 Hajime Hirao
Hajime Hirao Nandun Thellamurege
Nandun Thellamurege Xi Zhang
Xi Zhang- Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore, Singapore
The past decades have seen an explosive growth in the application of density functional theory (DFT) methods to molecular systems that are of interest in a variety of scientific fields. Owing to its balanced accuracy and efficiency, DFT plays particularly useful roles in the theoretical investigation of large molecules. Even for biological molecules such as proteins, DFT finds application in the form of, e.g., hybrid quantum mechanics and molecular mechanics (QM/MM), in which DFT may be used as a QM method to describe a higher prioritized region in the system, while a MM force field may be used to describe remaining atoms. Iron-containing molecules are particularly important targets of DFT calculations. From the viewpoint of chemistry, this is mainly because iron is abundant on earth, iron plays powerful (and often enigmatic) roles in enzyme catalysis, and iron thus has the great potential for biomimetic catalysis of chemically difficult transformations. In this paper, we present a brief overview of several recent applications of DFT to iron-containing non-heme synthetic complexes, heme-type cytochrome P450 enzymes, and non-heme iron enzymes, all of which are of particular interest in the field of bioinorganic chemistry. Emphasis will be placed on our own work.
Introduction
Density functional theory (DFT) has been playing increasingly important roles in many research activities of science and engineering in recent decades and has already become a mainstay for the quantum mechanical investigations of a broad range of complex molecular systems that are of interest in chemistry, biology, and physics (Parr and Yang, 1989; Kohn et al., 1996; Baerends and Gritsenko, 1997; Kohn, 1999; Koch and Holthausen, 2001; Zhao and Truhlar, 2008; Perdew et al., 2009; Burke, 2012; Cohen et al., 2012). DFT offers viable computational protocols with a good balance between accuracy and computational cost. This feature is particularly useful when one intends to investigate large molecular systems, to which the application of accurate ab initio methods may be difficult or even impossible. The availability of user-friendly software packages greatly assists in applying DFT calculations to individual specific problems.
In DFT, electronic energy E is expressed as a functional of electron density, viz.,
where T is the total kinetic energy of electrons, Vne the potential energy resulting from an external potential and Vee the electron–electron repulsion energy. Electron density ρ(r) for an N-electron system is defined as
where xi collectively denote spatial (ri) and spin (si) coordinates. In stark contrast to the wave function Ψ that depends on 3 × N (space) + N (spin) = 4N variables, ρ(r) contains only three spatial variables, implying that E may be obtained in a much more straightforward manner using ρ(r). In 1964, Hohenberg and Kohn proved that there is a one-to-one correspondence between the ground-state density and the external potential (Hohenberg and Kohn, 1964). They also showed that the variational principle holds for the ground-state energy. The variational principle and the Levy constrained-search formulation of DFT (Levy, 1979) ensure that E can be determined by minimizing it with respect to some N-representable trial electron densities.
Despite the fundamental importance of the Hohenberg–Kohn theorems, they do not provide explicit forms of the energy functionals in Equation 1 (except Vne). To proceed, one needs to know how T and Vee are expressed as functionals of ρ(r). A practical approach to this problem was proposed by Kohn and Sham (1965). Their approach attempts to describe the real electron density by way of non-interacting electrons that are described using a Slater determinant of molecular orbitals (MOs) [or Kohn–Sham (KS) orbitals]. When the density is treated this way, the total energy is written as
where Ts[ρ(r)] is the kinetic energy of non-interacting electrons, J[ρ(r)] the classical electron-electron repulsion energy (Hartree energy), and EXC[ρ(r)] the exchange-correlation energy [= (T − Ts) + (Vee − J)]. The first term Ts[ρ(r)], which accounts for a large portion of T[ρ(r)], may now be calculated using KS orbitals as in Hartree–Fock calculations.
Nevertheless the explicit form of EXC remains unknown. In practice, KS equations are solved by employing an approximate EXC; as such, the accuracy of DFT energy depends critically on the quality of EXC. So far, a number of exchange-correlation functionals have been developed, by either constraint satisfaction or semi empirical fitting (Perdew et al., 2005). EXC functionals are classified into the following five major levels (rungs) of “Jacob's Ladder” according to their types: namely, local spin density approximation (LSDA), generalized gradient approximation (GGA), meta-GGA, hyper-GGA, and generalized random phase approximation (Perdew and Schmidt, 2001). In the field of chemistry, Gaussian- or Slater-type atomic orbitals (AOs) are usually used as basis sets of KS orbitals, while GGA, meta-GGA, or hyper-GGA functionals are mainly used to approximate EXC. When KS equations are solved for periodic solids or nanomaterials, plane-wave basis sets and pseudopotentials are used often with LSDA or GGA functionals.
When AO-type localized basis sets are used, the maximum number of atoms that DFT can manage is normally around a few hundred. Nevertheless, DFT is useful in investigating even larger systems such as metalloenzymes that contain thousands of atoms or more, even though the application of DFT to an entire enzyme may be prohibitive. Two of the more practical approaches are (1) to extract relatively important active site atoms and then apply DFT to this active site model, and (2) to divide the system into a few layers first, and then apply different computational methods to individual layers in a hybrid manner to describe the entire system effectively. The latter hybrid calculation can be performed, e.g., by using a QM/MM method (Warshel and Levitt, 1976; Field et al., 1990; Maseras and Morokuma, 1995; Gao, 1996; Humbel et al., 1996; Svensson et al., 1996; Murphy et al., 2000; Cui et al., 2001; Friesner and Guallar, 2005; Nam et al., 2005; Riccardi et al., 2006; Vreven et al., 2006; Senn and Thiel, 2007a,b, 2009; Lin and Truhlar, 2007; Hu and Yang, 2008, 2009; Kamerlin et al., 2009; Chung et al., 2011a; Hirao and Morokuma, 2011a; Sameera and Maseras, 2012) There are three major schemes of QM/MM, i.e., mechanical-embedding (ME), electronic-embedding (EE), and polarizable-embedding (PE) schemes, which differ in how the electrostatic interaction between QM atoms and remaining atoms is treated (Senn and Thiel, 2007a,b, 2009). In ME-QM/MM, the electrostatic interaction is evaluated as the interaction between the gas-phase state of QM atoms and the point charges of MM atoms. Thus, the polarization effect of the QM electron density caused by MM point charges is not taken into account. The EE-QM/MM method can describe such QM polarization. PE-QM/MM further allows the MM atoms to be polarized, and thus the effect of mutual polarization between QM atoms and surrounding atoms can be taken into account. Many conventional QM/MM methods evaluate the total energy of the entire system in an additive manner. That is, the energy EQM/MM is given as
By contrast, the ONIOM (our own N-layer integrated MO molecular mechanics) method allows one to combine QM and MM (QM:MM) in a subtractive (extrapolative) fashion (Maseras and Morokuma, 1995), in which case the total energy is
where superscripts R and M denote real (large) and model (small) systems, respectively. It should be noted that the “MM region” in an additive QM/MM means the atoms outside the QM region. However, in a subtractive QM/MM, MM is applied to both the entire system and the small model system (i.e., “QM region”). Thus, even though both methods give more or less similar approximations to the total energy, the meanings of the individual subsystems, especially the subsystems to which MM is applied, could be somewhat different. In the subtractive ONIOM approach, two (or more) different levels of QM methods can also be naturally combined [i.e., ONIOM(QM:QM')], and such hybrid QM/QM methods are useful, e.g., in exploring the mechanisms of chemical reactions catalyzed by bulky transition-metal catalysts within a reasonable computational time.
With these theoretical frameworks and tools in hand, it is obvious that there could be an infinite number of potential research targets to be explored by DFT calculations. However, in this paper, we shall focus on Fe/O-containing molecules, or more specifically, synthetic non-heme iron complexes (Costas et al., 2000; Nam, 2007; Que, 2007; Que and Tolman, 2008; McDonald and Que, 2013; Nam et al., 2014), heme-type cytochrome P450 enzymes (Dawson and Sono, 1987; Sono et al., 1996; Guengerich, 2001, 2008; Denisov et al., 2005; Ortiz de Montellano, 2005, 2010; Sligar et al., 2005; Makris et al., 2006; Groves, 2006; Isin and Guengerich, 2007; Poulos, 2014), and non-heme iron enzymes (Feig and Lippard, 1994; Que and Ho, 1996; Wallar and Lipscomb, 1996; Hegg and Que, 1997; Lange and Que, 1998; Solomon et al., 2000, 2009; Merkx et al., 2001; Baik et al., 2003; Stubbe et al., 2003; Costas et al., 2004; Tshuva and Lippard, 2004; Abu-Omar et al., 2005; Bollinger and Krebs, 2006, 2007; Neidig et al., 2006; Krebs et al., 2007; Friedle et al., 2010; van der Donk et al., 2010).
Our motivation to study these iron-containing molecules comes from the remarkable catalytic machinery operating in iron enzymes. In fact, iron enzymes display diverse reactivity patterns, e.g., as oxygenases [monooxygenases (Equation 6) or dioxygenases (Equation 7)] or as peroxidases (Dawson and Sono, 1987; Wang et al., 2013a; Poulos, 2014).
However, the active intermediates of iron enzymes, such as compound I of P450, (Dawson and Sono, 1987; Sono et al., 1996; Schlichting et al., 2000; Davydov et al., 2001; Guengerich, 2001, 2008; Denisov et al., 2005; Ortiz de Montellano, 2005, 2010; Sligar et al., 2005; Groves, 2006; Makris et al., 2006; Isin and Guengerich, 2007; Rittle and Green, 2010; Poulos, 2014) intermediate J of taurine dioxygenase (TauD) (Krebs et al., 2007), intermediate Q of soluble methane monooxygenase (sMMO) (Shu et al., 1997; Solomon et al., 2000; Merkx et al., 2001; Baik et al., 2003; Bollinger and Krebs, 2006), and intermediate X of ribonucleotide reductase (RNR) (Solomon et al., 2000; Bollinger and Krebs, 2006), are often difficult to trap and spectroscopically characterize in normal turnover conditions. Theoretical DFT and DFT/MM calculations have useful roles to play in providing insights into the elusive aspects of iron enzymes such as the nature of short-lived intermediates and chemical reaction mechanisms.
Applications of DFT
Synthetic Iron Complexes
Reactivity patterns
Transition metals in enzymes or synthetic complexes enable otherwise difficult chemical transformations such as C–H activation (Groves et al., 1978; Ryabov, 1990; Shilov and Shul'pin, 1997; Shilov and Shteinman, 1999; Jia et al., 2001; Labinger and Bercaw, 2002; Lehnert et al., 2002; Yoshizawa, 2002; Kakiuchi and Chatani, 2003; Goldberg and Goldman, 2004; Godula and Sames, 2006; Groves, 2006, 2014; Siegbahn and Borowski, 2006; Bergman, 2007; Chen and White, 2007; Seregin and Gevorgyan, 2007; Davies and Manning, 2008; Balcells et al., 2010; Lyons and Sanford, 2010; Ackermann, 2011; Yamaguchi et al., 2012; Webb et al., 2013; Yosca et al., 2013). The activation or functionalization of C–H bonds of cheap and abundant substrates such as alkanes is one of the most important goals in transition-metal catalysis.
Iron-containing cytochrome P450 enzymes (P450s) are capable of activating C–H bonds (Groves, 1985), and thus the development of synthetic, biomimetic analogs that resemble P450s has been actively pursued (Lim et al., 2003; Rohde et al., 2003; Kaizer et al., 2004; Bukowski et al., 2005; Kim et al., 2005; Sastri et al., 2005; Bautz et al., 2006; Thibon et al., 2008; Yoon et al., 2009; Lee et al., 2010; Li et al., 2010, 2011; Cho et al., 2011a; Hohenberger et al., 2012). Theoretical studies of such biomimetic models may not only identify the key elements that determine their chemical reactivities, but may also provide insight into intermediates and reactivities of parent enzymes (Shaik et al., 2007a; de Visser et al., 2013). To date, DFT calculations have been applied extensively to various types of non-heme iron species (Scheme 1) (Bassan et al., 2002, 2005a,b; Roelfes et al., 2003; Decker and Solomon, 2005; Kumar et al., 2005; Quinonero et al., 2005; Berry et al., 2006; Bernasconi et al., 2007, 2011; de Visser, 2006, 2010; Hirao et al., 2006a, 2008a,b, 2011; Rohde et al., 2006; Decker et al., 2007; de Visser et al., 2007, 2011; Johansson et al., 2007; Noack and Siegbahn, 2007; Sastri et al., 2007; Sicking et al., 2007; Bernasconi and Baerends, 2008, 2013; Comba et al., 2008; Dhuri et al., 2008; Fiedler and Que, 2009; Klinker et al., 2009; Wang et al., 2009a, 2013b; Cho et al., 2010, 2012a, 2013; Geng et al., 2010; Chen et al., 2011; Chung et al., 2011b; Seo et al., 2011; Shaik et al., 2011; Vardhaman et al., 2011; Wong et al., 2011; Ye and Neese, 2011; Gonzalez-Ovalle et al., 2012; Gopakumar et al., 2012; Latifi et al., 2012; Mas-Ballesté et al., 2012; McDonald et al., 2012; Van Heuvelen et al., 2012; Ansari et al., 2013; Kim et al., 2013; Lee et al., 2013; Sahu et al., 2013; Tang et al., 2013; Ye et al., 2013; Hong et al., 2014; Sun et al., 2014). The intriguing reactivity patterns of these complexes are the result of active involvement of electrons in d-type MOs, which gives rise to multi-state scenarios (Shaik et al., 1998; Schröder et al., 2000; Schwarz, 2011).
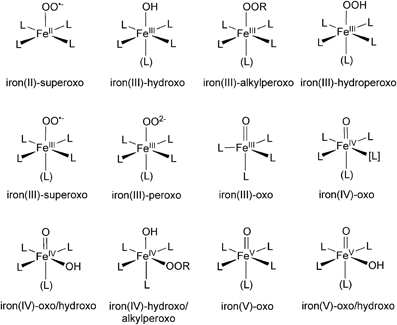
Scheme 1. Some of the (oxygen-bound) non-heme iron complexes studied by DFT calculations. L denotes atoms of non-oxygen ligands, but the use of the common symbol does not necessarily mean that the ligand atoms are equivalent.
When we study the chemical reactions of these non-heme iron complexes using DFT, a main goal is to obtain structural and energetic information for various transition states and intermediates on reaction pathways. In addition, by analyzing KS orbitals or some sort of transformed localized orbitals, one can gain valuable chemical insight into the electronic reorganization during the reactions. Of all the species in Scheme 1, iron(IV)-oxo complexes are particularly important in view of their resemblance to iron(IV)-oxo porphyrin π-cation radical species (compound I or Cpd I) of P450s. Figure 1A depicts the five important d-type orbitals of a non-heme iron(IV)-oxo complex supported by a 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (TMC) ligand and an acetonitrile axial ligand. Previous DFT studies identified typical electron-shift patterns for the reactions of non-heme iron(IV)-oxo complexes (Figure 1B) (Hirao et al., 2006a; Shaik et al., 2007a). In the lower-spin triplet state, the δ orbital is doubly occupied and each of the two π* orbitals is singly occupied in the initial stage. In the C–H bond activation step, an electron migrates from the substrate to a π* orbital. As a result, the formal oxidation state of iron becomes +3, with one of the two π* orbitals being singly occupied. By contrast, in the reaction of a higher-spin quintet state, an electron tends to transfer from the substrate C–H σ bond (σC−H) to the vacant σ*z2 orbital (denoted σ*2 in Figure 1), resulting again in a +3 Fe oxidation state. In this case, however, there is an increase in the number of unpaired electrons on the iron center. This gives rise to an enhanced exchange stabilization of the system, which results in a substantially lower energy barrier in the quintet state. Because the σ*2 orbital extends along the Fe–O axis, the charge transfer in the quintet state requires that the substrate should approach the iron(IV)-oxo moiety from just above the oxo unit to achieve the maximum overlap between σC−H and σ*2. Consequently, the transition state tends to have a markedly upright geometry, with the C, H, O, and Fe atoms aligned in a collinear configuration.
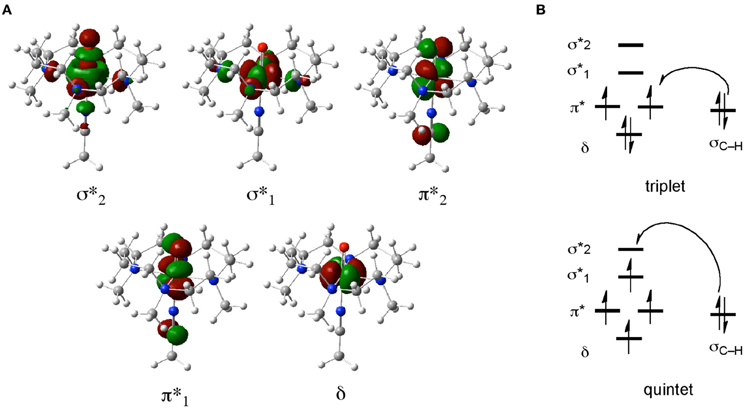
Figure 1. (A) Five key d-type MOs of the TMC iron(IV)-oxo complex. (B) Typical electron-shift patterns for the reactions of non-heme iron(IV)-oxo complexes.
Reactions involving O–O bond cleavage
Another interesting conundrum regarding synthetic iron(IV)-oxo species is how an iron(IV)-oxo species is itself produced from other species. In P450s, Cpd I is formed in the catalytic cycle when the O–O bond of the precursor intermediate, Cpd 0, undergoes heterolytic cleavage (Dawson and Sono, 1987; Sono et al., 1996; Guengerich, 2001, 2008; Denisov et al., 2005; Ortiz de Montellano, 2005, 2010; Sligar et al., 2005; Makris et al., 2006; Groves, 2006; Isin and Guengerich, 2007; Poulos, 2014). The O–O bond cleavage also constitutes a critical step in ferryl formation in non-heme enzymes (Krebs et al., 2007). Rohde et al. showed experimentally, that a synthetic TMC iron(IV)-oxo is formed at −40°C with 3 equivalents of H2O2 in acetonitrile (Rohde et al., 2003). More recently, Li et al. reported that a TMC iron(IV)-oxo species can be generated by reacting a TMC Fe(II) complex with a stoichiometric amount of H2O2 in acetonitrile in the presence of 2,6-lutidine (Scheme 2A) (Li et al., 2010). Interestingly, the addition of 2,6-lutidine accelerated the reaction rate and enhanced the reaction yield. It was postulated that the role of 2,6-lutidine is analogous to that played by a distal histidine in the formation of the Cpd I intermediate in heme peroxidases (Scheme 2B) (Poulos and Kraut, 1980; Newmyer and Ortiz de Montellano, 1995; Tanaka et al., 1997; Wirstam et al., 1999; Derat and Shaik, 2006; Chen et al., 2008; Poulos, 2014). The DFT calculations performed by Hirao et al. support this hypothesis (Path A in Scheme 2C) and provide additional insights into electronic details of the reaction mechanism (Scheme 3 and Figure 2A) (Hirao et al., 2011). If there is no base in the system, an acetonitrile molecule in the solvent could act as an acid-base catalyst in Path A. However, the calculated energy barrier for this acetonitrile-catalyzed reaction (39 kcal/mol) is too high for this reaction to proceed (Figure 2B). This is not in accordance with the fact that an iron(IV)-oxo can be produced even in the absence of 2,6-lutidine. In an attempt to find an alternative pathway, Hirao et al. examined Path B (Schemes 2C, 3), which is initiated by a homolytic O–O bond cleavage of H2O2. The energy barrier for this reaction was much lower (25.4 kcal/mol, see Figure 2B). In light of these results, they suggested that in the absence of 2,6-lutidine, the reaction follows Path B to afford the same iron(IV)-oxo product.
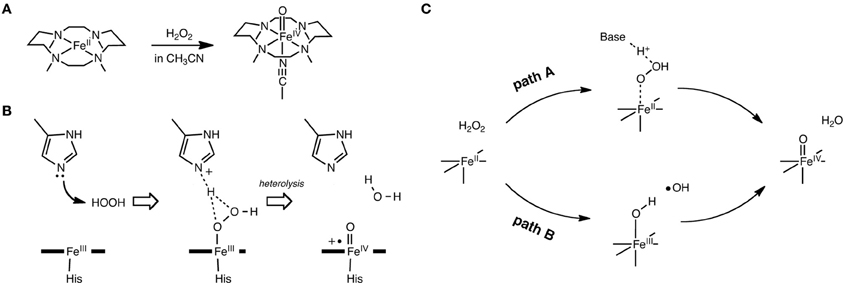
Scheme 2. (A) Iron(IV)-oxo formation from TMC Fe(II) and H2O2. (B) Cpd I formation in heme peroxidases. (C) The two pathways examined. Adapted from Hirao et al. (2011) with permission from the American Chemical Society.
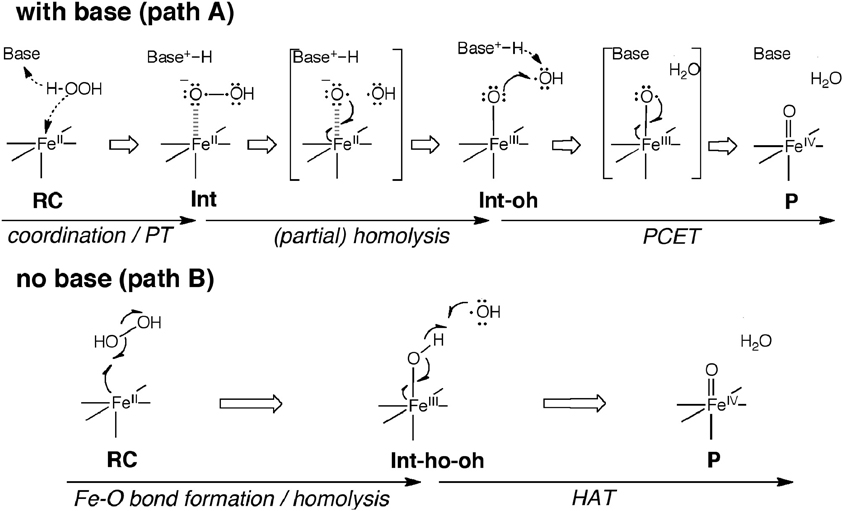
Scheme 3. Schematic illustration of electron reorganization during the reactions in Scheme 2C. Adapted from Hirao et al. (2011) with permission from the American Chemical Society.
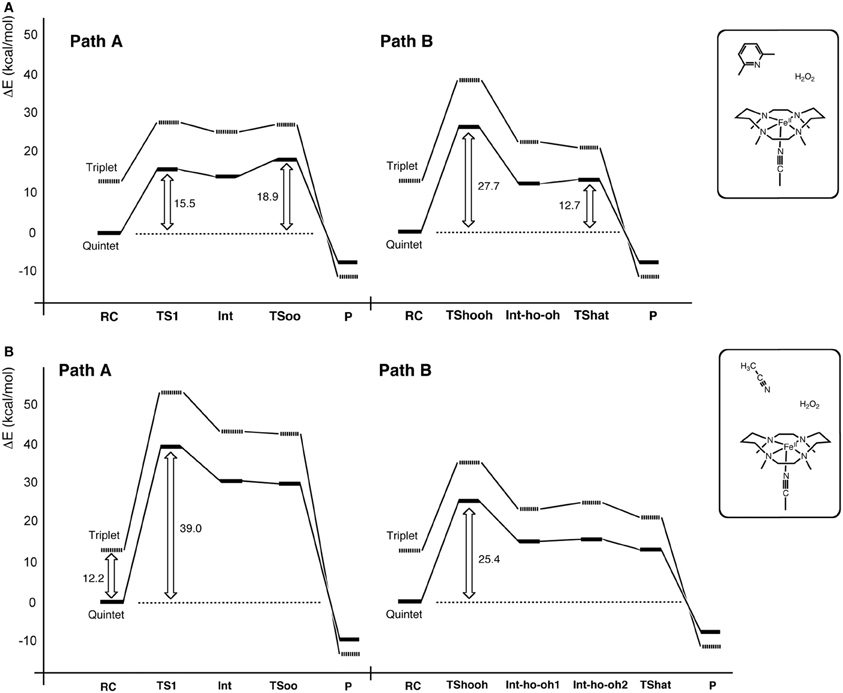
Figure 2. Energy profiles for the reactions in Scheme 2C in the presence (A) and absence (B) of 2,6-lutidine. Adapted from Hirao et al. (2011) with permission from the American Chemical Society.
As seen above, the mode of O–O bond cleavage in the reactions of synthetic non-heme complexes is a mechanistically intriguing aspect. In the above case, the mode was either (virtually) heterolytic or homolytic, depending on the presence of an acid-base catalyst. More specifically, these processes can be viewed essentially as O–O cleavage that gives OH− or OH•. Another type of O–O bond cleavage has been reported recently. Kim et al. found that a non-heme high-spin iron(III)-hydroperoxo species bearing a macrocyclic TMC ligand is capable of mediating sulfoxidation of thioanisole (Kim et al., 2013). Detailed DFT calculations and orbital analyses characterized the O–O bond cleavage as heterolytic; however, in this case, the high-spin Fe(III)OOH2+ species heterolytically splits into Fe(III)O+ and OH+. When the OH+ forms a bond with thioanisole, the proton of OH+ is donated back to the Fe(III)O+ moiety. All these events take place in a concerted fashion, and hence, an OH+ ion is not produced as an intermediate.
Ligand effects
The reactivity and many other properties of the iron (or more broadly, metal) center in enzymes and synthetic complexes are affected significantly by coordinating ligands. Therefore, the theoretical evaluation of ligand effects is an important task of computational chemistry. Myradalyyev et al. performed a comparative B3LYP DFT study of the interactions between the porphine, corrin, and TMC ligands (Scheme 4) and several metal ions (Cr2+, Mn2+, Fe2+, Co2+, Ni2+, and Cu2+) (Myradalyyev et al., 2013). The computationally determined ground spin states were in good agreement with the available experimental data (Figure 3A). The binding affinity was shown to increase in the order Mn2+ < Cr2+ ~ Fe2+ < Co2+ < Ni2+ < Cu2+ in all ligands (Figure 3B). The relative binding strength is determined by several factors, such as the total charge of the ligand, spin promotion in the complexation, electrostatic/Pauli interactions, and ligand-to-metal or metal-to-ligand charge transfer (Feixas et al., 2009).
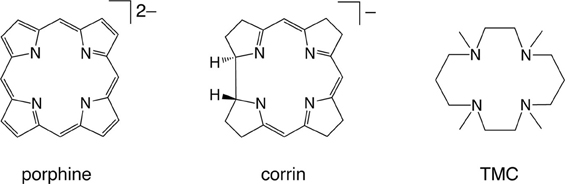
Scheme 4. The three ligands studied by Myradalyyev et al. Reprinted from Myradalyyev et al. (2013) with permission from Elsevier.
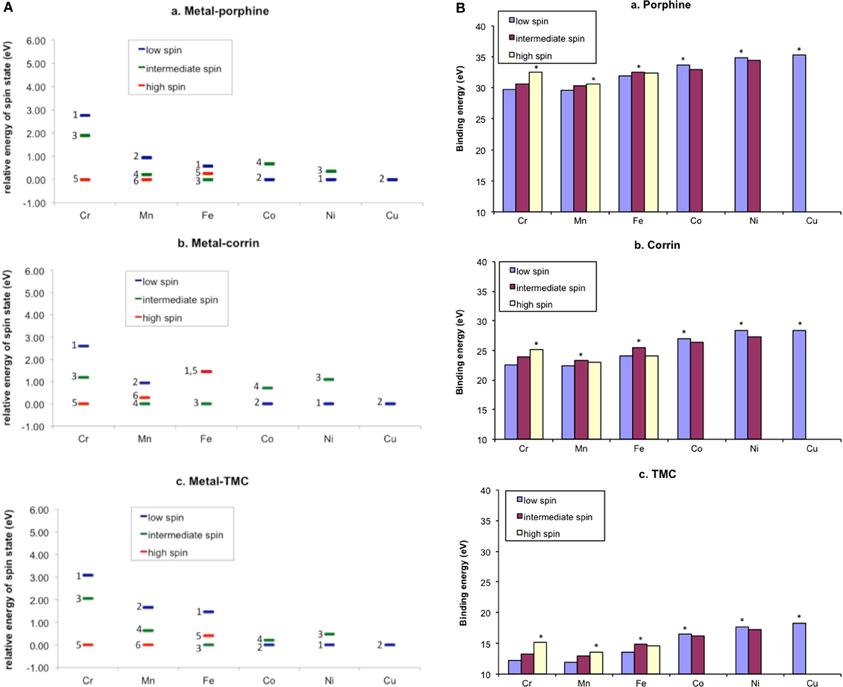
Figure 3. (A) Relative stability of various complexes metal(II)-ligand complexes in different spin states. (B) Binding energy of the complexes. Adapted from Myradalyyev et al. (2013) with permission from Elsevier.
Cytochrome P450 Enzymes
Reactive species of P450s
DFT and DFT/MM studies have also played vital roles in advancing our understanding of the electronic structure and reactivity of P450s. It is commonly accepted that Cpd I acts as the reactive species of P450-dependent oxidation reactions, and several previous experiments support this idea (Schlichting et al., 2000; Davydov et al., 2001; Rittle and Green, 2010). The electronic structure of Cpd I and the electron reorganization patterns of the substrate oxidation reactions of Cpd I have been discussed for several different spin states (Ogliaro et al., 2000a,b; Yoshizawa et al., 2001; de Visser et al., 2002; Himo and Siegbahn, 2003; Kamachi and Yoshizawa, 2003; Meunier et al., 2004; Hackett et al., 2005, 2007; Hirao et al., 2005; Shaik et al., 2005, 2007a,b, 2010a; Isobe et al., 2008, 2011, 2012; Shoji et al., 2008; Yamaguchi et al., 2009; Li et al., 2012; Rydberg et al., 2014). Computational studies to assess the reactivity of Cpd 0 have also been conducted (Ogliaro et al., 2002; Kamachi et al., 2003; Bach and Dmitrenko, 2006; Derat et al., 2006; Hirao et al., 2006b; Li et al., 2007). Overall, DFT calculations predicted that Cpd 0 is less reactive than Cpd I. Efforts to improve our understanding of the nature of P450 reactive species are still ongoing. Recently, Wang et al. examined sulfoxidation mediated by the Fe(III)(HOOH) complex of a P450 using DFT, and showed that the energy barrier is 5.3 kcal/mol, compared with 4.7 kcal/mol in the Cpd I reaction and 23.6 kcal/mol in the Cpd 0 reaction (Wang et al., 2013c). From these DFT results, they suggested that the Fe(III)(HOOH) complex could be an alternative reactive species for sulfoxidation.
Mechanism-based inactivation
The reactivity patterns of P450s are mechanistically interesting conundrums. P450s are also important enzymes in a practical sense, especially in the context of drug metabolism. The essential functions of P450s in Phase I drug metabolism are now well appreciated. An important feature of P450s in drug metabolism is their broad substrate specificity. Although >18,000 sequences of P450s have been identified thus far, humans have only 57, and drug metabolism is performed by only a few of these (Pelkonen et al., 2008; Guengerich, 2013; Poulos, 2014). As such, the inhibition of P450s through drug–drug interactions (DDIs) could adversely affect many metabolic reactions in the body.
DFT has begun to find applications in such practical issues of P450s. In particular, DFT can make significant contributions to the understanding of a special type of DDI, i.e., mechanism-based inactivation (MBI) (Zhou et al., 2005; Orr et al., 2012). There are two different types of MBI: quasi-irreversible and irreversible. In MBI, an inhibitor molecule first reacts with Cpd I to be converted to a metabolic intermediate (MI), and then the MI binds to the active site of a P450 quasi-irreversibly or irreversibly, resulting in the inhibition of the enzyme. In a quasi-irreversible MBI, the MI forms a coordination bond with the heme iron, whereas in an irreversible MBI, the MI forms a strong covalent bond with an amino acid residue or the porphyrin ligand. In either case, MBI involves a chemical reaction, which cannot be described by conventional docking simulations. Rather, one must use quantum mechanical methods such as DFT. In recent years, several research groups have reported DFT studies of P450 MBI (de Visser et al., 2004; Rydberg and Olsen, 2011; Hirao et al., 2012, 2013a,b; Taxak et al., 2012, 2013a,b).
Hirao et al. studied the irreversible MBI caused by terminal acetylenes (Figure 4) (Hirao et al., 2012). Their calculations showed that without the involvement of a water molecule, covalent bond formation between the ketene-type metabolic intermediate and the catalytically essential threonine residue is difficult, because the corresponding energy barrier is too high (>38 kcal/mol). However, when a water molecule was allowed to participate in the reaction, the barrier was reduced by about 20 kcal/mol. This result suggested that a water molecule plays a crucial role in “terminating” the enzymatic function of P450.
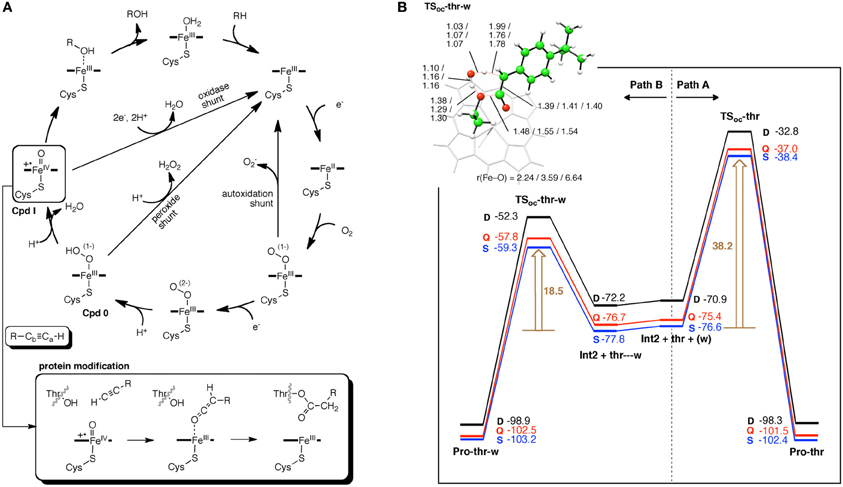
Figure 4. (A) Catalytic cycle of P450 (top) and a proposed mechanism of MBI caused by terminal acetylenes (bottom) (B) Energy diagrams (in kcal/mol) for the reactions of a ketene intermediate in the absence (A) and presence (B) of a water molecule. Adapted from Hirao et al. (2012) with permission from the American Chemical Society.
Hirao et al. also studied the mechanism of metabolic-intermediate formation in the quasi-irreversible MBI of P450 caused by 1,1-dialkylhydrazine (or unsymmetrical dimethylhydrazine, UDMH) (Hirao et al., 2013a). For this MBI, hydrazine is converted to an aminonitrene metabolic intermediate, which in turn binds to the heme iron. However, there are at least two possible mechanisms for this reaction (Scheme 5) (Ortiz de Montellano, 2005). In the first mechanism, two hydrogen atoms are successively abstracted from the terminal nitrogen by Cpd I or another ferryl species, Cpd II (Green et al., 2004). In the other mechanism, the first H-abstraction is followed by N-oxidation. Other mechanisms may also be possible in which the first step is not H-abstraction from a N–H bond. Hirao et al. examined four different pathways using DFT as shown in Figure 5A, and found that the preferred reaction involves H-abstraction from the N–H bond in the first step. After the H-abstraction, the substrate radical undergoes another H-abstraction from the same nitrogen, to yield an aminonitrene species. The DFT study also showed that the aminonitrene MI coordinates to the heme in a somewhat tilted orientation, and that aminonitrene binds to the heme more strongly than a water molecule.
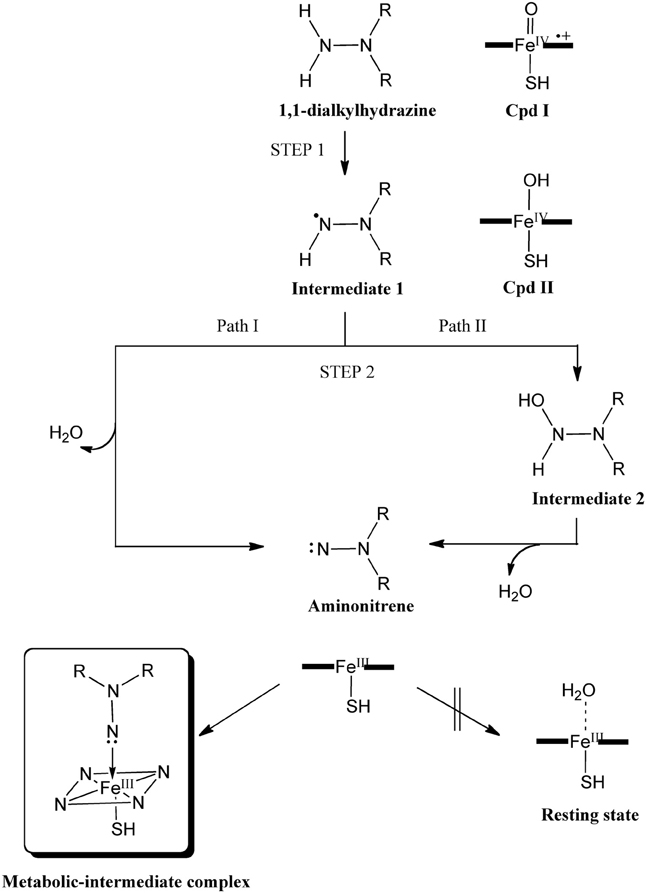
Scheme 5. Proposed mechanism of the MBI of P450 by UDMH. Adapted from Hirao et al. (2013a) with permission from Wiley-VCH.
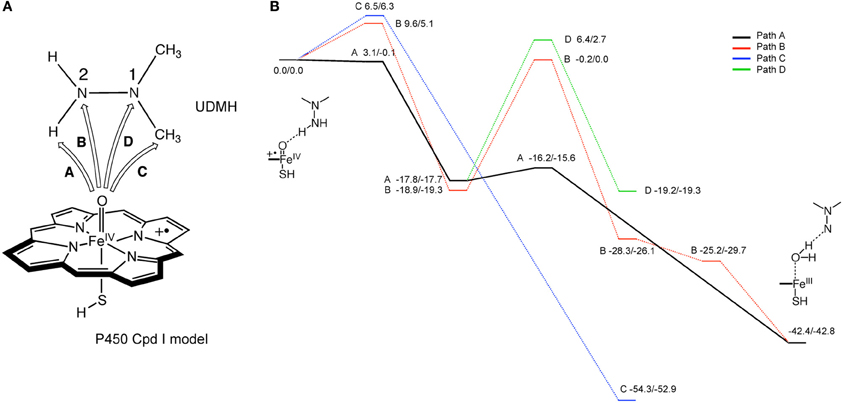
Figure 5. (A) Reaction pathways considered for the MI formation from dimethylhydrazine. (B) Energy profiles (kcal/mol) for the four pathways considered. Adapted from Hirao et al. (2013a) with permission from the Wiley-VCH.
More recently, Hirao et al. investigated part of the MBI process caused by amine-containing drugs (Hirao et al., 2013b). Amines are prone to form nitrosoalkane metabolic intermediates in P450-mediated metabolism, and the nitrosoalkanes form a quasi-irreversible metabolic-intermediate complex (MIC) to inhibit P450s (Scheme 6) (Hanson et al., 2010). In fact, many existing drugs contain an amine moiety, and thus this metabolic pathway presents a serious concern. The study conducted by Hirao et al. focused on the step from 4 to 7 in Scheme 6, and their comparative DFT study showed that the mechanisms involving H-abstraction from the O–H bond or from the N–H bond of 4 had particularly low energy barriers (paths A and B in Figure 6), suggesting that the reaction proceeds via either of these mechanisms. The mechanisms involving N-oxidation and H-abstraction from a C–H bond had much higher barriers. The coordination bonding in the MIC was also studied in detail. The N-bound form of a MIC was more stable than the O-bound form (Figure 7). Also, the ferrous MIC (1MIC(II)) had somewhat larger binding energy than the ferric MIC (2MIC(III)). This result was consistent with the fact that a ferrous MIC is formed in the MBI of amine-containing compounds.
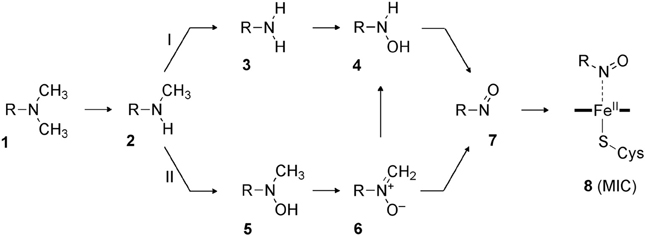
Scheme 6. Likely pathways of MIC formation starting from a tertiary amine (Hanson et al., 2010). Adapted from Hirao et al. (2013b).
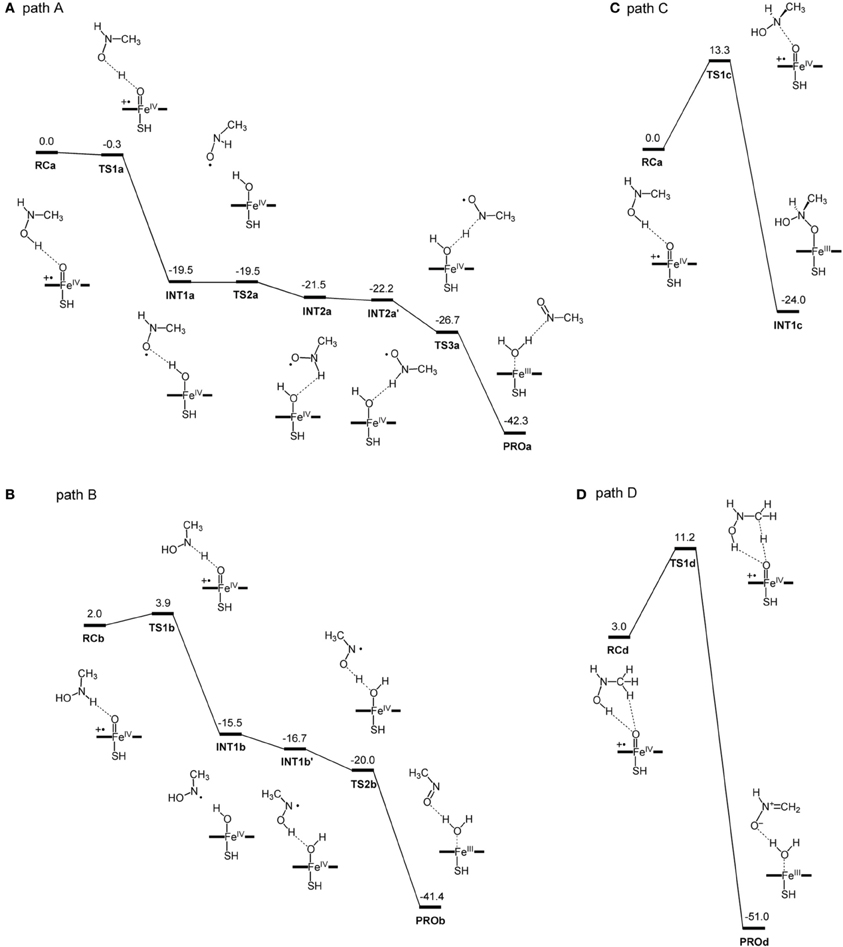
Figure 6. DFT-calculated reaction energy profiles (in kcal/mol) for paths (A–D), which are initiated by H-abstraction from the O–H bond (A), H-abstraction from the N–H bond (B), N-oxidation (C), and H-abstraction from the methyl group (D). Reprinted from Hirao et al. (2013b).
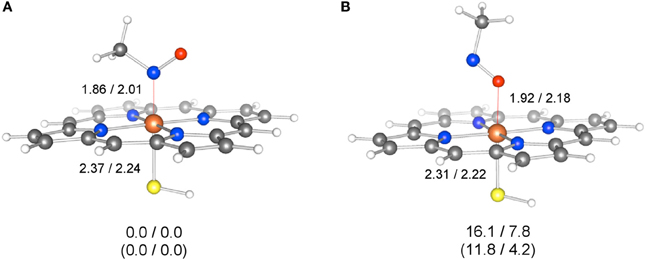
Figure 7. Geometries of N-bound (A) and O-bound (B) forms of 1MIC(II) and 2MIC(III), optimized at the M06/[SDD(Fe),6-31G*(other)] level. Key distances are given in Å. The values below the geometries are relative energies (kcal/mol) obtained at the M06(SCRF)/6-311+G(d,p) level (1MIC(II)/2MIC(III)), while the values in parentheses are relative energies obtained at the B3LYP(SCRF)/6-311+G(d,p) level. Reprinted from Hirao et al. (2013b).
Analyses of protein environmental effects
Another important aspect of P450s is the protein environmental effect on their active-site properties. P450s accommodate a heme cofactor and organic substrates in their active sites, thereby catalyzing chemically difficult reactions. There have been several QM/MM studies of P450s (Schöneboom et al., 2002, 2004, 2005; Guallar and Friesner, 2004; Schöneboom and Thiel, 2004; Lin et al., 2004; Altun and Thiel, 2005; Bathelt et al., 2005, 2008; Altun et al., 2006, 2007, 2008; Cohen et al., 2006; Harvey et al., 2006; Zheng et al., 2006; Fishelovitch et al., 2007; Cho et al., 2008, 2011b; Hirao et al., 2008c; Wang et al., 2008, 2009b; Porro et al., 2009; Tian and Friesner, 2009; Lai and Shaik, 2011; Lai et al., 2011; Schyman et al., 2011; Lonsdale et al., 2011, 2013; Krámos et al., 2012; Kwiecień et al., 2012; Usharani et al., 2012; Dumas et al., 2013; Li and Shaik, 2013; Li et al., 2013; van der Kamp and Mulholland, 2013; Krámos and Oláh, 2014), which demonstrated that the effect of the protein surroundings is often very important. To gain fundamental insights into the interaction between the active site and the surrounding atoms of P450s, Hirao and co-workers have performed energy decomposition analysis (EDA) studies (Hirao, 2011a; Thellamurege and Hirao, 2013, 2014).
Thellamurege and Hirao performed an DFT-based EDA study of the interaction between two key intermediates of P450 and a molecule of water (Figure 8) (Thellamurege and Hirao, 2013). They used two different types of EDA: the localized MO energy decomposition analysis (LMOEDA), which was developed by Su et al. and is implemented in GAMESS software (Schmidt et al., 1993; Gordon and Schmidt, 2005; Su and Li, 2009), and the EDA method, which is implemented in the Amsterdam Density Functional (ADF) program (Ziegler and Rauk, 1979a,b; ADF2012.01, 2012). In the LMOEDA, the total interaction energy is decomposed into electrostatic, exchange, repulsion, polarization, and dispersion components, whereas in the ADF-EDA, the total interaction energy is decomposed into electrostatic, Pauli repulsion, and orbital interaction terms. Such decomposition analyses provide chemically valuable insights (Kitaura and Morokuma, 1976; von Hopffgarten and Frenking, 2012). The LMOEDA and ADF-EDA were applied to two types of P450–H2O complexes. One is the resting state of P450, in which the water molecule is bound to the Fe(III) ion (Figure 8A). In the other complex, the oxo ligand of Cpd I forms a hydrogen bond with a water molecule (Figure 8B). The latter interaction had been suggested to reduce the activation barrier for the H-abstraction by P450 Cpd I (Altun et al., 2006; Kumar et al., 2011). Our EDA study revealed that the main driving force of the interaction in the resting state is the electrostatic energy. The polarization and exchange energies also play significant roles, but to a lesser extent. Compared with this metal–ligand interaction in the resting state, the interaction between Cpd I and H2O is weaker because it is viewed as a hydrogen bond. We observed that the electrostatic interaction plays a significant role here. Interestingly, polarization also contributes to a similar extent. This was different from the hydrogen bonding in the water dimer, where a larger contribution to the attractive interaction comes from the electrostatic term.
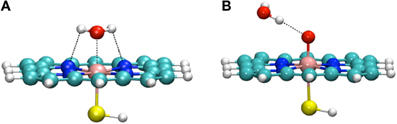
Figure 8. (A) The resting state of the P450 catalytic cycle (B) Cpd I interacting with a water molecule. Reprinted from Thellamurege and Hirao (2013).
Hirao performed an EDA of the protein environment effect within bacterial cytochrome P450cam Cpd I (Figure 9), using the ME and EE schemes of ONIOM(DFT:MM) (Hirao, 2011a). B3LYP/[SDD(Fe),6-31G*(others)] and AMBER94 were used as the QM and MM methods, respectively. The QM-MM non-bonding interaction energy was decomposed into electrostatic, van der Waals (vdW), and QM polarization terms. The EDA study demonstrated the particular importance of the electrostatic effect. The other effects were also large, but were less significant than the electrostatic effect. The electrostatic and vdW interaction energies were further decomposed into contributions from individual residues (Figure 10A). Positively charged nearby residues, especially those interacting with the two propionate groups of the heme (Arg112, Arg299, and His355), were shown to play particularly important roles in electrostatically stabilizing the active site. The electrostatic stabilization caused by each of these three residues was >100 kcal/mol. The protein environment also affects the spin distribution in the active site (Figure 10B). In the ONIOM-ME calculation, the protein electrostatic effect on the QM electronic state is not taken into account. With this method, significant amounts of spin populations were unnaturally localized on the propionate oxygen atoms. However, with the ONIOM-EE scheme, the unpaired electron shifted to the porphyrin ring.
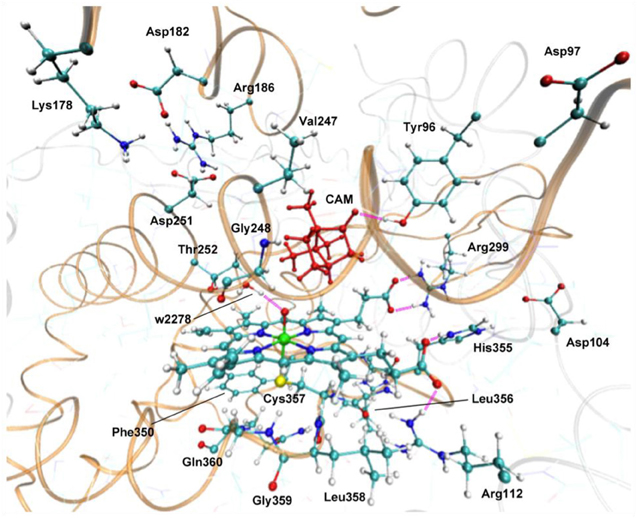
Figure 9. Active site of P450cam Cpd I. Reprinted from Hirao (2011a) with permission from the Chemical Society of Japan.
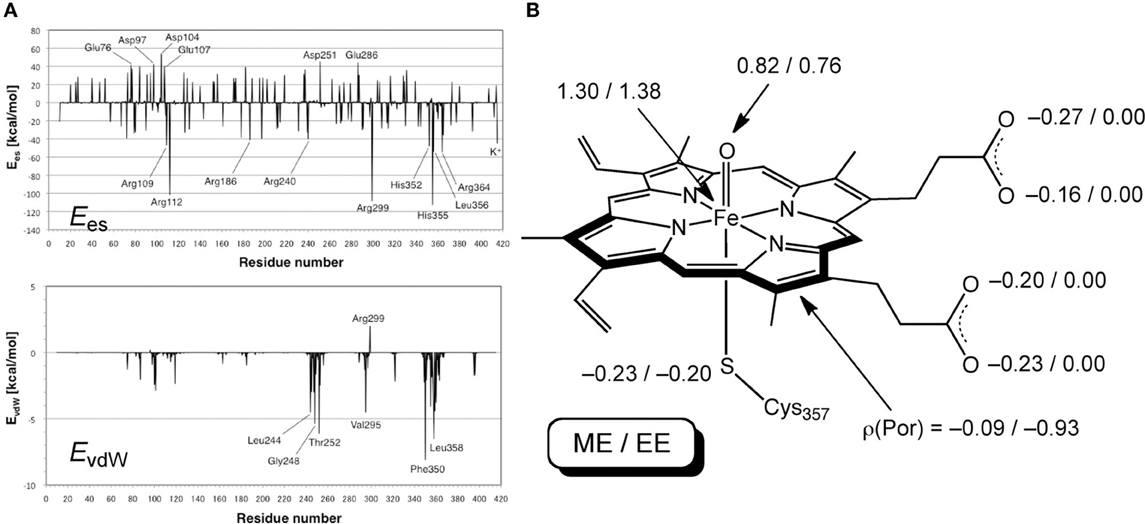
Figure 10. (A) Ees and EvdW of each residue. (B) Key atomic spin populations calculated by ONIOM-ME and ONIOM-EE calculations. Adapted from Hirao (2011a) with permission from the Chemical Society of Japan.
The EE scheme of QM/MM is often used because of its capability to describe the polarization of QM atoms in response to the atomic charges of surrounding atoms. However, in reality, the surrounding atoms will also undergo polarization. Such polarization can be described by a PE-QM/MM method. Thellamurege and Hirao performed ME-, EE-, and PE-QM/MM calculations for P450cam (Thellamurege and Hirao, 2014). An additive QM/MM method implemented in GAMESS was used, and the B3LYP/6-31G* method and the AMBER99/QP302 force fields (Thellamurege et al., 2013) were used for the QM and MM subsystems, respectively. This study again showed that the electrostatic interaction stabilizes the QM atoms most significantly. It was shown that when no charge adjustment was made for MM atoms at the QM-MM boundary, the MM point charges caused overpolarization of the QM density. Eliminating the charges of at least the MM atoms at the QM-MM boundary was deemed necessary to avoid overpolarization. Although the method of calculating the electrostatic interaction energy as well as the QM and MM methods used here were slightly different from those used in Hirao (2011a), the two studies yielded very similar residue-resolved electrostatic interaction energies (Figure 11). The MM polarization effect that was described by PE-QM/MM did not change the spin distribution within the QM atoms significantly (Figure 12). The dipole moments calculated by EE-QM/MM and PE-QM/MM were also very similar.
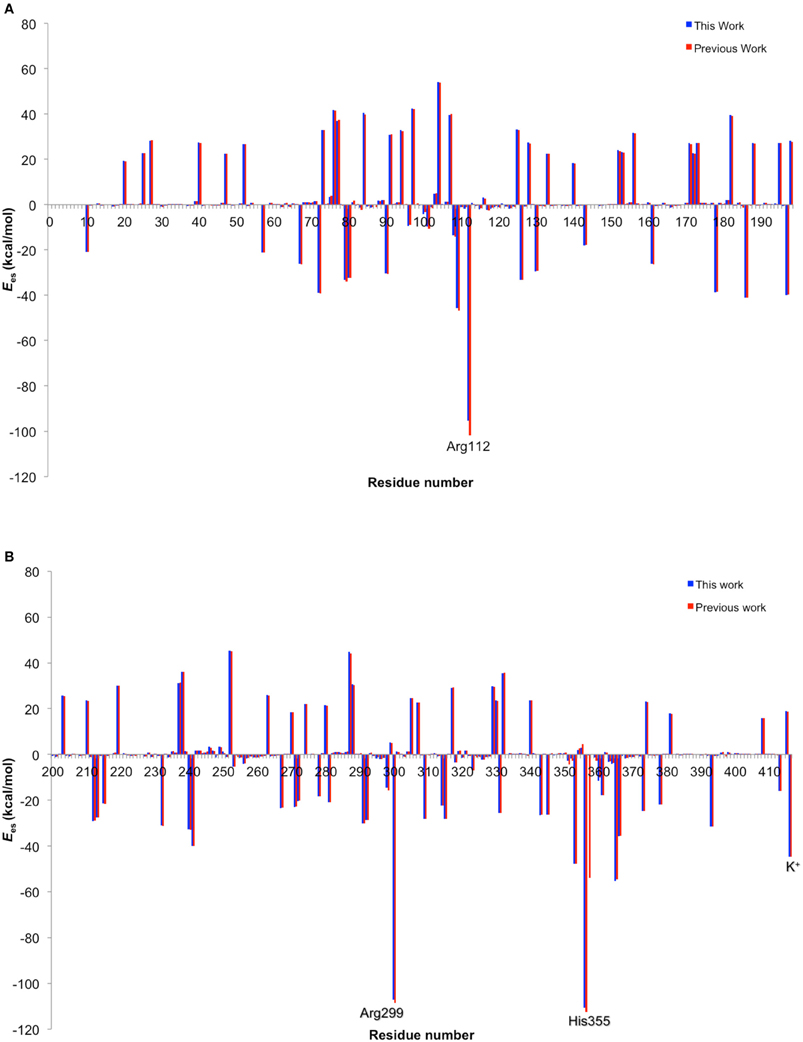
Figure 11. Comparison of electrostatic energy contributions from amino acid residues 10-199 (A) and 200-414 and K+ (#415) (B), obtained in this work with method 1 (blue) and in a previous work (red). Reprinted from Thellamurege and Hirao (2014) with permission from the American Chemical Society.
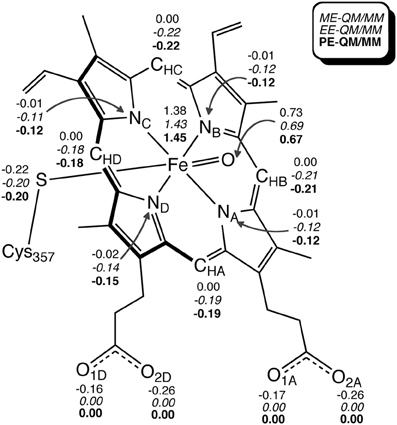
Figure 12. Key Mulliken atomic spin populations obtained with ME-QM/MM (gas-phase), EE-QM/MM, and PE-QM/MM. Reprinted from Thellamurege and Hirao (2014) with permission from the American Chemical Society.
Non-Heme Iron Enzymes
Myo-inositol monooxygenase
myo-Inositol monooxygenase (MIOX) is a non-heme diiron enzyme that is presumed to be involved in the pathogenesis of diabetic complications (Xing et al., 2006; Bollinger et al., 2009). MIOX catalyzes the first committed step in myo-Inositol catabolism, namely, the conversion of myo-inositol into D-glucuronate (Scheme 7). This conversion is initiated by H-abstraction from the C1 atom of myo-inositol by a ferric superoxide moiety. The reaction mechanism was investigated by Hirao and Morokuma using DFT and ONIOM(DFT:MM) calculations (Hirao and Morokuma, 2009). The calculations predicted that the barrier for the O–O bond cleavage is higher than that for the H-abstraction step. This explained why the experimentally determined kinetic isotope effect on the steady state turnover was close to unity.
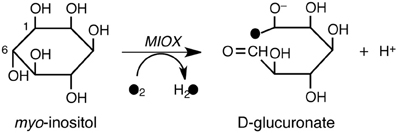
Scheme 7. MIOX-catalyzed conversion of myo-inositol into D-glucuronate. Reprinted from Hirao and Morokuma (2009) with permission from the American Chemical Society.
Hirao further performed an ONIOM-based EDA study of MIOX before (1) and after (2) the O2 binding to the iron (Scheme 8) (Hirao, 2011b). The ONIOM-EE calculation yielded an O2 binding energy of 3.5 kcal/mol, which might be somewhat underestimated (Siegbahn, 2006). For both intermediates, the electrostatic stabilization energy was the largest of all the decomposed energy terms. However, the difference in electrostatic stabilization between 1 and 2 was very small, indicating that the electrostatic protein effect may not make a significant contribution to the ferric-superoxide formation. Rather, the empirically estimated dispersion effect had a relatively large stabilizing effect on the O2 binding process.
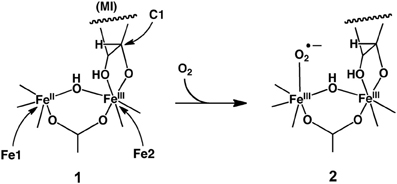
Scheme 8. O2 Binding in MIOX to form a ferric-superoxide species. Reprinted from Hirao (2011b) with permission from the American Chemical Society.
2-hydroxylethylphosphonate dioxygenase
Using DFT and ONIOM(DFT:MM) calculations, Hirao and Morokuma investigated the enzymatic reactions of a non-heme iron enzyme, 2-hydroxylethylphosphonate dioxygenase (HEPD) (Scheme 9) (Cicchillo et al., 2009; Whitteck et al., 2009, 2011; Hirao and Morokuma, 2010a, 2011b; Peck et al., 2011). Their DFT study of the HEPD-catalyzed reaction of 2-hydroxylethylphosphonate (2-HEP) substrate suggested that a radical intermediate should be formed in the late stage of the reaction (Hirao and Morokuma, 2010a). Such a radical intermediate was again observed in their ONIOM study (Int-rad in Scheme 10) (Hirao and Morokuma, 2011b). According to this scenario, the radical intermediate should have a sufficiently long lifetime to allow it to rotate about the P–C bond. As such, the radical should be able to attack the FeIII–OH from either face of the P–CH2• moiety. Whitteck et al. performed stereochemical experiments in which they introduced a deuterium atom into C1 of 2-HEP. Interestingly, the stereochemistry at C1 was lost in the hydroxymethylphosphonate product (Whitteck et al., 2011), which is consistent with our theoretically predicted radical-involving mechanism. The reaction energy profile for 1-HEP was mostly similar to that for 2-HEP (Figure 13), especially in the early phase of the reaction (Hirao and Morokuma, 2011b). However, at a late stage of the reaction, proton-coupled electron transfer was observed: after a homolytic P–C bond cleavage, an unpaired electron on the phosphorous transferred to the iron center to create an electron-deficient site, while there was proton transfer from the phosphate moiety to the hydroxyl group of ferric hydroxide. The formation of an electron-deficient site triggered the attack of the oxygen of acetate on the phosphorus, and P–O bond formation was accomplished.
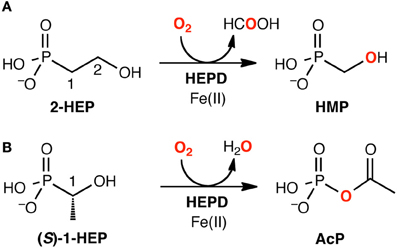
Scheme 9. HEPD-catalyzed reactions of (A) 2-HEP and (B) 1-HEP. Reprinted from Hirao and Morokuma (2011b) with permission from the American Chemical Society.
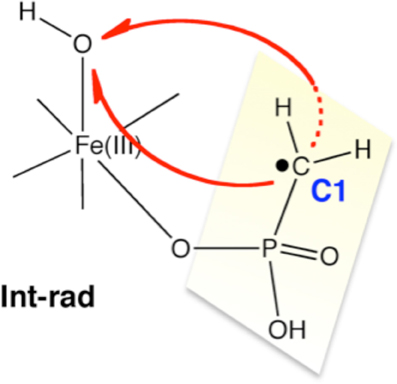
Scheme 10. Theoretically proposed radical intermediate. Reprinted from Hirao and Morokuma (2011b) with permission from the American Chemical Society.
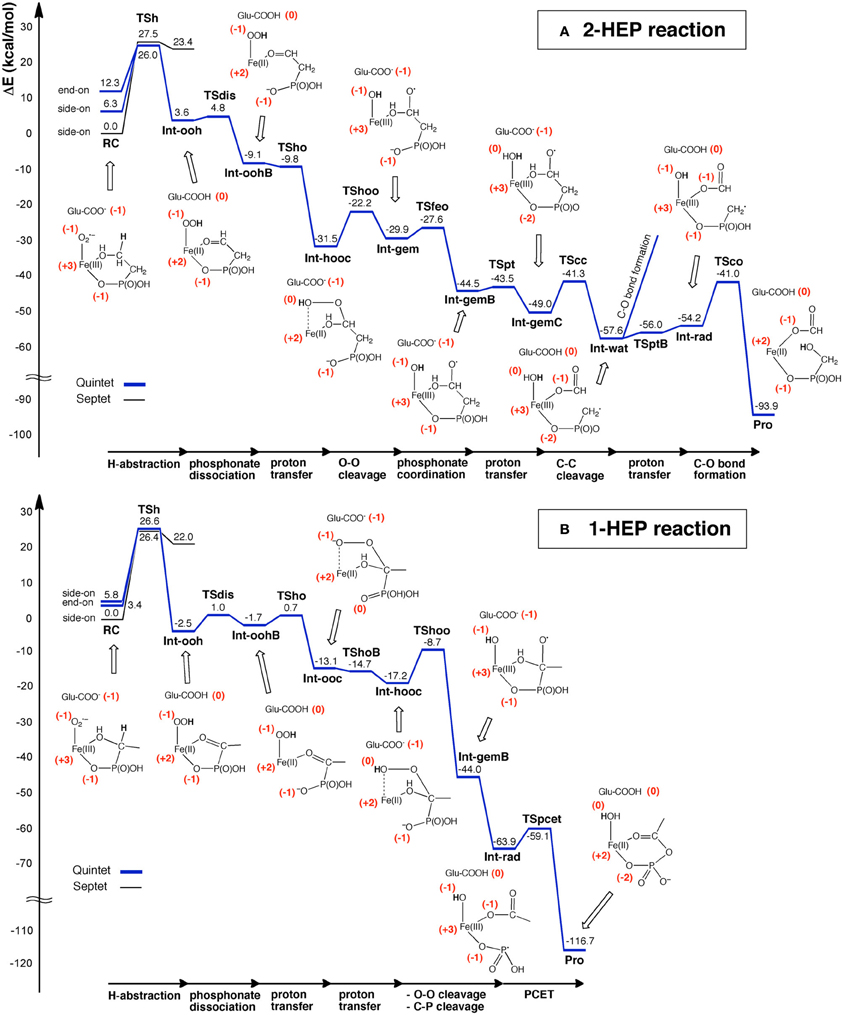
Figure 13. Energy profiles for the reactions of (A) 2-HEP and (B) 1-HEP, determined by ONIOM(DFT:MM) calculations. Reprinted from Hirao and Morokuma (2011b) with permission from the American Chemical Society.
Other Challenges
Although the usefulness of DFT calculations in studying iron-containing molecules has been well documented, there is a long way to go before DFT acquires the capability to describe iron-containing systems or any other transition-metal-containing systems perfectly (Harvey, 2006; Cramer and Truhlar, 2009).
First, reliable exchange-correlation functionals will need to be developed to describe accurately the spin-state ordering in various species. This goal is particularly important for studies of iron-containing molecules because multiple spin states often coexist within a narrow energy range. It is still not entirely clear which existing functional is most universally accurate for the description of iron-containing molecules in different oxidation states. For calibration purposes, comparisons of DFT with wave-function theories are often useful (Radoń and Broclawik, 2007; Swart, 2008; Chen et al., 2010, 2012; Vancoillie et al., 2010). Swart computed spin-state energies of several iron complexes that have an oxidation state of +2, +3, or +6, using CASPT2 and DFT methods, and suggested that the OPBE functional gives good results (Swart, 2008). Chen et al. applied CASPT2/MM to P450 and chloroperoxidase Cpd I, and showed that B3LYP/MM performed very well (Chen et al., 2010). Chen et al. also assessed the reliability of the B3LYP functional in calculating the spin-state ordering of the [(TMC)Fe(O2)]2+ species. To this end, they truncated the system to a smaller model ([(NH3)4Fe(O2)]2+) and applied the B3LYP and RCCSD(T) methods to the model. Their calculations showed that for most of the states, the relative energies obtained from these two methods were more or less similar (Chen et al., 2012).
In general, accurate evaluation of barrier heights of chemical reactions is a particularly important goal of DFT calculations, because barrier heights are directly linked to rate constants (Claeyssens et al., 2006; Llano and Gauld, 2010; Cho et al., 2012b). In many cases, DFT and DFT/MM provide reasonable trends in barrier heights at least qualitatively and are thus highly useful for mechanistic investigations. However, theoretically determined free energy barriers of reactions are often quite different from experimental ones. The reaction rate constant depends exponentially on the free energy barrier (according to the Eyring equation), and thus agreement of the rate constant between theory and experiment is even worse than that for the free energy. One of the major reasons for the error must be the insufficient accuracy of DFT functionals. Thus, DFT may not always be able to estimate reaction barriers accurately for a given spin state. In addition, the above-mentioned potentially poor descriptions of spin-state orderings could affect the accuracy of barrier heights, because there may be a switch in the spin state on a reaction pathway for iron-containing molecules. Reaction barriers are also influenced significantly by many weak interactions outside the reaction center, and in this respect, the dispersion effect is an important factor that has to be taken into account when improving DFT (Lynch and Truhlar, 2001; Zhao et al., 2004, 2005; Zhao and Truhlar, 2004; Grimme, 2006; Karton et al., 2008; Lonsdale et al., 2010, 2012; Siegbahn et al., 2010). Aside from the inherent problems of DFT, there are many other sources that make calculated free energy barriers of enzymatic reactions inaccurate (Llano and Gauld, 2010). For example, the effects of entropy (Hu and Yang, 2008; Lundberg et al., 2009) and tunneling (Cui and Karplus, 2002; Knapp and Klinman, 2002; Knapp et al., 2002; Hatcher et al., 2004; Olsson et al., 2004a,b; Tejero et al., 2006; Hammes-Schiffer and Soudackov, 2008; Hammes-Schiffer et al., 2008; Iyengar et al., 2008; Klinman, 2009; Phatak et al., 2012; Hammes-Schiffer, 2013) affect the rate constants of enzymatic reactions (or even synthetic complexes) profoundly. For realistic descriptions of enzymatic reactions, the reaction dynamics of iron enzymes may also be investigated actively using QM/MM (Lian et al., 2013).
The chemical reactions of iron enzymes often appear to be very simple. For example, a reaction may involve only the transfer of a hydrogen atom; however, the nature of the hydrogen transfer could differ critically. In some cases hydrogen may undergo “hydrogen atom transfer (HAT),” while in other cases, it may undergo “proton-coupled electron transfer (PCET).” Thermodynamic parameters such as redox potentials and pKa-values provide valuable insights into these ubiquitous processes; thus, theoretical evaluation of these quantities using DFT or DFT/MM is a key challenge (Kamerlin et al., 2009; Siegbahn and Blomberg, 2010; Hughes and Friesner, 2012; Castro and Bühl, 2014). Apart from the quantitative treatment of hydrogen-transfer processes, devising ways to derive chemical insight into the mechanistic complexities underlying these processes also presents an important goal (Shaik et al., 2010b,c; Usharani et al., 2013a,b).
The reactivity patterns of iron-containing enzymes or complexes cannot be easily generalized. For example, non-heme iron enzymes use not only iron(IV)-oxo or ferric-superoxide reactive species (Ye and Neese, 2009; Lundberg and Borowski, 2013), but also iron(III)-hydroxide to trigger a reaction (Knapp and Klinman, 2002; Knapp et al., 2002; Hatcher et al., 2004; Olsson et al., 2004a,b; Tejero et al., 2006; Hammes-Schiffer et al., 2008; Hammes-Schiffer and Soudackov, 2008; Iyengar et al., 2008; Klinman, 2009; Hirao and Morokuma, 2010b; Phatak et al., 2012; Hammes-Schiffer, 2013). DFT and DFT/MM investigations of intermediates and reactions of individual enzymes and synthetic complexes will continue to play valuable roles in their characterization. DFT-based spectroscopic studies of reaction intermediates will also provide us with crucial information on iron enzymes and complexes (Neese, 2006, 2009; Orio et al., 2009; Römelt et al., 2009; Chandrasekaran et al., 2011).
Summary
We have reviewed several recent applications of DFT to iron-containing synthetic complexes and enzymes, but not exhaustively. DFT provides insight into structural and spectroscopic features, chemical reaction mechanisms, etc., often with high reliability. Furthermore, DFT can be combined with other theoretical techniques, such as EDA schemes, to gain deeper insight into the nature of molecular interactions. Even for systems that contain thousands of atoms, we can still apply DFT by employing some hybrid approach (e.g., DFT/MM or DFT/DFT). Clearly, DFT is already a useful tool for the investigation of fundamental aspects of molecules. We believe that, in the future, DFT will become more accurate and play increasingly important roles, especially in practical areas such as catalyst design and drug design.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Nanyang Assistant Professorship and a MOE AcRF Tier 1 Grant (RG14/12).
References
Abu-Omar, M. M., Loaiza, A., and Hontzeas, N. (2005). Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 105, 2227–2252. doi: 10.1021/cr040653o
Ackermann, L. (2011). Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: mechanism and scope. Chem. Rev. 111, 1315–1345. doi: 10.1021/cr100412j
ADF2012.01. (2012). SCM Theoretical Chemistry. Amsterdam: Vrije Universiteit. Available online at: http://www.scm.com
Altun, A., Guallar, V., Friesner, R. A., Shaik, S., and Thiel, W. (2006). The effect of heme environment on the hydrogen abstraction reaction of camphor in P450cam catalysis: a QM/MM study. J. Am. Chem. Soc. 128, 3924–3925. doi: 10.1021/ja058196w
Altun, A., Kumar, D., Neese, F., and Thiel, W. (2008). Multireference ab initio quantum mechanics/molecular mechanics study on intermediates in the catalytic cycle of cytochrome P450cam. J. Phys. Chem. A 112, 12904–12910. doi: 10.1021/jp802092w
Altun, A., Shaik, S., and Thiel, W. (2007). What is the active species of cytochrome P450 during camphor hydroxylation? QM/MM studies of different electronic states of compound I and of reduced and oxidized iron–oxo intermediates. J. Am. Chem. Soc. 129, 8978–8987. doi: 10.1021/ja066847y
Altun, A., and Thiel, W. (2005). Combined quantum mechanical/molecular mechanical study on the pentacoordinated ferric and ferrous cytochrome P450cam complexes. J. Phys. Chem. B 109, 1268–1280. doi: 10.1021/jp0459108
Ansari, A., Kaushik, A., and Rajaraman, G. (2013). Mechanistic insights on the ortho-hydroxylation of aromatic compounds by non-heme iron complex: a computational case study on the comparative oxidative ability of ferric-hydroperoxo and high-valent FeIV=O and FeV=O intermediates. J. Am. Chem. Soc. 135, 4235–4249. doi: 10.1021/ja307077f
Bach, R. D., and Dmitrenko, O. (2006). The “somersault” mechanism for the P-450 hydroxylation of hydrocarbons. The intervention of transient inverted metastable hydroperoxides. J. Am. Chem. Soc. 128, 1474–1488. doi: 10.1021/ja052111+
Baerends, E. J., and Gritsenko, O. V. (1997). A quantum chemical view of density functional theory. J. Phys. Chem. A 101, 5383–5403. doi: 10.1021/jp9703768
Baik, M.-H., Newcomb, M., Friesner, R. A., and Lippard, S. J. (2003). Mechanistic studies on the hydroxylation of methane by methane monooxygenase. Chem. Rev. 103, 2385–2420. doi: 10.1021/cr950244f
Balcells, D., Clot, E., and Eisenstein, O. (2010). C-H bond activation in transition metal species from a computational perspective. Chem. Rev. 110, 749–823. doi: 10.1021/cr900315k
Bassan, A., Blomberg, M. R. A., Siegbahn, P. E. M., and Que, L. Jr. (2002). A density functional study of O–O bond cleavage for a biomimetic non-heme iron complex demonstrating an FeV-intermediate. J. Am. Chem. Soc. 124, 11056–11063. doi: 10.1021/ja026488g
Bassan, A., Blomberg, M. R. A., Siegbahn, P. E. M., and Que, L. Jr. (2005a). A density functional study of a biomimetic non-heme iron catalyst: insights into alkane hydroxylation by a formally HO-FeV=O oxidant. Chem. Eur. J. 11, 692–705. doi: 10.1002/chem.200400383
Bassan, A., Blomberg, M. R. A., Siegbahn, P. E. M., and Que, L. Jr. (2005b). Two faces of a biomimetic non-heme HO-FeV=O oxidant: olefin epoxidation versus cis-dihydroxylation. Angew. Chem. Int. Ed. Engl. 44, 2939–2941. doi: 10.1002/anie.200463072
Bathelt, C. M., Mulholland, A. J., and Harvey, J. N. (2008). QM/MM modeling of benzene hydroxylation in human cytochrome P450 2C9. J. Phys. Chem. A 112, 13149–13156. doi: 10.1021/jp8016908
Bathelt, C. M., Zurek, J., Mulholland, A. J., and Harvey, J. N. (2005). Electronic structure of compound I in human isoforms of cytochrome P450 from QM/MM modeling. J. Am. Chem. Soc. 127, 12900–12908. doi: 10.1021/ja0520924
Bautz, J., Bukowski, M. R., Kerscher, M., Stubna, A., Comba, P., Lienke, A., et al. (2006). Formation of an aqueous oxoiron(IV) complex at pH 2–6 from a nonheme iron(II) complex and H2O2. Angew. Chem. Int. Ed. Engl. 45, 5681–5684. doi: 10.1002/anie.200601134
Bergman, R. G. (2007). Organometallic chemistry: C–H activation. Nature 446, 391–393. doi: 10.1038/446391a
Bernasconi, L., and Baerends, E. J. (2008). The EDTA complex of oxidoiron(IV) as realisation of an optimal ligand environment for high activity of FeO2+. Eur. J. Inorg. Chem. 2008, 1672–1681. doi: 10.1002/ejic.200701135
Bernasconi, L., and Baerends, E. J. (2013). A frontier orbital study with ab initio molecular dynamics of the effects of solvation on chemical reactivity: solvent-induced orbital control in FeO-activated hydroxylation reactions. J. Am. Chem. Soc. 135, 8857–8867. doi: 10.1021/ja311144d
Bernasconi, L., Belanzoni, P., and Baerends, E. J. (2011). An abiotic analogue of the diiron(IV)oxo “diamond core” of soluble methane monooxygenase generated by direct activation of O2 in aqueous Fe(II)/EDTA solutions: thermodynamics and electronic structure. Phys. Chem. Chem. Phys. 13, 15272–15282. doi: 10.1039/c1cp21244c
Bernasconi, L., Louwerse, M. J., and Baerends, E. J. (2007). The role of equatorial and axial ligands in promoting the activity of non-heme oxidoiron(IV) catalysts in alkane hydroxylation. Eur. J. Inorg. Chem. 2007, 3023–3033. doi: 10.1002/ejic.200601238
Berry, J. F., Bill, E., Bothe, E., Neese, F., and Wieghardt, K. (2006). Octahedral non-heme oxo and non-oxo Fe(IV) complexes: an experimental/theoretical comparison. J. Am. Chem. Soc. 128, 13515–13528. doi: 10.1021/ja063590v
Bollinger, J. M. Jr., Diao, Y., Matthews, M. L., Xing, G., and Krebs, C. (2009). myo-Inositol oxygenase: a radical new pathway for O2 and C–H activation at a nonheme diiron cluster. Dalton Trans. 905–914. doi: 10.1039/b811885j
Bollinger, J. M. Jr., and Krebs, C. (2006). Stalking intermediates in oxygen activation by iron enzymes: motivation and method. J. Inorg. Biochem. 100, 586–605. doi: 10.1016/j.jinorgbio.2006.01.022
Bollinger, J. M. Jr., and Krebs, C. (2007). Enzymatic C–H activation by metal–superoxo intermediates. Curr. Opin. Chem. Biol. 11, 151–158. doi: 10.1016/j.cbpa.2007.02.037
Bukowski, M. R., Koehntop, K. D., Stubna, A., Bominaar, E. L., Halfen, J. A., Münck, E., et al. (2005). A thiolate-ligated nonheme oxoiron(IV) complex relevant to cytochrome P450. Science 310, 1000–1002. doi: 10.1126/science.1119092
Burke, K. (2012). Perspective on density functional theory. J. Chem. Phys. 136, 150901. doi: 10.1063/1.4704546
Castro, L., and Bühl, M. (2014). Calculations of one-electron redox potentials of oxoiron(IV) porphyrin complexes. J. Chem. Theory Comput. 10, 243–251. doi: 10.1021/ct400975w
Chandrasekaran, P., Stieber, S. C. E., Collins, T. J., Que, L. Jr., Neese, F., and Debeer, S. (2011). Prediction of high-valent iron K-edge absorption spectra by time-dependent density functional theory. Dalton Trans. 40, 11070–11079. doi: 10.1039/c1dt11331c
Chen, H., Cho, K.-B., Lai, W., Nam, W., and Shaik, S. (2012). Dioxygen activation by a non-heme iron(II) complex: theoretical study toward understanding ferric–superoxo complexes. J. Chem. Theory Comput. 8, 915–926. doi: 10.1021/ct300015y
Chen, H., Hirao, H., Derat, E., Schlichting, I., and Shaik, S. (2008). Quantum mechanical/molecular mechanical study on the mechanisms of compound I formation in the catalytic cycle of chloroperoxidase: an overview on heme enzymes. J. Phys. Chem. B 112, 9490–9500. doi: 10.1021/jp803010f
Chen, H., Lai, W. Z., Yao, J. N., and Shaik, S. (2011). Perferryl FeV–oxo nonheme complexes: do they have high-spin or low-spin ground states? J. Chem. Theory Comput. 7, 3049–3053. doi: 10.1021/ct200614g
Chen, H., Song, J. S., Lai, W. Z., Wu, W., and Shaik, S. (2010). Multiple low-lying states for compound I of P450cam and chloroperoxidase revealed from multireference ab initio QM/MM calculations. J. Chem. Theory Comput. 6, 940–953. doi: 10.1021/ct9006234
Chen, M. S., and White, M. C. (2007). A predictably selective aliphatic C–H oxidation reaction for complex molecule synthesis. Science 318, 783–787. doi: 10.1126/science.1148597
Cho, J., Jeon, S., Wilson, S. A., Liu, L. V., Kang, E. A., Braymer, J. J., et al. (2011a). Structure and reactivity of a mononuclear non-haem iron(III)–peroxo complex. Nature 478, 502–505. doi: 10.1038/nature10535
Cho, K.-B., Chen, H., Janardanan, D., de Visser, S. P., Shaik, S., and Nam, W. (2012a). Nonheme iron-oxo and -superoxo reactivities: O2 binding and spin inversion probability matter. Chem. Commun. 48, 2189–2191. doi: 10.1039/c2cc17610f
Cho, K.-B., Hirao, H., Chen, H., Carvajal, M. A., Cohen, S., Derat, E., et al. (2008). Compound I in heme thiolate enzymes: a comparative QM/MM study. J. Phys. Chem. A 112, 13128–13138. doi: 10.1021/jp806770y
Cho, K.-B., Kim, E. J., Seo, M. S., Shaik, S., and Nam, W. (2012b). Correlating DFT energy barriers to experiments in nonheme octhedral FeIV=O species. Chem. Eur. J. 18, 10444–10453. doi: 10.1002/chem.201200096
Cho, K.-B., Lai, W., Hamberg, M., Raman, C., and Shaik, S. (2011b). The reaction mechanism of allene oxide synthase: interplay of theoretical QM/MM calculations and experimental investigations. Arch. Biochem. Biophys. 507, 14–25. doi: 10.1016/j.abb.2010.07.016
Cho, K.-B., Shaik, S., and Nam, W. (2010). Theoretical predictions of a highly reactive non-heme Fe(IV)=O complex with a high-spin ground state. Chem. Commun. 46, 4511–4513. doi: 10.1039/c0cc00292e
Cho, K.-B., Wu, X., Lee, Y. M., Kwon, Y. H., Shaik, S., and Nam, W. (2013). Evidence for an alternative to the oxygen rebound mechanism in C–H bond activation by non-heme FeIVO complexes. J. Am. Chem. Soc. 134, 20222–20225. doi: 10.1021/ja308290r
Chung, L. W., Hirao, H., Li, X., and Morokuma, K. (2011a). The ONIOM method: its foundation and applications to metalloenzymes and photobiology. WIREs Comput. Mol. Sci. 2, 327–350. doi: 10.1002/wcms.85
Chung, L. W., Li, X., Hirao, H., and Morokuma, K. (2011b). Comparative reactivity of ferric-superoxo and ferryl-oxo species in heme and non-heme complexes. J. Am. Chem. Soc. 133, 20076–20079. doi: 10.1021/ja2084898
Cicchillo, R. M., Zhang, H., Blodgett, J. A. V., Whitteck, J. T., Li, G., Nair, S. K., et al. (2009). An unusual carbon–carbon bond cleavage reaction during phosphinothricin biosynthesis. Nature 459, 871–874. doi: 10.1038/nature07972
Claeyssens, F., Harvey, J. N., Manby, F. R., Mata, R. A., Mulholland, A. J., Ranaghan, K. E., et al. (2006). High-accuracy computation of reaction barriers in enzymes. Angew. Chem. Int. Ed. Engl. 45, 6856–6859. doi: 10.1002/anie.200602711
Cohen, A. J., Mori-Sánchez, P., and Yang, W. (2012). Challenges for density functinoal theory. Chem. Rev. 112, 289–320. doi: 10.1021/cr200107z
Cohen, S., Kumar, D., and Shaik, S. (2006). In silico design of a mutant of cytochrome P450 containing selenocysteine. J. Am. Chem. Soc. 128, 2649–2653. doi: 10.1021/ja056586c
Comba, P., Maurer, M., and Vadivelu, P. (2008). Oxidation of cyclohexane by a high-valent iron bispidine complex: a combined experimental and computational mechanistic study. J. Phys. Chem. A 112, 13028–13036. doi: 10.1021/jp8037895
Costas, M., Chen, K., and Que, L. Jr. (2000). Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 200–202, 517–544. doi: 10.1016/S0010-8545(00)00320-9
Costas, M., Mehn, M. P., Jensen, M. P., and Que, L. Jr. (2004). Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986. doi: 10.1021/cr020628n
Cramer, C. J., and Truhlar, D. G. (2009). Density functional theory for transition metals and transitionmetal chemistry. Phys. Chem. Chem. Phys. 11, 10757–10816. doi: 10.1039/b907148b
Cui, Q., Elstner, M., Kaxiras, E., Frauenheim, T., and Karplus, M. (2001). A QM/MM implementation of the self-consistent charge density functional tight binding (SCC-DFTB) method. J. Phys. Chem. B 105, 569–585. doi: 10.1021/jp0029109
Cui, Q., and Karplus, M. (2002). Quantum mechanics/molecular mechanics studies of triosephosphate isomerase-catalyzed reactions: effect of geometry and tunneling on proton-transfer rate constants. J. Am. Chem. Soc. 124, 3093–3124. doi: 10.1021/ja0118439
Davies, H. M. L., and Manning, J. R. (2008). Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 451, 417–424. doi: 10.1038/nature06485
Davydov, R., Makris, T. M., Kofman, V., Werst, D. E., Sligar, S. G., and Hoffman, B. M. (2001). Hydroxylation of camphor by reduced oxy-cytochrome P450cam: mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc. 123, 1403–1415. doi: 10.1021/ja003583l
Dawson, J. H., and Sono, M. (1987). Cytochrome P-450 and chloroperoxidase: thiolate-ligated heme enzymes. Spectroscopic determination of their active-site structures and mechanistic implications of thiolate ligation. Chem. Rev. 87, 1255–1276. doi: 10.1021/cr00081a015
de Visser, S. P. (2006). What factors influence the ratio of C-H hydroxylation versus C=C epoxidation by a nonheme cytochrome P450 biomimetic? J. Am. Chem. Soc. 128, 15809–15818. doi: 10.1021/ja065365j
de Visser, S. P. (2010). Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 132, 1087–1097. doi: 10.1021/ja908340j
de Visser, S. P., Kumar, D., and Shaik, S. (2004). How do aldehyde side products occur during alkene epoxidation by cytochrome P450? Theory reveals a state-specific multi-state scenario where the high-spin component leads to all side products. J. Inorg. Biochem. 98, 1183–1193. doi: 10.1016/j.jinorgbio.2004.01.015
de Visser, S. P., Latifi, R., Tahsini, L., and Nam, W. (2011). The axial ligand effect on aliphatic and aromatic hydroxylation by non-heme iron(IV)-oxo biomimetic complexes. Chem. Asian J. 6, 493–504. doi: 10.1002/asia.201000586
de Visser, S. P., Ogliaro, F., Sharma, P. K., and Shaik, S. (2002). What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 124, 11809–11826. doi: 10.1021/ja026872d
de Visser, S. P., Oh, K., Han, A. R., and Nam, W. (2007). Combined experimental and theoretical study on aromatic hydroxylation by mononuclear nonheme iron(IV)-oxo complexes. Inorg. Chem. 46, 4632–4641. doi: 10.1021/ic700462h
de Visser, S. P., Rohde, J. U., Lee, Y. M., Cho, J., and Nam, W. (2013). Intrinsic properties and reactivities of mononuclear nonheme iron–oxygen complexes bearing the tetramethylcyclam ligand. Coord. Chem. Rev. 257, 381–393. doi: 10.1016/j.ccr.2012.06.002
Decker, A., Rohde, J. U., Klinker, E. J., Wong, S. D., Que, L. Jr., and Solomon, E. I. (2007). Spectroscopic and quantum chemical studies on low-spin FeIV=O complexes: Fe-O bonding and its contributions to reactivity. J. Am. Chem. Soc. 129, 15983–15996. doi: 10.1021/ja074900s
Decker, A., and Solomon, E. I. (2005). Comparison of FeIV=O heme and non-heme species: electronic structures, bonding, and reactivities. Angew. Chem. Int. Ed. Engl. 44, 2252–2255. doi: 10.1002/anie.200462182
Denisov, I. G., Makris, T. M., Sligar, S. G., and Schlichting, I. (2005). Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2278. doi: 10.1021/cr0307143
Derat, E., Kumar, D., Hirao, H., and Shaik, S. (2006). Gauging the relative oxidative powers of compound I, ferric-hydroperoxide, and the ferric-hydrogen peroxide species of cytochrome P450 toward C-H hydroxylation of a radical clock substrate. J. Am. Chem. Soc. 128, 473–484. doi: 10.1021/ja056328f
Derat, E., and Shaik, S. (2006). The Poulos-Kraut mechanism of compound I formation in horseradish peroxidase: a QM/MM study. J. Phys. Chem. B 110, 10526–10533. doi: 10.1021/jp055412e
Dhuri, S. N., Mi, S. S., Lee, Y. M., Hirao, H., Wang, Y., Nam, W., et al. (2008). Experiment and theory reveal the fundamental difference between two-state and single-state reactivity patterns in nonheme FeIV=O versus RuIV=O oxidants. Angew. Chem. Int. Ed. Engl. 47, 3356–3359. doi: 10.1002/anie.200705880
Dumas, V. G., Defelipe, L. A., Petruk, A. A., Turjanski, A. G., and Marti, M. A. (2013). QM/MM study of the C—C coupling reaction mechanism of CYP121, an essential cytochrome p450 of Mycobacterium tuberculosis. Proteins. doi: 10.1002/prot.24474
Feig, A. L., and Lippard, S. J. (1994). Reactions of non-heme iron(II) centers with dioxygen in biology and chemistry. Chem. Rev. 94, 759–805. doi: 10.1021/cr00027a011
Feixas, F., Solà, M., and Swart, M. (2009). Chemical bonding and aromaticity in metalloporphyrins. Can. J. Chem. 87, 1063–1073. doi: 10.1139/V09-037
Fiedler, A. T., and Que, L. Jr. (2009). Reactivities of Fe(IV) complexes with oxo, hydroxo, and alkylperoxo ligands: an experimental and computational study. Inorg. Chem. 48, 11038–11047. doi: 10.1021/ic901391y
Field, M. J., Bash, P. A., and Karplus, M. (1990). A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 11, 700–733. doi: 10.1002/jcc.540110605
Fishelovitch, D., Hazan, C., Hirao, H., Wolfson, H. J., Nussinov, R., and Shaik, S. (2007). QM/MM study of the active species of the human cytochrome P450 3A4, and the influence thereof of the multiple substrate binding. J. Phys. Chem. B 111, 13822–13832. doi: 10.1021/jp076401j
Friedle, S., Reisner, E., and Lippard, S. J. (2010). Current challenges of modeling diiron enzyme active sites for dioxygenactivation by biomimetic synthetic complexes. Chem. Soc. Rev. 39, 2768–2779. doi: 10.1039/c003079c
Friesner, R. A., and Guallar, V. (2005). Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis. Annu. Rev. Phys. Chem. 56, 389–427. doi: 10.1146/annurev.physchem.55.091602.094410
Gao, J. (1996). Hybrid quantum and molecular mechanical simulations: an alternative avenue to solvent effects in organic chemistry. Acc. Chem. Res. 29, 298–305. doi: 10.1021/ar950140r
Geng, C., Ye, S., and Neese, F. (2010). Analysis of reaction channels for alkane hydroxylation by nonheme iron(IV)–oxo complexes. Angew. Chem. Int. Ed. Engl. 49, 5717–5720. doi: 10.1002/anie.201001850
Godula, K., and Sames, D. (2006). C-H bond functionalization in complex organic synthesis. Science 312, 67–72. doi: 10.1126/science.1114731
Goldberg, K. I., and Goldman, A. S. (2004). “Activation and functionalization of C–H bonds,” in ACS Symposium Series, Vol. 885 (Washington, DC: American Chemical Society). doi: 10.1021/bk-2004-0885
Gonzalez-Ovalle, L. E., Quesne, M. G., Kumar, D., Goldberg, D. P., and de Visser, S. P. (2012). Axial and equatorial ligand effects on biomimetic cysteine dioxygenase model complexes. Org. Biomol. Chem. 10, 5401–5409. doi: 10.1039/c2ob25406a
Gopakumar, G., Belanzoni, P., and Baerends, E. J. (2012). Hydroxylation catalysis by mononuclear and dinuclear iron oxo catalysts: a methane monooxygenase model system versus the Fenton reagent FeIVO(H2O)52+. Inorg. Chem. 51, 63–75. doi: 10.1021/ic200754w
Gordon, M. S., and Schmidt, M. W. (2005). “Advances in electronic structure theory: GAMESS a decade later,” in Theory and Applications of Computational Chemistry, eds C. E. Dykstra, K. S. Frenking, and G. E. Scuseria (Amsterdam: Elsevier).
Green, M. T., Dawson, J. H., and Gray, H. B. (2004). Oxoiron(IV) in chloroperoxidase compound II is basic: implications for P450 chemistry. Science 304, 1653–1656. doi: 10.1126/science.1096897
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799. doi: 10.1002/jcc.20495
Groves, J. T. (1985). Key elements of the chemistry of cytochrome P-450: the oxygen rebound mechanism. J. Chem. Educ. 62, 928–931. doi: 10.1021/ed062p928
Groves, J. T. (2006). High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 100, 434–447. doi: 10.1016/j.jinorgbio.2006.01.012
Groves, J. T. (2014). Enzymatic C–H bond activation: using push to get pull. Nat. Chem. 6, 89–91. doi: 10.1038/nchem.1855
Groves, J. T., McClusky, G. A., White, R. E., and Coon, M. J. (1978). Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun. 81, 154–160. doi: 10.1016/0006-291X(78)91643-1
Guallar, V., and Friesner, R. A. (2004). Cytochrome P450CAM enzymatic catalysis cycle: a quantum mechanics/molecular mechanics study. J. Am. Chem. Soc. 126, 8501–8508. doi: 10.1021/ja036123b
Guengerich, F. P. (2001). Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 14, 611–650. doi: 10.1021/tx0002583
Guengerich, F. P. (2008). Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 21, 70–83. doi: 10.1021/tx700079z
Guengerich, F. P. (2013). New trends in cytochrome P450 research at the half-century mark. J. Biol. Chem. 288, 17063–17064. doi: 10.1074/jbc.R113.466821
Hackett, J. C., Brueggemeier, R. W., and Hadad, C. M. (2005). The final catalytic step of cytochrome P450 aromatase: a density functional theory study. J. Am. Chem. Soc. 127, 5224–5237. doi: 10.1021/ja044716w
Hackett, J. C., Sanan, T. T., and Hadad, C. M. (2007). Oxidative dehalogenation of perhalogenated benzenes by cytochrome P450 compound I. Biochemistry 46, 5924–5940. doi: 10.1021/bi700365x
Hammes-Schiffer, S. (2013). Catalytic efficiency of enzymes: a theoretical analysis. Biochemistry 52, 2012–2020. doi: 10.1021/bi301515j
Hammes-Schiffer, S., Hatcher, E., Ishikita, H., Skone, J. H., and Soudackov, A. V. (2008). Theoretical studies of proton-coupled electron transfer: models and concepts relevant to bioenergetics. Coord. Chem. Rev. 252, 384–394. doi: 10.1016/j.ccr.2007.07.019
Hammes-Schiffer, S., and Soudackov, A. V. (2008). Proton-coupled electron transfer in solution, proteins, and electrochemistry. J. Phys. Chem. B 112, 14108–14123. doi: 10.1021/jp805876e
Hanson, K. L., VandenBrink, B. M., Babu, K. N., Allen, K. E., Nelson, W. L., and Kunze, K. L. (2010). Sequential metabolism of secondary alkyl amines to metabolic-intermediate complexes: opposing roles for the secondary hydroxylamine and primary amine metabolites of desipramine, (S)-fluoxetine, and N-desmethyldiltiazem. Drug Metab. Dispos. 38, 963–972. doi: 10.1124/dmd.110.032391
Harvey, J. N. (2006). On the accuracy of density functional theory in transition metal chemistry. Annu. Rep. Prog. Chem. Sect. C 102, 203–226. doi: 10.1039/b419105f
Harvey, J. N., Bathelt, C. M., and Mulholland, A. J. (2006). QM/MM modeling of compound I active species in cytochrome P450, cytochrome C peroxidase, and ascorbate peroxidase. J. Comput. Chem. 27, 1352–1362. doi: 10.1002/jcc.20446
Hatcher, E., Soudackov, A. V., and Hammes-Schiffer, S. (2004). Proton-coupled electron transfer in soybean lipoxygenase. J. Am. Chem. Soc. 126, 5763–5775. doi: 10.1021/ja039606o
Hegg, E. L., and Que, L. Jr. (1997). The 2-his-1-carboxylate facial triad—an emerging structural motif in mononuclear non-heme iron(II) enzymes. Eur. J. Biochem. 250, 625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x
Himo, F., and Siegbahn, P. E. M. (2003). Quantum chemical studies of radical-containing enzymes. Chem. Rev. 103, 2421–2456. doi: 10.1021/cr020436s
Hirao, H. (2011a). Energy decomposition analysis of the protein environmental effect: the case of cytochrome P450cam compound I. Chem. Lett. 40, 1179–1181. doi: 10.1246/cl.2011.1179
Hirao, H. (2011b). The effects of protein environment and dispersion on the formation of ferric-superoxide species in myo-inositol oxygenase (MIOX): a combined ONIOM(DFT:MM) and energy decomposition analysis. J. Phys. Chem. B 115, 11278–11285. doi: 10.1021/jp2057173
Hirao, H., and Morokuma, K. (2009). Insights into the (superoxo)Fe(III)Fe(III) intermediate and reaction mechanism of myo-inositol oxygenase: DFT and ONIOM(DFT:MM) study. J. Am. Chem. Soc. 131, 17206–17214. doi: 10.1021/ja905296w
Hirao, H., and Morokuma, K. (2010a). Ferric superoxide and ferric hydroxide are used in the catalytic mechanism of hydroxyethylphosphonate dioxygenase: a density functional theory investigation. J. Am. Chem. Soc. 132, 17901–17909. doi: 10.1021/ja108174d
Hirao, H., and Morokuma, K. (2010b). What is the real nature of ferrous soybean lipoxygenase-1? A new two-conformation model based on combined ONIOM(DFT:MM) and multireference configuration interaction characterization. J. Phys. Chem. Lett. 1, 901–906. doi: 10.1021/jz1001456
Hirao, H., and Morokuma, K. (2011a). Recent progress in the theoretical studies of structure, function, and reaction of biological molecules. Yakugaku Zasshi 131, 1151–1161. doi: 10.1248/yakushi.131.1151
Hirao, H., and Morokuma, K. (2011b). ONIOM(DFT:MM) study of 2-hydroxyethylphosphonate dioxygenase: what determines the destinies of different substrates? J. Am. Chem. Soc. 133, 14550–14553. doi: 10.1021/ja206222f
Hirao, H., Chen, H., Carvajal, M. A., Wang, Y., and Shaik, S. (2008b). Effect of external electric fields on the C-H bond activation reactivity of nonheme iron-oxo reagents. J. Am. Chem. Soc. 130, 3319–3327. doi: 10.1021/ja070903t
Hirao, H., Cheong, Z. H., and Wang, X. (2012). Pivotal role of water in terminating enzymatic function: a density functional theory study of the mechanism-based inactivation of cytochromes P450. J. Phys. Chem. B 116, 7787–7794. doi: 10.1021/jp302592d
Hirao, H., Cho, K.-B., and Shaik, S. (2008c). QM/MM theoretical study of the pentacoordinate Mn(III) and resting states of manganese-reconstituted cytochrome P450cam. J. Biol. Inorg. Chem. 13, 521–530. doi: 10.1007/s00775-007-0340-9
Hirao, H., Chuanprasit, P., Cheong, Y. Y., and Wang, X. (2013a). How is a metabolic intermediate formed in the mechanism-based inactivation of cytochrome P450 by using 1,1-dimethylhydrazine: hydrogen abstraction or N-nitrogen oxidation? Chem. Eur. J. 19, 7361–7369. doi: 10.1002/chem.201300689
Hirao, H., Kumar, D., Que, L. Jr., and Shaik, S. (2006a). Two-state reactivity in alkane hydroxylation by non-heme iron-oxo complexes. J. Am. Chem. Soc. 128, 8590–8606. doi: 10.1021/ja061609o
Hirao, H., Kumar, D., and Shaik, S. (2006b). On the identity and reactivity patterns of the “second oxidant” of the T252A mutant of cytochrome P450cam in the oxidation of 5-methylenenylcamphor. J. Inorg. Biochem. 100, 2054–2068. doi: 10.1016/j.jinorgbio.2006.09.001
Hirao, H., Kumar, D., Thiel, W., and Shaik, S. (2005). Two states and two more in the mechanisms of hydroxylation and epoxidation by cytochrome P450. J. Am. Chem. Soc. 127, 13007–13018. doi: 10.1021/ja053847+
Hirao, H., Li, F., Que, L. Jr., and Morokuma, K. (2011). Theoretical study of the mechanism of oxoiron(IV) formation from H2O2 and a nonheme iron(II) complex: O–O cleavage involving proton-coupled electron transfer. Inorg. Chem. 50, 6637–6648. doi: 10.1021/ic200522r
Hirao, H., Que, L. Jr., Nam, W., and Shaik, S. (2008a). A two-state reactivity rationale for counterintuitive axial ligand effects on the C-H activation reactivity of nonheme FeIV=O oxidants. Chem. Eur. J. 14, 1740–1756. doi: 10.1002/chem.200701739
Hirao, H., Thellamurege, N. M., Chuanprasit, P., and Xu, K. (2013b). Importance of H-abstraction in the final step of nitrosoalkane formation in the mechanism-based inactivation of cytochrome P450 by amine-containing drugs. Int. J. Mol. Sci. 14, 24692–24705. doi: 10.3390/ijms141224692
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous electron gas. Phys. Rev. B 136, 864–871. doi: 10.1103/PhysRev.136.B864
Hohenberger, J., Ray, K., and Meyer, K. (2012). The biology and chemistry of high-valent iron–oxo and iron–nitrido complexes. Nat. Commun. 3, 720. doi: 10.1038/ncomms1718
Hong, S., Lee, Y.-M., Cho, K.-B., Seo, M. S., Song, D., Yoon, J., et al. (2014). Conversion of high-spin iron(III)–alkylperoxo to iron(IV)–oxo species via O–O bond homolysis in nonheme iron models. Chem. Sci. 5, 156–162. doi: 10.1039/C3SC52236A
Hu, H., and Yang, W. (2008). Free energies of chemical reactions in solution and in enzymes with ab initio quantum mechanics/molecular mechanics methods. Annu. Rev. Phys. Chem. 59, 573–601. doi: 10.1146/annurev.physchem.59.032607.093618
Hu, H., and Yang, W. (2009). Development and application of ab initio QM/MM methods for mechanistic simulation of reactions in solution and in enzymes. J. Mol. Struct. Theochem 898, 17–30. doi: 10.1016/j.theochem.2008.12.025
Hughes, T. F., and Friesner, R. A. (2012). Development of accurate DFT methods for computing redox potentials of transition metal complexes: results for model complexes and application to cytochrome P450. J. Chem. Theory Comput. 8, 442–459. doi: 10.1021/ct2006693
Humbel, S., Sieber, S., and Morokuma, K. (1996). The IMOMO method: integration of different levels of molecular orbital approximations for geometry optimization of large systems: test for n-butane conformation and SN2 reaction: RCl+Cl−. J. Chem. Phys. 105, 1959–1967. doi: 10.1063/1.472065
Isin, E. M., and Guengerich, F. P. (2007). Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta 1770, 314–329. doi: 10.1016/j.bbagen.2006.07.003
Isobe, H., Nishihara, S., Shoji, M., Yamanaka, S., Shimada, J., Hagiwara, M., et al. (2008). Extended Hartree-Fock theory of chemical reactions. VIII. Hydroxylation reactions of chemical reactions by P450. Int. J. Quantum Chem. 108, 2991–3009. doi: 10.1002/qua.21874
Isobe, H., Yamaguchi, K., Okumura, M., and Shimada, J. (2012). Role of perferryl–oxo oxidant in alkane hydroxylation catalyzed by cytochrome P450: a hybrid density functional study. J. Phys. Chem. B 116, 4713–4730. doi: 10.1021/jp211184y
Isobe, H., Yamanaka, S., Okumura, M., Yamaguchi, K., and Shimada, J. (2011). Unique structural and electronic features of perferryl–oxo oxidant in cytochrome P450. J. Phys. Chem. B 115, 10730–10738. doi: 10.1021/jp206004y
Iyengar, S. S., Sumner, I., and Jakowski, J. (2008). Hydrogen tunneling in an enzyme active site: a quantum wavepacket dynamical perspective. J. Phys. Chem. B 112, 7601–7613. doi: 10.1021/jp7103215
Jia, C., Kitamura, T., and Fujiwara, Y. (2001). Catalytic functionalization of arenes and alkanes via C-H bond activation. Acc. Chem. Res. 34, 633–639. doi: 10.1021/ar000209h
Johansson, A. J., Blomberg, M. R. A., and Siegbahn, P. E. M. (2007). Quantum chemical modeling of the oxidation of dihydroanthracene by the biomimetic nonheme iron catalyst [(TMC)FeIV(O)]2+. J. Phys. Chem. C 111, 12397–12406. doi: 10.1021/jp0730444
Kaizer, J., Klinker, E. J., Oh, N. Y., Rohde, J. U., Song, W. J., Stubna, A., et al. (2004). Nonheme FeIVO complexes that can oxidize the C-H bonds of cyclohexane at room temperature. J. Am. Chem. Soc. 126, 472–473. doi: 10.1021/ja037288n
Kakiuchi, F., and Chatani, N. (2003). Catalytic methods for C-H bond functionalization: application in organic synthesis. Adv. Synth. Catal. 345, 1077–1101. doi: 10.1002/adsc.200303094
Kamachi, T., Shiota, Y., Ohta, T., and Yoshizawa, K. (2003). Does the hydroxo species of cytochrome P450 participate in olefein epoxidation with the main oxidant, compund I? Critisism from density functioal calculations. Bull. Chem. Soc. Jpn. 76, 721–732. doi: 10.1246/bcsj.76.721
Kamachi, T., and Yoshizawa, K. (2003). A theoretical study on the mechanism of camphor hydroxylation by compound I of cytochrome P450. J. Am. Chem. Soc. 125, 4652–4661. doi: 10.1021/ja0208862
Kamerlin, S. C. L., Haranczyk, M., and Warshel, A. (2009). Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B 113, 1253–1272. doi: 10.1021/jp8071712
Karton, A., Tarnopolsky, A., Lamère, J. F., Schatz, G. C., and Martin, J. M. L. (2008). Highly accurate first-principles benchmark data sets for the parametrization and validation of density functional and other approximate methods. Derivation of a robust, generally applicable, double-hybrid functional for thermochemistry and thermochemical kinetics. J. Phys. Chem. A, 112, 12868–12886. doi: 10.1021/jp801805p
Kim, S. O., Sastri, C. V., Seo, M. S., Kim, J., and Nam, W. (2005). Dioxygen activation and catalytic aerobic oxidation by a mononuclear nonheme iron(II) complex. J. Am. Chem. Soc., 127, 4178–4179. doi: 10.1021/ja043083i
Kim, Y. M., Cho, K.-B., Cho, J., Wang, B., Li, C., Shaik, S., et al. (2013). A mononuclear non-heme high-spin iron(III)–hydroperoxo complex as an active oxidant in sulfoxidation reactions. J. Am. Chem. Soc. 135, 8838–8841. doi: 10.1021/ja404152q
Kitaura, K., and Morokuma, K. (1976). A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 10, 325–340. doi: 10.1002/qua.560100211
Klinker, E. J., Shaik, S., Hirao, H., and Que, L. Jr. (2009). A two-state reactivity model explains unusual kinetic isotope effect patterns in C-H bond cleavage by nonheme oxoiron(IV) complexes. Angew. Chem. Int. Ed. Engl. 48, 1291–1295. doi: 10.1002/anie.200804029
Klinman, J. P. (2009). An integrated model for enzyme catalysis emerges from studies of hydrogen tunneling. Chem. Phys. Lett. 471, 179–193. doi: 10.1016/j.cplett.2009.01.038
Knapp, M. J., and Klinman, J. P. (2002). Environmentally coupled hydrogen tunneling. Eur. J. Biochem. 269, 3113–3121. doi: 10.1046/j.1432-1033.2002.03022.x
Knapp, M. J., Rickert, K., and Klinman, J. P. (2002). Temperature-dependent isotope effects in soybean lipoxygenase-1: correlating hydrogen tunneling with protein dynamics. J. Am. Chem. Soc. 124, 3865–3874. doi: 10.1021/ja012205t
Koch, W., and Holthausen, M. C. (2001). A Chemist's Guide to Density Functional Theory, 2nd Edn. New York, NY: Wiley-VCH. doi: 10.1002/3527600043
Kohn, W. (1999). Electronic structure of matter—wave functions and density functionals. Rev. Mod. Phys. 71, 1253–1266. doi: 10.1103/RevModPhys.71.1253
Kohn, W., Becke, A. D., and Parr, R. G. (1996). Density functional theory of electronic structure. J. Phys. Chem. 100, 12974–12980. doi: 10.1021/jp960669l
Kohn, W., and Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138. doi: 10.1103/PhysRev.140.A1133
Krámos, B., and Oláh, J. (2014). Enolization as an alternative proton delivery pathway in human aromatase (P450 19A1). J. Phys. Chem. B 118, 390–405. doi: 10.1021/jp407365x
Krámos, B. Z., Menyhárd, D. R. K., and Oláh, J. (2012). Direct hydride shift mechanism and stereoselectivity of P450nor confirmed by QM/MM calculations. J. Phys. Chem. B 116, 872–885. doi: 10.1021/jp2080918
Krebs, C., Fujimori, D. G., Walsh, C. T., and Bollinger, J. M. Jr. (2007). Non-heme Fe(IV)–oxo intermediates. Acc. Chem. Res. 40, 484–492. doi: 10.1021/ar700066p
Kumar, D., Altun, A., Shaik, S., and Thiel, W. (2011). Water as biocatalyst in cytochrome P450. Faraday Discuss. 148, 373–383. doi: 10.1039/c004950f
Kumar, D., Hirao, H., Que, L. Jr., and Shaik, S. (2005). Theoretical investigation of C-H hydroxylation by (N4Py)FeIV=O2+: an oxidant more powerful than P450? J. Am. Chem. Soc. 127, 8026–8802. doi: 10.1021/ja0512428
Kwiecień, R. A., Le Questel, J.-Y., Lebreton, J., Delaforge, M., André, F., Pihan, E., et al. (2012). Cytochrome P450-catalyzed degradation of nicotine: fundamental parameters determining hydroxylation by cytochrome P450 2A6 at the 5′-Carbon or the N-methyl carbon. J. Phys. Chem. B 116, 7827–7840. doi: 10.1021/jp304276v
Labinger, J. A., and Bercaw, J. E. (2002). Understanding and exploiting C–H bond activation. Nature 417, 507–514. doi: 10.1038/417507a
Lai, W., Chen, H., Cohen, S., and Shaik, S. (2011). Will P450cam hydroxylate or desaturate alkanes? QM and QM/MM studies. J. Phys. Chem. Lett. 2, 2229–2235. doi: 10.1021/jz2007534
Lai, W., and Shaik, S. (2011). Can ferric-superoxide act as a potential oxidant in P450cam? QM/MM investigation of hydroxylation, epoxidation, and sulfoxidation. J. Am. Chem. Soc. 133, 5444–5452. doi: 10.1021/ja111376n
Lange, S. J., and Que, L. Jr. (1998). Oxygen activating nonheme iron enzymes. Curr. Opin. Chem. Biol. 2, 159–172. doi: 10.1016/S1367-5931(98)80057-4
Latifi, R., Tahsini, L., Nam, W., and de Visser, S. P. (2012). Regioselectivity of aliphatic versus aromatic hydroxylation by a nonheme iron(II)-superoxo complex. Phys. Chem. Chem. Phys. 14, 2518–2524. doi: 10.1039/c2cp23352e
Lee, Y. M., Bang, S., Kim, Y. M., Cho, J., Hong, S., Nomura, T., et al. (2013). A mononuclear nonheme iron(III)–peroxo complex binding redox-inactive metal ions. Chem. Sci. 4, 3917–3923. doi: 10.1039/c3sc51864g
Lee, Y.-M., Hong, S., Morimoto, Y., Shin, W., Fukuzumi, S., and Nam, W. (2010). Dioxygen activation by a non-heme iron(II) complex: formation of an iron(IV)-oxo complex via C-H activation by a putative iron(III)-superoxo species. J. Am. Chem. Soc. 132, 10668–10670. doi: 10.1021/ja103903c
Lehnert, N., Neese, F., Ho, R. Y. N., Que, L. Jr., and Solomon, E. I. (2002). Electronic structure and reactivity of low-spin Fe(III)-hydroperoxo complexes: comparison to activated bleomycin. J. Am. Chem. Soc. 124, 10810–10822. doi: 10.1021/ja012621d
Levy, M. (1979). Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem. Proc. Natl. Acad. Sci. U.S.A. 76, 6062–6065. doi: 10.1073/pnas.76.12.6062
Li, C., and Shaik, S. (2013). How do perfluorinated alkanoic acids elicit cytochrome P450 to catalyze methane hydroxylation? An MD and QM/MM study. RSC Advances 3, 2995–3005. doi: 10.1039/c2ra22294a
Li, C., Zhang, L., Zhang, C., Hirao, H., Wu, W., and Shaik, S. (2007). Which oxidant is really responsible for sulfur oxidation by cytochrome P450? Angew. Chem. Int. Ed. Engl. 46, 8168–8170. doi: 10.1002/anie.200702867
Li, D., Huang, X., Lin, J., and Zhan, C.-G. (2013). Catalytic mechanism of cytochrome P450 for N-methylhydroxylation of nicotine: reaction pathways and regioselectivity of the enzymatic nicotine oxidation. Dalton Trans. 42, 3812–3820. doi: 10.1039/c2dt32106h
Li, D., Wang, Y., and Han, K. (2012). Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord. Chem. Rev. 256, 1137–1150. doi: 10.1016/j.ccr.2012.01.016
Li, F., England, J., and Que, L. Jr. (2010). Near-stoichiometric conversion of H2O2 to FeIV=O at a nonheme iron(II) center. Insights into the O-O bond cleavage step. J. Am. Chem. Soc. 132, 2134–2135. doi: 10.1021/ja9101908
Li, F., Meier, K. K., Cranswick, M. A., Chakrabarti, M., Van Heuvelen, K. M., Münck, E., et al. (2011). Characterization of a high-spin non-heme FeIII–OOH intermediate and its quantitative conversion to an FeIV-O complex. J. Am. Chem. Soc. 133, 7256–7259. doi: 10.1021/ja111742z
Lian, P., Li, J., Wang, D.-Q., and Wei, D.-Q. (2013). Car–Parrinello molecular dynamics/molecular mechanics (CPMD/MM) simulation study of coupling and uncoupling mechanisms of cytochrome P450cam. J. Phys. Chem. B 117, 7849–7856. doi: 10.1021/jp312107r
Lim, H. M., Rohde, J. U., Stubna, A., Bukowski, M. R., Costas, M., Ho, R. Y. N., et al. (2003). An FeIV=O complex of a tetradentate tripodal nonheme ligand. Proc. Natl. Acad. Sci. U.S.A. 100, 3665–3670. doi: 10.1073/pnas.0636830100
Lin, H., Schöneboom, J. C., Cohen, S., Shaik, S., and Thiel, W. (2004). QM/MM study of the product-enzyme complex in P450cam catalysis. J. Phys. Chem. B 108, 10083–10088. doi: 10.1021/jp0493632
Lin, H., and Truhlar, D. G. (2007). QM/MM: what have we learned, where are we, and where do we go from here? Theor. Chem. Acc. 117, 185–199. doi: 10.1007/s00214-006-0143-z
Llano, J., and Gauld, J. W. (2010). Mechanistics of enzyme catalysis: From small to large active-site models. Quantum Biochem. 643–666. doi: 10.1002/9783527629213.ch23
Lonsdale, R., Harvey, J. N., and Mulholland, A. J. (2010). Inclusion of dispersion effects significantly improves accuracy of calculated reaction barriers for cytochrome P450 catalyzed reactions. J. Phys. Chem. Lett. 1, 3232–3237. doi: 10.1021/jz101279n
Lonsdale, R., Harvey, J. N., and Mulholland, A. J. (2012). Effects of dispersion in density functional based quantum mechanical/molecular mechanical calculations on cytochrome P450 catalyzed reactions. J. Chem. Theory Comput. 8, 4637–4645. doi: 10.1021/ct300329h
Lonsdale, R., Houghton, K. T., Zurek, J., Bathelt, C. M., Foloppe, N., De Groot, M. J., et al. (2013). Quantum mechanics/molecular mechanics modeling of regioselectivity of drug metabolism in cytochrome P450 2C9. J. Am. Chem. Soc. 135, 8001–8015. doi: 10.1021/ja402016p
Lonsdale, R., Oláh, J., Mulholland, A. J., and Harvey, J. N. (2011). Does compound I vary significantly between isoforms of cytochrome P450? J. Am. Chem. Soc. 133, 15464–15474. doi: 10.1021/ja203157u
Lundberg, M., and Borowski, T. (2013). Oxoferryl species in mononuclear non-heme iron enzymes: biosynthesis, properties and reactivity from a theoretical perspective. Coord. Chem. Rev. 257, 277–289. doi: 10.1016/j.ccr.2012.03.047
Lundberg, M., Kawatsu, T., Vreven, T., Frisch, M. J., and Morokuma, K. (2009). Transition states in a protein environment - ONIOM QM:MM modeling of isopenicillin N synthesis. J. Chem. Theory Comput. 5, 222–234. doi: 10.1021/ct800457g
Lynch, B. J., and Truhlar, D. G. (2001). How well can hybrid density functional methods predict transition state geometries and barrier heights? J. Phys. Chem. A 105, 2936–2941. doi: 10.1021/jp004262z
Lyons, T. W., and Sanford, M. S. (2010). Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 110, 1147–1169. doi: 10.1021/cr900184e
Makris, T. M., von Koenig, K., Schlichting, I., and Sligar, S. G. (2006). The status of high-valent metal oxo complexes in the P450 cytochromes. J. Inorg. Biochem. 100, 507–518. doi: 10.1016/j.jinorgbio.2006.01.025
Mas-Ballesté, R., McDonald, A. R., Reed, D., Usharani, D., Schyman, P., Milko, P., et al. (2012). Intramolecular gas-phase reactions of synthetic nonheme oxoiron(IV) ions: proximity and spin-state reactivity rules. Chem. Eur. J. 18, 11747–11760. doi: 10.1002/chem.201200105
Maseras, F., and Morokuma, K. (1995). IMOMM: a new integrated ab initio + molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J. Comput. Chem. 16, 1170–1179. doi: 10.1002/jcc.540160911
McDonald, A. R., Guo, Y. S., Vu, V., Bominaar, E. L., Munck, E., and Que, L. Jr. (2012). A mononuclear carboxylate-rich oxoiron(IV) complex: a structural and functional mimic of TauD intermediate J. Chem. Sci. 3, 1680–1693. doi: 10.1039/c2sc01044e
McDonald, A. R., and Que, L. Jr. (2013). High-valent nonheme iron-oxo complexes: synthesis, structure, and spectroscopy. Coord. Chem. Rev. 257, 414–428. doi: 10.1016/j.ccr.2012.08.002
Merkx, M., Kopp, D. A., Sazinsky, M. H., Blazyk, J. L., Müller, J., and Lippard, S. J. (2001). Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: a tale of two irons and three proteins. Angew. Chem. Int. Ed. Engl. 40, 2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P
Meunier, B., de Visser, S. P., and Shaik, S. (2004). Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 104, 3947–3980. doi: 10.1021/cr020443g
Murphy, R. B., Philipp, D. M., and Friesner, R. A. (2000). A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments. J. Comput. Chem. 21, 1442–1457. doi: 10.1002/1096-987X(200012)21:16<1442::AID-JCC3>3.0.CO;2-O
Myradalyyev, S., Limpanuparb, T., Wang, X., and Hirao, H. (2013). Comparative computational analysis of binding energies between several divalent first-row transition metals (Cr2+, Mn2+, Fe2+, Co2+, Ni2+, and Cu2+) and ligands (porphine, corrin, and TMC). Polyhedron 52, 96–101. doi: 10.1016/j.poly.2012.11.018
Nam, K., Gao, J., and York, D. M. (2005). An efficient linear-scaling ewald method for long-range electrostatic interactions in combined QM/MM calculations. J. Chem. Theory Comput. 1, 2–13. doi: 10.1021/ct049941i
Nam, W. (2007). High-valent iron(IV)–oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc. Chem. Res. 40, 522–531. doi: 10.1021/ar700027f
Nam, W., Lee, Y.-M., and Fukuzumi, S. (2014). Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron(IV)-oxo complexes. Acc. Chem. Res. doi: 10.1021/ar400258p. [Epub ahead of print].
Neese, F. (2006). A critical evaluation of DFT, including time-dependent DFT, applied to bioinorganic chemistry. J. Biol. Inorg. Chem. 11, 702–711. doi: 10.1007/s00775-006-0138-1
Neese, F. (2009). Prediction of molecular properties and molecular spectroscopy with density functional theory: from fundamental theory to exchange-coupling. Coord. Chem. Rev. 253, 526–563. doi: 10.1016/j.ccr.2008.05.014
Neidig, M. L., Decker, A., Choroba, O. W., Huang, F., Kavana, M., Moran, G. R., et al. (2006). Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes. Proc. Natl. Acad. Sci. U.S.A. 103, 12966–12973. doi: 10.1073/pnas.0605067103
Newmyer, S. L., and Ortiz de Montellano, P. R. (1995). Horseradish peroxidase His-42→Ala, His-42→Val, and Phe-41→Ala mutants: histidine catalysis and control of substrate access to the heme iron. J. Biol. Chem. 270, 19430–19438.
Noack, H., and Siegbahn, P. E. M. (2007). Theoretical investigation on the oxidative chlorination performed by a biomimetic non-heme iron catalyst. J. Biol. Inorg. Chem. 12, 1151–1162. doi: 10.1007/s00775-007-0284-0
Ogliaro, F., Cohen, S., de Visser, S. P., and Shaik, S. (2000b). Medium polarization and hydrogen bonding effects on compound I of cytochrome P450: what kind of a radical is it really? J. Am. Chem. Soc. 122, 12892–12893. doi: 10.1021/ja005619f
Ogliaro, F., de Visser, S. P., Cohen, S., Sharma, P. K., and Shaik, S. (2002). Searching for the second oxidant in the catalytic cycle of cytochrome P450: a theoretical investigation of the iron(III)-hydroperoxo species and its epoxidation pathways. J. Am. Chem. Soc. 124, 2806–2817. doi: 10.1021/ja0171963
Ogliaro, F., Harris, N., Cohen, S., Filatov, M., de Visser, S. P., and Shaik, S. (2000a). A model “rebound” mechanism of hydroxylation by cytochrome P450: stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 122, 8977–8989. doi: 10.1021/ja991878x
Olsson, M. H. M., Siegbahn, P. E. M., and Warshel, A. (2004a). Simulating large nuclear quantum mechanical corrections in hydrogen atom transfer reactions in metalloenzymes. J. Biol. Inorg. Chem. 9, 96–99. doi: 10.1007/s00775-003-0503-2
Olsson, M. H. M., Siegbahn, P. E. M., and Warshel, A. (2004b). Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase. J. Am. Chem. Soc. 126, 2820–2828. doi: 10.1021/ja037233l
Orio, M., Pantazis, D. A., and Neese, F. (2009). Density functional theory. Photosynth. Res. 102, 443–453. doi: 10.1007/s11120-009-9404-8
Orr, S. T. M., Ripp, S. L., Ballard, T. E., Henderson, J. L., Scott, D. O., Obach, R. S., et al. (2012). Mechanism-based inactivation (MBI) of cytochrome P450 enzymes: structure–activity relationships and discovery strategies to mitigate drug–drug interaction risks. J. Med. Chem. 55, 4896–4933. doi: 10.1021/jm300065h
Ortiz de Montellano., P. R. (2005). Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd Edn. New York, NY: Kluwer Academic/Plenum Publishers. doi: 10.1007/b139087
Ortiz de Montellano, P. R. (2010). Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 110, 932–948. doi: 10.1021/cr9002193
Parr, R. G., and Yang, W. (1989). Density-Functional Theory of Atoms and Molecules. New York, NY: Oxford University Press.
Peck, S. C., Cooke, H. A., Cicchillo, R. M., Malova, P., Hammerschmidt, F., Nair, S. K., et al. (2011). Mechanism and substrate recognition of 2-hydroxyethylphosphonate dioxygenase. Biochemistry 50, 6598–6605. doi: 10.1021/bi200804r
Pelkonen, O., Turpeinen, M., Hakkola, J., Honkakoski, P., Hukkanen, J., and Raunio, H. (2008). Inhibition and induction of human cytochrome P450 enzymes: current status. Arch. Toxicol. 82, 667–715. doi: 10.1007/s00204-008-0332-8
Perdew, J. P., Ruzsinszky, A., Constantin, L. A., Sun, J., and Csonka, G. I. (2009). Some fundamental issues in ground-state density functional theory: a guide for the perplexed. J. Chem. Theory Comput. 5, 902–908. doi: 10.1021/ct800531s
Perdew, J. P., Ruzsinszky, A., Tao, J., Staroverov, V. N., Scuseria, G. E., and Csonka, G. I. (2005). Prescription for the design and selection of density functional approximations: more constraint satisfaction with fewer fits. J. Chem. Phys. 123, 062201. doi: 10.1063/1.1904565
Perdew, J. P., and Schmidt, K. (2001). Jacob's ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 577, 1–20. doi: 10.1063/1.1390175
Phatak, P., Sumner, I., and Iyengar, S. S. (2012). Gauging the flexibility of the active site in soybean lipoxygenase-1 (SLO-1) through an atom-centered density matrix propagation (ADMP) treatment that facilitates the sampling of rare events. J. Phys. Chem. B 116, 10145–10164. doi: 10.1021/jp3015047
Porro, C. S., Sutcliffe, M. J., and de Visser, S. P. (2009). Quantum mechanics/molecular mechanics studies on the sulfoxidation of dimethyl sulfide by compound I and compound 0 of cytochrome P450: which is the better oxidant? J. Phys. Chem. A. 113, 11635–11642. doi: 10.1021/jp9023926
Poulos, T. L., and Kraut, J. (1980). The stereochemistry of peroxidase catalysis. J. Biol. Chem. 255, 8199–8205.
Que, L. Jr. (2007). The road to non-heme oxoferryls and beyond. Acc. Chem. Res. 40, 493–500. doi: 10.1021/ar700024g
Que, L. Jr., and Ho, R. Y. N. (1996). Dioxygen activation by enzymes with mononuclear non-heme iron active sites. Chem. Rev. 96, 2607–2624. doi: 10.1021/cr960039f
Que, L. Jr., and Tolman, W. B. (2008). Biologically inspired oxidation catalysis. Nature 455, 333–340. doi: 10.1038/nature07371
Quinonero, D., Morokuma, K., Musaev, D. G., Mas-Ballesté, R., and Que, L. Jr. (2005). Metal-peroxo versus metal-oxo oxidants in non-heme iron-catalyzed olefin oxidations: computational and experimental studies on the effect of water. J. Am. Chem. Soc. 127, 6548–6549. doi: 10.1021/ja051062y
Radoń, M., and Broclawik, E. (2007). Peculiarities of the electronic structure of cytochrome P450 compound I: CASPT2 and DFT modeling. J. Chem. Theory Comput. 3, 728–734. doi: 10.1021/ct600363a
Riccardi, D., Schaefer, P., Yang, Y., Yu, H., Ghosh, N., Prat-Resina, X., et al. (2006). Development of effective quantum mechanical/molecular mechanical (QM/MM) methods for complex biological processes. J. Phys. Chem. B 110, 6458–6469. doi: 10.1021/jp056361o
Rittle, J., and Green, M. T. (2010). Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 330, 933–937. doi: 10.1126/science.1193478
Roelfes, G., Vrajmasu, V., Chen, K., Ho, R. Y. N., Rohde, J. U., Zondervan, C., et al. (2003). End-on and side-on peroxo derivatives of non-heme iron complexes with pentadentate ligands: models for putative intermediates in biological iron/dioxygen chemistry. Inorg. Chem. 42, 2639–2653. doi: 10.1021/ic034065p
Rohde, J. U., In, J. H., Lim, M. H., Brennessel, W. W., Bukowski, M. R., Stubna, A., et al Jr. (2003). Crystallographic and spectroscopic characterization of a nonheme Fe(IV)=O complex. Science 299, 1037–1039. doi: 10.1126/science.299.5609.1037
Rohde, J.-U., Stubna, A., Bominaar, E. L., Münck, E., Nam, W., and Que, L. Jr. (2006). Nonheme oxoiron(IV) complexes of tris(2-pyridylmethyl)amine with cis-monoanionic ligands. Inorg. Chem. 45, 6435–6445. doi: 10.1021/ic060740u
Römelt, M., Ye, S., and Neese, F. (2009). Calibration of modern density functional theory methods for the prediction of 57Fe Mössbauer isomer shifts: meta-GGA and double-hybrid functionals. Inorg. Chem. 48, 784–785. doi: 10.1021/ic801535v
Ryabov, A. D. (1990). Mechanisms of intramolecular activation of carbon-hydrogen bonds in transition-metal complexes. Chem. Rev. 90, 403–424. doi: 10.1021/cr00100a004
Rydberg, P., Jørgensen, F. S., and Olsen, L. (2014). Use of density functional theory in drug metabolism studies. Expert Opin. Drug Metab. Toxicol. 10, 215–227. doi: 10.1517/17425255.2014.864278
Rydberg, P., and Olsen, L. (2011). Do two different reaction mechanisms contribute to the hydroxylation of primary amines by cytochrome P450? J. Chem. Theory Comput. 7, 3399–3404. doi: 10.1021/ct200422p
Sahu, S., Widger, L. R., Quesne, M. G., de Visser, S. P., Matsumura, H., Moënne-Loccoz, P., et al. (2013). Secondary coordination sphere influence on the reactivity of nonheme iron(II) complexes: an experimental and DFT approach. J. Am. Chem. Soc. 135, 10590–10593. doi: 10.1021/ja402688t
Sameera, W. M. C., and Maseras, F. (2012). Transition metal catalysis by density functional theory and density functional theory/molecular mechanics. WIREs Comput. Mol. Sci. 2, 375–385. doi: 10.1002/wcms.1092
Sastri, C. V., Lee, J., Oh, K., Yoon, J. L., Jackson, T. A., Ray, K., et al. (2007). Axial ligand tuning of a nonheme iron(IV)–oxo unit for hydrogen atom abstraction. Proc. Natl. Acad. Sci. U.S.A. 104, 19181–19186. doi: 10.1073/pnas.0709471104
Sastri, C. V., Park, M. J., Ohta, T., Jackson, T. A., Stubna, A., Seo, M. S., et al. (2005). Axial ligand substituted nonheme FeIV=O complexes: observation of near-UV LMCT bands and Fe=O raman vibrations. J. Am. Chem. Soc. 127, 12494–12495. doi: 10.1021/ja0540573
Schlichting, I., Berendzen, J., Chu, K., Stock, A. M., Maves, S. A., Benson, D. E., et al. (2000). The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287, 1615–1622. doi: 10.1126/science.287.5458.1615
Schmidt, M. W., Baldridge, K. K., Boatz, J. A., Elbert, S. T., Gordon, M. S., Jensen, J. H., et al. (1993). General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363. doi: 10.1002/jcc.540141112
Schöneboom, J. C., Cohen, S., Lin, H., Shaik, S., and Thiel, W. (2004). Quantum mechanical/molecular mechanical investigation of the mechanism of C-H hydroxylation of camphor by cytochrome P450cam: theory supports a two-state rebound mechanism. J. Am. Chem. Soc. 126, 4017–4034. doi: 10.1021/ja039847w
Schöneboom, J. C., Lin, H., Reuter, N., Thiel, W., Cohen, S., Ogliaro, F., et al. (2002). The elusive oxidant species of cytochrome P450 enzymes: characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 124, 8142–8151. doi: 10.1021/ja026279w
Schöneboom, J. C., Neese, F., and Thiel, W. (2005). Toward identification of the compound I reactive intermediate in cytochrome P450 chemistry: a QM/MM study of its EPR and Mössbauer parameters. J. Am. Chem. Soc. 127, 5840–5853. doi: 10.1021/ja0424732
Schöneboom, J. C., and Thiel, W. (2004). The resting state of P450cam: a QM/MM study. J. Phys. Chem. B 108, 7468–7478. doi: 10.1021/jp049596t
Schröder, D., Shaik, S., and Schwarz, H. (2000). Two-state reactivity as a new concept in organometallic chemistry. Acc. Chem. Res. 33, 139–145. doi: 10.1021/ar990028j
Schwarz, H. (2011). Chemistry with methane: concepts rather than recipes. Angew. Chem. Int. Ed. Engl. 50, 10096–10115. doi: 10.1002/anie.201006424
Schyman, P., Lai, W. Z., Chen, H., Wang, Y., and Shaik, S. (2011). The directive of the protein: how does cytochrome P450 select the mechanism of dopamine formation? J. Am. Chem. Soc. 133, 7977–7984. doi: 10.1021/ja201665x
Senn, H. M., and Thiel, W. (2007a). QM/MM methods for biological systems. Top. Curr. Chem. 268, 173–290. doi: 10.1007/128_2006_084
Senn, H. M., and Thiel, W. (2007b). QM/MM studies of enzymes. Curr. Opin. Chem. Biol. 11, 182–187. doi: 10.1016/j.cbpa.2007.01.684
Senn, H. M., and Thiel, W. (2009). QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. Engl. 48, 1198–1229. doi: 10.1002/anie.200802019
Seo, M. S., Kim, N. H., Cho, K.-B., So, J. E., Park, S. K., Clémancey, M., et al. (2011). A mononuclear nonheme iron(IV)-oxo complex which is more reactive than cytochrome P450 model compound I. Chem. Sci. 2, 1039–1045. doi: 10.1039/c1sc00062d
Seregin, I. V., and Gevorgyan, V. (2007). Direct transition metal-catalyzed functionalization of heteroaromatic compounds. Chem. Soc. Rev. 36, 1173–1193. doi: 10.1039/b606984n
Shaik, S., Chen, H., and Janardanan, D. (2011). Exchange-enhanced reactivity in bond activation by metal–oxo enzymes and synthetic reagents. Nat. Chem. 3, 19–27. doi: 10.1038/nchem.943
Shaik, S., Cohen, S., Wang, Y., Chen, H., Kumar, D., and Thiel, W. (2010a). P450 enzymes: their structure, reactivity, and selectivity—modeled by QM/MM calculations. Chem. Rev. 110, 949–1017. doi: 10.1021/cr900121s
Shaik, S., Filatov, M., Schröder, D., and Schwarz, H. (1998). Electronic structure makes a difference: cytochrome P-450 mediated hydroxylations of hydrocarbons as a two-state reactivity paradigm. Chem. Eur. J. 4, 193–199.
Shaik, S., Hirao, H., and Kumar, D. (2007a). Reactivity of high-valent iron–oxo species in enzymes and synthetic reagents: a tale of many states. Acc. Chem. Res. 40, 532–542. doi: 10.1021/ar600042c
Shaik, S., Hirao, H., and Kumar, D. (2007b). Reactivity patterns of cytochrome P450 enzymes: multifunctionality of the active species, and the two states–two oxidants conundrum. Nat. Prod. Rep. 24, 533–552. doi: 10.1039/b604192m
Shaik, S., Kumar, D., de Visser, S. P., Altun, A., and Thiel, W. (2005). Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 105, 2279–2328. doi: 10.1021/cr030722j
Shaik, S., Lai, W., Chen, H., and Wang, Y. (2010c). The valence bond way: reactivity patterns of cytochrome P450 enzymes and synthetic analogs. Acc. Chem. Res. 43, 1154–1165. doi: 10.1021/ar100038u
Shaik, S., Wang, Y., Chen, H., Song, J. S. A., and Meir, R. (2010b). Valence bond modelling and density functional theory calculations of reactivity and mechanism of cytochrome P450 enzymes: thioether sulfoxidation. Faraday Discuss. 145, 49–70. doi: 10.1039/b906094d
Shilov, A. E., and Shteinman, A. A. (1999). Oxygen atom transfer into C-H bond in biological and model chemical systems. Mechanistic aspects. Acc. Chem. Res. 32, 763–771. doi: 10.1021/ar980009u
Shilov, A. E., and Shul'pin, G. B. (1997). Activation of C-H bonds by metal complexes. Chem. Rev. 97, 2879–2932. doi: 10.1021/cr9411886
Shoji, M., Isobe, H., Saito, T., Yabushita, H., Koizumi, K., Kitagawa, Y., et al. (2008). Theory of chemical bonds in metalloenzymes. VII. Hybrid-density functional theory studies on the electronic structures of P450. Int. J. Quantum Chem. 108, 631–650. doi: 10.1002/qua.21547
Shu, L., Nesheim, J. C., Kauffmann, K., Münck, E., Lipscomb, J. D., and Que, L. Jr. (1997). An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science 275, 515–518. doi: 10.1126/science.275.5299.515
Sicking, W., Korth, H.-G., Jansen, G., de Groot, H., and Sustmann, R. (2007). Hydrogen peroxide decomposition by a non-heme iron(III) catalase mimic: a DFT study. Chem. Eur. J. 13, 4230–4245. doi: 10.1002/chem.200601209
Siegbahn, P. E. M. (2006). The performance of hybrid DFT for mechanisms involving transition metal complexes in enzymes. J. Biol. Inorg. Chem. 11, 695–701. doi: 10.1007/s00775-006-0137-2
Siegbahn, P. E. M., and Blomberg, M. R. A. (2010). Quantum chemical studies of proton-coupled electron transfer in metalloenzymes. Chem. Rev. 110, 7040–7061. doi: 10.1021/cr100070p
Siegbahn, P. E. M., Blomberg, M. R. A., and Chen, S. L. (2010). Significant van der Waals effects in transition metal complexes. J. Chem. Theory Comput. 6, 2040–2044. doi: 10.1021/ct100213e
Siegbahn, P. E. M., and Borowski, T. (2006). Modeling enzymatic reactions involving transition metals. Acc. Chem. Res. 39, 729–738. doi: 10.1021/ar050123u
Sligar, S. G., Makris, T. M., and Denisov, I. G. (2005). Thirty years of microbial P450 monooxygenase research: peroxo-heme intermediates—the central bus station in heme oxygenase catalysis. Biochem. Biophys. Res. Commun. 338, 346–354. doi: 10.1016/j.bbrc.2005.08.094
Solomon, E. I., Brunold, T. C., Davis, M. I., Kemsley, J. N., Lee, S.-K., Lehnert, N., et al. (2000). Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 100, 235–350. doi: 10.1021/cr9900275
Solomon, E. I., Wong, S. D., Liu, L. V., Decker, A., and Chow, M. S. (2009). Peroxo and oxo intermediates in mononuclear nonheme iron enzymes and related active sites. Curr. Opin. Chem. Biol. 13, 99–113. doi: 10.1016/j.cbpa.2009.02.011
Sono, M., Roach, M. P., Coulter, E. D., and Dawson, J. H. (1996). Heme-containing oxygenases. Chem. Rev. 96, 2841–2887. doi: 10.1021/cr9500500
Stubbe, J., Nocera, D. G., Yee, C. S., and Chang, M. C. (2003). Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 103, 2167–2202. doi: 10.1021/cr020421u
Su, P., and Li, H. (2009). Energy decomposition analysis of covalent bonds and intermolecular interactions. J.Chem. Phys. 131, 014102. doi: 10.1063/1.3159673
Sun, X., Geng, C., Huo, R., Ryde, U., Bu, Y., and Li, J. (2014). Large equatorial ligand effects on C–H bond activation by nonheme iron(IV)-oxo complexes. J. Phys. Chem. B. 118, 1493–1500. doi: 10.1021/jp410727r
Svensson, M., Humbel, S., Froese, R. D. J., Matsubara, T., Sieber, S., and Morokuma, K. (1996). ONIOM: a multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J. Phys. Chem. 100, 19357–19363. doi: 10.1021/jp962071j
Swart, M. (2008). Accurate spin-state energies for iron complexes. J. Chem. Theory Comput. 4, 2057–2066. doi: 10.1021/ct800277a
Tanaka, M., Ishimori, K., Mukai, M., Kitagawa, T., and Morishima, I. (1997). Catalytic activities and structural properties of horseradish peroxidase distal His42→Glu or Gln mutant. Biochemistry 36, 9889–9898. doi: 10.1021/bi970906q
Tang, H., Guan, J., Liu, H., and Huang, X. (2013). Analysis of an alternative to the H-atom abstraction mechanism in methane C–H bond activation by nonheme iron(IV)-oxo oxidants. Dalton Trans. 42, 10260–10270. doi: 10.1039/c3dt50866h
Taxak, N., Desai, P. V., Patel, B., Mohutsky, M., Klimkowski, V. J., Gombar, V., et al. (2012). Metabolic-intermediate complex formation with cytochrome P450: theoretical studies in elucidating the reaction pathway for the generation of reactive nitroso intermediate. J. Comput. Chem. 33, 1740–1747. doi: 10.1002/jcc.23008
Taxak, N., Kalra, S., and Bharatam, P. V. (2013b). Mechanism-based inactivation of cytochromes by furan epoxide: unraveling the molecular mechanism. Inorg. Chem. 52, 13496–13508. doi: 10.1021/ic401907k
Taxak, N., Patel, B., and Bharatam, P. V. (2013a). Carbene generation by cytochromes and electronic structure of heme-iron-porphyrin-carbene complex: a quantum chemical study. Inorg. Chem. 52, 5097–5109. doi: 10.1021/ic400010d
Tejero, I., Garcia-Viloca, M., González-Lafont, Á., Lluch, J. M., and York, D. M. (2006). Enzyme dynamics and tunneling enhanced by compression in the hydrogen abstraction catalyzed by soybean lipoxygenase-1. J. Phys. Chem. B 110, 24708–24719. doi: 10.1021/jp066263i
Thellamurege, N., and Hirao, H. (2013). Water complexes of cytochrome P450: insights from energy decomposition analysis. Molecules 18, 6782–6791. doi: 10.3390/molecules18066782
Thellamurege, N. M., and Hirao, H. (2014). Effect of protein environment within cytochrome P450cam evaluated using a polarizable-embedding QM/MM method. J. Phys. Chem. B 118, 2084–2092. doi: 10.1021/jp412538n
Thellamurege, N. M., Si, D., Cui, F., Zhu, H., Lai, R., and Li, H. (2013). QuanPol: a full spectrum and seamless QM/MM program. J. Comput. Chem. 34, 2816–2833. doi: 10.1002/jcc.23435
Thibon, A., England, J., Martinho, M., Young, V. G. Jr., Frisch, J. R., Guillot, R., et al. (2008). Proton and reductant-assisted dioxygen activation by a nonheme iron(II) complex to form an oxoiron(IV) intermediate. Angew. Chem. Int. Ed. Engl. 47, 7064–7067. doi: 10.1002/anie.200801832
Tian, L., and Friesner, R. A. (2009). QM/MM simulation on P450 BM3 enzyme catalysis mechanism. J. Chem. Theory Comput. 5, 1421–1431. doi: 10.1021/ct900040n
Tshuva, E. Y., and Lippard, S. J. (2004). Synthetic models for non-heme carboxylate-bridged diiron metalloproteins: strategies and tactics. Chem. Rev. 104, 987–1012. doi: 10.1021/cr020622y
Usharani, D., Janardanan, D., Li, C., and Shaik, S. (2013a). A theory for bioinorganic chemical reactivity of oxometal complexes and analogous oxidants: the exchange and orbital-selection rules. Acc. Chem. Res. 46, 471–482. doi: 10.1021/ar300204y
Usharani, D., Lacy, D. C., Borovik, A. S., and Shaik, S. (2013b). Dichotomous hydrogen atom transfer vs proton-coupled electron transfer during activation of X–H bonds (X = C, N, O) by nonheme iron–oxo complexes of variable basicity. J. Am. Chem. Soc. 135, 17090–17104. doi: 10.1021/ja408073m
Usharani, D., Zazza, C., Lai, W., Chourasia, M., Waskell, L., and Shaik, S. (2012). A single-site mutation (F429H) converts the enzyme CYP 2B4 into a heme oxygenase: a QM/MM study. J. Am. Chem. Soc. 134, 4053–4056. doi: 10.1021/ja211905e
van der Donk, W. A., Krebs, C., and Bollinger, J. M. Jr. (2010). Substrate activation by iron superoxo intermediates. Curr. Opin. Struct. Biol. 20, 673–683. doi: 10.1016/j.sbi.2010.08.005
van der Kamp, M. W., and Mulholland, A. J. (2013). Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 52, 2708–2728. doi: 10.1021/bi400215w
Van Heuvelen, K. M., Fiedler, A. T., Shan, X., De Hont, R. F., Meier, K. K., Bominaar, E. L., et al. (2012). One-electron oxidation of an oxoiron(IV) complex to form an [O=FeV=NR]+ center. Proc. Natl. Acad. Sci. U.S.A. 109, 11933–11938. doi: 10.1073/pnas.1206457109
Vancoillie, S., Zhao, H., Radoń, M., and Pierloot, K. (2010). Performance of CASPT2 and DFT for relative spin-state energetics of heme models. J. Chem. Theory Comput. 6, 576–582. doi: 10.1021/ct900567c
Vardhaman, A. K., Sastri, C. V., Kumar, D., and de Visser, S. P. (2011). Nonheme ferric hydroperoxo intermediates are efficient oxidants of bromide oxidation. Chem. Commun. 47, 11044–11046. doi: 10.1039/c1cc13775a
von Hopffgarten, M., and Frenking, G. (2012). Energy decomposition analysis. WIREs Comput. Mol. Sci. 2, 43–62. doi: 10.1002/wcms.71
Vreven, T., Byun, K. S., Komáromi, I., Dapprich, S., Montgomery, J. A., Morokuma, K., et al. (2006). Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J. Chem. Theory Comput. 2, 815–826. doi: 10.1021/ct050289g
Wallar, B. J., and Lipscomb, J. D. (1996). Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev. 96, 2625–2657. doi: 10.1021/cr9500489
Wang, B., Li, C., Cho, K.-B., Nam, W., and Shaik, S. (2013c). The FeIII(H2O2) complex as a highly efficient oxidant in sulfoxidation reactions: revival of an underrated oxidant in cytochrome P450. J. Chem. Theory Comput. 9, 2519–2525. doi: 10.1021/ct400190f
Wang, C., Chang, W. C., Guo, Y., Huang, H., Peck, S. C., Pandelia, M. E., et al. (2013a). Evidence that the fosfomycin-producing epoxidase, HppE, is a non–heme-iron peroxidase. Science 342, 991–995. doi: 10.1126/science.1240373
Wang, Y., Chen, H., Makino, M., Shiro, Y., Nagano, S., Asamizu, S., et al. (2009b). Theoretical and experimental studies of the conversion of chromopyrrolic acid to an antitumor derivative by cytochrome P450 StaP: the catalytic role of water molecules. J. Am. Chem. Soc. 131, 6748–6762. doi: 10.1021/ja9003365
Wang, Y., Hirao, H., Chen, H., Onaka, H., Nagano, S., and Shaik, S. (2008). Electron transfer activation of chromopyrrolic acid by cytochrome P450 en route to the formation of an antitumor indolocarbazole derivative: theory supports experiment. J. Am. Chem. Soc. 130, 7170–7171. doi: 10.1021/ja711426y
Wang, Y., Janardanan, D., Usharani, D., Han, K., Que, L. Jr., and Shaik, S. (2013b). Nonheme iron oxidant formed in the presence of H2O2 and acetic acid is the cyclic ferric peracetate complex, not a perferryloxo complex. ACS Catal. 3, 1334–1341. doi: 10.1021/cs400134g
Wang, Y., Wang, Y., and Han, K. (2009a). Theoretical study of cyclohexane hydroxylation by three possible isomers of [FeIV(O)(R-TPEN)]2+: does the pentadentate ligand wrapping around the metal center differently lead to the different stability and reactivity? J. Biol. Inorg. Chem. 14, 533–545. doi: 10.1007/s00775-009-0468-x
Warshel, A., and Levitt, M. (1976). Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of reaction of lysozyme. J. Mol. Biol. 103, 227–249. doi: 10.1016/0022-2836(76)90311-9
Webb, J. R., Burgess, S. A., Cundari, T. R., and Gunnoe, T. B. (2013). Activation of carbon–hydrogen bonds and dihydrogen by 1,2-CH-addition across metal–heteroatom bonds. Dalton Trans. 42, 16646–16665. doi: 10.1039/c3dt52164h
Whitteck, J. T., Cicchillo, R. M., and van der Donk, W. A. (2009). Hydroperoxylation by hydroxyethylphosphonate dioxygenase. J. Am. Chem. Soc. 131, 16225–16232. doi: 10.1021/ja906238r
Whitteck, J. T., Malova, P., Peck, S. C., Cicchillo, R. M., Hammerschmidt, F., and van der Donk, W. A. (2011). On the stereochemistry of 2-hydroxyethylphosphonate dioxygenase. J. Am. Chem. Soc. 133, 4236–4239. doi: 10.1021/ja1113326
Wirstam, M., Blomberg, M. R., and Siegbahn, P. E. (1999). Reaction mechanism of compound I formation in heme peroxidases: a density functional theory study. J. Am. Chem. Soc. 121, 10178–10185. doi: 10.1021/ja991997c
Wong, S. D., Bell, C. B. III. Liu, L. V., Kwak, Y., England, J., Alp, E. E., et al. (2011). Nuclear resonance vibrational spectroscopy on the FeIV=O S=2 non-heme site in TMG3tren: experimentally calibrated insights into reactivity. Angew. Chem. Int. Ed. Engl. 50, 3215–3218. doi: 10.1002/anie.201007692
Xing, G., Diao, Y., Hoffart, L. M., Barr, E. W., Prabhu, K. S., Arner, R. J., et al. (2006). Evidence for C–H cleavage by an iron–superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 6130–6135. doi: 10.1073/pnas.0508473103
Yamaguchi, K., Yamanaka, S., Isobe, H., Shoji, M., Saito, T., Kitagawa, Y., et al. (2009). Theory of chemical bonds in metalloenzymes XIII: singlet and triplet diradical mechanisms of hydroxylations with iron-oxo species and P450 are revisited. Int. J. Quantum Chem 109, 3723–3744. doi: 10.1002/qua.22348
Yamaguchi, J., Yamaguchi, A. D., and Itami, K. (2012). C–H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed. Engl. 51, 8960–9009. doi: 10.1002/anie.201201666
Ye, S., and Neese, F. (2009). Quantum chemical studies of C-H activation reactions by high-valent nonheme iron centers. Curr. Opin. Chem. Biol. 13, 89–98. doi: 10.1016/j.cbpa.2009.02.007
Ye, S. F., Geng, C. Y., Shaik, S., and Neese, F. (2013). Electronic structure analysis of multistate reactivity in transition metal catalyzed reactions: the case of C–H bond activation by non-heme iron(IV)–oxo cores. Phys. Chem. Chem. Phys. 15, 8017–8030. doi: 10.1039/c3cp00080j
Ye, S. F., and Neese, F. (2011). Nonheme oxo-iron(IV) intermediates form an oxyl radical upon approaching the C–H bond activation transition state. Proc. Natl. Acad. Sci. U.S.A. 108, 1228–1233. doi: 10.1073/pnas.1008411108
Yoon, J., Wilson, S. A., Jang, Y. K., Seo, M. S., Nehru, K., Hedman, B., et al. (2009). Reactive intermediates in oxygenation reactions with mononuclear nonheme iron catalysts. Angew. Chem. Int. Ed. Engl. 48, 1257–1260. doi: 10.1002/anie.200802672
Yosca, T. H., Rittle, J., Krest, C. M., Onderko, E. L., Silakov, A., Calixto, J. C., et al. (2013). Iron(IV)hydroxide pKa and the role of thiolate ligation in C–H bond activation by cytochrome P450. Science 342, 825–829. doi: 10.1126/science.1244373
Yoshizawa, K. (2002). Theoretical study on kinetic isotope effects in the C-H bond activation of alkanes by iron-oxo species. Coord. Chem. Rev. 226, 251–259. doi: 10.1016/S0010-8545(01)00464-7
Yoshizawa, K., Kamachi, T., and Shiota, Y. (2001). A theoretical study of the dynamic behavior of alkane hydroxylation by a compound I model of cytochrome P450. J. Am. Chem. Soc. 123, 9806–9816. doi: 10.1021/ja010593t
Zhao, Y., González-García, N., and Truhlar, D. G. (2005). Benchmark database of barrier heights for heavy atom transfer, nucleophilic substitution, association, and unimolecular reactions and its use to test theoretical methods. J. Phys. Chem. A 109, 2012–2018. doi: 10.1021/jp045141s
Zhao, Y., Lynch, B. J., and Truhlar, D. G. (2004). Development and assessment of a new hybrid density functional model for thermochemical kinetics. J. Phys. Chem. A 108, 2715–2719. doi: 10.1021/jp049908s
Zhao, Y., and Truhlar, D. G. (2004). Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 108, 6908–6918. doi: 10.1021/jp048147q
Zhao, Y., and Truhlar, D. G. (2008). Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167. doi: 10.1021/ar700111a
Zheng, J., Wang, D., Thiel, W., and Shaik, S. (2006). QM/MM study of mechanisms for compound I formation in the catalytic cycle of cytochrome P450cam. J. Am. Chem. Soc. 128, 13204–13215. doi: 10.1021/ja063439l
Zhou, S. F., Chan, S. Y., Goh, B. C., Chan, E., Duan, W., Huang, M., et al. (2005). Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 44, 279–304. doi: 10.2165/00003088-200544030-00005
Ziegler, T., and Rauk, A. (1979a). A theoretical study of the ethylene-metal bond in complexes between Cu+, Ag+, Au+, Pt0, or Pt2+ and ethylene, based on the Hartree-Fock-Slater transition-state method. Inorg. Chem. 18, 1558–1565. doi: 10.1021/ic50196a034
Keywords: density functional theory, QMMM, iron-containing molecules, protein environment, enzyme reactions, catalysis
Citation: Hirao H, Thellamurege N and Zhang X (2014) Applications of density functional theory to iron-containing molecules of bioinorganic interest. Front. Chem. 2:14. doi: 10.3389/fchem.2014.00014
Received: 11 September 2013; Accepted: 10 March 2014;
Published online: 29 April 2014.
Edited by:
Hans Martin Senn, University of Glasgow, UKReviewed by:
Dimitrios A. Pantazis, Max Planck Institute for Chemical Energy Conversion, GermanyAhmet Altun, Fatih University, Turkey
Copyright © 2014 Hirao, Thellamurege and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hajime Hirao, Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore e-mail:aGlyYW9AbnR1LmVkdS5zZw==