasin A in AfChiB1 active site. Banyasin A (1b) (magenta carbon atoms) and the key interacting residues (gray carbon atoms) are shown in capped-sticks. Hydrogen bonds are displayed as black dashed lines, and non-polar hydrogens are removed for clarity.
The retrosynthetic analysis of banyasin A (1) is illustrated in Figure 3. The N-methyl carbamoyl modification of the Arg residue will be conducted as the final step of the synthesis. There are a number of possible positions to close the macrocyclic portion (2) of the molecule (Figure 2). We chose to close the cyclopeptide via macrolactamization between the alanine and amoa residues because it allowed versatility in the construction of the cyclization precursor (3). Because the absolute configuration of the 3-amino-2-methyl-5E-octenoic acid residue was not established, the incorporation of four possible diastereomers of the 3-amino-2-methyl-5E-octenoic acid residue to afford linear precursor 3 could be exerted in the late stage. Further disconnection of the linear peptide 3 revealed amoa unit (4) and a tetrapeptide equivalent, the latter intermediate being traced to the protected amino acids 5, 6, 7, and 8. (Figure 3).
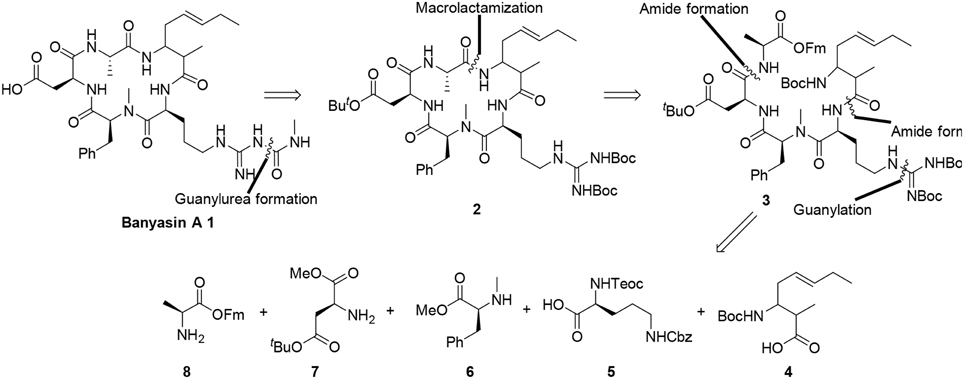
Figure 3. Retrosynthetic analysis of banyasin A (1).
Experimental procedure and compound characterization data are furnished in the Supplementary Material.
The synthesis commenced from crotylation of 3-benzyloxy-propionaldehyde (9) with Brown's (E)-(+)-crotyldiisopinocampheylborane, prepared from (+)-diisopinocampheyl(methoxy)borane, and yielded the anti-homoallylic alcohol 10 as a single diastereomer after silica gel chromatography (Figure 4). (Brown and Bhat, 1986) The homoallylic alcohol was then converted to its mesylate derivative, followed by exposure of the resulting mesylate to sodium azide in DMF furnished, with inversion of configuration, the corresponding azido derivative 11 in 59% overall yield from aldehyde 9. The azide moiety of 11 was reduced by the action of triphenylphosphine to afford the corresponding primary amine, which was then protected as a Boc carbamate (12) in 94% yield over two steps. Oxidative cleavage of the olefin of 12 using OsO4–NaIO4 in the presence of 2,6-lutidine afforded an aldehyde, which was further oxidized to the corresponding carboxylic acid by Pinnick oxidation. A subsequent methylation of the carboxylic group using iodomethane and potassium carbonate gave the methyl ester 13 in 72% yield over the three steps. Subsequent removal of the benzyl ether under hydrogenative conditions followed by Dess-Martin oxidation of the resultant primary alcohol gave rise to aldehyde 14 in 79% yield. The Kocienski-modified Julia olefination of aldehyde 14 with sulfone 15 in THF at −78°C delivered olefin 16 as an inseparable mixture of olefin isomers (4:1) in favor of the desired E configuration. (Blakemore et al., 1998). Subsequent saponification of the methyl ester gave 3-amino-2-methyl-5E-octenoic acid 4a in 97% yield. Further incorporation of 4a in the construction of the macrocycle reveled the undesired geometrical isomer could not be completely removed in the later stage of the synthesis. Bearing in mind the problems encountered with the formation of an inseparable mixture of olefin isomers, we required an alternative approach which could efficiently deliver 3-amino-2-methyl-5E-octenoic acid 4 in fairly high purity. Thus, the homoallylic alcohol 10 was protected as its tert-butyldimethylsilyl ether and the terminal alkene underwent a substrate-controlled dihydroxylation in the presence of osmium tetroxide to provide 18 as a diastereomeric mixture (3.7:1) of diols in 88% combined yields (Figure 5). This mixture was simply exposed to 2,2-dimethoxypropane under acidic conditions to furnish the corresponding acetonide, which was then subjected to hydrogenolysis of the benzyl ether to give rise to 19 in 86% yield for two steps. Dess-Martin oxidation of 19 provided crude aldehyde, which was treated with the anion derived from sulfone 15 and KHMDS. When the olefination was performed in 1,2-dimethoxyethane (DME) at −68°C, we obtained E/Z mixtures (13/1) of olefins 20 in 83% yield. Removal of the TBS group in 20 using TBAF and the resultant secondary hydroxy group engaged in Mitsunobu reactions using diphenylphosphoryl azide (DPPA) (Lal et al., 1977) as the nucleophile to give rise to azide 21 in 78% yield. Acid-catalyzed deprotection of the acetonide functionality of 21, followed by oxidative cleavage of the resulting diol with sodium periodate, followed by Pinnick oxidation (Bal et al., 1981) afforded the carboxylic acid 22a in 79% yield over three steps. As the absolute configuration of two stereogenic centers in the subunit 4 was not assigned in the original isolation work, our synthetic strategy grew from the desire to have a versatile approach to make all four possible diastereomers of 4, along with the defined stereochemistry of the disubstituted olefin component. With a practical route to 22a in hand, we synthesized the other three possible diastereomers (22b, 22c, and 22d) through the condensation of aldehyde 9 with Brown's (E)-(−)-crotyldiisopinocampheylborane, (Z)-(+)-crotyldiisopinocampheylborane and (Z)-(−)-crotyldiisopinocampheylborane, respectively. Azido acid 22a was then subjected to hydrogenation in the presence of the palladium on charcoal and the resulting amine was protected as its tert-butoxycarbonyl carbamate 4a.
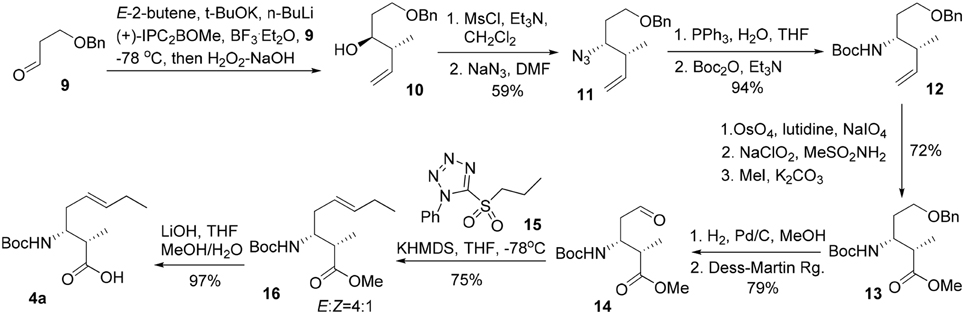
Figure 4. Initial route for the synthesis of 3-amino-2-methyl-5E-octenoic acid 4.
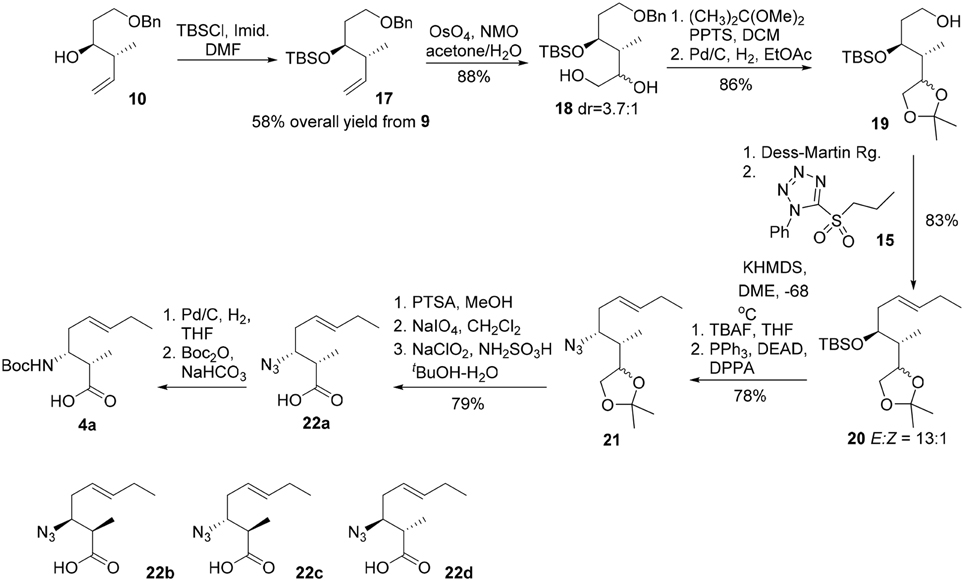
Figure 5. Revised route for the synthesis of 3-amino-2-methyl-5E-octenoic acid 4.
With the critical amoa fragment in hand, we then turned our attention to the assembly of the remaining amino acids to a tetrapeptide and further elaboration to the linear precursor 3. Thus, the 2-(trimethylsilyl)-ethoxycarbonyl (Teoc) protecting group was installed on the the alpha amino group of N-δ-Cbz-L-ornithine to afford the orthogonally protected ornithine 5 in 81% yield, which underwent a HATU-mediated coupling reaction with N-methyl-L-phenylalanine methyl ester (6) to provide dipeptide 24 in 59% yield (Figure 6) (Carpino, 1993). Saponification of the methyl ester of 24 produced the corresponding acid, which was then condensed with aspartate 7 in the presence of HATU and triethylamine to provide tripeptide 25 in 83% yield. Tripeptide 25 was submitted to catalytic hydrogenation to remove the benzyl protecting group, and the guanidine function was introduced by reaction with 1,3-di-Boc-2-(trifluoromethanesulfonyl)guanidine (26) (Feichtinger et al., 1998) to give 27 in 91% yield. Tripeptide 27 was further elongated to tetrapeptide 28 in 61% yield by a two step sequence, involving (1) hydrolysis of the methyl ester to its free acid, and (2) HATU-mediated condensation of the acid with L-alanine 9-fluorenylmethyl ester (8). This set the stage for the introduction of the 3-amino-2-methyl-5E-octenoic acid 4 to complete the synthesis of linear precursor 3. In the event, the Teoc protecting group of 28 was removed in one step using TBAF in THF to provide the corresponding free amine. Unfortunately, condensation of this amine with acid 4a using various coupling agents, including HATU, PyAOP, DCC and EDCI, turned out to be relatively slow and led to the formation of 3a with significant epimerization at the center adjacent to the newly formed amide carbonyl. In general, formation of 20–35% of undesired epimer could be observed during the coupling reaction depending on the reagent employed. We speculated that the conformation of the tetrapeptide 28 may significantly affect the reactivity of the amine and resulted in a slow condensation reaction. Despite this significant setback to our synthetic plan toward banyasin A, we sought that to install the 3-amino-2-methyl-5E-octenoic acid unit (4 or its masked form 22) to the peptide at earlier stage might avoid epimerization of the C2-methyl group. Gratifyingly, HATU-mediated coupling reaction of azido acid 22a with the amine derived from tripeptide 27 afforded 29a in 93% yield. That no epimerization had occurred during this coupling reaction was confirmed by synthesizing the corresponding epimeric material through the condensation of the same amine with azide acid 22c. Saponification of the methyl ester of 29a and subsequent condensation of the resultant acid with L-alanine 9-fluorenylmethyl ester (8) afforded 30a in 82% yield. Of the various reduction protocols that were examined for converting azido acid 30a into the corresponding amine, the Staudinger reduction with PMe3 in THF:H2O (7:1) proved the most successful. In the event, the Staudinger reduction of the azide and the cleavage of 9-fluorenylmethyl ester with aqueous NH4OH were carried out in a one-pot procedure to afford the resultant amino acid, which was immediately activated by diethyl cyanophosphonate (DEPC) (Yamada et al., 1973) in the presence of collidine to afford cyclopeptide 2a in 56% yield over two steps. Simultaneous removal of the tert-butyl ester and Boc-protecting group was achieved by treatment of 2a with trifluoroacetic acid in dichloromethane at room temperature. The guanidino group of the resultant cyclopeptide was mono-acylated with N-succinimidyl N-methylcarbamate (31) in the presence of DBU to afford banyasin A 1a in 66% yield. (Dixon et al., 2005) Having one diastereomer (1a), and a practical route to bayansin A in hand, we set out to pursue total syntheses of the other three possible diastereomers. This was readily achieved by following the same synthetic procedure as for 1a, by employing azido acids 22b, 22c, and 22d as key building blocks. These syntheses proceeded smoothly under the previous conditions to give rise to three diastereomers of banyasin A, 1b, 1c, and 1d in 28, 27, and 30% overall yield from tripeptide 27 (Figure 7).
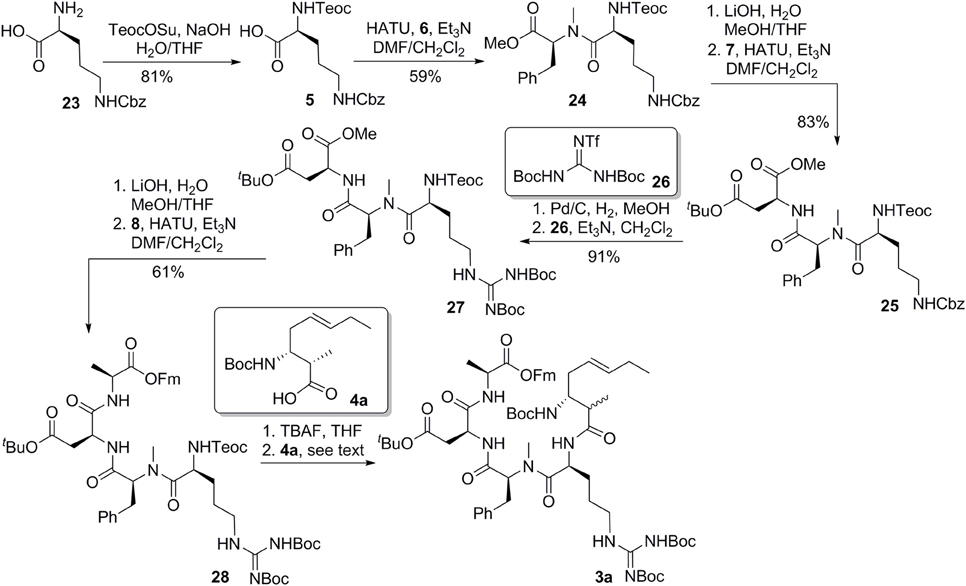
Figure 6. Synthesis of the linear precursor 3.
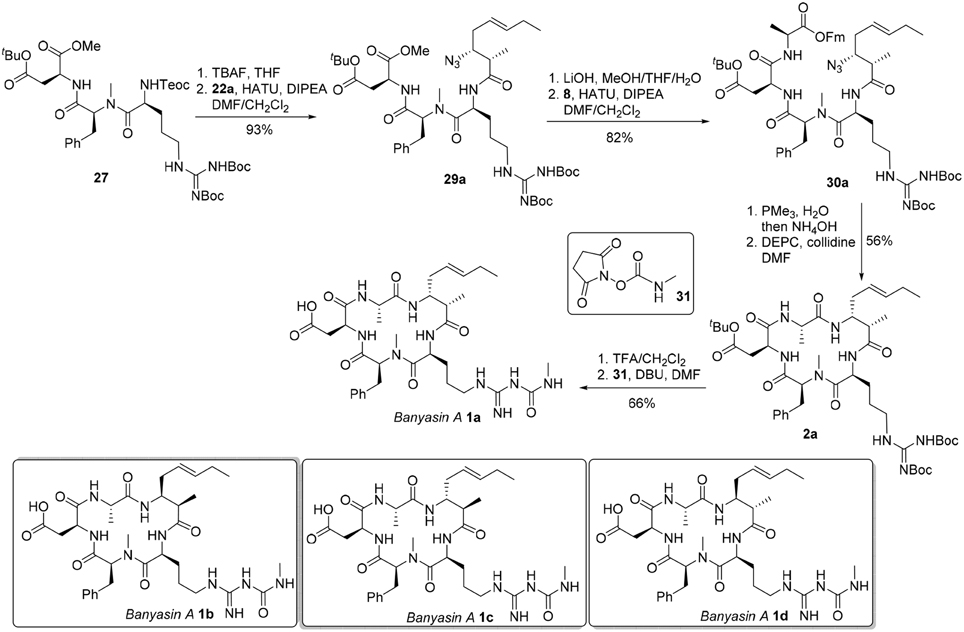
Figure 7. Completion of the total synthesis of banyasin A (1).
We have developed a convergent route for the synthesis of the proposed structure of banyasin A. The optical rotation measured for synthetic banyasin A 1a–d ([α]20D = −45.6 (c = 0.70, MeOH); [α]20D = −59.4 (c = 0.70, MeOH); [α]20D = −37.3 (c = 0.70, MeOH); and [α]20D = −53.1 (c = 0.70, MeOH); were identical or nearly identical to that reported for the natural product [α]20D = −45.3 (c = 0.7, MeOH). To our disappointment, both the 1H and 13C spectroscopic properties of each diastereomer 1a, 1b, 1c, and 1d were different from those of banyasin A (Figure 8 and Supplementary Material). It would appear that the error in the original assignment of the configuration of banyasin A lies somewhere in the remaining α-aminoacids. The extension of this chemistry toward the synthesis of additional diastereomers aiming to resolve the true structure of natural banyasin A and synthesis of novel analogs of banyasin for biological evaluation are underway in our laboratory.
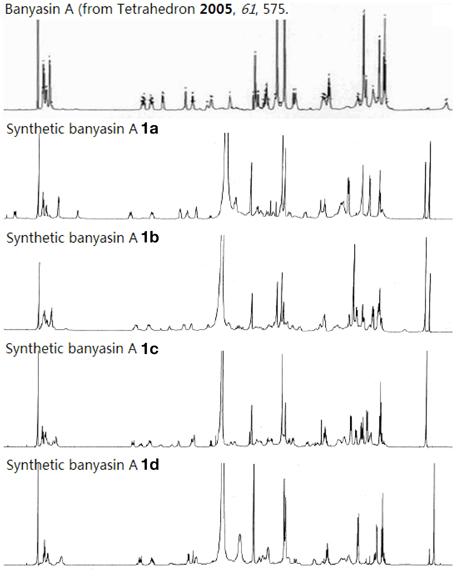
Figure 8. Comparison of 1H NMR of natural and synthetic banyasin A (1a–d).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge financial support from the National Natural Science Foundation of China (21272011, 21133002) and Shenzhen Science and Technology Development Fund (JCYJ20130329175740481); Hong Kong Research Grants Council (Projects: PolyU 5037/11P, 5020/12P; 5030/13P, 153035/14P); Fong Shu Fook Tong Foundation and Joyce M. Kuok Foundation. Part of this work was supported by grants from National Leading Research Lab (NLRL) program (2011-0028885) funded by the Ministry of Science, ICT and Future Planning (MSIP) and the National Research Foundation of Korea (NRF) (to SC). This work was also inspired by the international and interdisciplinary environments of the JSPS Asian CORE Program, “Asian Chemical Biology Initiative.”
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fchem.2015.00019/abstract
Andersen, O. A., Nathubhai, A., Dixon, M. J., Eggleston, I. M., and van Aalten, D. M. (2008). Structure-based dissection of the natural product cyclopentapeptide chitinase inhibitor argifin. Chem Biol. 15, 295–301. doi: 10.1016/j.chembiol.2008.02.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arai, N., Shiomi, K., Yamaguchi, Y., Masuma, R., Iwai, Y., Turberg, A., et al. (2000). Argadin, a new chitinase inhibitor, produced by clonostachys sp. fo-7314. Chem. Pharm. Bull. (Tokyo) 48, 1442–1446. doi: 10.1248/cpb.48.1442
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bal, B. S., Childers, W. E., and Pinnick, H. W. (1981). Oxidation of α,β-unsaturated aldehydes. Tetrahedron 37, 2091–2096.
Blakemore, P. R., Cole, W. J., Kocienski, P. J., and Morley, A. (1998). A stereoselective synthesis of trans-1,2-disubstituted alkenes based on the condensation of aldehydes with metallated 1-phenyl-1H-tetrazol-5-yl sulfones. Synlett. 1998, 26–28. doi: 10.1055/s-1998-1570
Brown, H. C., and Bhat, K. S. (1986). Enantiomeric Z- and E-crotyldiisopinocampheylboranes. Synthesis in high optical purity of all four possible stereoisomers of β-methylhomoallyl alcohols. J. Am. Chem. Soc. 108, 293–294. doi: 10.1021/ja00262a017
Carpino, L. A. (1993). 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 115, 4397–4398. doi: 10.1021/ja00063a082
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dai, L., Chen, B., Lei, H., Wang, Z., Liu, Y., Xu, Z., et al. (2012). Total synthesis and stereochemical revision of lagunamide A. Chem. Commun. 48, 8697–8699. doi: 10.1039/c2cc34187e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dixon, M. J., Andersen, O. A., van Aalten, D. M. F., and Eggleston, I. M. (2005). An efficient synthesis of argifin: a natural product chitinase inhibitor with chemotherapeutic potential. Bioorg. Med. Chem. Lett. 15, 4717–4721. doi: 10.1016/j.bmcl.2005.07.068
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dixon, M. J., Nathubhai, A., Andersen, O. A., van Aalten, D. M. F., and Eggleston, I. M. (2009). Solid-phase synthesis of cyclic peptide chitinase inhibitors: SAR of the argifin scaffold. Org. Biomol. Chem. 7, 259–268. doi: 10.1039/b815077j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Feichtinger, K., Zapf, C., Sings, H. L., and Goodman, M. (1998). Diprotected triflylguanidines: a new class of guanidinylation reagents. J. Org. Chem. 63, 3804–3805. doi: 10.1021/jo980425s
Gao, X., Liu, Y., Kwong, S., Xu, Z., and Ye, T. (2010). Total synthesis and stereochemical reassignment of bisebromoamide. Org. Lett. 12, 3018–3021. doi: 10.1021/ol101021v
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jokela, J., Herfindal, L., Wahlsten, M., Permi, P., Selheim, F., Vasconçelos, V., et al. (2010). A novel cyanobacterial nostocyclopeptide is a potent antitoxin against microcystins. ChemBioChem 11, 1594–1599. doi: 10.1002/cbic.201000179
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, H.-S., Santarsiero, B. D., Kim, H., Krunic, A., Shen, Q., Swanson, S. M., et al. (2012). Merocyclophanes A and B, antiproliferative cyclophanes from the cultured terrestrial Cyanobacterium Nostoc sp. Phytochemistry 79, 109–115. doi: 10.1016/j.phytochem.2012.03.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lal, B., Pramanik, B. N., Manhas, M. S., and Bose, A. K. (1977). Diphenylphosphoryl azide a novel reagent for the stereospecific synthesis of azides from alcohols. Tetrahedron Lett. 18, 1977–1980. doi: 10.1016/S0040-4039(01)83657-1
Lei, H., Yan, J., Yu, J., Liu, Y., Wang, Z., Xu, Z., et al. (2014). Total synthesis and stereochemical reassignment of mandelalide A. Angew. Chem. Int. Ed. 53, 6533–6537. doi: 10.1002/anie.201403542
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, J., Ma, X., Liu, Y., Wang, Z., Kwong, S., Ren, Q., et al. (2012). Total synthesis and stereochemical revision of burkholdac A. Synlett 19, 783–787. doi: 10.1055/s-0031-1290339
Niedermeyer, T. H. J. (2014). Biologically active natural substances isolated from cyanobacteria of the genus Nostoc. BIOspektrum 20, 151–153. doi: 10.1007/s12268-014-0420-7
Omura, S., Arai, N., Yamaguchi, Y., Masuma, R., Iwai, Y., Namikoshi, M., et al. (2000). Argifin, a new chitinase inhibitor, produced by gliocladium sp. FTD-0668. i. taxonomy, fermentation, and biological activities. J. Antibiot. (Tokyo) 53, 603–608. doi: 10.7164/antibiotics.53.603
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ploutno, A., and Carmeli, S. (2000). Nostocyclyne A, a Novel antimicrobial cyclophane from the cyanobacterium Nostoc sp. J. Nat. Prod. 63, 1524–1526. doi: 10.1021/np0002334
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ploutno, A., and Carmeli, S. (2002). Modified peptides from a water bloom of the cyanobacterium Nostoc sp. Tetrahedron 58, 9949–9957. doi: 10.1016/S0040-4020(02)01326-1
Pluotno, A., and Carmeli, S. (2005). Banyasin A and banyasides A and B, three novel modified peptides from a water bloom of the cyanobacterium Nostoc sp. Tetrahedron 61, 575–583. doi: 10.1016/j.tet.2004.11.016
Rao, F. V., Houston, D. R., Boot, R. G., Aerts, J. M. F. G., Hodkinson, M., Adams, D. J., et al. (2005). Specificity and affinity of natural product cyclopentapeptide inhibitors against A. fumigatus, human, and bacterial chitinases. Chem. Biol. 12, 65–76. doi: 10.1016/j.chembiol.2004.10.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rawat, D., and Bhargava, S. (2011). Bioactive compounds from Nostoc species. Curr. Res. Pharm. Sci. 2, 48–54.
Singh, R. K., Tiwari, S. P., Rai, A. K., and Mohapatra, T. M. (2011). Cyanobacteria: an emerging source for drug discovery. J. Antibiot. 64, 401–412. doi: 10.1038/ja.2011.21
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Trimurtulu, G., Ohtani, I., Patterson, G. M. L., Moore, R. E., Corbett, T. H., Valeriote, F. A., et al. (1994). Total structures of cryptophycins, potent antitumor depsipeptides from the blue-green alga Nostoc sp. Strain GSV 224. J. Am. Chem. Soc. 116, 4729–4737. doi: 10.1021/ja00090a020
Wang, M., Feng, X., Cai, L., Xu, Z., and Ye, T. (2012). Total synthesis and absolute configuration of nocardioazine B. Chem. Commun. 48, 4344–4346. doi: 10.1039/c2cc31025b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, Z., Peng, Y., and Ye, T. (2003). The total synthesis and stereochemical revision of yanucamide A. Org. Lett. 5, 2821–2824. doi: 10.1021/ol034803d
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Banyasin A, total synthesis, stereoselective, cyclopeptide, chitinase inhibitors
Citation: Gao X, Ren Q, Choi S, Xu Z and Ye T (2015) Total synthesis of the putative structure of the proposed Banyasin A. Front. Chem. 3:19. doi: 10.3389/fchem.2015.00019
Received: 12 January 2015; Paper pending published: 28 January 2015;
Accepted: 02 March 2015; Published: 17 March 2015.
Edited by:
Bastien Nay, Centre National de la Recherche Scientifique, FranceReviewed by:
Fang-Rong Chang, Kaohsiung Medical University, TaiwanCopyright © 2015 Gao, Ren, Choi, Xu and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengshuang Xu, Laboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, 518055, ChinaeHV6c0Bwa3Vzei5lZHUuY24=;
Tao Ye, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong, ChinadGFvLnllQHBvbHl1LmVkdS5oaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.