 Robert J. Nicholas
Robert J. Nicholas Michael A. McGuire
Michael A. McGuire Seok-Hee Hyun
Seok-Hee Hyun Madeline N. Cullison
Madeline N. Cullison David H. Thompson
David H. Thompson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. Eng., 27 June 2022
Sec. Microfluidic Engineering and Process Intensification
Volume 4 - 2022 | https://doi.org/10.3389/fceng.2022.877498
This article is part of the Research TopicMicrofluidic-Based Technologies for Sustainable Process IntensificationView all 6 articles
In an effort to strengthen the resiliency of supply chains for active pharmaceutical ingredients (API), continuous manufacturing processes may be optimized with respect to improved chemoselectivity, production rate, yield, and/or process intensity. We report an efficient two-step continuous flow synthesis of diazepam, an agent on the World Health Organization’s (WHO) list of essential medicines. Different conditions were rapidly screened in microfluidic chip reactors by varying residence times, temperatures, solvents, and ammonia sources to identify the best telescoped reaction conditions. We report a telescoped flow synthesis that uses two microreactors in series set to 0°C and 60°C, respectively, to produce a 96% yield of 91% pure diazepam within 15 min using an NH4Br/NH4OH solution in the second step. Diazepam of >98% purity was obtained after a single recrystallization.
The stability and reliability of drug supply chains are crucial for maintaining quality patient care. Unfortunately, there are multiple factors that contribute to supply chain fragility and rapid changes in market demand, leaving manufacturers unable to adequately respond with their current infrastructure and production processes (Srai et al., 2015). This liability is further compounded by the just-in-time inventory systems that are practiced by multiple pharmaceutical companies in order to keep waste and inventory costs low (Shukar et al., 2021). By encouraging the pharmaceutical industry to pursue continuous manufacturing (CM), the U.S. Food and Drug Administration (FDA) has focused attention on the supply chain issue to enable greater responsiveness, robustness, and higher product quality compared to batch processes (Burcham et al., 2018). In a striking recent example of the responsiveness possible, CM enabled the controlled encapsulation of mRNA in lipid nanoparticles at unprecedented speeds to successfully distribute millions of COVID-19 vaccines globally at the height of the pandemic (Ali et al., 2021).
Continuous flow synthesis of various active pharmaceutical ingredients (APIs) by academic and industrial groups has been well documented (Hopkin et al., 2010; Cole et al., 2017; Porta et al., 2016; Ziegler et al., 2018; Jaman et al., 2019; Chen et al., 2020). Other enabling technologies for end-to-end CM (Domokos et al., 2021) and design space mapping for scale-up (Diab and Gerogiorgis, 2020) are also growing rapidly. One API of interest is diazepam, a fast-acting anxiolytic, that is considered an essential medicine by the World Health Organization (World Health Organization, 2021) and is among the top 200 drugs prescribed in the U.S. (Fuentes et al., 2018). Unfortunately, diazepam is a drug that is very susceptible to shortages and may contribute to price and/or demand increase of other benzodiazepines when in short supply (Shtull-Leber et al., 2017). Developing a robust, scalable CM process for diazepam is timely given the upward trends in prescription rates (Bachhuber et al., 2016; Agarwal and Landon, 2019), making it a worthy target for process optimization.
Various publications document the continuous synthesis of diazepam in two steps: N-acylation and sequential substitution/cyclization reactions, starting from 5-chloro-2-(methylamino)benzophenone (1) (Figures 1A,B). Our group has reported high throughput experimentation to identify conditions for the synthesis of diazepam (Ewan et al., 2017). Using a toluene and methanol solvent system, we were able to achieve 70% pure diazepam using a microfluidic reactor system. Adamo et al. engineered a process as part of their reconfigurable manufacturing platform where they dissolved their reagents in NMP to produce 78% of crude target API (94% yield) according to HPLC analysis (Adamo et al., 2016). This same process was implemented in a subsequent publication by Rogers et al. describing the production of 2800 dose equivalents that met USP standards (Rogers et al., 2020). Collins et al. developed an automated platform called AutoSyn to synthesize diazepam in 96% yield and 81% purity (the other 19% being reaction intermediate 3b) (Collins et al., 2020). Bédard et al. focused on eliminating waste in their flow process to improve the E-factor (Bédard et al., 2017). While they were only able to obtain a 55% yield, they reduced the E-factor of the method first reported by Adamo et al. from 36 to 9 primarily by using 2-MeTHF, a solvent immiscible with water, instead of NMP to enable in-line aqueous extractions without needing extra solvent or solvent exchanges.
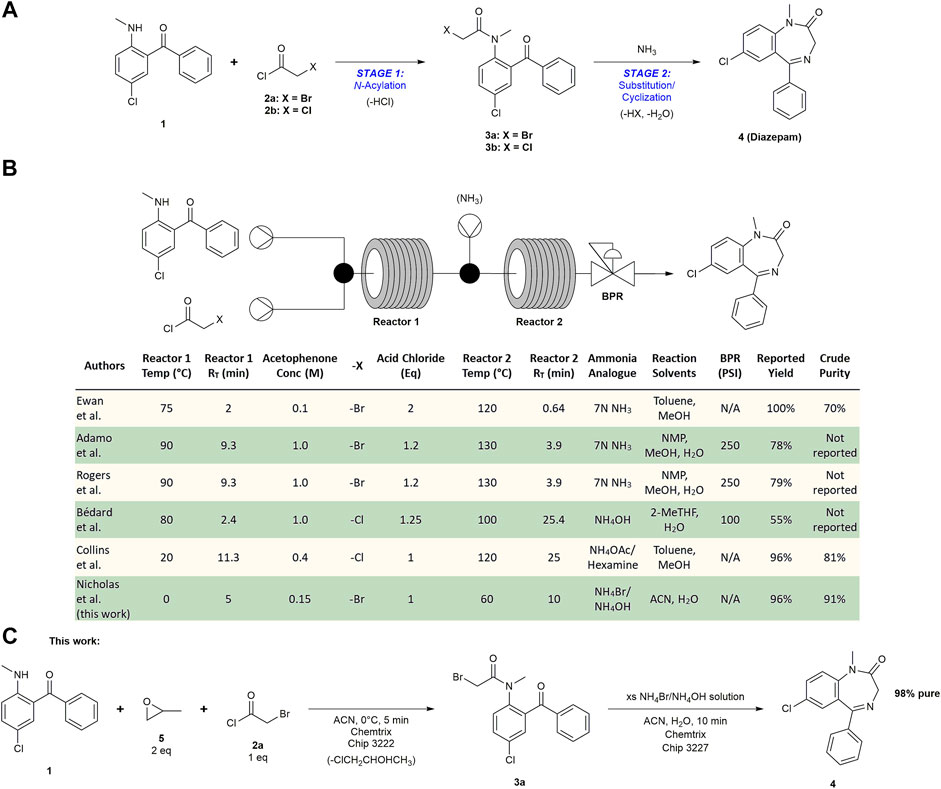
FIGURE 1. Summary of the transformation of 5-chloro-2-(methylamino)benzophenone to diazepam. (A) Scheme of the two-step synthesis of diazepam 4 from benzophenone 1. N-Acylation with an acid chloride occurs in Stage 1. Stage 2 includes a nucleophilic substitution with ammonia followed by an intramolecular cyclization. (B) Summary of relevant literature where authors performed a two-step diazepam synthesis in continuous flow. (C) Scheme of our optimized synthesis of diazepam in flow where we obtained highly pure API directly from the process stream (98% pure after a single recrystallization).
Given the wide range of yields and purities, we anticipated that there was potential to improve upon the published flow syntheses for diazepam. Ewan et al. and Collins et al. both report high yields; however, their impurity profiles indicate that intermediate 3a/b did not fully react and was present in their final products. Rogers et al. noted that the downstream liquid-liquid extraction work-up led to yield deterioration since the product adsorbed to the membranes of the in-line phase separators. We sought to maximize starting material and intermediate conversion to produce high quality API by increasing recovery from a simplified work-up. For this reason, we wanted to avoid high boiling protic solvents since many of them are miscible with water and require high energy inputs for their removal by distillation. While it was not a top priority, we also considered methods for improving the safety of the flow process. Amination reactions in flow are commonly performed with pressurized NH3 gas or 7N NH3 in MeOH at high temperatures. Ammonia vapors could escape from a high pressure reactor in the event of a valve or line failure (Feng et al., 1999), thus posing a significant manufacturing safety risk upon process upscaling.
With these considerations in mind, we sought to improve upon the reported methods by optimizing parameters such as retention time, temperature, solvent, and ammonia source using Chemtrix microfluidic chip reactors. We also evaluated methods for increasing reaction rates and improving chemoselectivity. The inclusion of propylene oxide as an HCl scavenger in Stage 1 improved the conversion of 1 into 3a instead of the less reactive side product 3b. We also discovered that saturated solutions of NH4Br dissolved in 30% NH4OH were able to convert 3a into 4 using moderate temperatures without a back pressure regulator (BPR) (Figure 1C). Use of the NH4Br/NH4OH blend produces NH3 ‘on demand’ via the common ion effect, thus obviating the need for a BPR that could serve as a process failure point. As a result of these efforts, we were able to develop a more efficient and simplified telescoped process that fully converts 1 into 91% pure diazepam in ∼96% yield before final recrystallization.
5-Chloro-2-(methylamino)benzophenone, propylene oxide, bromoacetyl chloride, 7N NH3/MeOH solution, triethylamine (TEA), N,N-diisopropylethylamine (DIPEA), 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU), 2,6-lutidine, Na2CO3, 28–30% NH4OH solution, NH4OAc, and NH4Br were purchased from MilliporeSigma and were used without further purification. Diazepam HPLC standard was also obtained from MilliporeSigma and used as supplied. Toluene, ethyl acetate (EtOAc), N-methyl pyrrolidone (NMP), and ethanol (EtOH) and HPLC grade acetonitrile (ACN) were supplied by Fisher Scientific and used as received.
HPLC chromatograms were acquired using an Agilent 1200 Series HPLC with a G1315B Diode Array Detector. Samples were analyzed on a ZORBAX Eclipse XDB-18 column (5 µm particle size, 4.6 mm × 150 mm) at 20°C. The mobile phase consisted of a 40:40:20 ACN:H2O:MeOH solvent system flowing at 0.5 mL/min. UPLC chromatograms were obtained using a Waters Acuity system fitted with a CORTECS C18+ column (1.6 µm particles, 2.1 × 100 mm) and an Acquity QDa Mass Detector. A 2.1 mm × 5 mm CORTECS VanGuard Pre-Column was installed preceding the analytical column. 1H and 13C NMR spectra were obtained using a Bruker Avance III-HD (500 MHz) instrument. Chemical shifts are reported using the residual solvent peak as reference. All microfluidics experiments were carried out using a Chemtrix Labtrix S1 system. Telescoped reactions used a Chemtrix Labtrix Start system as a second temperature-controlled stage. Each system was equipped with 3222 (5 μL), 3223 (10 μL), 3225 (10 μL), or 3227 (19.5 μL) reactor chips that had channel dimensions of 300 μm wide x 120 μm high.
Batch reactions were screened by adding 5-chloro-2-(methylamino)benzophenone (50 mg, 0.2035 mmol) to 4 mL vials with a stir bar and toluene or ACN (1.5 ml) as solvent. Solutions were allowed to stir in an ice bath (4°C), room temperature (23°C), or a heated oil bath (75°C). Once the solution temperature had equilibrated, 1 equivalent of base (TEA, DIPEA, DBU, or 2,6-lutidine) and 0, 1, 2, 4, or 8 equivalents of propylene oxide were added to solution. The reaction was initiated upon bromoacetyl chloride (32.1 mg, 0.2035 mmol) addition and allowed to stir for 15 min. Reactions were analyzed by TLC (2:1 petroleum ether:ethyl acetate) and HPLC.
Solutions of 5-chloro-2-(methylamino)benzophenone (0.6 M), propylene oxide (1.2 M), and bromoacetyl chloride (0.6 M) were dissolved in toluene and loaded into three 1 mL Hamilton gastight syringes. A fourth syringe containing neat ACN was used for quenching. The filled syringes were mounted on the Labtrix S1 system and connected to the first three and second to last inlets of the chip holder by FEP tubing (0.8 mm × 0.25 mm).
Reactor chip 3225 (10 μL) was used for Stage 1 evaluation. Each condition was allowed to equilibrate for three times the retention time before collection. Samples of 20 μL were collected in vials filled with 890 μL ACN and 100 μL of 0.25 M Na2CO3. Prior to UPLC analysis, samples were filtered through a 0.20 μm PTFE filter.
To optimize reagent stoichiometries, flow rates for the three reagents were initially set to 0.667 μL/min for 1.0 equivalents of bromoacetyl chloride. ACN flowed at a constant 1.00 μL/min throughout the run. As the bromoacetyl chloride equivalents increased by 0.02 increments, the flow rates for the benzophenone and propylene oxide streams decreased by 0.004 μL/min. The rate for bromoacetyl chloride increased by 0.008 μL/min with each condition. This trend was carried throughout the screen until the flowrate for the benzophenone and propylene oxide was 0.625 μL/min and the bromoacetyl chloride was 0.75 μL/min for 1.2 equivalents of acid chloride. Residence time and temperature were held constant at 5 min and 0°C, respectively.
Residence times were then tested between 1 and 10 min at 1 min increments. The flow rates for the starting reagents were each set to 3.33 μL/min for RT = 1 min and decreased equally to give the appropriate residence time in the reactor until 10 min. An ACN quench was connected to the second to last port to dilute the reactant mixture by 50% v/v. Temperature was kept at 0 °C unless stated otherwise.
Isolated intermediate 3a was then used in batch to scout preferred ammonia sources. Vials were charged with 3a (367 mg, 1.0 mmol) dissolved in 5 ml of solvent (toluene, ACN, or NMP) and different ammonia analogues: 7N NH3 in MeOH (2 ml, 14 mmol), 28–30% NH4OH (2 ml, ∼29.6 mmol), 6 M NH4OAc in water or MeOH (4 ml, 25.9 mmol), or a saturated solution of NH4Br/NH4OH (2 ml, 3.27 mmol) added to the vial. Reactions were performed at temperatures between room temperature and 100°C and allowed to react at various times between 15 min and overnight. Reactions were characterized by TLC or UPLC. Results are summarized in the Supplementary Material.
Intermediate 3a was dissolved in toluene to give a 0.20 M solution that was loaded into a 1 mL syringe. Ammonia solutions were loaded into a second syringe. Syringes were mounted on the S1 system and fitted with FEP tubing leading to the first two inlets of the chip holder. Each sample obtained was separated by equilibration period set for three times the retention time of the condition about to be sampled.
In trials testing 7N NH3 in MeOH, residence times (5–25 min) and temperatures (100, 120°C) were evaluated using reactor chip 3227 (19.5 μL). Flow rates for 3a, 7N NH3, and ACN quench were each set at 1.95 μL/min for 5 min down to 0.390 μL/min for 25 min. The system was fitted with a BPR set to 100 PSI. Solutions were collected in vials pre-filled with 225 μL ACN until 50 μL of product solution was collected and analyzed by UPLC.
The NH4Br/NH4OH solution was prepared by dissolving 5.1g NH4Br in 1.36 mL NH4OH solution and diluting to 12.5 mL with water. This solution and 3a in toluene were flowed at rates between 0.5 μL/min to 5 μL/min to achieve residence times of 1, 3, 5, or 10 min in reactor chip 3223 (10 μL) held at either 40 or 60°C. The process flowed until 50 μL of reaction mixture were dispensed into vials containing 500 μL ACN. Samples were immediately analyzed by UPLC and stored at −80°C.
For experiments using NH4OAc as an ammonia source, Stage 1 was fed solutions of 5-chloro-2-(methylamino)benzophenone (0.6 M), propylene oxide (1.2 M), and bromoacetyl chloride (0.6 M) in toluene at a rate of 0.667 μL/min (RT = 5 min). Reactor chip 3225 (10 μL, Stage 1) was cooled to 0 °C and the contents from this stage flowed into reactor chip 3227 (19.5 μL). A solution of 6 M NH4OAc dissolved in 100:0, 98:2, 95:5, or 90:10 MeOH:H2O (v/v) was added at the second inlet of Stage 2 at a rate of 1.9 μL/min. The reactor was heated to 120–160 °C using the Labtrix Start system. A BPR set to 100 PSI was added to the end of Stage 2. Fractions of 100 μL were collected for each solvent and temperature condition. A 10 μL aliquot of product was diluted in 100 μL ACN for UPLC analysis.
Our optimized telescoped process used ACN solutions of 5-chloro-2-(methylamino)benzophenone (0.15 M), propylene oxide (0.30 M), and bromoacetyl chloride (0.15 M) for Stage 1 and the NH4Br/NH4OH solution for Stage 2. Benzophenone and propylene oxide solutions mixed in an external T-mixer were fed into the first inlet of 3222 (5 μL) reactor chip where it mixed with the bromoacetyl chloride solution introduced via the second inlet. Flow rates for the three reagents in Stage 1 were 0.333 μL/min giving a residence time of 5 min. The output of this reactor was fed into a 3227 (19.5 μL) reactor chip held at 60°C. A NH4Br/NH4OH solution was flowed into the chip at 0.95 μL/min to produce a residence time of 10 min for Stage 2 (no BPR was installed). Fractions were collected for 1 h and analyzed by UPLC after diluting a 100 μL aliquot up to 1000 μL ACN. Diazepam concentrations in the final solutions were calculated based on a UPLC standard curve.
The optimized microfluidics system was converted into a larger scale setup using FEP tubing with an inner diameter of 1.016 mm. Solutions of 5-chloro-2-(methylamino)benzophenone (0.15 M), propylene oxide (0.30 M), and bromoacetyl chloride (0.15 M) were loaded into 6 mL syringes and positioned onto syringe pumps. The benzophenone and propylene oxide feeds were each pumped at a rate of 10 μL/min and mixed through a T-mixer. The mixed solution flowed into a second T-mixer where it would mix with the bromoacetyl chloride solution also being pumped at 10 μL/min. The reactor tubing for stage one (RT = 5 min, length = 18.5 cm) was submerged in an ice bath. The output of Stage 1 was fed into a final T-mixer immersed in a 60°C water bath where the NH4Br/NH4OH solution was introduced at 30 μL/min via a 10 ml syringe pump. The coiled tubing of Stage 2 (RT = 5 min, length = 18.5 cm) sat in the heated water bath and deposited the final solution into a 20 mL scintillation vial. Fractions were collected in 1 h increments. Solutions were worked up by EtOAc extraction and the solvent was removed in vacuo.
Our initial efforts were focused on developing a deeper understanding of both reactions. In the N-acylation step (Figure 2A), 1 will attack 2a to give 3a in a nucleophilic acyl substitution by the secondary amine of 1. An ammonium intermediate is formed with subsequent deprotonation of the intermediate by chloride to generate 3a and HCl as products. It is important to note that deprotonation by unreacted 1 competes with chloride in this process and acts as a buffer for the HCl byproduct in the reaction. In either case (Figure 2B), the nucleophilicity of 1 is decreased, thus impeding reaction progress. A common strategy to offset this limitation is to increase the equivalence of the amine; however, given our goals of maximizing conversion in flow, this was not an option as it would increase E-factor by requiring more starting material and a downstream purification that could negatively impact product yield.
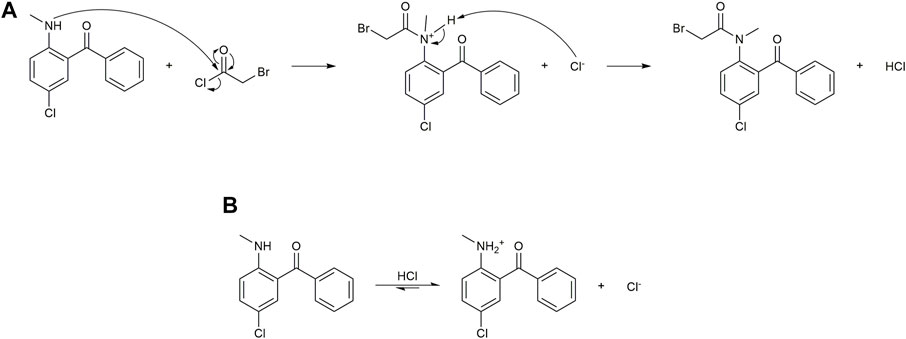
FIGURE 2. Microscopic steps associated with the N-acylation of Stage 1. (A) Mechanism of the intended reaction with 1 and 2a, generating HCl along with the desired intermediate 3a. (B) Unreacted 1 can be protonated by generated HCl, a strong electrolyte, or other protonated ammonium intermediate, thereby reducing the rate of the intended reaction.
We initially thought that adding a bulky base to the reaction could quench the generated HCl and facilitate the acylation. After evaluating a variety of bases (triethylamine, DIPEA, DBU, and 2,6-lutidine) in 15 min batch reactions at different temperatures, we found that they inhibited the formation of 3a and promoted the formation of 3b (Table 1). A plausible explanation for this is that under basic conditions, chloride nucleophilicity increases and substitutes at the α-bromide on 3a. When looking at temperature as a variable, we generally observed the formation of the chloride intermediate 3b at elevated temperatures in both the presence and absence of base. It is possible to drive 3b to diazepam as Bédard et al. and Collins et al. have shown; however, we have a preference for the bromide analogue 3a since the subsequent cyclization reaction is much more facile with this species. In fact, under a wide variety of conditions tested that fully consumed 3a, the 3b concentrations remained virtually unchanged in the reaction mixture. Since base addition did not improve conversion into the desired 3a intermediate, we then explored the effects of temperature and additives on reaction efficiency.
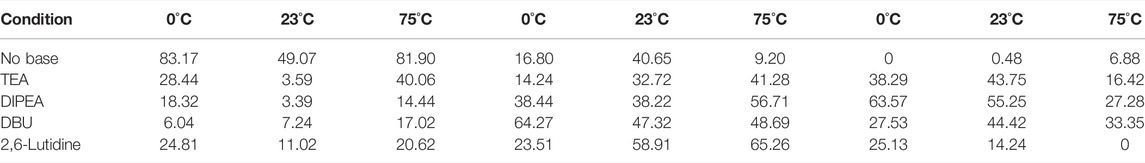
TABLE 1. Peak areas of key compounds as determined by HPLC. The control conditions where there was no base had the best conversion into 3a as opposed to any of the reactions with base present. Lower temperatures generally seem to improve conversion rates into 3a.
While investigating alternative methods to improve the N-acylation reaction, we began to explore the use of propylene oxide as a neutral HCl scavenger. Balaji and Dalal (2018) have reported rapid conversion of primary and secondary amines into amide derivatives using propylene oxide as an additive. Propylene oxide has been used specifically for the purpose of quenching HCl in reactions to form 1- and 2-chloropropanols (Phillips and Soulen, 1995); the chlorohydrin byproducts were benign spectators in those reactions as well (Dhaon, 1997). Although there were concerns about the use of this GHS08 agent in our process, analytical data showed that the vicinal aminopropanol byproducts were removed during final product recrystallization.
In Stage 1 batch reactions with different equivalents of propylene oxide, we found that the conversion of 1 to 3a was increased significantly. As shown in Figure 3, addition of one equivalent of propylene oxide led to a 3a yield improvement from 83% to 91% and a more complete conversion of 1 (8.6% vs. 1.2% unreacted 1 for the two trials). Notably, the amount of 3b side product also decreases slightly as the propylene oxide equivalents increased. Since the increased reaction rate with propylene oxide offered the opportunity to reduce the residence time of Stage 1 in flow while promoting better 3a vs. 3b selectivity, all subsequent experiments used two equivalents of propylene oxide as the best compromise between reaction yield vs. reagent consumed.
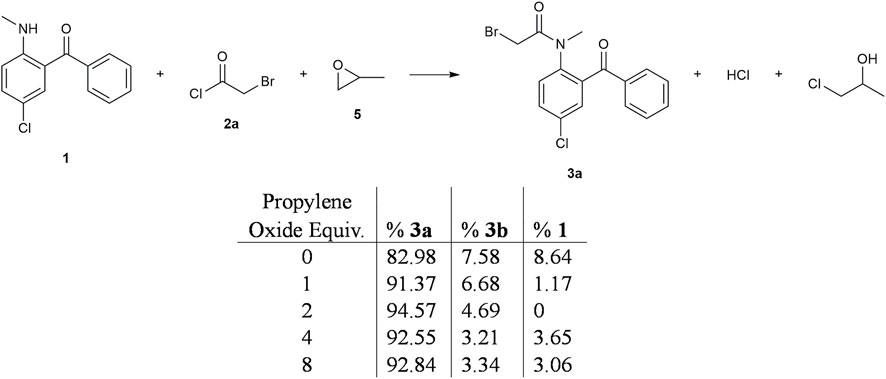
FIGURE 3. Summary of propylene oxide screen in batch. Propylene oxide (5) was added to the above reaction at different equivalencies. It was found that propylene oxide plays a role in converting starting material into the resulting amide more efficiently.
With the above conditions above in hand, we were able to translate the batch N-acylation reaction into flow using a Chemtrix Labtrix S1 system to operate at a milligram scale for flow rate, equivalence, and temperature optimization efforts. Solutions of 1, 5, and 2a were dissolved in toluene to give solutions of 0.6, 1.2, and 0.6 M, respectively, loaded into 1 mL syringes, and mounted on the S1 system. The flow rate of each reagent was adjusted to rapidly test different concentrations of 2a at increments of 0.02 equivalents between 1.0–1.2 equivalents. We initially conducted this experiment with a 10 min residence time; however, we observed that the N-acylation reaction reached completion for all trials tested between 1.0–1.1 equivalents (Supplementary Material), thus warranting a shorter retention time.
Next, we ran three trials of this optimization experiment with a RT = 5 min, where each trial was conducted on different days by different operators (Figure 4). We observed high conversions into intermediate 3a, with a majority of the starting material being consumed even using 1.0 equivalents of 1. Since the peak areas were unchanged as the amount of 2a increases, we concluded that the N-acylation reaction is reaching completion within 5 min under these conditions.
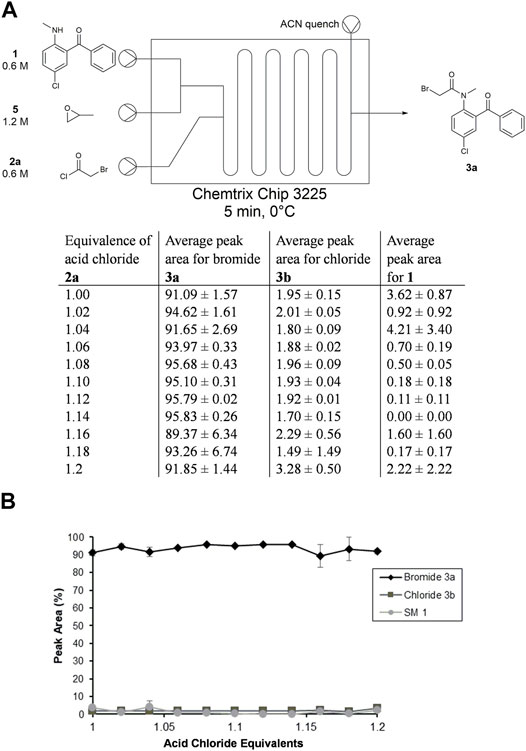
FIGURE 4. Summary of acid chloride stoichiometry optimization in toluene in Stage 1. (A) Setup for reaction screen in microfluidic reactor with tabulated results. (B) Plot of results with standard error from three trials.
We then sought to determine the optimal residence time for Stage 1 at this stoichiometry (Figure 5). Using the same flow setup as before, we were able to test 1–10 min residence times at 1-min intervals. We found that the N-acylation is fast at 0°C, such that the majority of starting material 1 is consumed within 1 min. There is a gradual increase in the amount of bromide intermediate 3a up to the ∼5-min mark; after this point the amount of product plateaus as time progresses. The amount of 1 present also reaches a minimum at 5 min; however, 3a slightly decreases at residence times ≥7 min due to formation of 3b or a reverse reaction that converts the intermediate back into starting material 1. Based on an average of three trials, the chloride intermediate 3b stays relatively constant at 2–3%. Given that the maximum conversion from 1 to 3a occurs at 5 min, we moved forward with a 1:1:2 ratio of 1:2a:5 and a RT = 5 min at 0°C as our optimized conditions for Stage 1.
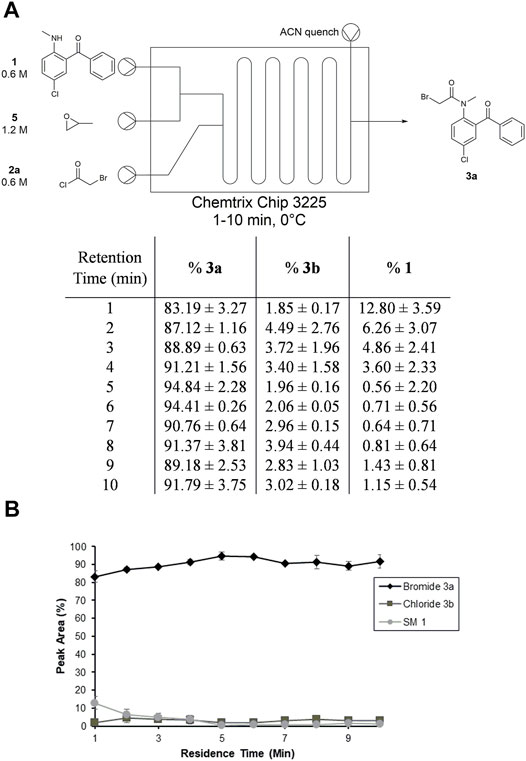
FIGURE 5. Summary of residence time optimization in toluene in Stage 1. (A) Setup for reaction screen in microfluidic reactor with tabulated results. (B) Plot of results with standard error from three trials.
The mechanism of the Stage 2 process (Figure 6) begins with a nucleophilic displacement of bromide from 3a by NH3 in an SN2 reaction that generates HBr as a byproduct. The α-amino intermediate then condenses with the ketone to form an imine to give diazepam. Since the HBr and H2O byproducts can promote hydrolysis of the amide bond of 3a to regenerate the starting material, it is essential for NH3 to displace the bromide and quickly cyclize to form diazepam. This transformation appears to be the rate-limiting step in the diazepam synthesis given the high temperatures reported in the previously cited literature.
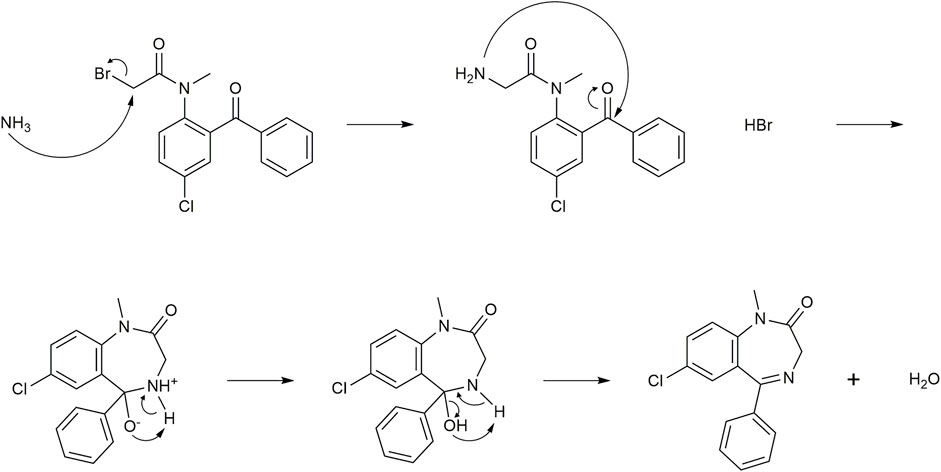
FIGURE 6. Mechanism associated with the substitution and subsequent cyclization in Stage 2. Ammonia displaces bromide before engaging in an intramolecular attack of the ketone to form a 7-membered ring. Loss of water from the intermediate oxonium ion leads to imine formation of diazepam along with HBr and H2O.
This analysis led us to identify the most favorable NH3 source given the variety of analogues described in the literature. Our first candidate was ammonia dissolved in methanol as used by Ewan et al., Adamo et al., and Rogers et al. We conducted Stage 2 batch reactions with isolated intermediate 3a and 7N NH3 in MeOH. Due to pressure built-up from the ammonia and low boiling MeOH, we conducted this reaction at 65°C. Although diazepam formation was observed, the presence of 1 was also detected by HPLC, indicating that these conditions can induce hydrolysis of the amide bond.
We then evaluated reactions in flow where we could reproduce the conditions as described by Ewan et al. and Rogers et al. We first set out to evaluate Stage 2 at different residence times from 5 to 25 min at 100°C and 120°C using a 0.2 M solution of 3a in toluene (Figure 7). A BPR was installed to maintain an internal pressure of 100 PSI. These trials were successful in producing diazepam; however, the material collected was slightly yellow, a byproduct formed during Stage 2 that was identified by UPLC as the hydrolysis product 1. This impurity was produced at various quantities depending on the flow conditions employed. Given this drawback and the need for a highly pressurized system, we were motivated to investigate alternative ammonia sources.
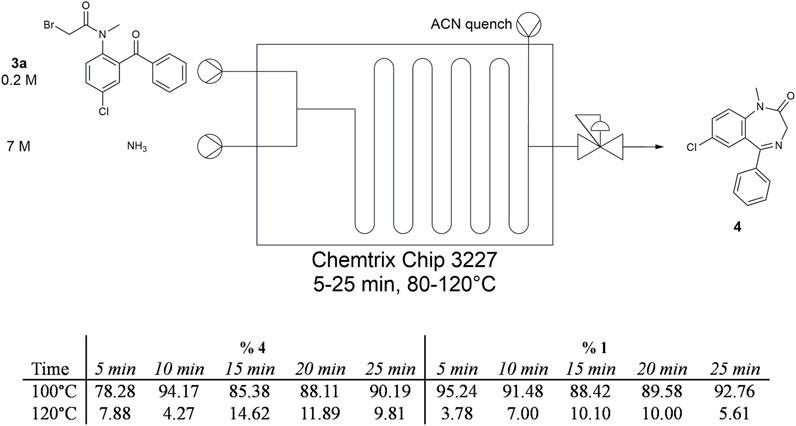
FIGURE 7. Summary of temperature and residence time trials for NH3 in Stage 2. Intermediate 3a was dissolved in toluene whereas NH3 was dissolved in MeOH. The BPR was set to 100 PSI.
Reactions conducted at 70°C with 3a and NH4OH dissolved in a toluene:ACN mixture also produced diazepam; however, a significant portion of 3a reverted back to benzophenone 1 based on UPLC analysis. While this had potential for being a viable NH3 source, especially with the work by Collins et al. supporting this reagent, we chose not to investigate this reagent further in flow due to the impact that the intermediate hydrolysis would have on the process E-factor.
We then pursued the use of an aqueous solution of NH4OAc in an effort to suppress the competition between hydrolysis and cyclization. By keeping the reaction closer to a neutral pH, we reasoned that the hydrolysis rate might be reduced enough to enable intermediate conversion into diazepam before the amide bond cleaves. We first carried out reactions with NH4OAc as the ammonia source in batch with a variety of solvents, including ACN, NMP, and toluene, in a one-pot reaction with benzophenone 1 as the starting reactant. When ACN was the solvent, we obtained a crude purity of 87% diazepam that could be readily crystallized to 95% pure API. Given the promise of the NH4OAc reagent, we moved on to flow trials.
For the flow experiments, we made a 6 M solution of NH4OAc in MeOH and mixed it with the Stage 1 product in Stage 2 while varying temperature between 120 and 160°C at 10°C intervals (Figure 8). With a fixed residence time of 5 min, we observed 50% or more diazepam in the crude product, although a significant amount of 1 and another prominent peak that eluted near the toluene peak was observed. UPLC-MS analysis revealed that the new peak had an m/z value of 346 without an accompanying M+2 peak, indicative of an acetate adduct arising from bromine displacement (Supplementary Material). Flash chromatographic isolation of this impurity and characterization by 1H and 13C NMR confirmed this structure (Supplementary Material). While acetate is not a particularly strong nucleophile, we suspected that the abundance of this anion in the reaction mixture could promote displacement at the halogenated α-carbon. In an effort to suppress the amount of the acetate adduct formed, we increased the water content in the 6 M solutions of NH4OAc using 2%, 5%, and 10% water in MeOH by volume; however, these efforts proved fruitless.
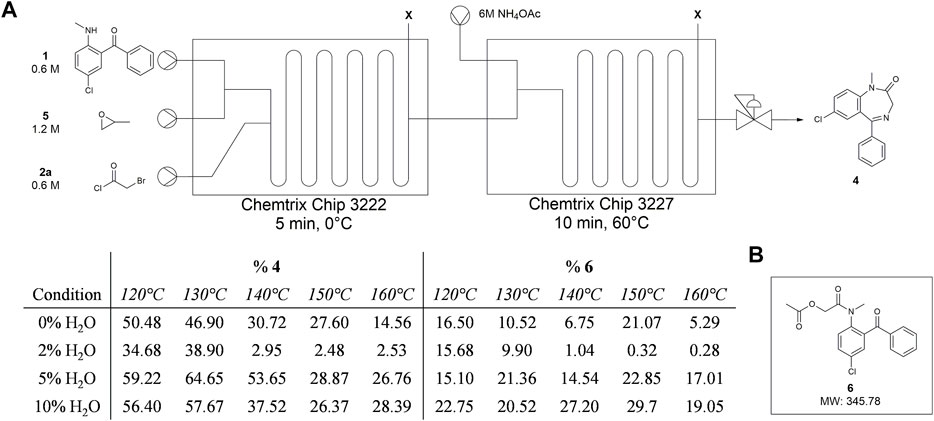
FIGURE 8. Summary of Stage 2 temperature and NH4OH solvent screen in telescoped synthesis of diazepam. (A) Setup for reaction screen in microfluidic reactor with tabulated results. Starting materials were dissolved in toluene. NH4OH was dissolved in MeOH with 0%, 2%, 5%, or 10% water to give 6M. BPR was set to 100 PSI. (B) Proposed impurity that arises from reaction conditions with NH4OAc.
The formation of the acetate adduct made us focus on the role the spectator ion plays in the reaction. Knowing that acetate has the ability to substitute the halogenated α-carbon, we decided to incorporate NH4Br in the case that if the common bromide ion were to act as a nucleophile, it would be a zero sum reaction. We dissolved the salt in an aqueous NH4OH solution to serve as our primary source of ammonia via a common ion effect to drive the formation of NH3 as shown in Figure 9. Since NH4Br dissociates into NH4+ and Br− ions and NH4OH is in equilibrium with NH4+ and OH−, NH4+ is a common ion, thus shifting the equilibrium of NH4OH towards NH3 and water as the NH3 is consumed in the displacement reaction.
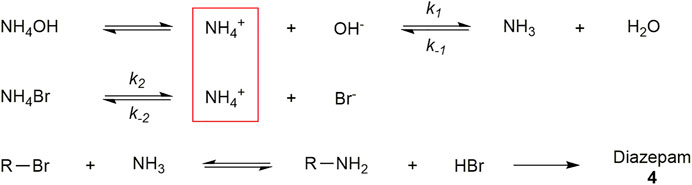
FIGURE 9. Relevant equilibria for the NH4Br/NH4OH ammonia source. Note that NH4Br and NH4OH will dissociate and share NH4+ as a common ion. Since NH4Br is a strong electrolyte, the higher concentration of NH4+ generated will drive equilibrium towards NH3. This higher equivalence of NH3 appears to drive the formation of 4 from the intermediate 3a. Constants: k-1 = 1.76*10–5. k2 = 25.5.
Preliminary batch trials with the NH4Br/NH4OH solution for cyclization demonstrated that we could achieve excellent conversion of 3a into diazepam within 2.5 h. UPLC of the crude revealed 73% and 90% diazepam at room temperature and 40°C, respectively. Performing a one-pot reaction from 1 to 4 generated 94% diazepam when using NH4Br/NH4OH in an overnight reaction. In all trials attempted, we observed little or no 1 byproduct and traces of 3b arising from Stage 1 in flow.
Subsequent flow experiments with NH4Br/NH4OH in Stage 2 employed 3a in ACN (Figure 10). We transitioned to this solvent since batch experiments revealed that toluene slows down the Stage 2 reaction, likely due to its immiscibility with water and its inability to dissolve NH4Br or NH4OH. During our residence time screening campaign, we realized that a BPR was no longer necessary for the reactor system, so it was removed. At 40°C, we were able to observe crude yield of diazepam as high as 61% after 10 min and the yield improved to 86% diazepam when the temperature was increased to 60°C.
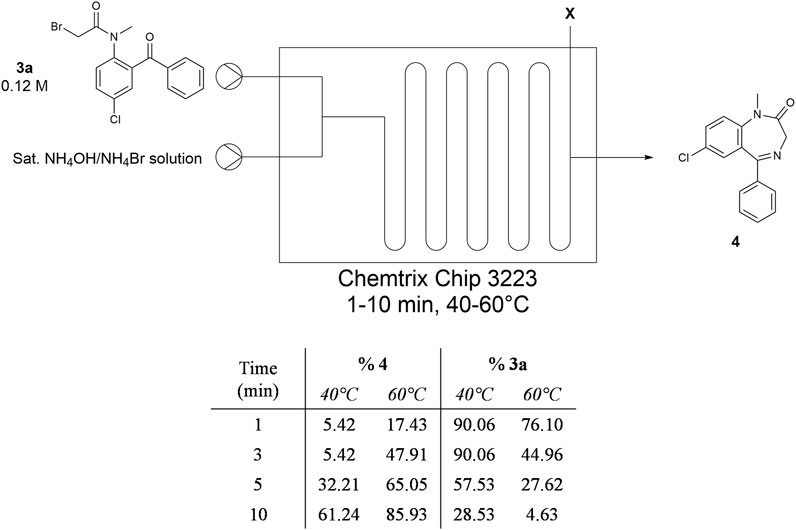
FIGURE 10. Summary of temperature and residence time trials using NH4OH/NH4Br solution in Stage 2. Intermediate 3a was dissolved in ACN, while NH4OH/NH4Br solution was dissolved in water.
With a promising ammonia source in hand, we moved forward with optimization of the telescoped sequence as shown in Figure 11A. The first reactor chip was set for a 5 min residence time at 0°C. As for Stage 2, we used a 19.5 μL 3227 reactor chip heated to 60°C with a residence time of 10 min. Early attempts of telescoping this reaction in flow saw varying degrees of success that were primarily dictated by the buildup of a dark brown precipitate in Stage 2 where the NH4Br/NH4OH solution mixes with the reactant solution in the narrow channels of the static mixer in the chip (Figure 11B). Both Ewan et al. and Bédard et al. reported a precipitate forming at a similar position. We were able to isolate enough of the precipitate to identify it as diazepam based on the similarity in UPLC retention times relative to diazepam standard. We infer from these findings that the diazepam is formed instantaneously in the mixing element and forms a salt precipitate with residual HCl that accumulates and fouls the reactor.
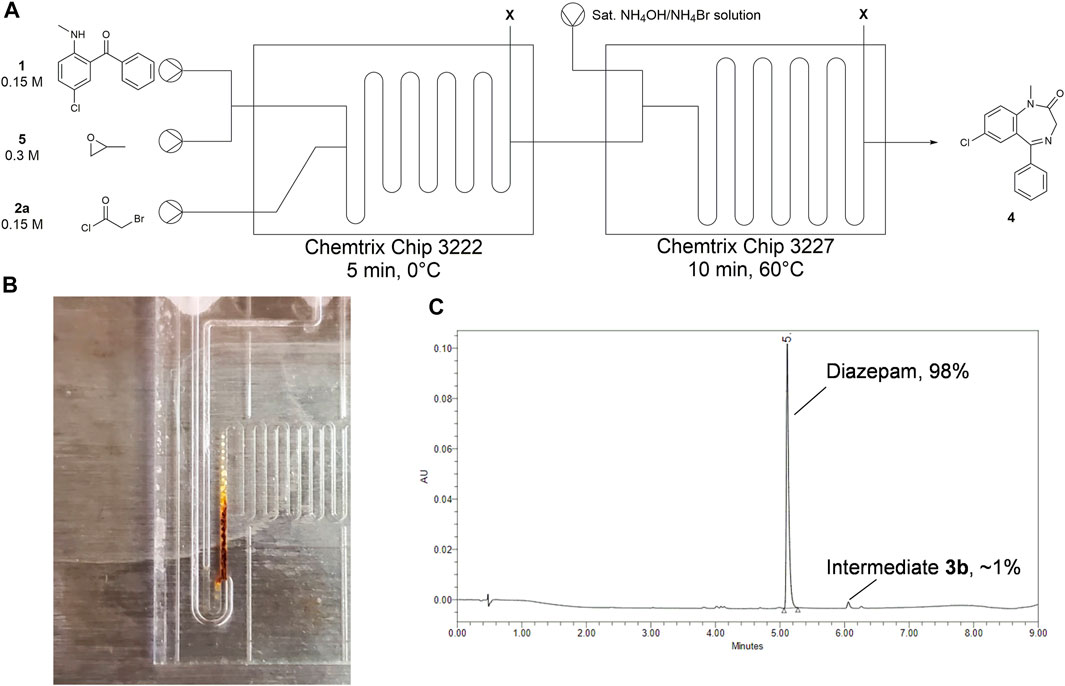
FIGURE 11. Summary of finalized telescoped flow process to synthesize diazepam in two-steps. (A) Setup for two-step reaction in microfluidic reactor with tabulated results. Starting materials were dissolved in ACN. The NH4OH/NH4Br solution was dissolved in water. (B) Precipitate thought to be diazepam crashes out at the point where the ammonia analogue enters the system. Sonication reduces the amount of precipitation that is formed here. (C) Chromatogram of a fraction collected over 60 min to give 30% diazepam with a purity of 98%. Retention times: 5.1 min = diazepam, 6.0 min = chloride intermediate 3b.
The solution Bédard et al. used to overcome this issue was sonication of the T-mixer where the ammonia source was added. We initially tried this remedy by submerging the second reactor into a bath sonicator heated to 60°C. This seemed to have alleviated our precipitation problem as our conversions substantially improved. Our best fraction from the telescoped trials without sonication was 86% diazepam. With sonication, we were able to consistently obtain 98% pure diazepam across multiple 30 min fractions (Figure 11C), although we still observed some precipitation buildup as before. Sonication also boosted conversion of the chloride impurity 3b that was inert under our previous conditions.
To demonstrate the viability of our conditions for upscaled production, we shifted operations away from microfluidic reactors to a system capable of higher flowrates and larger bore sizes to mitigate the issues stemming from the precipitation (Supplementary Material). Using the setup described in the Methods section and Supplementary Material, we were able to collect multiple fractions that had diazepam purities of ∼91% (>98% after a single recrystallization), while obtaining yields of ∼96%. These findings demonstrate that we are able to produce highly pure samples of diazepam in high yield using reaction conditions optimized in a telescoped microfluidic system.
To conclude, we have been able to build upon previous work in the literature to intensify the reaction rate in continuous flow to obtain ∼91% pure diazepam within the process stream (>98% after a single recrystallization). We have also demonstrated that this process retains a similar purity with increasing scales of operation. The benefit of having such a high purity product is the reduced solvent consumption for purification, reduced labor costs, and improved E-factor. Since diazepam is a member of a large family of 1,4-benzodiazepine derivatives, this work may serve as the foundation to synthesize other benzodiazepines in flow to further strengthen the supply chains of essential medicines.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
RN, MM, and MC explored conditions in batch and flow, interpreted results, and helped write the manuscript. S-HH obtained and processed HPLC/UPLC data. DT secured funding, designed experiments, interpreted experimental findings, and helped write the manuscript.
The authors gratefully acknowledge the financial support of the Purdue University—Department of Chemistry (RN and MC) and DARPA Contract HR0011-20-C-0199 (MM and S-HH).
DHT is a founder and board member of Continuity Pharma. Authors MAM and S-HH are employed by Continuity Pharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors gratefully acknowledge the NMR and MS support of the Purdue Center for Cancer Research grant CCSG CA23168.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fceng.2022.877498/full#supplementary-material
Adamo, A., Beingessner, R. L., Behnam, M., Chen, J., Jamison, T. F., Jensen, K. F., et al. (2016). On-Demand Continuous-Flow Production of Pharmaceuticals in a Compact, Reconfigurable System. Science 352, 61–67. doi:10.1126/science.aaf1337
Agarwal, S. D., and Landon, B. E. (2019). Patterns in Outpatient Benzodiazepine Prescribing in the United States. JAMA Netw. Open 2, e187399. doi:10.1001/jamanetworkopen.2018.7399
Ali, M. S., Hooshmand, N., El-Sayed, M., and Labouta, H. I. (2021). Microfluidics for Development of Lipid Nanoparticles: Paving the Way for Nucleic Acids to the Clinic. ACS Appl. Bio Mater. Ahead of Print. doi:10.1021/acsabm.1c00732
Bachhuber, M. A., Hennessy, S., Cunningham, C. O., and Starrels, J. L. (2016). Increasing Benzodiazepine Prescriptions and Overdose Mortality in the United States, 1996-2013. Am. J. Public Health 106, 686–688. doi:10.2105/AJPH.2016.303061
Balaji, B. S., and Dalal, N. (2018). An Expedient and Rapid Green Chemical Synthesis of N-Chloroacetanilides and Amides Using Acid Chlorides under Metal-free Neutral Conditions. Green Chem. Lett. Rev. 11, 552–558. doi:10.1080/17518253.2018.1545874
Bédard, A.-C., Longstreet, A. R., Britton, J., Wang, Y., Moriguchi, H., Hicklin, R. W., et al. (2017). Minimizing E-Factor in the Continuous-Flow Synthesis of Diazepam and Atropine. Bioorg. Med. Chem. 25, 6233–6241. doi:10.1016/j.bmc.2017.02.002
Burcham, C. L., Florence, A. J., and Johnson, M. D. (2018). Continuous Manufacturing in Pharmaceutical Process Development and Manufacturing. Annu. Rev. Chem. Biomol. Eng. 9, 253–281. doi:10.1146/annurev-chembioeng-060817-084355
Chen, Y., Glotz, G., Cantillo, D., and Kappe, C. O. (2020). Organophotocatalytic N‐Demethylation of Oxycodone Using Molecular Oxygen. Chem. Eur. J. 26, 2973–2979. doi:10.1002/chem.201905505
Cole, K. P., Groh, J. M., Johnson, M. D., Burcham, C. L., Campbell, B. M., Diseroad, W. D., et al. (2017). Kilogram-Scale Prexasertib Monolactate Monohydrate Synthesis under Continuous-Flow CGMP Conditions. Science 356, 1144–1150. doi:10.1126/science.aan0745
Collins, N., Stout, D., Lim, J.-P., Malerich, J. P., White, J. D., Madrid, P. B., et al. (2020). Fully Automated Chemical Synthesis: Toward the Universal Synthesizer. Org. Process Res. Dev. 24, 2064–2077. doi:10.1021/acs.oprd.0c00143
Diab, S., and Gerogiorgis, D. I. (2020). Design Space Identification and Visualization for Continuous Pharmaceutical Manufacturing. Pharmaceutics 12, 235. doi:10.3390/pharmaceutics12030235
Domokos, A., Nagy, B., Szilágyi, B., Marosi, G., and Nagy, Z. K. (2021). Integrated Continuous Pharmaceutical Technologies-A Review. Org. Process Res. Dev. 25, 721–739. doi:10.1021/acs.oprd.0c00504
Ewan, H. S., Iyer, K., Hyun, S.-H., Wleklinski, M., Cooks, R. G., and Thompson, D. H. (2017). Multistep Flow Synthesis of Diazepam Guided by Droplet-Accelerated Reaction Screening with Mechanistic Insights from Rapid Mass Spectrometry Analysis. Org. Process Res. Dev. 21, 1566–1570. doi:10.1021/acs.oprd.7b00218
Feng, Y., Xie, R., Wu, Z., and Marsh, K. N. (1999). Vapor–Liquid Equilibria for Ammonia + Methanol. J. Chem. Eng. Data 44 (3), 401–404. doi:10.1021/je980244n
Fuentes, A., Pineda, M., and Venkata, K. (2018). Comprehension of Top 200 Prescribed Drugs in the US as a Resource for Pharmacy Teaching, Training and Practice. Pharmacy 6, 43. doi:10.3390/pharmacy6020043
Hopkin, M. D., Baxendale, I. R., and Ley, S. V. (2010). A Flow-Based Synthesis of Imatinib: The API of Gleevec. Chem. Commun. 46, 2450–2452. doi:10.1039/C001550D
Jaman, Z., Sobreira, T. J. P., Mufti, A., Ferreira, C. R., Cooks, R. G., and Thompson, D. H. (2019). Rapid On-Demand Synthesis of Lomustine under Continuous Flow Conditions. Org. Process Res. Dev. 23, 334–341. doi:10.1021/acs.oprd.8b00387
Phillips, R. E., and Soulen, R. L. (1995). Propylene Oxide Addition to Hydrochloric Acid: A Textbook Error. J. Chem. Educ. 72, 624. doi:10.1021/ed072p624
Porta, R., Benaglia, M., and Puglisi, A. (2016). Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products. Org. Process Res. Dev. 20, 2–25. doi:10.1021/acs.oprd.5b00325
Rogers, L., Briggs, N., Achermann, R., Adamo, A., Azad, M., Brancazio, D., et al. (2020). Continuous Production of Five Active Pharmaceutical Ingredients in Flexible Plug-and-Play Modules: A Demonstration Campaign. Org. Process Res. Dev. 24, 2183–2196. doi:10.1021/acs.oprd.0c00208
Shtull-Leber, E., Silbergleit, R., and Meurer, W. J. (2017). Pre-Hospital Midazolam for Benzodiazepine-Treated Seizures before and after the Rapid Anticonvulsant Medication Prior to Arrival Trial: A National Observational Cohort Study. PLOS ONE 12, e0173539. doi:10.1371/journal.pone.0173539
Shukar, S., Zahoor, F., Hayat, K., Saeed, A., Gillani, A. H., Omer, S., et al. (2021). Drug Shortage: Causes, Impact, and Mitigation Strategies. Front. Pharmacol. 12, 693426. doi:10.3389/fphar.2021.693426
Srai, J. S., Badman, C., Krumme, M., Futran, M., and Johnston, C. (2015). Future Supply Chains Enabled by Continuous Processing-Opportunities Challenges May 20-21 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 104, 840–849. doi:10.1002/jps.24343
World Health Organization (2021). Model List of Essential Medicines 2021. Geneva, Switzerland: World Health Organization.
Keywords: diazepam, benzodiazepine, continuous syntheses, reaction scale-up, common ion effect, process optimization
Citation: Nicholas RJ, McGuire MA, Hyun S-H, Cullison MN and Thompson DH (2022) Development of an Efficient, High Purity Continuous Flow Synthesis of Diazepam. Front. Chem. Eng. 4:877498. doi: 10.3389/fceng.2022.877498
Received: 16 February 2022; Accepted: 26 May 2022;
Published: 27 June 2022.
Edited by:
Brahim Benyahia, Loughborough University, United KingdomReviewed by:
Dimitrios Gerogiorgis, University of Edinburgh, United KingdomCopyright © 2022 Nicholas, McGuire, Hyun, Cullison and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David H. Thompson, ZGF2ZXRob21AcHVyZHVlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.