 Thobile L. Mhlabeni
Thobile L. Mhlabeni Tamba Jamiru
Tamba Jamiru Washington Mhike
Washington Mhike- 1Polymer Technology Division, Department of Chemical, Metallurgical and Materials Engineering, Tshwane University of Technology, Pretoria, South Africa
- 2Department of Mechanical and Mechatronics Engineering, Tshwane University of Technology, Pretoria, South Africa
Blending wax with polyethylene (PE) has garnered significant attention in recent years due to its potential to enhance processing capabilities of polyethylene-based products. The miscibility of polyethylene and low molecular mass wax blends is a critical factor influencing various industrial applications. In this study, the theoretical framework of the miscibility of polyethylene/wax blends is discussed in terms of thermodynamic principles. The state-of-the-art of miscibility of polyethylene/wax blends is comprehensively examined, focusing on their thermophysical and rheological properties. It is shown that thermophysical properties assessed by differential scanning calorimetry and dynamic mechanical analysis can provide insights into thermal transitions affected by blend composition. Furthermore, the rheological properties of blends can provide insights into their microstructure, phase behaviour, and interaction between components. Literature search of the influence of wax compositions and temperature on the polyethene/wax compatibility was shown to be inconsistent. Also, studies on the miscibility of paraffin and Fischer-Tropsch (F-T) wax/polyethene blends is limited. However, the review suggests full miscibility of low molecular mass waxes and polyethylene in the melt and partial co-crystallisation in the solid state.
1 Introduction
Polyethylene (PE) is amongst the most utilized plastics worldwide. Its popularity and wide range of applications stem from its characteristics, which include a simple molecular structure consisting of repeating ethylene units that is held together by weak dispersion forces (Ashby, 2013; Ragaert et al., 2016). This simplicity in molecular structure facilitates its production through various polymerization processes, making it cost-effective and widely available. Polyethylene exhibit good mechanical properties, including excellent impact resistance making it ideal for applications that require durability and toughness, such as packaging materials (Sadiku, 2009; Ogah, 2012; Bayat et al., 2013). Despite its many advantages, the stiffness of polyethylene is relatively low. This is primarily because the glass transition temperature (Tg) of the amorphous regions in PE is extremely low (Cheremisinoff, 2001; Guzzi et al., 2023).
Calcium carbonate (CaCO3) is commonly used as an inexpensive inorganic filler in polyethylene compounds to increase its impact toughness and modulus and heat deflection temperature (Elleithy et al., 2011; Tanniru and Misra, 2005). However, due to their polar nature, when added to polymer matrices, the CaCO3 particles tend to form agglomerates instead of being uniformly dispersed (Hostomsky and Jones, 1991). Filler agglomeration can have negative effects on the properties and performance of the polymer composites. One significant disadvantage of using CaCO3 as a filler is that it negatively impacts the tensile strength and impact resilience properties of the solid material. Furthermore, the presence of CaCO3 clumps within the polyethylene matrix reduces the apparent maximum volume fraction of the filler, leading to increased melt viscosity (Dangtungee et al., 2005). Hence, processing additives are often added to ensure the necessary flexibility and adequate flow properties. The benefits of processing additives are mainly seen in the melt phase of the polymer resin using conventional processing techniques including extrusion, injection moulding, film blowing machines, and others.
Wax is well-known for its ability to improve polymer processability, particularly in thermoplastics e.g., polyethylene and polypropylene masterbatch processing (Gale, 1997). In polymer compounding, wax can act as both an internal and an external lubricant, reducing the melt viscosity and increasing the melt flow rate of polymer melt (King and Noël, 1972). Due to its lubricating effect, the industry and academia have eagerly adopted wax to either provide lubrication and/or provide physical modification during polyethylene compounding. For instance, polyethylene waxes are commercially available and have been used for their excellent dispersive and distributive mixing properties in highly filled CaCO3 compounds and pigment masterbatches (Gale, 1997). For these reasons, polyethylene waxes are also used for their ability to enhance lubricity and improve the softening point of hot melt adhesives (Deshmukh et al., 2010; Gale, 1997; Motooka et al., 1986). Polyethylene wax is derived from the polymerization of ethylene monomers following the same method as their parent polymers i.e., HDPE (Ciesińska et al., 2016). The excellent dispersive and distributive mixing properties of polyethene waxes have been attributed to their inherently high molecular masses and melt viscosities when compared to low molecular mass waxes such as paraffin and Fischer-Tropsch (F-T) waxes. F-T wax is a by-product of the coal chemical industry and can also be derived from natural gas. Paraffin wax is saturated hydrocarbons derived as by-products from the processing of petroleum oils. While polyethylene and paraffin wax are commercially available in different grades from the following producers: Clariant (Licowax), Dow chemical company (Insite™ catalyst), BASF (Luwax) and Mitsui (Excerex™ process) etc., F-T wax is mainly produced by Sasol, South Africa (Bennett, 1944).
Anecdotal evidence suggests that lower molecular mass waxes such as paraffin or FT wax, when used on their own, cannot serve as an effective alternative to polyethylene waxes. This is because lower molecular mass waxes tend to exacerbate die-drool (Lee, 2002; Musil and Zatloukal, 2014; Chaloupková and Zatloukal, 2009). For example, polyethylene wax exhibits a broader molecular mass distributions with an average molecular mass up to 10,000 g mol−1 while paraffin and F-T waxes may range from 200 to 1,000 g mol−1 (Ciesińska et al., 2016). In such cases, some studies attributed polymer melt die-lip drooling to poor thermodynamic compatibility between additives and the parent polymer (Koningsveld et al., 1974; Patterson and Robard, 1978). The lack of compatibility between additives and the polymer matrix strongly influences die-lip drooling in polymer extrusion. This is because when there is incompatibility between the polymer and additives, low viscosity additives can migrate to the die landings during the extrusion process. This leads to the accumulation of the oxidized extrudate. Hence in cases of poor compatibility between the paraffin or F-T wax and polymer, the wax may have a higher tendency to migrate to the metal surfaces and contribute to additive build-up. Therefore, it is imperative to consider the extent of compatibility between these low molecular mass waxes and base polymers, in efforts to use them as processing additives.
Sotomayor et al. (2014) demonstrated that adding up to 50 wt% soft paraffin wax significantly reduced the melt viscosity of HDPE by more than an order of magnitude during injecting moulding, as seen in Figure 1.
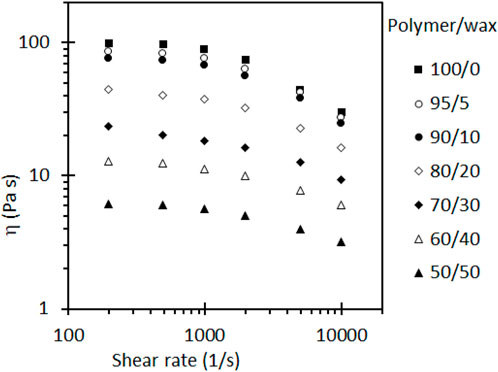
Figure 1. Viscosity versus shear rate of paraffin wax/HDPE blends at 160°C. Figure reproduced with permission (Sotomayor et al., 2014).
Several other studies successfully explored the use of various paraffin waxes as processing aids for linear low-density polyethylene (LLDPE), high-density polyethylene (HDPE) and ultrahigh-molecular-weight polyethylene (UHMWPE) as base polymers (Mpanza and Luyt, 2006; Esmaeilzade et al., 2022; Bakshi and Ghosh, 2022). Some studies investigated paraffin waxes mixed with different grades of polyethylene, however, for different applications (Salyer, 1996; Zalba et al., 2003; Krupa et al., 2007; Molefi et al., 2010; Mngomezulu et al., 2011). In all these studies, it was consistently found that due to the different lubrication effects, adding wax reduces the viscosity and improves the flow properties of highly entangled polyethylene chains. Furthermore, some studies investigating melt flow rates concluded that the presence of wax reduced the melt viscosity of polyethylene substantially thereby improving its melt flow rates (Krupa and Luyt, 2001a; Mpanza and Luyt, 2006). F-T wax was also found to improve the melt flow rate of polyethylene by reducing its viscosity, without compromising other thermal properties (Mpanza and Luyt, 2006a; Esmaeilzade et al., 2022; Gudiño Rivera et al., 2022). However, challenges encountered regarding compatibility, particularly when the molecular mass of polyethylene was high.
Empirical studies investigating the miscibility of various paraffin waxes or alkanes combined with polyethylene primarily employed thermal analysis. Thermal studies provide a rapid means of evaluating the miscibility/compatibility in polymer blends. The available literature indicates that, in both crystalline and molten states, alkanes/paraffin and F-T waxes are more compatible with LLDPE than with low-density polyethylene (LDPE) or high-density polyethylene (HDPE) (Krupa and Luyt, 2000; Krupa and Luyt, 2001a; Mpanza and Luyt, 2006a; Molefi et al., 2010; Chen and Wolcott, 2014; Chen and Wolcott, 2015; Gumede et al., 2017). Differential scanning calorimetry (DSC) results showed that LLDPE and wax may even be partially miscible in the crystalline phase (Krupa and Luyt, 2000). Miscibility of polyethylene and paraffin wax in the crystalline phase was confirmed by crystallisation analysis fractionation (CRYSTAF) which showed co-crystallisation of waxes with LLDPE but not with HDPE and LDPE (Luyt and Brüll, 2004). Thus, it can be inferred that a paraffin wax or F-T wax, with relatively high molecular mass, is likely to exhibit at least some partial miscibility with a low molecular mass linear low-density polyethylene.
Inconsistent results were found in the literature concerning the miscibility of wax with polyethylene. The literature revealed some partial solubility of some waxes in some polymers but not others. It is also possible that the polymers can be miscible with the waxes over certain composition ranges. For instance, one study reported one endothermic peak for LLDPE blended with EnHance, H1 and M3 waxes at wax contents up to only 10 wt%, while other studies observed similar endothermic responses with wax loadings up to 30 wt% (Djoković et al., 2003; Krupa and Luyt, 2001; Mpanza and Luyt, 2006; Mtshali et al., 2003). In these studies, partial miscibility was observed with further increase of wax concentration. Partial miscibility has also been associated with co-crystallisation. Thus, other studies have focussed on co-crystallisation phenomena to establish the extent of solid-state miscibility. Gumede et al. (2016) observed that the co-crystallisation phenomenon was associated with shifts to lower melting and crystallisation temperatures for the LLDPE-rich phase. This occurrence could imply either solubility of the LLDPE in the wax-containing melt, or co-crystallisation, or both. Based on these observations, exploration of the extent of miscibility of low molecular mass waxes with polyethylene for their use as processing additives is recommended.
2 Properties of processing additives
In polymer compounding, processing additives are substances used to improve the melt strength and flow properties of polymers. Some processing additives improve the general flow rate of the polymer melt by lowering the melt viscosity. Increasing the melt flow rate increases product output during polymer compounding. In most cases, processing additives also work by reducing the processing temperature. The reduction in processing temperature reduces polymer stress endurance in the barrel and energy consumption. Therefore, because of these features of processing additives, polymers are one of the most versatile, cost-efficient materials in the world. In fact, without processing additives, the general polymer products would not exist since many polymer materials are useless until they undergo processability modification process.
Processing additives can be categorised depending on their function and chemical nature. Some of the commonly used processing additives include fluoropolymers, heat stabilizers, lubricants, release and anti-slip agents, flow enhancers and viscosity reducers. These processing aids function uniquely from each other. For instance, heat stabilizers e.g., metal salt combinations function by preventing thermal degradation of polyvinyl chloride (PVC). They help to prevent unwanted chemical reactions, such as chain scission or crosslinking, which can lead to product defects. Heat stabilizers are mainly employed during PVC compounding. Fluoropolymer on the other hand, functions by building up on metal die surfaces thereby forming a slippery surface coating that increases the flow stability. Fluoropolymers are mostly used for copolymers of vinylidene fluoride and hexafluoropropylene. Lubricants are known to provide either internal or external lubrication effect for the general polyolefins. Lubricants are additives that reduce friction and help the polymer melt flow more easily. They are particularly useful in preventing sticking of the melt to the processing equipment. Release agents are used to prevent the adhesion of the polymer melt to the mould surfaces. They help in easing the release of the finished product. Flow enhancers or viscosity reducers, improve the flow properties of the polymer melt or lower the viscosity of the polymer melt by reducing intermolecular forces between polymer chains. This improves the processability of the polymer by allowing it to flow more easily during processing.
It is important to make a suitable selection of the appropriate processing additives to ensure synergy between the additive and polymer. Furthermore, it is crucial to achieve a good balance of the concentration and optimum processing temperature between additive and polymer. In many respects, the concentration of processing additives is typically added in lesser amounts, usually in the range of 0.1%–5% by mass, depending on the specific additive and application. They are often used in masterbatch formulations, which are highly concentrated mixtures of additives and/or pigments dispersed in a polymer carrier resin. In this application, the processing aid, typically a wax assists the break-down of particle agglomerates. This is achievable due to their known lubricating properties, which can reduce friction between particles while improving their rheological properties. Overall, processing additives such as wax play a critical role in polymer compounding by improving the processability and performance of polymers, ultimately leading to better product quality and production efficiency.
3 Polymer blends thermodynamic fundamentals
Achieving miscibility between polymer blend components can be a major challenge in the synthesis of polymer blends. The lack of miscibility can affect the final properties of the blend, prompting researchers to expend significant efforts into understanding how the two components interact. The solid-state morphology and physical properties of blends are often determined by miscibility in the melt phase. Therefore, studying the molten state of blends has become a key area of research (Hill et al., 1993; Vadalia et al., 1994). However, studying the thermodynamic equilibrium interactions in the molten state can be controversial due to the use of indirect methods and costly techniques that are not always accessible. As a result, researchers often rely on experiments carried out in the solid state to determine the extent of miscibility in polymer blends. Researchers have used various techniques such as modifying the chemical structure of the polymer components and adjusting processing conditions. By carefully controlling these factors, researchers can improve the extent miscibility of polymer blends and create new materials with unique properties. On the other hand, compatibilizers have also been adopted to reduce the interfacial tension between the two phases. Compatibilizers help achieve finer phase dispersion and improving adhesion between the polymer components. When the interfacial tension is reduced, the dispersed phase forms smaller, more uniformly distributed domains, which enhances the overall blend properties.
Generally, the Gibbs free energy (ΔG), entropy (ΔS), and enthalpy (ΔH) are fundamental thermodynamic parameters that govern the miscibility between the two components of a polymer blend. In polymer blends, the Gibbs free energy of mixing must be minimized for the blend to achieve miscibility. This minimization promotes a stable, homogeneous mixture of the components. When the Gibbs free energy of mixing is zero or negative, the blend is in thermodynamic equilibrium and the two components are completely miscible. In this system governing conditions must be fulfilled as shown in Equation 1:
Where ΔGmix is the Gibbs free energy of mixing, ΔSmix is the combinatorial entropy of mixing and ΔHmix is the enthalpy of mixing.
When the two components of a blend are completely miscible, there is usually a small enthalpy of mixing, which can be positive or negative, depending on the specific components and conditions. However, for all polymer systems for which the interaction forces are of the van der Waals type, the enthalpy of mixing is always positive. The entropy is the state of molecular disorder or randomness. When two components of a blend are completely miscible, the disorder of the system increases, and the entropy of the system increases. This leads to a decrease in the Gibbs free energy of the system, making it more favourable for the two components to be miscible. Overall, the thermodynamic parameters of Gibbs free energy, entropy, and enthalpy are important in understanding and predicting the miscibility of polymer blends. By controlling these parameters through adjustments to the chemical structure of the components or processing conditions, researchers can improve the miscibility of polymer blends and create new materials with desired properties.
In polymer blends, there are three distinct mixing states: miscible, immiscible, and partially miscible (Hill et al., 1993; Vadalia et al., 1994; Martínez-Salazar et al., 1996; Crist and Hill, 1997). A miscible blend is also referred to as a homogeneous blend. In this type of blend, the two or more polymer components mix well to an extent that it appears as a single-phase system. The blend exhibits zero interfacial tension between the components, and it displays average or deviated properties of the two components or a whole new set of properties. In contrast, an immiscible or heterogeneous blend consists of non-uniform and visibly distinct phases. The polymer components in this type of blend are incompatible due to high interfacial tension. A partially miscible blend is a blend that exhibits phase separation in a range of compositions while appearing as a single-phase system in another range. Phase separation is mostly observed in the intermediate region, such as a 50/50 composition. A phase-separated morphology typically evolves from dispersed droplets in one phase to co-continuous morphologies depending on the specific composition and interaction parameters. In the co-continuous morphology, both components form continuous networks that are interconnected and dispersed throughout the system, resulting in a bi-continuous phase structure. In the phase-separated morphology, the two components form separate domains or droplets within the blend. The partially miscible blends often exhibit unique properties that are different from either of the pure components or the miscible blends. The properties of the partially miscible blends can be tuned by adjusting the blend composition and processing conditions to control the morphology of the system (Ajitha and Thomas, 2020; Sinha Ray and Banerjee, 2022).
Figure 2 shows the three distinct mixing states, conditions required, and typical graphs observed for this state. The graphs relate Gibbs free energy of mixing as a function of volume fraction of a binary system showcasing different degrees of miscibility states. For thermodynamically miscible blends, the entropy of mixing is greater than the enthalpy of mixing and favours mixing condition shown in Equation 2:
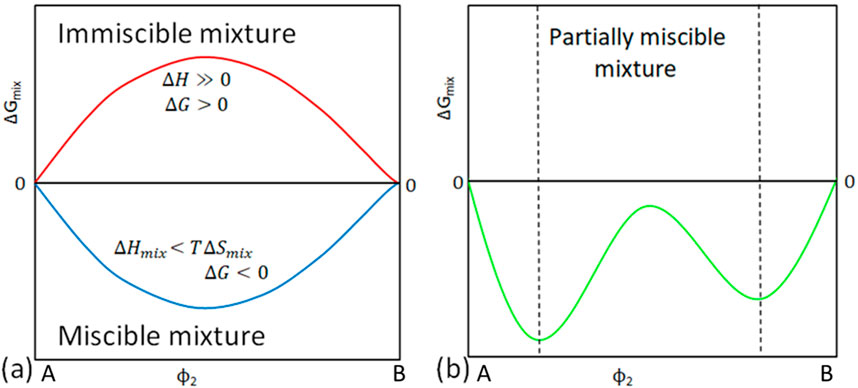
Figure 2. Variation of Gibbs energy of mixing, enthalpy of mixing and entropy of mixing with composition providing insights into the miscibility behaviour of a binary polymer blend. (A) blue line-miscible blend, (A) red line-immiscible blend and (B) green line-partially miscible blend. Figure adapted from (Higgins et al., 2010).
This condition is represented by the blue curve in Figure 2A. On the contrary, a polymer blend represented by the red curve does not satisfy the aforementioned conditions and is said to be immiscible. With this type of a blend, the enthalpy of mixing dominates and is the determining factor for miscibility, Equation 3:
One of the reasons for immiscibility in polymer blends is the significant difference in molecular masses of the components (Mishra et al., 2017; Cardinaels and Moldenaers, 2016). This is because the size difference between the polymer chains affects their ability to mix and form a homogenous blend. The reason is to be found in the reduced entropy of mixing relative to the entropy of mixing when the components are of comparable size. The entropy of mixing is not sufficient to overcome the positive enthalpy of mixing. This results in the formation of non-uniform, visibly distinct phases, i.e., the formation of a thermodynamically partially miscible blend. Another reason for immiscibility can be differences in the polarity or chemical nature of the polymer components. For example, chemically different polymers tend to be immiscible due to the differences in their intermolecular forces. Both cases are thermodynamically unfavourable because of the increase in free energy. Consequently, immiscible blends tend to phase separate over time as the system seeks to reach a more stable state, where the free energy is minimized (Mishra et al., 2017).
In a partially miscible polymer blend, shown in Figures 2A, B single phase can be observed at either end of the composition range (i.e., component A-rich phase or component B-rich phase) where the interaction between the two components is stronger. However, in the intermediate range, a two-phase morphology can be observed due to the presence of a balance between the interactions of the two components.
The Gibbs free energy of mixing curves obtained at a series of temperatures can be transformed to a temperature–composition phase diagram (Massalski and Laughlin, 2017). This summarizes the phase behaviour of a binary mixture. Figure 3 shows the typical temperature-composition symmetrical phase diagram for a regular polymer solution. This phase diagram shows the presence of a miscibility gap for a special polymer-diluent solution. Figure 3 also shows two areas of immiscibility whose coexistence curve limits are defined by the upper critical solution temperature (UCST) and lower critical solution temperature (LCST). These points of curvature define the critical temperature points. The LCST point is the temperature below which the mixture is completely miscible. Similarly, the UCST is the temperature above which the mixture is completely miscible. The outer boundary of the miscibility gap represents a single-phase region, where the polymer solution is stable and completely miscible. The metastable region is located between the spinodal and binodal curves and it is prone to fluctuations in composition and temperature. Within the metastable region, phase separation is governed by nucleation and growth processes. Finally, the unstable region between the spinodal lines indicates that any small fluctuation in composition can lead to phase separation by spinodal decomposition.
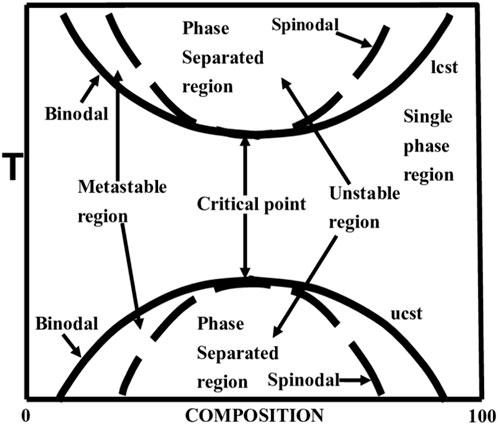
Figure 3. Phase diagrams of a regular polymer-diluent mixture represent of the phase behaviour of a mixture as a function of temperature and composition. For a regular polymer-diluent mixture, this includes the region of miscibility and binodal and spinodal curves with upper critical solution temperature (UCST) and lower critical solution temperature (LCST). Figure reproduced with permission (Robeson, 2014).
Flory and Huggins developed a theoretical framework for assessing the miscibility of polymer blends (Flory, 1941; Huggins, 1941). The Flory and Huggins theory considers parameters such as the difference in molecular mass, chemical structure, and interactions between the polymer chains in the blend. The Flory-Huggins equation is presented as:
where n1 and n2 are the moles of solvent and polymer present respectively; χ is the Flory-Huggins interaction parameter; R is the gas constant and T is the absolute temperature. ϕ1 and ϕ2 are the volume fractions of the solvent and the polymer respectively. They are defined in Equations 5a, 5b as follows:
where m is the ratio of the polymer molar volume to that of the solvent:
where M2 is the number average molecular mass of polymer, M1 is the molecular mass of the solvent and
The composition of the polymer solution can be better expressed in terms of volume fraction as shown in Equation 8:
where
The Flory-Huggins theory holds for UCST phase behaviour. The temperature dependent interaction parameter can be simplified by keeping only one temperature term:
The term A and B are constants and T is the absolute temperature. Furthermore, the theory predicts that the critical composition (ϕ2,c) and the critical interaction parameter (χc) can be determined by Equations 10, 11, respectively. The critical temperature (Tc) can be calculated using Equation 9 with known values of A and B.
McGuire et al. (1994) proposed two equations for locating the tie lines in the liquid coexistence region of UCST phase diagram. The equations relate the tie line compositions with the interaction parameter. These equations present a simple method to extrapolate the binodal curve:
where
In addition, for semi-crystalline polymer blends, the temperature dependent interaction parameter (χ) can be determined by measuring the melting point depression of the polymer in the presence of the solvent (McGuire et al., 1994). The melting point depression, Equation 16, is expressed as follows:
where
Understanding the miscibility state of a polymer blend is important for predicting its properties and performance in various applications. By controlling the mixing state, researchers can tailor the properties of the blend to meet specific application requirements.
4 Polymer blend miscibility and crystallinity
Linear low-density polyethylene is a semi-crystalline polymer. Therefore, polyethylene blends include crystallisable chains. The crystallisation process has a considerable influence on the ultimate morphology, thermal, optical and mechanical properties. Therefore, it is crucial to understand crystallinity in polyethylene blends for optimizing their properties for given specific applications. This subsection attempts to cover the important topics in polyethylene chain crystallisation which are directly connected to the present research work. It provides a brief overview of the fundamental concepts of crystallisation, factors influencing polymer crystallinity, experimental methods to determine crystallinity and polymer properties affected by crystallinity.
The extent of miscibility in polymer blends can be influenced by several factors including the molecular mass distribution, chemical composition of the components, composition ratio and temperature of the system. In polyethylene blends it has been found that factors such as differences in molecular structure i.e., degree of branching, and molecular mass are primary contributors towards immiscibility (Zhao and Choi, 2006). With respect to the case of the molecular structure, the wax primarily consists of linear alkanes, which are saturated hydrocarbons without any double bonds. On the other hand, LLDPE contains numerous short branches due to the incorporation of higher alpha-olefins as comonomers (Crist and Hill, 1997). These alpha-olefins introduce branches in the polymer chain, creating a more complex and branched structure compared to the linear structure of wax. Thus, due to the differences in molecular structure, subsequent packing arrangement, and melting behaviour, wax and polyethylene are generally not compatible with each other in the solid state. However, it is worth noting that compatibility in this instance can be influenced by factors such as composition, processing conditions, and type of wax used. Mpanza and Luyt investigated the influence of three different waxes blended with LLDPE and LDPE on separate studies (Mpanza and Luyt, 2006). The observation also reported that the type of wax blended with polyethylene plays a significant role in determining the levels of interaction, together with the amount of wax mixed with the polymer. Moreover, solid-liquid equilibrium is also observed for wax-polyethylene blends because of the significant difference in the melting points. In addition, co-crystallisation of the wax with the polymer was also observed (Hato and Luyt, 2007). It is worth noting that more complicated phase diagrams apply when the solvent used to dissolve the polymer is a binary mixture of two different compounds (Vadalia et al., 1994).
In many cases, crystallisation in polymer materials is induced by rapid cooling. However, there are other available processes including cold crystallisation i.e., annealing from the glassy state, re-crystallisation i.e., re-cooling after melting and chain orientation i.e., stretching of long chains to form crystals.
Polymers with linear chains such as HDPE and F-T waxes form a highly crystalline structure when compared to chain molecules with branches or bulky side groups. Consequently, high crystallinity is attributed to the highest order of chain packing in the absence of branches. Cooling rates are known to also influence crystallinity. For instance, faster cooling rates leads to the formation of smaller crystals due to enhanced nucleation. Conversely, slowly cooled crystallisation processes produce crystals that a have broader distribution of crystal size. Likewise, nucleating agents and plasticizers can influence the crystallisation process. In many instances, nucleating agents are added to provide a site or surface for secondary nucleation where, it acts as a pre-existing nucleus for the polymer melt. On the contrary, plasticizers tend to reduce polymer crystallinity. This is because plasticizers are often of relatively smaller size such that they occupy the space between chains. This occurrence obstructs polymer chain packing and crystal growth.
Crystallinity is an inherent property that has direct bearing on the polymer’s mechanical, thermal, barrier, optical properties. For example, thermal stability increases with the degree of crystallinity because it requires more heat energy to overcome intermolecular forces and melt the crystal structure. Hence, semicrystalline polymers such as polyethylene are processed well above their melting temperature. Moreover, a higher degree of crystallinity also increases mechanical strength and modulus. Crystallinity also influences the transparency of polymers, for example the opacity of a polymer increases with crystallinity. Many other physical properties, such as density, ductility, colour, phase transition temperature and yield strength are all highly dependent on the total crystallinity of the polymer. Normally, the standard analytical techniques utilised for the characterisation of crystallites include (1) optical microscopy which can reveal the nucleation and growth of the crystallites (Crist and Schultz, 2016), (2) electron microscopy which can show the morphology of a folded lamellar structure (Mandelkern, 2011), (3) differential scanning calorimetry (DSC) which measures the crystallisation and melting behaviour of the lamellar structure (Lorenzo et al., 2007), (4) atomic force microscopy which can be used to view the two or three-dimensional crystal structure of macromolecules (McPherson et al., 2000), (5) and wide and small angle x-ray scattering/diffraction of semicrystalline polymer that show sharp and broad peaks corresponding to the crystalline and amorphous regions respectively (shilpa kasargod nagaraj, 2016).
5 PE/wax: Thermophysical properties
The glass transition temperature (Tg) is a parameter used to probe miscibility in polymer blends, whether amorphous or semi-crystalline (Brostow et al., 2008; Kalogeras and Brostow, 2009). The Tg refers to the temperature at which a material transitions from a hard, brittle state to a more rubbery, viscous state. The Tg is sensitive to molecular interactions and mobility. When two polymers are miscible, their molecular interactions (e.g., Van der Waals forces, hydrogen bonding) and the resulting chain mobility affect the overall Tg. The presence of these interactions can shift the Tg compared to the pure components. The Tg can be determined using differential scanning calorimetry (DSC) and dynamic mechanical analysis (DMA) (Leyva-Porras et al., 2019; Li et al., 2009). In the study of blends, a single Tg, positioned in-between those of the two parent compounds, reflects full miscibility, while the observation of two distinct Tg’s (due to their respective components) reflects immiscibility (Kalogeras and Brostow, 2009; Thirtha et al., 2005).
Similarly, the DSC melting peak temperature (Tm), and crystallisation peak temperatures (Tc) have been used to evaluate the miscibility of semi-crystalline polymer materials (Krupa and Luyt, 2001a). As is the case with the glass transition, a single melting and crystallisation transition suggests the presence of a homogenous crystalline structure whereas two different peaks indicate immiscible crystal phases. On the other hand, shifts and alterations of the melting/crystalline peak have been associated with co-crystallisation and partial miscibility in the blend system (Gumede et al., 2016).
Most studies used comprehensive DSC analyses to determine the extent of compatibility in the wax/polyethylene blends systems. However, many of the blends were prepared for applications other than the one considered presently, i.e., the application as a processing additive. Table 1 summarises DSC data for a range of wax-polyethylene blends. It includes the effects of the components and the blend ratio on the crystallisation peak temperatures and enthalpy of melting. The studies considered both neat and oxidised paraffin and F-T waxes. Blends of these waxes with HDPE, LDPE and LLDPE were considered (Luyt and Brüll, 2004; Hato and Luyt, 2007; Molefi et al., 2010; Sotomayor et al., 2014; Mtshali et al., 2001; Mtshali et al., 2003; Krupa et al., 2007). Some of these studies compared the performance of a single wax with different polyethylene types (Luyt and Brüll, 2004; Hato and Luyt, 2007; Molefi et al., 2010; Mtshali et al., 2003). Other studies compared the influence of distinct types of wax blended with one type of polyethylene (Mtshali et al., 2003; Mpanza and Luyt, 2006a; Hato and Luyt, 2007).
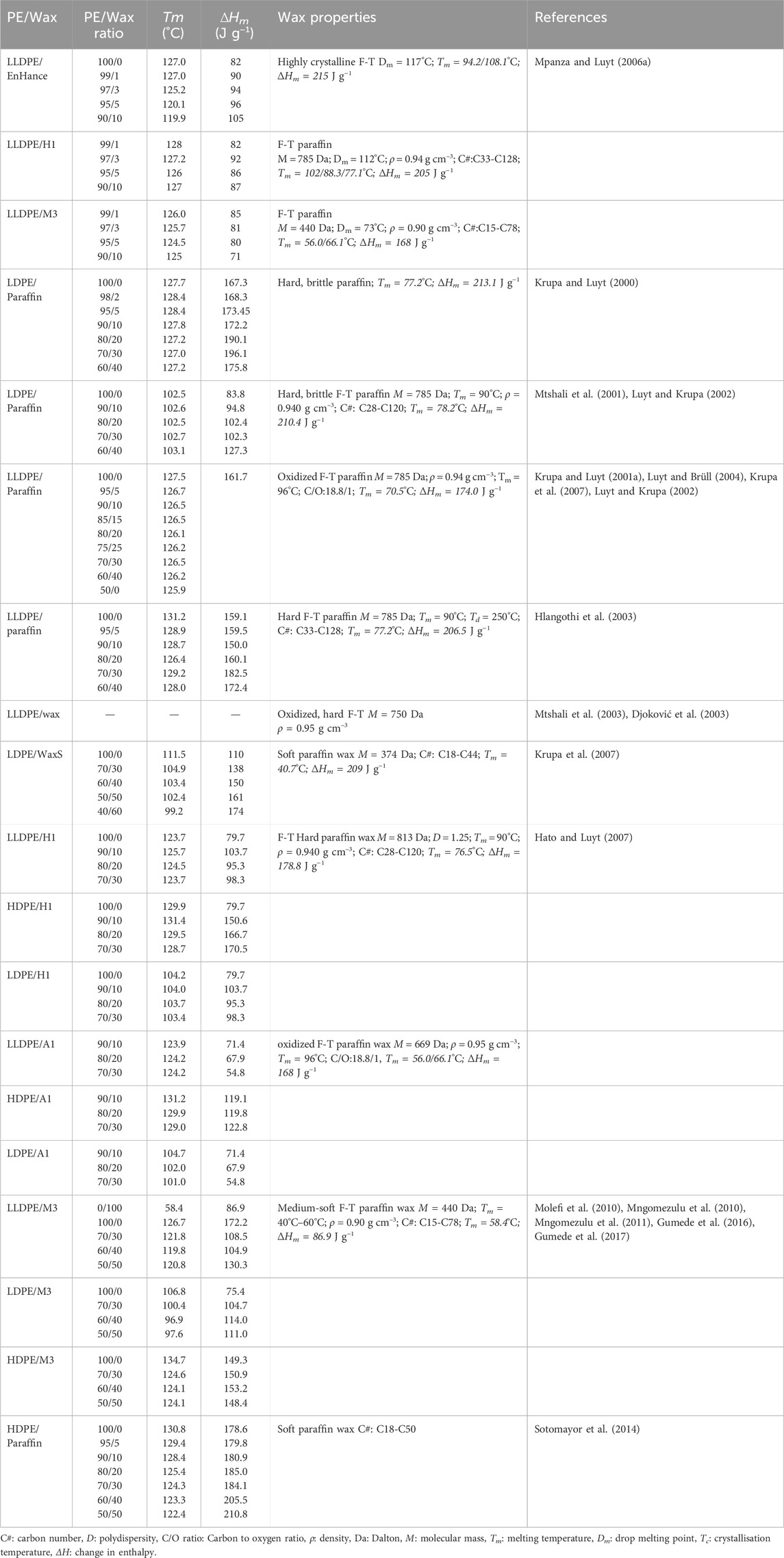
Table 1. Wax components used in polyethylene blend studies.
All these studies assumed that the presence of a single melting point temperature indicated complete miscibility. For instance, Molefi et al. (2010) blended M3 soft paraffin wax with LDPE, LLDPE and HDPE. In the study, they observed two well-separated crystallisation peaks in all the blends with wax contents of 30, 40 and 50 wt%. This result implied immiscibility of all the polyethylene with this wax. In mixtures of soft and hard paraffin waxes and LDPE, it was found that only the hard F-T wax co-crystallised with the LDPE (Krupa et al., 2007). Because of the strong distinction of the LDPE peak from the Wax S (the soft wax) peak, the total enthalpy of the mixture exceeded predictions based on the additive rule indicating a higher crystallinity of the polymer than expected (Krupa et al., 2007). However, there was strong peak overlap between the hard F-T wax and the same LDPE (Krupa et al., 2007). Sotomayor et al. (2014) also inferred incompatibility of HDPE with paraffin wax on the basis of dynamic mechanical test results, shown in Figure 4. The HDPE sample featured a unique tan δ peak located at −110°C. It corresponds to the α-relaxation, i.e., the glass transition temperature (Tg). The blends showed two peaks with one corresponding to the Tg of polyethylene and, another peak centred at −65°C corresponding to the Tg of the paraffin wax. This proved that the components were not miscible in the solid state.
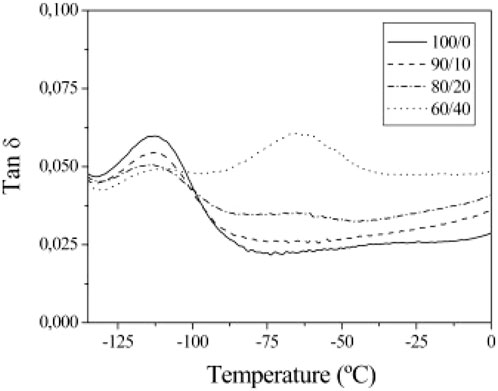
Figure 4. DMA loss tangent vs. temperature of HDPE/polyethylene wax blends. Reproduced with permission from (Sotomayor et al., 2014).
Some inconsistencies are noted in the literature with respect to the miscibility of wax and polyethylene as far as the composition of the blends is concerned. For instance Hato and Luyt investigated blends of two paraffin waxes, H1 and A1, with HDPE, LDPE, and LLDPE (2007). Complete solid-state miscibility of HDPE blends with both waxes up to 20 wax-% was found. The LDPE/H1 blend was only partially miscible whereas the LDPE/A1 blends were completely miscible up to 10 wt% wax. Complete miscibility was observed for all the LLDPE/A1 wax blends. However, in the LLDPE/H1 only partial miscibility was found at all wax contents. Moreover, in blends of cross-linked and uncross-linked low-density polyethylene (LDPE)/F-T wax, a single melting peak, belonging to LDPE phase, was found. This observation strongly suggested complete miscibility considering that the wax had three peaks in its pure state (Mtshali et al., 2001). In other studies, Mtshali et al. (2003) and Djoković et al. (2003) also observed similar mutual mixing for an oxidised F-T wax blended with both LDPE and LLDPE. Luyt and Brüll (2004) performed crystallisation analysis fractionation (CRYSTAF) and size exclusion chromatography coupled to FTIR (SEC-FTIR) on a series of HDPE-wax, LDPE/wax and LLDPE/wax blends. The CRYSTAF analysis shows little or no co-crystallisation of wax with HDPE and LDPE but provided strong indications of co-crystallisation with LLDPE. Furthermore, the co-elution of wax with LLDPE in SEC-FTIR analyses indicated some chemical interaction between the oxidized wax and LLDPE.
Focusing on blends of LLDPE and a variety of waxes, complete miscibility in the crystalline phase has been observed for LLDPE combined with oxidised Fischer-Tropsch wax or paraffin wax blends (Krupa and Luyt, 2001a; Luyt and Krupa, 2002). This behaviour was indicated by the presence of a single melting and crystallisation peak of an LLDPE-rich phase and the absence of three melting peaks due to the wax in the blends containing up to 50 wax %. When compared to neat F-T or paraffin wax/LLDPE blends only one endothermic peak in blends up to 30 wax% was observed. Surprisingly, these studies reported that the oxidised wax has virtually no influence on the melting and crystallisation temperature and the corresponding heat of melting. Figure 5 shows the heating and cooling DSC scans of the oxidised F-T wax/LLDPE blends. It appeared as if the oxygen-containing groups in oxidised wax caused the wax to interact differently with the LLDPE compared to the neat wax.
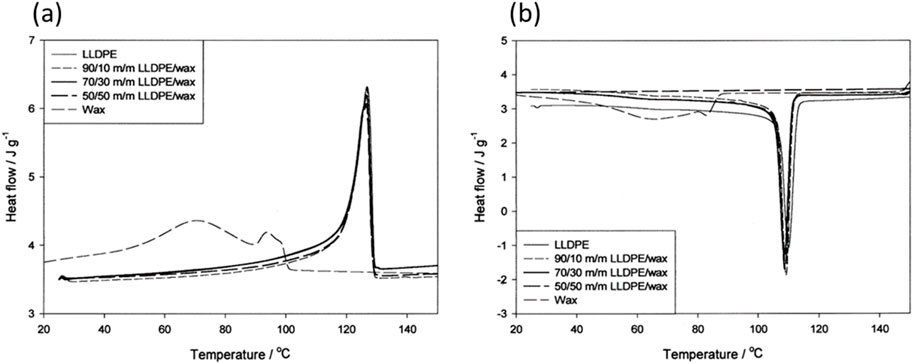
Figure 5. DSC (A) heating and (B) cooling curves of LLDPE, wax and different LLDPE/wax blends. Figure reproduced with permission (Krupa and Luyt, 2001a).
By blending LLDPE with oxidised Fischer–Tropsch wax, Mtshali et al. (2003) also showed that the wax and polyethylene chains do crystallise together, producing a single melting peak for compositions containing up to 30 wt% wax. However, with increasing wax, a melting peak associated with a wax-rich phase appears at lower temperatures, which shows the presence of crystal phase separation. This result indicated the occurrence of the distinct phase structure belonging to the wax. In contrast, Mpanza and Luyt (2006a) reported that only trivial amounts of wax can be dissolved in the LLDPE phase. The study compared three different waxes mixed with LLDPE and found partial miscibility up to 5 wt% wax.
Gumede et al. (2016) investigated the structure and thermal properties of linear low-density polyethylene (LLDPE)/medium soft paraffin wax blends with a range of sophisticated characterisation techniques. Small- and wide-angle X-ray scattering (SAXS) showed that the blends form a single phase in the melt. However, upon cooling from the melt, two crystalline phases, with depressed melting points, develop when more than 10 wt% wax is present. Gumede et al. (2016) attributed the higher melting point crystalline phase to less-branched LLDPE fractions. Standard DSC results, successive self-nucleation, and annealing (SSA) thermal fractionation and the detection of a new SAXS signal were attributed to the lamellar long period of the co-crystals. The results indicate that the lower melting point crystalline phase is a wax-rich phase constituted by co-crystals of extended chain wax and short linear sequences of highly branched LLDPE chains. The implication is that the LLDPE fractions are molecularly segregated as they crystallise upon cooling from the melt to form first the higher melting phase composed of exclusive linear LLDPE chains free of side branches. During this process, the wax molecules are expelled to the amorphous regions. Upon further cooling, the wax chains co-crystallise with the linear portions of highly branched LLDPE chains forming a wax-rich phase. Gumede et al. (2016) attributed this to the wax acting as an effective plasticizer for LLDPE, decreasing both its crystallisation and melting temperature.
6 PE/wax: Rheological properties
Correlations between rheological functions and molecular mass or composition or temperature etc. have been the subjects of interests (Utracki and Schlund, 1987). These correlations were used to discover a wide range of phase behaviours that directly influence the properties and ultimate applications of polyethylene blends. Furthermore, to also investigate the degree of miscibility within the polymer blends, many researchers have relied on these correlations (Grunberg and Nissan, 1949; Friedman and Porter, 1975; Utracki and Schlund, 1987). Miscibility studies for polyethylene/polyethylene blends employing rheology have been done (Utracki and Schlund, 1987; Müller et al., 1994; Kukaleva et al., 2003). Unfortunately, as a new member in polyethylene blends, there is limited information on the rheological properties and miscibility of PE/wax blends. Although, the rheological properties for polyethylene/polyethylene blends display complex behaviours, in general their characteristic responses might give helpful information on the internal structure of PE/wax blends.
Using rheology, the miscibility of PE/wax blends can be easily established if they exhibit single phase behaviour. For some selected miscible polymer blends, up to 50% blend ratio, the viscosity follows the popular log-additivity rule shown in Equation 17 (Robeson, 2007; Sotomayor et al., 2014). It expresses the logarithm of the viscosity (η) as mass fraction (wi) weighted mean over the logarithm of the viscosity of the pure blend components:
In this equation, the subscripts 1 and 2 correspond to the “diluent” and polymer, respectively. This model assumes that the blend components are completely miscible down to the molecular level, i.e., there is no phase separation in the molten state. It is a fully predictable model once the composition and viscosity of the pure components are known.
Partially miscible blends, i.e., emulsion-like systems tend to display more complex viscosity behaviour. In these cases, the viscosity will deviate from the log additive rule. Deviations from such systems can arise from many factors including polymer-polymer interaction, molecular mass distribution, chain conformation, chain entanglements, etc. The viscosity of such mixtures may show either positive or negative deviations from the log additive rule. Positive deviations are likely to arise from strong intermolecular interactions whereas negative deviations can be seen because of weakened interactions or the presence of a dilution factor. Regrettably, deviations from the log additive rule suggests failure to accurately predict the mixing behaviour of these typical polymer mixes. Hence, the development of alternative mixing rules to provide guidance and improve analysis of the phase structure of these blends is key.
To account for the excess viscosity, the log additive rule has been modified to include an excess viscosity term (∆ln (η)) represented in Equation 18:
The addition of an excess viscosity term provides a more accurate estimation of the viscosity behaviour in polymer blends that exhibit more complex viscosity behaviour (Grunberg and Nissan, 1949; Hind et al., 1960). The excess viscosity can be determined experimentally and by means of referencing to the predicted viscosity data using the log additive rule. The difference in viscosity from the measured and calculated can be used to predict the magnitude and direction of the excess viscosity term (Padding and Briels, 2002).
On the other hand, for homopolymers, the zero-shear viscosity as a function of molecular mass in polymer melts can be described by an empirical power-law relationship similar to the Mark-Houwink equation:
where ηo represents the zero-shear viscosity of the polymer melt, K is a proportionality constant, α is the power-law exponent that relates the viscosity to the molecular mass. M is the mass average molecular mass of the polymer defined by Equation 20:
The relationship in Equation 19 is divided into two separate regimens for homopolymers as shown in Figure 6.
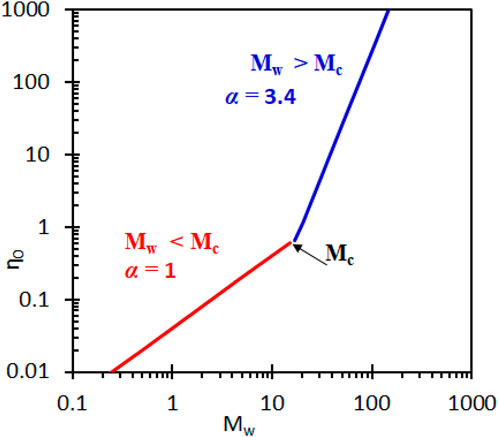
Figure 6. Illustration of zero-shear viscosity dependence on molecular mass of polymer melts. Figure adapted from (Padding and Briels, 2002).
Regime (1. red coloured) is below the critical molecular mass (Mc) below which the effect of entanglement on the polymer melt viscosity is insignificant. The zero-shear viscosity generally increases with increasing molecular mass given by Equation 21:
Regime (2. blue colour) is above the critical molecular mass above which the effect of entanglement on polymer melt viscosity is significant. Herein, the zero-shear viscosity-molecular mass relationship often deviates from the power-law behaviour observed below Mc and the relationship is given by:
The exponent takes on a universal value of α = 3.4. Based on Equation 22, Friedman and Porter have proposed that a combination of polymers of a similar chemical structure, but different molecular mass should obey the following mixing rule presented by Equation 23 (Friedman and Porter, 1975):
This model predicts a positive deviation from the log-additive rule for binary miscible molecular blends. It is important to note that the values of K and α are specific to each polymer system and can vary based on factors such as the polymer composition and structure, and experimental conditions. A few other models and theories which consider the interaction parameters have been proposed to model the non-linear dependence of viscosity on composition. The Lederer model can fit both positive and negative deviations from the log-linear blending rule described by Equation (24) (Lederer, 1931):
Other models that are able to predict deviations from the log-linear relationship include Grunberg and Nissan model, Equation 25 and Hind model Equation 26, models given below (Grunberg and Nissan, 1949; Hind et al., 1960).
Grunberg and Nissan:
Hind et al., model:
Figure 7A demonstrates that a similar linear approximation can be achieved in this composition range up to 50%. However, due to the entanglement of long-chain molecules in the melt, significantly higher viscosities are observed, as observed in Figure 7B. This is because only cooperative molecular motion is required (Fox and Flory, 1951).
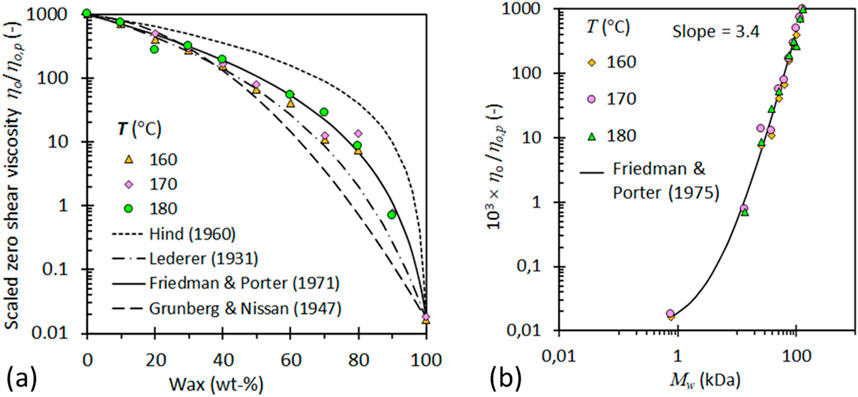
Figure 7. (A) Variation of the scaled zero-shear viscosity with wax content. Testing different predictive viscosity mixture rules (B) Zero-shear viscosity versus the calculated average molecular mass of the blends at various testing temperatures (Mhlabeni et al., 2023).
Other rheological methods used to evaluate the presence of morphological changes in polymer blends, associated with miscibility, include the Cole-Cole plots and Han plots (Han and Jhon, 1986; Han, 1988; Mohammadi et al., 2012; Agrawal et al., 2022; Bai et al., 2010). These plots are particularly relevant for blends characterised by a two-phase morphology or similar structural complexity. Both these plots are used to explore the presence of different internal structures viz homogenous and heterogenous (co-continuous and dispersed phase) structure. For the Cole-Cole plots, the plots are represented by a relationship between the real (η′) and the imaginary (η″) parts of the complex viscosity, whereas the Han Plots are plots of log G′ versus log G″ (Bai et al., 2010). A strong linear correlation and a single smooth semi-circular curve in the Han Plots and Cole-Cole plots, respectively, indicates good compatibility. Deviations from the above-mentioned plots suggests heterogeneity of the blend. Recently, these plots have been used to describe the viscoelastic properties of polyethylene/wax blends having a high degree of relaxation (Mhlabeni et al., 2023), shown in Figure 8. These plots demonstrate relaxation mechanism induced by composition and temperature changes. By analysing these plots for a polymer blend, researchers can gain a better understanding of the blend’s rheological behaviour. This enables adjustment to improve processing properties.
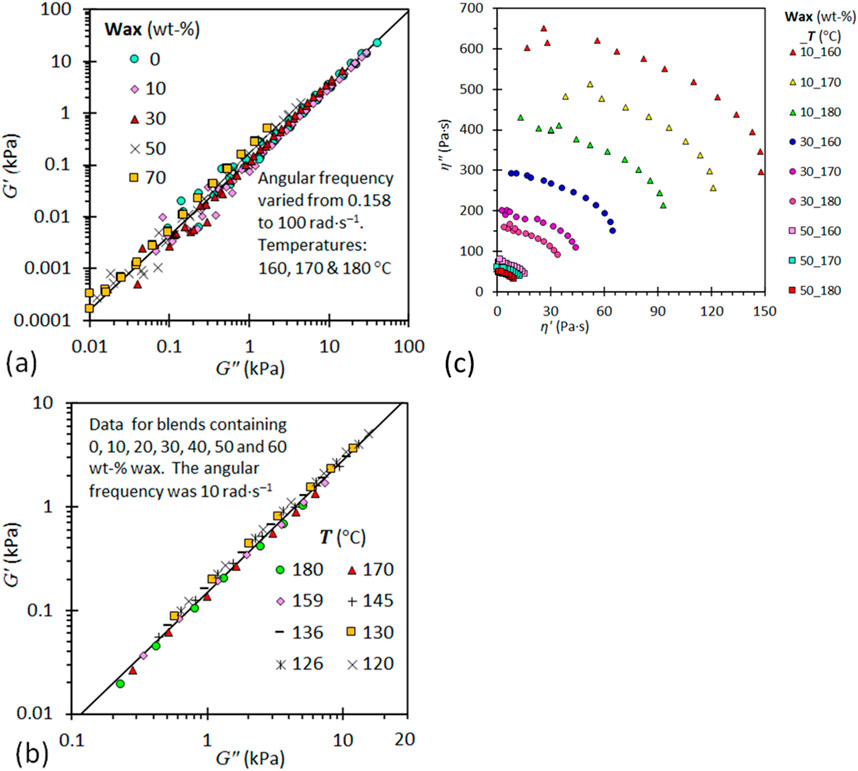
Figure 8. Plots of G′ versus G″ for LLDPE/wax blends, (A) Emphasizing the effect of blend composition at three different temperatures and five wax content levels and (B) Emphasizing the effect of temperature at seven different wax content levels at a fixed angular frequency of 10 rad s−1. (C) Cole–Cole plots of η″ against η′ for LLDPE/wax blends (Mhlabeni et al., 2023).
7 Conclusion and recommendations
The purpose of review was to explore studies on the miscibility of low molecular mass waxes blended with polyethylene’s for possible use as a processing additive in polyethylene masterbatch applications. This study focused on aspects that affect compatibility of polymer blends, in an effort to elucidate the interactions between polyethene and wax. The theoretical framework for the miscibility of polymer blends was discussed in terms of thermodynamic principles. The thermophysical and rheological properties of the blends are useful tool to characterize their miscibility. The study shows that, in both crystalline and the molten state, paraffin and F-T waxes are more compatible with LLDPE than with LDPE or HDPE. However, the discussion of the influence of wax composition on the blend compatibility/miscibility and crystallisation behaviour is not consistent. Moreover, research covering miscibility of low molecular mass paraffin or F-T wax/PE area is not extensive. On the other hand, experimental studies focusing on rheological properties have been relatively rare. Furthermore, the influence of composition and temperature have not been fully covered. Nonetheless, the available literature suggests full miscibility of the low molecular mass waxes and the polyethylene in the melt and partial co-crystallisation in the solid state. Moreover, in dynamic DSC scans, the near complete absence of a wax-like melting peak for the blends containing less than 10 wt% wax suggests complete miscibility at that concentration.
DSC analysis showed that polyethylene appeared as a dissimilar phase to the wax phase with increasing blend ratio however its melting point depression was seen. Therefore, it is unclear whether the insoluble polyethylene fraction was the short, branched chains due to the incorporation of higher alpha-olefins as comonomers or an entirely different fraction present in small quantities. Therefore, further work to study the structural and molecular mass distribution of the blends would be needed to identify this fraction, also accompanied by extra studies to elucidate the blends thermal degradation with time.
In conclusion, the versatile role of waxes as processing aids, encompassing a range of compositions, temperatures, rheological improvement, and lubrication, underscores their significance in polymer compounding. Areas for future research can expound on rheology and DSC measurements. The rheological methods, viz shear rate rheometry, can provide valuable insights into the rheological behaviour of the PE/wax blends. However, there are additional rheological methods that can complement this existing knowledge and elucidate the scope of the blends’ rheological properties. For example, further studies can incorporate methods such as controlled shear stress rheometry, creep and stress relaxation tests etc., to gain more comprehensive understanding about the internal molecular rearrangements and time-dependent behaviour of the blends. Furthermore, both polyethylene and these wax compounds belong to the same chemical family of saturated hydrocarbons. Amongst a wide range of n-alkane diluents, including waxes, mixed with polyethylene, the solubility of polyethylene increased with increasing molecular mass of the diluents. Perhaps, increasing the average molar mass and chain distribution of the wax could help to overcome the “poor of compatibility” limitation. Since, the required compatibility could be achieved by appropriate blending of high molar waxes with low molar mass LLDPE. Perhaps small additions (1-5 wt%) of low molecular mass LLDPE to wax, could yield high molar mass wax with a broader chain distribution to encourage better compatibility with the base polymer. The envisioned, higher average molar mass wax could reduce or even prevent die-lip drooling. The aim would be to moderately raise the melt viscosity and processing temperature of the wax to achieve longer residence time with the polymer matrices during high shear thermodynamic exposure of the extruders.
The compatibility of polyethylene remains a foundational but evolving topic. Another forward-looking perspective on specific research topics may involve highly entangled molecular chains such as in the combinations of ultra-high molecular weight polyethylene (UHMWPE) with wax. Future research can focus on techniques for molecular chain disentanglement, such as mechanical shear, controlled thermal processing, or chemical modification. These studies could also explore the role of compatibilizers in addressing interfacial adhesion challenges. The blending of polyolefin elastomers (POE), olefin block copolymers (OBC), and PE opens new avenues for achieving tailored mechanical properties such as toughness and flexibility. Future research could investigate the interplay between block structure, chain mobility, and phase behaviour in these systems. Understanding how these factors influence miscibility and the microstructure could lead to more precise control over blend properties. Finally, potential applications of these blends in cutting-edge industries such as advanced packaging, sustainable materials, and biomedical applications could be explored. The use of multifunctional nanofillers represent a new trend in polyolefin blends for various applications for the purpose of enhancing primary properties and/or creating new functionalities. Perhaps the paraffin or F-T wax/PE blends coupled with such nanofillers could exploit these properties adding value to existing applications.
Author contributions
TM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. TJ: Funding acquisition, Project administration, Resources, Supervision, Validation, Writing–review and editing. WM: Data curation, Funding acquisition, Project administration, Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Open access funding provided by Tshwane University of Technology. The authors received no direct funding for this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agrawal, P., Silva, M. H. A., Cavalcanti, S. N., Freitas, D. M. G., Araújo, J. P., Oliveira, A. D. B., et al. (2022). Rheological properties of high-density polyethylene/linear low-density polyethylene and high-density polyethylene/low-density polyethylene blends. Polym. Bull. 79, 2321–2343. doi:10.1007/s00289-021-03635-8
Ajitha, A. R., and Thomas, S. (2020). “Chapter 1 - introduction: Polymer blends, thermodynamics, miscibility, phase separation, and compatibilization,” in Compatibilization of polymer blends. Editors A. A.R, and S. THOMAS (Elsevier).
Ashby, M. F. (2013). “Chapter 15 - material profiles,” in Materials and the environment Second Edition (Boston: Butterworth-Heinemann).
Bai, L., Li, Y. M., Yang, W., and Yang, M.-B. (2010). Rheological behavior and mechanical properties of high-density polyethylene blends with different molecular weights. J. Appl. Polym. Sci. 118, 1356–1363. doi:10.1002/app.32329
Bakshi, A. K., and Ghosh, A. K. (2022). Processability and physico-mechanical properties of ultrahigh-molecular-weight polyethylene using low-molecular-weight olefin wax. Polym. Eng. and Sci. 62, 2335–2350. doi:10.1002/pen.26013
Bayat, M. H., Abdouss, M., and Javanbakht, M. (2013). Quantification of polyethylene wax in semibatch laboratory reactor and a study of different parameters on wax production as a by-product in the slurry ethylene polymerization. J. Appl. Polym. Sci. 127, 1027–1031. doi:10.1002/app.37916
Bennett, H. (1944). Commercial waxes, natural and synthetic, including properties, uses, methods of handling and formulas for making commercial wax compositions. Brooklyn, N. Y: Chemical publishing co., inc.
Brostow, W., Chiu, R., Kalogeras, I. M., and Vassilikou-Dova, A. (2008). Prediction of glass transition temperatures: binary blends and copolymers. Mater. Lett. 62, 3152–3155. doi:10.1016/j.matlet.2008.02.008
Cardinaels, R., and Moldenaers, P. (2016). Morphology development in immiscible polymer blends. Polym. Morphol., 348–373. doi:10.1002/9781118892756.ch19
Chaloupková, K., and Zatloukal, M. (2009). Effect of die design on die drool phenomenon for metallocene based LLDPE: Theoretical and experimental investigation. J. Appl. Polym. Sci. 111, 1728–1737. doi:10.1002/app.29146
Chen, F., and Wolcott, M. P. (2014). Miscibility studies of paraffin/polyethylene blends as form-stable phase change materials. Eur. Polym. J. 52, 44–52. doi:10.1016/j.eurpolymj.2013.09.027
Chen, F., and Wolcott, M. (2015). Polyethylene/paraffin binary composites for phase change material energy storage in building: a morphology, thermal properties, and paraffin leakage study. Sol. Energy Mater. Sol. Cells 137, 79–85. doi:10.1016/j.solmat.2015.01.010
Cheremisinoff, N. P. (2001). in Condensed encyclopedia of polymer engineering terms. Editor N. P. CHEREMISINOFF (Boston: Butterworth-Heinemann).
Ciesińska, W., Liszyńska, B., and Zieliński, J. (2016). Selected thermal properties of polyethylene waxes. J. Therm. Analysis Calorim. 125, 1439–1443. doi:10.1007/s10973-016-5706-1
Crist, B., and Hill, M. J. (1997). Recent developments in phase separation of polyolefin melt blends. J. Polym. Sci. Part B Polym. Phys. 35, 2329–2353. doi:10.1002/(sici)1099-0488(199710)35:14<2329::aid-polb12>3.0.co;2-e
Crist, B., and Schultz, J. M. (2016). Polymer spherulites: a critical review. Prog. Polym. Sci. 56, 1–63. doi:10.1016/j.progpolymsci.2015.11.006
Dangtungee, R., Yun, J., and Supaphol, P. (2005). Melt rheology and extrudate swell of calcium carbonate nanoparticle-filled isotactic polypropylene. Polym. Test. 24, 2–11. doi:10.1016/j.polymertesting.2004.08.006
Deshmukh, S. P., Parmar, M. B., Rao, A. C., and Wadhwa, V. (2010). Polymer- and wax-based monoconcentrate predispersed pigments in the colouring of plastics. Color. Technol. 126, 189–193. doi:10.1111/j.1478-4408.2010.00245.x
Djoković, V., Mtshali, T. N., and Luyt, A. S. (2003). The influence of wax content on the physical properties of low-density polyethylene–wax blends. Polym. Int. 52, 999–1004. doi:10.1002/pi.1180
Elleithy, R., Ali, I., Al-Haj Ali, M., and Al-Zahrani, S. (2011). High density polyethylene/micro calcium carbonate composites: a study of the morphological, thermal, and viscoelastic properties. J. Appl. Polym. Sci. 117, 2413–2421. doi:10.1002/app.32142
Esmaeilzade, R., Sharif, F., Rashedi, R., and Dordi Nejad, A. (2022). Morphology, phase diagram, and properties of high-density polyethylene/thermally treated waste polyethylene wax blends. J. Appl. Polym. Sci. 139, 51750. doi:10.1002/app.51750
Flory, P. J. (1941). Thermodynamics of high polymer solutions. J. Chem. Phys. 9, 660–661. doi:10.1063/1.1750971
Fox, T. G., and Flory, P. J. (1951). Further studies on the melt viscosity of polyisobutylene. J. Phys. Chem. 55, 221–234. doi:10.1021/j150485a010
Friedman, E. M., and Porter, R. S. (1975). Polymer viscosity-molecular weight distribution correlations via blending: for high molecular weight poly(dimethyl siloxanes) and for polystyrenes. Trans. Soc. Rheol. 19, 493–508. doi:10.1122/1.549382
Gale, M. (1997). Compounding with single-screw extruders. Adv. Polym. Technol. 16, 251–262. doi:10.1002/(sici)1098-2329(199711)16:4<251::aid-adv1>3.3.co;2-j
Grunberg, L., and Nissan, A. H. (1949). Mixture law for viscosity. Nature 164, 799–800. doi:10.1038/164799b0
Gudiño Rivera, J., Hernández Gámez, J. F., Borjas Ramos, J. J., Salinas Hernández, M., and Sánchez Martínez, D. I. (2022). Effects of residual wax content on the thermal, rheological, and mechanical properties of high-density polyethylene. Polym. Eng. and Sci. 62, 1867–1875. doi:10.1002/pen.25971
Gumede, T. P., Luyt, A. S., Pérez-Camargo, R. A., Iturrospe, A., Arbe, A., Zubitur, M., et al. (2016). Plasticization and cocrystallization in LLDPE/wax blends. J. Polym. Sci. Part B Polym. Phys. 54, 1469–1482. doi:10.1002/polb.24039
Gumede, T. P., Luyt, A. S., Pérez-Camargo, R. A., and Müller, A. J. (2017). The influence of paraffin wax addition on the isothermal crystallization of LLDPE. J. Appl. Polym. Sci. 134, 44398. doi:10.1002/app.44398
Guzzi, F., Parrotta, E., Zaccone, S., Limongi, T., Cuda, G., and Perozziello, G. (2023). “Chapter 4 - materials,” in Microfluidics for cellular applications. Editors G. PEROZZIELLO, U. KRÜHNE, and P. LUCIANI (Elsevier).
Han, C. D. (1988). Influence of molecular weight distribution on the linear viscoelastic properties of polymer blends. J. Appl. Polym. Sci. 35, 167–213. doi:10.1002/app.1988.070350115
Han, C. D., and Jhon, M. S. (1986). Correlations of the first normal stress difference with shear stress and of the storage modulus with loss modulus for homopolymers. J. Appl. Polym. Sci. 32, 3809–3840. doi:10.1002/app.1986.070320302
Hato, M. J., and Luyt, A. S. (2007). Thermal fractionation and properties of different polyethylene/wax blends. J. Appl. Polym. Sci. 104, 2225–2236. doi:10.1002/app.25494
Higgins, J., Lipson, J., and White, R. (2010). A simple approach to polymer mixture miscibility. Philosophical Trans. Ser. A, Math. Phys. Eng. Sci. 368, 1009–1025. doi:10.1098/rsta.2009.0215
Hill, M. J., Barham, P. J., and Van Ruiten, J. (1993). Liquid-liquid phase segregation in blends of a linear polyethylene with a series of octene copolymers of differing branch content. Polymer 34, 2975–2980. doi:10.1016/0032-3861(93)90623-i
Hind, R. K., Mclaughlin, E., and Ubbelohde, A. R. (1960). Structure and viscosity of liquids. Camphor + pyrene mixtures. Trans. Faraday Soc. 56, 328–330. doi:10.1039/tf9605600328
Hlangothi, S. P., Krupa, I., Djoković, V., and Luyt, A. S. (2003). Thermal and mechanical properties of cross-linked and uncross-linked linear low-density polyethylene–wax blends. Polym. Degrad. Stab. 79, 53–59. doi:10.1016/s0141-3910(02)00238-0
Hostomsky, J., and Jones, A. G. (1991). Calcium carbonate crystallization, agglomeration and form during continuous precipitation from solution. J. Phys. D Appl. Phys. 24, 165–170. doi:10.1088/0022-3727/24/2/012
Huggins, M. L. (1941). Solutions of long chain compounds. J. Chem. Phys. 9, 440. doi:10.1063/1.1750930
Kalogeras, I. M., and Brostow, W. (2009). Glass transition temperatures in binary polymer blends. J. Polym. Sci. Part B Polym. Phys. 47, 80–95. doi:10.1002/polb.21616
King, L. F., and Noël, F. (1972). Characterization of lubricants for polyvinyl chloride. Polym. Eng. and Sci. 12, 112–119. doi:10.1002/pen.760120207
Koningsveld, R., Kleintjens, L., and Schoffeleers, M. (1974). Thermodynamic aspects of polymer compatibility. Pure Appl. Chem. 39, 1–32. doi:10.1351/pac197439010001
Krupa, I., and Luyt, A. S. (2000). Thermal properties of uncross-linked and cross-linked LLDPE/wax blends. Polym. Degrad. Stab. 70, 111–117. doi:10.1016/s0141-3910(00)00097-5
Krupa, I., and Luyt, A. S. (2001). Thermal and mechanical properties of extruded LLDPE/wax blends. Polym. Degrad. Stab. 73, 157–161. doi:10.1016/s0141-3910(01)00082-9
Krupa, I., and Luyt, A. S. (2001a). Physical properties of blends of LLDPE and an oxidized paraffin wax. Polymer 42, 7285–7289. doi:10.1016/s0032-3861(01)00172-0
Krupa, I., Miková, G., and Luyt, A. S. (2007). Phase change materials based on low-density polyethylene/paraffin wax blends. Eur. Polym. J. 43, 4695–4705. doi:10.1016/j.eurpolymj.2007.08.022
Kukaleva, N., Simon, G. P., and Kosior, E. (2003). Modification of recycled high-density polyethylene by low-density and linear-low-density polyethylenes. Polym. Eng. and Sci. 43, 26–39. doi:10.1002/pen.10002
Lederer, E. (1931). Zur theorie der viskosität von flüssigkeiten. Beih. 34, 270–338. doi:10.1007/bf02556584
Lee, C. D. (2002). “Die-lip build-up in the extrusion of iighly filled low density polyethylene,” in Annual technical conference ANTEC, conference proceedings, 1.
Leyva-Porras, C., Cruz-Alcantar, P., Espinosa-Solís, V., Martínez-Guerra, E., Balderrama, C. I. P., Martínez, I. C., et al. (2019). Application of differential scanning calorimetry (DSC) and modulated differential scanning calorimetry (MDSC) in food and drug industries. Polym. (Basel) 12, 5. doi:10.3390/polym12010005
Li, J., Li, H., Wu, C., Ke, Y., Wang, D., Li, Q., et al. (2009). Morphologies, crystallinity and dynamic mechanical characterizations of polypropylene/polystyrene blends compatibilized with PP-g-PS copolymer: effect of the side chain length. Eur. Polym. J. 45, 2619–2628. doi:10.1016/j.eurpolymj.2009.06.002
Lorenzo, A. T., Arnal, M. L., Albuerne, J., and Müller, A. J. (2007). DSC isothermal polymer crystallization kinetics measurements and the use of the Avrami equation to fit the data: guidelines to avoid common problems. Polym. Test. 26, 222–231. doi:10.1016/j.polymertesting.2006.10.005
Luyt, A. S., and Brüll, R. (2004). Investigation of polyethylene-wax blends by CRYSTAF and SEC-FTIR. Polym. Bull. 52, 177–183. doi:10.1007/s00289-004-0274-0
Luyt, A. S., and Krupa, I. (2002). PE/wax blends: interesting observations. Macromol. Symp. 178, 109–116. doi:10.1002/1521-3900(200202)178:1<109::aid-masy109>3.0.co;2-u
Mandelkern, L. (2011). Crystallisation of polymers: volume 1, equilibrium concepts. New York: Cambridge University Press.
Martínez-Salazar, J., Alizadeh, A., Jiménez, J. J., and Plans, J. (1996). On the melting behaviour of polymer single crystals in a mixture with a compatible oligomer: 2. Polyethylene/paraffin. Polymer 37, 2367–2371. doi:10.1016/0032-3861(96)85347-x
Massalski, T. B., and Laughlin, D. E. (2017). “Phase diagrams,” in Reference module in materials science and materials engineering. Elsevier.
Mcguire, K. S., Laxminarayan, A., and Lloyd, D. R. (1994). A simple method of extrapolating the coexistence curve and predicting the melting point depression curve from cloud point data for polymer-diluent systems. Polymer 35, 4404–4407. doi:10.1016/0032-3861(94)90099-x
Mcpherson, A., Malkin, A. J., and Kuznetsov, Y. G. (2000). Atomic force microscopy in the study of macromolecular crystal growth. Annu. Rev. Biophysics Biomol. Struct. 29, 361–410. doi:10.1146/annurev.biophys.29.1.361
Mhlabeni, T., Ngobese, C., Ramjee, S., and Focke, W. (2023). Rheological characterization of linear low-density polyethylene–Fischer–Tropsch wax blends. J. Vinyl Addit. Technol. 29, 698–708. doi:10.1002/vnl.21984
Mishra, J., Tiwari, S. K., Abolhasani, M. M., Azimi, S., and Nayak, G. C. (2017). “2 - fundamental of polymer blends and its thermodynamics,” in Micro and nano fibrillar composites (MFCs and NFCs) from polymer blends. Editors R. K. MISHRA, S. THOMAS, and N. KALARIKKAL (India: Woodhead Publishing).
Mngomezulu, M. E., Luyt, A. S., and Krupa, I. (2010). Structure and properties of phase change materials based on HDPE, soft Fischer-Tropsch paraffin wax, and wood flour. J. Appl. Polym. Sci. 118, 1541–1551. doi:10.1002/app.32521
Mngomezulu, M. E., Luyt, A. S., and Krupa, I. (2011). Structure and properties of phase-change materials based on high-density polyethylene, hard Fischer–Tropsch paraffin wax, and wood flour. Polym. Compos. 32, 1155–1163. doi:10.1002/pc.21134
Mohammadi, M., Yousefi, A., and Ehsani, M. (2012). Thermorheological analysis of blend of high- and low-density polyethylenes. J. Polym. Res. 19, 9798. doi:10.1007/s10965-011-9798-9
Molefi, J. A., Luyt, A. S., and Krupa, I. (2010). Comparison of LDPE, LLDPE and HDPE as matrices for phase change materials based on a soft Fischer-Tropsch paraffin wax. Thermochim. Acta 500, 88–92. doi:10.1016/j.tca.2010.01.002
Motooka, M., Mantoku, H., and Takao, O. (1986) Process for producing stretched articles of ultrahigh-molecular-weight polyethylene, 06/755, 590.
Mpanza, H. S., and Luyt, A. S. (2006). Comparison of different waxes as processing agents for low-density polyethylene. Polym. Test. 25, 436–442. doi:10.1016/j.polymertesting.2006.01.008
Mpanza, H. S., and Luyt, A. S. (2006a). Influence of different waxes on the physical properties of linear low-density polyethylene. South Afr. J. Chem. 59, 48–54.
Mtshali, T. N., Krupa, I., and Luyt, A. S. (2001). The effect of cross-linking on the thermal properties of LDPE/wax blends. Thermochim. Acta 380, 47–54. doi:10.1016/s0040-6031(01)00636-0
Mtshali, T., Sittert, C. V., Djokovj, V., and Luyt, A. S. (2003). Binary mixtures of polyethylene and oxidized wax: dependency of thermal and mechanical properties upon mixing procedure. J. Appl. Polym. Sci. 89, 2446–2456. doi:10.1002/app.12466
Müller, A. J., Balsamo, V., Silva, F. D., Rosales, C. M., and Sáez, A. E. (1994). Shear and elongational behavior of linear low-density and low-density polyethylene blends from capillary rheometry. Polym. Eng. and Sci. 34, 1455–1463. doi:10.1002/pen.760341904
Musil, J., and Zatloukal, M. (2014). Historical review of die drool phenomenon in plastics extrusion. Polym. Rev. 54, 139–184. doi:10.1080/15583724.2013.860987
Ogah, O. (2012). The effects of linear low-density polyethylene (LLDPE) on the mechanical properties of high-density polyethylene (HDPE) film blends. Int. J. Eng. Manag. Sci. 3, 85–90.
Padding, J., and Briels, W. (2002). Time and length scales of polymer melts studied by coarse-grained molecular dynamics simulations. J. Chem. Phys. 117, 925–943. doi:10.1063/1.1481859
Patterson, D., and Robard, A. (1978). Thermodynamics of polymer compatibility. Macromolecules 11, 690–695. doi:10.1021/ma60064a015
Ragaert, K., Delva, L., Van Damme, N., Kuzmanovic, M., Hubo, S., and Cardon, L. (2016). Microstructural foundations of the strength and resilience of LLDPE artificial turf yarn. J. Appl. Polym. Sci. 133, 44080. doi:10.1002/app.44080
Robeson, L. (2014). Historical perspective of advances in the science and technology of polymer blends. Polymers 6, 1251–1265. doi:10.3390/polym6051251
Sadiku, E. (2009). “4 - automotive components composed of polyolefins,” in Polyolefin fibres. Editor S. C. O. UGBOLUE (Cambridge, England: Woodhead Publishing).
Salyer, I. O. (1996). Thermoplastic, moldable, non-exuding phase change materials. United states: University of Dayton: European patents.
Shilpa Kasargod Nagaraj, S. S.Nithin kundachira subramani and hatna and siddaramaiah (2016). Revisiting powder X-ray diffraction technique: a powerful tool to characterise polymers and their composite films. J. Material Sci. 4, 1–5. doi:10.4172/2321-6212.1000158
Sinha Ray, S., and Banerjee, R. (2022). “5 - fundamentals of polymer blend technology,” in Sustainable polylactide-based blends. Editors S. SINHA RAY, and R. BANERJEE (Elsevier).
Sotomayor, M. E., Krupa, I., Várez, A., and Levenfeld, B. (2014). Thermal and mechanical characterization of injection moulded high density polyethylene/paraffin wax blends as phase change materials. Renew. Energy 68, 140–145. doi:10.1016/j.renene.2014.01.036
Tanniru, M., and Misra, R. D. K. (2005). On enhanced impact strength of calcium carbonate-reinforced high-density polyethylene composites. Mater. Sci. Eng. A 405, 178–193. doi:10.1016/j.msea.2005.05.083
Thirtha, V., Lehman, R., and Nosker, T. (2005). Glass transition phenomena in melt-processed polystyrene/polypropylene blends. Polym. Eng. and Sci. 45, 1187–1193. doi:10.1002/pen.20387
Utracki, L. A., and Schlund, B. (1987). Linear low density polyethylenes and their blends: Part 2. Shear flow of LLDPE's. Polym. Eng. and Sci. 27, 367–379. doi:10.1002/pen.760270510
Vadalia, H. C., Lee, H. K., Myerson, A. S., and Levon, K. (1994). Thermally induced phase separation in ternary crystallizable polymer solutions. J. Membr. Sci. 89, 37–50. doi:10.1016/0376-7388(93)e0207-z
Zalba, B., Marı́N, J. M., Cabeza, L. F., and Mehling, H. (2003). Review on thermal energy storage with phase change: materials, heat transfer analysis and applications. Appl. Therm. Eng. 23, 251–283. doi:10.1016/s1359-4311(02)00192-8
Keywords: polyethylene, wax, blends, miscibility, rheology
Citation: Mhlabeni TL, Jamiru T and Mhike W (2024) Thermophysical, and rheological insights of polyethylene/wax blends. Front. Chem. Eng. 6:1507921. doi: 10.3389/fceng.2024.1507921
Received: 08 October 2024; Accepted: 25 November 2024;
Published: 13 December 2024.
Edited by:
Yongrong Yang, Zhejiang University, ChinaCopyright © 2024 Mhlabeni, Jamiru and Mhike. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thobile L. Mhlabeni, TWhsYWJlbmlUTEB0dXQuYWMuemE=